

Abstract
The enzyme nitrogenase couples adenosine triphosphate (ATP) hydrolysis to the multi-electron reduction of atmospheric dinitrogen into ammonia. Despite extensive research, the mechanistic details of ATP-dependent energy transduction and dinitrogen reduction by nitrogenase are not well understood, requiring new strategies to monitor its structural dynamics during catalytic action. Here we report cryogenic electron microscopy structures of the nitrogenase complex prepared under enzymatic turnover conditions. We observe that asymmetry governs all aspects of nitrogenase mechanism including ATP hydrolysis, protein-protein interactions, and catalysis. Conformational changes near the catalytic iron-molybdenum cofactor are correlated with the nucleotide-hydrolysis state of the enzyme.
One-Sentence Summary:
High-resolution structures of the nitrogenase complex provide insight into complex asymmetry and cofactor environment under turnover conditions.
Reduced forms of nitrogen are essential for the biosynthesis of amino- and nucleic acids as well as the production of fertilizers and many commodity chemicals (1). As the only enzyme capable of nitrogen fixation, nitrogenase catalyzes the eight-electron reduction of atmospheric nitrogen (N2) and protons (H+) into ammonia (NH3) and hydrogen (H2) (2, 3) (Fig. 1A). Nitrogenase is a two-component enzyme, which, in its most common form, consists of the iron protein (FeP, a γ2 homodimer) and the molybdenum-iron protein (MoFeP, an α2β2 heterotetramer) (Fig. 1B) (4, 5). Nitrogenase is distinct from most redox enzymes in its requirement for adenosine triphosphate (ATP) hydrolysis to enable the successive transfer of electrons and protons for substrate reduction (6, 7). The coupling of ATP hydrolysis to electron transfer (ET) is mediated by FeP, which forms a specific, nucleotide-dependent complex with MoFeP, and hydrolyzes two ATP molecules for the transfer of an electron to MoFeP (4–7). This part of nitrogenase catalysis is termed the “FeP cycle” (Fig. 1A) (2, 8). In the “MoFeP cycle” (Fig. 1A) (2, 8), the electrons from FeP are received by the P-cluster (an [8Fe:7S] complex) of MoFeP and relayed to the iron-molybdenum cofactor (FeMoco, a [7Fe:9S:C:Mo]-homocitrate complex) where N2 binding and reduction occur. Specific ATP-dependent interactions between FeP and MoFeP are necessary not only for interprotein ET, but also for gated ET between the P-cluster and FeMoco through long-distance conformational perturbations, the nature of which are not understood (6, 7).
Fig. 1. Catalytic cycle for nitrogenase and its structural characterization under non-turnover and turnover conditions.
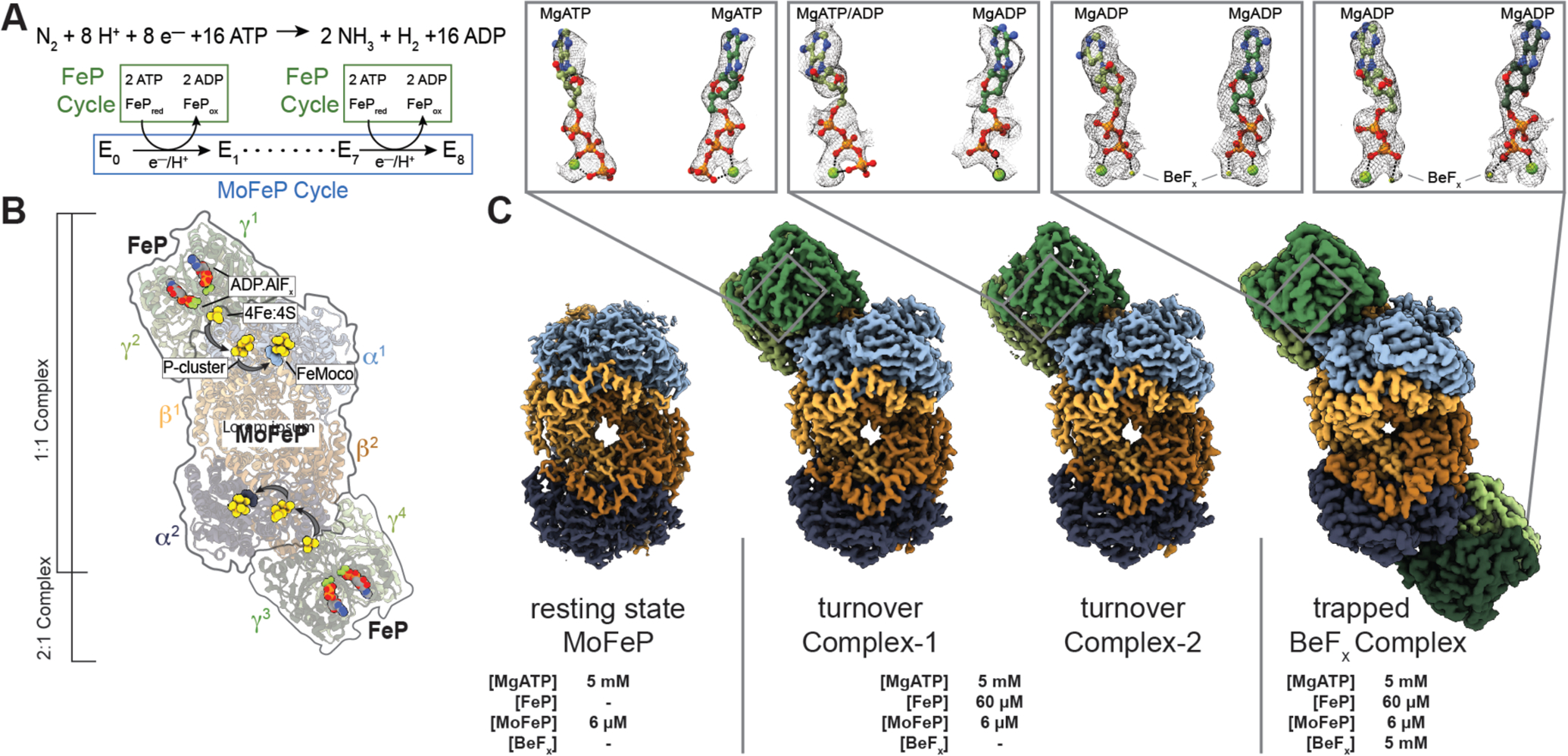
(A) Chemical reaction catalyzed by nitrogenase. There are eight FeP cycles in each MoFeP cycle. (B) Crystal structure (PDB ID: 1M34) (10) of the 2:1 FeP:MoFeP complex stabilized by MgADP.AlFx, showing the relative positions of the individual FeP (γ1 and γ2 and γ3 and γ4) and MoFeP (α1β1α2β2) subunits, the nucleotides, and the metalloclusters. The FeP subunits are shown in dark green (γ1 or γ3) and light green (γ2 or γ4), and the MoFeP subunits are highlighted in light blue (α1), dark blue (α2), light orange (β1) and dark orange (β2). Black arrows indicate the path of electron flow. (C) CryoEM maps of resting state MoFeP (rsMoFeP; ~1.8 Å resolution) obtained in the absence of FeP, and the turnover Complex-1 (t/oComplex-1; ~2.3 Å resolution) and turnover Complex-2 (t/oComplex-2; ~2.3 Å resolution) obtained under turnover conditions, and BeFx trapped FeP-MoFeP complex (~2.4 Å resolution). Coloring scheme for the subunits is the same as in (B). CryoEM maps for nucleotides are shown as a gray mesh and contoured at the following levels: rsMoFeP – 0.008, t/oComplex-1 – 0.17, t/oComplex-2 – 0.075. Magnified views of the nucleotides bound to the nitrogenase complexes and their corresponding cryoEM densities are shown in boxes.
