

Abstract
Indolizine-carbaldehydes with the easily modifiable carbaldehyde group are important synthetic targets as versatile precursors for distinct indolizines. However, the efficient one-pot construction of trisubstituted indolizine-2-carbaldehydes represents a long-standing challenge. Herein, we report an unprecedented recyclable stereoauxiliary aminocatalytic approach via aminosugars derived from biomass, which enable the efficient one-pot synthesis of desired trisubstituted indolizine-2-carbaldehydes via [3+2] annulations of acyl pyridines and α,β-unsaturated aldehyde. Compared to the steric shielding effect from α-anomer, a stereoauxiliary effect favored by β-anomer of D-glucosamine is supported by control experiments. Furthermore, polymeric chitosan containing predominantly β-D-anhydroglucosamine units also shows excellent catalytic performance in aqueous solutions for the conversion of various substrates, large-scale synthesis and catalytic cycling experiments. Thus, our approach advances the existing methodologies by providing a rich library of indolizine-2-aldehydes. In addition, it delivers an efficient protocol for a set of late-stage diversification and targeted modifications of bioactive molecules or drugs, as showcased with 1,2,3-trisubstituted indolizine-2-carbaldehydes.
Subject terms: Catalyst synthesis, Sustainability, Organocatalysis
Indolizine-2-carbaldehydes are important precursors for indolizine synthesis, however, their efficient construction remains challenging. Here, a recyclable stereoauxiliary aminocatalyzed strategy for one-pot synthesis of 1,2,3-trisubstituted indolizine-2-carbaldehydes is reported, via [3+2] annulations of acyl pyridines and α,β-unsaturated aldehydes.
Introduction
Indolizines, an important group of N-heterocyclic compounds1, play a pivotal role in various fields ranging from pharmaceutics (Fig. 1a)2–4 to material science5 and chemical synthesis6–10. Thus, significant efforts have been made and remarkable progress has been achieved in the synthesis of such type of scaffolds11. Four representative strategies are known for the efficient preparation of indolizines, which include Scholtz reaction12,13, Tschitschibabin reaction14,15, pyridinium N-methylides16,17, and cyclization of alkynes with heteroaromatic compounds18–20. Recently, multi-step synthesis strategies for the preparation of indolizine-carbaldehydes have been reported21–23 and the easily modifiable aldehyde group in pyrrole ring makes indolizine-2-carbaldehydes versatile building blocks (Fig. 1b)22,23. One-pot synthesis and synthetic modifications of indolizine-2-carbaldehydes, however, were rarely studied, probably due to the lack of efficient synthetic strategies (Fig. 1b). In particular, such an one-pot synthetic strategy would be highly attractive and desired among synthetic and medicinal chemists22,23.
Fig. 1. Indolizine.

a Pharmaceuticals derived from indolizine. b Traditional approaches for indolizine-carbaldehydes. c Our design: iminium ion/enamine tandem sequence strategy for trisubstituted indolizine-2-carbaldehydes. d This work: Unprecedented stereoauxiliary aminocatalysis with iminium ion/enamine strategy for the preparation of 1,2,3-trisubstituted indolizine-2-carbaldehydes via one-pot reaction.
The [3+2] annulations of α,β-unsaturated aldehydes and 2-acetylpyridine is a pivotal step for the one-pot construction of indolizine-2-carbaldehydes. Generally, 2-acetylpyridine easily reacts with the carbonyl group of α,β-unsaturated aldehydes24, and 2-acetylpyridine activated by metal-based Lewis acid could attack the β-position of α,β-unsaturated aldehydes with the presence of a secondary aminocatalyst25. These challenges have hampered the development of [3+2] cyclization of 2-acetylpyridine and α,β-unsaturated aldehydes. Inspired by the two-component Baylis–Hillmann reaction26,27, an acetic acid-catalyzed method for the one-pot preparation of desired indolizine-2-carbaldehydes was first time reported in 2021 as a state-of-the-art method28. This is the only one-pot synthesis of indolizine-2-carbaldehydes reported to date, and the reaction was carried out in acetic acid as catalyst and solvent to improve the efficiency29. A generalized strategy to overcome the harsh reaction conditions for broader scope of indolizine-2-carbaldehydes with even higher efficiency via [3+2] cyclization is still highly desired.
