

Abstract
The NIMH “Fast-Fail” approach seeks to improve too-often misleading early phase drug development methods by incorporating biomarker-based “Proof-of-Mechanism” (POM) testing in Phase-IIa. This first comprehensive application of “Fast-Fail” evaluated the potential of kappa opioid receptor (KOR) antagonism for treating anhedonia with a POM study determining if robust target engagement favorably impacts brain circuitry hypothesized to mediate clinical effects. This 8-week, double-blind, placebo-controlled, randomized (selective KOR antagonist [JNJ-67953964 10 mg] [N=45] and placebo [N=44]), parallel-group trial including patients with anhedonia and a mood/anxiety disorder was carried out in 6 academic centers. JNJ-67953964 significantly increased fMRI ventral striatum activation during reward anticipation (primary outcome) compared with placebo (Baseline-Adjusted Means: JNJ-67953964 0.72[S.D.=0.67]; Placebo 0.33[S.D.=0.68], F(1,86)=5.58; p<0.01; Effect-Size=0.58[95% Confidence-Interval 0.13–0.99]). JNJ-67953964, generally well-tolerated, was not associated with any serious adverse events. This study supports proceeding with assessment of the clinical impact of target engagement and serves as a model for implementing “Fast-Fail”.
INTRODUCTION:
Clinical trials document the limited effectiveness of currently available medications for mood and anxiety spectrum disorders and speak to the need for new treatments.1–4 Advances in research provide unprecedented opportunities to meet this pressing need. Yet, the growing costs and failure-rate associated with CNS drug development have led many pharmaceutical companies to cease investing in CNS drugs, especially psychiatric medications.5–7
In response to this alarming trend, the NIMH developed the New Experimental Medicine Studies: Fast-Fail Trials Program (https://www.nimh.nih.gov/research-priorities/research-initiatives/fast-fast-fail-trials.shtml) to target one factor identified as a key contributor to drug development failures: early phase drug development methods that are notoriously unreliable and mislead companies into pursuing extremely costly unsuccessful Phase-III studies.5–7 The central contribution of “Fast-Fail” is to include in all drug development efforts a rigorous determination of whether engaging the target of interest has the intended neurobiological effects. The assumption is that, by testing a very specific “proof-of-mechanism” (POM) hypothesis, the development process has less vulnerability to bias and non-specific effects.7,8 Establishing POM decreases the likelihood that positive effects found in subsequent trials with clinical outcomes are due largely to factors other than the direct neural effects mediated by target engagement.8 It also increases the confidence that negative results in Phase III trials are interpretable as indicating that achieving the hypothesized effect on brain function does not lead to the expected clinical effect. As a result, establishing POM is set as a precondition for proceeding to carry out such trials which would otherwise be prohibitively risky and potentially uninterpretable.
Carrying out such POM studies requires a drug that is specific for the target, safe, and produces high target engagement, ideally established for a specific dose range with ligand-based positron emission tomography (PET).8 Most critically, POM studies require the use of biomarkers, which are assumed to be closer to pathophysiology and drug therapeutic mechanisms than clinical endpoints.5,9,10 This allows for smaller Phase II studies that more reliably predict Phase-III outcomes.5,9,10 Additionally, under the “Fast-Fail” approach, use of the NIMH Research Domain Criteria Project (RDoC) framework is encouraged. Unlike the Diagnostic and Statistical Manual (DSM) diagnostic system, RDoC is neuroscience-based, thereby increasing the likelihood that the neural circuitry relevant to the condition of interest has been established and biomarkers exist allowing confirmation that target engagement has the anticipated neural impact.8,11
The first comprehensive application of this approach is presented here. Under the auspices of the NIMH New Experimental Medicine Studies: Fast-Fail Trials in Mood and Anxiety Spectrum Disorders (FAST-MAS) Program, we assessed the potential of kappa opioid receptor (KOR) antagonism to have brain effects consistent with therapeutic benefit for anhedonia, a symptom of major depressive disorder (MDD) that cuts across traditional DSM as well as medical diagnoses.12 “Anhedonia” can be defined in various ways and is not considered a diagnosis. Anhedonia, as broadly construed, is represented in several reward-related RDoC Constructs (“Reward Responsiveness”, “Reward Learning”, and “Reward Valuation”).11,13 Preclinical data strongly suggest that KOR antagonism will affect reward-related brain circuitry (particularly, ventral striatum) in a manner that could improve reward-associated function and reverse anhedonic symptoms and behaviors.13–21 We selected and evaluated JNJ-67953964 (previously CERC-501 and LY2456302), a high-affinity, selective KOR antagonist with favorable pharmacologic and safety profiles based on (1) completed pre-clinical toxicology and human single ascending dose and multiple ascending dose studies22,23 and (2) PET evidence that it robustly engages KOR at tolerable doses24.
We based our decision to use reward-related activation in the ventral striatum as our primary target on the compelling preclinical literature indicating that KOR antagonism would release inhibition on dopamine (DA) neurons, increase nucleus accumbens function, and prevent the development of anhedonic-like states.13,25 Evidence suggests that KOR stimulation inhibits dopamine release in the striatum (nucleus accumbens) and induces a negative mood state.15 Further, KOR agonists decrease phasic dopamine release in the nucleus accumbens and increase intracranial self-stimulation (an anhedonia model), whereas KOR antagonists have the opposite effect.14,17–19 In light of evidence of positive correlations between reward-related dopamine release (as assessed by PET) and blood oxygen level dependent (BOLD) activation (as assessed by functional magnetic resonance imaging, fMRI),26–27 reward-related BOLD activation in the ventral striatum was selected as the primary outcome variable (Figure 1).
Figure 1.
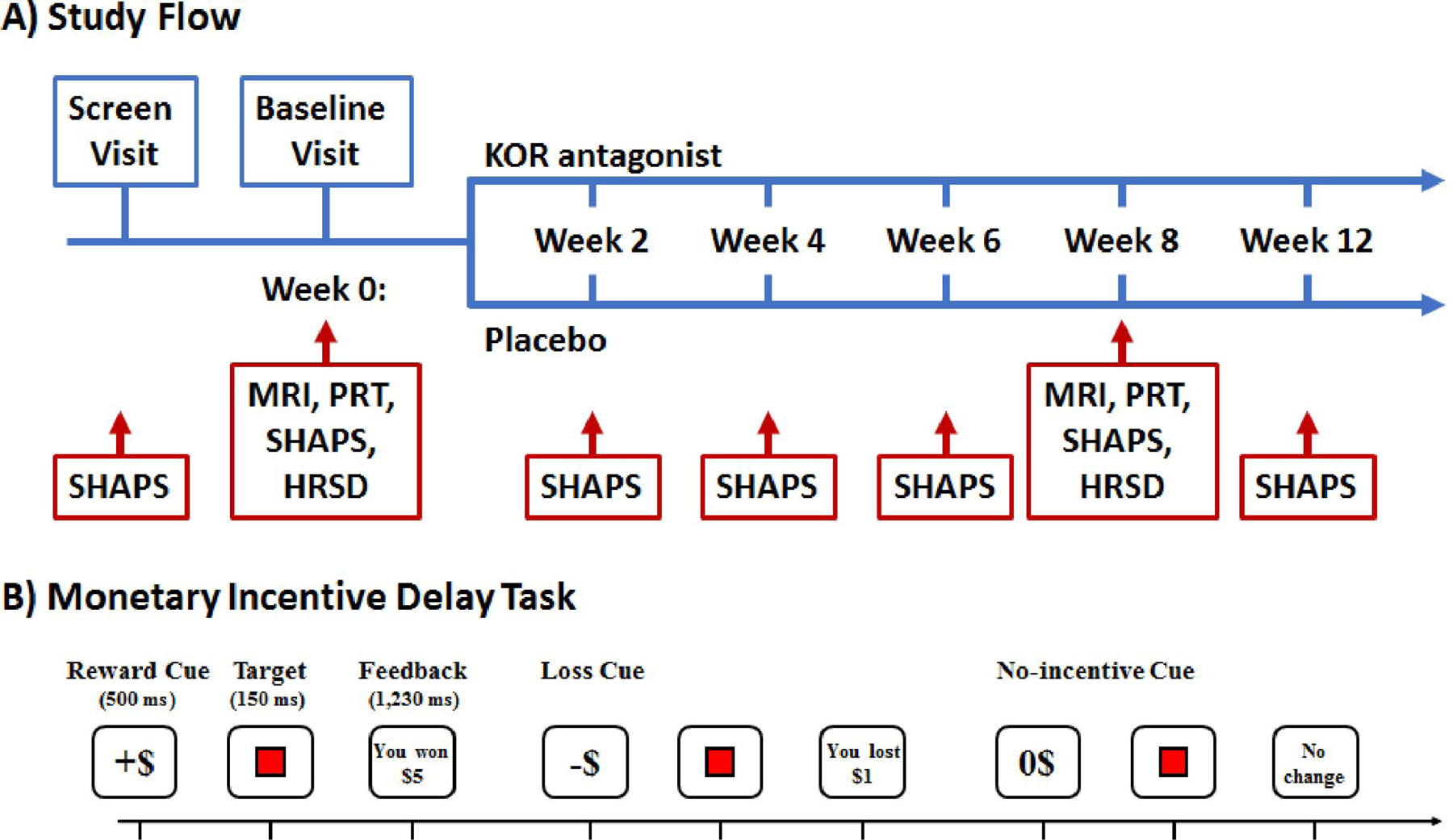
(A) Summary of Study Flow. Within 30 days following screening patients returned for a baseline visit, which included administration of the Snaith-Hamilton Pleasure Scale (SHAPS)28–31, which assesses anhedonic symptoms, Hamilton Depression and Anxiety Rating Scales32–33, MRI, EEG, and the Probabilistic Reward Task (PRT)34–40. Patients were then randomized to JNJ-67953964 (10 mg) or placebo (1:1 ratio) for eight weeks. After 8 weeks of double-blind treatment, patients underwent MRI, EEG, PRT, SHAPS and anxiety and depression scales and treatment was discontinued. At the final visit (week 12), patients were assessed for possible adverse effects. The primary outcome measure was ventral striatal activation during reward anticipation assessed with fMRI during the Monetary Incentive Delay (MID) Task.41–51 Secondary measures were the SHAPS and response bias in the PRT. The PRT is a computerized task that objectively measures participants’ ability to modulate behavior as a function of reinforcement history.34
(B) Trial Structure of the Monetary Incentive Delay (MID) Task. During fMRI, participants performed four runs of the MID (24 trials/run). For each trial (6 s), participants were presented with one of 3 possible cue shapes for 500 ms which signaled whether the upcoming trial had the potential for monetary gain (n=40; denoted by +$), potential for monetary losses (n=40; denoted by −$), or were no-incentive trials (n=40; denoted by 0$). Trial types were pseudo-randomly ordered within each run. After 2250–3750 ms, a red square target was presented for 150 ms. Participants were instructed that: for reward trials, they could win money if they responded quickly to the target; for penalty trials, they could avoid losing money if they responded quickly to the target; and for no-incentive trials there would be no monetary change, but they should still respond quickly to the target. Subjects received feedback 2400–3900 ms following target presentation. To standardize task difficulty, the 66th percentile of reaction times collected during a practice session was used to determine wins/penalties.
RESULTS:
Participants
163 subjects were screened and 94 met eligibility criteria (Extended Data Figure 1). Of these, three withdrew consent prior to baseline and two were unable to complete baseline procedures. Thus, 89 subjects were randomized, 45 to JNJ-67953964 and 44 to placebo. These 89 subjects constituted the “Intention-to-Treat” (ITT) population. There were no significant differences (p<0.05) between groups on any demographic or baseline variables in the ITT population (Table 1). For baseline characteristics of the ITT, “As-Treated”, “Per-Protocol”, and Completer populations see Supplementary Tables 1 and 2.
Table 1.
