

Abstract
Twelve populations of Escherichia coli B all lost d-ribose catabolic function during 2,000 generations of evolution in glucose minimal medium. We sought to identify the population genetic processes and molecular genetic events that caused these rapid and parallel losses. Seven independent Rbs− mutants were isolated, and their competitive fitnesses were measured relative to that of their Rbs+ progenitor. These Rbs− mutants were all about 1 to 2% more fit than the progenitor. A fluctuation test revealed an unusually high rate, about 5 × 10−5 per cell generation, of mutation from Rbs+ to Rbs−, which contributed to rapid fixation. At the molecular level, the loss of ribose catabolic function involved the deletion of part or all of the ribose operon (rbs genes). The physical extent of the deletion varied between mutants, but each deletion was associated with an IS150 element located immediately upstream of the rbs operon. The deletions apparently involved transposition into various locations within the rbs operon; recombination between the new IS150 copy and the one upstream of the rbs operon then led to the deletion of the intervening sequence. To confirm that the beneficial fitness effect was caused by deletion of the rbs operon (and not some undetected mutation elsewhere), we used P1 transduction to restore the functional rbs operon to two Rbs− mutants, and we constructed another Rbs− strain by gene replacement with a deletion not involving IS150. All three of these new constructs confirmed that Rbs− mutants have a competitive advantage relative to their Rbs+ counterparts in glucose minimal medium. The rapid and parallel evolutionary losses of ribose catabolic function thus involved both (i) an unusually high mutation rate, such that Rbs− mutants appeared repeatedly in all populations, and (ii) a selective advantage in glucose minimal medium that drove these mutants to fixation.
We and others have been studying the dynamics of phenotypic and genomic changes in 12 populations of Escherichia coli B while they have evolved in, and adapted to, a minimal glucose medium for more than 20,000 generations (8, 23, 24, 30, 33, 35, 38). As part of this research, the performances of the derived lines and the common ancestor were quantified on a large set of diverse substrates to determine if losses of catabolic function accumulated during evolution in an environment in which glucose was the sole carbon source (8). We observed a general decline in catabolic niche breadth, which was largely attributable to subtle reductions in function on several carbon sources. However, absolute losses of function were rare, with one conspicuous exception: all 12 populations lost the ancestral ability to catabolize d-ribose. In this study, we examined the mechanisms responsible for these parallel losses, including both population genetic processes and the underlying molecular genetic events.
Parallel evolution of a trait across multiple lineages is often used as an indicator that the change is adaptive and has been shaped by natural selection (9, 18, 19, 29, 34). Therefore, we hypothesized that the loss of the ability to use ribose may have improved fitness in the glucose-limited environment. The alternative explanation—that mutations accumulated in genes affecting ribose function, but without those mutations enhancing fitness on glucose—seemed less likely on two grounds. First, other catabolic functions also experienced relaxed selection but did not exhibit such losses. Second, given typical mutation rates and the duration of the evolution experiment (12, 38), we would expect the accumulation of neutral mutations to cause sporadic but not parallel losses of function in the replicate populations.
In this paper, we report experiments that examined (i) the phenotypic dynamics of loss of ribose function in each population, (ii) the underlying rate of mutation from Rbs+ to Rbs−, (iii) the affected loci and mutational mechanism responsible for the losses of ribose function, and (iv) the effects of the losses on competitive fitness in glucose medium. We used these data to examine quantitatively the contributions of mutation and selection to the evolutionary dynamics that drive this systematic loss of function.
MATERIALS AND METHODS
Long-term evolution experiment and culture media.
The design of the long-term evolution experiment is described in detail elsewhere (24). Briefly, 12 populations evolved from a single clone of E. coli B (strain Bc251 T6r Strr rm111 ara (see [22]) also http://myxo.css.msu.edu/ecoli/strainsource.html). This ancestor is unable to grow on arabinose (Ara−) and was used to found six lines; the other six lines were founded with a spontaneous Ara+ mutant of the ancestor. The Ara marker is neutral in the experimental environment (24), which consists of daily 1:100 dilution into fresh Davis minimal broth supplemented with limiting glucose at 25 μg/ml. The 100-fold dilution and regrowth allows ∼6.6 (=log2 100) generations per day. The 12 populations have been evolving under the same conditions for more than 20,000 generations (3,000 days); the experiments described here focus on the first 2,000 generations, during which time all 12 populations lost catabolic function on d-ribose. Every 500 generations, samples from the populations were stored in glycerol at −80°C, where they are available for study at any time.
Two other media used in this study are tetrazolium-ribose (TR) indicator agar and minimal-ribose (MR) selective agar. Rbs+ and Rbs− cells produce pink and red colonies, respectively, on TR agar, while only Rbs+ cells can form colonies on MR agar.
Phenotypic screening.
The loss of d-ribose function was initially discovered in experiments with Biolog microtiter plates used to detect changes in catabolic functions during the evolution experiment (8). These plates contain different carbon sources in each of 95 wells plus a tetrazolium indicator dye. Three clones from each of the 12 population samples stored at generation 2000 were evaluated using these plates; all but 1 of the 36 clones (the exception being a clone from population Ara+2) showed almost no growth after 48 h in the well containing d-ribose (8).
