

Abstract
Tourette Syndrome (TS) is a neuropsychiatric disorder thought to involve a reduction of basal ganglia (BG) interneurons and malfunctioning of the BG circuitry. However, whether interneurons fail to develop or are lost postnatally remains unknown. To investigate the pathophysiology of early development in TS, induced pluripotent stem cell (iPSC)-derived BG organoids from TS patients and healthy controls were compared on multiple levels of measurement and analysis. BG organoids from TS individuals manifested an impaired medial ganglionic eminence fate and a decreased differentiation of cholinergic and GABAergic interneurons. Transcriptome analyses revealed organoid mispatterning in TS, with a preference for dorsolateral at the expense of ventromedial fates. Our results point to altered expression of GLI transcription factors downstream of the Sonic Hedgehog signaling pathway with cilia disruption at the earliest stages of BG organoid differentiation as a potential mechanism for the BG mispatterning in TS. This study uncovers early neurodevelopmental underpinnings of TS neuropathological deficits using organoids as a model system.
Introduction
The ventral telencephalon gives rise to the basal ganglia (BG), a subpallial brain region responsible for the storage and execution of motor programs (1). The BG are implicated in many movement disorders, including Tourette Syndrome (TS) (2–4), a childhood-onset neuropsychiatric disorder characterized by involuntary motor and vocal tics. Although considered to be neurodevelopmental in origin, its etiology and pathophysiology is unknown. Imaging studies have previously shown abnormalities in the BG of TS individuals, including differences in size and morphology (5). In addition, postmortem immunocytochemical analyses showed that the number of multiple subtypes of interneurons, both GABAergic and cholinergic, was reduced by approximately 50% in striatal tissue of adult postmortem TS patients as compared to controls (6, 7), a finding confirmed by transcriptome analyses (8). In the BG, interneurons exert feed-forward inhibition upon medium spiny neurons and are crucial regulators of cortico-striatal excitability and proper BG function (9). Therefore, deficits in GABAergic and cholinergic interneurons suggest decreased or altered inhibition within the striatum, which is ultimately thought to underlie involuntary motor actions in the form of tics. However, it is not known whether the interneuron deficit in TS is the consequence of developmental abnormalities, as suggested by the disorder’s early age of onset, or whether they occur later in life. Additionally, the molecular mechanisms behind these abnormalities are poorly understood. Barriers to answering these questions include the limited availability of animal model systems that fully recapitulate TS and the challenge in capturing the dynamics of brain development using neuropathological tissue.
Here we have conducted a study to elucidate developmental differences between control (CT) and TS individuals utilizing the human iPSC-derived brain organoid system (10–14). Forebrain interneurons are generated in the basal forebrain, primarily in the medial and caudal ganglionic eminences (MGE and CGE, respectively) as well as the preoptic area (POA) in the anterior hypothalamus, and subsequently migrate widely throughout the forebrain during development (15). The POA is a source for cholinergic neurons, neuropeptide Y (NPY+), reelin (RELN+) and calbindin (CB+) interneurons; cholinergic neurons also derive from Fgf8-expressing cells of the rostral patterning center (16). The MGE is a source for parvalbumin (PV+), somatostatin (SST+), nitric oxide synthase (NOS)+ and cholinergic interneurons; and the CGE is a source for vasoactive intestinal peptide (VIP+), RELN+ and calretinin (CR+) interneurons. Lateral ganglionic eminence (LGE), on the other hand, is a source for striatal medium spiny neurons (MSN) (15). We first generated BG-like organoids recapitulating early human ventral telencephalic development to explore altered BG development in TS, as well as to reveal potential underlying mechanisms. Transcriptome and immunocytochemical analyses show that TS-derived BG organoids, as compared to CT-derived organoids, manifest a loss of NKX2.1+ precursor cells at earlier stages followed by a decrease in number of GABAergic and cholinergic interneurons at later stages. We attribute these cellular alterations to a fundamental mispatterning of the BG organoids in TS individuals. We reveal disruptions in the expression of key components of the cilia-dependent Sonic Hedgehog (SHH) signaling pathway at the earliest stages of BG organoid development, which could be – at least in part – responsible for ventral telencephalic mispatterning and manifestation of these cellular phenotypes. Our data implicate the SHH pathway and cilia in the etiology of TS, providing a potential mechanism for previously described neuronal phenotypes in postmortem TS brain tissue.
Materials /Subjects and Methods
Subject Participants
Five Tourette Syndrome (TS) participants (Supplementary Table 1) were recruited at the Yale Child Study Center, based on patient availability. Severity of TS symptoms was assessed using the YGTSS (Yale Global Tic Severity Scale), the SCID (Structured Clinical Interview), and the PUTS (Premonitory Urge for Tic Scale). The criteria for inclusion were a confirmed diagnosis of TS based on clinical evaluation and the results of the diagnostic assessment (17). Participants were excluded if they had other psychiatric diagnoses, with the exception of Obsessive-Compulsive Disorder and Attention Deficit Hyperactivity Disorder, which do not represent exclusionary criteria because of frequent co-morbidity with TS. The unaffected control participants (CT) were selected from a larger pool of participants recruited in conjunction with other research projects (see Supplementary Table 1 for subject information).
All participants donated a punch skin biopsy. Informed consent was obtained from each subject according to the regulations of the Institutional Review Board and Yale Center for Clinical Investigation at Yale University. Primary cultures of fibroblasts were derived from the biopsy using standard explant procedures (18) and iPSC lines were generated using an episomal reprogramming method (19), with the exception of three control lines, generated by the retroviral method (Supplementary Table 1). All iPSC lines used in this study were characterized using three sets of quality control criteria: (a) morphology; (b) immuno-cytochemical expression of pluripotency markers, and (c) semiquantitative RT-PCR of pluripotency marker genes and downregulation of exogenous reprogramming factors.
Induced Pluripotent Stem Cells (iPSCs)
Two to three iPSC lines were used for each of the five TS subjects (12 lines total). One to two iPSC lines were used for each of 12 healthy CT subjects (n=14 lines total); 11 CT subjects were used for the comparison with the TS patients; an additional CT subject was utilized only for the initial stages of protocol development (Supplementary Table 1). Representative data for iPSC characterization and quality control for TS individuals TS02-03, TS03-03 and TS08-03 are shown in Supplementary Figure 1.
Neuronal Differentiation
Telencephalic neural organoids patterned toward basal ganglia development (BG organoids) were generated by a modification of previously described methods (10, 20). The outline of the protocol is shown in Figure 1A. Briefly, iPSCs were aggregated to generate embryoid bodies, which were cultured in suspension from day 2 to 7 in serum-free medium supplemented with fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF) (10ng/ml each). The neural aggregates form lumen-filled rosettes, which were manually harvested and cultured from day 8 to 11 in the presence of the SHH agonist purmorphamine (PUR, 2 μM) and fibroblast growth factor 8 (FGF8, 200ng/ml). Day 12 is deemed terminal differentiation day 0 (TD0), when growth media were changed into differentiation-promoting media. Upon entry into TD0, the organoids were cultured for one week (day 12 to 19) with PUR, 1μM; FGF8, 100ng/ml, retinoic acid (RA, 500ng/ml) and bone morphogenic protein 9 (BMP9, 10ng/ml) (21, 22) to promote differentiation.
Figure 1: Characterization of ventral telencephalic or basal ganglia (BG) organoids in control (CT) iPSC lines.