Study Subject Demographic and Baseline Data for ITT Population
| Variable | JNJ-67953964 N=45 | Placebo N=44 | Total N=89 |
|---|---|---|---|
| Mean Age in Years (SD) | 40.7 (13.3) | 38.2 (13.0) | 39.5 (13.2) |
| Gender - %Female | 64.4 | 61.4 | 62.9 |
| Race | |||
| %Caucasian | 70.5 | 65.1 | 67.8 |
| %African American | 22.7 | 18.6 | 20.7 |
| %Asian | 2.3 | 4.7 | 3.4 |
| %American Indian/Alaskan Native | 0.0 | 2.3 | 1.1 |
| %More Than One Race | 4.5 | 9.3 | 6.9 |
| Ethnicity - %Hispanic Origin | 11.6 | 11.6 | 11.6 |
| Mean BMI (SD) | 29.4 (6.4) | 28.0 (5.9) | 28.7 (6.2) |
| Mean Weight (lbs) (SD) | 180.9 (43.7) | 180.3 (40.6) | 180.6 (41.9) |
| Mean Baseline fMRI Ventral Striatal Activation in MID Task in Anticipation of Gain_Contrasted with No-incentive Trials (SD)** | 0.63 (0.9) (N=44) | 0.64 (0.8) (N=44) | 0.63 (0.8) (N=88) |
| Mean Maximum Baseline fMRI Ventral Striatal Activation in MID Task (SD) in Anticipation of Gain Contrasted with No-Incentive Trials (SD) | 2.66 (1.2) (N=44) | 2.73 (1.2) (N=44) | 2.70 (1.2) (N=88) |
| Mean Baseline fMRI Ventral Striatal Activation in MID Task in Anticipation of Loss Contrasted with No-incentive Trials (SD) | 0.29 (0.8) (N=44) | 0.36 (0.7) (N=44) | 0.33 (0.7) (N=88) |
| Mean Maximum Baseline fMRI Ventral Striatal Activation in MID Task (SD) in Anticipation of Loss Contrasted with No-incentive Trials (SD) | 2.15 (1.2) (N=44) | 2.23 (0.9) (N=44) | 2.19 (1.0) (N=88) |
| Mean Baseline PRT Change in Response Bias from Block 1 to Block 2 (SD)* | 0.02 (0.2) (N=42) | 0.05 (0.2) (N=44) | 0.04 (0.2) (N=86) |
| Mean Baseline SHAPS (SD)* | 36.4 (8.5) (N=44) | 33.4 (5.9) (N=44) | 34.9 (7.4) (N=88) |
| Mean Baseline PRT Response Bias (averaged across blocks) (SD) | 0.108 (0.027) (N=24) | 0.113 (0.025) (N=31) | 0.111(0.026) (N=55) |
| Mean Baseline EEfRT (SD) | 0.35 (0.2) (N=42) | 0.38 (0.2) (N=41) | 0.36 (0.2) (N=83) |
| Mean Baseline TEPS Anticipatory Subscore (SD) | 29.3 (5.7) (N=44) | 29.5 (5.6) (N=44) | 29.4 (5.7) (N=88) |
| Means Baseline TEPS Consummatory Subscore (SD) | 26.3 (4.4) (N=44) | 26.1 (4.4) (N=44) | 26.2 (4.4) (N=88) |
| Mean Baseline VAS Anhedonia (SD) | 2.93 (2.1) (N=44) | 3.59 (2.2) (N=44) | 3.26 (2.2) (N=88) |
| Mean Baseline Resting State EEG Delta Current Density in Rostral Anterior Cingulate (SD) | 73.0 (97.1) (N=43) | 75.2 (51.6) (N=38) | 74.0 (78.6) (N=81) |
| Mean Baseline HAM-D (SD) | 16.3 (5.2) | 14.8 (5.9) | 15.6 (5.6) |
| Mean Baseline HAM-A (SD) | 16.0 (5.8) | 15.1 (6.6) | 15.5 (6.2) |
| Mean Baseline CGI-S (SD) | 3.9 (0.6) | 4.0 (0.5) | 3.9 (0.5) |
| Mean Baseline CPFQ (SD) | 27.2 (6.4) (N=44) | 25.4 (5.7) (N=44) | 26.3 (6.1) (N=44) |
Note: No variables different between groups at p<0.05 level;
Number of subjects is 45 in the JNJ-67953964 and 44 in the Placebo group unless otherwise stated in the table;
When the N’s are less than 45/44 it was due to missing baseline data for that variable;
A priori specified primary outcome measure;
A priori specified secondary outcome measure.
“As-Treated” and “Per-Protocol” Population.
Two JNJ-67953964 subjects were randomized but did not receive treatment and one placebo subject received the wrong treatment, resulting in 43 subjects receiving the assigned treatment in both groups. These 86 individuals constituted the “As-Treated” population and the “Per-Protocol” population as there were no major protocol deviations.
Dropouts and the Completer Population.
The “Completer” population consisted of 33 JNJ-67953964 subjects and 35 placebo subjects. Ten JNJ-67953964 subjects dropped out. Two exited the study after double-blind treatment but before post-treatment follow-up. Eight placebo subjects dropped out during double-blind treatment and one exited between the end of double-blind treatment and post-treatment follow-up (for reasons for dropout, see Supplement).
POM/Efficacy
Planned Statistical Analyses.
We carried out efficacy analyses in the ITT population and performed one-sided statistical tests using a p value threshold of 5% for statistical significance. Analyses consisted of mixed effects models including mean-centered baseline values, age, study site, and sex as covariates. We computed JNJ-67953964 vs placebo group effect-sizes using Hedges’ g.52
Primary Outcome.
Relative to placebo, JNJ-67953964 led to statistically significantly greater mean fMRI ventral striatal activation during anticipation of gain vs. no incentive trials in the MID task (F(1,86)=5.58; p<0.01; Hedges’ g=0.58, 95% Confidence Interval for effect-size=[0.13, 0.99]; Table 2 and Figure 2). Findings providing confirmatory support for this significant effect are that the between-group effect was statistically significant: 1) with imputed results based on 20 imputations for missing data (p<0.02); 2) in the “Per-Protocol” and “As-Treated” populations (p<0.04); 3) in the “Completer” population (p<0.05); and 4) in analysis of the maximum fMRI ventral striatal activation during the MID in anticipation of gain in the ITT population with and without imputation for missing data (p<0.02), in the “Per-Protocol” and “As-Treated” populations (p<0.02), and in the “Completer” population (p<0.025). We did not find a statistically significant site effect for the primary outcome measure.
Table 2.
Efficacy Results Based on Mixed Effects Models in ITT Population Baseline Corrected Means (SD) at End of Double-Blind Treatment Period
| Variable | JNJ-67953964 | Placebo | N | p | Effect Size (Hedges’ g) |
|---|---|---|---|---|---|
| Primary Outcome Measure | |||||
| Mean fMRI Ventral Striatal Activation in MID Task in Anticipation of Gain Contrasted with No-incentive Trials | 0.72 (0.67) | 0.33(0.68) | 88 | 0.0095 | 0.58 |
| Secondary Outcome Measures | |||||
| Mean SHAPS | 30.8 (3.7) | 32.4 (3.6) | 88 | 0.0345 | 0.44 |
| PRT Change in Response Bias from Block 1 to Block 2 | 0.059 (0.15) | 0.066 (0.15) | 67 | >0.10 | 0.10 |
| Exploratory Outcome Measures | |||||
| Maximum fMRI Ventral Striatal Activation in MID Task in Anticipation of Gain Contrasted with No-incentive Trials | 2.84 (0.86) | 2.36(0.86) | 88 | 0.012 | 0.55 |
| Mean fMRI Ventral Striatal Activation in MID Task in Anticipation of Loss Contrasted with No-incentive Trials | 0.73 (0.6) | 0.07 (0.6) | 88 | <0.001 | 1.12 |
| Maximum fMRI Ventral Striatal Activation in MID Task in Anticipation of Loss Contrasted with No-incentive Trials | 2.73 (0.8) | 2.18 (0.8) | 88 | 0.0035 | 0.66 |
| Mean PRT Response Bias | 0.153 (0.013) | 0.070 (0.018) | 67 | 0.030 | 0.49 |
| TEPS Anticipatory Subscale | 32.8 (5.5) | 32.6 (5.4) | 88 | >0.10 | 0.03 |
| TEPS Consummatory Subscale | 29.3 (4.2) | 27.1 (4.2) | 88 | 0.017 | 0.51 |
| EEfRT | 0.41 (0.11) | 0.41 (0.11) | 83 | >0.10 | 0.00 |
| VAS Anhedonia | 4.2 (1.5) | 4.6 (1.5) | 88 | >0.10 | 0.30 |
| Resting State EEG Delta Current Density in Rostral Anterior Cingulate | 62.0 (77.9) | 81.2 (81.1) | 81 | >0.10 | 0.24 |
| HAM-D | 10.8 (4.0) | 11.1 (3.9) | 89 | >0.10 | 0.09 |
| HAM-A | 11.0 (4.2) | 10.6 (4.4) | 89 | >0.10 | 0.09 |
| CGI-I | 3.27 (0.7) | 3.20 (0.6) | 89 | >0.10 | 0.08 |
| CGI-S | 3.28 (0.5) | 3.32 (0.5) | 89 | >0.10 | 0.08 |
| CPFQ | 21.1 (4.2) | 21.1 (4.2) | 88 | >0.10 | 0.00 |
Means are baseline corrected derived from mixed effects models; SHAPS = Snaith Hamilton Pleasure Scale; TEPS=Temporal Experience of Pleasure Scale; EEfRT=Effort Expenditure for Rewards Task; VAS=Visual Analogue Scale; HAM-D=Hamilton Depression Rating Scale; HAM-A=Hamilton Anxiety Rating Scale; CGI-I=Clinical Global Impression of Improvement; CGI-S=Clinical Global Impression of Severity; CPFQ=Cognitive and Physical Functioning Questionnaire
Figure 2:
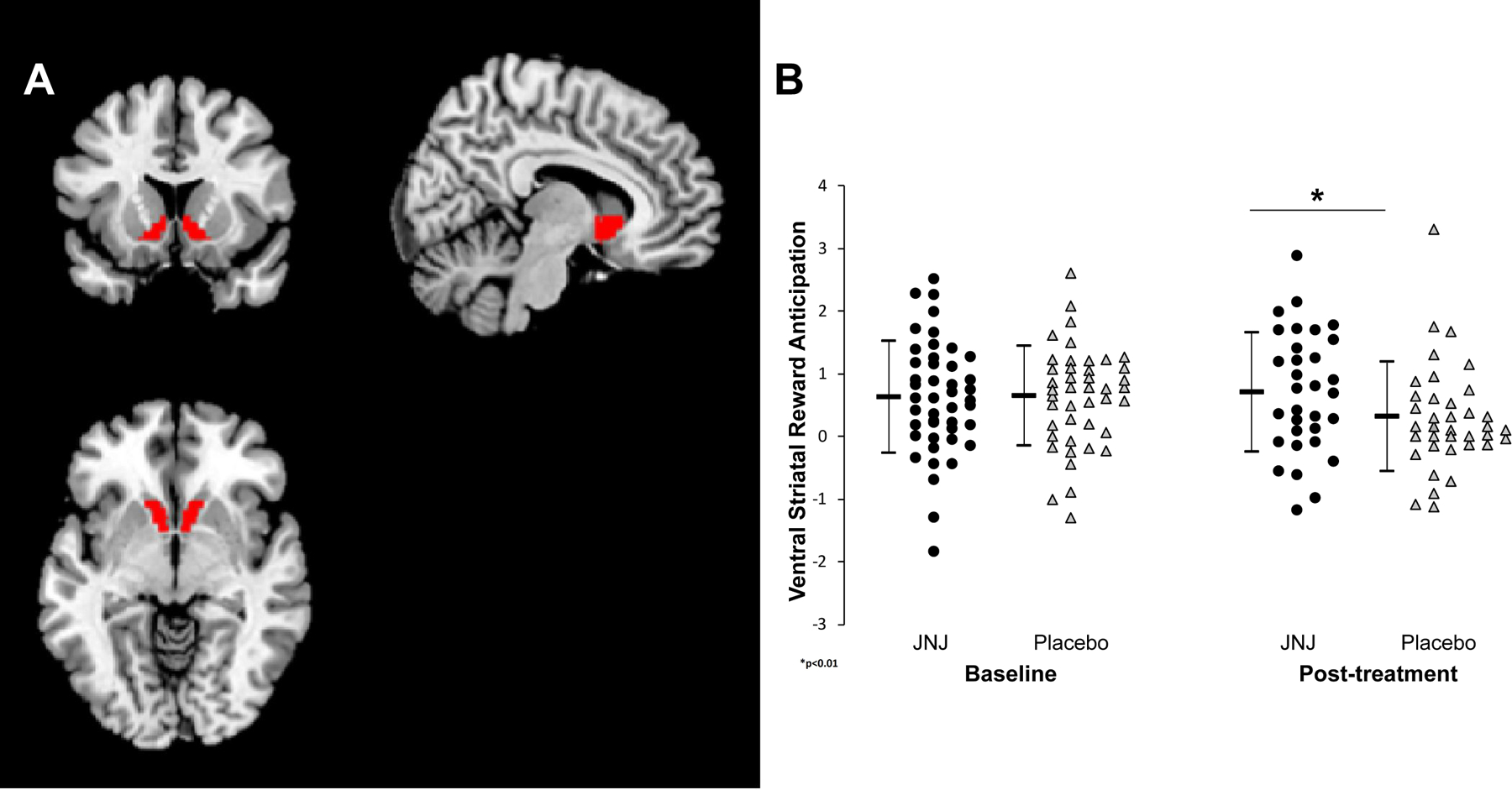
a. Location of ventral striatal ROI based on the Harvard-Oxford Subcortical Atlas. b. Mean signal intensity during reward anticipation within the ventral striatal ROI pre- and post-treatment with JNJ-67953964 and placebo. Error bars represent 95% confidence intervals surrounding the mean signal intensity.
Secondary Outcomes.
Pre-specified analyses included group comparison on mean SHAPS score and change in PRT response bias from Block1 to Block2 carried out in the ITT population.