Although the dynamic coupling between ATP hydrolysis and ET ultimately drives N2 fixation, it also creates experimental challenges for a mechanistic understanding of nitrogenase. First, substrates and inhibitors can only bind reduced forms of FeMoco, whose generation requires continuous turnover conditions that include ATP and reduced FeP (2, 3). Because MoFeP is also an inherent hydrogenase, such reduced forms of FeMoco promptly return to the resting state through H2 evolution upon termination of ATP hydrolysis (8, 9), rendering substrate-bound states of FeMoco too fleeting for structural characterization. Second, the necessity of continuous ATP hydrolysis for catalysis leads to a heterogeneous distribution of nitrogenase sub-states which are difficult to interrogate experimentally. These sub-states differ not only in the FeP-MoFeP complexation state and the extent of ATP hydrolysis within the FeP cycle, but also in the oxidation states of the metal cofactors and the reaction intermediates on FeMoco (E0-E8) within the MoFeP cycle (Fig. 1A–B).
Previous crystallographic studies have provided detailed views of the nucleotide-dependent conformations of the FeP-MoFeP complexes and outlined the path of ET in nitrogenase (Fig. 1B) (10, 11). Yet, these static views—obtained under non-turnover conditions and constrained by crystal lattice—showed no variation in the structure of MoFeP, providing little insight into the mechanism of ATP/FeP-mediated redox events within this protein. Recent crystallographic studies of MoFeP and its vanadium analog, VFe-protein, revealed possible modes of ligand interactions with FeMoco and showed that the cofactor is capable of undergoing compositional changes (12–14). However, these structural snapshots were obtained using crystals that formed hours to days after the last enzymatic turnover reaction and their catalytic relevance remains an open question. In parallel, extensive freeze-quench spectroscopic studies of nitrogenase have characterized substrate- and intermediate-bound states of FeMoco (15, 16). Yet, these methods do not report on the atomic structure of the cofactor as a whole and provide little information regarding how the local cofactor-ligand interactions are linked to global ATP/FeP-dependent structural dynamics of MoFeP. Consequently, we lack a detailed understanding of why and how ATP hydrolysis is used to drive N2 fixation and how catalysis at FeMoco proceeds. Clearly, new experimental approaches are needed to structurally interrogate nitrogenase during catalysis at or near atomic resolution to simultaneously characterize the ATP-dependent FeP-MoFeP interactions and the associated structural dynamics within each protein.
We sought to overcome some limitations of prior studies by using cryogenic electron microscopy (cryoEM) to directly visualize nitrogenase under enzymatic turnover conditions. We prepared cryoEM samples of Azotobacter vinelandii nitrogenase under high-electron flux turnover conditions (see Supplementary Methods), which contained a 10-fold molar excess of FeP over MoFeP and low ionic strength (≤25 mM NaCl) to favor the formation of electrostatically driven FeP-MoFeP complexes. We included sufficient MgATP and dithionite (5 mM each) to ensure that they are not depleted during turnover while minimizing background electron scattering. Turnover samples were prepared anaerobically under a N2 atmosphere in an anaerobic glovebox and flash frozen in liquid N2 within 15 s following initiation of turnover. For purposes of rapid manual freezing of cryoEM grids, these turnover samples were removed from the glovebox, thawed, applied to grids, blotted, and frozen within <15 s. The ~ 30-s total preparation period was sufficiently long to ensure that steady-state catalytic conditions were reached but short enough such that there was still remaining reductant and MgATP in solution (see Supplemental Methods). We collected a large cryoEM dataset (>15K movies) which yielded >4.5 million usable particles (figs. S1, S2; table S1). Through exhaustive 2- and 3-D classification and refinement, we isolated free MoFeP (~60%) and FeP particles (~5%) as well as FeP-MoFeP nitrogenase assemblies (~35%) from this heterogeneous mixture. We determined the structures of two conformationally distinct 1:1 FeP:MoFeP complexes under turnover conditions, designated “t/oComplex-1” and “ t/oComplex-2”, at ~2.3 Å resolution (Fig. 1C). Both complexes contain intact FeP and reduced, all-ferrous P-clusters (vide infra), indicating the lack of potential oxidation or damage by O2 to the clusters. As a reference, we also obtained a ~1.8-Å resolution cryoEM structure of resting-state MoFeP, termed “rsMoFeP”, using the same turnover conditions but in the absence of FeP (Fig 1C; fig. S3, table S2).