To characterize the dynamics of the loss of ribose catabolic function, samples of each of the evolving populations from generations 500, 1000, and 2000 were spread on TR indicator agar, which allowed the relative frequencies of the ancestral Rbs+ and derived Rbs− types to be estimated with greater precision. The declining frequency of the Rbs+ type was confirmed by plating samples onto MR agar.
Direct measurement of the mutation rate from Rbs+ to Rbs−.
In preliminary experiments, we observed that the ancestral Rbs+ strain generated many Rbs− mutants. We then performed a Luria-Delbrück fluctuation test (21, 26, 27, 36) to estimate the mutation rate among 56 replicate cultures. Independent cultures were each founded from a small number (∼50) of cells of the Rbs+ ancestor in flasks containing 10 ml of the same glucose-supplemented Davis minimal medium. After 24 h, each culture was diluted, several hundred cells were spread on TR agar, and the Rbs+ and Rbs− colonies were counted. These data yielded the total number of cells and the frequency of Rbs− mutants in each culture, from which the rate of mutation from Rbs+ to Rbs− was estimated (21, 27, 36) (further details are provided in Results).
Fitness assays.
The protocol used for estimating the relative fitness of two strains during direct competition is described in detail elsewhere (15, 24) and summarized here. We estimated the fitness of seven independently isolated Rbs− mutants, each relative to that of the Rbs+ ancestor (their progenitor) and with fivefold replication. Equal culture volumes of the mutant and the ancestor were mixed and diluted in flasks containing fresh medium. These cultures were propagated by daily serial transfer over the course of 6 days (as opposed to a 1-day competition) to better detect small fitness differences. Initial and final (day 6) densities of both types were enumerated on indicator agar. Fitness is expressed as the ratio of the realized population growth rates for the mutant and the ancestor while they competed for the common pool of nutrients. The relative fitness of a mutant will be >1.0 if it grows more quickly than does the ancestor and <1.0 if the mutant grows more slowly.
DNA handling.
Genomic DNA was prepared from 3-ml cultures using standard methods (31). DNA fragments used as probes were cold labeled, and hybridizations were performed with the digoxigenin-labeling and detection kit sold by Roche. All hybridizations and washings were done at 68°C under high-stringency conditions.
PCR experiments.
The sequences adjacent to IS150 were cloned from HincII-digested genomic DNA of the ancestor by inverse PCR. Genomic DNA of the ancestor was digested with HincII, and the resulting fragments were separated on a 0.8% agarose gel with PstI- and HindIII-digested lambda DNA used as size markers. The gel fraction containing the expected insertion element (IS) was cut, and DNA was purified using a modification of the Geneclean kit of Bio-101 (6). The fragments were self-ligated with T4 DNA ligase (Roche) at 5 to 10 μg/ml, and the ligated mixtures were used as templates in PCR experiments. Primers used for inverse PCR to amplify sequences adjacent to IS150 were G5, 5′ GAT CCT GTA ACC ATC ATC AG 3′, and G6, 5′ CTG AAG GAT GCT GTT ACG G 3′. Both sequences lie near the IS150 extremities and are directed outward. All PCRs were performed with Expand Taq DNA polymerase (Roche) according to the manufacturer's recommendations. The PCR product containing the IS150-adjacent sequences was cloned into the pCRII-Topo vector (Invitrogen), which contains no HincII and two EcoRI restriction sites located on either side of the inserted DNA in the multiple cloning sites. After transformation of E. coli TOP10 competent cells (Invitrogen), the plasmid content of white colonies was digested with EcoRI and HincII, giving rise to three fragments. One fragment contained only the vector sequence, while each of the others contained one of the two IS150-adjacent sequences. The inverse PCR was performed with HincII-digested DNA, such that a single HincII restriction site was present in the PCR product; in this particular case, there were no EcoRI restriction sites in the product. The two IS150-adjacent sequences were used as probes in hybridization experiments and they were also sequenced.
PCR to characterize mutations in the ribose operon was also performed using the Expand Taq polymerase (Roche) according to the manufacturer's recommendations. The primers were G76, 5′ TGC CGG ATG ATG GAA ACC TC 3′, and G77, 5′ GAT GGC CTT CTT CAT GCA GG 3′. Sequences were obtained by following the method of Sanger et al. (32) with an ABI automated sequencer. Sequences of the different PCR products were obtained using primers G5 and G6, and they were compared with the databases using the BLAST program (2).
Restoration of Rbs+ phenotype by transduction.
P1 transduction (28) was used to restore the functional rbs operon to two of the seven Rbs− mutants of the ancestor. Each Rbs+ transductant was then competed against its corresponding Rbs− mutant, with sixfold replication.
Construction of a non-IS150-mediated deletion in the rbs operon.
A deletion of 5,563 bp of the rbs operon was constructed in the ancestor strain without involving the upstream IS150 element. This deletion includes about 90% of rbsA, all of rbsC, rbsB, rbsK, and rbsR, and about 20% of yieO (which lies downstream of the rbs operon).