SHAPS.
A significant group effect emerged reflecting significantly lower baseline-adjusted mean SHAPS score after treatment with JNJ-67953964 than Placebo (F(1,86)=3.35; p=0.0345; Hedges’ g=0.44, 95% Confidence Interval=[0.012, 0.86]; Table 2 and Figure 3a)). Confirmatory support for this significant effect includes that the between-group effect was statistically significant: 1) in the “Per-Protocol” and “As-Treated” populations (p<0.025); and 2) in the “Completer” population (p<0.05). We found a significant site effect for the SHAPS (p<0.03) (Supplementary Table 3).
Figure 3:
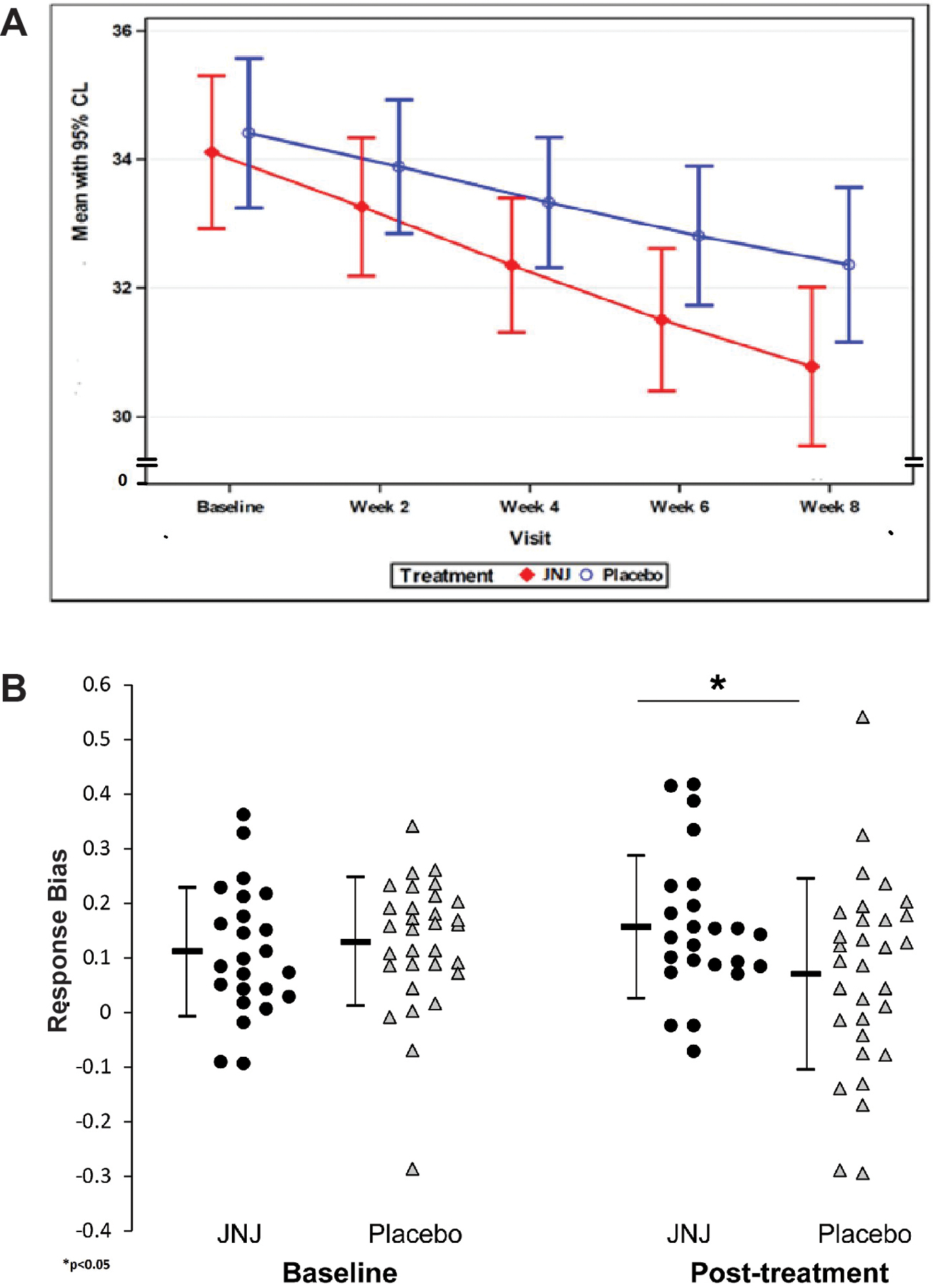
a. Effects of study drug vs placebo on mean SHAPS score (intent-to-treat population). b. Effects of study drug vs placebo on mean PRT response bias (intent-to-treat population). Error bars represent 95% confidence intervals surrounding the means.
PRT.
67 subjects passed a priori defined quality control check (performed blindly to drug randomization; see Supplement), and were included in analyses. Groups did not differ in the rich-to-lean reward ratio (F(1,53)=0.42; p=0.53) or discriminability (F(1,53)=5.72; p=0.20), indicating that they were exposed to a similar reinforcement schedule and task difficulty. Contrary to our hypothesis, the Block(1,2) x Treatment Arm(JNJ-67953964, PLA) x Time(Baseline, Treatment Week 8) analysis of response bias was not significant (F(1,53)=0.5, p=0.48). However, the Treatment Arm x Time interaction was significant (F(1,53)=3.44, p=0.030; Hedges’ g=0.49, 95% Confidence Interval=[0.45, 0.54]) driven by higher post-treatment response bias with JNJ-67953964 relative to placebo (Table 2 and Figure 3b).
Exploratory Outcomes.
Statistically significant group effects emerged for the mean and maximum fMRI ventral striatal activation during anticipation of loss contrasted with no-incentive cue trials. Relative to placebo, JNJ-67953964 was associated with greater mean (F(1,86)=11.7; p<0.001; Hedges’ g=1.12, 95% Confidence Interval=[0.96, 1.21]; Table 2) and maximum (p<0.004; Hedges’ g=0.66, 95% Confidence Interval= [0.51, 0.85]) ventral striatal activation. These effects were also statistically significant in the “Per-Protocol”/”As-Treated” (mean p<0.001; maximum p<0.008), and “Completer” (mean p<0.001; maximum p<0.012) populations.
Statistically significantly greater enhancement of the Consummatory Subscale of the Temporal Experience of Pleasure Scale (TEPS) was also found with JNJ-67953964 vs Placebo (F(1,86)=4.3; p<0.02; Hedges’ g=0.51, 95% Confidence Interval=[−0.37, 1.41]; Table 2) but only in the ITT population.
No significant between group differences were found for any of the other exploratory variables studied (Table 2).
Additional Post-Hoc Exploratory Analyses
Exploratory analysis indicated that the degree of change in ventral striatal activation with treatment was significantly inversely correlated with baseline ventral striatal activation (r=−0.49; p<0.0001). This relationship was somewhat larger in JNJ-67953964 (r=−0.60; p<0.0002) than placebo (r=−0.50; p<0.001) subjects. Logistic regression analysis indicated that baseline ventral striatal activation significantly predicted, at the individual level, which subjects would be responders (defined based on a median-split of the change in mean ventral striatal activation) (Wald Chi-Square 12.4 p<0.0004; 76% accuracy in predicting responder status).
See Supplementary Table 3 for results of exploratory analyses of site-by-time and site-by-treatment arm-by-time interactions for all variables.
Additionally, we assessed the relatedness of our primary outcome with the key secondary outcomes. We found that the change in our primary outcome with treatment was significantly correlated with the change in SHAPS score (r=0.2; p<0.05) and associated with the change in the Block1-to-Block2 difference in PRT response bias at a trend level (r=0.23; p<0.071).
Finally, we computed the treatment effect-size for our primary outcome separately for the JNJ-67953964 and placebo groups. This measure increased from baseline with JNJ-67953964 with an effect-size of 0.30 (Hedges’ g) and decreased from baseline with placebo with an effect-size of 0.52.
Safety
JNJ-67953964 was not associated with any serious adverse events and was generally well tolerated. No subjects discontinued participation due to medication-related side-effects other than worsening of depression or anxiety. More than mild side-effects occurring at least 5% more frequently with JNJ-67953964 than placebo were pruritis (11.1%), depression exacerbation (6.7%), and rash (6.7%) (Table 3). There were no clinically meaningful changes in physical examination, vital signs, electrocardiogram (ECG), or laboratory tests.
Table 3.
Adverse Effects of More Than Mild Severity
| Side Effect | JNJ-67953964 N=45 | Placebo N=44 | ||
|---|---|---|---|---|
| 1N | 2% | 1N | 2% | |
| Headache | 5 | 11.1% | 4 | 9.1% |
| Pruritus | 5 | 11.1% | 1 | 2.3% |
| Anxiety | 3 | 6.7% | 2 | 4.5% |
| Insomnia | 3 | 6.7% | 2 | 4.5% |
| Suicidal ideation | 3 | 6.7% | 2 | 4.5% |
| Diarrhea | 1 | 2.2% | 3 | 6.8% |
| Constipation | 2 | 4.4% | 1 | 2.3% |
| Depression | 3 | 6.7% | 0 | 0.0% |
| Dizziness | 2 | 4.4% | 1 | 2.3% |
| Fatigue | 1 | 2.2% | 2 | 4.5% |
| Rash | 3 | 6.7% | 0 | 0.0% |
| Restlessness | 1 | 2.2% | 2 | 4.5% |
| Vision blurred | 2 | 4.4% | 1 | 2.3% |
| Arthralgia | 1 | 2.2% | 1 | 2.3% |
| Disturbance in attention | 1 | 2.2% | 1 | 2.3% |
| Dry mouth | 1 | 2.2% | 1 | 2.3% |
| Nausea | 0 | 0.0% | 2 | 4.5% |
| Panic attack | 1 | 2.2% | 1 | 2.3% |
| Pollakiuria | 2 | 4.4% | 0 | 0.0% |
| Pyrexia | 0 | 0.0% | 2 | 4.5% |
| Back pain | 0 | 0.0% | 1 | 2.3% |
| Blepharitis | 1 | 2.2% | 0 | 0.0% |
| Chest pain | 1 | 2.2% | 0 | 0.0% |
| Coordination abnormal | 0 | 0.0% | 1 | 2.3% |
| Costochondritis | 1 | 2.2% | 0 | 0.0% |
| Cough | 0 | 0.0% | 1 | 2.3% |
| Dry skin | 0 | 0.0% | 1 | 2.3% |
| Dysuria | 1 | 2.2% | 0 | 0.0% |
| Eye pruritus | 0 | 0.0% | 1 | 2.3% |
| Fall | 0 | 0.0% | 1 | 2.3% |
| Hyperhidrosis | 0 | 0.0% | 1 | 2.3% |
| Hypersomnia | 0 | 0.0% | 1 | 2.3% |
| Irritability | 1 | 2.2% | 0 | 0.0% |
| Libido decreased | 0 | 0.0% | 1 | 2.3% |
| Muscle twitching | 1 | 2.2% | 0 | 0.0% |
| Musculoskeletal chest pain | 0 | 0.0% | 1 | 2.3% |
| Nasopharyngitis | 0 | 0.0% | 1 | 2.3% |
| Night sweats | 0 | 0.0% | 1 | 2.3% |
| Palpitations | 0 | 0.0% | 1 | 2.3% |
| Self-injurious ideation | 1 | 2.2% | 0 | 0.0% |
| Tendon rupture | 1 | 2.2% | 0 | 0.0% |
| Toothache | 0 | 0.0% | 1 | 2.3% |
| Upper respiratory tract infection | 0 | 0.0% | 1 | 2.3% |
| Viral infection | 1 | 2.2% | 0 | 0.0% |
N = number of subjects experiencing this event at least once (not total number of events)
% = percent that experienced this event within each treatment group
DISCUSSION
The present results establish that a dose of a medication documented to have robust KOR antagonism based on PET imaging24 has the hypothesized effect on neural function, thereby establishing POM that engaging this target has an effect on a brain function implicated in hedonic responses. This, in turn, suggests that JNJ-67953964 has potential as a specific anhedonia therapy.
The strength of the conclusion is reinforced by the fact that a significant JNJ-67953964 vs placebo effect was evident in all measures assessing ventral striatal activity. In addition to the statistically significant effect observed with the primary outcome measure, we found significant effects vs placebo on maximum ventral striatal activation in anticipation of gains and mean and maximum activation in anticipation of losses.
Notably, while there was an increase in the primary outcome measure in the JNJ-67953964 group with treatment, a substantial amount of the JNJ-67953964 vs placebo difference was driven by a decrease over time in the placebo group. This impression was verified in post-hoc exploratory analysis. However, while the decrease from baseline in the placebo group (Hedges’ g=0.52) was larger than the increase in the JNJ-67953964 group, the effect-size for the increase with treatment in the JNJ-6795364 group was non-trivial in magnitude (Hedges’ g=0.30). Our findings are consistent with prior evidence of diminished ventral striatal response to anticipation of reward with repeated testing. For example, the degree of activation in anticipation of reward in the MID decreased with repeated testing in the same session in 26/29 anhedonic subjects.53 Further, healthy controls experienced a decrease (not statistically significant) in ventral striatal activation in anticipation of reward across two test sessions separated by an average of 48 days.54 In order to mitigate the possible effects of such adaptation, our primary analysis involved a direct comparison between the JNJ-67953964 and placebo groups. When viewed in the context of prior findings,53,54 our results imply that JNJ-67953964 led to an increase in ventral striatal activation with an effect-size of 0.30 in spite of possible habituation effects stemming from repeated administration (as seen in the placebo group).