Given the C2 symmetry of MoFeP and the large separation (>65 Å) between the nearest clusters from the symmetry-related αβ subunits, it has long been assumed that the two αβ halves of MoFeP function independently from one another (2, 8). In support of this assumption, the crystal structures of various FeP-MoFeP complexes in different nucleotide-bound states largely possess a 2:1 FeP:MoFeP stoichiometry (Fig. 1B) (10, 11, 17). Unexpectedly, our turnover samples did not contain any particles that could be assigned to a 2:1 FeP:MoFeP complex (Fig. 1C; fig. S1). We considered the possibility that the exclusive observation of 1:1 complexes in our turnover samples could arise from an experimental artifact such as protein degradation or increase in ionic strength during cryoEM grid preparation. Therefore, we prepared a second set of turnover samples as above that also included 5 mM beryllium fluoride (BeFx), which is known to arrest ATP hydrolysis in a transition-like state to yield quasi-irreversible, solution-stable 2:1 and 1:1 FeP-MoFeP complexes (18). Accordingly, our cryoEM samples contained a large fraction of 2:1 FeP:MoFeP complexes alongside 1:1 species, but no detectable free MoFeP particles (figs. S4, S5; table S3). We determined the structure of the MgADP.BeFx-bound 2:1 FeP:MoFeP at ~2.4 Å resolution and found it to be isostructural to the crystal structure of the related MgADP.AlFx-bound 2:1 FeP:MoFeP complex (PDB ID: 1M34; Fig. 1C, fig. S5). These observations affirm that our cryoEM samples contain intact proteins and operate under native turnover conditions, in turn indicating that the 1:1 FeP:MoFeP stoichiometry is the predominant nitrogenase assembly state during catalysis.
Prior work using pre-steady-state kinetics measurements revealed that the extents of interprotein ET and ATP hydrolysis were approximately half of what would be expected if there were two independent FeP binding sites on MoFeP (19). Originally, such half-reactivity was attributed to either partial inactivity of FeP molecules (19) or to the possible existence of an alternative interaction mode between FeP and MoFeP (20). Recent studies favored a model of negative cooperativity within a 2:1 FeP:MoFeP complex, whereby one of the bound FeP molecules suppresses ATP hydrolysis by the other bound FeP and the redox activity of the opposite αβ half of MoFeP (21, 22). Our cryoEM structures instead suggest that half-reactivity and negative cooperativity in nitrogenase arise from MoFeP binding to only one FeP molecule at a time during turnover.
t/oComplex-1 and t/oComplex-2 were distinguished during cryoEM data processing based primarily on the structural variability of the FeP components. Thus, we first examined whether these differences are associated with the ATP-hydrolysis state of the two complexes. Earlier crystal structures identified at least three nucleotide-state-dependent FeP-MoFeP docking geometries (DG1, DG2 and DG3) and led to the hypothesis that FeP moves in a unidirectional fashion across the MoFeP surface during turnover (Fig. 2A) (6, 11). The DG1 state predominates in the absence of nucleotides but is also populated in the presence of ATP (23) and corresponds to an electrostatically guided encounter complex wherein FeP largely interacts with the β-subunit of MoFeP. DG2 is the activated nitrogenase complex in which ATP hydrolysis is coupled to interprotein ET, with FeP occupying the quasi-symmetric surface of MoFeP shared between α and β subunits (Figs. 1B, 2A) (10, 11). Finally, DG3 is formed by ADP-bound FeP and primarily utilizes the α-subunit surface of MoFeP (11). The cryoEM analysis of our turnover samples revealed that t/oComplex-1 and t/oComplex-2 were exclusively in DG2 (Fig. 2A), implying that this configuration has a higher stability and/or longer residence time relative to DG1 and DG3. Characteristic of a DG2 configuration, both complexes feature extensive interactions between FeP and MoFeP (buried surface areas >3600 Å2) and a short [4Fe:4S]-to-P-cluster edge-to-edge distance of ~15 Å, primed for rapid interprotein ET (Fig. 2B). In t/oComplex-1, both FeP γ subunits (γ1 and γ2) are occupied by ATP molecules with clear densities for the γ-phosphate groups and associated Mg2+ ions (Fig. 1C). By contrast, the γ1 subunit of t/oComplex-2 features an ATP molecule with weak density for the γ-phosphate, whereas the γ2 subunit is ADP-bound and the γ-phosphate is completely absent from the nucleotide binding pocket, indicative of asymmetry in ATP hydrolysis (Fig. 1C). This observation is consistent with the crystal structure of a mixed-nucleotide FeP-MoFeP complex, in which AMPPCP (a non-hydrolyzable ATP analog) and ADP were selectively bound to the γ1 and γ2 subunits, respectively (24). The differences in the nucleotide occupancies of t/oComplex-1 and t/oComplex-2 are reflected in their distinct FeP conformations (Fig. 2C), which is further corroborated by a principal component analysis of available FeP structures (fig. S6). Collectively, we infer that (1) our cryoEM structures report on active turnover states from the standpoint of ATP hydrolysis in FeP, (2) the hydrolysis of two ATP molecules in each FeP cycle occurs in a stepwise fashion, and (3) t/oComplex-1 and t/oComplex-2 correspond, respectively, to pre- and mid-ATP hydrolysis states of the nitrogenase complex that are populated along the catalytic reaction coordinate.
