

Abstract
Metals can play key roles in stabilizing protein structures, but ensuring their proper incorporation is a challenge when a metalloprotein is overexpressed in a non-native cellular environment. Here, we have used computational protein design tools to redesign cytochrome b562 (cyt b562), which relies on the binding of its heme cofactor to achieve its proper fold, into a stable, heme-free protein with high structural similarity. The resulting protein, ApoCyt, features only four mutations and no metal-ligand or covalent bonds, yet has improved stability over cyt b562. Mutagenesis studies and X-ray crystal structures reveal that the increase in stability is due to the computationally prescribed mutations, which stabilize the protein fold through a combination of hydrophobic packing interactions, hydrogen bonds, and cation-p interactions. Upon installation of the relevant mutations, ApoCyt is capable of assembling into previously reported, cytochrome-based trimeric and tetrameric assemblies, demonstrating that ApoCyt retains the structure and assembly properties of cyt b562. The successful design of ApoCyt therefore enables further functional diversification of cytochrome-based assemblies and demonstrates that structural metal cofactors can be replaced by a small number of well-designed, non-covalent interactions.
Graphical Abstract
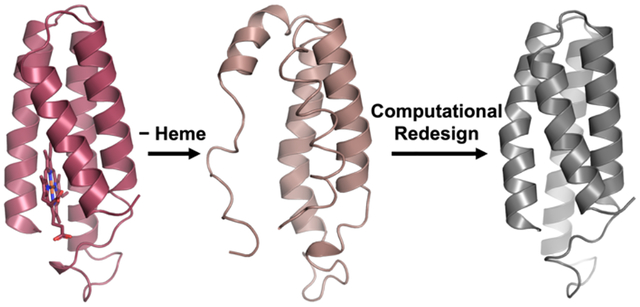
Introduction
For proteins to perform their functions, they must fold into their proper, three-dimensional structures. However, the tertiary folds of proteins are often only marginally stable.1–2 Thus, nature commonly uses metal ions to further augment the stabilities of proteins as well as to endow them with functional properties.3 Indeed it is estimated that nearly half of all natural proteins bind at a metal ion or cofactor,4 which are often essential for the proper folding and stability of the protein.2, 5 For example, zinc finger domains, which are ubiquitous in DNA-binding proteins, are inherently unstructured and can only achieve their b-hairpin or Greek key folds upon binding a ZnII ion.6–7 Similarly, the heme cofactor, essential for small-molecule binding,8 redox,9 and signaling functions,10 is often also crucial for the stabilization of protein hosts. Heme proteins such as the oft-studied four-helix bundle cytochrome b562 (cyt b562) frequently lose their well-defined tertiary structures upon the removal of the cofactor (Figure 1).11–12
Figure 1.
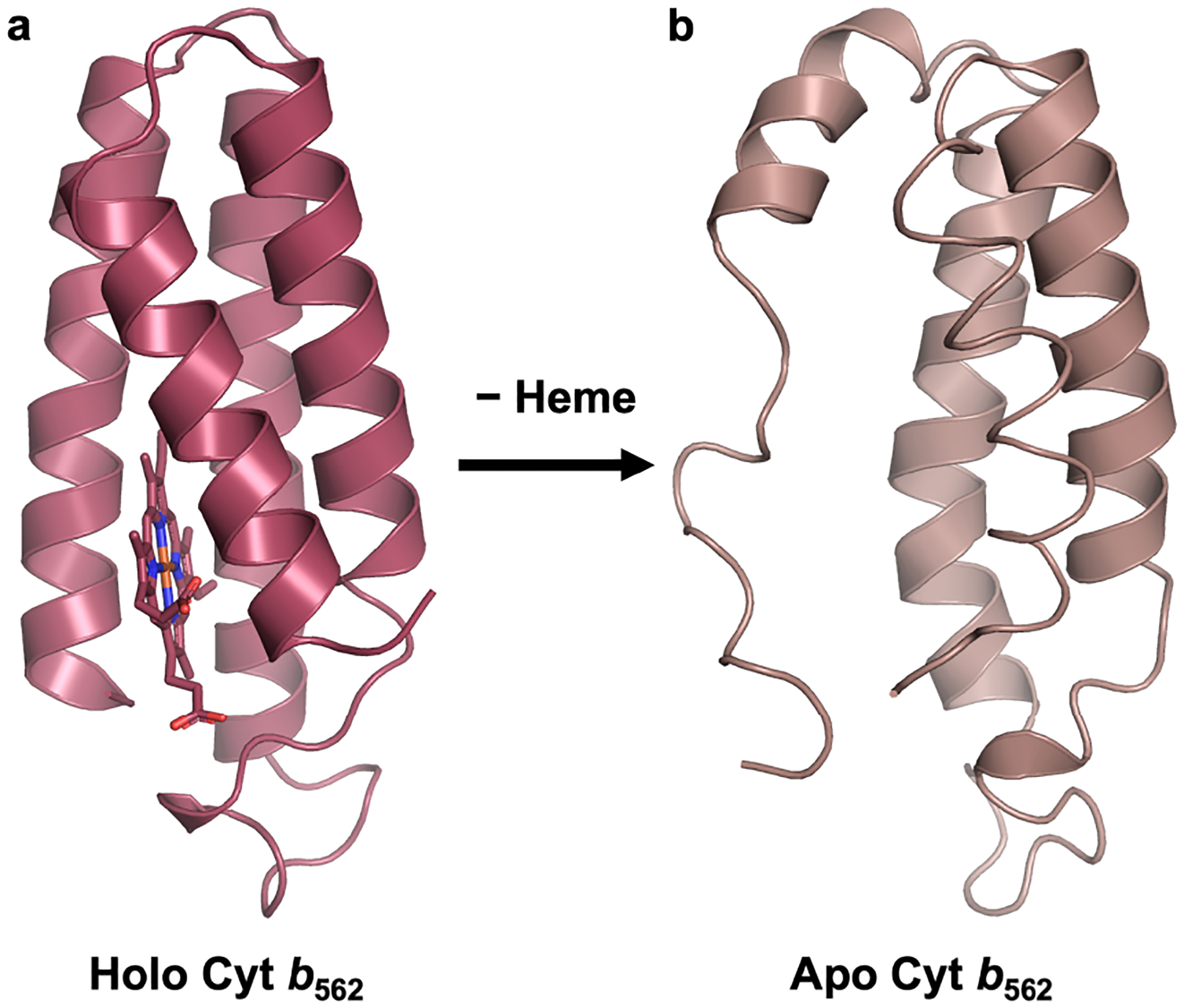
(a) Structure of cyt b562 (PDB ID: 256B) (b) Solution NMR structure (PDB ID: 1APC) of apo cyt b562.
In the cell, the incorporation of metal cofactors into proteins relies on a complex set of factors, including the protein’s affinity and selectivity for the metal cofactor, metal concentrations in the relevant cellular compartment, and, in many cases, biosynthetic machinery that actively mediates metal incorporation.13–18 As a result, controlling the metallation of a protein outside its natural cellular environment or host organism can be challenging, and indeed, recombinant overexpression of metalloproteins can often lead to mismetallation or isolation of the protein in the apo (i.e., metal-free) form.13, 19–20 This can complicate applications of industrially relevant enzymes whose metal centers solely serve a structural role, such as alcohol dehydrogenases3, 21 and a-amylases.22–24 The replacement of structural metal binding sites with non-covalent, stabilizing interactions can therefore facilitate biotechnological applications of such proteins by simplifying their expression conditions,22 while also enabling their expression in different organisms and different cellular environments.
Over the past two decades, computational protein design has emerged as a powerful approach to enhance the stability of protein folds, often without reliance on structural metal centers or covalent crosslinks.1 Indeed, early computational protein design studies reported forming stable, metal-independent variants of proteins with structural metal centers. For example, the Zn finger domain has been used as a backbone template to test both rational and computational design strategies, yielding stable, zinc finger-like folds that are independent of the metal ion.25–26 Similarly, the utility of the RosettaDesign algorithm in designing stable protein folds was validated through the complete redesign of the Zn metalloenzyme procarboxypeptidase into a metal-free protein that was over 10 kcal/mol more stable than the wild-type metalloprotein.27–28
More recently, targeted approaches to protein stabilization have been developed using the Rosetta macromolecular modeling suite.29–31 Such approaches have focused on enhancing the stability of folded proteins but have not been used to replace structural metal cofactors or redesign disordered states into stable proteins. Given the challenges of controlling metal cofactor incorporation in non-native cellular environments, a targeted approach that can redesign unstable apo proteins into stable proteins – while conserving native structural features – can expand the applications and expression conditions of proteins with structural metal centers, while also providing fundamental insights into protein folding and metal-protein interactions in general.
Along these lines, an experimental approach to screening for protein stability based on protease resistance has been used to select for stable, heme-free cyt b562 variants.32 This approach, though, introduced biases in the selection of the protease cleave site and yielded only one variant (referred to as cyt-RIL in this manuscript) that was characterized by x-ray crystallography. Cyt-RIL also had only marginally increased stability compared to wild-type, apo cyt b562.
In addition to serving as a model protein for the study of cofactor-dependent protein folding and protein engineering,33–36 cyt b562 and its isostructural variant cyt cb562 (with engineered c-type heme-protein covalent links37) have been used as versatile building blocks for a wide variety of designed metal-templated protein assemblies,38 including small oligomers,39–41 cages,42 and extended assemblies.43–44 These assemblies have served as starting points for engineering functions such as selective metal coordination,45–46 allostery,47–48 catalysis,49 and small-molecule binding and encapsulation.42 However, further functional diversification of cyt cb562-based assemblies has been impeded by the heme cofactor, which results in a high optical/electronic background that can complicate enzymatic assays and obscures spectroscopic investigations of engineered metal centers. The c-type covalent attachment of the heme cofactor also requires specialized cellular machinery in the bacterial periplasmic space, thereby preventing the expression of cyt cb562 in different cellular environments or host organisms.