Whatever the source of the decrease in striatal activation in anticipation of reward in patients administered placebo, based on the a priori planned approach of assessing for statistical significance in a JNJ-67953964 vs placebo comparison, this study robustly establishes POM for KOR antagonism. This provides a rational basis for proceeding with development of this target and carrying out larger trials powered for the use of clinical endpoints to determine the clinical impact of engaging this target with JNJ-67953964.
According to the “Fast-Fail” framework, establishing POM for KOR antagonism achieves two critical objectives for future studies with clinical endpoints:
it increases the likelihood that positive outcomes reflect drug effects on the brain circuitry hypothesized to mediate therapeutic effects, thereby decreasing the likelihood that they reflect non-specific effects or bias to which such studies are relatively vulnerable; and
it ensures that negative results are interpretable, indicating that achieving the hypothesized effect on brain function does not lead to the expected clinical effect. Otherwise, such negative outcomes could reflect insufficient dosage to engage hypothesized circuitry, or that Phase-II findings were false positives reflecting non-specific effects/bias.
It is critical to appreciate the priority placed on our neuroimaging-based primary outcome in determining the promise of KOR antagonism. “Fast-Fail” proposes to focus on biomarker-based outcomes in Phase-II to assess, as directly as possible, the circuitry hypothesized to mediate treatment effects. Biomarkers, being closer to the direct biological effects of the drug than clinical measures, are assumed to be associated with effect-sizes that are sufficiently large to be detected with studies of the size typically carried out in Phase-II. According to our model, effects seen on clinical symptom outcomes are unreliable with regard to informing on whether the drug has the hypothesized brain effect. Having a significant effect on the primary, biomarker-based outcome is sufficient for proceeding to trials with symptom measures. Significant effects on all of the symptom measures (SHAPS, TEPS, and VAS) but not on the primary neuroimaging-based outcome would not support proceeding. At the same time, having supportive significant effects on symptom measures is of value and increases confidence in the likelihood of success of development. However, the presence of such effects is not a precondition for meeting “Go” criteria for drug development because such effects are likely to be too small to be detected in this type of study.
It is also important to note that we cannot cite examples that demonstrate the effectiveness of this “Fast-Fail” framework. This effort is the first to ever attempt implementing the “Fast-Fail” approach, which speaks to the high significance of our study and sets the stage for the first time for carrying out the Phase 3 studies needed to determine the utility of “Fast-Fail”.
The methods of this study also serve as a model for implementing “Fast-Fail” early phase methodology and specifically establishes “Fast’Fail” methods for the development of anhedonia treatments. It should be noted that in carrying out “Fast-Fail” POM studies, failure to establish POM may not indicate that engaging the target does not impact the relevant neural circuitry. It could also reflect insufficient sensitivity of the outcome measure. This is a key “Fast-Fail” challenge given the limited number of POM study-suitable outcome measures with well characterized statistical properties. In this regard, our effort establishes the POM study utility of the MID fMRI measure. Lastly, our findings generally support the feasibility of implementing the “Fast-Fail” approach.
This study brings us closer to our original vision of how “Fast-Fail” methods should be implemented in research studies and ultimately applied in clinical practice. This vision includes that biomarkers would be used to select and effectively phenotype patients. We originally intended to select patients on the basis of ventral striatal activation but it was felt that insufficient work had been completed characterizing the statistical properties of our fMRI measure to allow us to do so. However, the results of our exploratory analysis indicating that baseline mean ventral striatal activation in anticipation of gains was a significant predictor of both the change in this variable with treatment and with responder status suggest the promise of using fMRI mean ventral striatal activation in anticipation of rewards for selection of subjects who are likely to respond to KOR antagonist therapy. As such, we anticipate that this study and additional studies carried out with measures of ventral striatal activation in appropriately selected subject populations will make it possible to select patients using these measures for “Fast-Fail” studies of potential therapies for anhedonia and, ultimately, for optimizing clinical practice.
Evidence that the effects on neural circuitry were accompanied by effects on clinical measures of anhedonia further reinforces the promise of KOR antagonism as an anhedonia therapy. Statistically significantly greater improvement was found with JNJ-67953964 compared with placebo on the SHAPS, (a priori specified secondary outcome) as well as the consummatory subscale of the TEPS (exploratory outcome). At the same time, there was some divergence in outcomes in terms of a lack of significant effects on some exploratory measures of reward-related function (VAS anhedonia, EEfRT, and TEPS anticipatory subscale). Although the divergence in outcomes could indicate limited replicability of the effect on the primary outcome measure with clinical and behavioral measures, another possible explanation is that our study was underpowered to detect the effects on these measures, which were hypothesized to be smaller than that of the neuroimaging measure for which we estimated power and sample size. Another possibility is that our reward-related measures might assess various aspects of reward-related function which are differentially impacted by KOR antagonism. Collectively, these findings suggest that the observed brain effect may mediate clinically meaningful anhedonic effects but that further work is needed to determine the relationship between such brain effects and clinical and behavioral measures of anhedonia.
Although there was a statistically significant effect on the key secondary clinical outcome measure, the SHAPS, the size of this effect is of uncertain clinical significance. We are aware of no accepted, standard means for determining clinical significance or clinical effect-size with the SHAPS. However, the effect-size (Hedges’ g=0.44) would be considered just less than medium-sized when compared against the most commonly used benchmarks (small=0.2; medium=0.5; large=0.8).55
In terms of the behavioral measures, we did not find a significant effect on the pre-planned behavioral secondary analysis (Treatment Arm x Block x Time interaction for PRT response bias).56–57 However, we did find a significant Treatment Arm x Time interaction which also implies that the groups differed in how strongly their behavior was modulated by rewards. Factors related to why we did not find a significant effect for the planned PRT analysis are reviewed in the Supplement.
An important consideration with respect to the PRT analyses is that approximately 25% of the participants failed the a priori defined quality control evaluation. This study was among the first that implemented the PRT in a multi-site randomized controlled trial. Although standardization and training across sites was implemented, it was evidently challenging to maintain high reliability across sites and across years of a complex RCT. This rate of data loss is similar to what emerged in another large, multi-site RCT (EMBARC), but was significantly greater than prior large, multi-site studies using the PRT.58 It is clear that the significant data loss for the PRT decreased the available power for PRT analyses and is a limitation. Accordingly, further work is needed to limit data loss associated with this behavioral measure in the context of multi-site studies.
Nonetheless, it was the case that we found significant effects of JNJ-67953964 across all 3 units of analysis – brain circuitry, behavior, and self-report. Notably, the results of exploratory analysis indicating that the change with treatment in our primary outcome (fMRI measure) was statistically significantly correlated with the change in a self-report measure (the SHAPS) and associated at a trend level with our behavioral key secondary outcome (the PRT), supports that KOR antagonism had a coherent effect on measures of anhedonia across units of analysis and increases confidence in the likelihood of success of development.
A comparison of the relative treatment effect-sizes seen across various measures of anhedonia supports a key principle of the “Fast-Fail” approach: that biomarkers should be associated with greater effect-sizes when used as treatment outcome measures than clinical and behavioral measures.
Specifically, we found that the effect-size associated with the primary neuroimaging measure (Hedges’ g=0.58) was greater than the effect-size associated with a behavioral measure (Hedges’ g=0.49) and the key secondary self-report measure (Hedges’ g=0.44). As a result, this study provides an example supporting the notion that the use of biomarkers in early phase studies could increase power thereby allowing smaller studies which are more likely to be reliable and replicated.
It is notable that the largest drug vs placebo treatment effect-size was seen with ventral striatal activity in anticipation of loss. While this may seem inconsistent with an intervention that improves anhedonia, it is consistent with prior work. Several MID studies have reported robust fMRI ventral striatal activation during anticipation of losses or aversive events and this activation correlated with ventral striatal activation occurring during anticipation of gains.54,59–60 These observations led to the hypothesis that ventral striatal activation reflects the “motivational relevance of an upcoming event” and is not specific to anticipation of reward.59 Taken in this context, the findings suggest the hypothesis that anhedonia may be best thought of as loss of “motivational relevance” rather than impairment specific to reward function.
Another notable finding of this study is that KOR antagonism led to improvement in measures of anhedonia but did not improve broad measures of depression (HAM-D) or anxiety (HAM-A). This suggests that, although anhedonia is a core feature of MDD, it is possible to distinguish effects of anhedonia from antidepressant effects. A limitation to this conclusion, however, is that this study was not designed to nor capable of rigorously determining the effects of KOR antagonism on depression. There was no minimum depression severity required for participation, nor were subjects required to have MDD to participate (20% had an anxiety disorder or PTSD and not MDD). The initial depression and anxiety severity in this study was not high, decreasing the possibility for HAM-D and HAM-A improvement and likely decreasing treatment effect-sizes. Also, the anhedonia factor on the HAM-D only includes several of the items from this scale, diminishing the likelihood that a treatment effect will be found with this measure due to a therapy that specifically improves anhedonia. Nonetheless, the effect-sizes seen for anhedonia measures are so much greater than the depression and anxiety effect-sizes that it seems reasonable to conclude that KOR antagonism likely has a specific therapeutic effect on anhedonia and not a broad effect on depression and anxiety and that it is possible to distinguish anhedonic effects from more general antidepressant/anxiolytic effects. As such, this study opens the door to developing treatments specifically targeting anhedonia.
Lastly, this study had a number of limitations not mentioned above. We did not ask subjects whether they believed they had received study medication or placebo. As a result, we could not assess whether subjects were unblinded. We also had limited ability to examine interaction effects. For example, because of the number of covariates and sites relative to the sample size we did not plan to examine site-by-time and site-by-treatment arm-by-time interactions (post-hoc analysis results appear in Supplementary Table 3). Further, we had a dropout rate of approximately 25%, slightly higher than anticipated (20%), which limited the power available for completer analyses.
Still, this study is notable because it represents the first successful implementation of the NIMH “Fast-Fail” treatment development approach. It is hoped that it will set the stage for future applications of “Fast-Fail”which has the potential to lead to more efficient treatment development thereby facilitating an increase in investment in developing much-needed psychiatric therapies.
METHODS:
Detailed information regarding study methods appears in the accompanying Life Sciences Reporting Summary.
Patient Population
Patients were eligible for enrollment if they were 21 to 65 years of age and had clinically significant anhedonia as defined by a Snaith Hamilton Pleasure Scale (SHAPS)28 score of at least 20 (as assessed using dimensional scoring guidelines)29. The use of this cutoff was based on a receiver-operating curve analysis carried out with the SHAPS score for discriminating a Montgomery-Åsberg depression rating scale (MADRS) anhedonia item score of greater than 4/6 (considered clinically significant) vs 4 or less.28 In addition, subjects had to currently meet the Diagnostic and Statistical Manual version 4 text revision (DSM-IV TR) diagnostic criteria for Major Depressive Disorder (MDD), Bipolar I or II Depressed, Generalized Anxiety Disorder, Social Phobia, Panic Disorder, or Post Traumatic Stress Disorder based on the Mini-International Neuropsychiatric Interview for DSM 4 (MINI)).61–62
We sought to include a cross-diagnostic sample of patients with anhedonia in order to be consistent with the NIMH RDoC dimensional approach to classification of mental disorders.8 Because anhedonia is a core symptom of MDD and one of its diagnostic criteria, we were concerned that without constraining enrollment we might exclusively find and enroll subjects with anhedonia occurring in the setting of MDD. Lacking prior studies attempting to enroll a similar population as a guide, we estimated that it would be feasible to recruit at least 33% of the subjects who had an anxiety disorder without current MDD based on the MINI. Over the course of the study we found recruiting subjects with anxiety disorders without current MDD more challenging than initially anticipated; as a result, with approval of the NIMH Data Safety Monitoring Board and Institutional Review Boards, we decreased the target to 20%.