Briefly, a 648-bp PCR product using primers G267 (5′ AGT CAG GAT TAA ACT GTG GGT 3′) and G266 (5′ ATC GCG AGT ATA GAT GCC AG 3′) was cloned into the pCRII-Topo vector. This fragment contains rbsD. Next to this first PCR product, 3′ to it and in the correct orientation according to the rbs operon, a second PCR product of 662 bp was inserted. This product was obtained with primers G268 (5′ GGT AAA CTG CGT CGA CAT AG 3′) and G269 (5′ GTT CTT GGC GGC GTG CTG 3′), and it is internal to yieO. The resulting plasmid thus contains a 5,563-bp deletion allele of the rbs operon, the two PCR products representing its flanking sequences. This insert was isolated from the pCRII-Topo vector as a 1,310-bp SacI-XhoI fragment and was inserted into a suicide plasmid cut with SacI-SalI. This suicide plasmid (D. Schneider, unpublished data) is derived from pCVD442 (11), one difference being the replacement of bla, conferring ampicillin resistance, by cat, which confers chloramphenicol resistance. This suicide plasmid contains the replication origin of R6K, which cannot function in E. coli B owing to the absence of the Pir protein needed for its replication. It also carries sacB, encoding levane sucrase, which is toxic for many gram-negative bacteria in the presence of sucrose.
The specific replacement of the rbs deletion allele into the ancestral strain was performed as described generally by Donnenberg and Kaper (11). Briefly, the suicide plasmid carrying the deletion allele was introduced into the ancestor, and chloramphenicol-resistant colonies were selected, which indicates the integration of the entire plasmid into the chromosome. Several colonies were serially diluted and plated onto sucrose-containing agar. Sucrose-resistant colonies appeared, indicating excision of the plasmid. These clones were also checked for loss of the plasmid by testing for chloramphenicol sensitivity. Among the plasmid-excised clones, those carrying the deletion allele of rbs were identified by their inability to grow on minimal ribose agar, and these were confirmed by PCR and hybridization experiments. One such clone, called GBE127, was then used in competition experiments against the Rbs+ ancestor, with 10-fold replication.
RESULTS
Differences in ribose utilization in E. coli K-12 and B strains.
E. coli K-12 is able to catabolize d-ribose, and it has a single functional rbs operon located at 83 min on the chromosome (25). The particular strain of E. coli B with which we began the long-term evolution experiment is also able to grow on ribose. However, other widely used strains of E. coli B are unable to catabolize ribose and have two defective rbs operons located at 2 and 83 min (1, 25). Possible explanations for the differences between E. coli B strains in ribose function are considered in Discussion.
Ribose catabolism is rapidly lost from evolving populations.
Figure 1 shows the dynamics of loss of the d-ribose catabolic phenotype in 12 experimental populations. After only 500 generations, Rbs− mutants had taken over 7 of the 12 populations, and by generation 2000 the Rbs− phenotype had become fixed, or nearly so, in all 12 of the populations. Previous studies indicated that several beneficial mutations swept through each of these populations in this interval, and these mutations conferred fitness gains of about 10% each (23, 24). The rapidity and reproducibility with which the losses of ribose catabolic function arose suggested to us that the Rbs− mutations might therefore represent one of these strongly beneficial mutations.
FIG. 1.

Frequency of Rbs− cells over time in the 12 evolving populations. At generation 2000 all the populations contained between 97 and 100% Rbs− cells.
Rate of mutation from Rbs+ to Rbs− is very high.
An alternative explanation for the rapid parallel losses of ribose function is that this character is genetically unstable. This alternative was suggested by the unexpected finding that occasional spontaneous Rbs− mutants were observed when ancestral cells were plated on TR indicator agar, which led us to perform a fluctuation test to measure the ancestor's rate of mutation from Rbs+ to Rbs−. Among 56 independent cultures (each inoculated from ∼50 cells), the average total population size was 4.2 × 108 cells and the average frequency of Rbs− mutants was 0.000512. We employed a local computer program (P. J. Gerrish, Los Alamos National Laboratory) (see also reference 35) to find the mutation rate consistent with these data. This program generates expected Luria-Delbrück distributions according to the method of Ma et al. (27) and then utilizes maximum likelihood to refine the mutation rate estimate (36) and calculates the 95% confidence interval for the mutation rate based upon the theoretical variance of the maximum likelihood estimate. This procedure generated a mutation rate of 8.56 × 10−5 per cell per generation, with an upper confidence limit of 1.22 × 10−4 and a lower bound of 5.99 × 10−5. However, this program does not take into account the fitness benefit that we measured for the Rbs− mutants over their Rbs+ progenitor (see below), which would lead to a slight overestimation of mutation rate. Using numerical simulations consistent with the median method of Lea and Coulson (21), and allowing the mutants to replicate with the measured selective advantage, we calculated a rate of mutation from Rbs+ to Rbs− of 5.4 × 10−5 per cell per generation, which is our best estimate of the true rate.
In E. coli, the typical mutation rate per base pair has been estimated to be about 5 × 10−10 (12). The entire rbs operon is approximately 7,000 bp; extrapolating this mutation rate to the operon as a whole yields a mutation rate of about 3.5 × 10−6, and only a small fraction of these point mutations would be expected to eliminate ribose catabolic function. Evidently, the actual rate of mutation from Rbs+ to Rbs− is unusually high, which implies that an unusual mutational mechanism affects one or more of the loci required for d-ribose catabolism. This high mutation rate appears restricted to this particular locus, because previous estimates of the mutation rate of these populations that used other loci have been substantially lower (35).
Rbs− mutants are slightly more fit than their progenitor.