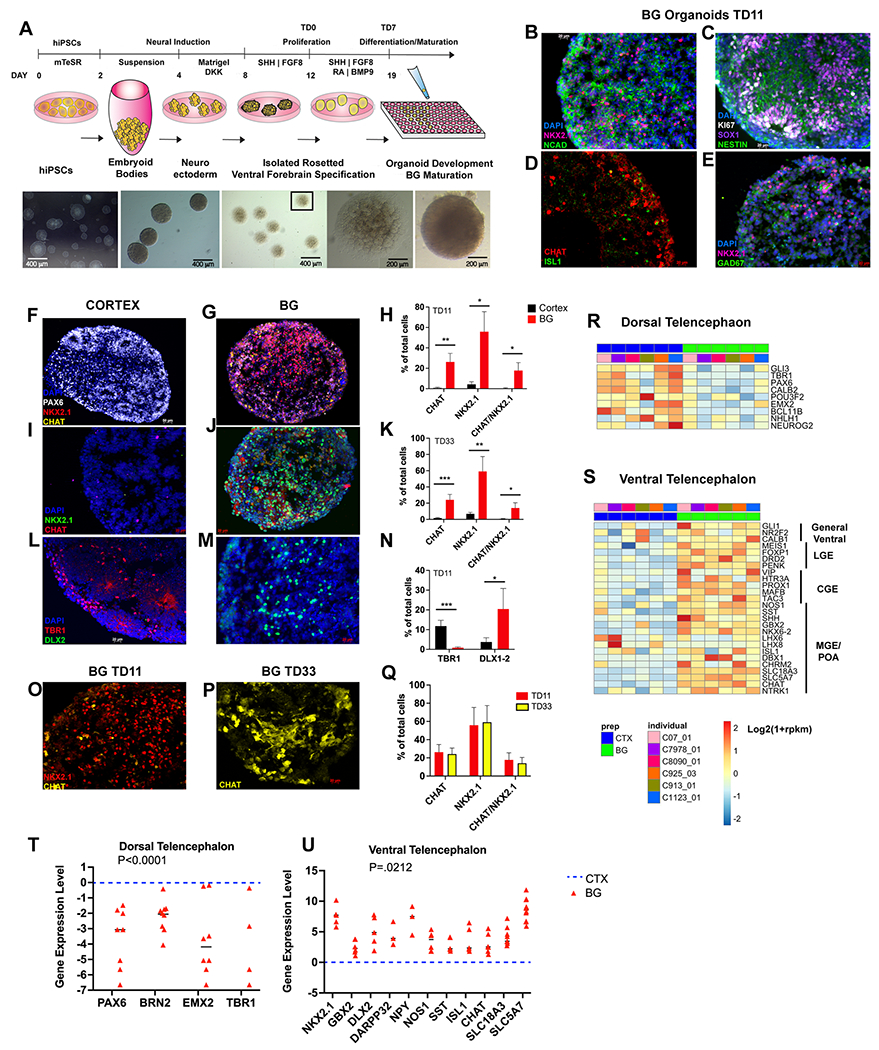
(a), Schematic of the BG organoid protocol. (b-e), Immunostaining at TD11, showing increased expression of markers of the ventral telencephalon in BG organoids. (f-n), Immunostaining and stereological unbiased quantification of cells expressing dorsal (PAX6, TBR1) and ventral (NKX2.1, CHAT, DLX1-2) telencephalic gene markers comparing cortical (CTX) and BG organoids at TD11 and TD33. Counts were obtained from 4 organoids and 2 serial sections each per iPSC line per condition (n= 3 CT iPSC lines). *p <0.05, ** p <0.01, *** p <0.005 by unpaired t-test when comparing BG to CTX organoids. (o-q), NKX2.1/CHAT robust expression in BG organoids across time. (r, s), Heatmaps from bulk RNA-seq analyses showing the relative expression levels of a curated set of dorsal (r) and ventral (s) telencephalic gene markers in CTX and BG organoids (P<0.05; n=6 CT iPSC lines). Log2(1+rpkm) values were scaled in each row. (t, u), Relative gene expression levels by RT-qPCR in BG (red) and CTX (blue) organoids at TD14 (n=6-8 cortical; n=6-8 BG iPSC lines). Datapoints are plotted using a Log2 scale. P values were calculated using a two-way ANOVA between CTX and BG organoids in each gene set. Significance was set at P=0.05 (Dorsal Telencephalon: P<0.0001; Ventral Telencephalon: P=0.0212). CTX is used as a reference and is shown as a dotted line in the graphs; all quantifications were normalized to the housekeeping gene GAPDH.
RNA Isolation, RT-qPCR and Bulk RNA Sequencing Experiments
RNA was extracted from 16-20 randomly-chosen BG organoids at TD0, TD14 or TD30 in vitro (10). To perform RT-qPCR, total RNA was isolated using the PicoPure RNA isolation kit (Applied Bioscience, Beverly Hills, CA, USA), and cDNA was synthesized utilizing the SuperScript III First-Strand Synthesis Kit and random hexamers (Invitrogen) using 100 ng of total RNA. The cDNA was amplified using Power SYBR Green PCR Master Mix (Applied Biosystems) on a StepOnePlus Real-Time PCR System (Applied Biosystems). Data were normalized to GAPDH housekeeping gene expression. A Two-Way ANOVA and post hoc test was used to establish statistical significance of differential gene expression. Primer sequences used for RT-qPCR experiments are reported in Supplementary Table 2. For RNA sequencing experiments, HiSeq paired-end, 100 bp RNA sequencing (with rRNA depletion) was conducted on a NovaSeq platform to a sequencing depth of 40 million reads per sample.
Immunostaining and Stereology
Organoids were harvested at TD14 or TD30 in vitro. Samples were processed as previously described (20). Organoids were collected and fixed in 4% PFA for 3 hours, incubated in 25% sucrose overnight, embedded in OCT at room temperature and stored at −80 degrees Celsius. Embedded samples were cryo-sectioned at 12 μm on glass slides. Cryo-sections were first incubated at room temperature for 1 hour in blocking solution (10% donkey serum/0.1% Triton X-100 in PBS). Samples were then incubated overnight at 4°C with primary antibodies in diluted blocking buffer (5% donkey serum/0.05% Triton X-100 in PBS). After three washes in PBS/0.1% Triton X-100, sections were treated for 1 hour at room temperature with secondary antibodies (1:500, Invitrogen, Waltham, MA, USA or Jackson Laboratories, Bar Harbor, ME, USA) in diluted blocking buffer. Primary antibody are as follows: PAX6 (mouse, BD Bioscience, Franklin Lakes, NJ, USA); N-cadherin (mouse, BD Bioscience); TBR1 (rabbit, Abcam, Waltham, MA, USA); SOX1 (goat, R&D System, Minneapolis, MN, USA); DLX-1/2 (rabbit, gift of Yuri Morozov, Yale University, New Haven, CT); NKX2.1 (mouse, Thermo Scientific, Waltham, MA, USA); BRN2 (goat, Santa Cruz, Dallas, TX, USA); Ki67 (rabbit, Vector, Newark, CA, USA); pH3 (rat, Sigma, St. Louis, MO, USA); CHAT (goat, Chemicon, St. Louis, MO, USA); NOS (rabbit, Millipore, St. Louis, MO, USA); SST (rat, Chemicon).
For image acquisition, an ApoTome-equipped Axiovert 200M with Axiovision 4.5 software was used (White Plains, NY, USA). Stereological analysis was performed on a Carl Zeiss Axioskop 2 Mot Plus using Stereoinvestigator software (MicroBright-Field). Per individual, two organoids and four serial sections per organoid were used. The optical fractionator probe (100x oil-immersion objective) was utilized to count the nuclear profiles of stained cells by superimposing a counting grid (180μm by 170μm) and using a counting box of 50μm by 50μm by 10μm. The percentage of positive cells was calculated relative to total DAPI+ cells.
Western Blotting
Western blot experiments were conducted as previously described (20). Organoids were washed with cold PBS, lysed with Lysis-M, EDTA-free buffer (Roche, Basel, Switzerland, Cat. 04 791 964 001), which was supplemented with proteases (Roche) and phosphatase inhibitor cocktail tablets (Roche). Samples were then centrifuged for 15 minutes at 14000 rpm at 4°C and the supernatant was obtained. Total protein was quantified using the Qubit™ Protein Assay Kit (Invitrogen) and 40 μg total protein was loaded per lane into TGX stain-free 4-15% precast gels (BioRad, Hercules, CA, USA). After SDS gel electrophoresis, gels were transferred to nitrocellulose membranes (BioRad). Signal was visualized using SuperSignal West Pico Chemioluminescent Substrate (Thermo Scientific) and quantified using ImageJ software. Primary antibodies were as follows: GLI1 (rabbit, Abcam); GLI2 (rabbit, Millipore); GLI3 (rabbit, Abcam); GAPDH (mouse, Millipore). Protein signal for each band was internally normalized to the GAPDH expression in each lane and quantified via ImageJ software.
Transcriptome Analysis by RNA Sequencing
We used STAR (23) to map the RNA-seq reads to the human genome (hg38) and the GencodeV34 for transcriptome annotation. The mapped reads were sorted by coordinates in BAM file format using SAMtools (24). Gene expression in counts was estimated using the RSubread function featureCounts (25). Gene expression in RPKM was estimated using the edgeR function rpkm (26). All the heatmaps were plotted using log2(1+rpkm) values scaled at row level. Differential expression analyses were performed by using edgeR (26) functions glmFit and glmLRT and using trended dispersions as dispersion input. Lowly expressed genes were filtered out before analysis by using edgeR function filterByExpr. An FDR-corrected p-value cut-off of 0.05 was used for all the DGE tests. For GO term and canonical pathway enrichment analysis, we used the ConsensusPathDB tool (27). GO term enrichment analyses were performed at GO term level 4 category to avoid redundancy. For TS vs CT comparison at TD30, GO term enrichment analyses at all GO term level categories (2–5) were also performed.
Correlation analysis between the transcriptome of BG organoids and the fetal brain
A gene expression read counts table for human developmental transcriptome was downloaded from http://development.psychencode.org/ (28). The top 200 differentially expressed genes between the fetal cortex and ventral telencephalon/striatum (8-21 PCW) were obtained by edgeR. Taking into account these top DEGs, Spearman’s correlations were performed between the BG organoids and the fetal brain cortex and striatum regions (8-21 PCW). Correlations were performed based on log2(1+cpm) values.