Subjects underwent medical and psychiatric history, physical examination, laboratory testing, and MINI assessment and, on this basis were excluded if they: were expected to require any hospitalization during the course of the study; had a history of a psychotic disorder, current manic or mixed episode, autism spectrum disorders, mental retardation; met DSM-IV-TR criteria for substance abuse within the last 3 months or substance dependence within the last 6 months; had a history of unstable or untreated serious medical condition; had active suicidal intent or plan, or history of attempt within the past 3 months based on physician evaluation and the Columbia Suicide Severity Rating Scale (C-SSRS);63 used any medication with significant central nervous system effects including antidepressants, antipsychotics, anxiolytics, anticonvulsants, mood stabilizing agents, muscle relaxants, centrally acting anti-histaminergics, stimulants or insomnia medications within 5 half-lives of baseline or at any time during the study; used any medication that is primarily metabolized by Cytochrome P450 2C8 within 14 days of baseline or at any time during the study; had any contraindications to the MRI procedures; had a positive urine drug screen at any time during the study; used any investigational medication within 3 months; had a history of gastric disease (including peptic ulcer disease, gastritis, upper GI bleeding, or any GI precancerous condition), had current clinically evident GI complaints; had a positive urea breath test (exclusionary in the first half of the study, after which approval was obtained from FDA, the NIMH DSMB, and the relevant Institutional Review Boards to drop this requirement); current use of a proton pump inhibitor or histamine 2 blocker, or a history of chronic non-steroidal anti-inflammatory drugs use; history of use of Salvia divinorum or use of Salvia divinorum at any time during the study; any smoking of cigarettes or use of other nicotine containing products within the last month or at any time during the study; or were pregnant or lactating.
Trial Design
The trial was conducted at six centers in the United States (Duke University, Yale University, Icahn School of Medicine at Mount Sinai, Baylor College of Medicine, Indiana University, and Case Western Reserve University) from September 2015 through October 2017. The study consisted of a randomized, double-blind, placebo-controlled, parallel-group, 8-week trial. Subjects were randomized to JNJ-6795396410 mg or placebo in a 1:1 ratio administered as identical-appearing tablets. Qualifying subjects were provided the next container among sequentially numbered containers at each site through which the randomization sequence, established by the coordinating center, was implemented. All site personnel were blinded to the randomization sequence and the contents of the containers. The 10 mg dosage of JNJ-6795396410 was chosen for use in this study because: 1) a PET study demonstrated dose-dependent kappa opioid receptor occupancy and at approximate peak occupancy (2.5 hours post-dose) brain kappa opioid receptors were almost saturated with single doses of 10 mg or more (96.7% receptor occupancy) with a trough occupancy >60%;24 and 2) 10 mg was the highest dosage where completed toxicity studies supported carrying out a trial as long as 8 weeks of daily dosing in humans.
Assessments
POM/Efficacy Assessments:
fMRI during the Monetary Incentive Delay (MID) Task
The primary outcome measure was the magnitude of fMRI determined ventral striatal (including nucleus accumbens) activation during anticipation of rewards in the MID task.42 This measure was chosen as a means of testing Proof of Mechanism for KOR antagonism because: it is a measure of function in brain reward circuitry found to be modulated by kappa opioid antagonism in preclinical work;14,17–19 and it differentiated depressed patients from healthy controls.54
The MID fMRI was obtained at baseline and at the end (week 8) of double-blind therapy and was carried out based on previous designs.42 It was administered in five task runs each consisting of 24 trials. For each trial, subjects were presented with one of three possible cues for 500 msec, followed by a fixation crosshair on a computer screen. These cues signaled whether the upcoming trial had the potential for monetary gain (n=40 denoted by “+$”), potential for monetary losses (n=40; denoted by “−$”), or that no there was no possibility for monetary gain or loss (n=40; denoted by “0$”). Subjects were instructed that on incentive trials, they could either gain or avoid losing money by pressing button when presented with a red square target. On non-incentive trials, subjects were instructed to still press the button as soon as the target appeared. Trial types were pseudo-randomly ordered within each run. Duration of fixation following presentation of the cue was jittered between 2250 and 3750 ms, and the target was displayed for a period of 150 msec. 2400 – 3900 msec after target offset, subjects were notified as to how much money they had gained or lost on that trial.
Prior to testing, subjects engaged in a training and practice run in the scanner. Task difficulty (i.e., maximum allowable reaction time for both gain and loss trials) was titrated based on reaction times collected during the practice session. Separate gain and loss reaction time standards were established to achieve approximately 70% success in each incentivized trial type.
MRI Scan Acquisition:
All scans were conducted on research-dedicated 3.0 Tesla MRI scanners running the latest software version using an advanced 32-channel RF headcoil. However, the manufacturer and model type of the MRI scanners varied across sites including 3 Siemens Trios, 1 Siemens Verio, 1 Siemens Skyra and a GE MR750. MR-compatible video projection system with vision correction lenses, high-quality headphones and a button box were used for fMRI task presentation and response recording. The MRI acquisition sequence consisted of 15 sec of Localizer followed by Gradient-echo echo-planer fMRI scans, axial, TR/TE: 2000/30 ms, flip angle: 70 deg, FOV: 25.6 cm, matrix: 64x64, 32 axial slices, acceleration factor = 2, voxel size: 4x4x4 mm, 137 fMRI time points + 4 dummy scans at the beginning (total 141 points/TRs), total scan time 4 min 42 seconds (141 x 2 s) for each of five fMRI runs.
fMRI data processing was carried out using FEAT (FMRI Expert Analysis Tool) Version 6.00, part of FSL (FMRIB’s Software Library, www.fmrib.ox.ac.uk/fsl). Registration to high resolution structural and/or standard space images was carried out using FLIRT.64–65 Registration from high resolution structural to standard space was then further refined using FNIRT nonlinear registration.66 We employed the MNI152 normalization template. The following pre-statistics processing was applied: motion correction using MCFLIRT;65 slice-timing correction using Fourier-space time-series phase-shifting; non-brain removal using BET;67 spatial smoothing using a Gaussian kernel of FWHM 5mm; grand-mean intensity normalisation of the entire 4D dataset by a single multiplicative factor; high-pass temporal filtering (Gaussian-weighted least-squares straight line fitting, with sigma=45.0s). Time-series statistical analysis was carried out using FILM with local autocorrelation correction.68 Higher-level analysis was carried out using a fixed effects model, by forcing the random effects variance to zero in FLAME (FMRIB’s Local Analysis of Mixed Effects).69–70 The primary contrast of interest was averaged activation during reward anticipation (time points from onset of reward type cue to onset of target cue), for the contrast cued reward > cued non-reward. The primary outcome measure was obtained for an a priori specified bilateral non-thresholded ventral striatal area mask, defined by the Harvard-Oxford Subcortical Atlas and involved an a priori contrast using GLM of averaged z-statistic of all voxels within the region of interest. As exploratory outcomes we also computed the maximum activation during reward anticipation (time points from onset of reward type cue to onset of target cue), for the contrast cued reward > cued non-reward, and mean and maximum activation during loss anticipation (time points from onset of loss type cue to onset of target cue), for the contrast cued loss > cued non-reward, for the a priori specified ventral striatal area mask.
We instituted a set of Quality Control procedures in order to standardize fMRI methods across sites. First, all sites were provided with the same E-Prime files for running the MID fMRI protocol and written materials outlining detailed fMRI methods. We also presented the fMRI methods in detail several times to all sites. In addition, our fMRI leads (Drs. Song and Smoski) provided one-on-one consultation to site fMRI personnel. We also required all sites to obtain and upload agar phantom scans to our central fMRI data analysis site (Dr. Allen Song’s laboratory at Duke) both to qualify to begin to enroll subjects and regularly throughout the study. Specifications for the agar phantom scans and their review and analysis were carried out as in the FIRST-BIRN multi-site fMRI study quality assurance protocol.59 The phantom scan raw data were reviewed and estimates of Signal-to-Noise-Ratio (SNR) and Signal-to-Fluctuation-Noise-Ratio (SFNR) were generated. The signal image was the voxel-by-voxel average across all of the images. The Fluctuation noise image was the standard deviation of the residuals resulting from detrending the time series across all images for each voxel using a second-order polynomial. The SNFR was then generated by computing the voxel-by-voxel ratio of the signal image and the temporal fluctuation image. A summary value of the SNFR was also computed taking the average of the SNFR across a 21x21 voxel region of interest in the center of the SNFR image. The SNR computation started with creating separate sums of the odd and even numbered images and taking the difference between these two sums. The SNR is the ratio of the average of this difference image taken over a 21x21 voxel region of interest in the center of the image to the square root of the variance of this difference within the same 21x21 voxel region divided by 198 time points. To compute the Percent Fluctuation and Drift, first the average across images within a 21x21 voxel region of interest in the center of the image was computed generating a time series of average intensity. Next, the time series was fit with a second-order polynomial. The Percent Fluctuation was computed as 100 times the standard deviation of the residuals of this fit divided by the average intensity. The Percent Drift was generated by dividing the difference between the maximum and minimum fit value by the average signal intensity and multiplying by 100.
The submitted scans were reviewed for artifacts and fluctuations over time and the statistics derived from these scans were assessed for the degree of deviation from the mean values. Artifacts, systematic drift or substantial deviation based on the judgment of the MRI team reviewer led to contact with the site and implementation of a plan for correcting the identified problems. Lastly, we made onsite help available to the sites and this was required for one of the sites to be able to successfully implement the procedures and meet our criteria for standardization.
Snaith Hamilton Pleasure Scale (SHAPS)
The Snaith-Hamilton Pleasure Scale (SHAPS)28 was used to screen subjects, was a secondary outcome measure, and was obtained at every visit. It is a 14-item questionnaire used to assess anhedonia covering four domains of hedonic experience: interest/pastimes, social interaction, sensory experience, and food/drink. It asks participants to agree or disagree with statements of hedonic response in pleasurable situations (e.g., “I would enjoy my favorite television or radio program”) based on their experience in the “last few days”. Four responses are possible: Strongly disagree, Disagree, Agree, or Strongly agree with higher scores indicating greater pleasure capacity. A total score can be derived by summing the responses to each item. Items answered with “strongly agree” are coded as “1”, while a “strongly disagree” response was assigned a score of “4.” Therefore, scores on the SHAPS can range from 14 to 56, with higher scores corresponding to higher levels of anhedonia.29 This scale has shown adequate overall psychometric properties including convergent validity,28,30–31 discriminant validity,29 and test-retest reliability.29 Another important consideration that supports the use of the SHAPS in this study is that it is the only anhedonia measure found to significantly improve with the administration of treatments in clinical trials.72–73
Probabilistic Reward Task (PRT)
The Probabilistic Reward Task (PRT) was a secondary outcome measure obtained at baseline and after 8 weeks of double-blind treatment. The PRT was designed to objectively assess the propensity to modulate behavior as a function of reinforcement history and has been found to reflect reward-related function in multiple independent samples.34–37,56,57 Participants completed two blocks of 100 trials where they determined whether a briefly presented mouth on a cartoon face was ‘long’ or ‘short’, and reported their decision by pressing one of two corresponding keys on a computer keyboard (‘z’ or ‘/’). Importantly, the brief presentation time (100 ms) and the minimal difference in length between the two target stimuli (11.5 vs. 13 mm) make it difficult for participants to distinguish the stimuli. Moreover, an asymmetrical reinforcement ratio is implemented across the two blocks so that one of the two stimuli (the ‘rich’ stimulus) is consistently rewarded (“Correct!! You Won 20 Cents”) three times more frequently than the ‘lean’ stimulus (30 vs. 10 times per block). Reinforcement allocation and key assignments were counterbalanced across participants. Participants were instructed to respond as quickly and accurately as possible to maximize monetary rewards, and that not all correct responses were followed by rewards. Owing to the asymmetrical reinforcement schedule, performance in the PRT can be decomposed into response bias (log b) and discriminability (log d), which were computed as:
To allow calculations in cases with a zero in one cell of the formula, 0.5 was added to every cell of the detection matrix.58
Diminished preference for the “rich” stimulus (a decrease in response bias) has been found in individuals with increased depressive symptoms,57 current MDD,35,56,57 particularly those with elevated anhedonic symptoms56–57 or melancholic depression,38 as well as in youth reporting anhedonia across various DSM diagnoses.39 Before response bias and discriminability scores were computed, quality control checks were performed blind to drug randomization using a priori defined cutoffs used in recent PRT studies (Supplement).37
The change in response bias from Block 1 to Block 2 served as a secondary outcome measure, which was tested by evaluating the 3-way interaction of Treatment Arm (JNJ, PLA) by Block (1, 2) by Time (Baseline, Treatment Week 8). This measure captures the total amount of reward learning across blocks. As an exploratory outcome we also evaluated the Treatment Arm (JNJ, PLA) x Time (Baseline, Treatment Week 8) effect, which is computed as part of the analysis of the Treatment Group by Block by Time interaction. This effect probed changes in overall response bias (i.e., averaged across both blocks) as a function of treatment and might be especially sensitive for designs (such as in the current study) involving only two blocks of the PRT, which might not allow response bias to grow throughout the blocks as strongly as in prior studies that used three blocks.40,56,57
Resting state delta EEG current density in the rostral anterior cingulate.
