

Abstract
The colony-forming cyanobacteria Trichodesmium spp. are considered one of the most important nitrogen-fixing genera in the warm, low nutrient ocean. Despite this central biogeochemical role, many questions about their evolution, physiology, and trophic interactions remain unanswered. To address these questions, we describe Trichodesmium pangenomic potential via significantly improved genomic assemblies from two isolates and 15 new >50% complete Trichodesmium metagenome-assembled genomes from hand-picked, Trichodesmium colonies spanning the Atlantic Ocean. Phylogenomics identified ~four N2 fixing clades of Trichodesmium across the transect, with T. thiebautii dominating the colony-specific reads. Pangenomic analyses showed that all T. thiebautii MAGs are enriched in COG defense mechanisms and encode a vertically inherited Type III-B Clustered Regularly Interspaced Short Palindromic Repeats and associated protein-based immunity system (CRISPR-Cas). Surprisingly, this CRISPR-Cas system was absent in all T. erythraeum genomes, vertically inherited by T. thiebautii, and correlated with increased signatures of horizontal gene transfer. Additionally, the system was expressed in metaproteomic and transcriptomic datasets and CRISPR spacer sequences with 100% identical hits to field-assembled, putative phage genome fragments were identified. While the currently CO2-limited T. erythraeum is expected to be a ‘winner’ of anthropogenic climate change, their genomic dearth of known phage resistance mechanisms, compared to T. thiebautii, could put this outcome in question. Thus, the clear demarcation of T. thiebautii maintaining CRISPR-Cas systems, while T. erythraeum does not, identifies Trichodesmium as an ecologically important CRISPR-Cas model system, and highlights the need for more research on phage-Trichodesmium interactions.
Subject terms: Water microbiology, Biogeochemistry
Introduction
Low bioavailable concentrations of nitrogen can limit primary productivity in many oceanic euphotic zones (e.g. [1]). In the warm, oligotrophic open ocean, these low nitrogen concentrations select for nitrogen-fixing organisms that can efficiently convert atmospheric N2 to bioavailable forms [2]. While our understanding of nitrogen-fixing organisms in the oceans is evolving to include non-autotrophic diazotrophs and other unexpected physiologies (e.g. [3–6]), the filamentous, colony-forming cyanobacterium Trichodesmium is still considered a critical oceanic nitrogen fixer [3, 7].
Mariners have known filamentous Trichodesmium spp. as ‘sea-saw dust’ for hundreds of years because of the massive surface blooms they can form resembling small, water-suspended wood shavings [7]. Trichodesmium filaments can aggregate in natural communities, forming 1–4 mm colonies of essentially two morphologies (i.e., radial tufts or spherical puffs; [8]) that are visible to the naked eye and thus aided these early observations. Fitting with this long-term recognition, scientists defined six morphologically described species in the late 1800s [9]. Still, oceanographers did not recognize their central role in N2 fixation until the 1960s (e.g. [2, 3]). Researchers now know that Trichodesmium has a wide distribution in the tropics and subtropics [3, 10] and, even though some appear to have lost N2 fixation capabilities [4], the genus is still an essential source of bioavailable N to the oligotrophic oceans [7, 11]. Thus, while Trichodesmium species names have existed for >100 years, experiments to understand their evolution, genomic potential, and ecological impact are still active research areas.
Members of the Trichodesmium genus are closely related with large, repeat rich genomes [12, 13]. Yet, enrichment strains and field samples can show surprising morphological and physiological character variability (i.e., pigmentation, cell size, trichome shape, growth rate, N2 fixation rate, or colony structure) and abundance differences (e.g. [8, 14–16]). For example, marker gene phylogenetics shows four clades of Trichodesmium [8, 14, 17], with the best bootstrap support defining the Trichodesmium thiebautii and Trichodesmium erythraeum-enriched clades I and III, respectively [8]. Recent metagenomic work from the Red Sea has shown that some single gene metrics for Trichodesmium (i.e., hetR gene) have led to misidentification of clades due to paralogous copies in certain genomes [13], but this work also supports the importance of the two major clades of Trichodesmium. Additionally, morphological and molecular fieldwork shows that members of these same two clades are commonly observed, although T. thiebautii-containing clade I is typically more abundant throughout the water column (e.g. [4, 10, 18, 19]). Thus, while there are six classically defined Trichodesmium species, T. thiebautii clade I typically dominates field populations. Despite this recognition, the internal and external factors causing the numerical dominance of T. thiebautii are poorly defined.
Herein we used metapangenomics and metaproteomics of enrichment cultures and hand-picked colonies spanning the Atlantic Ocean to define genomic features predicted to impact Trichodesmium population dynamics. Our efforts showed that predicted mobile genetic element immunity (i.e., against phage and mobile plasmids; MGE) is a defining feature of T. thiebautii, as all clade members maintain and express a conserved Type III-B CRISPR-Cas system [20].
Materials and methods
Trichodesmium colony collection
Trichodesmium colonies were collected with a hand-towed line (~150 ft) 130-µm Sea Gear plankton net on February 8th thru March 11th, 2018, during the R/V Atlantis TriCoLim cruise (AT39-05) that transected from Cape Verde to Puerto Rico (Fig. 1). Colonies were rapidly removed from the cod end and picked into tuft and puff morphologies with sterile plastic disposable Pasteur pipettes into 50 ml of sterilized seawater. These morphology-segregated samples were sequentially washed two times with sterile, local seawater, gently filtered down onto 5-µm polycarbonate membranes, and rapidly frozen in liquid N2.
Fig. 1. Map of the 2018 trans-Atlantic TriCoLim Cruise.
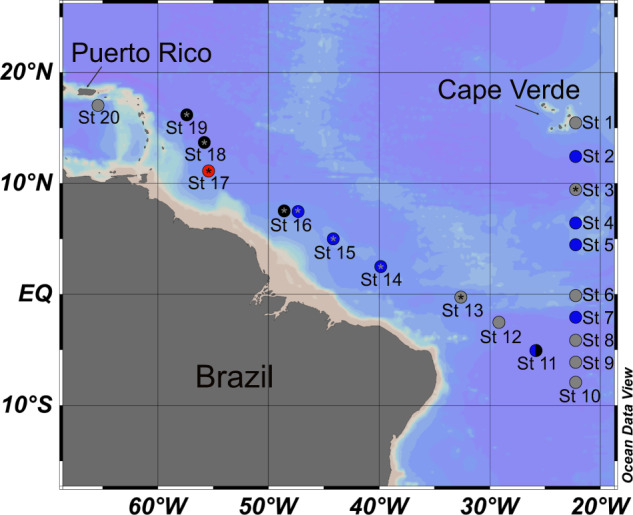
Color of the station location indicates hand-picked Trichodesmium colony morphology, specifically puff (blue), tuft (black), combined (blue and black), not hand-picked (red), no metagenomic data (gray) and metaproteomic data (asterisk).