Each of the evolved populations carries many mutations, including several that are beneficial (23, 24, 30, 33). It would therefore be inappropriate to use genotypes from these populations to measure the effect of the Rbs− mutation on fitness, because they contain confounding mutations. Hence, we performed competition experiments between seven spontaneous Rbs− mutants of the ancestor and their Rbs+ progenitor to quantify the effects of the Rbs− mutations on fitness in the glucose minimal medium. All seven Rbs− mutants were fitter than the Rbs+ ancestor (Fig. 2), with an average advantage of 1.4% ± 0.4% (95% confidence interval). Loss of d-ribose catabolic function therefore confers a small, but consistent, selective advantage in the environment that prevailed during the evolution experiment. Evidently, both selection for loss of function and a high underlying mutation rate contributed to the rapid and parallel evolution of the Rbs− phenotype in the 12 experimental populations.
FIG. 2.

Fitnesses of seven spontaneous Rbs− mutants relative to their progenitor, as measured by competition experiments. Error bars show 95% confidence intervals based on fivefold replication for each mutant.
An IS150 element adjacent to the rbs operon mediated deletions during the evolution experiment.
Given the unusually high rate of mutation from Rbs+ to Rbs−, we sought to identify the loci affected and the molecular basis of the mutations that were fixed during the experimental evolution. Restriction fragment length polymorphism analysis (30, 33) of HincII-digested genomic DNA from the ancestor and from clones sampled at generation 10000 using IS150 as a probe revealed parallel losses in all 12 populations of a 2.7-kb fragment that contained an IS150 element. The regions adjacent to this element were cloned from HincII-digested genomic DNA of the ancestor and then sequenced. The IS150 element was found to be located immediately upstream of the rbs operon (20) that is involved in ribose utilization (Fig. 3). The IS150 insertion also led to a 3-bp duplication of the target site.
FIG. 3.

Losses of d-ribose catabolism in evolving populations caused by deletions in the rbs operon. (A) Map of the rbs operon in E. coli B ancestor, based on the genome sequence for E. coli K-12 (4). Arrows show the directions of gene transcription. An IS150 element is located upstream of the rbs operon. Thick lines indicate the left and right adjacent sequences used as probes. The primers used in PCR experiments (G5, G6, G76, and G77) and for constructing strain GBE127 (G266, G267, G268, and G269) are also shown, as are relevant HincII restriction sites. (B) The deletions in clones isolated from 11 of the 12 populations are shown as horizontal lines. Population Ara+2 is not depicted because an Rbs+ minority clone was sampled (see text). The right adjacent sequence of IS150 did not hybridize with any of the Rbs− clones, whereas the left adjacent sequence hybridized in every case with a HincII fragment, whose size is shown in the column labeled Hyb. The sizes of the PCR products obtained from these clones using primers G76 and G77 are given in the column labeled PCR. The sizes, in the ancestral strain, of the hybridizing HincII fragment and PCR product are indicated at the bottom.
The two sequences adjacent to IS150 were then used as digoxigenin-labeled probes with HincII-digested genomic DNA isolated from one clone randomly chosen from each of the 12 evolving populations at generation 2000. Eleven of the 12 evolved clones were Rbs− while the clone from population Ara+2 happened to belong to the Rbs+ minority (∼1.5%) still present in that population. The control hybridization with HincII-digested genomic DNA of the ancestor revealed, as expected, a 2.7-kb signal using both probes. The same pattern was also observed in hybridizations with genomic DNA from the one evolved Rbs+ clone; the rbs operon of this atypical clone also showed no differences from the ancestor and no movement of the IS150 element. By contrast, in hybridizations with all 11 Rbs− evolved clones, the left IS150-adjacent sequence hybridized with one HincII fragment, which ranged in size from 2.0 to 4.2 kb; no hybridization signal was detected with these clones using the right IS150-adjacent sequence as a probe (Fig. 3). These results indicated the presence of deletions affecting at least part of the rbs operon in the Rbs− clones.
To characterize the extent of the deletion in the 11 Rbs− clones, the endpoints were PCR amplified using two primers, one located upstream of the IS150 insertion and one at the beginning of yieO, a gene of unknown function located immediately downstream of the rbs operon (Fig. 3). (Expression of yieO has recently been shown to have some role in ςs regulation and homocysteine production, and the gene has been designated hsrA [17].) PCR products obtained ranged from 2.1 to 7.4 kb depending on the clones and these were sequenced using primers G5 and G6. In each case, ∼700 bp of sequence were obtained with each primer; comparison with the E. coli K-12 genome sequence (4) allowed precise mapping of the deletion endpoints in or near the rbs operon (Fig. 3). In all cases, the left end of the deletion coincided exactly with the right end of IS150, which strongly implicated the involvement of this element in the rearrangement. In 7 of the 11 clones, the right deletion endpoints were located within yieO, although the precise location of the endpoints within yieO varied among the clones. These deletions thus removed the entire rbs operon. In the remaining four cases, the right endpoint of the deletion fell twice within rbsR (the last gene of the operon), once within rbsB, and once within rbsA. Even the smallest deletion therefore included the promoter region, the first gene of the operon (rbsD), and part of rbsA.
In two populations, Ara−1 and Ara+1, we also examined the extent of the deletions in clones that were isolated as early as generation 500 and as late as generation 10000. In each population, the same hybridization pattern was detected over time, indicating that no further changes in the extent of the deletion occurred after it first arose.
Similar deletions of the rbs operon occur in spontaneous Rbs− mutants.