Curation of reference human fetal ventral telencephalic genes
We curated a set of reference genes reported to be enriched in fetal ventral telencephalon in various studies (Supplementary Table 3). Firstly, we retrieved the top 500 genes that are upregulated in the fetal ventral vs dorsal telencephalon (PCWs 8-12) from the BrainSpan developmental transcriptome dataset using the differential search tool at https://www.brainspan.org/rnaseq/search/index.html (Supplementary Table 3a). Additionally, we obtained the genes that were reported to be the MGE, LGE or CGE cluster markers by scRNA-seq analyses of the human fetal ventral telencephalon (Supplementary Table 3b) (29). We also included multiple genes that have been well characterized as ventral telencephalon markers by molecular assays (Supplementary Table 3c).
Results
Generation of Basal Ganglia-like Organoids
To obtain neural organoids enriched in ventral telencephalic precursors, neural rosettes generated from 7 genetically distinct CT iPSC lines were cultured free-floating in serum-free medium supplemented with FGF2 and EGF. From day 8 to 11, organoids were treated with the SHH agonist purmorphamine and FGF8 and from day 12 (TD0) onward were cultured in media promoting cell differentiation (see Methods and Figure 1a). Cells were harvested at TD0, TD11-14 and TD30 for analysis.
Initial immunocytochemical analyses of the BG organoids at TD11 showed protein expression profiles that are consistent with ventral telencephalic commitment. NKX2.1+ cells are expressed throughout the MGE and POA and are common progenitors for various types of interneurons derived from these regions, most prominently PV, SST and cholinergic interneuron subtypes, as well as for globus pallidus projection neurons (30–34) . Within each organoid, neuronal cells expressed NKX2.1 with the neuroepithelial marker N-Cadherin (Figure 1b) and the cholinergic synthetic enzyme CHAT (Figure 1d), consistent with an MGE/POA fate (31, 35), whereas progenitors expressed the radial glial protein NESTIN and the pan-neural transcription factor (TF) SOX1, as well as the cell cycle protein Ki67, consistent with their active proliferative stage (Figure 1c). In a subset of cells, NKX2.1+ colocalized with the GABAergic synthetic enzyme GAD67 (Figure 1e) highlighting their potential to develop into GABAergic interneurons and projection neurons.
We next compared cell type-specific markers in BG and cortical (CTX) organoids at TD11 and TD33 using unbiased stereological analyses. We found that, at both time points, over 55% of the BG organoids’ cells expressed NKX2.1 and over 20% expressed the cholinergic synthetic enzyme (CHAT). In contrast, only 4% of the CTX organoids’ cells expressed NKX2.1 and less than 2% expressed CHAT (Figure 1f–k). In addition, nearly 20% of cells in BG organoids co-expressed NKX2.1 and CHAT at both TD11 and TD33, indicating differentiation into a cholinergic fate. Instead, NKX2.1 and CHAT were co-expressed in less than 1% of cells in CTX organoids (Figure 1f–k). Further, 20% of BG organoids’ cells expressed DLX1-2, whereas only 3% of the cells expressed DLX1-2 in CTX organoids (Figure 1l–n). Conversely, the number of cells that were positive for PAX6 and TBR1, TFs expressed in cortical progenitors and neurons, respectively, were decreased in BG organoids (Figure 1f–g, l–n). These data provide evidence for a ventral telencephalic phenotype in the BG organoids at both early and more mature time points.
To further confirm the ventral identity of our BG organoid preparations, we compared their transcriptome to that of CTX organoids. We performed bulk RNA-sequencing (RNA-seq) on CTX and BG organoids at TD14, differentiated in parallel from 6 genetically distinct CT iPSC lines. We detected a total of 4694 differentially expressed genes (DEGs) between CTX and BG organoids (FDR-corrected p-value ≤ 0.05; n=6 iPSC lines, Supplementary Table 4). The relative expression levels of selected DEGs are shown in Figure 1r–s. The data show that the expression of canonical cortical genes, such as PAX6, TBR1, BRN2/POU3F2 and EMX2, was higher in CTX organoids when compared to their BG counterparts (Figure 1r), whereas the expression of canonical ventral telencephalic genes was higher in BG organoids (Figure 1s), including genes broadly expressed in all ventral telencephalic progenitors (GLI1, SHH), as well as genes specific for LGE (FOXP1, DRD2, PENK) (36), CGE (PROX1,VIP) (37, 38), and MGE/POA (NOS/SST/CHAT) (37, 38).
Real-time qPCR analyses confirmed appropriate ventral forebrain gene expression in the BG organoids (Figure 1t,u). For instance, TFs expressed in cortical progenitors (PAX6, BRN2, EMX2) and neurons (TBR1) were significantly downregulated in the BG organoids (Figure 1t), whereas genes expressed in LGE-derived medium spiny neurons (DARPP32), MGE-derived interneurons (NXK2.1, NOS-1, NPY, SST) and cholinergic interneurons (ISL1, CHAT, SLC18A3, SLC5A7), were significantly upregulated in BG organoid preparations (Figure 1u). Overall, the data indicate that progenitors and neurons resembling LGE, MGE and POA fates are represented within the BG organoids (6–8).
We then correlated BG organoids’ transcriptional profile with that of the human fetal telencephalon (8-21 post-conceptional age in weeks, PCW) obtained from the BrainSpan dataset (28). The BG organoids correlated best with the fetal ventral telencephalon, particularly the ganglionic eminences of 8-9 PCW (Supplementary Figure 3).
Finally, neurons within BG organoids developed a more mature morphology upon long-term culture. After six months in vitro (TD180-TD200), organoids were transduced with a SYN-GFP lentivirus, in which the GFP is driven by the human Synapsin promoter (39) and visualized via two-photon microscopy (Supplementary Figure 4). We observed robust neurite extensions as well as the development of putative dendritic spines (n=4 organoids, from 2 biologically different iPSC lines). Organoids survived up to 6 months in 5 biological replicates and up to a year in 2 biological replicates (data not shown).
Modeling Tourette Syndrome using BG organoids: progenitor stage
We utilized our BG organoid protocol (Figure 1a) to compare the neurodevelopment of the ventral telencephalon between TS individuals (n=5) and healthy CT (n=11, Supplementary Table 1d). To accomplish this, we recruited five individuals with TS (1 female and 4 males) and generated iPSC lines (Supplementary Table 1b). The five TS cases, all adults, had current Yale Global Tic Severity Scale (YGTSS) scores that ranged between 11 and 30. BG organoids from two different iPSC lines derived from each of the 5 TS probands were compared to those derived from 11 adult CT individuals (2 females and 9 males) (Supplementary Table 1d). CT and TS BG organoids were cultured in parallel to minimize batch effects.
We first examined an intermediate organoid stage, TD14, when both progenitors and post-mitotic cells are identifiable. Immunostaining for NKX2.1 and PAX6, two TFs expressed by progenitors of the MGE and dorsal cortex, respectively, revealed that BG organoids from the TS individuals had decreased NKX2.1 together with an increase in PAX6 protein expression when compared to controls (Figure 2a and Supplementary Figure 5).
Figure 2: Decreased ventral and increased dorsal patterning in TS BG organoids at TD14.