We therefore set out to computationally redesign cyt b562 into a heme-free version while maintaining its native structure, stability, and ability to assemble into previously designed oligomeric or extended architectures. In our design approach, we aimed to stabilize the protein on as small a design footprint as possible, targeting a minimal number of residues in close proximity to the heme-binding pocket rather than redesigning the entire protein core. We also chose to evaluate designs not just on the basis of the Rosetta score function, but also by their structural alignment with the target fold. The resulting variant, termed ApoCyt, features no covalent crosslinks or metal binding sites and differs from cyt b562 by only four mutations, yet it has improved thermodynamic stability. Furthermore, variants of ApoCyt retain the self-assembly properties of cyt cb562, as evidenced by the formation of a previously reported trimeric50 and tetrameric51 assemblies. The success of the design strategy demonstrates that computational protein design tools can be used in a minimally perturbative manner to replace structural metal cofactors with non-covalent interactions.
Materials and Methods
General Methods.
All reagents were purchased from commercial sources and used without further purification.
Computational Design of ApoCyt.
To design a stable, heme-free variant of cyt cb562, an initial heme-free model of the protein was first obtained by deleting the heme cofactor from the crystal structure of cyt cb562 (PDB ID 2BC5) in PyMol. This model then served as the starting point for the computational design protocol, which was performed using the PyRosetta interface52 to Rosetta3.53 The protocol consisted of 2 rounds of sequence design with the PackRotamersMover function and minimization with the FastRelax function. A void penalty was added to the default Rosetta full atom score function (ref2015) that penalizes buried void spaces to help push the PackRotamersMover to fill the empty heme pocket. This void penalty is by default only active during side chain packing and not during the FastRelax steps (https://www.rosettacommons.org/docs/latest/rosetta_basics/scoring/VoidsPenaltyEnergy). A total of 200 iterations of the entire design protocol were performed, yielding 200 total designs. Structural alignments of the designs to the backbone of cyt cb562 were performed using the CA_rmsd function in PyRosetta. The Python scripts used for protein design and structural alignments are included below. The 200 designs were ranked by both their ref2015 scores (Table S1) and the Helix 3 aC root-mean-square deviations (rmsd) between the designs and the cyt cb562 backbone (Table S2), and the top-ranking design by Helix 3 aC rmsd was chosen for experimental characterization.
Protein Mutagenesis.
The genes encoding ApoCyt and its related variants, as well as ApoCyt-TriCyt3, were obtained as previously described54 or by a modified protocol in which the corresponding cyt cb562 gene was first amplified with mutagenic primers, then ligated into the pET20b(+) plasmid via Gibson Assembly. The mutated PCR product or Gibson Assembly product was transformed into Escherichia coli (E. coli) XL1-Blue cells and plated onto LB/agar plates containing 100 mg/L ampicillin overnight at 37 °C. Single colonies were picked and used to inoculate cultures containing ~5 mL LB media and 100 mg/L ampicillin, which were again allowed to grow overnight. Finally, the plasmid was extracted from the cultures using a Miniprep kit (Qiagen or NEB) and sequenced (Eton Bioscience).
All constructs were designed and expressed without the addition of any purification tags, except for the ApoCyt-TriCyt3 variant that was used for crystallography, which contained a 6xHis tag. ApoCyt-TriCyt3 with and without a 6xHis tag displayed highly similar Sedimentation velocity/analytical ultracentrifugation SV-AUC profiles (Figure S7). Amino acid sequences of all proteins used are shown in Table S3.
General Protocol for Protein Expression and Purification.
The plasmid of the relevant cytochrome variant was transformed into E. coli BL21(DE3) cells and plated onto LB/agar plates containing 100 mg/L ampicillin overnight at 37 °C. A single colony was picked and used to inoculate a starter culture containing ~5 mL LB media and 100 mg/L ampicillin, which was again allowed to grow overnight. 2.8-L flasks containing 1 L of LB media and 100 mg/L ampicillin were then inoculated with the starter culture and shaken at 200 rpm at 37 °C. Variants were typically expressed in a total of 3 to 6 L of cell culture. Cells were allowed to grow to OD600 = 0.8 (typically 6–7 h), upon which IPTG was added to a final concentration of 0.5 mM, and the cultures were allowed to continue to shake at 200 rpm at 37 °C overnight. The next morning, the cells were pelleted by centrifugation at 5,000 rpm for 10 min. If cell pellets were not immediately lysed and purified, they were stored at −80 °C.
To purify the cytochrome variants, the cell pellet was allowed to de-thaw if necessary, then was resuspended in ~75 mL Milli-Q filtered water and lysed by sonication. The pH of the resulting cell lysate was adjusted to 4.0 with acetic acid, and the lysate was centrifuged at 10,000 rpm for 20 min to pellet cellular debris and precipitate. The supernatant was collected, passed through a 0.2 μm filter, and loaded onto a high-S cartridge column pre-equilibrated in 20 mM NaOAc pH 4.0 buffer using a BioLogic DuoFlow FPLC workstation (BioRad). The protein was eluted using a linear gradient of 0–1 M NaCl, and fractions were analyzed by (sodium dodecyl sulfate-Polyacrylamide gel electrophoresis (SDS-PAGE). Fractions that contained the desired protein were pooled, concentrated if necessary using a 3 kDa Amicon membrane, and dialyzed into a buffer solution containing 20 mM NaPi (pH 8.0). The solution was then loaded onto a high-Q cartridge column pre-equilibrated in the phosphate buffer via the FPLC workstation and eluted using a linear gradient of 0–500 mM NaCl (the proteins typically eluted early, and some variants did not stick to the column at all and were collected in the flow-through). Fractions were again analyzed by SDS-PAGE, and fractions that contained pure protein were pooled, incubated with 5 mM EDTA for ~1 h, and exhaustively dialyzed into chloride-free buffer solution containing 20 mM NaPi (pH 7.4). Finally, the protein was concentrated down to 1–4 mM using a 3-kDa Amicon ultracel centrifugal filter. The exact concentration was determined by measuring the absorbance at 280 nm with an Agilent Cary 60 UV-Visible spectrometer and using the extinction coefficient at 280 nm predicted by the Expasy ProtParam tool. This stock was stored at 4 °C and used within 2 months.
Purification of ApoCyt-TriCyt3 With 6xHis Tag.
Protein expression was performed as for variants without the 6xHis tag. The resulting cell pellet was allowed to de-thaw if necessary, then was resuspended in a ~200 mL buffer solution containing 50 mM NaPi (pH 8.0) and 300 mM NaCl, and lysed by sonication. The lysate was centrifuged at 10,000 rpm for 20 min and loaded onto a gravity flow column with Ni-NTA resin preequilibrated in the same phosphate buffer. The column was washed with 200 mL of phosphate buffer, then two rounds of 200 mL of phosphate buffer + 50 mM imidazole, before the protein was eluted with two rounds of 200 mL of phosphate buffer + 150 mM imidazole. All wash and imidazole fractions were collected separately and analyzed by SDS-PAGE. The purified protein in the 150 mM imidazole fractions was incubated with 5 mM EDTA for ~1 h, exhaustively dialyzed into a buffer solution containing 20 mM MOPS (pH 7.4), concentrated down to 4.6 mM with a 3-kDa Amicon Ultracel centrifugal filter (the concentration was determined by absorbance at 280 nm), and used within 1 month.
Circular Dichroism (CD) Spectroscopy.
All CD spectra were taken on an Aviv model 215 CD spectrometer in chloride-free buffer solution of 20 mM NaPi (pH 7.4) in a 2-mm pathlength quartz cuvette. CD samples without guanidine hydrochloride (GuHCl) were prepared by diluting a concentrated protein stock solution in chloride-free phosphate buffer into additional chloride-free phosphate buffer to a final protein concentration of 10 μM. For chemical denaturation studies, stock solutions of GuHCl in chloride-free phosphate buffer were first prepared, and their GuHCl concentrations were determined from refractive indices measured on an Abbe-3L refractometer (Milton Roy, Rochester, NY). CD samples were then prepared by adding the concentrated protein stock solution to the GuHCl stock solutions to a final protein concentration of 10 μM and a total volume of 400 μL. CD spectra were collected from 185–260 nm with 0.5 nm intervals and an averaging time of 1 s at 25 °C. Thermal denaturation studies were performed in a temperature range of 25–95 °C with 2 °C intervals and a 15 s temperature equilibration time between steps. For both the thermal and chemical denaturation studies, protein unfolding was monitored at 222 nm. Thermal and chemical denaturation curves were fit to a two-state folding model using established procedures,55 and first-order baseline corrections were applied when appropriate, as has been done in previous cytochrome studies.11, 37
Analytical Ultracentrifugation.
Samples contained 60 μM (ApoCyt-TriCyt3) or 200 μM (ApoCyt-RIDC1) protein monomer in a 10 mM MOPS (pH 7.4) buffer solution. Sedimentation velocity measurements were made on a Beckman XL-A instrument using an An-60 Ti rotor. For both samples, 500–1000 scans were obtained at 25 °C at 41,000 rpm. The data were processed using the SEDFIT56 software with the following parameters: buffer density = [0.99901] g/mL; buffer viscosity = [0.0104] poise; partial specific volume = [0.7313]
Protein Structure Determination.