During the last decades, aminocatalysis via iminium ion or enamine has emerged as an important approach for the construction of various C–C bonds30–36. Herein, we propose an aminocatalysis mode via iminium ion/enamine tandem sequence that could efficiently overcome the reaction energy barriers for Michael reaction and aldol reaction for the construction of 1,2,3-trisubstituted indolizine-2-carbaldehydes (Fig. 1c). In particular, carbohydrates as the most abundant and renewable biomass with native chiral backbones have been widely utilized as carbohydrate-derived ligands for enantioselective reactions37–39, whereas aminocatalyst derived from amino sugars has received less attention so far40,41. Inspired by our recently work on anomeric stereoauxiliary cleavage of the C−N bond of glucosamine for the efficient preparation of imidazo[1,5-a]pyridines42, we discovered a novel sustainable aminocatalysis strategy via recyclable stereoauxiliary combined with iminium ion/enamine tandem sequence as potential synthesis strategy (Fig. 1d). D-glucosamine and even the polymeric chitosan containing mostly β-D-anhydroglucosamine units as building blocks representing one of the most abundant and renewable biobased compounds43, were first time utilized as attractive stereoauxiliary aminocatalysts for the one-pot efficient synthesis of 1,2,3-trisubstituted indolizine-2-carbaldehydes via [3+2] cyclization. This new approach largely expands the scope of readily accessible indolizine-2-carbaldehydes relative to existing state-of-the-art methods.
Results and discussion
Reaction development
We initiated our studies using cinnamaldehyde (1a) and 2-acetylpyridine (2a) as substrates to evaluate the envisioned aminocatalyzed [3+2] cyclization reaction for the synthesis of desired 1-methyl-3-phenylindolizine-2-carbaldehyde (4) (see Supplementary Note 1 and Supplementary Method 1, 2). In addition, Brønsted acid (2 equiv.) was used to hinder the deprotonation of the methyl group of 2a (Supplementary Tables 1, 2)29, while lithium cations were used to improve the catalytic performance of the cyclization reaction (Supplementary Table 3)25,44. Bronsted acid, e.g., Li+, could help to activate the carbonyl group in the iminium formation and/or in the intramolecular cyclization, with the release of water. At the outset without catalyst, the reaction was tested with a trace yield of product 4 with a mixture of 1a (0.20 mmol), 2a (2.5 equiv.), LiSO3CF3 (3.0 equiv.) and acetic acid (2.0 equiv.) in CF3CH2OH (0.90 mL) for 18 h under Ar atmosphere (Fig. 2) (Supplementary Table 4, 5). We also examined various widely-used representative aminocatalysts and ligands derived from amino acids (Fig. 2). By using (S)-(-)-α, α-Diphenyl-2-pyrrolidinemethanol (3a)45, (S)-(-)-α, α-Diphenylprolinoltrimethylsilyl ether (3b)45, L-proline (3c)30, (5S)-(-)-5-Benzyl-2,2,3-trimethylimidazolidin-4-one (3d)31, N-(tert-butoxycarbonyl)-L-valine (3e)46 and glycine (3f)47, as catalysts, only low yields of product 4 were achieved.
Fig. 2. Optimization of the aminocatalyzed [3+2] annulations for indolizine-2-aldehyde.

a1a (0.2 mmol), 2a (2.5 equiv.), aminocatalyst (20 mol%), LiSO3CF3 (3.0 equiv.), AcOH (2.0 equiv.), CF3CH2OH (0.9 mL), Ar, 18 h, 80 °C. bYields were determined by 1H-NMR analysis with CH2Br2 as internal standard. Chitosan has a degree of deacetylation of 97.96%.