Fig. 2. Structural details of nitrogenase complexes characterized under turnover conditions.
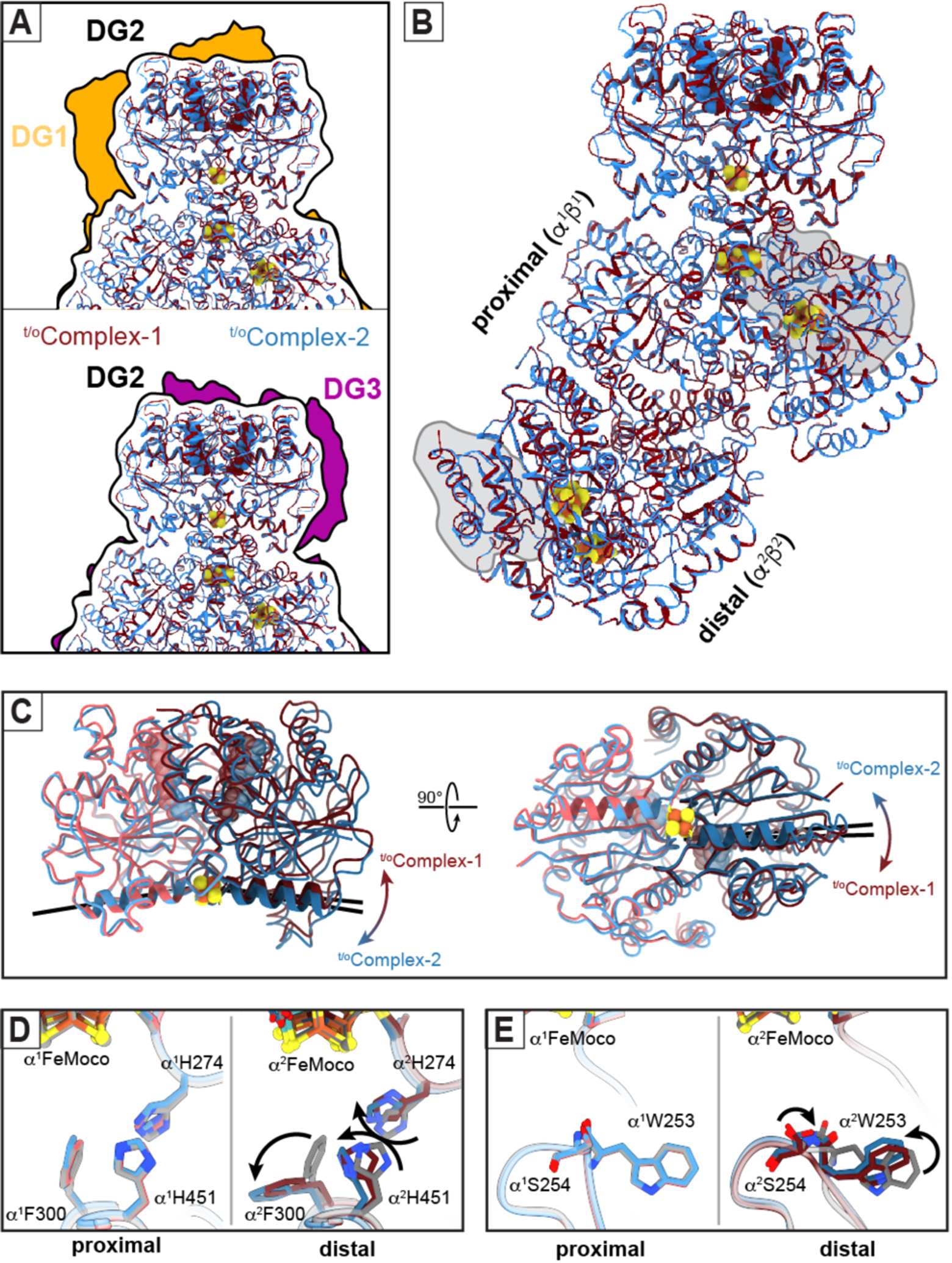
(A) Comparison of the FeP:MoFeP docking geometry (DG) in t/oComplex-1 (maroon) and t/oComplex-2 (blue) observed in the DG2 configuration (black outline) compared to DG1 (gold; PDB ID: 2AFH) and DG3 (purple; PDB ID: 2AFI) configurations characterized by X-ray crystallography (11). (B) Structural overlay of t/oComplex-1 and t/oComplex-2, indicating that the only large-scale conformational changes are observed in FeP. “Proximal” and “distal” refer to the α1β1 and α2β2 halves of MoFeP bound and not bound to FeP, respectively. The αIII domains of MoFeP are outlined in gray. (C) Structural overlay of the FeP components in t/oComplex-1 and t/oComplex-2, highlighting the nucleotide-dependent conformational differences (hinging – left, twisting - right) motions of the γ1 and γ2 subunits relative to one another during ATP hydrolysis. The axes of the γ100’s helices (residues γ98-γ112) that radiate from [4Fe:4S] cluster are shown as black lines to illustrate these conformational differences. (D,E) Residues in the vicinity of FeMoco (D, αH274, αF300, αH451; E, αW253) that have undergone conformational changes in the distal subunit (α2) of MoFeP during turnover. Movements between the resting-state (rsMoFeP) (gray) and the turnover structures (maroon and blue) are indicated with arrows.