Crystals of ApoCyt and ApoCyt-TriCyt3 were obtained by sitting-drop vapor diffusion at room temperature. The sitting drop contained 1 μL of protein and 1 μL of precipitant solution, while the reservoir contained 200 μL of precipitant solution. Experimental growth conditions are shown in Table S4. Crystals appeared within 1–2 days and were harvested after ~1 week. Prior to freezing, crystals were briefly soaked in perfluoro polyether (Hampton) for cryoprotection. Diffraction data was collected at 100 K at SSRL beamline 9–3 (ApoCyt) and a Bruker Apex II homesource at the UCSD crystallography facility (ApoCyt-TriCyt3). Diffraction data was processed with iMosflm57 for ApoCyt and the Bruker Apex4 crystallography software suite for ApoCyt-TriCyt3. Molecular replacement was performed using Phaser-MR58 with a single chain of TriCyt3 as the search model for ApoCyt-TriCyt3 and a single chain of ApoCyt-TriCyt3 as the search model for ApoCyt. Models were refined with a combination of automated refinement with phenix.refine59 and manual refinement and water placement with Coot.60 X-ray data collection and refinement statistics are given in Table S5. Though no metal was added to the ApoCyt-TriCyt3 crystallization solution, the crystal structure showed a metal bound at the TriCyt3 His6 metal-binding site, possibly from trace metal in the crystallization solutions and buffers. Though a Zn(II) ion was modeled, the identity of the metal ion could not be determined. Structural alignments of the crystal structures with cyt cb562 and the ApoCyt design model were performed in PyMol (The PyMOL Molecular Graphics System, Version 2.4.0 Schrödinger, LLC.).
Results
Computational Design of ApoCyt:
To design a stable, heme-free variant of cyt b562, we first generated a model of the protein without the heme cofactor. We then performed 200 iterations of two cycles of sequence design and energy minimization using the Rosetta53 PackRotamers and FastRelax algorithms, respectively (Figure 2a). In the sequence design stages, only five residues – at positions 3, 7, 98, 101, and 102 – were allowed to vary. These residues reside on Helices 1 and 4 of the protein and surround the heme binding pocket. It is important to note that most protein assemblies that have been designed using cyt cb562 as a building block feature protein-protein interfaces along Helix 2 (residues 22–43) and/or Helix 3 (residues 56–81).39–40, 50 We therefore reasoned that, by limiting the sequence design space to residues around the heme binding pocket on Helices 1 and 4, we would minimize the risk of disrupting the remainder of the protein structure, and in particular, the conformations of Helices 2 and 3. We also implemented a void penalty during the sequence design stages which penalizes buried void spaces, helping guide the PackRotamers algorithm to make mutations that fill in the void space left by the absence of the heme cofactor.
Figure 2.
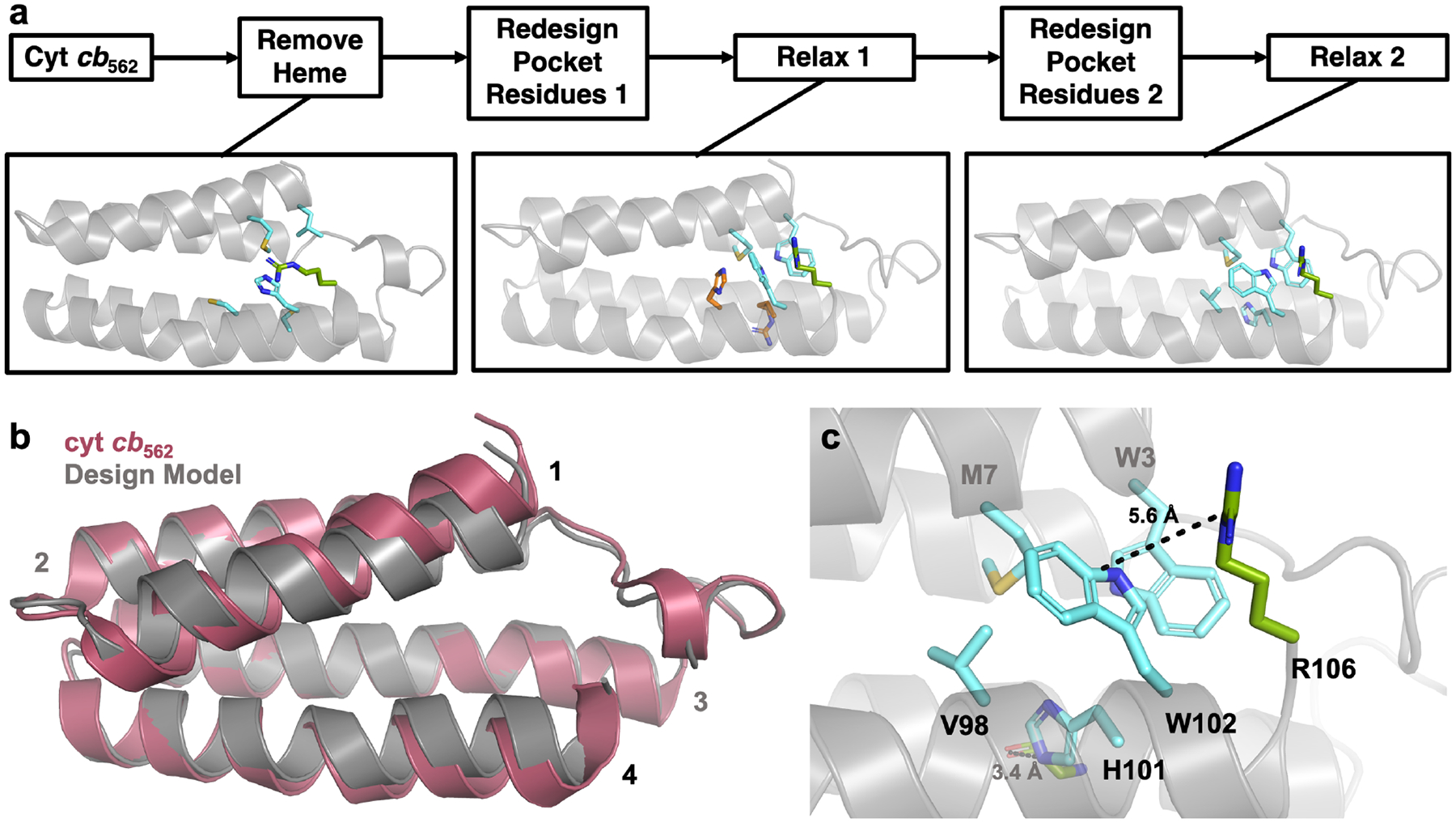
(a) Computational design process to obtain a stable, heme-free cyt b562 variant. Residues that change in the design round 2 compared to the round 1 are highlighted in orange, whereas mutated residues that do not change in round 2 are shown in cyan. The native R106 residue which forms cation-p interactions with designed residue W102 is shown in green. (b) Alignment of the ApoCyt design model with cyt cb562. (c) Computationally designed residues that fill in the heme pocket in the ApoCyt design model.
Following each round of sequence design, we performed an all-atom energy minimization. The two cycles of sequence design and energy minimization enabled potential backbone movements to be considered in the design process – any backbone movements resulting from the first round of design were subjected to a second round of design, which might lead to a more optimized sequence for that backbone conformation. The all-atom energy minimizations also meant that, in principle, any backbone distortions that resulted from the design process would be reflected in the final design models. This enabled the 200 final designs to be evaluated not just by the Rosetta score (which does not always predict the most stable proteins from a library of possible designs), but also by the backbone alignment of each design to the wild-type cyt cb562. In most designs, Helices 1 and 4 moved closer together, likely to help fill the empty heme pocket (Figure 2b). We therefore reasoned that the backbone alignment of Helix 3 to that of wild-type cyt cb562 might predict which designs were most likely to preserve the remainder of the cytochrome fold and its self-assembly properties. After ranking all 200 final designs by both Rosetta score (Table S1, Figure S1) and Helix 3 backbone root-mean-square deviation (rmsd) compared to wild-type cyt cb562 (Table S2, Figure S2), we therefore chose the top-ranking design by Helix 3 rmsd, hereafter referred to as ApoCyt, for experimental characterization (REU = −357.225, rmsd = 0.25520). ApoCyt differs from cyt b562 by only four mutations, W3, V98, H101, and W102. The computed Met in position 7 is coincidentally the native residue in cyt cb562. As expected, the redesigned heme pocket is primarily filled with the bulky hydrophobic residues W3, M7, and W102 (Figure 2c). Helix 4 is further stabilized by a predicted H-bond, between H101 and the backbone carbonyl of G64.
Structure of Wild-Type ApoCyt:
To structurally validate our design, we recombinantly expressed ApoCyt in Escherichia coli (E. coli) and determined its X-ray crystal structure at 1.69 Å resolution. ApoCyt crystallized in the P21 space group with four protein monomers (termed A, B, C, and D) in the asymmetric unit (Figure S3). All four copies of ApoCyt are structurally very similar to one another, with pairwise rmsd between all aC’s ranging from 0.39 to 0.78 Å (Figure S4). The ApoCyt monomers also align fairly well with the design model (Figure 3, Figure S5), with aC rmsd values ranging from 1.1 to 1.3 Å. In particular, there is good agreement in the Helix 3 rmsd values between the ApoCyt monomers and the design model (0.25–0.65 Å), as well as between the ApoCyt monomers and cyt cb562 (0.23–0.37 Å), which is particularly important for the self-assembly/oligomerization properties of cyt cb562 (vide infra).
Figure 3.