DNA isolation and sequencing
High-quality DNA was isolated from ~50 frozen colonies per station via Qiagen DNeasy Powersoil Kit (Germantown, MD) using the manufacturer’s protocol with the following exceptions. DNA quality and quantity was determined via NanoDrop UV-Vis spectrophotometer and Qubit Fluorometer, respectively (ThermoFisher; Waltham, MA) and twelve TriCoLim samples were 150 PE Illumina sequenced by Novogene (Sacramento, CA) to a final depth of 25 Gbps. DNA was isolated from frozen T. thiebautii H94 samples using the same protocol as above and was sequenced via 250PE Illumina MiSeq at the USC Epigenome Center (1.8 Gbps total) because the original assembly in [12] was poor quality. Raw reads are available at NCBI’s SRA under the BioProject PRJNA828267.
Isolate and field MAG assembly
Both the field samples and T. thiebautii H94 reads were run through similar assembly pipelines, but H94 was assembled on KBase (https://www.kbase.us), while the former were on a Linux server. The quality of reads was checked with FastQC v0.11.2 [21] and trimmed to enhance stats using Trimmomatic v0.33 [22]. MAGs were assembled de novo using metaSPAdes v3.12.0 [23] for H94 and MEGAHIT v1.2.6 [24] for the TriCoLim samples. These programs were chosen because each yielded the best assembly stats. Binning of contigs was performed via MaxBin2 v2.2.4, quality was checked with CheckM v1.1.3 [25], and phylogenetic placement of the MAGs was determined with GTDB-tk v1.3.0 [26]. Field MAGs were dereplicated using fastANI [27] with a cutoff of 98.5% ID. Dereplicated bins >50% CheckM complete were hand refined in Anvi’o v7 [28, 29] until the contamination level was below 5%. T. erythraeum strain 2175 was downloaded from NCBI and hand-refined in Anvi’o [28, 29] to remove contaminating contigs using the TriCoLim reads, and its final genome stats were determined with CheckM [25].
Phylogenomics
Higher quality Trichodesmium MAGs (>50% complete; Supplementary Table 1) and nearest relative genomes downloaded from the NCBI Assembly page were run through the program GToTree v1.6.12 to define a initial guide tree based on 251 cyanobacterial core protein Hidden Markov Models [30, 31]. Alignment and partition files from GToTree were piped to IQtree v2.1.4-beta in ModelFinder optimality mode (models LG + F + R10) with 1000 ultrafast bootstraps to generate the phylogenomic tree [32]. The tree was visualized and edited in the interactive Tree of Life (ITol; [33]).
Metapangenomics
The metapangenomic pipeline in Anvi’o was used to characterize shared and distinct blast-defined gene clusters (GCs; mcl-inflation 10) in the MAGs and determine if these GCs were represented in the TriCoLim reads [28, 29]. Briefly, this pipeline creates a contig database for all MAGs that was then annotated with prodigal [34], COGs [35], PFAMS [36], KOFAM [37] and KEGG [38]. Reads were recruited to contigs in the Anvi’o database with TriCoLim read samples using bowtie2 v 2.4.1 [39], matching Sequence Alignment Map (SAMs) were converted to Binary Alignment Maps (BAMs) with SAMtools v1.11 [40], and BAMs were profiled across the TriCoLim read sets using Anvi’o to determine environmental auxiliary and environmental core genes (EAG and ECG, respectively). The reads were square root normalized to compress the results and allow one to visually see the presence of T. erythraeum in the recruitment heat map. COG categories per 100 kb was determined by exporting the annotation from Anvi’o, determining the COG categories per MAG, summing those results per clade, and then analyzing and graphing in R v 4.0.3 (2020-10-10) with R Studio v1.4.1103 [41]. Differences between COG category counts per clades were tested for statistical significance using ANOVA in the R package rstatix [42]. The same general pipeline was used to determine single gene copy per clade and toxin:antitoxin GCs per clade. The Anvi’o summary data was converted into a matrix via the scripts in [43] and used with the R package micropan [44] to generate Heaps’ law alpha value and genome fluidity estimates.
CRISPR-Cas analyses
We scanned all Trichodesmium assemblies and those from their nearest relatives with CRISPRCasTyper [45]. This tool aids in the sometimes difficult task of identifying and typing CRISPR arrays and disparate cas loci based on the currently defined 44 subtypes and variants [20]. Because many of the MAGs were fragmented, CRISPR-Cas system portions on other contigs are shown with double slashes if (1) the pieces were found on the edges of their located contigs, and (2) the associated cas genes are still predicted to be part of the subtype III-B, I-D, or III-D systems defined in [20]. Additional annotations for accessory genes (purple) and hypothetical genes (gray) were determined by CRISPRCasFinder, and BLAST [46–48].
We used clustal in the program Geneious Prime (Biomatters, San Diego, CA) to align Cas10 protein sequences from all genomes, and RaxML v8 to generate the maximum likelihood phylogenetic tree with 100 bootstraps [49].
Virome assembly
We screened for putative phage genome, prophage or plasmid fragments in TriCoLim and enrichment culture assemblies using the virsorter2, DRAM-V, and checkV pipelines for viruses and metaplasmidSPAdes for plasmids [50–53]. The contig sequences supplied from these efforts were used to generate custom blast databases [48] that were subsequently BLAST screened with Trichodesmium spacers defined above and grouped with FastANI [27].
Proteome analysis of Trichodesmium enriched field samples
The raw proteome spectra were collected from Trichodesmium colony samples published in a prior work [54] and newly analyzed for this study. Specifically, raw spectra were searched with the Sequest algorithm implemented in Proteome Discoverer 2.2 using a custom-built Trichodesmium genomic database. To avoid inflation of the sequence database and later misinterpretation of phylogenetic signals, only one version of any identical/redundant protein sequences were included in the database, with the possible phylogenetic attributions for the redundant proteins noted in downstream phylogenetic analyses. Sequest mass tolerances were set to ±10ppm (parent) and ±0.6 Dalton (fragment). Fixed Cysteine modification of +57.022, and variable N-terminal acetylation of +42 and methionine modification of +16 were included. Protein identifications were made with Peptide Prophet in Scaffold (Proteome Software) at the 80% peptide threshold minimum, resulting in an estimated peptide false discovery rate (FDR) of 1.5% and an estimated protein FDR of 0.0%. Relative protein abundances are reported as normalized total exclusive spectral counts, so only spectra corresponding to a specific peptide for a given protein were considered. This avoids the problem of overlapping peptides in the phylogenetic analysis. The values are normalized to total spectral counts identified in each sample. The peptides identified for the CRISPR-Cas proteins were further checked using the Metatryp 2.0 tool (www.metatryp.whoi.edu) [55] to ensure phylogenetic specificity of the signals.