We obtained resting state, eyes-closed quantitative 32 channel EEG (QEEG) in order to provide an additional, exploratory circuit-based measure of hedonic function. From these data we computed current density using Low Resolution Electromagnetic Magnetic Tomography (LORETA) based on evidence that using this method, greater resting EEG delta (1.5–6 Hz) current density (i.e., lower brain activity) in the rostral anterior cingulate is correlated with higher anhedonia scores among healthy individuals.74 The recording electrodes were located according to the Modified International 10–20 System. Right infra-orbital and left outer canthus electrodes were used to monitor eye movements. Data were obtained with a referential montage employing a linked-ear reference using a digitization rate of 256 Hz. Electrode impedances were maintained at below 5 kOhm during the recordings and standard square-wave and bio-calibrations were performed. Data collection occurred for 20 minutes (10 minutes with eyes closed and 10 minutes with eyes opened) following calibration with filter settings of 0.5 and 70 Hz. Manual epoch-by-epoch artifacting was carried out by the central Duke QEEG Core blinded to subject and study time point. QEEG analysis was only carried out if a minimum of 30 seconds of waking, artifact-free data were available.
Effort Expenditure for Rewards Task (EEfRT)
The Effort-Expenditure for Rewards Task (EEfRT),75 an exploratory behavioral measure for the study was intended to assess the motivation to pursue rewards, one important dimension of reward-related function. It has been reported to be correlated with several anhedonia scales.75 The EEfRT task is a multi-trial game in which participants are given an opportunity on each trial to choose between two different task difficulty levels in order to obtain monetary rewards. For additional information about EEfRT methods see Supplementary Information.
Visual Analogue Scale (VAS) of Anhedonia
The VAS-Anhedonia is a standard VAS assessment of anhedonia severity which is included because it provides a global anhedonia indicator which takes very little time to obtain and which was found to be sensitive to change with treatment in a prior placebo-controlled trial in alcohol dependent subjects.72 The test consists of making a rating on a 100 mm scale in response to the directive: “Make a mark on the line below that indicates how much pleasure you experience from food, sexual behavior, and meeting friends”. At the left end of the scale is the anchor “No Pleasure” and at the right end of the scale is the anchor “Extreme Pleasure.”
Temporal Experience of Pleasure Scale (TEPS)
We also included the Temporal Experience of Pleasure Scale (TEPS) as an exploratory measure because it provides different information about reward-related function than the SHAPS and has been found to be correlated with activation in the key circuits of interest (nucleus accumbens and putamen) in Monetary Incentive Delay Task-related fMRI.76 The TEPS is an 18-item self-report measurement of anticipatory (10 items) and consummatory (eight items) components of anhedonia which consist of a series of statements that must be rated according to how accurate they are for the individual.76
The Hamilton Rating Scale for Depression (HAM-D)
The Hamilton Rating Scale for Depression (HAM-D) 17-item version was included in exploratory analysis to provide confirmatory support for changes in depression severity with treatment.32 This interviewer-administered semi-structured interview is one of the most widely used instruments in depression treatment studie.
The Hamilton Rating Scale for Anxiety (HAM-A)
Similarly, the Hamilton Rating Scale for Anxiety (HAM-A) is a rating scale designed to measure the severity of anxiety symptoms.33 It is widely used in both clinical and research settings. The scale consists of 14 items, each defined by a series of symptoms, and measures both psychic anxiety (mental agitation and psychological distress) and somatic anxiety (physical complaints related to anxiety). It has been demonstrated to acceptable reliability, validity and sensitivity to change.33 Each item is scored on a scale of 0 (not present) to 4 (severe), with a total score range of 0–56, where 9–16 indicates mild severity, 18–24 mild to moderate severity and 25–30 moderate to and greater than 30 indicates severe depression. This instrument was included in secondary analysis to provide confirmatory support for changes in anxiety severity with treatment.
The Cognitive and Physical Functioning Questionnaire (CPFQ)
The Cognitive and Physical Functioning Questionnaire (CPFQ) is a 7-item self-report instrument including in exploratory analysis intended to be a brief scale for measuring cognitive and executive dysfunction in patients with mood and anxiety disorders.77 This scale has demonstrated to have strong internal consistency, good temporal stability and sensitivity to change with treatment.77
Clinical Global Impression (CGI)
The Clinical Global Impression - Severity (CGI-S) is a widely administered clinician rated global measure of subject overall illness severity. Subjects are rated on a 1–7 scale where 1 corresponds to “Normal, Not at All Ill”, 2 is “Borderline Mentally Ill”, the anchor for 3 is “Mildly Ill”, the anchor for 4 is “Moderately Ill”, 5 is “Markedly Ill”, 6 is “Severely Ill”, and 7 is “Among the Most Extremely Ill Patients”. The Clinical Global Impression - Improvement (CGI-I) is a widely administered clinician rated global measure of the degree of improvement from the initial assessment in subject overall illness severity. Subjects are rated on a 1–7 scale where 1 corresponds to “Very Much Improved”, 2 is “Much Improved”, the anchor for 3 is “Minimally Improved”, the anchor for 4 is “No Change”, 5 is “Minimally Worse”, 6 is “Much Worse”, and 7 is “Very Much Worse”. Both the CGI-S and CGI-I were administered at all visits. During the course of the trial subjects for whom the CGI-I is greater than 5 were removed from the study and appropriate care given, for safety purposes.
Safety Assessments
Safety assessments carried out at all visits included clinical evaluation of adverse events, adverse effects assessment with the Patient Reported Inventory of Side-Effects (PRISE), vital signs (height, weight, blood pressure, and pulse), urine drug screen, complete blood count with differential, electrolytes, comprehensive metabolic panel including liver function tests, thyroid function tests, urinalysis, CGI-I, and ECG. Physical examination was carried out at baseline and the end of double-blind treatment. Beta-HCG serum pregnancy test was obtained during screening. Suicidality was assessed at every visit using the Columbia Suicide Severity Rating Scale (CSSRS).63 Tests for assessment of gastric adverse events were obtained at baseline, week 4, and week 8 of double-blind treatment and included gastrin, and pepsinogen I/II levels. These gastric tests were obtained for the first half of the study based on an FDA recommendation for monitoring, after which approval was obtained from FDA, the NIMH DSMB, and the relevant Institutional Review Boards to drop this requirement.
Oversight
The trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and was approved by the relevant institutional review boards. Written informed consent was provided by the patients or their legal representatives. Data were collected and analyzed by the investigators and interpreted by all authors. The first draft of the manuscript was prepared by the first author. All the authors approved subsequent drafts and agreed to submit the manuscript for publication. The authors had full access to the trial data and vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol. The trial was governed by a committee of site investigators and NIMH Program Officers.
Outcomes
Primary Outcome
The primary outcome for evaluating “Proof of Mechanism” that engaging the target (kappa opioid antagonism) had the hypothesized effect on reward-related brain function, was the baseline corrected fMRI activation at the end of 8 weeks of double-blind treatment in an a priori bilateral non-thresholded ventral striatal area mask, defined by the Harvard-Oxford Subcortical Atlas, during anticipation of monetary gain in the Monetary Incentive Delay (MID) Task contrasted with neutral (non-gain/non-loss) trials.
Owing to the experimental medicine approach taken, the driving factor leading to the choice of the primary outcome measure was the neurobiological target. Accordingly, based on the compelling preclinical literature available to us when we designed the study indicating that KOR antagonism releases inhibition on DA neurons and increases nucleus accumbens function,14–21 we chose reward-related activation in the nucleus accumbens as our primary target. Having committed to this mechanism and target, we sought an fMRI paradigm that would reliably engage the nucleus accumbens in response to rewards. Based on the early work of Knutson41,43–45 and others,46,47 the use of the monetary incentive delay (MID) task in anticipation of gains was selected. We note that, although lack of replications exist,42,48 prior studies in both MDD and psychiatrically healthy samples available to us when the study was designed have linked reward-related ventral striatal activation and anhedonic symptoms,74 and this relationship has been replicated in more recent studies.49–50 Further, recent meta-analyses of fMRI data collected among healthy controls have confirmed that the ventral striatum (nucleus accumbens) is reliably recruited during reward anticipation.51,78
This choice was further supported by the fact that there was data from a prior treatment study (open-label escitalopram administered to 15 MDD patients and 15 controls) where outcome was assessed with fMRI determined ventral striatum activation during anticipation of gains and losses in the MID.54 That study provided a set of findings that differed for ventral striatal activation in anticipation of gains and losses but overall suggested the promise of both measures. Findings suggesting the relative utility of ventral striatal activation in anticipation of loss are that the baseline Beck Depression Inventory (BDI) anhedonia item score was significantly related to ventral striatal loss anticipation and not gain and that escitalopram led to a significant change in ventral striatal loss anticipation-related activity not gain. However, a number of findings supported our choice to employ ventral striatal activation in anticipation of gains in our study. These include: (1) depressed subjects had a significantly smaller ventral striatal activation in anticipation of gain than the control group before treatment with escitalopram (p=0.025) but not after treatment; (2) there was a trend for a group*time interaction with treatment with ventral striatal activation in anticipation of gain (p<0.07) in this study with only 15 subjects per group; and (3) there was a trend for the decrease in BDI score over treatment to be correlated with the increase in ventral striatal activation in anticipation of gain (r=0.54, p=0.058).
Secondary Outcomes
A priori specified secondary outcomes included:
A clinical measure of anhedonia, the SHAPS
A behavioral measure of reward-related function, the change in response bias from block 1 to block 2 in the PRT (evaluated by testing the Treatment Arm x Block x Time interaction).
Exploratory Outcomes
All other outcomes were included in exploratory analyses including the PRT group x time interaction, Resting State Delta EEG Current Density in the Rostral Anterior Cingulate, TEPS, EEfRT, VAS-A, HAM-D, HAM-A, CPFQ, and CGI.
Statistical Analysis
Sample Size
We powered this study to detect an effect-size of 0.5. Our capacity to estimate the expected effect-size on our primary outcome measure was limited because there has been only 1 treatment study carried out with this measure to date.54 This prior study involved open-label escitalopram treatment of 15 patients with MDD and 15 controls. Pre-to-post treatment effects and the group-by-time interaction suggest an effect-size of 0.88 or higher with our primary outcome measure. It was understood that estimates of power based on the prior treatment study were likely overly optimistic because it was an open-label treatment study and only included 15 depressed subjects. As a result, we assumed a lower effect-size of 0.50. On this basis we planned to enroll 90 total subjects with the conservative expectation that we could have incomplete data on up to 20% of subjects due to drop out or loss of data due to factors such as poor scan quality. This would make available at least 72 subjects for analyses which provides 80% power to detect an effect-size of 0.5 at the alpha=0.05 level in a one-tailed test of significance.
Efficacy Analyses:
We planned to carry out efficacy analyses in the “Intention-to-Treat” (ITT) population and to perform one-sided statistical tests using a p value threshold of 5% level for statistical significance. Analyses consisted of mixed effects models including baseline values, age, study site, and sex as covariates. The mixed effects models utilized a random intercept and fixed slopes model with compound symmetry structure and employed a maximum likelihood estimation approach.
Site was included as an independent variable with analysis carried out employing centering based on Kraemer and Blasey79 in order to account for variability among the study sites. Because of the number of covariates and sites relative to the sample size, examination of site x time and site x treatment arm x time interactions were not pre-planned analyses.
The rationale for including baseline as a covariate in mixed effects models is included in the Supplementary Information.
For each variable, the baseline adjusted mean was computed for both treatment groups. We also computed JNJ-67953964 vs placebo group effect-sizes (Hedges’ g), which were calculated as (ME – MC) / SD pooled, where ME represents the adjusted mean of experimental treatment, MC represents the adjusted mean of the comparison treatment, and SD pooled represents pooling of the standard deviations from within both groups. Hedges’ g is similar to Cohen’s d except that it employs a sample-size weighted pooled standard deviation whereas Cohen’s d employs the pooled standard deviation.52,55 As a result, Hedges’ g is believed to be a less biased measure of effect-size with groups of unequal size and in limited size data sets, which is why it was chosen for use in this study.52
Multiple imputations (MI) were used in this study to account for the missing data to verify the results for the primary endpoint for ITT and as treated populations. The “Per Protocol” analyses did not employ MI. We did multiple imputations of missing data using the SAS Procedure PROC MI employing the Markov chain Monte Carlo method with a single chain carrying out 20 imputations with a seed of 788 employing the same covariates as our mixed models analysis. We then analyzed the complete data sets using proc MIXED, and then we analyzed the output from the two previous steps using PROC MIANALYZE. The hypothesized missing mechanism was “Missing At Random”. We employed a “Missing At Random” model despite the fact that the reasons for discontinuation are known because the reasons for discontinuation do not suggest additional variables that could be included in the multiple imputation models that are likely to be predictive of missingness/discontinuation.80 The reasons for discontinuation, outlined in the Supplement, reflect a relatively even distribution across a number of different discontinuation reasons.
In addition to the above analyses we carried out a set of post-hoc exploratory analyses to follow-up on the findings of the planned analyses. In order to evaluate the capacity to determine the degree with which response to treatment can be predicted from the baseline value of our primary outcome we carried out an exploratory correlation analysis. Moreover, in order to determine the extent to which response to treatment can be predicted at the individual level, we carried out a logistic regression analysis determining the extent to which baseline ventral striatal activation predicted which subjects would be responders in terms of ventral striatal activation while controlling for treatment arm. Response was defined based on a median split on the change in ventral striatal activation with treatment.