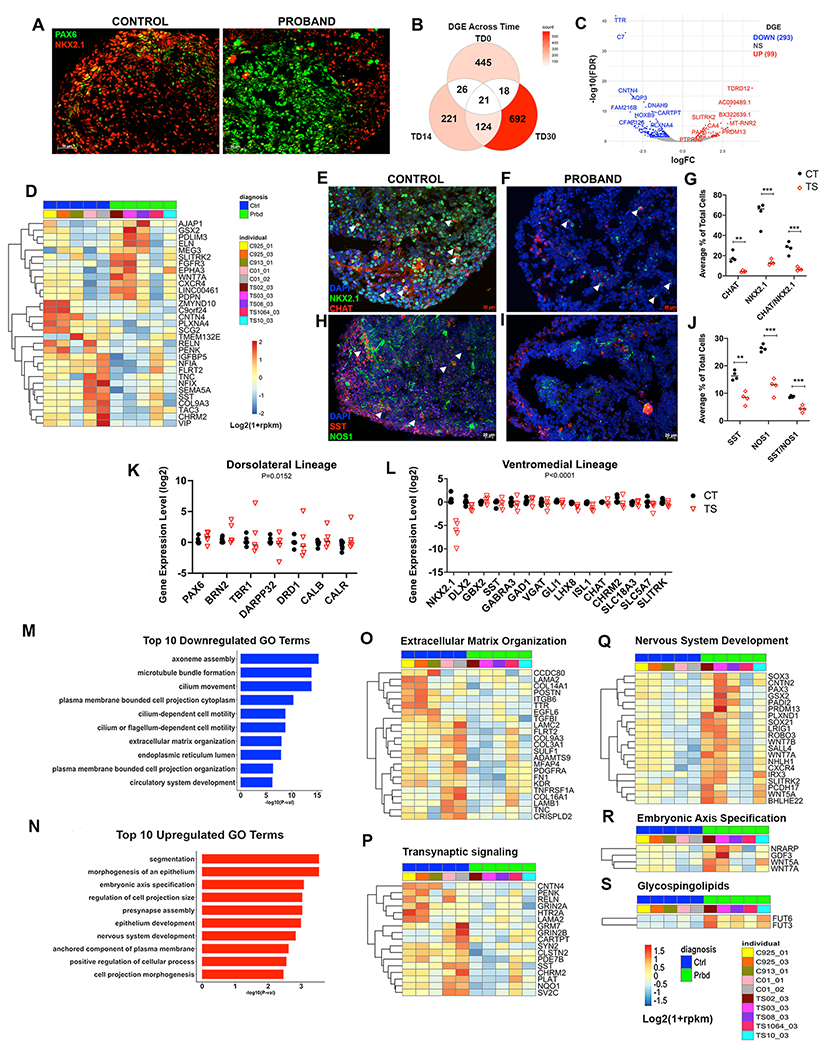
(a), Representative immunostaining shows reduced NKX2.1 and increased PAX6 expression in TS-derived BG organoids; for additional lines see Supplementary Figure 5. (b), Venn diagram showing numbers of DEGs between TS and CT organoids across TD0, TD14 and TD30 by bulk RNA-seq. (c), TS vs CT DEGs for TD14 shown by volcano plot with significance by FDR-corrected p value (FDR) on Y axis and fold change (log2FC) on X axis. Top significant up- and down-regulated genes were labeled. n=5 CT; n=5 TS. (d), Heatmap showing relative expression of a reference set of ventral gene markers (i.e., ventrally upregulated genes in human fetal brains, see methods and Supplementary Table 3a–c) that are also TS vs CT DEGs in BG organoids at TD14 (P<0.05). Log2(1+rpkm) values were scaled in each row. (e-j), Immunostaining and unbiased stereological quantification comparing numbers of NKX2.1+ progenitors and CHAT+, SST+, or NOS+ interneurons in CT vs TS BG organoids. (g,j), ** p <0.01, *** p <0.001 by unpaired t-test when comparing TS vs CT organoids (n= 4 TS and 4 CT iPSC lines, 4 organoids per individual, 2 sections each). (k,l), RT-qPCR at TD14 showing that TS BG organoids (red triangles) exhibit loss of NKX2.1+, GABA and cholinergic gene expression, and increased expression of LGE and dorsal cortex markers as compared to CT (black circles). Data points are plotted using a Log2 scale. Significance between TS vs CT organoids in each gene set by two-way ANOVA; n=5 TS individuals (2 clones each = 10 iPSC lines); n=11 CT individuals/iPSC lines. (m), Top 10 significantly downregulated GO terms (P<0.05) for the DEGs presented in panel (b). (n), Top 10 significantly upregulated GO terms (P<0.05) for the DEGs presented in panel (b). (o-p), Heatmaps showing expression of the DEGs associated with downregulated GO terms: (o) Extracellular Matrix Organization; (p) Trans-Synaptic signaling. (q-s), Heatmaps showing expression of the DEGs associated with upregulated GO terms: (q) Nervous System Development; (r) Embryonic Axis Specification; and (s) canonical pathway term Glycosphingolipid Synthesis. RNA sequencing was analyzed at TD14 for all heatmaps. All heatmaps show log2(1+rpkm) values, scaled in each row.
We next examined the BG organoids’ transcriptome by RNA-seq and analyzed DGE between the 5 TS lines and 5 CT lines (see Supplementary Table 5). Samples were submitted from different batches of organoid preparations; however, no batch effect was observed (Supplementary Figure 2). Across 3 different time points – TD0, TD14 and TD30 – a total of 1547 genes were differentially expressed between CT and TS organoids. The highest number of DEGs were seen at the earliest (TD0, 510 DEGs) and latest (TD30, 855 DEGs) time points (Figure 2b), with very little overlap between DEGs across times. Interestingly, one of the top down-DEGs at TD14 is CNTN4 (Figure 2c), a gene duplicated in rare cases of TS, along with its homolog CNTN6 (40, 41).
To assess whether these DEGs include genes that may be critical for ventral vs dorsal telencephalic patterning, we curated a set of reference genes that have been reported to be enriched in the human fetal ventral vs dorsal telencephalon, using the BrainSpan developmental transcriptome dataset and a recent scRNA-seq analysis of the fetal ventral telencephalon (29) (see Methods). Among these ventral telencephalic genes, approximately half were downregulated in TS vs CT organoids at TD14, whereas the rest were upregulated (Figure 2d). Interestingly, canonical MGE/CGE genes marking ventromedial BG fate (SST, CHRM2, VIP and RELN) were downregulated in TS organoids, whereas genes marking dorsolateral precursor identities in the basal ganglia (GSX2) or in the cortex (WNT7a) were upregulated (Figure 2d). Within the BG, GSX2 shows stronger expression in the dorsolateral LGE than in the MGE (42, 43).
Next, RT-qPCR was performed on an additional subset of regional canonical markers at TD14. This revealed an overall upregulation of LGE and MSN gene expression (i.g, DARPP32 and DRD1) (Figure 2k), as well as an overall reduction in MGE/POA identity, (i.g, TFs such as NKX2.1, DLX2, LHX8 and ISL1) across all 5 TS individuals (Figure 2l). Additionally, a reduction in receptors and transporters for GABAergic (GABRA3, GAD1, VGAT) and cholinergic (SLC18A3, SLC5A7, CHAT) interneurons and projection neurons – both derivatives of the MGE and POA – was observed (Figure 2l). Thus, among basal ganglia precursors, dorsolateral cell fates appear overrepresented in TS BG organoids as compared to CT, at the expense of ventromedial fates. Taken together, the immunocytochemistry, RNA-seq and RT-qPCR data suggest that at TD14, we observe the emergence of a potential impairment of ventromedial patterning in TS BG organoids.
NKX2.1+ progenitor cells of the MGE and POA are common progenitors for PV, SST and cholinergic interneuron subtypes (30–33). Unbiased stereological analyses comparing BG organoids derived from 4 CT individuals and 4 TS individuals revealed that NKX2.1+ cells accounted for ~65-70% of the total cell number in CT and only ~15% of the cells in TS (Figure 2e–g). Next, we assessed numbers of cholinergic interneurons, which express CHAT and are mostly derived from ventral progenitors of the POA and adjacent regions expressing LHX8 and NKX2.1 (44–46). While CHAT+ cells were ~20% of total cells in the CT individuals, they represented ~5% of all cells (P= 0.0017) in TS individuals (Figure 2e–g). As expected, similar decreases were obtained for neurons co-expressing CHAT and NKX2.1 (25% CHAT+/NKX2.1+ in CT, and ~10% CHAT+/NKX2.1+ in TS individuals, P=0.0005, Figure 2e–g). The reduction in CHAT+ cells in TS organoids at the protein level is consistent with reduction in the expression of CHAT and other markers for cholinergic progenitors and neurons (NKX2.1, LXH8, ISL1, CHRM2, SLC18A3, SLC5A7) observed at the RNA level (Figure 2l).
We also examined GABAergic interneuron subtypes co-expressing SST and NOS1 which also originate from the MGE (15). SST+ cells represented about ~18-20% of total cells in BG organoids from CT individuals and were reduced by 50% in TS individuals (Figure 2h–j). Similarly, NOS1+ interneurons were reduced by 50% across the same CT and TS lines (Figure 2h–j). Finally, double positive NOS1/SST+ cells were ~9% of total cells in CT BG organoids but only ~4% in TS organoids (Figure 2h–j) at TD14. Overall, these data suggest significant decreases in the production of SST+/NOS+ GABAergic as well as cholinergic interneurons in TS BG organoids when compared to CT.
Functional annotation enrichment analyses on TS vs CT DEGs (Supplementary Table 6) revealed an enrichment for cilia development and related subcellular components (axoneme, flagella and microtubule bundle formation) among downregulated DGEs (Figure 2m). Cilia are required for SHH signaling, which is upstream from NKX2.1 expression and MGE/POA development (47–50), revealing a remarkable functional convergence with disrupted ventromedial development in TS organoids. Additional GO categories enriched in downregulated DEGs at TD14 were Extracellular Matrix Organization (Figure 2o) and Trans-Synaptic Signaling (Figure 2p).
In contrast, GO terms related to Segmentation, Nervous System Development, and Embryonic Axis Specification were enriched among upregulated DEGs (Figure 2n). Some of the genes associated with these categories included genes expressed in dorsolateral BG, notably, GSX2, SLITRK2, and PAX3, as well as genes for ligands of the WNT signaling pathway (WNT5a, WNT7a), which are involved in dorsal CNS development (51) (Figure 2q). These enrichment analyses agree with the postulate that there is a potential mispatterning away from the typical ventral telencephalic fate in the TS-derived BG organoids.
Modeling Tourette Syndrome using BG organoids: neuronal stage
To evaluate how the cellular phenotypes found at TD14 evolved with time, we looked further into RNA-seq data at TD30. Intersecting TD30 DEGs with the reference fetal ventral telencephalic gene lists derived from bulk RNA-seq (Figure 3d) and scRNA-seq datasets (Figure 3e) revealed that most of the reference ventral telencephalic genes were downregulated in TS BG organoids. These included genes that were consistently downregulated both at TD14 and TD30 (SST, CNTN4), and additional ventromedial genes downregulated at TD30, such as SIX3, OTX2, GAD1, and multiple DLX genes.
Figure 3: Bulk RNA sequencing at TD30 show consistent disruption in ventral telencephalic development in TS.