Transcriptome read recruiting
Trichodesmium colony metatranscriptomes were downloaded from NCBI SRA (projects PRJNA381915 and PRJNA374879) and mapped against all cas genes from H94 and cas10 from MAG T. thiebautii Indian Ocean that is a representative of cluster 3. Read quality was checked with FastQC v0.11.2 [21], trimmed with Trimmomatic v0.33 [22], recruited to cas genes with Bowtie v 2.4.1, converted to BAMs with Samtools v1.11, and profiled in Anvi’o v7.0. Average read depth values were normalized with the constitutive Trichodesmium gene rotA, as in [56]. The data were visualized in R v 4.0.3 (2020-10-10) with R Studio v1.4.1103.
Results and discussion
All N2-fixingTrichodesmium genomes are “low” protein-encoding
Past work with a handful of Trichodesmium isolates shows that their genomes are low protein-encoding (i.e., ~63%) and enriched in selfish DNA elements;[12] however, these observations have never been studied systematically across the genus. To address this, we assembled 15 new Trichodesmium MAGs from our hand-picked colonies obtained on the 2018 Atlantic Ocean spanning TriCoLim cruise (Fig. 1) and compared them to previously published isolate genomes [12] and three MAGs from a Tara Oceans analysis [4]. Two of our previously published USC Trichodesmium Culture Collection (USCTCC) genomes, T. thiebautii H94 and T. erythraeum 2175, were significantly improved by MiSeq resequencing the former and Anvi’o refining both using reads from TriCoLim. Supplementary Table 1 lists the final refined CheckM [25] statistics for all MAGs and genomes. Similar to a prior three isolate comparison [12], Supplementary Table 2 shows that higher-quality Trichodesmium MAGs have low GC% and much lower coding (~64%) than the bacterial average of ~90% [57], and are large, with an average length of ~6.5 ± 0.9 MB, and relatively gene sparse, only encoding for an average of ~5396 ± 784 proteins.
There are four N2-fixing clades of Trichodesmium
To improve Trichodesmium cladistics, we performed phylogenomics using 251 conserved core genes from Trichodesmium genomes and MAGs >50% complete. The resulting tree in Fig. 2A shows that the N2-fixing members of the genus are divided into four major clades and suggests that T. thiebautii assembled from the Atlantic (Clade A) are phylogenomically different from those from other basins (Clade B). However, our read recruiting and metaproteomics (see below) indicate that genomes with high identity to T. thiebautii B are also present and active in the Atlantic Ocean, but they did not assemble with high quality from TriCoLim samples (e.g., Supplementary Table 1; St11_bin2_1_1 and St14_bin2_1 are >98% ANI with T. thiebautii B H94 and MAG_Trichodesmium_thiebautii_Indian). Lastly, while Delmont has shown that a distinct branch of Trichodesmium are non-N2-fixers [4], BLAST searches of the TriCoLim MAGs with T. erythraeum IMS101 nif genes confirmed that all of these MAGs encode for diazotrophy (i.e., when missing in a MAG annotation, nif genes fragments were consistently found at the end of contigs).
Fig. 2. Phylogenomic tree of Trichodesmium and nearest relatives and COG enrichment per 100Kb of Trichodesmium clades.

In (A), TriColim bins are named by station, USCTCC strains are in bold, and names beginning in “MAG” are from (4). Trichodesmium MAGs >80% complete are color coded by clade (i.e.), T. thiebautii (Blue), T. erythraeum A (Black), T. erythraeum B (Red), while MAGs < 80% are shown in gray. Other Trichodesmium MAGs in Supplementary Table 1 were excluded from the tree due to low completion values. B shows the normalized and summed quantity of COG categories for each MAG per clade. Significantly different categories determined by ANOVA of the mean are denoted above the bracket (p < 0.05 = *, <0.01 = **, <0.001 = ***, <0.0001 = ****).
Since there are no published isolate genomes for T. thiebautii clade A and T. erythraeum clade B, reconciling past species designations and predicting their physiological and morphological characters was not directly possible. However, we attempted to place these MAGs in context with previously isolated strains [8, 58] by comparing 16S-23S internal transcribed spacer (ITS) gene sequences via blast. The isolate ITS hits ranged from 86–100% ID to our MAGs and old strain/clade designations from Hynes et al (8) generally tracked well with our cladistics. However, the high identity hits (>99.5%) allowed us to make three conclusions. (1) T. erythraeum strains (6-1, 6-2, 6-5), that formed a weak subgroup in a prior analysis [8], are members of T. erythraeum clade B, and thus we predict this group has phycoerythrobilin-rich red cells ~6.5–9.5 µm in diameter that can form colonies or loose aggregates. (2) Trichodesmium contortum with larger diameter cells (~20–30 µm) and bright red coloration [8] is likely a member of T. erythraeum clade A. (3) The ITS does not contain enough information to determine if previous Trichodesmium hildebrandtii, Trichodesmium tenue, and Trichodesmium spiralis isolates are in either T. thiebautii clade A or B. Because of this taxonomic uncertainty, hereafter we forgo using other Trichodesmium species names and simply use the broad clade designations (i.e., T. erythraeum A & B and T. thiebautii A & B).
Trichodesmium genomes have many paralogous genes dominated by predicted mobile genetic elements
To understand broad-level genome evolution in the genus, we explored copy number enriched gene families in Trichodesmium genomes and MAGs. Our results show many paralogous GCs shared by all Trichodesmium genomes, with some found in very high copy numbers per clade. Interestingly, each clade’s top ten duplicated GCs are similar but not 100% identical in sequence or copy number (Supplementary Table 3). Thus, our data corroborate a prior finding that Trichodesmium genomes are repeat-rich [12] and show that these duplications are commonplace in situ. Annotation of these paralogous GCs shows that they are enriched (~78%) in “selfish DNA elements” like transposases, retrons, or group II introns. Since there is evidence that “selfish DNA elements” can be involved in bacterial genome re-arrangement or niche adaptation [59, 60], these results suggest that transposition or other related duplication generating processes may be important evolutionary mechanisms in Trichodesmium.
T. thiebautii MAGs are enriched in specific clusters of orthologous genes (COG) compared to T. erythraeum
To begin to understand the selective pressures driving speciation in the genus, we next characterized the genomic potential of Trichodesmium in a phylogenomic context. At first glance, the average gene number per genome is greater in T. thiebautii than T. erythraeum (Supplementary Tables 2). As shown in Fig. 2B, many of the COG categories per 100 kb in each clade are statistically indistinguishable by ANOVA. However, there are five enriched categories in T. thiebautii A, T. thiebautii B, or both. These include Lipid Metabolism (I), Intracellular trafficking (U), Defense Mechanisms (V), Mobilome (X), and genes not categorized by COG. While most of these COG enrichments were dominated by transposases, repeat-filled genes, or hypothetical genes, the Lipid Metabolism and Defense genes had many informative annotations. Closer inspection of the Lipid Metabolism genes showed that T. thiebautii A has increased acyl-carrier proteins, many of which appear to be involved in polyketide synthases or annotated with multiple functions. These findings suggest increased secondary metabolite production in this clade. However, despite the interest in understanding how Trichodesmium acquires Fe (e.g. [16, 61],), none of these clusters are predicted to encode siderophores. Lastly, the T. thiebautii Defense category was enriched in putative toxin-antitoxin proteins [62] antiphage systems [63], and CRISPR-Cas genes [20], suggesting phage interactions are a selective pressure in the two T. thiebautii clades.