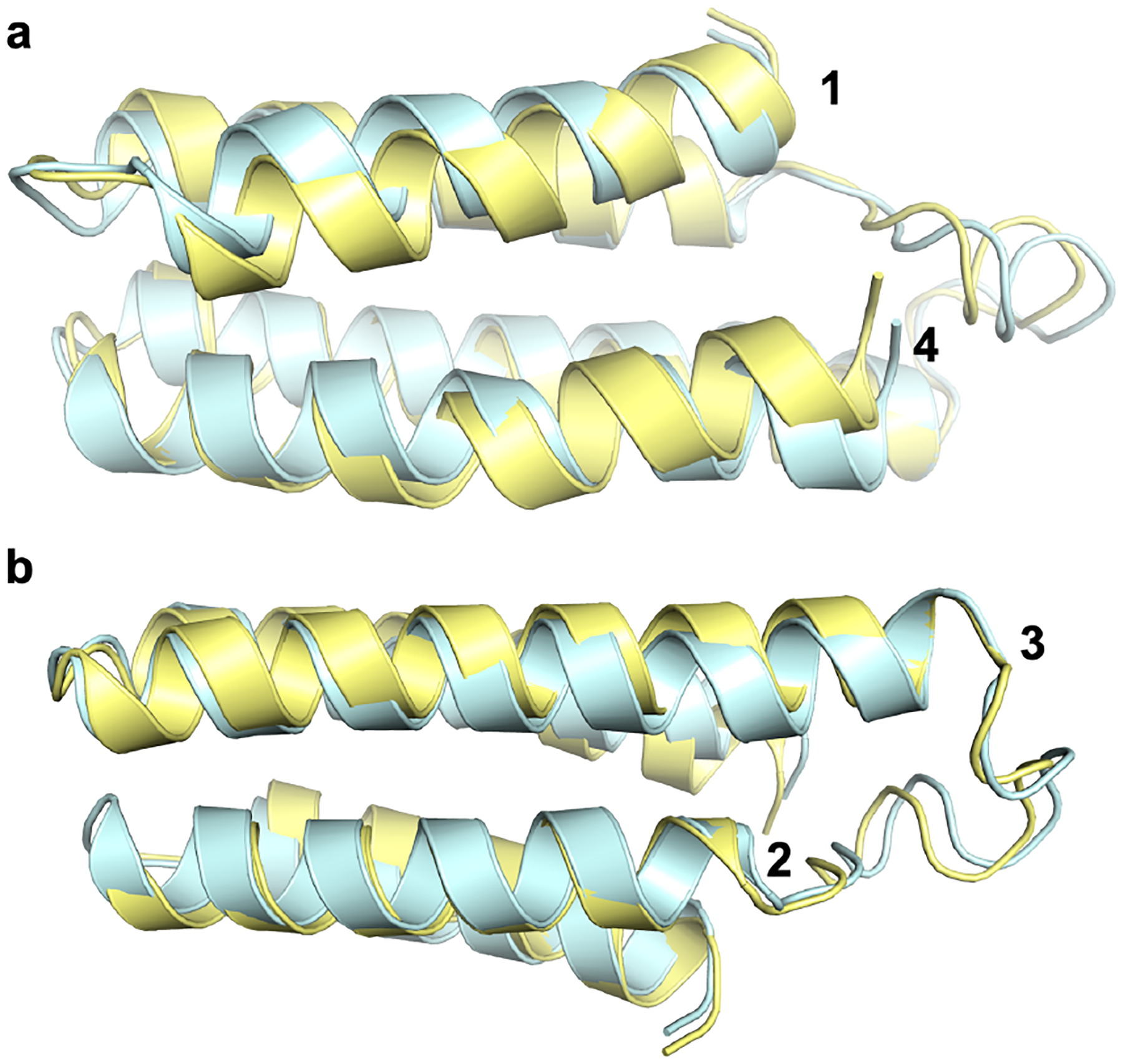
C-a alignment of ApoCyt monomer A (yellow) to the design model (cyan), focusing on (a) Helices 1 and 4 and (b) Helices 2 and 3.
While the overall backbone structure of ApoCyt is close to the designed model, there are variations in the sidechain orientations of reengineered residues in the heme pocket, which are affected by crystal packing interactions (Figure 4a–b). These variations can be categorized into two conformations, adopted by Monomers A and C (Conformation 1) and Monomers B and D (Conformation 2). In Conformation 1, residues W102 and R106 are in close proximity to one another and form intramonomer cation-p interactions (Figure 4b–c). By contrast, the same sidechains in Conformation 2 are splayed apart, which enables them to sandwich the W102 sidechain from Monomers A or C and form intermonomer contacts (Figure 4b, d). In addition, the redesigned H101 sidechain forms a H-bond to the backbone carbonyl of G64 in conformation 1 (Figure 4c), whereas it alternates between forming a H-bond to G64 or to the indole ring of W3 in conformation 2 (Figure 4d). In both conformations, the orientations of W3 and V98 are largely the same. The redesigned residues thus replace the heme cofactor with a combination of hydrophobic packing, H-bonding, and cation-p interactions (Figure S6–S7). Meanwhile, core residues within 4 Å of the heme cofactor in cyt cb562 that were not redesigned align well between ApoCyt and cyt cb562 (all-atom rmsd of 1.36–1.43 Å), further demonstrating that the structure of the remainder of the protein has been conserved (Figure S8).
Figure 4.
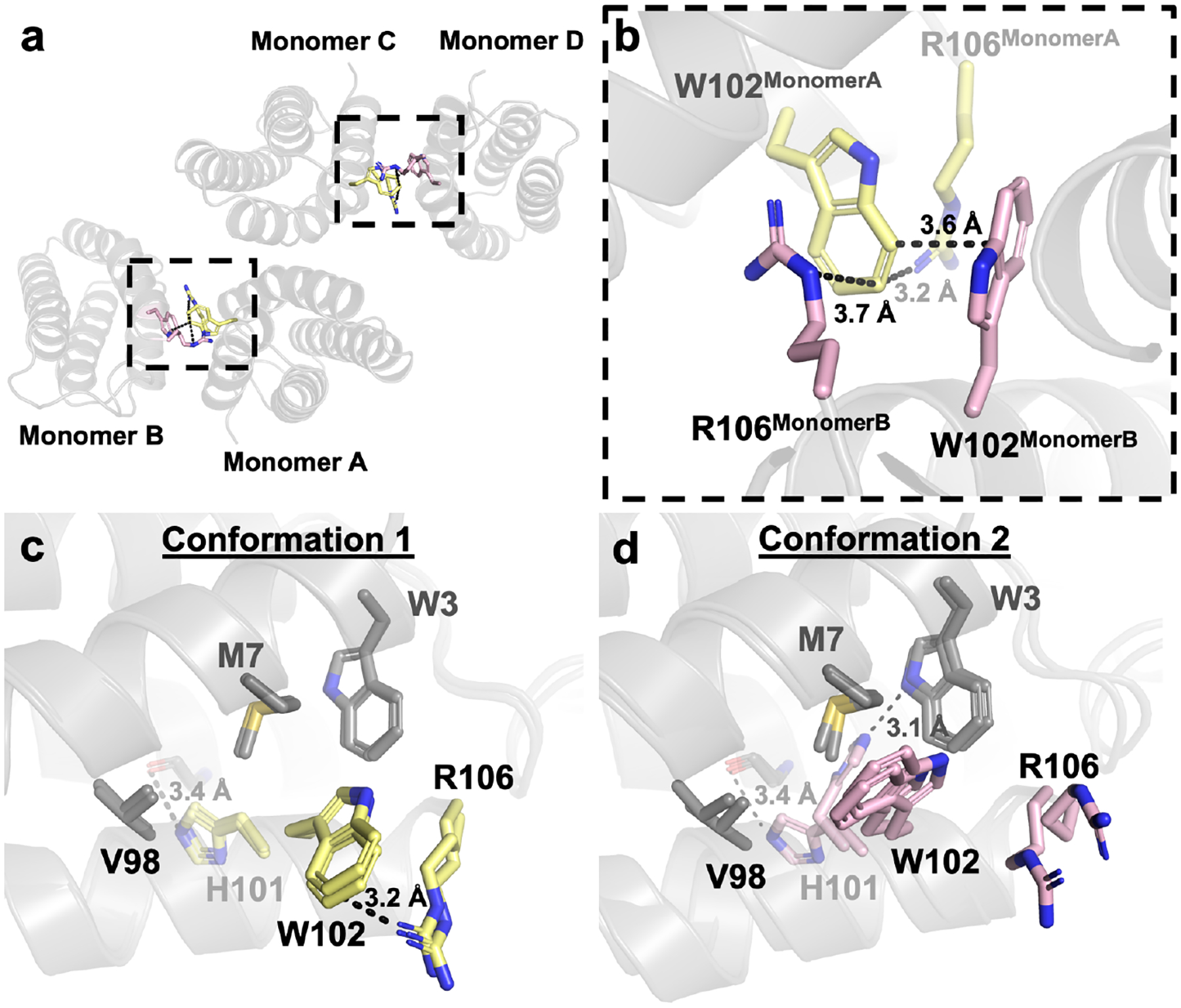
(a) Crystallographic interface of ApoCyt. (b) Cation-p interactions between W102 and R106 that help stabilize the crystallographic interface. (c) Conformation 1 of the redesigned ApoCyt heme pocket. (d) Conformation 2 of the redesigned heme pocket. Residues that vary between the two conformations are highlighted in yellow and magenta, respectively.
The multiple orientations of H101, W102, and R106 suggest that there is considerable sidechain mobility in the ApoCyt heme pocket, likely giving rise to nearly isoenergetic conformations populated in solution. This is further supported by the B-factors of the redesigned region of Helix 4, which are higher than elsewhere on the protein (Figure S9). Structural alignment of the redesigned residues from the computed model to those of the four monomers in the crystal structure indicates that there are slight differences in the orientations of the hydrophobic residues W3, M7, and W102 (Figure 5a), suggesting a slight “overpacking” of the heme pocket during the design process. The design model also does not predict the possibility of H101 forming a H-bond to W3 (Figure 5b), and the cation-p interaction between W102 and R106 sidechains in Conformation 1 brings these residues closer together than predicted (Figure 5c). However, these inaccuracies are not surprising given that the design process only attempts to predict a single conformational state and does not account for the influence of crystal packing interactions. Regardless, the design model correctly predicts the involvement of the buried H101 residue in a H-bond and the cation-p interactions between W102 and R106.