Various sustainable amino sugars and their derivatives, including D-glucosamine hydrochloride (3g), D-mannosamine hydrochloride (3h), N-acetyl-D-glucosamine hydrochloride (3i), 1,3,4,6-tetra-O-acetyl-2-amino-2-deoxy-β-D-glucopyranose hydrochloride (3j), 1,3,4,6-tetra-O-acetyl-2-amino-2-deoxy-α-D-glucopyranose hydrochloride (3k) and chitosan were used as aminocatalysts under the same conditions (Fig. 2). Surprisingly, 97% yield of 4 was achieved by using catalyst 3j (see Supplementary Note 2, Supplementary Fig. 1), while 3k only achieved 53% yield of 4. In comparison, lower yields of 4 were obtained with 3g-3i, 3k and chitosan. Based on all these results, 3j was taken as the optimal aminocatalyst for further synthesis. In addition to amine-containing catalysts showing the central function for the efficient reaction, acetic acid plays an important role. Without acetic acid (Supplementary Table 2), the yield of 4 decreased obviously from 97 to 44%48. As well, the amount of LiSO3CF3 (2 equiv.) and 2-acetylpyridine (1.5 equiv.), reaction time (12 h) and reaction temperature (25 and 50 °C) also affected the yields (Supplementary Table 1, entries 13-17). Furthermore, in order to exclude the Lewis-acid catalytic pathway through acetic acid29, a mixture of 1a (0.2 mmol), 2a (2.5 equiv.) and NaOAc (3.0 equiv.) in acetic acid (0.9 mL) was tested (Supplementary Table 4, entries 8). As a result, only 2% of 4 was obtained, which further demonstrates the higher catalytic activity of our aminocatalysis protocol.
Substrate scope
With the optimized reaction conditions in hand, we next probed the scope of various α,β-unsaturated aldehydes using 2-acetylpyridine as a representative heteroaryl ketone (Fig. 3a) (see Supplementary Method 3 and Supplementary Note 3). A series of α,β-unsaturated aldehydes, including those with electron-donating or -withdrawing groups at different positions (ortho, meta or para), delivered the corresponding products 4-13 under General procedure A. An array of valuable products 4-8 were efficiently accessed with this stereoauxiliary aminocatalyzed protocol. Notably, in our system, a substrate with an electron-donating methoxy group at ortho position (6, 95%) could even achieve a higher yield than those at para position (5, 63%). Surprisingly, a native valuable substrate from Gliricidia sepium with a hydroxyl group and a methoxy group was smoothly transformed into a value-added indolizine-2-aldehyde with a moderate yield (7, 63%). As well, an important substrate for the detection of catechins was also tolerant under this method with a 46% yield (8) under General procedure B. In addition, a variety of valuable functional groups at diverse positions, such as fluoro (9), chloro (10), bromo (11, 12), and nitro moiety (13), were well compatible with the standard conditions. Particularly, the sensitive (E)-3-(furan-2-yl)acrylaldehyde was also tolerated in our protocol under General procedure C and was successfully transformed into the desired product (14). Moreover, aliphatic α,β-unsaturated aldehyde was also well compatible under the optimal conditions (15).
Fig. 3. Scope of substrates for the synthesis of indolizine-2-carbaldehydes.

a Scope of aldehydes. b Scope of the heteroaryl ketones. Unless otherwise specified, all products were prepared with General procedure A: α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketones (2.5 equiv.), catalyst 3j (20 mol%), AcOH (2.0 equiv.) and LiSO3CF3 in CF3CH2OH (0.9 mL) for 18 h at 80 °C under Ar atmosphere. aGeneral procedure B: α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketones (2.5 equiv.), catalyst 3j (20 mol%), and LiSO3CF3 in AcOH : CF3CH2OH (0.4 : 0.5 mL) for 36 h at 80 °C under Ar atmosphere. bGeneral procedure C: α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketones (2.5 equiv.), catalyst 3j (20 mol%), AcOH (4.0 equiv.) and LiSO3CF3 in CF3CH2OH (0.9 mL) for 42 h at r.t.. cGeneral procedure D: α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketones (2.5 equiv.), catalyst 3j (20 mol%), AcOH (2.0 equiv.) and LiSO3CF3 in CF3CH2OH (0.9 mL) for 36 h at 80 °C under Ar atmosphere. Yields are those of isolated products.
