

Abstract
Ferroptosis is an iron-dependent cell death with the accumulation of lipid peroxidation and dysfunction of antioxidant systems. As the critical regulator, glutathione peroxidase 4 (GPX4) has been demonstrated to be down-regulated in amyotrophic lateral sclerosis (ALS). However, the mechanism of ferroptosis in ALS remains unclear. In this research, bioinformatics analysis revealed a high correlation between ALS, ferroptosis, and Speedy/RINGO cell cycle regulator family member A (SPY1). Lipid peroxidation of ferroptosis in hSOD1G93A cells and mice was generated by TFR1-imported excess free iron, decreased GSH, mitochondrial membrane dysfunction, upregulated ALOX15, and inactivation of GCH1, GPX4. SPY1 is a “cyclin-like” protein that has been proved to enhance the viability of hSOD1G93A cells by inhibiting DNA damage. In our study, the decreased expression of SPY1 in ALS was resulted from unprecedented ubiquitination degradation mediated by MDM2 (a nuclear-localized E3 ubiquitin ligase). Further, SPY1 was identified as a novel ferroptosis suppressor via alleviating lipid peroxidation produced by dysregulated GCH1/BH4 axis (a resistance axis of ferroptosis) and transferrin receptor protein 1 (TFR1)-induced iron. Additionally, neuron-specific overexpression of SPY1 significantly delayed the occurrence and prolonged the survival in ALS transgenic mice through the above two pathways. These results suggest that SPY1 is a novel target for both ferroptosis and ALS.
Subject terms: Neuroscience, Gene regulation
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that involves upper and lower motor neurons (MNs). The manifestation characterized by progressive muscle weakness and muscle atrophy frequently ends in respiratory failure. Until now, edaravone and riluzole are the only drugs approved by FDA but with limited effect. Mutations in genes such as superoxide dismutase 1 (SOD1), C9ORF72, TDP43, FUS/TLS, OPTN, TBK1, GRN, NEK1, and C21ORF2 contribute to the etiology [1]. The pathogenesis has been revealed to be related to oxidative stress, mitochondrial dysfunction, hyperexcitability, impaired protein homeostasis, axonopathy, aberrant RNA metabolism, dysregulated vesicle transport, impaired DNA repair, nuclear export, and glial dysfunction [2]. Mutant hSOD1G93A, the earliest and most widely used model, presents the complex pathology of ALS [3]. Although lots of mechanisms have been studied, there is still a lack of effective treatment implying that current mechanisms can not well interpret the reasons for occurrence and development of ALS.
Unlike other programmed death, ferroptosis is an iron-dependent cell death with accumulation of lipid peroxidation and dysfunction of antioxidant systems [4]. The main recognized endogenous mechanisms against lipid peroxidation consist of glutathione peroxidase 4 (GPX4), ferroptosis suppressor protein 1 (FSP1), and tetrahydrobiopterin (BH4) restrictively synthesized by GTP cyclohydrolase-1 (GCH1) [5]. Interestingly, iron metabolism disorder is a common feature of almost all neurodegenerative diseases confirmed by blood samples, imaging, and histopathology [6]. As a strong oxidant, abnormally accumulated iron will generate highly reactive hydroxyl radicals, especially through the Fenton reaction [7]. However, the administration of antioxidants alone cannot delay the development of ALS [8]. In recent years, various iron chelators have been shown to prolong the survival in SOD1-mutant transgenic mice [9, 10]. Deferoxamine (DFO), a ferroptosis inhibitor, once was used as an iron chelator to be tested in neurodegenerative diseases such as Huntington’s disease (HD) and Parkinson’s disease (PD) [11, 12]. Moreover, deferiprone has already entered the phase II clinical trial of ALS (NCT03293069). On the contrary, edaravone has displayed restraint for ferroptosis through reducing lipid peroxidation and the same as CuII(ATSM), which is effective in the initial clinical trials of ALS [13, 14]. As an essential regulator of ferroptosis, deletion of GPX4 has been found to induce degeneration of MNs and summarize the main characteristics of ALS [15]. Furthermore, the down-regulated expression of GPX4 in ALS mice and patients has been validated, and the course can be delayed by overexpression of GPX4 [16–18]. Nevertheless, systematic research on the mechanism of ferroptosis in ALS remains blank compared with other neurodegenerative diseases. And whether there are other mechanisms independent of GPX4 involved is still unknown.
Speedy/RINGO cell cycle regulator family member A (SPY1) is a highly conserved “cyclin-like” protein, previously considered to be associated with several cancers [19–22]. It contributes to CDK2 activation without promoting CDK2 phosphorylation, by inducing a conformation change of the CDK2 T-loop that obstructs the substrate-binding cleft prior to kinase activation [23]. Functions majorly include cell proliferation, chromosome depolymerization, apoptosis, cell cycle checkpoint activation, and DNA damage response (DDR). Intersections between DDR and ferroptosis have been found in different tumors. The silencing of DDR crucial molecules such as ATM provides a robust protective phenotype for ferroptosis [24]. P53 has been continuously demonstrated to affect ferroptosis, and its classical upstream E3 ubiquitin-protein ligase MDM2 also regulates ferroptosis in a p53-independent manner [25, 26]. In the previous study, we first showed that the expression of SPY1 has a significant decrease in ALS. Furthermore, overexpression of SPY1 can increase the viability of hSOD1G93A cells by inhibiting the activation of ATM, ATR, and P53 in DDR [27]. However, an expression correlation between SPY1 and the ferroptosis suppressor GCH1 was found in the preliminary bioinformatics analysis of this study. It is still unclear whether there is a regulatory relationship between SPY1 and ferroptosis, and whether they will affect the progression of ALS.
In this research, we identified multiple mechanisms of ferroptosis in pathogenesis of ALS and clarified MDM2 accounting for ubiquitination degradation of SPY1. Furthermore, we confirmed that SPY1 was a novel regulator to inhibit ferroptosis by regulating GCH1/BH4 and transferrin receptor protein 1 (TFR1). These results provide new ideas for exploring the pathogenesis and treatment in ALS.
Materials and methods
Acquisition of dataset and data processing
The expression profiling by array was derived from series GSE7493 of the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. Total RNA was extracted from embryonic MNs isolated from the ventral spinal cord of e14 mutant SOD1G93A overexpressing rat embryos compared to wildtype SOD1 overexpressing embryos, then followed by a p75-antibody purification in combination with magnetic beads [28]. The probe ID was annotated by GPL1355 (Affymetrix Rat Genome 230 2.0 Array).
The transcriptome data contained 4 mutations (MU) and 5 wildtypes (WT) and was divided into 4 highs (High) and 4 lows (Low) according to the expression of SPY1. The original expression matrix was normalized with log2 and processed by R.
The ferroptosis dataset was downloaded from FerrDb (http://www.datjar.com:40013/bt2104/) which collects published drivers, suppressors, markers, inducers, inhibitors, and diseases associated with ferroptosis [29].
Analysis for functional enrichment and differential expression
The normalized expression matrix was analyzed by Gene set enrichment analysis (GSEA) Linux 4.1.0 software using the ferroptosis dataset to obtain the gene list, and then processed with the GSEA package in RStudio.
The limma package was used to identify the differential expressed genes (DEGs) in MU vs. WT and SPY1 High vs. Low. The cut-off criteria for statistical significance were |logFC| ≥ 1 and p < 0.05. Since there were four probes representing SPY1, in two of which logFC > 0 while logFC < 0 in the other. To avoid omission, we took the average value of normalized data in these two groups respectively followed by differential expression analysis. The volcano plot using the ggplot2 package with geom point function displayed DEGs in MU vs. WT, while the heatmap using complex heatmap package was for SPY1 High vs. Low.
DAVID v6.8 (http://david.ncifcrf.gov) provided DEGs in SPY1 High vs. Low with the gene ontology (GO) analysis, including annotation for biological process (BP), molecular function (MF), and cellular component (CC) [30].
Pearson analysis was tested to confirm the correlation of DEGs.
Reagents and antibodies
The reagents diluted according to the manufacturer’s instructions were as follows: z-VAD-fmk (Zvad, Promega, G7231), Necrostatin-1 (Nec-1, Sigma-Aldrich, N9037), Deferoxamine (DFO, Sigma-Aldrich, D9533), Ferrostatin-1 (Fer-1, Sigma-Aldrich, SML0583), RSL3 (Selleck, S8155), Erastin (Selleck, S7242), MG-132 (Enzo Life Science, BML-P102), pepstatin A (Sigma-Aldrich, P5318), E-64d (Sigma-Aldrich, E8640), (6 R)-5,6,7,8-tetrahydro-L-Biopterin (BH4, Cayman, 81880).
The antibodies diluted according to manufacturer’s were as follows: rabbit anti-ALOX15 (Abcam, ab244205), rabbit anti-FSP1 (Invitrogen, PA5-103183), rabbit anti-GDF15 (Abcam, ab105738), rabbit anti-GCH1 (Invitrogen, PA5-103865), rabbit anti-GPX4 (Abcam, ab125066), rabbit anti-SPY1 (Invitrogen, PA1-16959), rabbit anti-MDM2 (Proteintech, 17914-1-AP), rabbit anti-NEDD4 (Abcam, ab240753), rabbit anti-SKP2 (Abcam, ab183039), rabbit anti-TFR1 (Abcam, ab84036), rabbit anti-P53 (Proteintech, 10442-1-AP), rabbit anti-SP1 (Abcam, ab227383), rabbit anti-4-HNE (Abcam, ab46545), mouse anti-SMI32 (Santa Cruz, sc-133165), rabbit anti-Ubiquitin (Proteintech, 10201-2-AP), mouse anti-FLAG (Sigma-Aldrich, F1804), mouse anti-HA (Sigma-Aldrich, H3663), mouse anti-His (Abcam, ab18184), mouse anti-β-actin (Sigma-Aldrich, A5316), IRDye 800CW goat anti-rabbit IgG (Li-COR, 926-32210), IRDye 800CW goat anti-mouse IgG (Li-COR, 926-32211), HRP-labeled goat anti-rabbit IgG (Beyotime, A0208), HRP-labeled goat anti-mouse IgG (Beyotime, A0216), Alexa Fluor 488 labeled Goat anti-mouse IgG (ZSGB-BIO, ZF-0512), Alexa Fluor 594 labeled Goat anti-rabbit IgG (ZSGB-BIO, ZF-0516).