One-quarter of all Trichodesmium MAGs have shared gene clusters
To explore differences between Trichodesmium clades, we examined specific gene cluster presence/absence, annotated function, and detection in our TriCoLim reads. This effort allowed us to determine if the functionalities enriched or depleted in each clade in Fig. 2B were caused by distinct, new gene clusters, paralogous duplications, or deletions. Based on BLAST clustering, there are 1454 single and paralogous GCs in the conservative Trichodesmium core found in all genomes (Fig. 3A). Thus, approximately 1/4 of each genome is conserved core gene content. The total pangenome count was 10,054 genes. Pangenome modeling with the R package micropan [44] obtained Heap’s power law alpha estimates of ~1 for all Trichodesmium MAGs together and slightly >1 for T. thiebautii and T. erythraeum MAGs individually, indicating that these pangenomes are either “completely” sampled with this dataset (i.e., closed) or slowly growing logarithmically [64].
Fig. 3. N2-fixing Trichodesmium Metapangenomic visualization.

A shows blast-defined conserved gene clusters (GCs) in a MAG as filled colored rings (blue for all T. thiebautii, red for T. erythraeum B, and black for T. erythraeum A). Lighter fill colors indicate that those GCs are missing from that MAG. Singleton GCs (i.e., appearing in only one MAG) are mostly shown between 9 and 11 o’clock in the diagram. The innermost rings of the diagram indicate number of contributing genomes to a GC, single copy genes (SCG), number of genes in a GC, and max number of paralogs. Continuing outwards, if the GC has annotation it is marked in green, while if it does not it is white. The outermost two rings show whether a GC is environmentally core (green) or auxiliary (red; i.e., the redder the color, the less commonly the GC was observed in TriCoLim reads) and GC homogeneity (i.e., high homogeneity = all green fill). Clear groupings of clade specific auxiliary gene clusters (AGC) are labeled on the edge of the diagram. B shows BLAST ANI clustering at the top and the ANI heat map in red from 96 to 100. The black heat map shows square root normalized read recruiting to each MAG from TriCoLim (Blacker bars = higher relative read recruiting). C shows statistical analysis of singleton GCs per clade with ANOVA statistical support shown above the brackets (p < 0.05 = *, 0.01 = **).
Others have argued that genome fluidity (ϕ), a metric of genome dissimilarity, is a better method for estimating the likelihood of identifying new genes as more genomes in a taxonomic group are sequenced [65, 66]. We determined the MAG genome fluidity values for all Trichodesmium (ϕ = 0.303 ± 0.10) and the major clades of T. thiebautii (ϕ = 0.24 ± 0.04), and T. erythraeum (ϕ = 0.18 ± 0.03). Strict interpretation of these data suggests a 30% chance of identifying new genes as more Trichodesmium genomes are sequenced—again fitting with a growing/open pangenome. Additionally, our data predicts that the likelihood of discovering new genes is higher as more T. thiebautii are sequenced compared to T. erythraeum. While it is important to note that these ϕ values will likely improve with increased numbers of genomes in each clade [65], the data are consistent with the T. thiebautii pangenome being more ‘open’ than T. erythraeum with the former likely experiencing increased horizontal gene transfer (HGT).
T. thiebautii dominated the read recruiting regardless of colony type, and intra-clade average nucleotide identity (ANI) of the MAGs was very high (Fig. 3B; black and red heat maps, respectively). Thus, in situ quantification of each MAG was not possible because of likely random read recruiting among high ANI genomes [67]. However, since this issue would likely only underestimate the abundances of each clade, we report that T. thiebautii MAGs were recruiting at least 1–2 orders of magnitude more reads than T. erythraeum from TriCoLim colonies.
Trichodesmium auxiliary gene content and genomic average nucleotide identity (ANI) recapitulate the phylogenomic signal
While the predicted Trichodesmium core N2-fixing genome makes up ~1/4 of the genes, many auxiliary GCs are also detected. As shown in Fig. 3A, some auxiliary GCs were only found in one genome (i.e., singletons), while others associate with specific clades. The environmentally accessory genes (EAGs; i.e., not found in situ) to environmentally core genes (ECGs; i.e., found in situ) ratio shown on the outer ring indicates that many, but not all of these auxiliary GC bins, are commonly detected in Atlantic Ocean Trichodesmium colonies. Coloring the rings of Fig. 3A by phylogenomic group shows that the auxiliary gene content, average nucleotide identity (ANI; Fig. 3B), and phylogenomics of core genes (Fig. 2A) give the same relationships between Trichodesmium clades. Additionally, statistical analysis of singleton genes shows an uneven distribution in the genus, with T. thiebautii genomes maintaining significantly more (Fig. 3C). These empirical data are consistent with the genome fluidity results above and suggest mechanisms that increase novel gene content, like horizontal gene transfer, are more common in T. thiebautii.
We next took the GCs in each clade-specific bin, highlighted in Fig. 3A, to characterize enriched functionalities (Supplementary Fig. 1). The largest groups of clade-specific genes are found in the primary division between T. thiebautii AB and T. erythraeum AB, where the former shares 313 GCs and the latter shares 315, respectively. Percentage normalized COG analyses of these conserved GC bins showed four things: (1) non-COG categorized GCs dominate those found in all bins (ranging from ~44 to 79%), and 18–75% of these non-categorized GCs are hypotheticals, (2) the Tery-AB bin has much more COG diversity than the similarly sized Thieb-AB bin (Supplementary Fig. 1), (3) Thieb-AB, Thieb-B, and Tery-B bins are enriched in mobilome sequences (~10% of the bin’s GCs), while in Tery-A and Tery-AB, the mobilome GCs only account for ~5% of GCs, and (4) the Thieb-AB bin has a higher percentage of Defense COGs. While our data show that specific CRISPR-related gene duplications are common in Trichodesmium MAGs (Duplicated GCs; Supplementary Table 3), the Thieb-specific bin is enriched in CRISPR-Cas immunity genes.
T. thiebautii encodes a complete Type III-B CRISPR-Cas system, while T. erythraeum does not
The expanded, Trichodesmium-nearest relative phylogenomic tree in Fig. 4A shows cas gene detection in the lineage. These data demonstrate that the non-N2 fixing Trichodesmium, T. thiebautii, Hydrocoleum and Okeania MAGs all encode CRISPRs. In contrast, none of the 10 T. erythraeum MAGs analyzed here have them. Additionally, we could not identify any cas10 gene hits in any T. erythraeum MAGs in our dataset or from NCBI [13]. Finally, the observation that the T. erythraeum IMS101 genome is closed [12] and completely missing the CRISPR-Cas system, supports its absence from T. erythraeum in general.