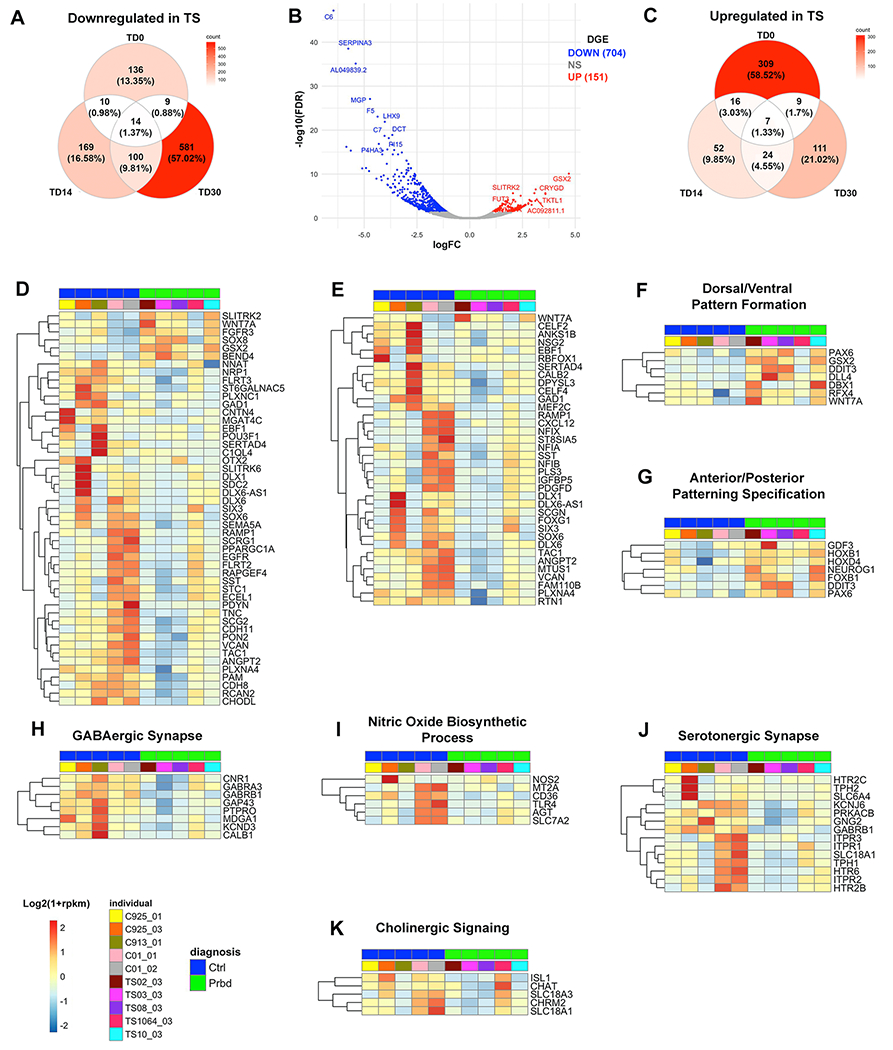
(a,c), Venn diagrams showing numbers of downregulated (a) and upregulated (c) TS vs CT DEGs in organoids across TD0, TD14 and TD30. (b), Volcano plot for TS vs CT DEGs at TD30. Top significant up- and down-regulated genes were labeled (n=5 CT; n=5 TS). (d,e), Heatmaps showing relative expression of a reference set of ventral gene markers [i.e., ventrally upregulated genes in human fetal ganglionic eminences by bulk RNA-seq (d) or scRNA-seq (e), see methods and Supplementary Table 3a] that are also DEGs in TS vs CT BG organoids at TD30 (P<0.05). Log2(1+rpkm) values were scaled in each row. (f,g), Heatmap of gene expression for the DEGs associated with the upregulated Dorsal/Ventral Pattern Formation and Anterior/Posterior Pattern Specification GO terms (P<0.05). (h-j), Heatmaps of gene expression for the DEGs associated with selected downregulated GO terms (P<0.05): (h), GABAergic Synapse; (i), Nitric Oxide Biosynthetic Process; (j), Serotonergic Synapse. (k); Manually curated list of genes associated with Cholinergic Signaling (n=5 TS; n=5 CT). All heatmaps show log2(1+rpkm) values which were scaled in each row.
GO terms related to Spinal Cord Development and D-V Pattern Formation were enriched among upregulated DEGs (Supplementary Figure 6 and Supplementary Table 6). Upregulated DEGs in intersection, including PAX6, NEUROG1, and WNT7A, all expressed in the developing cortex, and GSX2 and DBX1, expressed in the LGE and adjacent pallial regions (42, 52) (Figure 3f,g) confirmed a dorsal fate preference in TS BG organoids already evidenced at TD14. Remarkably, GSX2 is the top upregulated gene in TS (Figure 3b).
Top GO terms enriched among downregulated DEGs were related to Extracellular Matrix Organization and Cell Motility (Supplementary Figure 6c). The genes in intersection included collagen and laminin genes involved in basement membrane development (53) (Supplementary Figure 7a) as well as genes necessary for cellular migration and axon guidance (54, 55) (Supplementary Figure 7b).
Additional GO terms enriched among downregulated DEGs in TS organoids at TD30 were related to synaptic signaling, and particularly Serotonergic (e.g. HTR2A. HTR2C, HTR6) nitric oxide, and GABAergic signaling (Supplementary Figure 6d, Figure 3h–j, and Supplementary Table 6g–h). Finally, genes expressed in cholinergic synapses were also downregulated in TS (Figure 3k). These data agree with the decreased number of GABAergic, cholinergic and NOS+ interneurons in TS-derived BG organoids.
DEGs in Tourette Syndrome organoids align with Adult Human Tourette Syndrome Post-Mortem Data
We next determined the extent to which transcriptome abnormalities found in TS BG organoids overlap with those found in the TS adult basal ganglia, specifically the caudate/putamen (8). From the post-mortem DGE dataset, 20 downregulated genes were of particular interest because they reflected interneuron abnormalities in the disease (see Table 1 in Lennington et al (8)); of these 20 manually curated DEGs, 6 were found to be also downregulated in TS vs CT BG organoids at TD30, with CHRM2, a cholinergic receptor, being downregulated at both TD0 and TD30 time points (Table 1).
Table 1: DGE overlap between TS BG organoids and adult human TS postmortem data.
Manually curated gene list selected from DEGs associated with both postmortem striatum from TS individuals and TS BG organoids at TD14 and TD30. Shown are selected genes expressed within cholinergic (CHRM2, NTRK1), GABAergic (GAD1, GABRA3), SST), and serotonergic (HTR2C) neurons. FC, log2fold change in gene expression between TS and controls; FDR, false discovery rate corrected p value.
| Gene | Gene Name | Strategy | FC | FDR | P Value |
|---|---|---|---|---|---|
| CHRM2 | Cholinergic receptor, muscarinic 2 | Postmortem | −1.99 | 4.32E-04 | |
| Organoid - TD14 | −1.49 | 0.038 | 0.0005 | ||
| Organoid - TD30 | −1.44 | 0.008 | 0.0001 | ||
| NTRK1 | Neurotrophic tyrosine kinase receptor, type1 | Postmortem | −1.91 | 1.17E-03 | |
| Organoid - TD14 | 0.17 | 0.999 - NS | 0.676 | ||
| Organoid - TD30 | −1.66 | 0.05 | 0.001 | ||
| GAD1 | Glutamate decarboxylase 1 (brain, 67 kDa) | Postmortem | −1.64 | 1.21E-02 | |
| Organoid - TD14 | 0.14 | 0.999 - NS | 0.722 | ||
| Organoid - TD30 | −1.61 | 0.002 | 4.25E-05 | ||
| GABRA3 | Gamma-aminobutyric acid A receptor, alpha 3 | Postmortem | −1.96 | 5.85E-04 | |
| Organoid - TD14 | −0.93 | 0.128 - NS | 0.003 | ||
| Organoid - TD30 | −1.32 | 0.0159 | 0.0004 | ||
| SST | Somatostatin | Postmortem | −1.59 | 4.42E-03 | |
| Organoid - TD14 | −0.93 | 0.021 | 0.0002 | ||
| Organoid - TD30 | −2.07 | 6.69E-07 | 2.88E-09 | ||
| HTR2C | 5-hydroxytryptamine (serotonin) receptor 2C | Postmortem | −1.62 | 7.44E-03 | |
| Organoid - TD14 | −0.51 | 0.999 - NS | 0.289 | ||
| Organoid - TD30 | −2.31 | 0.0001 | 1.07E-06 |
We also performed a more systematic comparison of TS vs CT DGE between the BG organoids and the post-mortem BG. We identified 108 genes that were differentially expressed in both organoid and post-mortem analyses (see Methods; Supplementary Figure 8). Many of these genes showed differential expression in the same direction across the two systems, including SST, cholinergic genes (CHRM2 and ISLR), GABRA3, and the serotonin receptor HTR2C.