Cell culture and model establishment of ALS
NSC34, the motor neuronal cell line (Cedarlane Laboratories, Vancouver, Canada), is a hybrid of embryonic mouse spinal cord motor neuron and neuroblastoma cell line. It is characterized by extension of neuritis, generation of action potentials, expression of neurofilament proteins and choline acetyl-transferase, synthesis and storage of acetylcholine, and induction of twitching in co-cultured muscle cells [31]. The ALS cell model was established by stably transfection of mutant hSOD1G93A in NSC34, while the control was transfected with wild-type hSOD1 (WT) or empty puromycin lentivirus vector (EV).
SHSY5Y (human neuroblastoma), Hela (human cervical cancer), and HEK293T (human embryonic kidney) cells were obtained from the Cell Bank of Chinese Academy of Sciences.
The cells have been authenticated by short tandem repeat (STR) profiling and tested for mycoplasma contamination. The cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented by 10% fetal bovine serum (FBS, Natocor) and 100 U/mL of penicillin-streptomycin at 37 °C with 5% CO2.
Measurement of cell viability and cytotoxicity
The cell viability was evaluated by the cell counting kit-8 (CCK8, Beyotime, C0038) according to the manufacturer’s instructions. The absorbance was assessed at 450 nm by a microplate reader (BioTek Instruments, Winooski, VT, USA). Similarly, LDH cytotoxicity assay kit (Beyotime, C0017) was used to detect cell cytotoxicity in cells, and the absorbance was assessed at 490 nm by a microplate reader with multiscan spectrum (Enspire, PE, USA).
Measurement of lipid peroxidation, labile iron pool, and GSH
Cells were orderly stained with C11-BODIPY 581/591 fluorescence probe (1 μM, Invitrogen, C10445) and Hoechst 33342 (Invitrogen, H1399) for 30 min. The extent of lipid peroxidation was represented as the ratio of GFP(484/510 nm) to RFP (581/610 nm) fluorescence detected by the inverted fluorescence microscope (EVOS FL, AMG, USA) and quantified by ImageJ. Fluorescence was also measured by channel 488 Grn using the multiparameter flow cytometer (A60-Universal, Apogee, Britain), and the data were analyzed by FlowJo v10.8.1.
Accompanied by DFO chelating iron, Calcein-AM (1 μM, Invitrogen, C3099) was used to assess the labile iron pool (LIP). After incubation of Calcein-AM for 15 min, the fluorescence intensity at 496 nm (excitation wavelength) and 520 nm (emission wavelength) of initial and after iron chelation were measured by the microplate reader (Enspire, PE, USA) were recorded, respectively. The discrepancy between the latter and the former was indicated as the relative content of LIP.
The ratio of GSH/GSSG was tested by GSH and GSSG Assay Kit (Beyotime, S0053) according to the manufacturer’s instructions.
Morphology of cell and mitochondria
The cell morphology was observed using an inverted fluorescence microscope (EVOS FL, AMG, USA). Transmission electron microscopy (HITACHI-H7650) was used to examine the morphological changes of mitochondria in cells and spinal cord tissues.
Immunoblotting and immunoprecipitation
The cleaned cells and tissues were lysed by RIPA (Beyotime, P0013B) with the addition of PMSF (Beyotime, ST506) for 1 h at 4 °C. For immunoprecipitation, protein A + G magnetic beads (Beyotime, P2108) were incubated to remove nonspecific binding for 4 h at 4 °C. After incubation with antibodies overnight at 4 °C, the immunoprecipitates were separated by binding with beads for another 2 h at 4 °C and adequate washing. After concentration determination by the BCA protein assay kit (Beyotime, P0012), equal amounts of protein were transferred to the nitrocellulose membrane by 4–20% SDS-PAGE and transmembrane. Having finished blocking, the membrane was incubated with specific primary antibodies overnight at 4 °C. Subsequently, the rewarmed membrane was incubated with corresponding IRDye800-conjugated secondary antibodies for 1 h at room temperature. The visualization of blots was detected by the Odyssey infrared imaging system (Li-COR Biotechnology, Lincoln, NE, USA) and quantified by ImageJ.
In vivo and in vitro ubiquitination assay
For in vivo ubiquitination assay, cells were co-transfected with SPY1-Flag, MDM2-HA, and Ub-His for 24 h. Subsequently, cells were incubated with MG132 (10 μM) for 4 h to block proteasome degradation. For in vitro ubiquitination assay, SPY1-Flag (1 μg), MDM2-HA (1 μg), and Ub-His (2 μg) proteins which were expressed and purified in E. coli Rosetta (DE3) were incubated with E1 (50 nM) and E2 (500 nM) in a 30 μl ubiquitination assay buffer (20 mM HEPES, pH 7.2, 5 mM MgCl2, 0.1 mM DTT, and 1 mM ATP) at 30 °C for 2 h. Subsequent immunoprecipitation and immunoblotting experiments were performed as described previously using anti-ubiquitin and anti-His antibodies.
Plasmids, small-interfering RNA, and transfection
The SPY1 overexpression plasmid, empty vector with Flag-tag (SPY1-Flag and Flag), the SKP2, NEDD4, MDM2, GCH1, TFR1, P53 overexpression plasmids, empty vector with HA-tag (SKP2-HA, NEDD4-HA, MDM2-HA, GCH1-HA, TFR1-HA, P53-HA, and HA), and the Ubiquitin with His-tag (Ub-His) were purchased from Genscript (Nanjing, China). The designed plasmids expressing different domains of SPY1, wildtype of SP1, and SP1(S59A) were synthesized by Genscript (Nanjing, China). The small-interfering RNA targeting MDM2, SPY1, GCH1 (SiMDM2, SiSPY1, and SiGCH1), and control (SiNC) were synthesized by GenePharma (Shanghai, China). The siRNA sequences used were as follows:
SiMDM2, 5’-GCC AUU GCU UUU GAA GUU AUU-3’ and 5’-UAA CUU CAA AAG CAA UGG CUU-3’.
SiSPY1, 5’-CCA GAG GGC CUU GUC UAA UTT-3’ and 5’-AUU AGA CAA GGC CCU CUG GTT-3’.
SiGCH1, 5’-UAC AAG UAC CUA ACU UUA CUU-3’ and 5’-GUA AAG UUA GGU ACU UGU AAU-3’.
SiNC, 5’-UUC UCC GAA CGU GUC ACG UTT-3’ and 5’-ACG UGA CAC GUU CGG AGA ATT-3’.
The plasmids and siRNA (3 μg) were used to transiently transfect cells assisted by Lipofectamine2000 (Invitrogen, 11668030). Briefly, incubated with transfection for 4-6 h, cells were maintained in low-serum culture for 48 h to accomplish interfering expression for subsequent experiments.
Detection of mRNA level
The mRNA levels of target genes were detected by quantitative real-time polymerase chain reaction (qRT-PCR) as previously described [32]. The mouse primer sequences used in this study were as follows:
ALOX15, 5′-GACACTTGGTGGCTGAGGTCTT-3′ (forward) and 5′-TCTCTGAGATCAGGTCGCTCCT-3′ (reverse).
FSP1, 5′-GCGACCTTCAAGGACAACTTCC-3′ (forward) and 5′-GCCAGGATAAGATGTGAGAAGGG-3′ (reverse).
GDF15, 5′-AGCCGAGAGGACTCGAACTCAG-3′ (forward) and 5′-GGTTGACGCGGAGTAGCAGCT-3′ (reverse).
GCH1, 5′-AGCAAGTCCTTGGTCTCAGTAAAC-3′ (forward) and 5′-ACCGCAATCTGTTTGGTGAGGC-3′ (reverse).
GPX4, 5′-CCTCTGCTGCAAGAGCCTCCC-3′ (forward) and 5′-CTTATCCAGGCAGACCATGTGC-3′ (reverse).
SPY1, 5′-AAGGGACCAACTCTGGGACAGA-3′ (forward) and 5′-ACAGCTCCACTGTGATGCACAG-3′ (reverse).
TFR1, 5′-GAAGTCCAGTGTGGGAACAGGT-3′ (forward) and 5′-CAACCACTCAGTGGCACCAACA-3′ (reverse).
DMT1, 5′-TTGCAGCGAGACTTGGAGTGGT-3′ (forward) and 5′-GCTGAGCCAATGACTTCCTGCA-3′ (reverse).
FPN1, 5′-TTGGCAGGTGCTAGAAGGAAG-3′ (forward) and 5′-CAGGCCGGACATGACTTGTA-3′ (reverse).
β-actin, 5′-CCAGCCTTCCTTCTTGGGTAT-3′ (forward) and 5′-TGCTGGAAGGTGGACAGTGAG-3′ (reverse).
Prediction of E3 ligase for SPY1 ubiquitination
UbiBrowser (http://ubibrowser.ncpsb.org) is an integrated bioinformatics platform to predict and present the proteome-wide human E3-substrate interaction network based on multiple types of heterogeneous biological evidence including homology E3-substrate interaction, enriched domain and GO term pair, protein interaction network loop, and inferred E3 recognition consensus motif [33]. On account of the high homology and conservation, SPY1 was regarded as a substrate to predict the interacted E3 ligase using the UbiBrowser.
Measurement of mitochondrial lipid peroxidation and membrane potential
Cells were treated with 3-[4-(Perylenylphenylphosphino) phenoxy] propyltriphenylphosphoniumio-dide (MitoPeDPP, 0.1 μM, Dojindo, M466) for 30 min at 37 °C. The fluorescence of mitochondrial lipid peroxidation was measured by channel 488 Grn using the multiparameter flow cytometer (A60-Universal, Apogee, Britain), And analyzed by FlowJo v10.8.1.