Fig. 4. Presence of Trichodesmium CRISPR systems in a phylogenomic context and Cas10 phylogenetics.

Presence or absence of CRISPR-Cas genes in a Trichodesmium MAGs and their nearest relatives phylogenomic context (A) and a maximum likelihood tree of their Cas10 protein sequences (B). In (A), the color-coded, directional shapes on the right represent detected Cas genes (yellow), CRISPR arrays (black), Cas accessory genes (purple) and hypothetical genes (gray) that were annotated by as described in the Materials and Methods. Lighter color indicates lower confidence in the annotation. Double-slashes are contig break positions near the annotated CRISPR-Cas systems, indicating that some clusters are fragmented due to breaks in the assemblies. Gene lengths are not drawn to scale. In (B), the color coding corresponds to T. thiebautii (Blue) and other relatives (Black). Cas10 protein sequences are in Supplementary Table 12.
As the cas10 gene is diagnostic for the Type III-B CRISPR predicted to be encoded by T. thiebautii and can show significant sequence variation [68], we performed phylogenetic analyses of Cas10 protein sequences to explore the origins of this system in the lineage. The Cas10 maximum likelihood phylogeny shown in Fig. 4B suggests two-to-three Type III-B systems in T. thiebautii. Additionally, this tree indicates that these systems are likely ancestral because the phylogeny of each of the three distinct sequence clusters is roughly congruent with the phylogenomic signal shown in Fig. 4A. However, careful comparison of both trees shows that all three Type III-B Cas10 protein clusters are not conserved in every T. thiebautii assembly. BLASTN searches confirmed that the missing cas10 genes were present at contig breaks in our T. thiebautii MAGs (i.e., St18_bin1, St19_bin1, and St16_bin2_tuft) corresponding to clusters 1 & 2 in Fig. 4B. The most straightforward interpretation of these data is that most T. thiebautii assemblies do not have cluster 3, and perhaps it is currently disappearing from the Trichodesmium pangenome. Fittingly, cluster 3 is undetectable from our best-assembled MAG, T. thiebautii H94 isolate genome (566 contigs). Thus, while there is variation in III-B CRISPR-Cas system copy number in T. thiebautii, its loss in T. erythraeum is a phylogenetically constrained defining difference between these major Trichodesmium clades.
Generally speaking, CRISPR-Cas systems protect the cell from mobile genetic elements (MGEs; phage and mobile plasmids) via a sequence-based, targeted genome degradation [69, 70]. Many different CRISPR-Cas systems that vary in gene content and recognition molecule (RNA vs. DNA) have been described [20]. That said, while all CRISPR-Cas variants appear to provide memory-driven immunity against MGEs, the Type III-B subtype, predicted in numerically abundant T. thiebautii clades, requires active RNA transcription for function, can use other CRISPR arrays in addition to its own, and provides better protection against phage protospacer mutagenic evasion [71].
Mechanistically, Type III-B CRISPR-Cas systems operate in three steps (1) adaptation: recognition and incorporation of transcribed 30–50 bp protospacers (i.e., DNA or RNA sequences of invading MGEs; typically mediated by Cas1 or Cas1-reverse transcriptase (RT) fusion proteins, respectively [72, 73]) into CRISPR arrays as spacer sequence DNA “memories” of past attacks, (2) expression: spacer RNAs are expressed as precursor CRISPR RNA (crRNA), and (3) interference: sequence-specific crRNAs guides interfere with invading phage or plasmids by the action of the Cas10 protein [68, 69]. The absence of a Cas1-RT fusion protein in T. thiebautii suggests that the primary adaptation targets for this system are transcribed DNA MGEs. In contrast, an HD superfamily nuclease domain in T. thiebautii Cas10 proteins indicates that the interference step is likely cleaving both RNA and transcribed DNA [68, 72, 74]. Importantly, these DNA spacer sequences also provide ‘fingerprints’ of past MGE attacks that link phage/plasmid sequences with the CRISPR-Cas system containing host (e.g. [75–77]).
Predicted phage genome fragments assembled from TriCoLim 100% match T. thiebautii CRISPR spacers
We next sought to identify predicted MGEs from colony assemblies and determine if they matched T. thiebautii spacer sequences. While metaplasmidSPAdes identified several putative plasmids in enrichment and field samples (data not shown), none matched any T. thiebautii spacers. We also could not detect phage particles/genomes from the enrichment MAGs; however, the TriCoLim assemblies revealed 1000 s of putative phage genome fragments with contigs sizes ranging from 1000 s to >100kbps (data not shown).
Next, we asked whether these phage genomes sequences matched the T. thiebautii spacers. This effort identified seven 100% ID hits and 29 more with ANI > 90% covering ≥93% of the spacer (Supplementary Table 4). We conservatively picked the latter ID and coverage level because Type III-B crisper systems can function with mismatches, a feature that requires phage to delete ‘whole’ spacer-protospacer targets from their genomes to escape degradation [78, 79]. Unfortunately, these spacer-matching putative phage DNA fragments only ranged from 1755 to 5628 bps and were thus too small to identify the phage. All spacer-matching contigs were categorized as virstorter2 category 2 (i.e., likely phage DNA) and contained many predicted hypothetical viral genes, while one also is expected to encode a transposase (Supplementary Table 5). None of the fragments drew hits from known phage sequences in Genbank or IMG or any other gene. It is noteworthy that many of these putative phage genome fragments were detected multiple times from independently assembled TriCoLim stations (Supplementary Table 5; fastANI groupings), suggesting that some consistent phage particles were present across the transect. As past research shows that high phage relatedness selects for CRISPR-Cas systems [69, 80, 81], these data suggest that the T. thiebautii CRISPR-Cas system is defending against a relatively conserved phage group.
T. thiebautii CRISPR-Cas systems are expressed in situ
Identifying conserved Trichodesmium spacers and putative phage genome fragments suggests that the CRISPR-Cas systems are active in the field. To verify, we screened our TriCoLim Trichodesmium colony metaproteomics dataset for in situ Cas protein expression (protein identifications are provided as Supplementary Tables 6–8). In this re-analysis, we identified 3498 proteins and 68058 peptides. After binning the detected proteins by clade, T. thiebautii proteins were >10x more abundant across the transect than either T. erythraeum clades (Fig. 5A—Red, Pink, Blue, and Cyan filled colored bars, respectively). This protein detection dataset agrees well with our metagenomic read mapping in Fig. 3B, where T. thiebautii consistently dominated the sequencing reads. Major metabolic proteins such as the photosystems and nitrogenase were detected in high levels across the transect (Fig. 5B) and originated mainly from T. thiebautii proteins. Non-COG categorized proteins were ×50 more abundant across the transect than even these core metabolic functions, consistent with this being one of the most enriched categories in the T. thiebautii assemblies. Additionally, the in situ expression of these non-categorized proteins suggests that they are required for environmental growth and highlights the importance of characterizing them further.