We then performed the PCR assays used to identify deletion endpoints in evolved clones to characterize the seven spontaneous Rbs− mutants whose fitnesses we had measured. In all seven mutants, the loss of d-ribose catabolism was caused by deletions of various sizes within and near the rbs operon. All had the same left endpoint of their deletion adjacent to the IS150 element; three had the right endpoint of the deletion in the rbsK gene, two in yieO, and one each in rbsA and rbsC. We reported in a previous section that these seven mutations all had beneficial effects on competitive performance, in each case increasing fitness by about 1 to 2% in glucose minimal medium. Thus, different underlying mutations conferred similar selective benefits, with the commonality being that the rbs operon was rendered nonfunctional by a deletion beginning adjacent to the IS150 element located just upstream of this operon. We also performed an experiment to measure the rate of reversion of the Rbs− mutants back to the Rbs+ state. However, no Rbs+ revertants were observed, consistent with the rbs operon having been partially or completely deleted.
Restoration of the functional rbs operon reduces fitness.
To confirm that the deletions in the rbs operon (and not some hypothetical mutation elsewhere) caused the observed fitness advantage of the Rbs− mutants, we used P1 transduction to restore the functional rbs operon to two of the Rbs− mutants. These two mutants differed in the extent of the deletion of the rbs operon: one lacked only part of the rbs operon, whereas the other lacked the entire operon plus part of yieO. In both cases, the restored functional operon significantly reduced fitness relative to the corresponding Rbs− mutant (P < 0.01 in each case, based on six replicate competitions). Thus, restoration of the functional rbs operon counteracts the beneficial effect caused by its earlier deletion.
Non-IS150-mediated deletion of the rbs operon also increases fitness.
As a final test of the relationship between ribose catabolic function and competitive fitness in glucose-limited medium, we constructed strain GBE127. In this strain, unlike others we examined, the promoter region and first gene of the operon, rbsD, are intact, but the rest of the genes in the rbs operon have been deleted. Like all seven spontaneous deletions (Fig. 2), this constructed deletion also enhanced competitive fitness, in this case by 2.1% (P < 0.0001 based on 10 replicate competitions). Therefore, neither the upstream IS150 element nor the promoter region needs to be involved in the deletion mutation in order to confer the beneficial fitness effect.
DISCUSSION
Twelve populations of E. coli B experienced rapid and parallel losses of d-ribose catabolic function during evolution in glucose minimal medium (Fig. 1). We showed that these losses were caused by similar deletions of the rbs operon; in all cases, one end of the deletion was immediately adjacent to an IS150 element located just upstream of the operon, whereas the other endpoint varied (Fig. 3). We further showed that mutations from Rbs+ to Rbs− occurred at an unusually high rate, that these mutations were caused by deletions of the rbs operon similar to those during the evolution experiment, and that Rbs− mutations consistently conferred a small but significant competitive advantage in glucose minimal medium (Fig. 2).
It may be instructive to examine the logic that led us to identify the evolutionary forces responsible for the loss of ribose function. The rapid and parallel evolution of the Rbs− phenotype initially suggested to us that this change was adaptive. Repeatable change across lineages is widely taken as evidence of adaptation by natural selection (9, 18, 19, 29, 34). Furthermore, the fact that this loss of function occurred when adaptation to the new environment was most rapid (23, 24) reinforced our view that the loss was beneficial for the bacteria in glucose minimal medium. However, our observation that Rbs− mutants were readily isolated from the ancestor led us to question our preconceptions and focus on the alternative possibility that the losses of ribose catabolic function might be caused by a hypermutable locus of some sort. Indeed, a fluctuation test confirmed this possibility. We then performed competition experiments between the spontaneous Rbs− mutants and their Rbs+ progenitor, which showed that the mutants were also fitter than their progenitor in the glucose minimal medium. Evidently, both selection for the loss of ribose catabolic function and its hypermutable genetic basis contributed to the rapid and parallel phenotypic evolution that we observed.
The finding that all seven independent spontaneous Rbs− mutants had a competitive advantage strongly suggested that the beneficial effect was caused by the rbs deletion, as opposed to some hypothetical secondary mutations that arose during the fluctuation test in which the mutants were generated. It is generally accepted that only a very small fraction of all mutations are beneficial in any particular environment, with the vast majority being either deleterious or neutral (13, 16). Thus, it is highly unlikely that secondary mutations would produce any improvement in fitness.
Nevertheless, to confirm absolutely that the rbs deletions per se were responsible for the fitness benefits observed in glucose minimal medium, we performed two additional types of strain constructions and corresponding competition experiments. First, we used P1 transduction to restore a functional rbs operon to two of the Rbs− mutants. This restoration reduced their competitive fitness, which indicates that the deletion of the rbs operon itself was beneficial in the glucose environment. Second, we constructed de novo a deletion of most of the rbs operon that did not involve the upstream IS150 element or the rbs promoter region, and we used homologous gene replacement to introduce this into our progenitor. This deletion also produced a small but significant competitive advantage. Thus, three different genetic approaches—spontaneous mutation, transduction, and gene replacement—all demonstrate that the deletion of the rbs operon causes a beneficial effect in minimal glucose medium.
Molecular basis of the high rate of mutation from Rbs+ to Rbs−.