Mitochondrial membrane potential (MMP) was detected through incubation with the JC-1 fluorescence probe (5 μM, Abcam, ab141387) but followed by the analysis using a flow cytometer (FC500, Beckmancoulter, USA). JC-1 aggregates showed double-positive for FL1 (488/525 nm) and FL2 (488/585 nm) suggesting the normal MMP, and monomers showed single-positive for FL1 implying the decrease of MMP. The data were analyzed by FlowJo v10.8.1.
Measurement of BH4
BH4 and oxidized biopterins (BH2) were evaluated by high-performance liquid chromatography (HPLC) followed by electrochemical and fluorescent detection respectively as described previously [34].
Experimental animals and behavioral evaluation
Mutant hSOD1G93A overexpressing (B6SJL-Tg-SOD1G93A-1Gur/J) transgenic mice purchased from Jackson Laboratory (Stock no. 002726; Bar Harbor, ME, USA) were bred with non-transgenic C57/BL6 mice to continue species. The genotypes were determined by PCR which is blind in the study. The littermates not expressing hSOD1G93Awere regarded as wild-type controls (WT). All experimental animals were conducted according to the protocol approved by the Harbin Medical University Experimental Animal Research Ethics Committee. The size of each group (n = 10, randomly assigned) is based on the preclinical study guidelines recommended by the ALS Institute of Therapeutic Development.
The model of hSOD1G93A mice with overexpressed SPY1 was established by intracerebroventricular injection of recombinant viruses targeting neuron-specific expression. In brief, the adeno-associated virus 9 (AAV9) carrying neuronal-specific promoter and overexpressed SPY1 (AAV9-hSyn-SPY1-mCherry) and control (AAV9-hSyn-mCherry) purchased from Genechem (Shanghai, China) were injected (108 TU/ml, 5 μl) into the lateral ventricle of hSOD1G93A mice at 60 d. The injection point is 1.5 mm beside the anterior fontanelle, 1.1 mm in the direction of posterior fontanelle and 2.0 mm deep. Then their expression was verified by fluorescence of cherry at 90 d.
The evaluation of mice behavior consisted of weight changes, rotarod retention, onset time, and survival period as Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND described. Body weight and rotarod retention were measured every other week. The onset was determined by the occurrence of rotating retention for less than 180 s. And the endpoint of mice defined as being unable to stand upright within 30 s after lying on one side was recorded.
Immunohistochemistry and immunofluorescence
Tissues of gastrocnemius muscle and lumbar spinal cord isolated from mice at 150 d were used for immunohistochemistry and immunofluorescence as described previously [35]. Briefly, the frozen section (8 μm) of gastrocnemius was stained with hematoxylin and eosin (H&E). The paraffin section (5 μm) of the spinal cord was stained orderly with specific primary and HRP-labeled secondary antibodies, then visualized by the DAB horseradish peroxidase color development kit (Beyotime, P0202) for immunohistochemistry. The images were obtained using the microscope (Leica, Wetzlar, Germany) and quantified by Image-Pro Plus. For immunofluorescence, sections were incubated with fluorescence-labeled secondary antibodies followed by the antifade mounting medium with DAPI (Beyotime, P0131). The images were captured using the inverted fluorescence microscope (Leica, Wetzlar, Germany).
Statistical analysis
All the data were tested for normal distribution. Based on at least three independent experiments, data were presented as mean ± standard deviation (SD). Variance between groups was similar as assessed by Bartlett’s test for one-way ANOVA. The differences between two groups were determined by the student’s t-test. The differences between multiple groups were analyzed by one-way or two-way ANOVA followed by Tukey’s post hoc tests. p < 0.05 was considered statistically significant. The analysis of Pearson correlation and Kaplan-Meier survival were performed using the software of GraphPad Prism 8 (San Diego, CA, USA).
Results
Transcriptome data of primary rat neurons reveals an association between ALS, ferroptosis, and SPY1
For the preliminary screening of related mechanism and target genes in ALS, GSE7493 (including 4 MU and 5 WT) from the GEO database was selected for analysis. Firstly, the transcriptome data from embryonic motor neurons of SOD1G93A rats was performed standardization to reduce the impact of individual deviations (Fig. 1A). Next, all 16324 genes were enriched by the GSEA tool using the ferroptosis-related gene set downloaded from FerrDb. The ALS transcriptome data showed significant enrichment in ferroptosis (NES = 1.26, p = 0.046) (Fig. 1B). To identify the target genes, gene differential expression analysis was performed. A total of 417 differentially expressed genes (DEGs) were screened. Among them, SPY1 (LogFC = −1.175, p = 0.028) was confirmed to be down-regulated (Fig. 1C) as previously detected in mice and cells [27]. Subsequently, to identify potential effector genes of SPY1 involved in ALS, the transcriptome data were divided into SPY1 high and low expression groups. 215 up-regulated genes and 205 down-regulated genes (p < 0.05, |LogFC| ≥ 1) were screened (Fig. 1D). Through further intersection with DEGs in MU vs. WT, SPY1 High vs. Low, and ferroptosis-related gene set, the GTP cyclohydrolase GCH1 was the only gene to link to ALS, ferroptosis, and SPY1 (Fig. 1E). Moreover, the DEGs in SPY1 High vs. Low were functionally annotated for biological process, cell composition, and molecular function by David, indicating that GTPase activity was also associated with SPY1 expression (Fig. 1F). Finally, the direct positive correlation between SPY1 and GCH1 was identified by Pearson correlation analysis (r = 0.7387, p = 0.0230) (Fig. 1G). These bioinformatic results suggest that SPY1 may play a role in affecting ferroptosis in ALS through GCH1.
Fig. 1. The association between ALS, ferroptosis, and SPY1 is uncovered in the transcriptome data of primary rat neurons.

The transcriptome data of GSE7493 abstracted from embryonic MNs in the SOD1G93A mutant model of ALS was downloaded for analysis. A Standardization with log2 for the data, including 4 wildtypes (WT) and 5 mutants (MU). B GSEA for ferroptosis in MU vs WT (NES = 1.26, p = 0.046). C Differential expression analysis in MU vs WT containing 193 up-regulated genes and 224 down-regulated genes (p < 0.05, |logFC | ≥ 1). D The data were divided into groups of 4 lows and 4 highs based on the expression of SPY1. The heatmap exhibited the top 20 DEGs in SPY1 High vs. Low (p < 0.05, |logFC | ≥ 1). E The Venn diagram overlapped 417 DEGs in MU vs. WT, 420 DEGs in SPY1 High vs. Low, and 259 ferroptosis-related genes. F DEGs in SPY1 High vs Low were performed enrichment in the GO terms of CC, BP, and MF via David. The bar chart showed the top 5 of each term sorted by gene ratio (p < 0.05). G Pearson correlation analysis was performed on the expression of SPY1 and GCH1 (r = 0.7387, p = 0.0230). Statistical analysis by Student’s t-test.
Ferroptosis is involved in the cell model of ALS
Although reversing the down-regulation of GPX4 can effectively delay the course of ALS [16–18], the existence of ferroptosis in ALS still needs more verification. As an in vitro cell line for MNs in vitro, the NSC34 cells were chosen to validate the results of bioinformatics analysis. ALS model was successfully established by stably transfecting hSOD1G93A. For preliminary judgment on cell death, the CCK8 method was used to observe the response to reagents. After 2 h of incubation with 2 μM RSL3 (a ferroptosis inducer that inhibits GPX4), the ferroptosis in NSC34 cells could be specifically controlled by DFO and Fer-1 (classical ferroptosis inhibitor), but unchanged with Zvad (apoptosis inhibitor for caspase) and Nec-1 (necroptosis inhibitor for RIPK1) (Fig. 2A). H2O2 administration has been widely used as a substitute model for ALS to mimic exogenous oxidative stress in apoptosis and DNA damage studies [36, 37]. Conversely, Zvad and Nec-1 exhibited protective effects on cells with H2O2-induced apoptosis, whereas ferroptosis inhibitor Fer-1 had no effect (Fig. 2B). Nevertheless, inhibitors of both apoptosis and ferroptosis enhanced the viability of hSOD1G93A cells (Fig. 2C). Furthermore, combining these two inhibitors showed notable higher viability than any other combinations or any individual reagent (Fig. 2C). These results indicate a contribution of both apoptosis and ferroptosis to ALS.
Fig. 2. Evidence of ferroptosis is found in the ALS cell model.

Cells were incubated with Zvad (20 μM), Nec-1 (50 μM), DFO (100 μM), Fer-1 (10 μM), and vehicle for 24 h. The cell viability was assessed by CCK8 assay. A The bar chart showed different inhibitors’ effects on the viability of NSC34 cells with induction of 2 μM RSL3 for 2 h (n = 6). B Different inhibitors’ effects on the oxidative stress model mediated by administration of H2O2 (200 μM) for 24 h in NSC34 cells (n = 6). C The effect of each alone or any two of four inhibitors combined on hSOD1G93A and WT cells (n = 6). D, E C11-BODIPY immunofluorescence staining was displayed in representative microscopy images. Scale bar, 100 μM. Fluorescence which shifts from red to green in response to oxidation, was quantified by the ratio of GFP (484/510 nm) to RFP (581/610 nm) using ImageJ, indicating the extent of lipid peroxidation (n = 3). F Electron microscopy images exhibited morphological changes in mitochondria. Scale bar, 1 μM. G The relative content of LIP was assessed to compare with hSOD1G93A and WT cells using Calcein-AM and DFO assay (n = 6). H The GSH and GSSG Assay Kit were used for the detection of GSH in hSOD1G93A and WT cells. The relative content was represented by the ratio of GSH/GSSG (n = 6). I The relative mRNA level of ALOX15, FSP1, GDF15, GCH1, and GPX4 in hSOD1G93A cells compared with control was measured by qRT-PCR (n = 6). J, K The protein level of ALOX15, FSP1, GDF15, GCH1 and GPX4 was tested by Western Blotting (n = 3). Values represent mean ± SD. Statistical analysis by one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
For systematical research, the generally accepted features including iron accumulation, lipid peroxidation, and GSH consumption involved in ferroptosis were tested [4]. Firstly, obvious lipid peroxidation was observed in hSOD1G93A compared with WT as measured by the C11-BODIPY fluorescent probe, and it could be limited by ferroptosis inhibitors (Fig. 2D, E). As illustrated in photos of Electron microscopy, hSOD1G93A resulted in the same mitochondrial reduction, increase of membrane density, and ridge destruction as ferroptosis induction (Fig. 2F). Besides, the elevated LIP was affirmed by the Calcein AM test (Fig. 2G). And with the detection kit, an abnormal decrease of GSH also appeared in hSOD1G93A compared with control (Fig. 2H). Further, expression of the screened ferroptosis DEGs and critical genes in the current research was verified by qRT-PCR and Western blots. The mutant led to the up-regulated ALOX15 and GDF15 (respectively a driver and a marker of ferroptosis), down-regulated GCH1 and GPX4, and unaltered FSP1 (Figs. 1C and 2I–K). These results systematically prove ferroptosis involved in ALS and relevant features.