Figure 5.
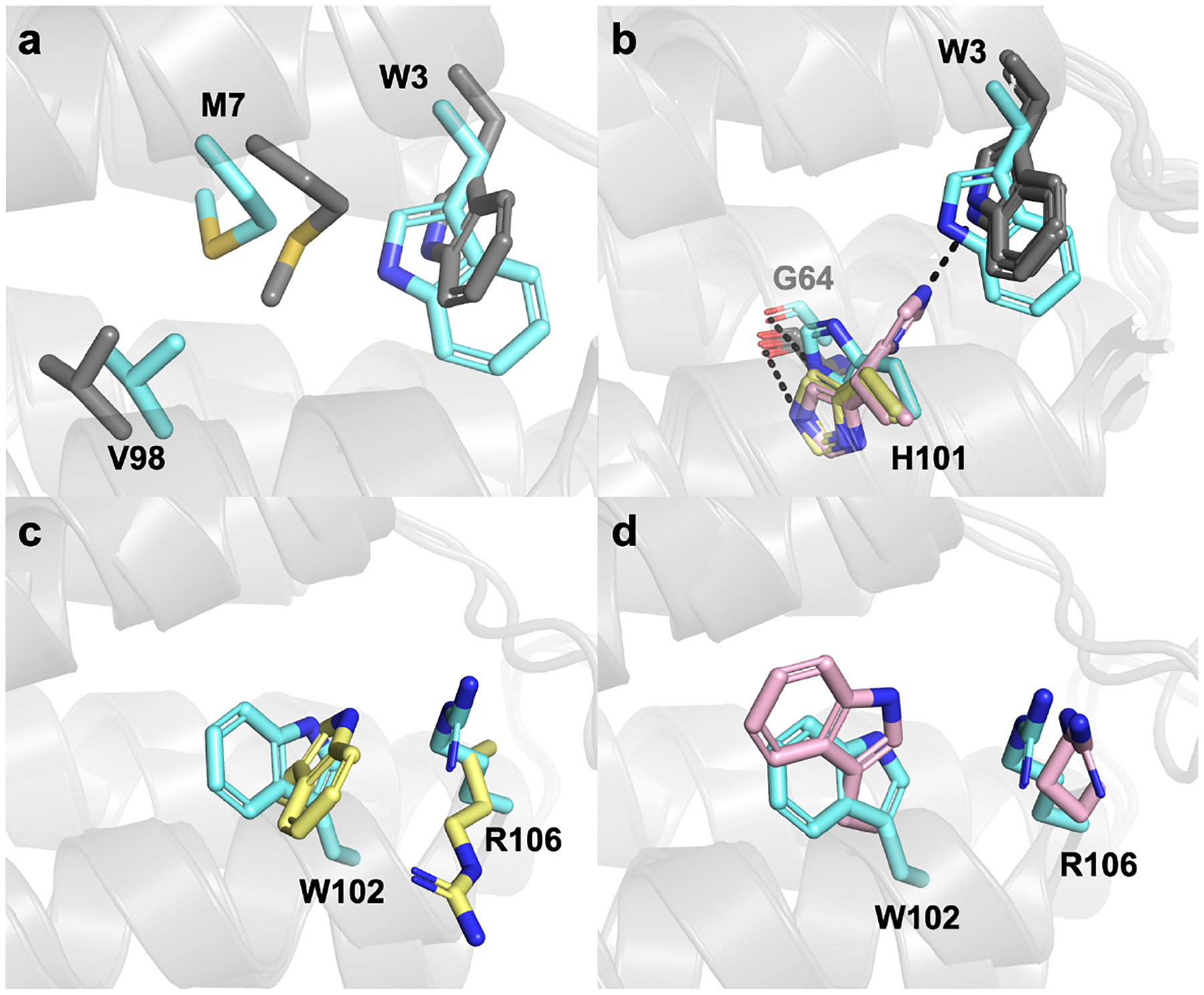
Structural alignments of the design model (cyan) to the ApoCyt crystal structure. (a) Alignment of the design model to Monomer A (grey), focusing on residues that are structurally the same across different ApoCyt monomers. (b) Alignment of the design model to Monomers A and C (Conformation 1, yellow) and Monomers B and D (Conformation 2, pink) highlighting the alternate conformations of H101. (c) Comparison of residues 102 and 106 in the design model to Conformation 1. (d) Comparison of residues 102 and 106 in the design model to Conformation 2.
Stability of Wild-Type ApoCyt:
To investigate how the stability of ApoCyt compared to its predecessors cyt b562 and cyt cb562, we used circular dichroism (CD) spectroscopy. The CD spectrum of ApoCyt is characteristic of a highly a-helical protein and very similar to that of cyt cb562, albeit with a decrease in mean residue ellipticity (MRE) of the a-helical peak at 222 nm (Figure 6a). Estimation of the secondary structures of cyt cb562 and ApoCyt using the K2D algorithm61 on the DichroWeb server62 yields a-helical contents of 100% and 87%, respectively, with the remaining 13% of ApoCyt predicted to be random coil (Table S6, Figure S10–S11). This decrease in a-helical content may be attributed to the increased structural fluxionality of ApoCyt compared to cyt cb562. Meanwhile, the previously reported heme-free variant of cyt b562, cyt-RIL32, exhibits a considerably greater decrease in MRE (Figure 6a) and only 60% a-helical content, suggesting an even greater degree of flexibility (Table S6, Figure S12).
Figure 6.
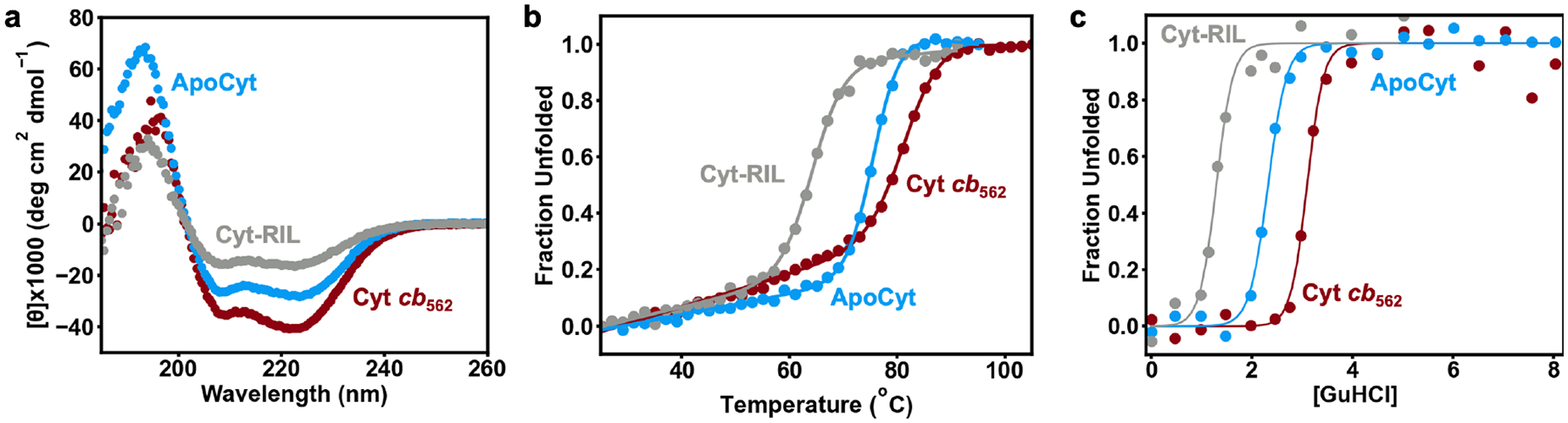
(a) CD spectra, (b) thermal denaturation curves, (c) and GuHCl denaturation curves of cyt cb562, ApoCyt, and Cyt-RIL. All CD experiments were performed with 10 μM protein solutions at pH 7.4.
We next evaluated the stability of ApoCyt through thermal and chemical denaturation studies. The thermal unfolding of ApoCyt is cooperative with a midpoint (Tm) of 75 °C (Figure 6b), which is considerably higher than those reported for apo cyt b562 (Tm = 52 °C),11 holo cyt b562 (Tm = 67 °C),11 and cyt-RIL (Tm = 63 °C),32 and approximates that for cyt cb562 (Tm = 79 °C).37 Titrations with guanidine hydrochloride (GuHCl) similarly show that while ApoCyt (DG°folding = −31.5 ± 2.7 kJ/mol) does not achieve the chemical stability of cyt cb562 (DG°folding = −47.4 ± 11.8 kJ/mol)37, likely due to the lack of covalent links in ApoCyt, it is more stable than holo cyt b562 (DGfolding° = −27.6 ± 2.1 kJ/mol)11 despite the loss of the heme cofactor and associated heme-protein coordination bonds. ApoCyt is also substantially more stable than cyt-RIL (DG°folding = −19.4 ± 4.5 kJ/mol) and apo cyt b562 (DG°folding = −13.4 ± 2.1 kJ/mol)11 (Figure 6c).