Fig. 5. Trichodesmium protein abundances across the TriCoLim transect.

A Protein abundance data sorted into Trichodesmium phylogenetic groups. Proteins were normalized across each sample, then sorted into the respective phylogenetic group and summed. Error bars indicate the standard deviation of the averaged biological replicates. Quantitation is displayed as normalized spectral counts (see “Methods”). Core and T. thiebautii proteins are much more abundant than those derived from T. erythraeum. B Protein abundance data sorted by biological function. Again, proteins were normalized across each sample, then sorted by COG function and summed. Selected, highly abundant functions are shown. C Summed normalized protein data for CRISPR-Cas and toxin/antitoxin proteins across the TriCoLim transect. D Nonsignificant correlation of CRISPR-Cas protein abundances versus T. thiebautii total protein abundance (p = 0.88).
Proteins involved in cellular defense, including toxin/antitoxin proteins (i.e., the toxin components of RelE and MazEF and the antitoxin component of ParD) and the CRISPR-Cas system, were identified in across the transect (Fig. 5C). The CRISPR-Cas proteins did not correlate with total T. thiebautii protein abundance, suggesting that the former are not constitutively expressed (i.e., as a function of biomass) and are instead under some regulatory control. The CRISPR proteins identified included Cas10 and Cas7, and their phylogenetic assignment at the peptide level corresponded to T. thiebautii species and were assigned to Cas10 clusters 1 & 2 from Fig. 4B. Specifically, we identified peptides that were identical to those in T. thiebautii MAGS H94, St18_bin1, St16_bin2, and MAG T. thiebautii Indian Ocean, indicating that these species were contributing to CRISPR-Cas protein production in situ (Supp. Table 8). These data also show that Thieb-B clade members are present and active in the Atlantic Ocean.
Research shows that CRISPR-Cas adaptation (i.e., protospacer incorporation into spacer arrays) requires Cas1 or Cas2 to respond to new MGE threats [20, 73]. Thus the absence of these proteins in our metaproteome could suggest that the T. thiebautii CRISPR-Cas system is not actively adapting to new phage and is perhaps performing alternative functions in the cell independent of viral immunity [82, 83]. Three observations argue against this supposition. (1) self-targeting spacers (i.e., matching alternative sites in the MAG) were not identified, suggesting that interference-based gene regulation is not occurring (e.g. [82–84]). (2) most of the spacers detected in each MAG are distinct from those in other MAGs, suggesting that “rapid” adaptation occurs in T. thiebautii [75, 85], (3) read recruiting from Pacific Ocean Trichodesmium community data collected by others [86] shows that all annotated T. thiebautii H94 cas genes are expressed (including cas1 and cas2), and they appear to have diel periodicity (Supplementary Fig, 2; gene targets and results in Supp. Tables 9–12). Thus, while we cannot exclude alternative CRISPR functions in T. thiebautii, our data strongly suggest CRISPR-Cas mediated phage immunity is commonplace in the clade.
Conclusion
We show that the conserved maintenance of a functional CRISPR-Cas system in T. thiebautii is a defining speciation difference between the major clades of Trichodesmium. This conservation combined with singleton gene enrichment suggests that immunity allows the recipients of transduced genes to survive and thereby increase their genetic diversity (as noted in other systems [87, 88]). More research is needed to determine if CRISPR-Cas immunity conservation in T. thiebautii is the cause or the result of their numerical dominance over T. erythraeum. Because we are only just beginning to address how MGE selection maintains CRISPR-Cas systems in global populations [80], our findings also highlight the importance of future Trichodesmium virome studies. For example, will T. erythraeum be a climate change winner as predicted [15, 89] or will increased phage infectivity reduce their future expansion?
Supplementary Information
Acknowledgements
We would like to thank the captain and crew of the RV Atlantis for their essential role in sample collection and providing a safe and efficient platform for marine microbiology, and JL Weissman and Murat Eren for insightful conversations and suggestions. This work was funded by NSF grants OCE 1657757 and OCE 1851222 to DAH, FXF, and EAW, OCE 1850719 to MAS, discretionary USC funds, and BIO2125191 to EAW.
Author contributions
The data for this manuscript were collected by EAW, NAH, YZ, YF, AEC, JS, and DAH and analyzed by EAW, NAH, EDG, YZ, MDL, and MAS. The manuscript was written by EAW, NAH, DAH, AEC, YZ, MAS, MDL, and FXF and the research was directed by EAW.
Data availability
Our sequences files are accessible from the National Center for Biotechnology Information (NCBI; BioProject PRJNA828267), Sequence Read Archive accession numbers SRR19658988 through SRR19658999. MAG Assemblies are available at NCBI Assembly page with accessions SAMN29146503 through SAMN29146503. The mass spectrometry proteomics data were originally deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the identifier PXD016225 and can be accessed at 10.6019/PXD016225 [90, 91]. The data is also available at BCO-DMO (https://www.bco-dmo.org/dataset/787078).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s43705-023-00214-y.
References
- 1.Moore CM, Mills MM, Arrigo KR, Berman-Frank I, Bopp L, Boyd PW, et al. Processes and patterns of oceanic nutrient limitation. Nat Geosci. 2013;6:701–10. doi: 10.1038/ngeo1765. [DOI] [Google Scholar]
- 2.Sohm JA, Webb EA, Capone DG. Emerging patterns of marine nitrogen fixation. Nat Rev Microbiol. 2011;9:499–508. doi: 10.1038/nrmicro2594. [DOI] [PubMed] [Google Scholar]
- 3.Zehr JP, Capone DG. Changing perspectives in marine nitrogen fixation. Science. 2020;368:eaay9514. [DOI] [PubMed]
- 4.Delmont TO. Discovery of nondiazotrophic Trichodesmium species abundant and widespread in the open ocean. Proc Natl Acad Sci USA. 2021;118:e2112355118. doi: 10.1073/pnas.2112355118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bombar D, Kubo KAT, Robidart J, Carter BJ, Zehr JP. Non‐cyanobacterial nifH phylotypes in the North Pacific Subtropical Gyre detected by flow‐cytometry cell sorting. Env Microbiol Rep. 2013;5:705–15. doi: 10.1111/1758-2229.12070. [DOI] [PubMed] [Google Scholar]
- 6.Kubo KAT, Karamchandani M, Capone DG, Zehr JP. The paradox of marine heterotrophic nitrogen fixation: abundances of heterotrophic diazotrophs do not account for nitrogen fixation rates in the Eastern Tropical South Pacific. Environ Microbiol. 2014;16:3095–114. doi: 10.1111/1462-2920.12346. [DOI] [PubMed] [Google Scholar]
- 7.Capone D, Zehr J, Paerl H, Bergman B, Carpenter E. Trichodesmium, a globally significant marine cyanobacterium. Science. 1997;276:1221. doi: 10.1126/science.276.5316.1221. [DOI] [Google Scholar]
- 8.Hynes AM, Webb EA, Doney SC, Waterbury JB. Comparison of cultured Trichodesmium (Cyanophyceae) with species characterized from the field. J Phycol. 2012;48:196–210. doi: 10.1111/j.1529-8817.2011.01096.x. [DOI] [PubMed] [Google Scholar]
- 9.Webb EA, Foster R, Villareal TA, Waterbury JB, Zehr J. Genus Trichodesmium. Bergey’s Manual of Systematics of Archaea and Bacteria. 2022.