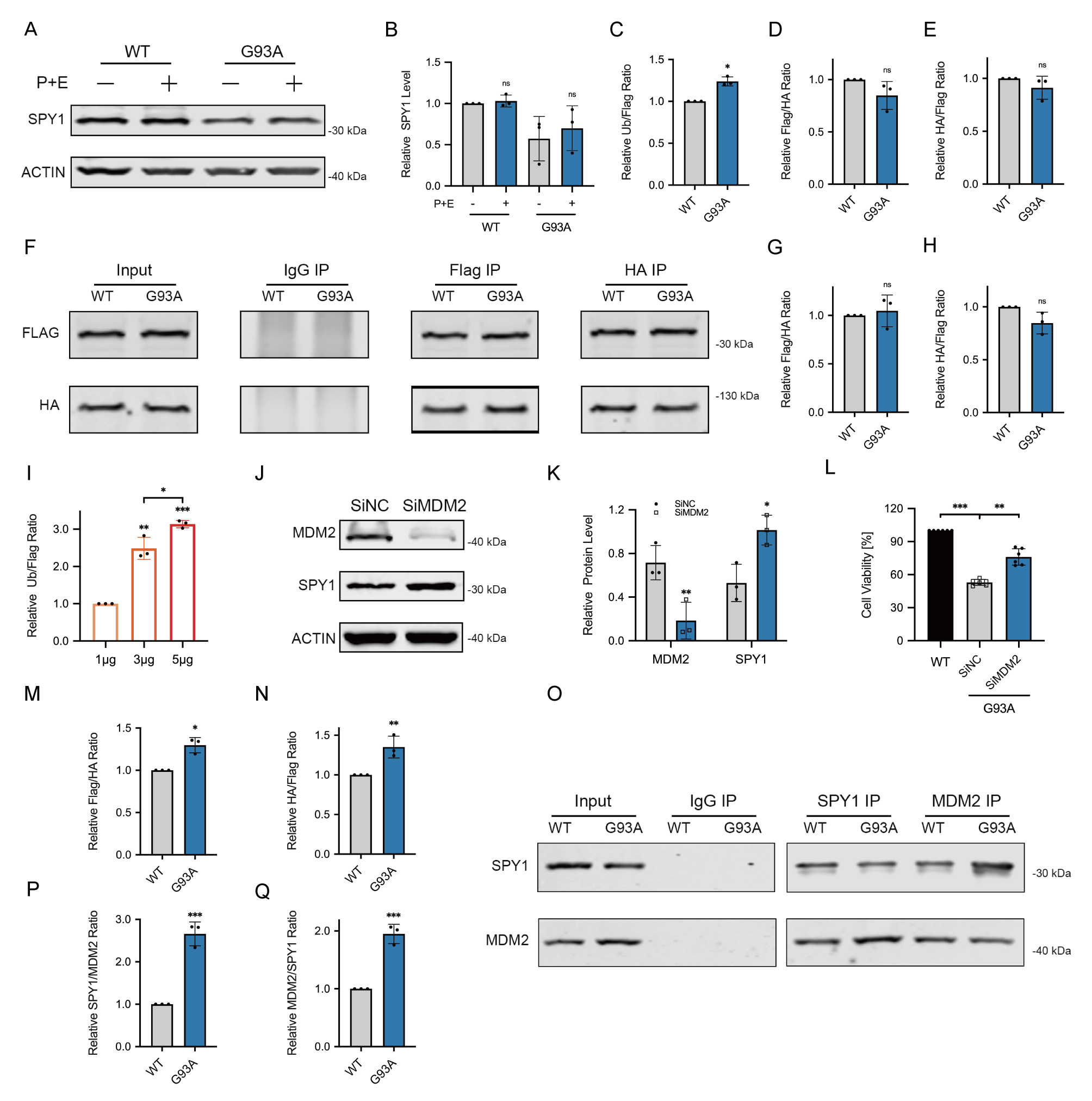
MDM2 degrades SPY1 through ubiquitination in ALS
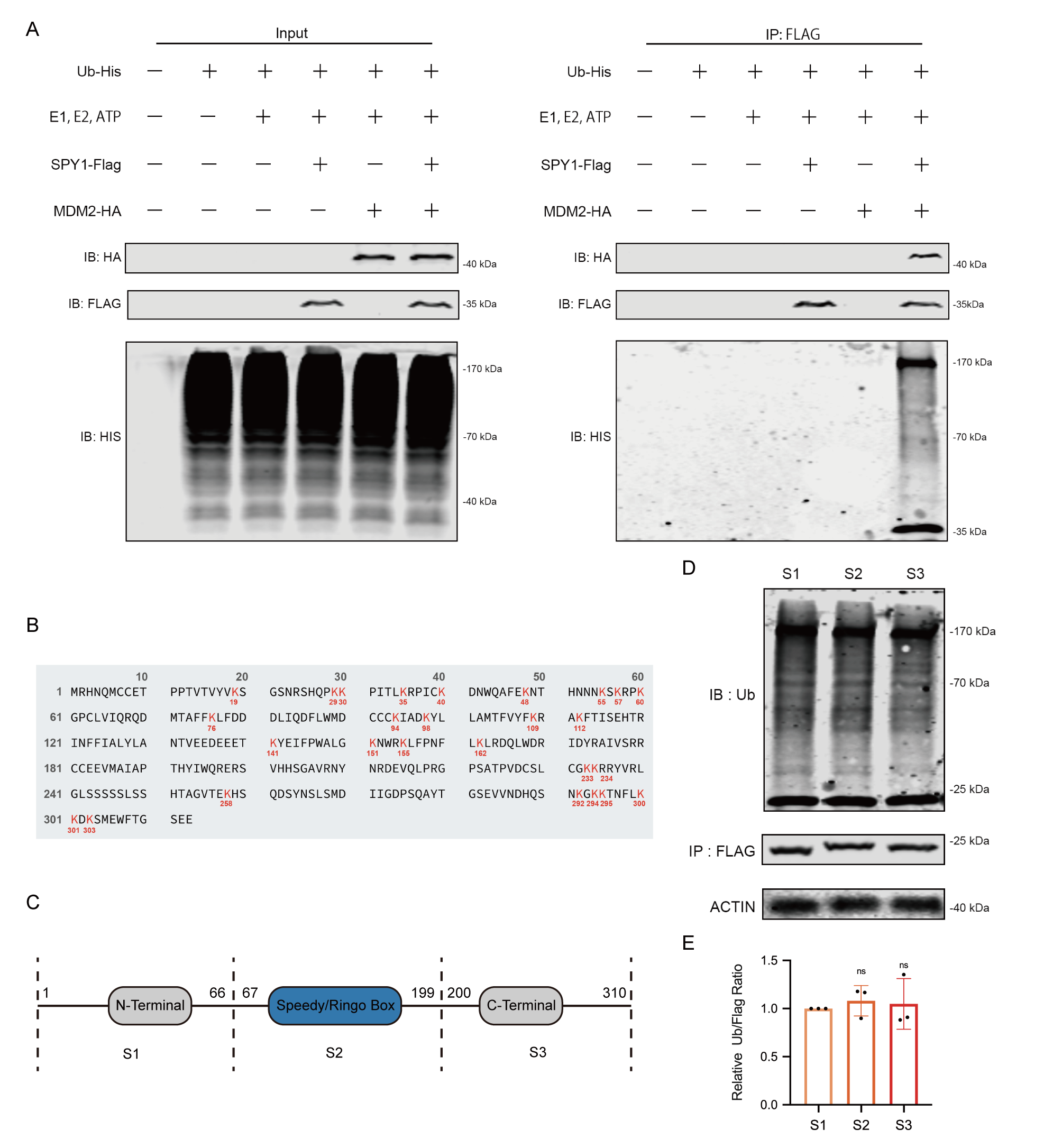
To explore the reason for the decrease of SPY1 in ALS, transcriptional and translational levels were detected by qRT-PCR and Western blots showing that the decreased proportion of mRNA was not as obvious as protein (approximately 46.3% vs. 83.8%) (Fig. 3A–C). Proteins are considered as important functional executors. Ubiquitin-proteasome system and autophagy-lysosome pathway are the major mechanisms for protein degradation. In further test, the expression of SPY1 was raised by administration of MG132 (proteasome inhibitor) rather than pepstatin A with E64D (PE, autophagy-lysosome inhibitor) (Figs. 3D, E and S1A, B). Both E3 ligase SKP2 and NEDD4 have been reported to interact with SPY1 through ubiquitination [38, 39]. As expected, the ubiquitination level of SPY1 was significantly elevated in hSOD1G93A (Figs. 3F and S1C). However, there was no significant increase of interaction between SPY1 and SKP2 or NEDD4 detected by co-immunoprecipitation (CoIP) (Figs. 3G and S1D–H). To find the true protein that mediated SPY1 ubiquitination in ALS, the sequence of amino acids was adopted to predict the E3 ligase with corresponding combined structure through the UbiBrowser. Among the results, MDM2 obtained the highest score of prediction (Fig. 3H). Strikingly, both SPY1 and its ubiquitination were increased in a dose-dependent manner with transfection of MDM2, and the degradation of SPY1 was inhibited in knockdown experiment. (Figs. 3I–K and S1I–K). In addition, knockdown of MDM2 enhanced the viability of hSOD1G93A cells (Fig. S1L). More exciting was that CoIP confirmed the enhanced exogenous and endogenous interaction between SPY1 and MDM2 in hSOD1G93A compared with WT (Figs. 3L and S1M–Q). The fact that MDM2 mediates ubiquitination of SPY1 was also verified in an in vitro assay (Fig. S2A). To locate specific sites among the 27 potential lysine ubiquitination sites, we examined the ubiquitination levels of the three structural domains of SPY1 separately but with no significant difference (Fig. S2B–E). Additionally, the expression of MDM2 in hSOD1G93A was significantly increased along with the unchanged NEDD4 and SKP2 (Fig. 3M, N). These results clarify that the elevated MDM2 mediates ubiquitination of SPY1, which mainly leads to decreased expression in ALS.