Mechanism of mispatterning in Tourette Syndrome
To understand the mechanisms of the deficit in ventral telencephalic development observed in the TS BG organoids and their potential involvement in the etiology of TS, we focused on the earliest time point (TD0), and on the SHH pathway because of its central involvement in ventral patterning in the CNS.
In the absence of SHH signaling, the truncated forms of the TFs GLI2 and GLI3 repress SHH target genes. In the ventral telencephalon, the SHH ligand acts through the Patched 1 (PTCH1) receptor to de-repress Smoothened (SMO) within cilia (47, 49, 50) resulting in translocation of the GLI1 TF to the nucleus and activation of SHH downstream target genes, primarily NKX2.1, ensuing MGE fate (Figure 4a). We therefore asked whether altered SHH signaling through any of these components could explain the organoid mispatterning phenotype observed in TS -derived BG organoids.
Figure 4: Upregulation of GLI genes and repressor proteins at TD0 in TS-derived BG organoids.
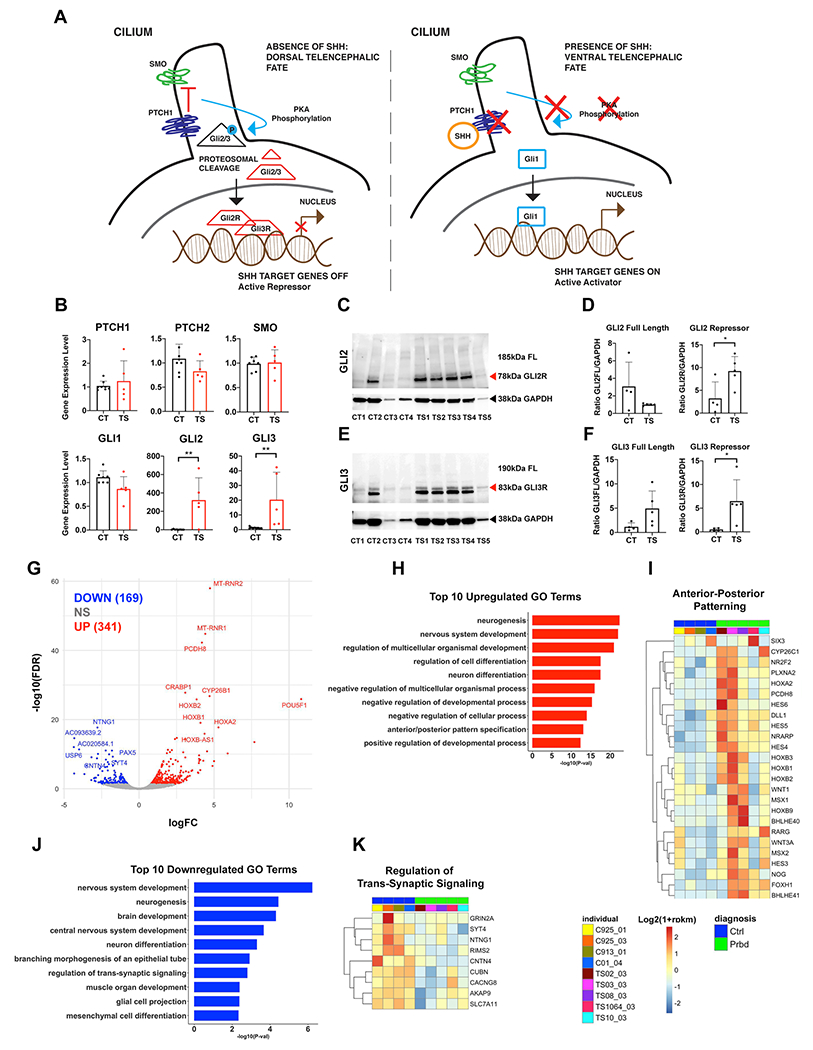
(a), Schematic diagram of SHH signaling in dorsal and ventral contexts. (b), qPCR examining expression levels of key SHH signaling players in BG organoids at TD0. (c-f), Western blots for GLI2 and GLI3 repressor proteins in CT- and TS-derived BG organoids. See Supplementary Figure 9 for additional western blots with full length (FL) GLI2 and GLI3 protein expression. Protein bands were quantified by ImageJ software (n=4 CT; n=5 TS iPSC lines). CT lines 1-4 are as follows: 7978-01 #1; RDH925-01 #10; RDH913-01 #1; 01-04 #28. TS lines 1-5 are as follows: TS 02-03 #4; TS 03-03 #12; TS 08-03 #7; TS 10-03 #22; TS 1064-03 #12. (g) Volcano plots of TS vs CT DGE at TD0. Topmost significant up and downregulated genes were labeled. n=5 CT; n=5 TS. (h), Top 10 GO terms enrichment for upregulated DGE (P<0.05) at TD0. (i), Heatmaps of expression levels for DEGs associated with Anterior-Posterior Patterning. (j), Top 10 GO terms enrichment for downregulated DGE (P<0.05) at TD0. (k), Heatmaps of expression levels for the DEGs associated with Regulation of Trans-Synaptic Signaling. For all TD0 RNA sequencing, n=5 TS; n=4 CT. All heatmaps show log2(1+rpkm) values that were scaled in each row.
We first assessed the expression level of molecules related to SHH signaling at TD0, which marks the 4th day post-exposure to SHH. While mRNA expression level of the surface SHH receptors PTCH1, PTCH2 and SMO or their downstream gene GLI1 were not significantly altered, BG organoids from all 5 TS patients showed increased expression levels of the GLI2 and GLI3 genes by qPCR (Figure 4b). To examine levels of the repressor forms of the GLI2 and GLI3 proteins, we performed western blots on organoids from the TS and CT cohorts (Figure 4c–f). Protein levels for the truncated repressor forms of GLI2 (~78 kDa) (Figure 4c,d) and GLI3 (~83 kDa) (Figure 4e,f and Supplementary Figure 9) were significantly increased in TS vs CT organoids. The expression of GLI2 and GLI3 proteins in CT are expectedly low, in line with typical ventral telencephalic development.
Anterior/Posterior Pattern Specification is among the top 10 GO annotations for upregulated DGE at TD0 (Figure 4h,i), with several HOX and WNT genes in intersection, which are among the top overexpressed genes in TS vs CT at this time point (Figure 4g,i). These genes are mostly expressed in the dorsal neural tube. Hence, the increase in GLI2 and GLI3 mRNA and repressor proteins, as well as the upregulation of dorsal neural tube genes in TS BG organoids, suggest that the dorsal fate preference observed in TS organoids at TD14 and TD30 is already present at TD0 and potentially linked with dysregulation of the SHH pathway.
To further explore the mechanisms of mispatterning we revisited cilia-related functional annotation for TD14 RNA-seq data (Figure 2m). Expression levels of genes associated with cilia-related GO terms were displayed in a single heatmap (Figure 5a). While variability in the expression of cilia-related genes existed across CT organoids, cilia-related genes were largely downregulated in TS. We then performed immunostaining in BG organoids for AC3 (Figure 5b), a protein localized in cilia. We found that while cilia were distributed broadly throughout the BG organoids in CT, cilia were located mostly on the apical side of cells abutting the lumens in TS BG organoids (Figure 5b). Lumens are a general morphological feature typical of dorsal fate in organoids, and they are not seen in CT BG organoids under normal conditions. Thus, the different cytoarchitecture of organoids and different cilia location represent additional evidence of altered ventral fate in TS organoids.
Figure 5: Development of cilia in TS.