The asymmetry present both in ATP hydrolysis and FeP-MoFeP interactions has important implications for the timing of ET events during catalysis. Compared to a concerted process, stepwise nucleotide hydrolysis by FeP would be expected to increase the lifetime of the activated DG2 complex and provide additional conformational states for orchestrating the multistep redox reactions that occur at FeMoco and the P-cluster (6, 24). Similarly, an alternating docking mechanism between FeP and MoFeP (as imposed by negative cooperativity between the αβ halves) would effectively slow down successive ET steps to a given αβ subunit, providing sufficient time for anticipated protein and metallocluster rearrangements during N2 reduction. This is consistent with the suggestion of Thorneley and Lowe that the slow kinetics of nitrogenase (kturnover ≈ 1 s−1) governed by FeP-MoFeP interactions may be a mechanistic imperative to favor N 2 fixation over the competing but less demanding H+ reduction (8).
Having analyzed t/oComplex-1 and t/oComplex-2 in the context of the FeP cycle, we next investigated if they exhibited any conformational changes in their MoFeP components that may be correlated with the ATP hydrolysis state. In both t/oComplex-1 and t/oComplex-2, the P-clusters are in their fully reduced, all-ferrous (PN) forms (fig. S7), and the protein backbone arrangements of MoFeP’s in both complexes are essentially indistinguishable from one another as well as from those in rsMoFeP and previously determined MoFeP crystal structures (Cα RMSD: 0.2 Å; Fig. 2B; fig. S8; table S4). We observed no large-scale conformational changes that could account for mechanical coupling and negative cooperativity between symmetry-related FeP docking surfaces on MoFeP, implicating the involvement of a dynamic allosteric mechanism (e.g., FeP-induced changes in MoFeP conformational entropy) (fig. S9) (25). Yet, a detailed inspection of the structures revealed that the FeP-free (i.e., “distal”) α2β2 half in both t/oComplex-1 and t/oComplex-2 possessed several features that distinguish it from the FeP-bound (i.e., “proximal”) α1β1 half and rsMoFeP.
First, there are several conserved residues (α2Trp253, α2His274, α2Phe300, α2His451) (26) in the vicinity of the distal FeMoco that adopt non-resting-state conformations (Figs. 2D,E). αTrp253 is particular in its cis-peptide bond to αSer254 and its position in a proposed substrate access channel from the protein surface to FeMoco (27, 28). The observed conformational flip in α2Trp253 leads to the diversion of this putative channel to an alternate face of FeMoco (Fig. 2E, fig. S10). α2His274, α2Phe300 and α2His451 sidechains appear to have undergone a concerted motion compared to their resting state (Fig. 2D), whereby α2His274 and α2Phe300 assume a similar configuration as that seen in the low-pH crystal structure of MoFeP (fig. S11)(29). This αHis274 configuration was proposed to form a water-bridged H-bond to a protonated belt sulfur (S5A) of FeMoco (29). Along these lines, the observed rearrangement of the α2His274 sidechain in the turnover complexes could be envisioned to stabilize a protonated FeMoco intermediate and/or to increase the reduction potential of the cofactor, thus promoting its reduction by the P-cluster.
Second, the cryoEM densities surrounding the α2His442 and homocitrate ligands to the Mo center of the distal FeMoco are considerably less well defined compared to their counterparts in the proximal αβ half and the residues in the vicinity (fig. S12) and cannot be modeled with the resting-state configurations of these ligands (Fig. 3). The reduction in map density is particularly pronounced for t/oComplex-2 (i.e., mid-ATP-hydrolysis) compared to t/oComplex-1 (i.e., pre-ATP-hydrolysis). These observations suggest that α2His442 and homocitrate are mobile during turnover and that Mo undergoes changes in inner-sphere coordination in a way that is correlated with the nucleotide hydrolysis state of FeP bound to the opposing αβ half of MoFeP. The substitution of Mo with V or Fe in alternative nitrogenases, the replacement of homocitrate with citrate, and alterations in H-bonding to homocitrate have been shown to substantially diminish N2 reduction activity and alter substrate specificity (2, 30–32). Indeed, the direct involvement of the Mo center in N2 reduction has been proposed early on (2, 33), although recent experimental findings have shifted the focus to the central Fe centers of FeMoco as being the primary sites for substrate activation (15, 16). Our cryo-EM observations provide evidence that the Mo-homocitrate moiety may also participate in dynamic structural transformations that accompany catalysis at FeMoco.
Fig. 3. Changes in the FeMoco environment observed during catalytic turnover.