We also carried out exploratory post-hoc correlation analyses controlling for treatment arm to determine the degree of relatedness of our primary outcome measure with the key secondary outcome measures the SHAPS and PRT change from Block1 to Block 2 in Response Bias.
Lastly, we carried out an exploratory post-hoc analysis of the magnitude of the change in mean ventral striatal activation in anticipation of gains in response to treatment separately in the JNJ-67953964 and placebo groups. This was performed to better delineate the nature of the between-group effects found with this measure. This analysis consisted of determining the effect-sizes (Hedges g) for the change in our primary outcome measure for the JNJ-67953964 and placebo groups. We evaluated effect-sizes rather than assessing for statistical significance because our study was not powered to demonstrate statistical significance in such analyses, each of which includes approximately half of the total sample size and, unlike statistical significance, effect-size is not dependent upon sample size. Further, an important consideration with respect to this analysis is that we assumed a priori that ventral striatal activation in anticipation of gains might tend to decrease over time. As a result, our plan was to compare the drug group with the placebo group and not carry out within group statistical significance testing because we were aware of the possibility that repeating the test after 8 weeks in the population studied might be accompanied by an adaptation effect which would be manifested in a tendency towards a diminished response in the second test session in both groups. The comparison vs placebo was intended to mitigate this contingency. The reasons we believed there was a possibility that ventral striatal activation might decrease in the second test session vs the first are outlined in the Discussion section.
No interim analyses were planned or carried out.
DATA AVAILABILITY
Study data have been posted to the NIH/NIMH Data Archive and are accessible at the following address: NDAHelp@mail.nih.gov
Extended Data
Extended Data Figure 1.
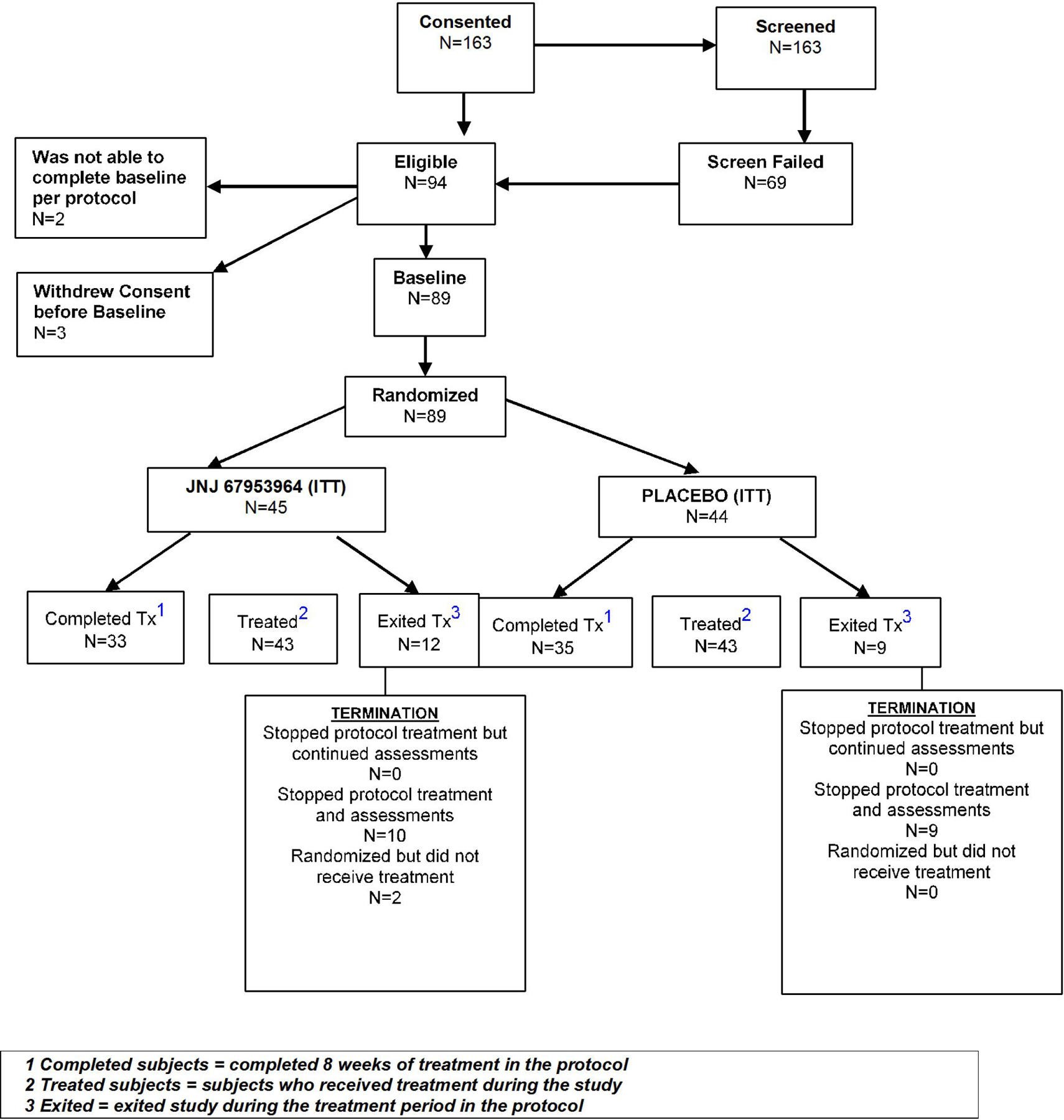
Consort Diagram
Supplementary Material
ACKNOWLEDGMENTS:
This project was supported by Contract HHS-N271–2012-000006-I from the National Institute of Mental Health awarded to ADK. DAP was partially supported by R37 MH068376. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the United States Government.
COMPETING INTERESTS:
A. Krystal: Consultant: Adare, Eisai, Ferring, Galderma, Idorsia, Jazz, Janssen, Takeda, Merck, Neurocrine, Pernix, Physician’s Seal, Evecxia, Sage
Research Support: NIH, Janssen, Jazz. Axsome, Reveal Biosensors
D. Pizzagalli: Consulting: Akili Interactive Labs, BlackThorn Therapeutics, Boehringer Ingelheim, Posit Science and Takeda Pharmaceuticals. Honoraria: Alkermes. Dr. Pizzagalli has a financial interest in BlackThorn Therapeutics, which has licensed the copyright to the Probabilistic Reward Task through Harvard University. Dr. Pizzagalli’s interests were reviewed and are managed by McLean Hospital and Partners HealthCare in accordance with their conflict of interest policies. No funding from these entities was used to support the current work, and all views expressed are solely those of the authors.
S. Mathew: Consultant: Allergan, Alkermes, Greenwich Biosciences; Clexio Biosciences; Intra-Cellular Therapies; Janssen; Perception Neuroscience; Sage Therapeutics; Signant Health; Seelos Therapeutics; Research Support: Biohaven, Janssen, NIH, NeuroRx, VistaGen Therapeutics; Drug from Biohaven for NIMH funded study. Support from the Michael E. Debakey VA Medical Center, Houston, TX for use of resources and facilities.
G. Sanacora: Consulting: Allergan, Alkermes,, AstraZeneca, Avanier Pharmaceuticals, Axsome Therapeutics Biohaven Pharmaceuticals, Boehringer Ingelheim International GmbH, Bristol-Myers Squibb, Clexio Biosciences, Hoffman La-Roche, Intra-Cellular Therapies, Janssen, Merck, Naurex, Navitor Pharmaceuticals, Novartis, Noven Pharmaceuticals, Otsuka, Praxis Therapeutics, Sage Pharmaceuticals, Servier Pharmaceuticals, Taisho Pharmaceuticals, Teva, Valeant, and Vistagen therapeutics.
Research Funding: AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson, Hoffman La-Roche, Merck, Naurex, and Servier.
Equity Interest: Biohaven Pharmaceuticals.
Patent Royalties: Biohaven.
J Murrough: Consultation: Boehreinger Ingelheim, Sage Therapeutics, Novartis, Allergan, Fortress Biotech, Janssen Research and Development, Medavante-Prophase, and Global Medical Education (GME);
Research Support: Avanir Pharmaceuticals, Inc.
R. Keefe: Consultant, Speaker, or Advisory Board Member: Abbvie, Acadia, Aeglea, Akebia, Akili, Alkermes, Allergan, ArmaGen, Astellas, Avanir, Axovant, Biogen, Boehringer-Ingelheim, Cerecor, CoMentis, Critical Path Institute, FORUM, Gammon Howard & Zeszotarski, Global Medical Education (GME), GW Pharmaceuticals, Intracellular Therapeutics, Janssen, Kempharm, Lundbeck, Lysogene, MedScape, Mentis Cura, Merck, Merrakris Therapetics, Minerva Neurosciences Inc., Mitsubishi, Montana State University, Monteris, Moscow Research Institute of Psychiatry, Neuralstem, Neuronix, Novartis, NY State Office of Mental Health, Orygen, Otsuka, Paradigm Testing, Percept Solutions, Pfizer, Pharm-Olam, Regenix Bio, Reviva, Roche, Sangamo, Sanofi, SOBI, Six Degrees Medical, Sunovion, Takeda, Targacept, Teague Rotenstreich Stanaland Fox & Holt, Thrombosis Research Institute, University of Moscow, University of Southern California, University of Texas Southwest Medical Center, WebMD, and Wilson Therapeutics.
Research Funding: The National Institute of Mental Health and Boehringer-Ingelheim.
Royalties: Versions of the BAC testing battery, the MATRICS Battery (BACS Symbol Coding), and the Virtual Reality Functional Capacity Assessment Tool (VRFCAT).
Shareholder: VeraSci and Sengenix.
S.H. Lisanby: Dr. Sarah H. Lisanby contributed to this article while at Duke University, prior to joining NIMH. The views expressed are her own and do not necessarily represent the views of the National Institutes of Health or the United States Government. Dr. Lisanby is a co-inventor on a patent for TMS technology, unrelated to this manuscript
J. Nurnberger: Research Funding: Janssen and Assurex
A. Song: Consulting: N/A Research Support: NIH, GE Healthcare
W. Goodman: Consulting: Biohaven Pharmaceuticals;
Research Funding: NIH, Simons Foundation and Biohaven Pharmaceuticals Device Donation: Medtronic
A. Goddard: Consultant: UpToDate, Biohaven Pharmaceuticals, Almatica Pharma Inc. Research Funding: National Network of Depression Centers.
M. Smoski: None
R. Weiner: None.
S. Szabo: Consultant: Otsuka Pharmaceuticals, Neurocrine Biosciences, Jazz Pharmaceuticals, Teva Pharmaceuticals, Centers of Psychiatric Excellence, Continuous Precision Medicine; Research Support: Otsuka Pharmaceuticals, Veteran’s Affairs Grant IK2CX001397.
K. Gao: Advisory Board: Sunovion, Otsuka Pharmaceuticals; Research Funding: AstraZeneca, Cleveland Foundation, Brain and Behavioral Research Foundation; Speaker: AstraZeneca, Pfizer, Sunovion.
W. Potter: Advisory Board/Consultant: Takeda, Lilly, Praxis, Astellas, Otsuka and Noven: DSMB: Agene-Bio; Stock Ownership: Merck.