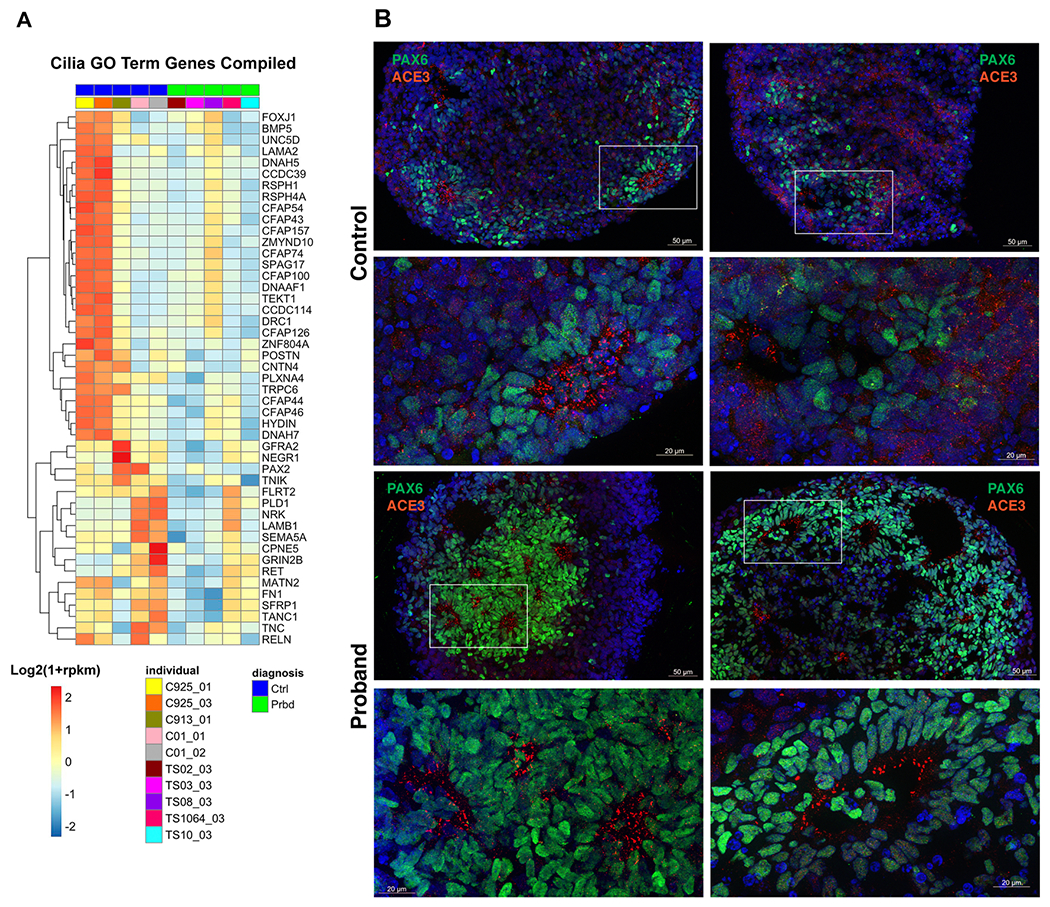
(a) Heatmap of gene expression levels for the DEGs associated with the union of all ciliary GO terms from Figure 2m at TD14 [axoneme assembly, cilium movement, cilium-dependent cell motility, plasma membrane bounded cell projection organization]. All heatmaps show Log2(1+rpkm) values scaled in each row. For all TD14 RNA sequencing, n=5 TS; n=5 CT. (b) Immunostaining for the AC3 cilia protein (red) and the radial glial progenitor cell marker PAX6 in CT and TS organoids show differences in the location of cilia between the 2 groups. Corresponding low and high magnification images in each row, where white boxes show the location of high magnification images. Note higher levels of PAX6 staining in TS organoids, in consistence with data shown in Fig. 2a.
Discussion
Our work models Tourette Syndrome using an organoid approach and demonstrates a mispatterning of the ventral telencephalon in TS BG organoids, with a relative lack of ventromedial progenitors (MGE/POA) accompanied by enhanced dorsolateral (LGE and cortex) fates. This results in developmental loss of MGE-derived interneurons, and potentially globus pallidus projection neurons, suggesting that the interneuron loss noted in the TS adult postmortem BG is a potential consequence of an inherent tendency of the TS BG organoids to undergo different regional specification. These findings contribute to our understanding of TS etiology and offer a developmental lens to the pathologies currently associated with TS.
Previous reports have pointed to the BG as a source of neural impairment and the origin of symptom in TS. Tic release elicits prominent activity in the BG (56, 57) and neuroimaging data suggest decreases in striatal volume in TS patients (5, 58). Moreover, there are decreases in interneuron mRNA and proteins in the postmortem TS striatum, including cholinergic (6, 7), parvalbumin (6, 7) and SST/NOS/NPY subtypes (8), suggesting loss and/or dysfunction of interneurons in adult TS patients. However, whether these findings are the consequences of early developmental disruptions or are acquired postnatally has remained unknown.
The current study is based on a cohort of unremitted adult TS individuals with variable tic severity. We observed that the MGE was less represented in the TS BG organoids when compared to CT, with interneuron deficiency on both RNA and protein levels, including the NOS, SST GABAergic as well as cholinergic interneurons. This loss included receptors, transporters, biosynthetic enzymes, and downstream transducers specific to interneuron subpopulations. We also observed deficits in serotonergic gene expression, which is intriguing given that serotonergic signaling has been previously implicated in TS (59–62). When comparing DGE of the TS BG organoids with the DGE of adult TS postmortem tissue, we found convergence across the same neuronal categories – GABAergic, cholinergic and serotonergic. This overlap both underscores the role of these neuronal populations in TS and confirms an early developmental origin for these findings.
Alongside the blunted MGE patterning and maturation there was an enhanced dorsolateral gene expression in TS BG organoids, including transcripts normally expressed in the cortex (PAX6, WNT7A, POU3F2, TBR1), and the LGE (GSX2, a top upregulated gene in TS organoids). We suggest that the underlying mechanism for this mispatterning of the ventral telencephalon in TS is the increases in GLI2 and GLI3 gene products, which, in their processed forms, repress SHH activity downstream of receptor activation and GLI1 expression, and typically specify dorsal territories of the brain (63). A proof of concept of this hypothesis is provided by one of the TS individuals utilized in this study, case TS1064-03. This individual (TS5 on Figure 4c,e western blots) exhibited the least GLI2R and GLI3R protein overexpression across the TS cohort, and, at the same time, the mildest reduction in ventromedial gene expression compared to the other 4 TS individuals (Figures 2d and 3d,e) as well as the least reduction in GABAergic, serotonergic and cholinergic neuron gene expression (Figure 3h–k) . Interestingly, case TS1064-03 is also the individual with mildest current adult tic severity in our cohort (YGTSS of 11, Supplementary Table 1c). This suggests that the degree of GLI2/3 repressor over-expression may dictate the degree of BG mispatterning in TS individuals, and that this disruption is related to tic severity. Another potential mechanism for the decrease in MGE fate is reduced FGF8 signaling, as loss of function FGF8 mutations in mouse result in decreased NKX2.1 expression (64).
Several cilia-related genes were downregulated in TS organoids. Cilia – into which the SHH-responsive transmembrane receptors are imbedded – are known to facilitate the SHH signaling cascade. GLI2 and GLI3 accumulate in cilia within minutes upon SHH stimulation (65, 66), suggesting that cilia dysfunction might lead to their altered trafficking, processing and nuclear localization (67, 68), contributing to BG mispatterning during BG development. While this is the first report to implicate cilia in TS etiology, primary cilia have been implicated in Fragile X syndrome (69) and several other neurological and systemic disorders.
In summary, our findings delineate a likely pathophysiology for TS. The decreased ventral patterning and MGE development, likely due to altered GLI2/3 protein expression, offers a potential explanation for the reduced interneuron generation, and accounts for their loss seen in the TS post-mortem BG. We anticipate that the degree of this mispatterning might vary across individuals with TS, as seen from the range of phenotypes observed in this study. These data illustrate the importance of a developmental human model system to detect convergent pathophysiology, and offer a bioassay for investigation of other disorders affecting the BG.
Supplementary Material
Acknowledgments
We wish to thank the participants who donated samples and time to our study.
We want to thank Jeremy Schreiner and Livia Tomasini for technical assistance and Scott Norton for guidance in performing the transcriptome analyses.
We thank Michael Higley and Riccardo Parra for use of the dual photon microscope. We are grateful to Drs. Christopher Pittenger and Nenad Sestan for comments and suggestions on an earlier version of this work.
We acknowledge the Yale Center for Clinical Investigation for clinical support in obtaining the biopsy specimens, the Yale Stem Cell Center for the generation of the iPSC lines, and the Yale Center for Genome Analysis for library preparation and sequencing. We thank Dr. Pamela Ventola, Dr. Katarzyna Chawarska and Dr. Kevin Pelphrey for help with recruitment of control subjects.