The same class of molecular events led to the loss of ribose function in the evolved lines and spontaneous Rbs− mutants. A total of 18 Rbs− genotypes (11 evolved lines and 7 spontaneous mutants) all showed deletions in which one endpoint was located precisely at the end of an IS150 element that was inserted upstream of the rbs operon. The extent of the deletion varied among the genotypes, but it always encompassed the promoter region and first gene (rbsD) and in some cases included all six genes in the rbs operon plus part of an adjacent gene of unknown function (yieO) (17). The fact that one endpoint of the deletion was always precisely located at the end of this IS150 element suggests that the mechanism of deletion involved first the transposition of an IS150 element (either the one upstream of rbs or any other) into the site corresponding to the other endpoint and in the same orientation as the one upstream of rbs. This transposition was presumably then followed by a recombination event between the new IS150 and the one upstream of rbs, thereby causing deletion of the intervening region. Whether the transposition and recombination events occurred simultaneously or successively is unknown; the fact that none of the spontaneous Rbs− mutants showed a simple transposition that inactivated the rbs operon (without the associated deletion) suggests that the two events occurred in the same cell generation. Analysis of the nucleotide sequences at the right endpoints of the different deletions indicated no homology with the end of IS150 corresponding to the left deletion endpoint, which further suggests rearrangements associated with an initial transposition event. Examination of the nucleotide sequence of the presumptive target sites of the IS150 transpositions in the different genotypes failed to reveal any obvious preference for its insertion.
All IS-associated mutations have been studied (30, 33) in 2 of the 12 evolving populations, in part to identify candidate mutations that may have contributed to their adaptation. In both of these focal populations, the rbs deletions were already present at generation 500, when adaptation was most rapid (24), and they remained present in all clones sampled in later generations. By performing restriction fragment length polymorphism analyses using IS elements as probes with EcoRV-digested genomic DNA extracted from clones sampled over time, Papadopoulos et al. (30) found several other mutations that had been fixed in these focal populations. By characterizing these mutations at the sequence level, Schneider et al. (33) showed that they also involved IS-mediated events, including transpositions, inversions, and deletions; they further showed that none of the mutations in the two focal populations involved the same genes. However, Papadopoulos et al. (30) did not detect the parallel rbs deletions that we have described here, for the following reason: EcoRV cuts at the right end of IS150, whereas the probe they used corresponded to the left end of IS150, so that the type of rearrangement involved in the rbs operon (deletions at the right end of IS150) went undetected. Based on our analysis of HincII-digested genomic DNA of the evolved clones, the rbs deletions are the only IS-associated mutations that were fixed in the two focal populations but not detected by Papadopoulos et al. (30).
Some other studies have reported that certain strains of E. coli B are Rbs− (1, 25), whereas our founding strain was clearly Rbs+ but predisposed genetically to become Rbs−. One plausible explanation for the reports that some other B strains are Rbs− is that they lost the ribose catabolic function during propagation in the laboratory, much as we have observed in our experimental lines. Under this hypothesis, the ancestor of all B strains would have had two rbs operons, a nonfunctional one at 2 min and a functional one at 83 min, with deletions independently arising in different B lineages at different points in time. However, other sources of genetic instability may also contribute to such differences. For example, a transposable element containing the rbs operon was found in one experiment, and the operon at 2 min appears to have been generated by a duplication of the operon at 83 min, suggesting that the operon has been mobile in E. coli B (1).
IS and other transposable elements generate a substantial fraction of the mutations in bacteria, and they are therefore important evolutionary factors (5). Nonetheless, there are conflicting views about their costs and benefits and the balance of forces that maintain these elements in populations. One view emphasizes that a much higher proportion of mutations are deleterious than are beneficial, infers that transposable elements impose a burden on adaptation by substantially increasing the overall mutation rate, and concludes that active elements can thus be maintained only if horizontal gene transfer allows them to exist as genomic parasites. Another view emphasizes that transposable elements may, on balance, be adaptive to an evolving population just as mutator alleles are under certain conditions. Like mutators, transposable elements may spread in asexual populations by “hitchhiking” along with the occassional beneficial mutations that are produced by their activities (5, 7, 10, 35, 37). Our findings indicate the high mutation rate that can result locally from the presence of one IS element in a particular gene region. If these deletions occurred near any essential gene, then the load created by the IS element would be equal to the mutation rate, implying a weak but nontrivial selection coefficient of about 5 × 10−5 against that one element alone. Our results also show that some IS-mediated mutations are beneficial and promote the adaptation of an evolving population. Interestingly, and in contrast to another evolution experiment in which point mutations and IS transpositions generated functionally equivalent beneficial mutations (39), in our study all of the many spontaneous beneficial disruptions of the ribose function were associated with IS activity. This difference may arise because the mutations in our study caused the loss of gene function, which occurs readily by IS-mediated mutations, whereas the mutations in the earlier study involved a more subtle change in gene regulation.
It is clear that deletions of part or all of the rbs operon are beneficial to E. coli B in glucose minimal medium and that the IS150 element located immediately upstream of the operon plays a role in generating those deletions. However, the physiological basis for the benefit that accrues is unclear. In all 18 spontaneous deletions we examined, the promoter region and first gene (rbsD) of the operon were eliminated, suggesting that it was silencing of the operon that provided the selective advantage. The constructed rbs deletion in strain GBE127 retains the promoter region and rbsD, and this strain obtains a similar benefit. Also, the fact that similar benefits accrued whether the deleted region was 2 or 7 kb implies that neither energetic savings associated with chromosomal replication nor conformational changes in the chromosome can account for the beneficial effect. Taking all these considerations together, it appears likely that the beneficial effect involves the elimination of the ribose-catabolic function per se.