Fig. 3. The decrease of SPY1 in ALS is mediated by MDM2.

A, B Western blots and quantification for comparing the protein level of SPY1 in the cells of EV, WT, and hSOD1G93A (n = 3). C The relative mRNA level of SPY1 was assessed by qRT-PCR (n = 6). D, E MG132 (25 μM) was added into hSOD1G93A and WT cells for 6 h to evaluate the effect on expression of SPY1 (n = 3). F After transfection with SPY1-Flag for 24 h and incubation with MG132 (10 μM) for 4 h in cells of hSOD1G93A and WT, the supernatants of cell lysates were immunoprecipitated with anti-Flag and immunoblotted with anti-ubiquitin. G After transfection with SPY1-Flag and SKP2-HA for 24 h, the supernatants of cell lysates were immunoprecipitated with Flag (left) or HA (right) antibodies and immunoblotted with HA (lower panel) or Flag (upper panel) antibodies. H UbiBrowser was used to predict E3 ligase interacted with SPY1 as substrate. The prediction score of interaction was expressed from high to low by connecting lines from thick to fine. I, J Western blots and quantification for changes of SPY1 in NSC34 cells transfected with different doses of MDM2-HA (n = 3). K Alteration in ubiquitination of SPY1 with the increase of transfected MDM2. IP, anti-Flag. IB, anti-ubiquitin. L After transfection with SPY1-Flag and MDM2-HA for 24 h, immunoprecipitated with Flag (left) or HA (right) antibodies and immunoblotted with HA (lower panel) or Flag (upper panel) antibodies. M, N Western blots and quantification for NEDD4, SKP2, and MDM2 in the cells of EV, WT, and hSOD1G93A (n = 3). Values represent mean ± SD. Statistical analysis by one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
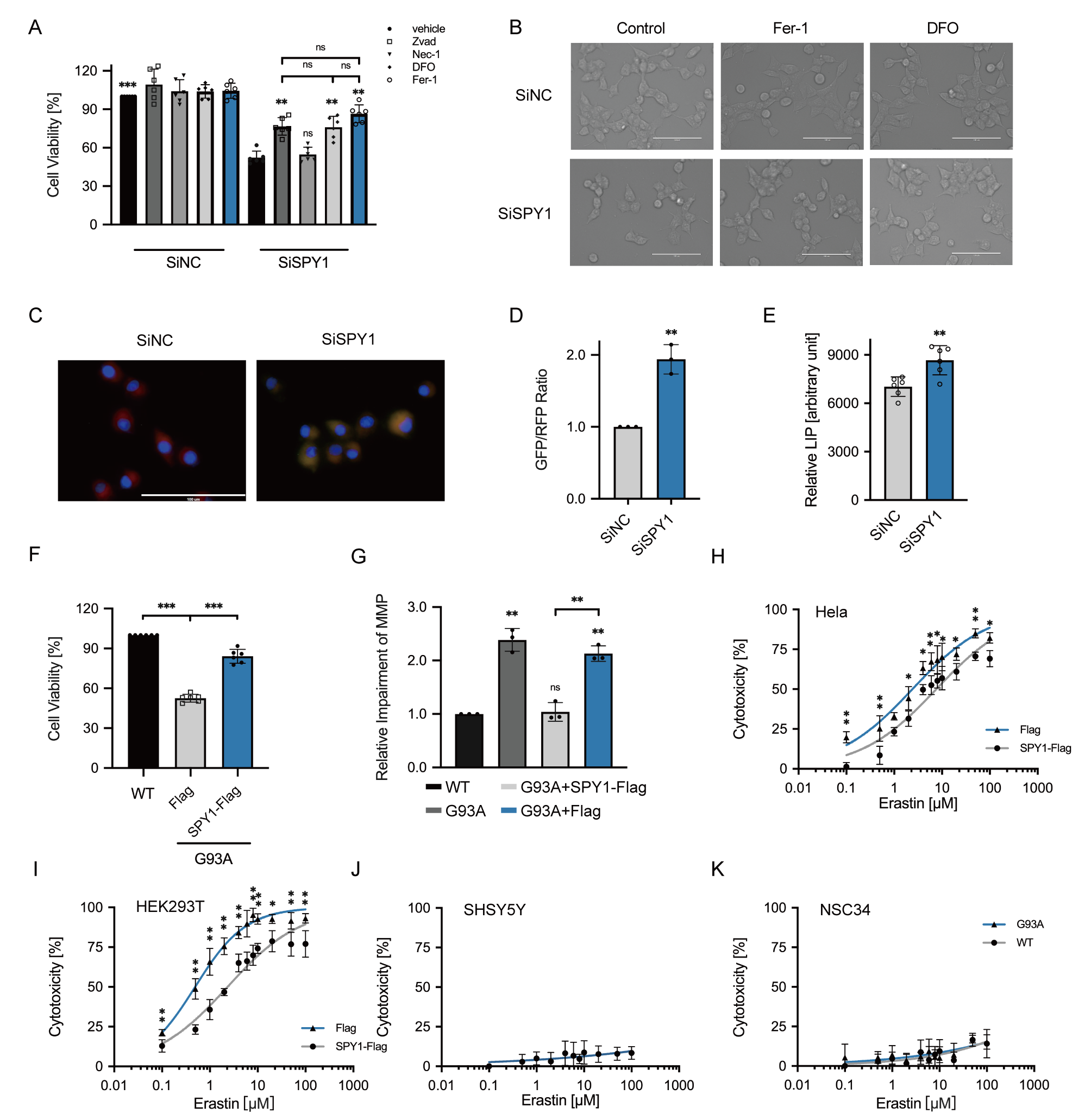
Overexpression of SPY1 inhibits ferroptosis in ALS
To identify the relationship between SPY1 and ferroptosis, the overexpression and knockdown of SPY1 were constructed respectively in hSOD1G93A and NSC34 cells (Fig. 4A). Previous experiments have shown the rescue of SPY1 to the apoptosis of hSOD1G93A cells [27]. The decrease of viability caused by knockdown of SPY1 showed a comparative improvement by ferroptosis and apoptosis inhibitors (Fig. S3A, B). Knockdown of SPY1 also led to lipid peroxidation and overloaded labile iron (Fig. S3C–E). By contrast, overexpression was verified to enhance the resistance to RSL3-induced ferroptosis in NSC34 cells (EC50 = 5.873 vs. 2.058 µM) and also reduce the death of hSOD1G93A cells(Figs. 4B, C and S3F). And the reduction of lipid peroxidation and free iron were dose-dependent on the transfection of SPY1 in hSOD1G93A cells (Fig. 4D–F). In spite of increased GSH, it did not correlate with the transfection dosage (Fig. 4G). The protection to mitochondrial function by overexpression of SPY1 was performed via reducing the fluorescence of mitochondrial lipid peroxidation and alleviating the decline of mitochondrial membrane potential (MMP) (Figs. 4H, I and S3G). These results conclude that SPY1 is an effective inhibitor of ferroptosis in ALS. Additionally, its’ resistance to RSL3-induced and Erastin (another ferroptosis inducer that inhibits xCT for reverse transport of cystine and glutamate)-induced ferroptosis also occurs in other tumor cells (Figs. 4J–L and S3H, I). However, SHSY5Y cells and NSC34-derived WT and hSOD1G93A cells were not sensitive to Erastin (Fig. S3J, K).
Fig. 4. Overexpressed SPY1 resists ferroptosis in the cell model of ALS.