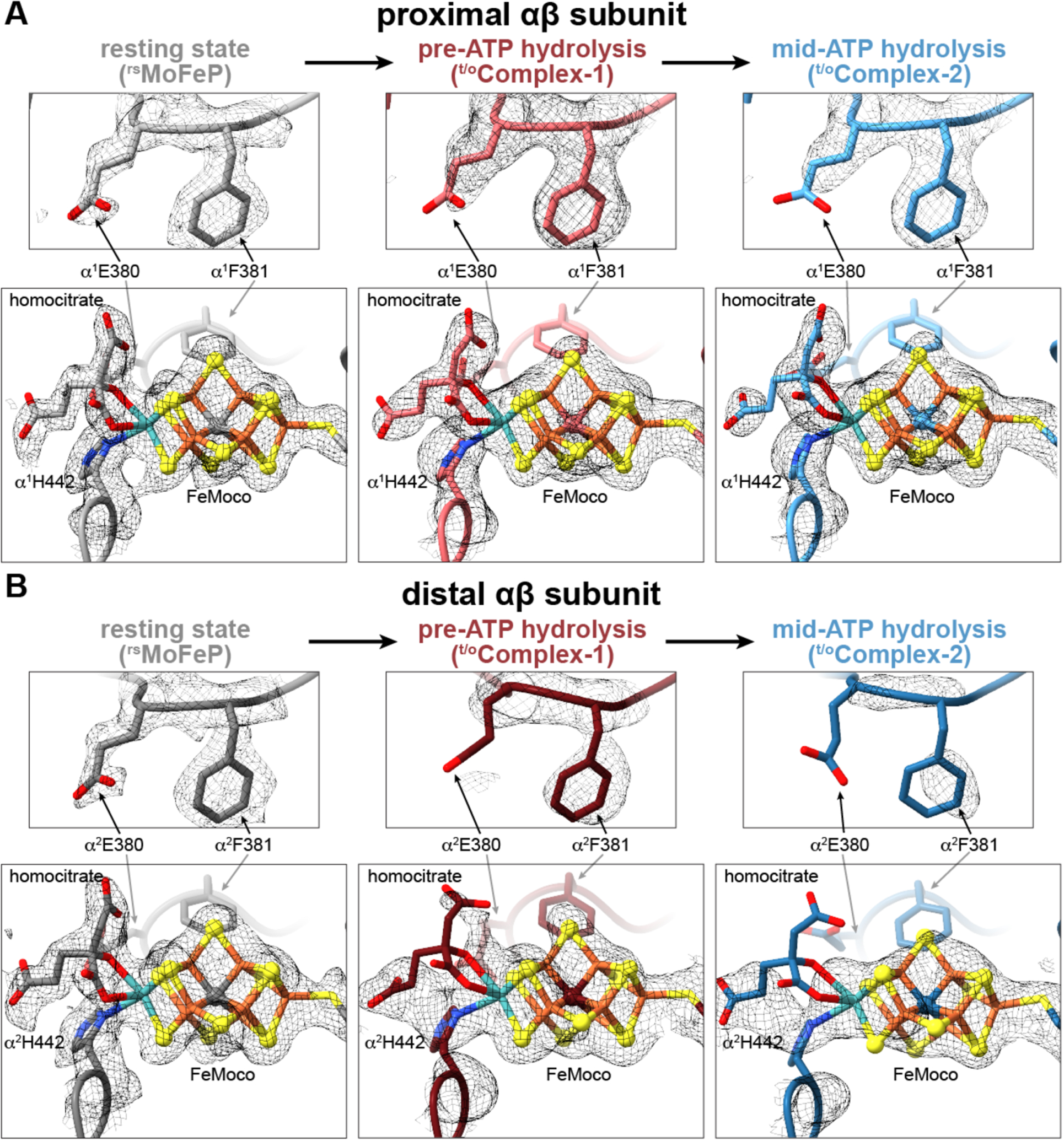
(A, B) Views of FeMoco and the nearby residues αE380 and αF381 in the proximal (A) and distal (B) αβ halves of MoFeP in rsMoFeP (gray), t/oComplex-1 (maroon), and t/oComplex-2 (blue) structures. CryoEM maps for each individual structure are shown as a gray mesh and contoured at the following levels: rsMoFeP – 0.008, t/oComplex-1 – 0.065, t/oComplex-2 – 0.075.
Third, portions of a large domain in the distal αβ subunit comprising residues α25–48 and α2378–403 (particularly in t/oComplex-2) possess increased mobility compared to the rest of the MoFeP (fig. S13). This so-called αIII domain forms a lid above FeMoco and includes residues αGlu380 and αPhe381 that form close contacts with FeMoco (Figs. 2B, 3). In resting state MoFeP, αIII is well-ordered, αGlu380 forms water-bridged H-bonds to the Mo-ligated αHis442 sidechain and homocitrate, and αPhe381 is in van der Waals contact with the labile belt sulfide S2B of FeMoco (Fig. 3A). By contrast, in the distal αβ halves of t/oComplex-1 and t/oComplex-2, the cryoEM densities for αIII and sidechains of α2Glu380 and α2Phe381 sidechains are either diffuse or entirely missing (Fig. 3B, fig. S13). This movement is likely coupled to the dynamics of the Mo ligands and FeMoco as a whole. αIII has also been shown to undergo major structural rearrangements associated with the insertion of FeMoco into MoFeP (34). Furthermore, αIII displays some of the highest temperature factors in MoFeP crystal structures and is positioned away from lattice contacts (table S5) (10, 11, 24, 29, 35), implying that it is inherently more flexible than other parts of MoFeP. Combined with this observation, t/oComplex-1 and t/oComplex-2 structures point to a possible role of αIII mobility in nitrogenase catalysis. Notably, αIII abuts the docking surface of MoFeP for MgADP-bound FeP in DG3 and may lie in the trajectory of FeP moving directionally across the MoFeP surface during ATP hydrolysis (11). Thus, a dynamic αIII domain could also provide a direct mechanical conduit between FeP and the proximal FeMoco, further linking the timing of nucleotide-dependent FeP-MoFeP interactions to redox transformations at FeMoco. The increased mobility of the αIII domain and the lack of a capping FeP could also allow more efficient substrate/molecular access to or exit from FeMoco.