- 10.Rouco M, Warren HJ, McGillicuddy DJ, Waterbury JB, Dyhrman ST. Trichodesmium sp. clade distributions in the western North Atlantic Ocean. Limnol Oceanogr. 2014;59:1899–909. doi: 10.4319/lo.2014.59.6.1899. [DOI] [Google Scholar]
- 11.Karl D, Letelier R, Tupas L, Dore J, Christian J, Hebel D. The role of nitrogen fixation in biogeochemical cycling in the subtropical North Pacific Ocean. Nature. 1997;388:533–8. doi: 10.1038/41474. [DOI] [Google Scholar]
- 12.Walworth N, Pfreundt U, Nelson WC, Mincer T, Heidelberg JF, Fu F, et al. Trichodesmium genome maintains abundant, widespread noncoding DNA in situ, despite oligotrophic lifestyle. Proc National Acad Sci. 2015;112:4251–6. doi: 10.1073/pnas.1422332112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koedooder C, Landou E, Zhang F, Wang S, Basu S, Berman-Frank I, et al. Metagenomes of red sea subpopulations challenge the use of marker genes and morphology to assess Trichodesmium Diversity. Front Microbiol. 2022;13:879970. [DOI] [PMC free article] [PubMed]
- 14.Janson S, Bergman B, Carpenter E, Giovannoni S, Vergin K. Genetic analysis of natural populations of the marine diazotrophic cyanobacterium Trichodesmium. FEMS Microbiol Ecol. 1999;30:65. doi: 10.1111/j.1574-6941.1999.tb00635.x. [DOI] [Google Scholar]
- 15.Hutchins DA, Fu F-X, Webb EA, Walworth N, Tagliabue A. Taxon-specific response of marine nitrogen fixers to elevated carbon dioxide concentrations. Nat Geosci. 2013;6:790–5. doi: 10.1038/ngeo1858. [DOI] [Google Scholar]
- 16.Chappell PD, Webb EA. A molecular assessment of the iron stress response in the two phylogenetic clades of Trichodesmium. Environ Microbiol. 2010;12:13–27. doi: 10.1111/j.1462-2920.2009.02026.x. [DOI] [PubMed] [Google Scholar]
- 17.Lundgren P, Soderback E, Singer A, Carpenter E, Bergman B. Katagnymene: Characterization of a novel marine diazotroph. J Phycol. 2001;37:1052–62. doi: 10.1046/j.1529-8817.2001.00192.x. [DOI] [Google Scholar]
- 18.Rouco M, Haley ST, Alexander H, Wilson ST, Karl DM, Dyhrman ST. Variable depth distribution of Trichodesmium clades in the North Pacific Ocean. Env Microbiol Rep. 2016;8:1058–66. doi: 10.1111/1758-2229.12488. [DOI] [PubMed] [Google Scholar]
- 19.Chappell PD, Moffett JW, Hynes AM, Webb EA. Molecular evidence of iron limitation and availability in the global diazotroph. Trichodesmium. ISME J. 2012;6:1728–39. doi: 10.1038/ismej.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. doi: 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babraham Bioinformatics -, FastQC A. Quality control tool for high throughput sequence data https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ Accessed 11 Sep 2020
- 22.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 25.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90 K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:7200. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: an advanced analysis and visualization platform for’omics data. PeerJ. 2015;3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, et al. Community-led, integrated, reproducible multi-omics with Anvi’o. Nat Microbiol. 2021;6:3–6. doi: 10.1038/s41564-020-00834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee MD. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics. 2019;35:4162–4. doi: 10.1093/bioinformatics/btz188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLOS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. Bmc Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021;49:D274–D281. doi: 10.1093/nar/gkaa1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2019;28:27. doi: 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.r-project.org/index.html. Accessed 1 Apr 2022.
- 42.Kassambara A. rstatix: Pipe-friendly framework for basic statistical tests. R package version 06 0. 2020.
- 43.Moulana A, Anderson RE, Fortunato CS, Huber JA. Selection Is a Significant Driver of Gene Gain and Loss in the Pangenome of the Bacterial Genus Sulfurovum in Geographically Distinct Deep-Sea Hydrothermal Vents. MSystems. 2020;5:e00673–19. [DOI] [PMC free article] [PubMed]
- 44.Snipen L, Liland KH. micropan: an R-package for microbial pan-genomics. BMC Bioinformatics. 2015;16:79. doi: 10.1186/s12859-015-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russel J, Pinilla-Redondo R, Mayo-Muñoz D, Shah SA, Sørensen SJ. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas Loci. The CRISPR Journal. 2020;3:462–9. doi: 10.1089/crispr.2020.0059. [DOI] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 47.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–W251. doi: 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST + : architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 2021;9:37. doi: 10.1186/s40168-020-00990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020;48:8883–8900. doi: 10.1093/nar/gkaa621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antipov D, Raiko M, Lapidus A, Pevzner PA. Plasmid detection and assembly in genomic and metagenomic data sets. Genome Res. 2019;29:961–8. doi: 10.1101/gr.241299.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides N. CheckV: assessing the quality of metagenome-assembled viral genomes. Nat Biotechnol. 2021;39:578–85. [DOI] [PMC free article] [PubMed]
- 54.Held NA, Webb EA, McIlvin MM, Hutchins DA, Cohen NR, Moran DM, et al. Co-occurrence of Fe and P stress in natural populations of the marine diazotroph. Trichodesmium. Biogeosci. 2020;17:2537–51. doi: 10.5194/bg-17-2537-2020. [DOI] [Google Scholar]
- 55.Saunders JK, Gaylord DA, Held NA, Symmonds N, Dupont CL, Shepherd A, et al. METATRYP v 2.0: metaproteomic least common ancestor analysis for taxonomic inference using specialized sequence assemblies—standalone software and web servers for marine microorganisms and Coronaviruses. J Proteome Res. 2020;19:4718–29. doi: 10.1021/acs.jproteome.0c00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orchard ED, Webb EA, Dyhrman ST. Molecular analysis of the phosphorus starvation response in Trichodesmium spp. Environ Microbiol. 2009;11:2400–11. doi: 10.1111/j.1462-2920.2009.01968.x. [DOI] [PubMed] [Google Scholar]
- 57.Giovannoni SJ, Thrash JC, Temperton B. Implications of streamlining theory for microbial ecology. ISMEJ. 2014;8:1553–65. doi: 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orcutt KM, Orcutt KM, Rasmussen U, Rasmussen U, Webb EA, Webb EA, et al. Characterization of Trichodesmium spp. by genetic techniques. App Environ Microbiol. 2002;68:2236–45. doi: 10.1128/AEM.68.5.2236-2245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pyle AM. Group II intron self-splicing. Ann Rev Biophys. 2016;45:183–205. doi: 10.1146/annurev-biophys-062215-011149. [DOI] [PubMed] [Google Scholar]