A The effect of SPY1-Flag or SiSPY1 transfection in hSOD1G93A or NSC34 cells was evaluated by Western blotting. B Microscopic images were used to show response to RSL3 (2 μM for 2 h) of NSC34 cells with SPY1-Flag and Flag. Scale bar, 100 μM. C The effect of overexpressed SPY1 on ferroptosis induction was assessed by LDH assay in NSC34 cells with the treatment of diverse dosages of RSL3 for 2 h (n = 6). D, E Images and quantification of C11-BODIPY fluorescence exhibited the level of lipid oxidation with the increase of transfected SPY1-Flag in hSOD1G93A cells (n = 3). Scale bar, 100 μM. F, G The effect of increased SPY1 on relative LIP and GSH was shown (n = 6). H The lipid peroxidation of inner mitochondria membrane was measured by MitoPeDPP immunofluorescence staining followed by flow cytometry (channel 488, n = 3). I The changes in mitochondrial membrane potential caused by mutant SOD1 and overexpressed SPY1 were detected by JC-1 fluorescence staining followed by flow cytometry. J–L The effect of overexpressed SPY1 on RSL3-induced ferroptosis in SHSY5Y, Hela, and HEK293T cells was measured by LDH assay (n = 6). Values represent mean ± SD. Statistical analysis by one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
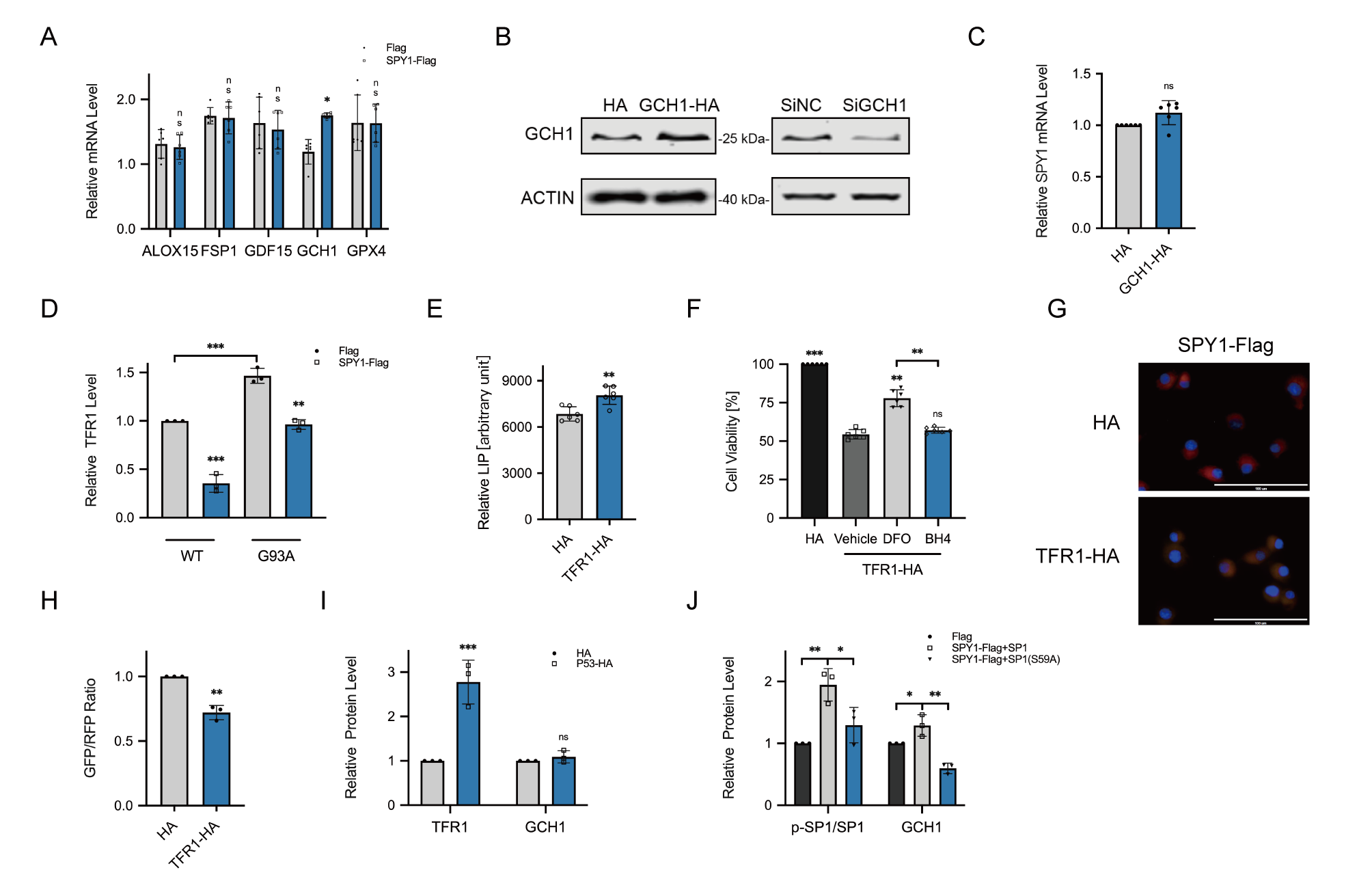
SPY1 resists ferroptosis by regulating GCH1/BH4 and TFR1
In order to further explore the mechanism of SPY1 regulating ferroptosis, the mRNA of vital genes altered in ALS was firstly screened in cells with overexpressed SPY1 and control. With the addition of the expression correlation analysis and functional annotation in SPY1 High vs. Low, the up-regulated ferroptosis suppressor GCH1 was speculated to be responsible for resistance to ferroptosis (Figs. 1D–G and S4A). Subsequently, the translational level of GCH1 was confirmed up-regulated in a dose-dependent manner with overexpressed SPY1 transfection (Fig. 5A, B). Plasmids for overexpression and knockdown of GCH1 were established (Fig. S4B). Besides, overexpression of GCH1 was unable to affect SPY1 inversely (Fig. S4C). GCH1 is a rate-limiting enzyme for the synthesis of BH4 which was significantly elevated by overexpression of SPY1 as well and turned decreased after the knockdown of GCH1 (Fig. 5C). Similarly, changes in viability and lipid oxidation induced by overexpressed SPY1 were reversed with knockdown of GCH1 (Fig. 5D–F). However, the LIP was found irrelevant to expression of GCH1 (Fig. 5G). Although administration of BH4 and overexpression of GCH1 protected most viability and lipid peroxidation in SPY1-knockdown cells (Fig. 5H, I). It is known that free iron plays an important role in generating lipid peroxidation, the combination of DFO and BH4 was casually tested and unexpectedly displayed an additional amelioration(Fig. 5H, I).
Fig. 5. SPY1 inhibits ferroptosis through regulatory of GCH1/BH4 and TFR1.

A, B Western blots and quantification for changes of GCH1 in hSOD1G93A cells transfected with different doses of SPY1-Flag (n = 3). C The effect of transfected SPY1-Flag and SiGCH1 on relative content of BH4 was shown (n = 6). D–G The effect of knockdown GCH1 on viability, lipid oxidation, and LIP in overexpressed SPY1 cells compared with control (in D and G, n = 6; in F, n = 3). Scale bar, 100 μM. H The viability was measured in SiSPY1 cells with transfected GCH1-Flag and treatment of BH4 (50 μM) and DFO (100 μM) for 24 h (n = 6). I The extent of lipid peroxidation was assessed by C11-BODIPY fluorescence staining followed by flow cytometry (channel 488). J The relative mRNA of TFR1, DMT1 and FPN1 in SPY1-Flag cells compared with control (n = 6). K Western blots for TFR1 in WT and hSOD1G93A cells with overexpressed SPY1 or control were shown. L After sequential transfection with P53-HA and control and incubation with DFO (100 μM), Fer-1 (10 μM), Zvad (20 μM), and vehicle for 24 h. The viability of SPY1-Flag cells was assessed by CCK8 assay (n = 6). M Western blots for P53, TFR1, and GCH1 in SPY1-Flag cells with overexpressed P53 and control were shown. N Western blots were shown for expression of p-SP1, SP1, and GCH1 in SPY1-Flag cells with overexpressed SP1(S59A) and wild type. Values represent mean ± SD. Statistical analysis by one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
So as to seek the modulation of labile iron independent on GCH1/BH4 axis, the transcriptional level of iron transport-related genes was detected in cells of overexpressing SPY1. Compared with control, the mRNA of TFR1 responsible for transporting iron into cells was increased along with unchanged DMT1 and FPN1 which were respectively accounting for intracellular iron to release and export (Fig. 5J). Consistent with mRNA, the protein level exhibited that overexpression of SPY1 lowered the abnormally elevated TFR1 in hSOD1G93A (Figs. 5K and S4D). Moreover, the LIP restored by TFR1 led to lipid peroxidation and cell death of overexpressed SPY1 cells (Fig. S4E–H).
As a multi-functional transcription factor, P53 has been demonstrated to be downregulated by SPY1 in our previous study [27]. To determine whether SPY1’s resistance to ferroptosis is mediated by regulating P53, we examined the effects of overexpressing P53 in SPY1-Flag cells on viability and related proteins. It was found that the inverse cell death caused by overexpressed P53 could be rescued by apoptosis inhibitors as well as by ferroptosis inhibitors, especially DFO implying a link between P53 and the transport of free iron (Fig. 5L). And the expression of TFR1 rather than GCH1 was upregulated with the overexpression of p53 (Figs. 5M and S4I). Studies have proposed that SP1 is likely to be a transcriptional activator of GCH1, and the phosphorylation of SP1 mediated by CDK2 can activate downstream transcriptional activity [40]. After expressing the mutation of SP1 (S59A), we found that SPY1 could activate GCH1 by upregulating the phosphorylation level of SP1 (Figs. 5N and S4J). Taken together, the restraint of SPY1 on ferroptosis attributes to inhibiting lipid peroxidation by regulating GCH1/BH4 axis and TFR1 expression.
SPY1 protects mouse model of ALS by inhibiting neuronal ferroptosis
To demonstrate the impact of SPY1-overexpression on the progression of ALS in vivo, the recombinant virus targeting neuron-specific expression was injected into the lateral ventricle of hSOD1G93A mice (Fig. 6A). Progressive muscle weakness and atrophy are the main manifestations of ALS. To evaluate the status of muscles, rotarod test and body weight were monitored during the course. The decline of exercise capacity was postponed in hSOD1G93A mice with SPY1-overexpression (Fig. 6B). And the occurrence of weight loss was also delayed for approximately 2 weeks (Fig. 6C). The degree of muscle weakness and muscle atrophy are dominant factors affecting the onset and survival of ALS. Compared with control, the overexpression of SPY1 retarded the onset (128.90 ± 8.17 vs. 120.40 ± 7.28 d) and prolonged the survival (149.90 ± 12.72 vs. 136.80 ± 7.98 d) (Fig. 6D, E).
Fig. 6. SPY1 plays a protective role in hSOD1G93A mice by resisting neuronal ferroptosis.