Based on the available structures, it is not obvious 1) how FeP docking induces conformational changes in the distal αβ subunit of MoFeP over a distance of ≥80 Å or 2) which states of the MoFeP cycle (i.e., E0-E8) the observed nitrogenase complexes correspond to. It is safe to assume, however, that all copies of FeMoco in these complexes represent the more stable or longer-lived of the many catalytic intermediates present in the turnover solution. The density maps for the proximal FeMoco’s are essentially identical to those for the resting-state cofactor (Fig. 3). Thus, an assignment of E0 for the proximal cofactors is plausible, although E1-E4 are also reasonable as these states have been proposed as hydride-bound FeMoco intermediates (15, 16), which may be structurally indistinguishable from E0 at the current resolution of these cryoEM maps. At the same time, the obvious deviations between the densities of distal and proximal FeMoco’s indicate that the distal cofactors in t/oComplex-1 and t/oComplex-2 might represent an E x state or a mixture of Ex states that are different from the proximal cofactors and previous crystal structures solved in the resting state of FeMoco. It is thus possible that this structure represents a more advanced state in the MoFeP cycle (i.e., ≥E1) than the proximal FeMoco’s, and involves the participation of the Mo center. We also note that the overall map density for the distal FeMoco in t/oComplex-2 is less well-defined than in those in rsMoFeP, t/oComplex-1 or the proximal subunit in t/oComplex-2, which may further indicate conformational changes in the cofactor during turnover such as the displacement of belt sulfurs or ligand binding (12–14). In light of the asymmetry between the FeMoco’s in the two αβ halves, it is tempting to propose a “ping-pong”-type mechanism in which the cofactors proceed through each of the eight catalytic steps in an alternating fashion. This scenario would assign a dual role to FeP: 1) to deliver an electron to one αβ subunit of MoFeP and 2) to suppress FeP binding to the opposite αβ subunit while priming it for catalytic transformations through long-distance activation of electron, H+ and/or substrate access pathways to the distal FeMoco. Establishing whether such a mechanism is operative will require future studies, but our current work illustrates that it is possible to characterize nitrogenase under turnover conditions via cryoEM at near-atomic resolution, representing a critical step toward understanding the mechanism of this enigmatic enzyme in full structural detail.
Supplementary Material
Acknowledgments:
We thank K. Corbett, S. Narehood, J. Figueroa, and R. Subramanian for critical discussions, and members of the Tezcan and Herzik Labs for their assistance. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health grant R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. We also think members of UCSD’s CryoEM facility, the Stanford-Slac Cryo-EM Center (S2C2), and UCSD’s Physics Computing Facility for help in data collection, data processing, and computational support.
Funding:
National Institutes of Health grant R01-GM099813 (F.A.T.)
National Institutes of Health grant R35-GM138206 (M.A.H.)
National Institutes of Health grant T32-GM008326 (H.L.R., HPMN)
NASA grant 80NSSC18M0093 (F.A.T. and H.L.R., ENIGMA: Evolution of Nanomachines in Geospheres and Microbial Ancestors, NASA Astrobiology Institute Cycle 8)
Searle Scholars Program (M.A.H.)
CryoEM experiments were conducted at UCSD’s CryoEM Facilities as well as the Stanford-SLAC Cryo-EM Center (S2C2) supported by the NIH Common Fund Transformative High-Resolution Cryoelectron Microscopy program (U24 GM129541)
Footnotes
Data and materials availability:
Structural models have been deposited in the Protein Data Bank (PDB) with accession codes 7UT6 (rsMoFeP, C1 symmetry), 7UT7 (rsMoFeP, C2 symmetry), 7UT8 ( t/oComplex-1), 7UT9 (t/oComplex-2), and 7UTA (BeFx-trapped complex). The corresponding cryoEM maps are available at the Electron Microscopy Data Bank (www.ebi.ac.uk/emdb/) with accession codes EMD-26756 (rsMoFeP, C1 symmetry), EMD-26757 (rsMoFeP, C2 symmetry), EMD-26758 (t/oComplex-1 consensus MoFeP), EMD-26759 (t/oComplex-1 locally refined FeP), EMD-26760 (t/oComplex-1), EMD-26761 (t/oComplex-2 consensus MoFeP), EMD-26762 (t/oComplex-1 locally refined FeP), EMD-26763 (t/oComplex-2), and EMD-26764 (BeFx-trapped complex). All other data are available in the main text or the supplementary materials.
References and Notes
- 1.Smil V, Global population and the nitrogen cycle. Sci. Amer 277, 76–81 (1997). [Google Scholar]
- 2.Burgess BK, Lowe DJ, Mechanism of molybdenum nitrogenase. Chem. Rev 96, 2983–3012 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Thorneley R, Lowe D, Nitrogenase: substrate binding and activation. J. Biol. Inorg. Chem 1, 576–580 (1996). [Google Scholar]
- 4.Rees DC et al. , Structural basis of biological nitrogen fixation. Phil. Trans. Roy. Soc. A 363, 971–984 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Einsle O, Rees DC, Structural enzymology of nitrogenase enzymes. Chem. Rev 120, 4969–5004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rutledge HL, Tezcan FA, Electron transfer in nitrogenase. Chem. Rev 120, 5158–5193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seefeldt LC et al. , Energy transduction in nitrogenase. Acc. Chem. Res 51, 2179–2186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowe D, Thorneley R, The mechanism of Klebsiella pneumoniae nitrogenase action. Pre-steady-state kinetics of H2 formation. Biochem. J 224, 877–886 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorneley RN, Lowe D, The mechanism of Klebsiella pneumoniae nitrogenase action. Pre-steady-state kinetics of an enzyme-bound intermediate in N2 reduction and of NH3 formation. Biochem. J 224, 887–894 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmid B, Einsle O, Chiu H-J, Willing A, Yoshida M, Howard JB, Rees DC, Biochemical and structural characterization of the crosslinked complex of nitrogenase: Comparison to the ADP-AlF4 stabilized structure. Biochemistry 41, 15557–15565 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Tezcan FA et al. , Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science 309, 1377–1380 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC, Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science 345, 1620–1623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sippel D et al. , A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 359, 1484–1489 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Kang W, Lee CC, Jasniewski AJ, Ribbe MW, Hu Y, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase. Science 368, 1381–1385 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev 114, 4041–4062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seefeldt LC et al. , Reduction of substrates by nitrogenases. Chem. Rev 