- 60.Pfreundt U, Hess WR. Sequential splicing of a group II twintron in the marine cyanobacterium Trichodesmium. Sci Rep. 2015;5:16829. doi: 10.1038/srep16829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basu S, Gledhill M, Beer D, de, Matondkar SGP, Shaked Y. Colonies of marine cyanobacteria Trichodesmium interact with associated bacteria to acquire iron from dust. Commun Biol. 2019;2:1–8. doi: 10.1038/s42003-019-0534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Unterholzner SJ, Poppenberger B, Rozhon W. Toxin–antitoxin systems: Biology, identification, and application. Mob Genet Elements. 2013;3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359:eaar4120. doi: 10.1126/science.aar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opi Microbiol. 2008;11:472–7. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 65.Kislyuk AO, Haegeman B, Bergman NH, Weitz JS. Genomic fluidity: an integrative view of gene diversity within microbial populations. BMC Genomics. 2011;12:32. doi: 10.1186/1471-2164-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andreani NA, Hesse E, Vos M. Prokaryote genome fluidity is dependent on effective population size. ISME J. 2017;11:1719–21. doi: 10.1038/ismej.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evans JT, Denef VJ. To dereplicate or not to dereplicate? mSphere. 2020;5:e00971–19. [DOI] [PMC free article] [PubMed]
- 68.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classification of CRISPR–Cas systems. Nature Reviews Microbiology. 2015;13:722–36. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Westra ER, Dowling AJ, Broniewski JM, van Houte S. Evolution and ecology of CRISPR. Annu. Rev. Ecol. Evol. 2016;47:307–31. doi: 10.1146/annurev-ecolsys-121415-032428. [DOI] [Google Scholar]
- 70.Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- 71.Silas S, Lucas-Elio P, Jackson SA, Aroca-Crevillén A, Hansen LL, Fineran PC, et al. Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. eLife. 2017;6:e27601. [DOI] [PMC free article] [PubMed]
- 72.Kolesnik MV, Fedorova I, Karneyeva KA, Artamonova DN, Severinov KV. Type III CRISPR-Cas systems: deciphering the most complex prokaryotic immune system. Biochem (Mosc) 2021;86:1301–14. doi: 10.1134/S0006297921100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Artamonova D, Karneyeva K, Medvedeva S, Klimuk E, Kolesnik M, Yasinskaya A, et al. Spacer acquisition by Type III CRISPR–Cas system during bacteriophage infection of Thermus thermophilus. Nucleic Acids Res. 2020;48:9787–803. doi: 10.1093/nar/gkaa685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Estrella MA, Kuo F-T, Bailey S, RNA-activated DNA. cleavage by the Type III-B CRISPR–Cas effector complex. Genes Dev. 2016;30:460–70. doi: 10.1101/gad.273722.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–50. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- 76.Roux S, Hawley AK, Torres Beltran M, Scofield M, Schwientek P, Stepanauskas R, et al. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife. 2014;3:e03125. doi: 10.7554/eLife.03125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martínez Arbas S, Narayanasamy S, Herold M, Lebrun LA, Hoopmann MR, Li S, et al. Roles of bacteriophages, plasmids and CRISPR immunity in microbial community dynamics revealed using time-series integrated meta-omics. Nat Microbiol. 2021;6:123–35. doi: 10.1038/s41564-020-00794-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pyenson NC, Gayvert K, Varble A, Elemento O, Marraffini LA. Broad targeting specificity during bacterial type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe. 2017;22:343–.e3. doi: 10.1016/j.chom.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Watson BNJ, Steens JA, Staals RHJ, Westra ER, van Houte S. Coevolution between bacterial CRISPR-Cas systems and their bacteriophages. Cell Host Microbe. 2021;29:715–25. doi: 10.1016/j.chom.2021.03.018. [DOI] [PubMed] [Google Scholar]
- 80.Westra ER, Levin BR. It is unclear how important CRISPR-Cas systems are for protecting natural populations of bacteria against infections by mobile genetic elements. PNAS. 2020;117:27777–85. doi: 10.1073/pnas.1915966117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paul BG, Eren AM. Eco-evolutionary significance of domesticated retroelements in microbial genomes. Mobile DNA. 2022;13:6. doi: 10.1186/s13100-022-00262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ratner HK, Sampson TR, Weiss DS. I can see CRISPR now, even when phage are gone: a view on alternative CRISPR-Cas functions from the prokaryotic envelope. Curr Opin Infect Dis. 2015;28:267–74. doi: 10.1097/QCO.0000000000000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Westra ER, Buckling A, Fineran PC. CRISPR–Cas systems: beyond adaptive immunity. Nat Rev Microbiol. 2014;12:317–26. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 84.Wimmer F, Beisel CL. CRISPR-Cas systems and the paradox of self-targeting spacers. Front Microbiol. 2020;10:3078. [DOI] [PMC free article] [PubMed]
- 85.Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol. 2008;10:200–7. doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- 86.Frischkorn KR, Haley ST, Dyhrman ST. Coordinated gene expression between Trichodesmium and its microbiome over day–night cycles in the North Pacific Subtropical Gyre. ISMEJ. 2018;37:1. doi: 10.1038/s41396-017-0041-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watson BNJ, Staals RHJ, Fineran PC. CRISPR-Cas-mediated phage resistance enhances horizontal gene transfer by transduction. mBio 2018;9:e02406-17. [DOI] [PMC free article] [PubMed]
- 88.Varble A, Meaden S, Barrangou R, Westra ER, Marraffini LA. Recombination between phages and CRISPR − cas loci facilitates horizontal gene transfer in Staphylococci. Nat Microbiol. 2019;4:956–63. doi: 10.1038/s41564-019-0400-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gradoville MR, White AE, Böttjer D, Church MJ, Letelier RM. Diversity trumps acidification: Lack of evidence for carbon dioxide enhancement of Trichodesmium community nitrogen or carbon fixation at Station ALOHA. Limnol Oceanogr. 2014;59:645–59.
- 90.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Held NA, Saito MA. Trichodesmium field metaproteomes. 2019. PRIDE.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our sequences files are accessible from the National Center for Biotechnology Information (NCBI; BioProject PRJNA828267), Sequence Read Archive accession numbers SRR19658988 through SRR19658999. MAG Assemblies are available at NCBI Assembly page with accessions SAMN29146503 through SAMN29146503. The mass spectrometry proteomics data were originally deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the identifier PXD016225 and can be accessed at 10.6019/PXD016225 [90, 91]. The data is also available at BCO-DMO (https://www.bco-dmo.org/dataset/787078).