hSOD1G93A transgenic mice at 60 d were injected with AAV9-hSyn-SPY1-mCherry and AAV9-hSyn-mCherry. A The overexpressed SPY1 in green MNs of lumbar spinal cord stained with SMI32 was verified by autofluorescence of mCherry at 90 d. Scale bar, 50 μM. B, C The decreased body weight and rotarod test were recorded (n = 10). D, E Kaplan–Meier survival analysis for probability of onset and survival (n = 10). F Images of immunohistochemical staining in end-stage mice. First column for the gastrocnemius muscle stained with H&E. The residue for the lumbar spinal cord tissue immunohistochemically stained with SMI32, TFR1, and 4-HNE. Scale bar, 50 μM. G–I Quantification for the number of SMI32 positive cells and relative density of TFR1 and 4-HNE (n = 3). J Electron microscopy for mitochondrial changes of the lumbar spinal cord in hSOD1G93A mice with overexpression of SPY1 and control. Scale bar, 1 μM. K, L Western blots and quantification for the effect of mutant SOD1 and overexpressed SPY1 on the expression of GCH1 in lumbar spinal cord (n = 3). Values represent mean ± SD. Statistical analysis by one-way or two-way ANOVA followed by Tukey’s post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, tissues of gastrocnemius and lumbar spinal cord from mice at 150 d were performed histochemical staining to elucidate the protective mechanism of SPY1. It was shown that SPY1 significantly delayed the neurogenic atrophy in hSOD1G93A mice corresponding to the rotarod test (Fig. 6F). As a specific marker of MNs, SMI32 staining in the anterior horn of the spinal cord revealed that SPY1 reduced the loss of MNs (Fig. 6F, G). The increased expression of TFR1 itself is strong evidence of iron accumulation which was found in hSOD1G93A and corrected by SPY1 (Fig. 6F, H) [7]. Additionally, immunohistochemical results of 4-HNE, a marker of lipid oxidation, showed that the initially elevated level of lipid peroxidation was decreased in mice overexpressing SPY1 (Fig. 6F, I). Moreover, SPY1 maintained healthier mitochondria with relative normal morphology (Fig. 6J). Having verified the expression of TFR1 by immunohistochemistry, GCH1 was proved down-regulated in hSOD1G93A mice and its activation by overexpressed SPY1 (Fig. 6K, L). These results obtained from hSOD1G93A mice are consistent with those in vitro, emphasizing that SPY1 prolongs survival by inhibiting neuronal ferroptosis in ALS.
Discussion
The death of motor neurons in ALS has always been controversial. Previous studies on the hSOD1G93A model mainly focused on apoptosis. Whereas embryonic stem-cell-derived MNs co-cultured with sporadic ALS (sALS) astrocytes were found to be triggered to degenerate through necroptosis rather than apoptosis dependent on caspases [41]. This distinction may be related to the uncertain mechanism of sALS astrocytes used, because the silencing of SOD1 or TDP-43 exhibited no effects on the loss of MNs, which indicates quite different from traditional models. Moreover, there seems to be more controversy about necroptosis in ALS. Another study holding the same view detected the expression of RIPK1, RIPK3, and MLKL (main components of necroptosis) in ALS patients and mouse models with mutant OPTN and SOD1, and concluded that the limitation of RIPK3 or RIPK1 protected ALS [42]. While diverse results were shown in the other three studies [43–45]. In particular, RIPK3 and MLKL have recently been proved undetected in central nervous system tissues of healthy or hSOD1G93A mice [16]. Consistent with our study, the RIPK1 inhibitor Nec-1 displayed no effects on hSOD1G93A cells, so we temporarily do not support the existence of necroptosis especially in hSOD1G93A model. In terms of ferroptosis in our research, its correlation with ALS was firstly concluded by the enrichment of bioinformatics analysis. Same as Dixon’s results in HT-1080 cells [4], Fer-1 exhibited no effect on NSC34 cells with H2O2 administered which is usually used to study apoptosis. Similarly, apoptosis inhibitor has no impact on induced ferroptosis. Both inhibitors showed a comparable specificity and obvious protection in the model of hSOD1G93A. Meanwhile, compared with hSOD1G93A, it is suggested that the oxidative stress model H2O2 still cannot fully replace the mutation. Furthermore, the lipid peroxidation in hSOD1G93A could be alleviated by ferroptosis inhibitors. Hence we reasonably infer that ferroptosis exists in ALS.
As for current results, lipid peroxidation involved in mutant hSOD1G93A-induced ferroptosis attributes to excess free iron imported by TFR1, decreased GSH, mitochondrial membrane dysfunction, upregulated ALOX15, and inactivation of GCH1, GPX4. The mRNA of DMT1 and TFR1 has been shown upregulated in SH-SY5Y cells with hSOD1G93A [7]. And similar results were also found in the cervical spinal cord of SOD1G37R mice at end-stage [46]. Despite focusing on the role of SPY1, the only detected expression of TFR1 is still enhanced in accordance with previous studies. Besides being involved in the Fenton reaction, the catalyzer iron bound with upregulated lipoxygenase ALOX15, which is a marker and also a driver of ferroptosis, promotes the stereo-specific peroxidation of free and esterified polyunsaturated fatty acids generating a spectrum of bioactive lipid mediators [47]. Accompanied by the decline of GPX4 consistent with preceding research [16–18], the new resistance factor GCH1 was verified downregulated demonstrating a unique dysfunctional mechanism of protecting phosphatidylcholine phospholipids through two polyunsaturated fatty acyl chains and increasing the level of coenzyme Q10 that is independent of the GSH/GPX4 system [48]. However, we have also detected the upregulation of ferroptosis suppressor GDF15 which seems not ought to emerge. The suppressive role was derived from a study in which its knockout can promote Erastin-induced ferroptosis by decreasing xCT expression [49]. But it has been shown in ALS that xCT expression is enriched in microglia compared to total mouse spinal cord and absent from motor neurons [50]. So the upregulated GDF15 by the mutation of hSODG93A can not rescue the various features of ferroptosis, even including functional defects in xCT/GSH system and still needs functional study in ALS. In addition, it is noted that Erastin can not result in death of hSOD1G93A or WT cells. Indeed, it is consistent with a previous study in which the resistance to Erastin in NSC34 and SHSY5Y cells is considered to be associated with the intact function of the xCT system [51]. Considering that Erastin still fails to induce ferroptosis in hSOD1G93A cells with functional defects in xCT/GSH system, presumably there is some unknown resistance property to Erastin in NSC34 cells. But this does not affect the results of those altered ferroptosis characteristics caused by the mutation of hSOD1G93A and the inhibition by SPY1.
It has been reported that the proteasome degrades SPY1 in U2OS cells [21]. Having excluded the autophagy-lysosome pathway, our experiments demonstrate the ubiquitin-proteasome system for SPY1 to degrade in MNs. CoIP has assisted us to rule out the possibility that the increased ubiquitination is mediated by NEDD4 and SKP2. This is probably because of no changes in the expression of both in ALS compared with WT. Although different domains of SPY1 were used to detect ubiquitination levels in order to narrow the search for specific ubiquitination sites, the insignificant results may suggest the presence of polyubiquitination sites in SPY1 and still need further study.
Via data analysis and validation in vitro and in vivo experiments, our study is the first to find the relationship between SPY1 and ferroptosis. SPY1 was regarded as an apoptosis suppressor by inhibiting p53-mediated DNA damage in previous studies [27, 52]. In the part of bioinformatics analysis, the DEGs of SPY1 High vs. Low were significantly enriched in negative regulation of apoptosis, which also suggests relatively reliability of data. In vitro, a similar protective effect was found in SPY1 knockdown cells with specific apoptosis and ferroptosis inhibitors. Especially the effective Fer-1 has no impact on H2O2-induced apoptosis and DNA damage, implying additional ferroptosis involved in SPY1 knockdown. Therefore, we do not exclude that SPY1 has other protective effects on ALS while resisting ferroptosis. In accordance with the reasonable screening of transcriptome data and verification in experiments, we confirm SPY1 as a new upstream activator of GCH1/BH4 pathway to control most of the lipid peroxidation in ALS. However, the reduced level of free iron by SPY1 was not mediated through GCH1. In addition, the combination of DFO and BH4 had a stronger effect on cell viability and lipid peroxidation, suggesting that the decrease of partial lipid peroxidation produced by free iron is also an important mechanism for SPY1 to resist ferroptosis. Among the genes related to iron transport, the downregulated TFR1 was detected to lessen excess labile iron. Considering the resistance of SPY1 to GPX4 inhibitor RSL3 in NSC34 and different tumor cells, we believe that SPY1 and downstream pathways may be effective protectors against ferroptosis caused by GPX4 deletion. Besides, there may be no direct regulatory relationship between SPY1 and the level of GSH, but improved lipid peroxidation reduces the consumption of partial GSH to resist ferroptosis induced by Erastin in other tumors.
SPY1/CDK2 complex, the unique binding motivates CDK2 phosphokinase activity, which is likely to affect the status of transcription factors through phosphorylation [23]. Chromatin immunoprecipitation assays have revealed that proteins that bind in vivo to the endogenous GCH1 proximal promoter actively transcribing GCH1 within living PC12 cells, including SP1 [53, 54]. The phosphorylation of SP1 mediated by CDK2 has been reported to activate downstream CTalpha transcription [40]. Moreover, our previous experiments have confirmed the interaction between SPY1 and CDK2 in hSOD1G93A cells [27]. In our study, functional enrichment of the gene SPY1 in ALS showed a positive regulation of transcription which also lays a foundation for SPY1 to regulate the expression of GCH1 by phosphorylating SP1. In addition, inhibition or knockdown of USP7 enhanced the ubiquitination of p53 along with the decreased levels of p53, which led to down-regulation of TFR1 [55]. Our results also demonstrate that SPY1 regulates TFR1 through the inhibition of P53.
Conclusion
In conclusion, our results demonstrate that ferroptosis is involved in the pathogenesis of ALS. The decreased SPY1 in ALS is due to the ubiquitination mediated by interaction with MDM2. Furthermore, SPY1 inhibits ferroptosis by regulating GCH1/BH4 axis and TFR1 to delay the occurrence and progression of ALS.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author contributions
DW and HF contributed to conception of the study and manuscript preparation. SY performed the bioinformatics analysis. DW, WL, DH, HW, and MG performed the cell experiment. YW, CC, CZ, XS, XT, WZ, LH, and DZ helped perform the mice experiment and revision of manuscript.
Funding
This work was supported by Major research project of National Natural Science Foundation of China (Project No. 91849133) and General project of National Natural Science Foundation of China (Project No. 81571227 and Project No. 82271450).
Data availability
All data are available from the corresponding author upon reasonable request. Supplementary figures and original Western blots images are available at Supplementary Materials.
Competing interests
The authors declare no competing interests.
Ethics approval
Animal studies were approved by the Ethics Committee of Harbin Medical University.
Footnotes
Edited by M Piacentini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-022-01089-7.