Footnotes
Trial Register: ClinicalTrials.gov
Registration Number: NCT02218736
REFERENCES:
- 1.Denys D, de Geus F. Predictors of pharmacotherapy response in anxiety disorders. Curr Psychiatry Rep 7(4), 252–7 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163(11),1905–17 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Insel TR, Wang PS. The STAR*D trial: revealing the need for better treatments. Psychiatr Serv 60(11), 1466–7 (2009) [DOI] [PubMed] [Google Scholar]
- 4.National Advisory Mental Health Workgroup. From Discovery to Cure: Accelerating the Development of New and Personalized Interventions for Mental Illness Washington, DC: NIMH. (2010). [Google Scholar]
- 5.Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9(3), 203–214 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Kesselheim AS, Hwang TJ, Franklin JM Two decades of new drug development for central nervous system disorders. Nat. Rev. Drug Discov 14, 815–816 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Brady LS, Insel TR. Translating discoveries into medicine: psychiatric drug development in 2011. Neuropsychopharmacology 37(1), 281–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krystal AD, Pizzagalli DA, Matthew SJ, Sanacora G, Keege R, Song A, Calabrese J, Goddard A, Goodman W, Lisanby SH, Smoski M, Weiner RD, Iosifescu D, Nurnberger J, Szabo S, Murrough J, Shekhar A, Potter W. The First implementation of the NIMH FAST-FAIL approach to psychiatric drug development. Nature Reviews Drug Discovery 18(1), 82–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedemann K Biomarkers in development of psychotropic drugs. Dialogues Clin. Neurosci 13, 225–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borsook D, Becerra L, Fava M. Use of functional imaging across clinical phases in CNS drug development. Transl Psychiatry 16;3:e282, (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Sanislow C, Wang P. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 167(7), 748–51 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Whitton AE, Treadway MT, Pizzagalli DA. Reward processing dysfunction in major depression, bipolar disorder and schizophrenia. Curr Opin Psychiatry 28(1), 7–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlezon WA Jr, Krystal AD. Kappa-opioid antagonists for psychiatric disorders: From bench to clinical trials. Depress Anxiety 33(10), 895–906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlezon WA Jr, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther 316(1), 440–7 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Bruijnzeel AW. Kappa-Opioid receptor signaling and brain reward function. Brain Res Rev 62(1), 127–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA. Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology 62(1), 167–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology (Berl) 210(2), 241–52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maisonneuve IM, Archer S, Glick SD. U50,488, a kappa opioid receptor agonist, attenuates cocaine-induced increases in extracellular dopamine in the nucleus accumbens of rats. Neurosci Lett 181(1–2), 57–60 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Muschamp JW, Van’t Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, Meloni EG, Carlezon WA Jr. Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci 31(8), 3095–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA Jr. The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry 1;64(11), 982–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 210(2), 121–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, Forster BM, Wong CJ, Li X, Crile RS, Shaw DB, Sahr AE, Adams BL, Quimby SJ, Diaz N, Jimenez A, Pedregal C, Mitch CH, Knopp KL, Anderson WH, Cramer JW, McKinzie DL. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 77C, 131–144 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, Rorick-Kehn L, Weller M, Stoltz RR, Royalty J, Tauscher-Wisniewski S. Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol 54(9), 968–78 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Zheng MQ, Nabulsi N, Kim SJ, Tomasi G, Lin SF, Mitch C, Quimby S, Barth V, Rash K, Masters J, Navarro A, Seest E, Morris ED, Carson RE, Huang Y. Synthesis and evaluation of 11C-LY2795050 as a k-opioid receptor antagonist radiotracer for PET imaging. J Nucl Med 54(3), 455–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlezon WA Jr, Béguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther 123(3), 334–43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knutson B, Gibbs SE. Linking nucleus accumbens dopamine and blood oxygenation. Psychopharmacology (Berl) 191(3), 813–22 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Schott BH, Minuzzi L, Krebs RM, Elmenhorst D, Lang M, Winz OH, Seidenbecher CI, Coenen HH, Heinze HJ, Zilles K, Düzel E, Bauer A. Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward-related ventral striatal dopamine release. J Neurosci 28(52), 14311–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith–Hamilton Pleasure Scale. Br J Psychiatry 167, 99–103 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Franken IH, Rassin E, Muris P. The assessment of anhedonia in clinical and non-clinical populations: further validation of the Snaith-Hamilton Pleasure Scale (SHAPS). J Affective Disorders 99, 83–89 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Nakonezny PA, Morris DW, Greer TL, Byerly MJ, Carmody TJ, Grannemann BD, Bernstein IH, Trivedi MH. Evaluation of anhedonia with the Snaith-Hamilton Pleasure Scale (SHAPS) in adult outpatients with major depressive disorder. J Psychiatr Res 65, 124–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakonezny PA, Carmody TJ, Morris DW, Kurian BT, Trivedi MH. Psychometric evaluation of the Snaith-Hamilton pleasure scale in adult outpatients with major depressive disorder. Int Clin Psychopharmacol 25(6), 328–33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamilton M Development of a rating scale for primary depressive illness. Br J Soc Psychol 6, 278–296 (1967). [DOI] [PubMed] [Google Scholar]
- 33.Maier W, Buller R, Philipp M, Heuser I. The Hamilton Anxiety Scale: reliability, validity and sensitivity to change in anxiety and depressive disorders. J Affect Disord 14(1), 61–8 (1988). [DOI] [PubMed] [Google Scholar]
- 34.Pizzagalli DA, Jahn AL, O’Shea JP. Toward an objective characterization of an anhedonic phenotype: a signal-detection approach. Biol Psychiatry 57(4), 319–27 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pizzagalli DA, Evins AE, Schetter EC, Frank MJ, Pajtas PE, Santesso DL, Culhane M. Single dose of a dopamine agonist impairs reinforcement learning in humans: behavioral evidence from a laboratory-based measure of reward responsiveness. Psychopharmacology (Berl) 196(2), 221–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pizzagalli DA, Iosifescu D, Hallett LA, Ratner KG, Fava M. Reduced hedonic capacity in major depressive disorder: evidence from a probabilistic reward task. J Psychiatr Res 43(1), 76–87 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santesso DL, Dillon DG, Birk JL, Holmes AJ, Goetz E, Bogdan R, Pizzagalli DA. Individual differences in reinforcement learning: behavioral, electrophysiological, and neuroimaging correlates. Neuroimage 42(2), 807–16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, et al. Deficits in sustaining reward responses in subsyndromal and syndromal major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1045–52 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Fletcher K, et al. Anhedonia in melancholic and non-melancholic depressive disorders. J. Affect. Disord 184, 81–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris BH, Bylsma LM, Yaroslavsky I, Kovacs M & Rottenberg J Reward learning in pediatric depression and anxiety: preliminary findings in a high-risk sample. Depress. Anxiety 32, 373–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knutson B, Westdorp A, Kaiser E, Hommer D. FMRI visualization of brain activity during a monetary incentive delay task. Neuroimage 12(1):20–7 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH. Neural responses to monetary incentives in major depression. Biol Psychiatry 63(7), 686–92 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Neuroreport 12(17), 3683–7 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Knutson B, Adams CM, Fong GW, Hommer D. Anticipation of increasing monetary reward selectively recruits nucleus accumbens. J Neurosci 21(16), RC159 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knutson B, Wimmer GE, Kuhnen CM, Winkielman P. Nucleus accumbens activation mediates the influence of reward cues on financial risk taking. Neuroreport 19(5), 509–13 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Carlson JM, Foti D, Mujica-Parodi LR, Harmon-Jones E, Hajcak G. Ventral striatal and medial prefrontal BOLD activation is correlated with reward-related electrocortical activity: a combined ERP and fMRI study. Neuroimage 57(4), 1608–16 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Dillon DG, Holmes AJ, Jahn AL, Bogdan R, Wald LL, Pizzagalli DA. Dissociation of neural regions associated with anticipatory versus consummatory phases of incentive processing. Psychophysiology 45(1), 36–49 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry 58(11), 843–53 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Keller J, Young CB, Kelley E, Prater K, Levitin DJ, Menon V. Trait anhedonia is associated with reduced reactivity and connectivity of mesolimbic and paralimbic reward pathways. J Psychiatr Res 47(10), 1319–28 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Zhang CY, Huang J, Wang Y, Yan C, Li K, Zeng YW, Jin Z, Cheung EFC, Su L, Chan RCK. Improving motivation through real-time fMRI-based self-regulation of the nucleus accumbens. Neuropsychology 32(6), 764–776 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Oldham S, Murawski C, Fornito A, Youssef G, Yücel M, Lorenzetti V. The anticipation and outcome phases of reward and loss processing: A neuroimaging meta-analysis of the monetary incentive delay task. Hum Brain Mapp 39(8), 3398–3418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hedges L Distribution Theory for Glass’s Estimator of Effect Size and Related Estimators. Journal of Educational Statistics 6(2), 107–128 (1981). [Google Scholar]
- 53.Green IW, Pizzagalli DA, Admon R, Kumar P. Anhedonia modulates the effects of positive mood induction on reward-related brain activation. Neuroimage 2019. Jun;193:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoy M, Schlagenhauf F, Sterzer P, Bermpohl F, Hägele C, Suchotzki K, Schmack K, Wrase J, Ricken R, Knutson B, Adli M, Bauer M, Heinz A, Ströhle A. Hyporeactivity of ventral striatum towards incentive stimuli in unmedicated depressed patients normalizes after treatment with escitalopram. J Psychopharmacol 26(5), 677–88 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Cohen J Statistical Power Analysis for the Behavioral Sciences. Routledge (1988).
- 56.Vrieze E, Pizzagalli DA, Demyttenaere K, Hompes T, Sienaert P, de Boer P, Schmidt M, Claes S. Reduced reward learning predicts outcome in Major Depressive Disorder. Biol Psychiatry 73(7), 639–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaiser RH, Treadway MT, Wooten DW, Kumar P, Goer F, Murray L, Beltzer M, Pechtel P, Whitton A, Cohen AL, Alpert NM, El Fakhri G, Normandin MD, Pizzagalli DA. Frontostriatal and dopamine markers of individual differences in reinforcement learning: A Multi-modal investigation. Cereb Cortex 2018. Dec 1;28(12):4281–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webb CA, Dillon DG, Pechtel P, Goer FK, Murray L, Huys QJ, Fava M, McGrath PJ, Weissman M, Parsey R, Kurian BT, Adams P, Weyandt S, Trombello JM, Grannemann B, Cooper CM, Deldin P, Tenke C, Trivedi M, Bruder G, Pizzagalli DA. Neural correlates of three promising endophenotypes of depression: Evidence from the EMBARC study. Neuropsychopharmacology 41(2), 454–63 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jensen J, McIntosh AR, Crawley AP, Mikulis DJ, Remington G, Kapur S. Direct activation of the ventral striatum in anticipation of aversive stimuli. Neuron 40,1251–1257 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Carter RM, McInnes JJ, Huettel SA, Adcock RA. Activation in the VTA and Nucleus Accumbens increases in anticipation of both gains and losses. Frontiers in Behav Neurosci 3,1–15 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS ONLY REFERENCES
- 61.American Psychiatric Association (2000). Diagnostic and statistical manual of mental disorders (4th ed., Text Revision). Washington, DC. (2009). [Google Scholar]
- 62.Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, Hergueta T, Baer R, Dunbar GC. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59 Suppl 20, 22–33 (1998). [PubMed] [Google Scholar]
- 63.Posner K Columbia-Suicide Severity Rating Scale (C-SSRS) http://www.cssrs.columbia.edu (2011).
- 64.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17(2), 825–41 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Med Image Anal 5(2), 143–56 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Andersson JLR, Jenkinson M, Smith S. Non-linear registration, aka Spatial normalisation FMRIB technical report TR07JA2 FMRIB Centre, Oxford, United Kingdom. 2007. [Google Scholar]
- 67.Smith SM. Fast robust automated brain extraction. Human Brain Mapping, 17(3), 143–155 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woolrich MW, Ripley BD, Brady M, & Smith SM Temporal Autocorrelation in Univariate Linear Modeling of FMRI Data. NeuroImage, 14(6), 1370–1386 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Woolrich MW, Behrens TEJ, Beckmann CF, Jenkinson M, & Smith SM Multilevel linear modelling for FMRI group analysis using Bayesian inference. NeuroImage, 21(4), 1732–1747 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, Beckmann C, Jenkinson M, Smith SM. Bayesian analysis of neuroimaging data in FSL. Neuroimage 45(1 Suppl), S173–86 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Friedman L, Glover GH. Report on a multicenter fMRI quality assurance protocol. J Magn Reson Imaging 23(6), 827–39 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Di Giannantonio M, Martinotti G. Anhedonia and major depression: the role of agomelatine. Eur Neuropsychopharmacol 22 Suppl 3, S505–10 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Martinotti G, Andreoli S, Reina D, Di Nicola M, Ortolani I, Tedeschi D, Fanella F, Pozzi G, Iannoni E, D’Iddio S, Prof LJ. Acetyl-l-Carnitine in the treatment of anhedonia, melancholic and negative symptoms in alcohol dependent subjects. Prog Neuropsychopharmacol Biol Psychiatry 35(4), 953–8 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Wacker J, Dillon DG, Pizzagalli DA. The role of the nucleus accumbens and rostral anterior cingulate cortex in anhedonia: integration of resting EEG, fMRI, and volumetric techniques. Neuroimage 46(1), 327–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Treadway MT, Buckholtz JW, Schwartzman AN, Lambert WE, Zald DH. Worth the ‘EEfRT’? The effort expenditure for rewards task as an objective measure of motivation and anhedonia. PLoS One 12;4(8), e6598 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gard DE, Gard-Germans M, Kring AM, Oliver JP. Anticipatory and consummatory components of the experience of pleasure: A scale development study. J Research in Personality 40, 1086–1102 (2006). [Google Scholar]
- 77.Fava M, Iosifescu DV, Pedrelli P, Baer L. Reliability and validity of the Massachusetts general hospital cognitive and physical functioning questionnaire. Psychother Psychosom 78(2), 91–7 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Wilson RP, Colizzi M, Bossong MG, Allen P, Kempton M; MTAC, Bhattacharyya S. The Neural Substrate of Reward Anticipation in Health: A Meta-Analysis of fMRI Findings in the Monetary Incentive Delay Task. Neuropsychol Rev 28(4), 496–506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kraemer HC, Blasey CM. Centering in regression analyses: a strategy to prevent errors in Statistical inference. Int J Methods Psychiatr Res 13(3),141–51 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azur MJ, Stuart EA, Frangakis C, Leaf PJ. Multiple imputation by chained equations: what is it and how does it work? Int J Methods Psychiatr Res 20(1), 40–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Study data have been posted to the NIH/NIMH Data Archive and are accessible at the following address: NDAHelp@mail.nih.gov