Quantitative analysis of the contributions of mutation and selection to the evolution of the Rbs− phenotype.
Both positive selection for loss of the rbs operon and its underlying mutability contributed to the evolutionary losses of ribose-catabolic function in the 12 experimental populations. Here, we examined mathematically their contributions as well as their interplay with one another and with selection at other loci.
We considered first the expected time course if selection had favored the loss of this function, but without any hypermutable basis. For simplicity, we assumed the mutation is just common enough that there is no waiting time for the mutation to appear, and then we calculated the time for the mutant genotype to increase from one cell to 50% of the total population. In our calculation, we used the 1.4% selective advantage (s), which we measured as the average of seven spontaneous Rbs− mutants, and the effective population size (Ne = 3 × 107) that prevailed during the evolution experiment (24). The ratio (R) of genotypes changes in a log-linear fashion under constant selection (14, 24). We expressed bacterial generations using a log2 transformation of the daily dilution and regrowth. Thus, the time in generations (g) for a single mutant to increase to 50% (a final ratio of 1) of the population was calculated as follows:
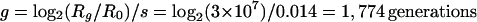 |
In fact, however, the actual frequency of mutants reached 50% much sooner than this in most of the experimental populations (Fig. 1).
Next we calculated the time required for the Rbs− mutants to achieve a 50% frequency, assuming they increased by recurring mutation only, without the benefit of any selection. In that case, the frequency (p) of the Rbs+ type should have decayed exponentially from an initial frequency of 1. Given the estimated mutation rate (μ), of 5.4 × 10−5 per cell per generation, the time for the progenitor to decline to 50% (and the mutant to reach 50%) was calculated as follows:
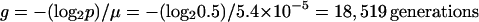 |
The actual spread of Rbs− mutants was much faster than this in all populations (Fig. 1).
Finally, we calculated the approximate time required for the mutant to reach a 50% frequency, given both its observed selective advantage and its high mutation rate. To do so, we noted that mutants should have been present after the first day of the evolution experiment in the same average frequency as in the fluctuation test, which was about 0.000512. We then used the fact that s ≫ μ to deduce that selection will drive the subsequent increase in the frequency of mutants, so that we could apply the first equation above to calculate the time to reach 50% (a final ratio of 1), given an initial ratio of 0.000512. We obtained the estimated time as follows:
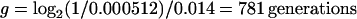 |
This last calculation gives better agreement with the observed spread of Rbs− mutants than does either calculation that ignores the contribution of mutation or selection. To summarize, the expected times required for Rbs− mutants to reach a frequency of 50% in a population were found to be 18,519 generations under the mutation accumulation process (μ = 5.4 × 10−5 per generation), 1,774 generations under the selection process (s = 0.014 per generation), and 781 generations under the selection-plus-mutation process.
Two features of the dynamics of the Rbs− mutants that are not explained by these simple models are (i) the pronounced variability in the frequency of Rbs− mutants among the replicate populations at generations 500 and 1000 and (ii) the temporary reversals in the frequency of Rbs− mutants in a few populations during that interval (Fig. 1). Both features are understood by realizing that selection was simultaneously acting on mutations at other loci (3, 13, 16), including some mutations that were much more beneficial than were the rbs deletions. Each population experienced several selective sweeps by beneficial mutations during these 2,000 generations, and the underlying mutations conferred fitness advantages, on average, of about 10% (23, 24). Given the asexual nature of the evolving populations, the short-term fate of Rbs− mutants in any population would depend on how long it took for one of the highly beneficial mutants to appear in a Rbs− clone and whether an even more beneficial mutation appeared in a Rbs+ clone, which could cause a reversal owing to clonal interference (16). Thus, variation among populations in the dynamics of the Rbs− mutants is expected from the stochastic appearance of beneficial mutations at other loci, which leads to divergence in the genetic linkage among beneficial alleles across the replicate populations.
ACKNOWLEDGMENTS
We thank N. Hajela for technical assistance and P. Gerrish, D. Rozen, and M. Stanek for discussion.
This research was supported by grants from the NSF to V.S.C. (DEB-9801538) and R.E.L. (DEB-9981397) and by grants from the French CNRS and CEA to M.B.