References
- 1.Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108:822–42. doi: 10.1016/j.neuron.2020.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Prim. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 3.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 4.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28:2029–44. doi: 10.1038/s41418-021-00814-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petillon C, Hergesheimer R, Puy H, Corcia P, Vourc’h P, Andres C, et al. The relevancy of data regarding the metabolism of iron to our understanding of deregulated mechanisms in ALS; Hypotheses and Pitfalls. Front Neurosci. 2019;12:1031. doi: 10.3389/fnins.2018.01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadzhieva M, Kirches E, Wilisch-Neumann A, Pachow D, Wallesch M, Schoenfeld P, et al. Dysregulation of iron protein expression in the hSOD1G93A model of amyotrophic lateral sclerosis. Neuroscience. 2013;230:94–101. doi: 10.1016/j.neuroscience.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Orrell RW, Lane RJ, Ross M. Antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2007;1:CD002829. [DOI] [PMC free article] [PubMed]
- 9.Hadzhieva M, Kirches E, Mawrin C. Review: iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol Appl Neurobiol. 2014;40:240–57. doi: 10.1111/nan.12096. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Zhang X, Chen S, Zhang X, Zhang S, Youdium M, et al. Prevention of motor neuron degeneration by novel iron chelators in SOD1(hSOD1G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener Dis. 2011;8:310–21. doi: 10.1159/000323469. [DOI] [PubMed] [Google Scholar]
- 11.Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–97. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Xie W, Qu S, Pan T, Wang X, Le W. Neuroprotection by iron chelator against proteasome inhibitor-induced nigral degeneration. Biochem Biophys Res Commun. 2005;333:544–9. doi: 10.1016/j.bbrc.2005.05.150. [DOI] [PubMed] [Google Scholar]
- 13.Homma T, Kobayashi S, Sato H, Fujii J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp Cell Res. 2019;384:111592. doi: 10.1016/j.yexcr.2019.111592. [DOI] [PubMed] [Google Scholar]
- 14.Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, et al. CuII (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharm. 2020;177:656–67. doi: 10.1111/bph.14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–106. doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. 2022;29:1187–98. doi: 10.1038/s41418-021-00910-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuo T, Adachi-Tominari K, Sano O, Kamei T, Nogami M, Ogi K, et al. Involvement of ferroptosis in human motor neuron cell death. Biochem Biophys Res Commun. 2021;566:24–29. doi: 10.1016/j.bbrc.2021.05.095. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Na R, Danae McLane K, Thompson CS, Gao J, Wang X, et al. Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1hSOD1G93A mice. Sci Rep. 2021;11:12890. doi: 10.1038/s41598-021-92369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fifield BA, Qemo I, Kirou E, Cardiff RD, Porter LA. The atypical cyclin-like protein Spy1 overrides p53-mediated tumour suppression and promotes susceptibility to breast tumourigenesis. Breast Cancer Res. 2019;21:140. doi: 10.1186/s13058-019-1211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golipour A, Myers D, Seagroves T, Murphy D, Evan GI, Donoghue DJ, et al. The Spy1/RINGO family represents a novel mechanism regulating mammary growth and tumorigenesis. Cancer Res. 2008;68:3591–3600. doi: 10.1158/0008-5472.CAN-07-6453. [DOI] [PubMed] [Google Scholar]
- 21.Porter LA, Dellinger RW, Tynan JA, Barnes EA, Kong M, Lenormand JL, et al. Human Speedy: a novel cell cycle regulator that enhances proliferation through activation of Cdk2. J Cell Biol. 2002;157:357–66. doi: 10.1083/jcb.200109045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lubanska D, Market-Velker BA, deCarvalho AC, Mikkelsen T, Fidalgo da Silva E, Porter LA. The cyclin-like protein Spy1 regulates growth and division characteristics of the CD133+ population in human glioma. Cancer Cell. 2014;25:64–76. doi: 10.1016/j.ccr.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 23.McGrath DA, Fifield BA, Marceau AH, Tripathi S, Porter LA, Rubin SM. Structural basis of divergent cyclin-dependent kinase activation by Spy1/RINGO proteins. EMBO J. 2017;36:2251–62. doi: 10.15252/embj.201796905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27:1008–22. doi: 10.1038/s41418-019-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020;34:526–43. doi: 10.1101/gad.334219.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XD, Zhu MW, Shan D, Wang SY, Yin X, Yang YQ, et al. Spy1, a unique cell cycle regulator, alters viability in ALS motor neurons and cell lines in response to mutant SOD1-induced DNA damage. DNA Repair. 2019;74:51–62. doi: 10.1016/j.dnarep.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Lobsiger CS, Boillée S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci USA. 2007;104:7319–26. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou N, Bao J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database (Oxf) 2020;2020:baaa021. doi: 10.1093/database/baaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier O, Böhm J, Dahm M, Brück S, Beyer C, Johann S. Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem Int. 2013;62:1029–38. doi: 10.1016/j.neuint.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Yang YQ, Zheng YH, Zhang CT, Liang WW, Wang SY, Wang XD, et al. Wild-type p53-induced phosphatase 1 down-regulation promotes apoptosis by activating the DNA damage-response pathway in amyotrophic lateral sclerosis. Neurobiol Dis. 2020;134:104648. doi: 10.1016/j.nbd.2019.104648. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z, et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat Commun. 2017;8:347. doi: 10.1038/s41467-017-00299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNeill E, Stylianou E, Crabtree MJ, Harrington-Kandt R, Kolb AL, Diotallevi M, et al. Regulation of mycobacterial infection by macrophage Gch1 and tetrahydrobiopterin. Nat Commun. 2018;9:5409. doi: 10.1038/s41467-018-07714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cong C, Liang W, Zhang C, Wang Y, Yang Y, Wang X, et al. PAK4 suppresses motor neuron degeneration in hSOD1hSOD1G93A -linked amyotrophic lateral sclerosis cell and rat models. Cell Prolif. 2021;54:e13003. doi: 10.1111/cpr.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda N, Shah SV, Endonuclease-induced DNA. damage and cell death in oxidant injury to renal tubular epithelial cells. J Clin Invest. 1992;90:2593–7. doi: 10.1172/JCI116154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 38.Dinarina A, Santamaria PG, Nebreda AR. Cell cycle regulation of the mammalian CDK activator RINGO/Speedy A. FEBS Lett. 2009;583:2772–8. doi: 10.1016/j.febslet.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 39.Al Sorkhy M, Craig R, Market B, Ard R, Porter LA. The cyclin-dependent kinase activator, Spy1A, is targeted for degradation by the ubiquitin ligase NEDD4. J Biol Chem. 2009;284:2617–27. doi: 10.1074/jbc.M804847200. [DOI] [PubMed] [Google Scholar]
- 40.Banchio C, Schang LM, Vance DE. Phosphorylation of SP1 by cyclin-dependent kinase 2 modulates the role of SP1 in CTP:phosphocholine cytidylyltransferase alpha regulation during the S phase of the cell cycle. J Biol Chem. 2004;279:40220–6. doi: 10.1074/jbc.M406468200. [DOI] [PubMed] [Google Scholar]
- 41.Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–8. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–8. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dominguez S, Varfolomeev E, Brendza R, Stark K, Tea J, Imperio J, et al. Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS. Cell Death Differ. 2021;28:915–31. doi: 10.1038/s41418-020-00625-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dermentzaki G, Politi KA, Lu L, Mishra V, Pérez-Torres EJ, Sosunov AA, et al. Deletion of Ripk3 prevents motor neuron death in vitro but not in vivo. eNeuro. 2019;6:ENEURO.0308-18.2018. [DOI] [PMC free article] [PubMed]
- 45.Wang T, Perera ND, Chiam MDF, Cuic B, Wanniarachchillage N, Tomas D, et al. Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ. 2020;27:1728–39. doi: 10.1038/s41418-019-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:610–9. doi: 10.1523/JNEUROSCI.5443-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perry SC, Kalyanaraman C, Tourdot BE, Conrad WS, Akinkugbe O, Freedman JC, et al. 15-Lipoxygenase-1 biosynthesis of 7 S,14S-diHDHA implicates 15-lipoxygenase-2 in biosynthesis of resolvin D5. J Lipid Res. 2020;61:1087–103. doi: 10.1194/jlr.RA120000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53. doi: 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Qiao L, Bian Y, Sun X. GDF15 knockdown promotes Erastin-induced ferroptosis by decreasing SLC7A11 expression. Biochem Biophys Res Commun. 2020;526:293–9. doi: 10.1016/j.bbrc.2020.03.079. [DOI] [PubMed] [Google Scholar]
- 50.Mesci P, Zaïdi S, Lobsiger CS, Millecamps S, Escartin C, Seilhean D, et al. System xC- is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain. 2015;138:53–68. doi: 10.1093/brain/awu312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez AM, Mirkovic J, Stanisz ZA, Patwari FS, Yang WS. NSC-34 motor neuron-like cells are sensitized to ferroptosis upon differentiation. FEBS Open Bio. 2019;9:582–93. doi: 10.1002/2211-5463.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McAndrew CW, Gastwirt RF, Donoghue DJ. The atypical CDK activator Spy1 regulates the intrinsic DNA damage response and is dependent upon p53 to inhibit apoptosis. Cell Cycle. 2009;8:66–75. doi: 10.4161/cc.8.1.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapatos G, Vunnava P, Wu Y. Protein kinase A-dependent recruitment of RNA polymerase II, C/EBP beta and NF-Y to the rat GTP cyclohydrolase I proximal promoter occurs without alterations in histone acetylation. J Neurochem. 2007;101:1119–33. doi: 10.1111/j.1471-4159.2007.04486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandran NS, Vunnava P, Wu Y, Kapatos G. Specificity proteins SP1 and Sp3 interact with the rat GTP cyclohydrolase I proximal promoter to regulate transcription. J Neurochem. 2008;104:1233–48. doi: 10.1111/j.1471-4159.2007.05054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang JJ, Luo XJ, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic Biol Med. 2021;162:339–52. doi: 10.1016/j.freeradbiomed.2020.10.307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding author upon reasonable request. Supplementary figures and original Western blots images are available at Supplementary Materials.