The recruitment and production of iPSC lines for control subjects were supported by the following grants: MH087879, MH089176, and MH109648 from the National Institutes of Health, and by the Simons Foundation. The recruitment and production of iPSC lines for TS subjects were supported by MH118453 from the National Institutes of Health, by the NARSAD-Brain and Behavior Research Fund and by the Tourette Association of America.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References:
- 1.Eblen F, Graybiel AM. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J Neurosci. 1995;15(9):5999–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Maia TV, Marsh R, Colibazzi T, Gerber A, Peterson BS. The neural circuits that generate tics in Tourette’s syndrome. Am J Psychiatry. 2011;168(12):1326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. TINS. 1995;18:63–4. [PubMed] [Google Scholar]
- 4.Bloch MH, Leckman JF, Zhu H, Peterson BS. Caudate volumes in childhood predict symptom severity in adults with Tourette syndrome. Neurology. 2005;65(8):1253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson BS, Thomas P, Kane MJ, Scahill L, Zhang H, Bronen R, et al. Basal Ganglia volumes in patients with Gilles de la Tourette syndrome. Arch Gen Psychiatry. 2003;60(4):415–24. [DOI] [PubMed] [Google Scholar]
- 6.Kalanithi PS, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, et al. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102(37):13307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kataoka Y, Kalanithi PS, Grantz H, Schwartz ML, Saper C, Leckman JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518(3):277–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lennington JB, Coppola G, Kataoka-Sasaki Y, Fernandez TV, Palejev D, Li Y, et al. Transcriptome Analysis of the Human Striatum in Tourette Syndrome. Biol Psychiatry. 2016;79(5):372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawaguchi Y. Physiological, morphological and histochemical characterization of three classess of interneurons in rat neostriatum. JNeurosci. 1993;13:4908–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109(31):12770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A. 2015;112(51):15672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545(7652):48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marton RM, Pasca SP. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020;30(2):133–43. [DOI] [PubMed] [Google Scholar]
- 15.Lim L, Mi D, Llorca A, Marin O. Development and Functional Diversification of Cortical Interneurons. Neuron. 2018;100(2):294–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoch RV, Clarke JA, Rubenstein JL. Fgf signaling controls the telencephalic distribution of Fgf-expressing progenitors generated in the rostral patterning center. Neural Dev. 2015;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bloch MH, Peterson BS, Scahill L, Otka J, Katsovich L, Zhang H, et al. Adulthood outcome of tic and obsessive-compulsive symptom severity in children with Tourette syndrome. Arch Pediatr Adolesc Med. 2006;160(1):65–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc. 2008;3(7):1180–6. [DOI] [PubMed] [Google Scholar]
- 19.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8(5):409–12. [DOI] [PubMed] [Google Scholar]
- 20.Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162(2):375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez-Coviella I, Berse B, Krauss R, Thies RS, Blusztajn JK. Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science. 2000;289(5477):313–6. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Coviella I, Follettie MT, Mellott TJ, Kovacheva VP, Slack BE, Diesl V, et al. Bone morphogenetic protein 9 induces the transcriptome of basal forebrain cholinergic neurons. Proc Natl Acad Sci U S A. 2005;102(19):6984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39(Database issue):D712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Y, Wang M, Mi D, Lu T, Wang B, Dong H, et al. Mouse and human share conserved transcriptional programs for interneuron development. Science. 2021;374(6573):eabj6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nobrega-Pereira S, Kessaris N, Du T, Kimura S, Anderson SA, Marin O. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59(5):733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126(15):3359–70. [DOI] [PubMed] [Google Scholar]
- 32.Corbin JG, Rutlin M, Gaiano N, Fishell G. Combinatorial function of the homeodomain proteins Nkx2.1 and Gsh2 in ventral telencephalic patterning. Development. 2003;130(20):4895–906. [DOI] [PubMed] [Google Scholar]
- 33.Magno L, Barry C, Schmidt-Hieber C, Theodotou P, Hausser M, Kessaris N. NKX2-1 Is Required in the Embryonic Septum for Cholinergic System Development, Learning, and Memory. Cell reports. 2017;20(7):1572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silberberg SN, Taher L, Lindtner S, Sandberg M, Nord AS, Vogt D, et al. Subpallial Enhancer Transgenic Lines: a Data and Tool Resource to Study Transcriptional Regulation of GABAergic Cell Fate. Neuron. 2016;92(1):59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luccardini C, Hennekinne L, Viou L, Yanagida M, Murakami F, Kessaris N, et al. N-cadherin sustains motility and polarity of future cortical interneurons during tangential migration. J Neurosci. 2013;33(46):18149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rataj-Baniowska M, Niewiadomska-Cimicka A, Paschaki M, Szyszka-Niagolov M, Carramolino L, Torres M, et al. Retinoic Acid Receptor beta Controls Development of Striatonigral Projection Neurons through FGF-Dependent and Meis1-Dependent Mechanisms. J Neurosci. 2015;35(43):14467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long JE, Cobos I, Potter GB, Rubenstein JL. Dlx1&2 and Mash1 transcription factors control MGE and CGE patterning and differentiation through parallel and overlapping pathways. Cereb Cortex. 2009;19 Suppl 1:i96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandberg M, Flandin P, Silberberg S, Su-Feher L, Price JD, Hu JS, et al. Transcriptional Networks Controlled by NKX2-1 in the Development of Forebrain GABAergic Neurons. Neuron. 2016;91(6):1260–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott BB, Lois C. Generation of tissue-specific transgenic birds with lentiviral vectors. Proc Natl Acad Sci U S A. 2005;102(45):16443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassett AS, Scherer SW. Copy Number Variation in Tourette Syndrome. Neuron. 2017;94(6):1041–3. [DOI] [PubMed] [Google Scholar]
- 41.Huang AY, Yu D, Davis LK, Sul JH, Tsetsos F, Ramensky V, et al. Rare Copy Number Variants in NRXN1 and CNTN6 Increase Risk for Tourette Syndrome. Neuron. 2017;94(6):1101–11 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin S, Madhavan M, Waclaw RR, Nakafuku M, Campbell K. Characterization of a new Gsx2-cre line in the developing mouse telencephalon. Genesis. 2016;54(10):542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yun K, Potter S, Rubenstein JL. Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development. 2001;128(2):193–205. [DOI] [PubMed] [Google Scholar]
- 44.Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506(1):16–29. [DOI] [PubMed] [Google Scholar]
- 45.Manabe T, Tatsumi K, Inoue M, Makinodan M, Yamauchi T, Makinodan E, et al. L3/Lhx8 is a pivotal factor for cholinergic differentiation of murine embryonic stem cells. Cell Death Differ. 2007;14(6):1080–5. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Y, Marin O, Hermesz E, Powell A, Flames N, Palkovits M, et al. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci U S A. 2003;100(15):9005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol. 2010;88(8):633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andreu-Cervera A, Anselme I, Karam A, Laclef C, Catala M, Schneider-Maunoury S. The Ciliopathy Gene Ftm/Rpgrip1l Controls Mouse Forebrain Patterning via Region-Specific Modulation of Hedgehog/Gli Signaling. J Neurosci. 2019;39(13):2398–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andreu-Cervera A, Catala M, Schneider-Maunoury S. Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol Dis. 2021;150:105236. [DOI] [PubMed] [Google Scholar]
- 50.Park SM, Jang HJ, Lee JH. Roles of Primary Cilia in the Developing Brain. Frontiers in cellular neuroscience. 2019;13:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, et al. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279(1):155–68. [DOI] [PubMed] [Google Scholar]
- 52.Arai Y, Cwetsch AW, Coppola E, Cipriani S, Nishihara H, Kanki H, et al. Evolutionary Gain of Dbx1 Expression Drives Subplate Identity in the Cerebral Cortex. Cell reports. 2019;29(3):645–58 e5. [DOI] [PubMed] [Google Scholar]
- 53.Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab. 2017;37(10):3300–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hartwig C, Veske A, Krejcova S, Rosenberger G, Finckh U. Plexin B3 promotes neurite outgrowth, interacts homophilically, and interacts with Rin. BMC Neurosci. 2005;6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu B, Chen C, Xue G, Moyzis RK, Dong Q, Chen C, et al. The SEMA5A gene is associated with hippocampal volume, and their interaction is associated with performance on Raven’s Progressive Matrices. NeuroImage. 2014;88:181–7. [DOI] [PubMed] [Google Scholar]
- 56.Bloch MH, Leckman JF. Clinical course of Tourette syndrome. Journal of psychosomatic research. 2009;67(6):497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leckman JF, Peterson B. The pathogenesis of Tourette’s syndrome: role od epigenetic factors active in early CNS development. Biol Psychiatry. 1993;34:425–7. [DOI] [PubMed] [Google Scholar]
- 58.Peterson B, Riddle MA, Cohen DJ, Katz LD, Smith JC, Hardin MT, et al. Reduced basal ganglia volumes in Tourette’s syndrome using three-dimensional reconstruction techniques from magnetic resonance images. Neurology. 1993;43(5):941–9. [DOI] [PubMed] [Google Scholar]
- 59.Heinz A, Knable MB, Wolf SS, Jones DW, Gorey JG, Hyde TM, et al. Tourette’s syndrome: [I-123]beta-CIT SPECT correlates of vocal tic severity. Neurology. 1998;51(4):1069–74. [DOI] [PubMed] [Google Scholar]
- 60.Muller-Vahl KR, Meyer GJ, Knapp WH, Emrich HM, Gielow P, Brucke T, et al. Serotonin transporter binding in Tourette Syndrome. Neurosci Lett. 2005;385(2):120–5. [DOI] [PubMed] [Google Scholar]
- 61.Wong DF, Brasic JR, Singer HS, Schretlen DJ, Kuwabara H, Zhou Y, et al. Mechanisms of dopaminergic and serotonergic neurotransmission in Tourette syndrome: clues from an in vivo neurochemistry study with PET. Neuropsychopharmacology. 2008;33(6):1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muller-Vahl KR, Szejko N, Wilke F, Jakubovski E, Geworski L, Bengel F, et al. Serotonin transporter binding is increased in Tourette syndrome with Obsessive Compulsive Disorder. Scientific reports. 2019;9(1):972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rallu M, Corbin JG, Fishell G. Parsing the prosencephalon. Nat Rev Neurosci. 2002;3(12):943–51. [DOI] [PubMed] [Google Scholar]
- 64.Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, et al. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133(9):1831–44. [DOI] [PubMed] [Google Scholar]
- 65.Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol Cell Biol. 2010;30(8):1910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A. 2009;106(51):21666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Wang C, Wu C, Cao T, Xu G, Meng Q, et al. PKA-mediated Gli2 and Gli3 phosphorylation is inhibited by Hedgehog signaling in cilia and reduced in Talpid3 mutant. Dev Biol. 2017;429(1):147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee B, Panda S, Lee HY. Primary Ciliary Deficits in the Dentate Gyrus of Fragile X Syndrome. Stem cell reports. 2020;15(2):454–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.