REFERENCES
- 1.Abou-Sabé, Pilla M J, Hazuda D, Ninfa A. Evolution of the d-ribose operon of Escherichia coli B/r. J Bacteriol. 1982;150:762–769. doi: 10.1128/jb.150.2.762-769.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul S, Stephen F, Madden T L, Schaffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atwood K C, Schneider L K, Ryan F J. Periodic selection in Escherichia coli. Proc Natl Acad Sci USA. 1951;37:146–155. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blattner F R, Plunkett III G, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, Gregor J, Davis N W, Kirkpatrick H A, Goeden M A, Rose D J, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 5.Blot M. Transposable elements and adaptation of host bacteria. Genetica. 1994;93:5–12. doi: 10.1007/BF01435235. [DOI] [PubMed] [Google Scholar]
- 6.Boyle J S, Lew A M. An inexpensive alternative to glassmilk for DNA purification. Trends Genet. 1995;11:8. doi: 10.1016/s0168-9525(00)88977-5. [DOI] [PubMed] [Google Scholar]
- 7.Chao L, Vargas C, Spear B B, Cox E C. Transposable elements as mutator genes in evolution. Nature. 1983;303:633–635. doi: 10.1038/303633a0. [DOI] [PubMed] [Google Scholar]
- 8.Cooper V S, Lenski R E. The population genetics of ecological specialization in evolving Escherichia coli populations. Nature. 2000;407:736–739. doi: 10.1038/35037572. [DOI] [PubMed] [Google Scholar]
- 9.Cunningham C W, Jeng K, Husti J, Badgett M, Molineux I J, Hillis D M, Bull J J. Parallel molecular evolution of deletions and nonsense mutations in bacteriophage T7. Mol Biol Evol. 1997;14:113–116. doi: 10.1093/oxfordjournals.molbev.a025697. [DOI] [PubMed] [Google Scholar]
- 10.De Visser J A G M, Zeyl C W, Gerrish P J, Blanchard J L, Lenski R E. Diminishing returns from mutation supply rate in asexual populations. Science. 1999;283:404–406. doi: 10.1126/science.283.5400.404. [DOI] [PubMed] [Google Scholar]
- 11.Donnenberg M S, Kaper J B. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drake J W. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci USA. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dykhuizen D E. Experimental studies of natural selection in bacteria. Annu Rev Ecol Syst. 1990;21:378–398. [Google Scholar]
- 14.Dykhuizen D E, Hartl D L. Selection in chemostats. Microbiol Rev. 1983;47:150–168. doi: 10.1128/mr.47.2.150-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elena S F, Lenski R E. Test of synergistic interactions among deleterious mutations in bacteria. Nature. 1997;390:395–398. doi: 10.1038/37108. [DOI] [PubMed] [Google Scholar]
- 16.Gerrish P J, Lenski R E. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;102–103:127–144. [PubMed] [Google Scholar]
- 17.Goodrich-Blair H, Kolter R. Homocysteine thiolactone is a positive effector of ςs levels in Escherichia coli. FEMS Microbiol Lett. 2000;185:117–121. doi: 10.1111/j.1574-6968.2000.tb09048.x. [DOI] [PubMed] [Google Scholar]
- 18.Harvey P H, Pagel M D. The comparative method in evolutionary biology. Oxford, United Kingdom: Oxford University Press; 1991. [Google Scholar]
- 19.Huey R B, Gilchrist G W, Carson M L, Berrigan D, Serra L. Rapid evolution of a geographic cline in size in an introduced fly. Science. 2000;287:308–309. doi: 10.1126/science.287.5451.308. [DOI] [PubMed] [Google Scholar]
- 20.Iida A, Harayama S, Iino T, Hazelbauer G L. Molecular cloning and characterization of genes required for ribose transport and utilization in Escherichia coli K-12. J Bacteriol. 1984;158:674–682. doi: 10.1128/jb.158.2.674-682.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lea D E, Coulson C A. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 22.Lederberg S. Genetics of host-controlled restriction and modification of deoxyribonucleic acid in Escherichia coli. J Bacteriol. 1966;91:1029–1036. doi: 10.1128/jb.91.3.1029-1036.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenski R E, Travisano M. Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proc Natl Acad Sci USA. 1994;91:6808–6814. doi: 10.1073/pnas.91.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenski R E, Rose M R, Simpson S C, Tadler S C. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am Nat. 1991;138:1315–1341. [Google Scholar]
- 25.Lin E C C. Dissimilatory pathways for sugars, polyols, and carboxylates. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 307–342. [Google Scholar]
- 26.Luria S E, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma W T, Sandri G H, Sarkar S. Analysis of the Luria-Delbrück distribution using discrete convolution powers. J Appl Prob. 1992;29:254–267. [Google Scholar]
- 28.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 29.Nakatsu C H, Korona R, Lenski R E, de Bruijn F J, Marsh T L, Forney L J. Parallel and divergent genotypic evolution in experimental populations of Ralstonia sp. J Bacteriol. 1998;180:4325–4331. doi: 10.1128/jb.180.17.4325-4331.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papadopoulos D, Schneider D, Meier-Eiss J, Arber W, Lenski R E, Blot M. Genomic evolution during a 10,000-generation experiment with bacteria. Proc Natl Acad Sci USA. 1999;96:3807–3812. doi: 10.1073/pnas.96.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 32.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider D, Duperchy E, Coursange E, Lenski R E, Blot M. Long-term experimental evolution in Escherichia coli. IX. Characterization of IS-mediated mutations and rearrangements. Genetics. 2000;156:477–488. doi: 10.1093/genetics/156.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simpson G G. The major features of evolution. New York, N.Y: Columbia University Press; 1953. [Google Scholar]
- 35.Sniegowski P D, Gerrish P J, Lenski R E. Evolution of high mutation rates in experimental populations of Escherichia coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- 36.Stewart F M. Fluctuation tests: how reliable are the estimates of mutation rates? Genetics. 1994;137:1139–1146. doi: 10.1093/genetics/137.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taddei F, Radman M, Maynard Smith J, Toupance B, Gouyon P H, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- 38.Travisano M, Vasi F, Lenski R E. Long-term experimental evolution in Escherichia coli. III. Variation among replicate populations in correlated responses to novel environments. Evolution. 1995;49:189–200. doi: 10.1111/j.1558-5646.1995.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 39.Treves D S, Manning S, Adams J. Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol. 1998;15:789–797. doi: 10.1093/oxfordjournals.molbev.a025984. [DOI] [PubMed] [Google Scholar]