120, 5082–5106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiu H-J et al. , MgATP-bound and nucleotide-free structures of a nitrogenase protein complex between the Leu 127Δ-Fe-protein and the MoFe-protein. Biochemistry 40, 641–650 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Clarke TA, Yousafzai FK, Eady RR, Klebsiella pneumoniae Nitrogenase: Formation and Stability of Putative Beryllium Fluoride−ADP Transition State Complexes. Biochemistry 38, 9906–9913 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Ashby GA, Thorneley RNF, Nitrogenase of Klebsiella pneumoniae. Kinetic studies on the Fe protein involving reduction by sodium dithionite, the binding of MgADP and a conformation change that alters the reactivity of the 4Fe-4S centre. Biochem. J 246, 455–465 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke Thomas A., Fairhurst S, Lowe David J., Watmough Nicholas J., Eady Robert R., Electron transfer and half-reactivity in nitrogenase. Biochem. J 39, 201–206 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Danyal K et al. , Negative cooperativity in the nitrogenase Fe protein electron delivery cycle. Proc. Natl. Acad. Sci. USA 113, E5783–E5791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Q et al. , Mechanical coupling in the nitrogenase complex. PLoS Comp. Biol 17, e1008719 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owens CP, Katz FE, Carter CH, Luca MA, Tezcan FA, Evidence for functionally relevant encounter complexes in nitrogenase catalysis. J. Am. Chem. Soc 137, 12704–12712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tezcan FA, Kaiser JT, Howard JB, Rees DC, Structural evidence for asymmetrical nucleotide interactions in nitrogenase. J. Am. Chem. Soc 137, 146–149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motlagh HN, Wrabl JO, Li J, Hilser VJ, The ensemble nature of allostery. Nature 508, 331–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howard JB, Kechris KJ, Rees DC, Glazer AN, Multiple amino acid sequence alignment nitrogenase component 1: insights into phylogenetics and structure-function relationships. PloS One 8, e72751 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Igarashi RY, Seefeldt LC, Nitrogen fixation: the mechanism of the Mo-dependent nitrogenase. Critc. Rev. Biochem. Mol. Biol 38, 351–384 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Morrison CN, Hoy JA, Zhang L, Einsle O, Rees DC, Substrate pathways in the nitrogenase MoFe protein by experimental identification of small molecule binding sites. Biochemistry 54, 2052–2060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison CN, Spatzal T, Rees DC, Reversible protonated resting state of the nitrogenase active site. J. Am. Chem. Soc 139, 10856–10862 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eady RR, Structure−Function Relationships of Alternative Nitrogenases. Chem. Rev 96, 3013–3030 (1996). [DOI] [PubMed] [Google Scholar]
- 31.McLean PA, Dixon RA, Requirement of nifV gene for production of wild-type nitrogenase enzyme in Klebsiella pneumoniae. Nature 292, 655–656 (1981). [DOI] [PubMed] [Google Scholar]
- 32.Durrant MC, Francis A, Lowe DJ, Newton WE, Fisher K, Evidence for a dynamic role for homocitrate during nitrogen fixation: the effect of substitution at the α-Lys426 position in MoFe-protein of Azotobacter vinelandii. Biochem. J 397, 261–270 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickett CJ, The Chatt cycle and the mechanism of enzymic reduction of molecular nitrogen. J. Biol. Inorg. Chem 1, 601–606 (1996). [Google Scholar]
- 34.Schmid B et al. , Structure of a cofactor-deficient nitrogenase MoFe protein. Science 296, 352–356 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Einsle O et al. , Nitrogenase MoFe-protein at 1.16 Å resolution: a central ligand in the FeMo-cofactor. Science 297, 1696–1700 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Rutledge HL et al. , Redox-Dependent Metastability of the Nitrogenase P-Cluster. J. Am. Chem. Soc 141, 10091–10098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen HPM, McGuire KL, Cook BD, Herzik MA Jr., Manual Blot-and-Plunge Freezing of Biological Specimens for Single-Particle Cryogenic Electron Microscopy. J. Vis. Exp, (2022). [DOI] [PubMed] [Google Scholar]
- 38.Chan JM, Christiansen J, Dean DR, Seefeldt LC, Spectroscopic evidence for changes in the redox state of the nitrogenase P-Cluster during turnover. Biochemistry 38, 5779–5785 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Morrison CN, Kaiser JT, Rees DC, Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08A° resolution: comparison with the Azotobacter vinelandii MoFe protein. Acta. Cryst. D 71, 274–282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O, Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334, 940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayhew SG, The redox potential of dithionite and SO2− from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem 85, 535–547 (1978) [DOI] [PubMed] [Google Scholar]
- 42.Kimanius D, Dong L, Sharov G, Nakane T, Scheres SHW, New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J 478, 4169–4185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng SQ et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Zivanov J, Nakane T, Scheres SHW, Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner T et al. , SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol 2, 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Punjani A, Fleet DJ, 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol 213, 107702 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Sanchez-Garcia R et al. , DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol 4, 874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan YZ et al. , Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Igarashi RY, Seefeldt LC, Nitrogen fixation: the mechanism of the Mo-dependent nitrogenase. Crit. Rev. Biochem. Mol. Biol 38, 351–384 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Chovancova E et al. , CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol 8, e1002708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structural models have been deposited in the Protein Data Bank (PDB) with accession codes 7UT6 (rsMoFeP, C1 symmetry), 7UT7 (rsMoFeP, C2 symmetry), 7UT8 ( t/oComplex-1), 7UT9 (t/oComplex-2), and 7UTA (BeFx-trapped complex). The corresponding cryoEM maps are available at the Electron Microscopy Data Bank (www.ebi.ac.uk/emdb/) with accession codes EMD-26756 (rsMoFeP, C1 symmetry), EMD-26757 (rsMoFeP, C2 symmetry), EMD-26758 (t/oComplex-1 consensus MoFeP), EMD-26759 (t/oComplex-1 locally refined FeP), EMD-26760 (t/oComplex-1), EMD-26761 (t/oComplex-2 consensus MoFeP), EMD-26762 (t/oComplex-1 locally refined FeP), EMD-26763 (t/oComplex-2), and EMD-26764 (BeFx-trapped complex). All other data are available in the main text or the supplementary materials.