To examine the contributions of the redesigned residues to the stabilization of ApoCyt, we generated ApoCyt variants with one of the following mutations: W3A, M7A, H101A, W102A, or R106A (Figure 7a). All five ApoCyt variants display a substantial decrease in alpha-helical content (Figure 7b, S13–17, Table S6) and DGfolding° (Figure 7c, Table 1), demonstrating that the Rosetta-prescribed mutations are necessary for the stability and proper folding of ApoCyt. A summary of the folding free energy changes of all protein variants discussed here is given in Table 1. Of the variants, the W3A and W102A mutations result in the greatest loss of stability, highlighting the importance of hydrophobic packing interactions in filling the void space left by the removal of the heme cofactor. These variants also display a significant decrease in their m-values, whereas the other Ala variants do not. As the m-value correlates with the change in solvent accessible surface area of the protein during unfolding,63 ApoCyt-A3 and ApoCyt-A7 may exist in a partially denatured state, whereas the other Ala variants may exist in a more compact, molten globular state. Meanwhile, the H101A and R106A mutations also lead to a considerable loss of stability, suggesting that the hydrogen bond and cation-π interactions that H101 and R106 form with nearby residues also play an important role in stabilizing the protein.
Figure 7.
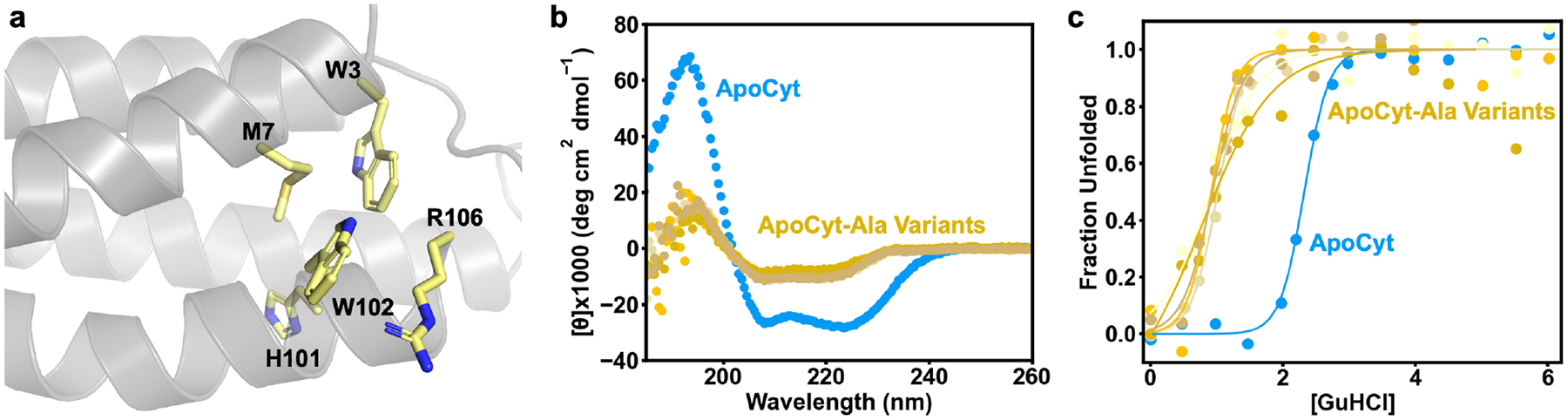
(a) Residues mutated to Ala to explore contributions to ApoCyt stability of the Rosetta-prescribed mutations. (b) CD spectra and (c) GuHCl denaturation curves of ApoCyt and ApoCyt variants with single Ala point mutations of designed residues.
Table 1.
Stabilities of cytochrome variants discussed in this paper. Values for cyt b562 are from ref. 11 and were reported at pH 7.0. [GuHCl]1/2 and m values were not reported for cyt b562.
| Variant | ΔG° (kJ/mol) | [GuHCl]1/2 (M) | m (kJ mol−1 M−1) |
|---|---|---|---|
| apo cyt b562 | −13.4 ± 2.1 | - | - |
| holo cyt b562 | −27.6 ± 2.1 | - | - |
| cyt cb562 | −47.4 ± 11.8 | 3.10 ± 0.04 | 15.29 ± 3.81 |
| cyt-RIL | −19.4 ± 4.5 | 1.29 ± 0.04 | 15.00 ± 3.42 |
| ApoCyt | −31.5 ± 2.7 | 2.32 ± 0.02 | 13.56 ± 1.14 |
| ApoCyt-A3 | −6.2 ± 1.9 | 0.91 ± 0.13 | 6.86 ± 1.80 |
| ApoCyt-A7 | −12.9 ± 2.2 | 1.02 ± 0.04 | 12.67 ± 2.09 |
| ApoCyt-A101 | −12.7 ± 3.1 | 0.92 ± 0.06 | 13.82 ± 3.28 |
| ApoCyt-A102 | −3.6 ± 3.6 | 0.77 ± 0.60 | 4.63 ± 2.92 |
| ApoCyt-A106 | −10.4 ± 2.3 | 0.94 ± 0.06 | 11.07 ± 2.36 |
Assembly of Oligomeric ApoCyt Variants:
We have previously shown that diverse oligomers can be obtained from the cyt cb562 building block by the installation of metal-binding residues and complementary non-covalent interactions on Helices 2 and 3 (Figure 8a). To explore whether ApoCyt could serve as an alternative, heme-free building block to cyt cb562, we generated two ApoCyt variants. ApoCyt-TriCyt3, contains metal-binding residues H73 and H77, as well as fifteen surface mutations (shown in magenta sticks in Figure 8a) that form hydrophobic packing and H-bond interactions which lead to the formation of a metal-independent trimer, TriCyt3.50 The other variant, ApoCyt-RIDC1, features metal binding residues H63, D74, H73, and H77 that enable the formation of a Zn(II)-dependent tetramer,39 as well as six surface mutations which improve the yield of tetramer by forming a tightly-packed hydrophobic core.51 Sedimentation velocity/analytical ultracentrifugation (SV-AUC) measurements of ApoCyt-TriCyt3 and ApoCyt-RIDC1 revealed the formation of single species with sedimentation coefficients corresponding to that of that of a trimer and tetramer, respectively (Figure 8 b–c), thus demonstrating that ApoCyt is capable of forming the same protein oligomers as cyt cb562.
Figure 8.
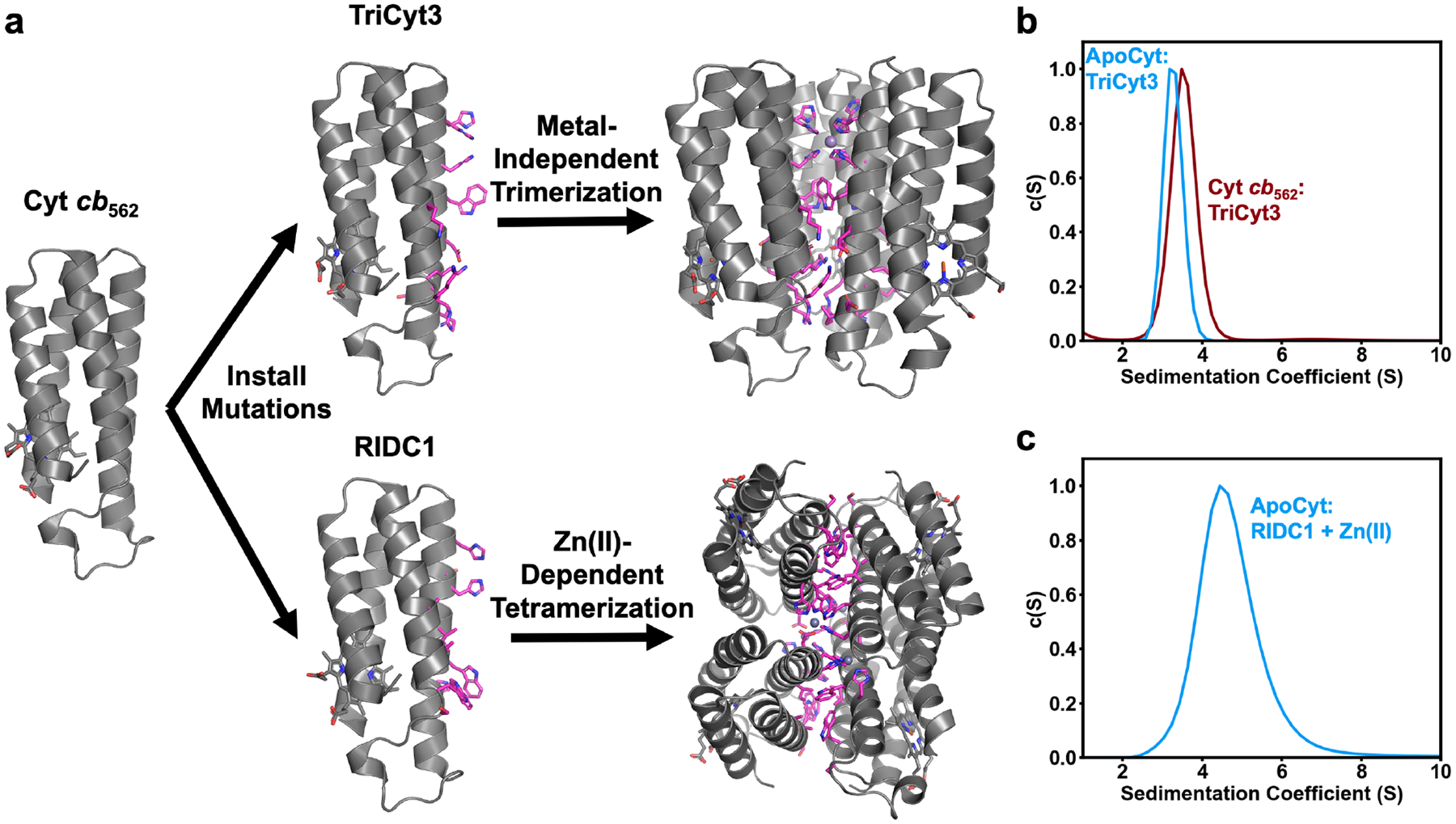
(a) Generation of TriCyt3 and RIDC1 oligomers from the cyt cb562 building block. (b) SV-AUC profiles of TriCyt3 with ApoCyt and cyt cb562. (c) SV-AUC profile of ApoCyt-RIDC1 + Zn(II).
To examine the oligomerization of ApoCyt in greater structural detail, we determined a 2.6-Å resolution crystal structure of ApoCyt-TriCyt3. Corroborating the AUC results, ApoCyt-TriCyt3 forms a trimeric assembly with high structural similarity to TriCyt3 (Figure 9a). Indeed, key interactions along the TriCyt3 interface, including the hexa-His metal binding site, Trp T-stack, and rings of H-bonding and hydrophobic residues, are preserved in ApoCyt-TriCyt3 (Figure 9b–e). Thus, ApoCyt-TriCyt3 assembles in the same manner as TriCyt3, suggesting that ApoCyt can serve as an alternative building block for the de novo design of protein assemblies.
Figure 9.
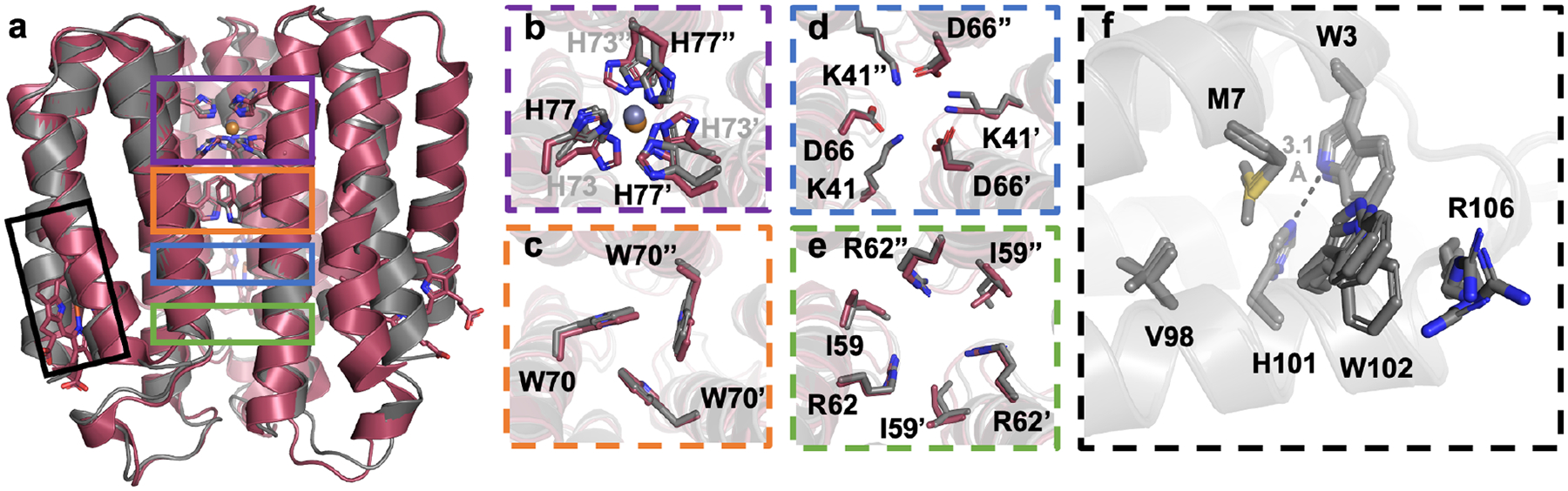
(a) Structural alignment of ApoCyt-TriCyt3 (gray) and cyt cb562:TriCyt3 (maroon). (b-e) Key TriCyt3 interactions that are preserved in ApoCyt-TriCyt3. (f) Structural alignment of the redesigned heme pockets.
Unlike wild-type ApoCyt, ApoCyt-TriCyt3 crystallized in a C21 spacegroup with three protein monomers in the asymmetric unit. These monomers do not form lattice contacts involving the engineered residues (Figure S19), providing the opportunity to examine the local environment of the redesigned heme pocket without the influence of intermonomer interactions. An inspection of the redesigned heme pockets in all monomers indicated that they adopted a single conformation (Figure 9f). As in wild-type ApoCyt, W3, M7, and W102 fill in the heme pocket and W102 and R106 form a cation-p interaction. Interestingly, H101 forms a H-bond with W3 in all three ApoCyt-TriCyt3 chains; this conformation of H101 was only seen in one of the four chains of wild-type ApoCyt. Thus, H101 appears to be able to toggle between two H-bonding states – either forming a H-bond to G64 or W3 – and the conformation it adopts may depend on the oligomeric state of the protein.
Conclusion
We have demonstrated that a targeted computational design approach can be used to stabilize cyt b562 in the absence of its structural heme cofactor. The redesigned protein, ApoCyt, is slightly more stable than holo cyt b562, despite lacking all of the extensive hydrophobic and metal coordination interactions associated with the heme. Because the vacant heme pocket in ApoCyt is stabilized without disrupting the structure of the rest of the protein, ApoCyt retains the self-assembly properties of the native cytochrome, as evidenced by its ability to form the TriCyt3 trimer and RIDC1 tetramer. The successful design of ApoCyt provides an example of how backbone alignments of computationally designed protein models to the target structure can complement energy functions for model evaluation. The minimal computational design approach described here may be a useful tool for redesigning proteins with structural metal cofactors to improve the ease of their expression and application in a wider range of cellular environments than was previously possible. Furthermore, the ease with which the large hydrophobic heme pocket can be re-designed opens up the possibility of repurposing such pockets as hosts for selective binding of organic guests.
Supplementary Material
Acknowledgements
This work was funded by National Institutes of Health grant R01-12948080 (to F.A.T.). A.M.H. was supported by the Molecular Biophysics Training Grant, NIH Grant T32 GM008326. U. M. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 502051463. Crystallographic data were collected at Stanford Synchrotron Radiation Lightsource (SSRL) and the Crystallography Facility of the University of California, San Diego.
Footnotes
Supporting Information
Tables S1–S6 and Figures S1–S19 detailing Rosetta calculations and predictions, experimental conditions and data statistics for crystallography, secondary structure contents of ApoCyt variants, overview of ApoCyt and ApoCyt-TriCyt3 crystal lattices, structural alignments, surface representations of proteins, B-factor plots, simulated and experimental CD spectra and SV-AUC profiles of ApoCyt-TriCyt3 (PDF).
The authors declare no competing financial interest.
Accession Codes. Coordinate and structure factor files have been deposited into the RCSB databank under the following PDB IDs: 8DEN (ApoCyt), 8DEL (ApoCyt-TriCyt3).
References
- 1.Goldenzweig A; Fleishman SJ, Principles of Protein Stability and Their Application in Computational Design. Annu. Rev. Biochem 2018, 87, 105–129. [DOI] [PubMed] [Google Scholar]
- 2.Arnold FH; Zhang JH, Metal-Mediated Protein Stabilization. Trends Biotechnol. 1994, 12 (5), 189–192. [DOI] [PubMed] [Google Scholar]
- 3.Holm RH; Kennepohl P; Solomon EI, Structural and functional aspects of metal sites in biology. Chem. Rev 1996, 96 (7), 2239–2314. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y; Yeung N; Sieracki N; Marshall NM, Design of functional metalloproteins. Nature 2009, 460 (7257), 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wittung-Stafshede P, Role of cofactors in protein folding. Acc. Chem. Res 2002, 35 (4), 201–208. [DOI] [PubMed] [Google Scholar]
- 6.Frankel AD; Berg JM; Pabo CO, Metal-dependent folding of a single zinc finger from transcription factor IIIA. Proc. Natl. Acad. Sci. U.S.A 1987, 84 (14), 4841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parraga G; Horvath SJ; Eisen A; Taylor WE; Hood L; Young ET; Klevit RE, Zinc-Dependent Structure of a Single-Finger Domain of Yeast Adr1. Science 1988, 241 (4872), 1489–1492. [DOI] [PubMed] [Google Scholar]
- 8.Shaanan B, Structure of Human Oxyhemoglobin at 2.1 a Resolution. J. Mol. Biol 1983, 171 (1), 31–59. [DOI] [PubMed] [Google Scholar]
- 9.Meunier B; de Visser SP; Shaik S, Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev 2004, 104 (9), 3947–3980. [DOI] [PubMed] [Google Scholar]
- 10.Nebert DW; Dalton TP, The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat. Rev. Cancer 2006, 6 (12), 947–960. [DOI] [PubMed] [Google Scholar]
- 11.Feng YQ; Sligar SG, Effect of Heme Binding on the Structure and Stability of Escherichia-Coli Apocytochrome-B562. Biochemistry 1991, 30 (42), 10150–10155. [DOI] [PubMed] [Google Scholar]
- 12.Feng Y; Sligar SG; Wand AJ, Solution structure of apocytochrome b562. Nat. Struct. Biol 1994, 1 (1), 30–5. [DOI] [PubMed] [Google Scholar]
- 13.Cotruvo JA; Stubbe J, Meta nation and mismetallation of iron and manganese proteins in vitro and in vivo: the class I ribonucleotide reductases as a case study. Metallomics 2012, 4 (10), 1020–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tottey S; Waldron KJ; Firbank SJ; Reale B; Bessant C; Sato K; Cheek TR; Gray J; Banfield MJ; Dennison C; Robinson NJ, Protein-folding location can regulate manganese-binding versus copper- or zinc-binding. Nature 2008, 455 (7216), 1138–U17. [DOI] [PubMed] [Google Scholar]
- 15.Naranuntarat A; Jensen LT; Pazicni S; Penner-Hahn JE; Culotta VC, The Interaction of Mitochondrial Iron with Manganese Superoxide Dismutase. J. Biol. Chem 2009, 284 (34), 22633–22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu YL; Fay AW; Lee CC; Yoshizawa J; Ribbe MW, Assembly of nitrogenase MoFe protein. Biochemistry 2008, 47 (13), 3973–3981. [DOI] [PubMed] [Google Scholar]
- 17.Lill R; Muhlenhoff U, Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem 2008, 77, 669–700. [DOI] [PubMed] [Google Scholar]
- 18.Sanders C; Turkarslan S; Lee DW; Daldal F, Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 2010, 18 (6), 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maret W, Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2 (2), 117–125. [DOI] [PubMed] [Google Scholar]
- 20.Nar H; Huber R; Messerschmidt A; Filippou AC; Barth M; Jaquinod M; Vandekamp M; Canters GW, Characterization and Crystal-Structure of Zinc Azurin, a by-Product of Heterologous Expression in Escherichia-Coli of Pseudomonas-Aeruginosa Copper Azurin. Eur. J. Biochem 1992, 205 (3), 1123–1129. [DOI] [PubMed] [Google Scholar]
- 21.Magonet E; Hayen P; Delforge D; Delaive E; Remacle J, Importance of the Structural Zinc Atom for the Stability of Yeast Alcohol-Dehydrogenase. Biochem. J 1992, 287, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson TH; Tan XQ; Frey G; Callen W; Cabell M; Lam D; Macomber J; Short JM; Robertson DE; Miller C, A novel, high performance enzyme for starch liquefaction - Discovery and optimization of a low pH, thermostable alpha-amylase. J. Biol. Chem 2002, 277 (29), 26501–26507. [DOI] [PubMed] [Google Scholar]
- 23.Linden A; Mayans O; Meyer-Klaucke W; Antranikian G; Wilmanns M, Differential regulation of a hyperthermophilic alpha-amylase with a novel (Ca,Zn) two-metal center by zinc. J. Biol. Chem 2003, 278 (11), 9875–9884. [DOI] [PubMed] [Google Scholar]
- 24.Mehta D; Satyanarayana T, Bacterial and Archaeal alpha-Amylases: Diversity and Amelioration of the Desirable Characteristics for Industrial Applications. Front. Microbiol 2016, 7, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Struthers MD; Cheng RP; Imperiali B, Design of a monomeric 23-residue polypeptide with defined tertiary structure. Science 1996, 271 (5247), 342–345. [DOI] [PubMed] [Google Scholar]
- 26.Dahiyat BI; Mayo SL, De novo protein design: Fully automated sequence selection. Science 1997, 278 (5335), 82–87. [DOI] [PubMed] [Google Scholar]
- 27.Dantas G; Kuhlman B; Callender D; Wong M; Baker D, A large scale test of computational protein design: Folding and stability of nine completely redesigned globular proteins. J. Mol. Biol 2003, 332 (2), 449–460. [DOI] [PubMed] [Google Scholar]
- 28.Dantas G; Corrent C; Reichow SL; Havranek JJ; Eletr ZM; Isern NG; Kuhlman B; Varani G; Merritt EA; Baker D, High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J. Mol. Biol 2007, 366 (4), 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borgo B; Havranek JJ, Automated selection of stabilizing mutations in designed and natural proteins. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (5), 1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korkegian A; Black ME; Baker D; Stoddard BL, Computational thermostabilization of an enzyme. Science 2005, 308 (5723), 857–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldenzweig A; Goldsmith M; Hill SE; Gertman O; Laurino P; Ashani Y; Dym O; Unger T; Albeck S; Prilusky J; Lieberman RL; Aharoni A; Silman I; Sussman JL; Tawfik DS; Fleishman SJ, Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63 (2), 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu R; Takei J; Knowlton JR; Andrykovitch M; Pei WH; Kajava AV; Steinbach PJ; Ji XH; Bai YW, Redesign of a four-helix bundle protein by phage display coupled with proteolysis and structural characterization by NMR and X-ray crystallography. J. Mol. Biol 2002, 323 (2), 253–262. [DOI] [PubMed] [Google Scholar]
- 33.WittungStafshede P; Gray HB; Winkler JR, Rapid formation of a four-helix bundle. Cytochrome b(562) folding triggered by electron transfer. J. Am. Chem. Soc 1997, 119 (40), 9562–9563. [Google Scholar]
- 34.Wittung-Stafshede P; Lee JC; Winkler JR; Gray HB, Cytochrome b562 folding triggered by electron transfer: approaching the speed limit for formation of a four-helix-bundle protein. Proc. Natl. Acad. Sci. U.S.A 1999, 96 (12), 6587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Springs SL; Bass SE; McLendon GL, Cytochrome b(562) variants: A library for examining redox potential evolution. Biochemistry 2000, 39 (20), 6075–6082. [DOI] [PubMed] [Google Scholar]
- 36.Garcia P; Bruix M; Rico M; Ciofi-Baffoni S; Banci L; Ramachandra Shastry MC; Roder H; de Lumley Woodyear T; Johnson CM; Fersht AR; Barker PD, Effects of heme on the structure of the denatured state and folding kinetics of cytochrome b562. J. Mol. Biol 2005, 346 (1), 331–44. [DOI] [PubMed] [Google Scholar]
- 37.Faraone-Mennella J; Tezcan FA; Gray HB; Winkler JR, Stability and folding kinetics of structurally characterized cytochrome c-b(562). Biochemistry 2006, 45 (35), 10504–10511. [DOI] [PubMed] [Google Scholar]
- 38.Salgado EN; Radford RJ; Tezcan FA, Metal-Directed Protein Self-Assembly. Acc. Chem. Res 2010, 43 (5), 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgado EN; Faraone-Mennella J; Tezcan FA, Controlling protein-protein interactions through metal coordination: Assembly of a 16-helix bundle protein. J. Am. Chem. Soc 2007, 129 (44), 13374–13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salgado EN; Lewis RA; Mossin S; Rheingold AL; Tezcan FA, Control of Protein Oligomerization Symmetry by Metal Coordination: C-2 and C-3 Symmetrical Assemblies through Cu-II and Ni-II Coordination. Inorg. Chem 2009, 48 (7), 2726–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rittle J; Field MJ; Green MT; Tezcan FA, An efficient, step-economical strategy for the design of functional metalloproteins. Nat. Chem 2019, 11 (5), 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golub E; Subramanian RH; Esselborn J; Alberstein RG; Bailey JB; Chiong JA; Yan XD; Booth T; Baker TS; Tezcan FA, Constructing protein polyhedra via orthogonal chemical interactions. Nature 2020, 578 (7793), 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brodin JD; Ambroggio XI; Tang CY; Parent KN; Baker TS; Tezcan FA, Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat. Chem 2012, 4 (5), 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brodin JD; Smith SJ; Carr JR; Tezcan FA, Designed, Helical Protein Nanotubes with Variable Diameters from a Single Building Block. J. Am. Chem. Soc 2015, 137 (33), 10468–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina-Morales A; Perez A; Brodin JD; Tezcan FA, In Vitro and Cellular Self-Assembly of a Zn-Binding Protein Cryptand via Templated Disulfide Bonds. J. Am. Chem. Soc 2013, 135 (32), 12013–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi TS; Tezcan FA, Overcoming universal restrictions on metal selectivity by protein design. Nature 2022, 603 (7901), 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Churchfield LA; Medina-Morales A; Brodin JD; Perez A; Tezcan FA, De Novo Design of an Allosteric Metalloprotein Assembly with Strained Disulfide Bonds. J. Am. Chem. Soc 2016, 138 (40), 13163–13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Churchfield LA; Alberstein RG; Williamson LM; Tezcan FA, Determining the Structural and Energetic Basis of Allostery in a De Novo Designed Metalloprotein Assembly. J. Am. Chem. Soc 2018, 140 (31), 10043–10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song WJ; Tezcan FA, A designed supramolecular protein assembly with in vivo enzymatic activity. Science 2014, 346 (6216), 1525–1528. [DOI] [PubMed] [Google Scholar]
- 50.Kakkis A; Gagnon D; Esselborn J; Britt RD; Tezcan FA, Metal-Templated Design of Chemically Switchable Protein Assemblies with High-Affinity Coordination Sites. Angew. Chem. Int. Ed 2020, 59 (49), 21940–21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salgado EN; Ambroggio XI; Brodin JD; Lewis RA; Kuhlman B; Tezcan FA, Metal templated design of protein interfaces. Proc. Natl. Acad. Sci. U.S.A 2010, 107 (5), 1827–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaudhury S; Lyskov S; Gray JJ, PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26 (5), 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman K; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban YEA; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popovic Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P, Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Meth. Enzymol 2011, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu HT; Naismith JH, An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greenfield NJ, Determination of the folding of proteins as a function of denaturants, osmolytes or ligands using circular dichroism. Nat. Protoc 2006, 1 (6), 2733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuck P, Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J 2000, 78 (3), 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey S, The Ccp4 Suite - Programs for Protein Crystallography. Acta Crystallogr. D 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- 58.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr. D 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andrade MA; Chacon P; Merelo JJ; Moran F, Evaluation of Secondary Structure of Proteins from Uv Circular-Dichroism Spectra Using an Unsupervised Learning Neural-Network. Protein Eng. 1993, 6 (4), 383–390. [DOI] [PubMed] [Google Scholar]
- 62.Miles AJ; Ramalli SG; Wallace BA, DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31 (1), 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Myers JK; Pace CN; Scholtz JM, Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4 (10), 2138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.