We further explored various heteroaryl ketones in combination with cinnamaldehyde under General procedure A (Fig. 3b). Di(pyridin-2-yl)methanone and pyridin-2-yl(pyridin-4-yl)methanone were well compatible under the conditions and smoothly achieved yields of 95% (16), 83% (17) and 85% (18), respectively. Diverse aromatic pyridine ketones, including those having electron-donating or -withdrawing groups at distinct positions (ortho, meta, or para) were efficiently transformed into corresponding products (19-24). Various valuable functional groups at distinct positions (meta or para), including methoxy (21), trifluoromethyl (22), bromo (23) and dibromo (24), were well tolerated under the optimized condition. Cyclopentyl(pyridin-2-yl)methanone efficiently delivered desired product (25). The structure of 19 was further confirmed by single-crystal X-ray crystallographic analysis (see Supplementary Fig. 2, Supplementary Table 6), and those of other products in Fig. 3 were assigned by analogy. It is worth noting that ethyl 3-oxo-3-(pyridin-2-yl)propanoate (26) and also 1-(3-methylpyrazin-2-yl)ethan-1-one (27) were successfully transformed into desired products under General procedure D.
Late-stage synthetic applications
On indolizines with important biological activities, the modifiable aldehyde group on the backbone is attractive for late-stage transformations into versatile value-added products. Until recently, such valuable indolizine-2-carbaldehydes were obtianed in 6-step reaction sequences with complex conditions or 2-step reaction sequences with rare and expensive feedstocks (Fig. 4a)22,23. Compared with these previous protocols via carboxylation and reduction for the desired products, we efficiently achieved the synthesis of a group of value-added 1,2,3-trisubstituted indolizine-2-carbaldehydes in a one-pot reaction via aminocatalyzed [3+2] cyclization reaction. A group of important bioactive molecules or drugs was used for our protocol (Fig. 4b) (see Supplementary Method 4). Surprisingly, an important fluvastatin intermediate was first time accessed by our protocol for the preparation of value-added indolizine-2-carbaldehyde (28). As well, (E)-3-(4-hydroxy-3-methoxyphenyl)acrylaldehyde from Gliricidia sepium was also tolerant under the optimal conditions, which led to 3-(4-hydroxy-3-methoxyphenyl)-1-(pyridin-2-yl)indolizine-2-carbaldehyde (29) with 79% yield. Interestingly, (E)-3-(4-(dimethylamino)phenyl) acrylaldehyde that is often used to detect catechins49 was also smoothly transformed into 3-(4-(dimethylamino)phenyl)-1-(pyridin-2-yl)indolizine-2-carbaldehyde (30, 81%). Furthermore, obtained indolizine-2-carbaldehydes could be readily diversified during late-stage modifications, thus providing more complex molecules in an efficient manner (Fig. 4c) (see Supplementary Method 5). For example, 3-(4-bromophenyl)-1-(pyridin-4-yl)indolizine-2-carbaldehyde (17) underwent successful reduction (31), arylation (32), condensation (33) or dehydration [5+1] annulations (34), to showcase the synthetic diversifications on 1,2,3-trisubstituted indolizine-2-carbaldehydes.
Fig. 4. Synthetic applications.

a Representative previous methods for 3-dimethylaminoindolizine-2-aldehyde. b Late-stage selective modifications of bioactive molecules and drugs. c Late-stage diversification. aYields are those of isolated products. bReaction for 42 h in AcOH : CF3CH2OH (0.45 : 0.45 mL). cReaction for 42 h in AcOH : CF3CH2OH (0.4 : 0.5 mL).
Many organocatalyzed reactions still require high catalyst loadings (20-30 mol%), while organocatalysts are difficult to separate, recycle and reuse32. Therefore, a recyclable aminocatalyst for desired indolizine-2-aldehydes is in high demand. Notably, our anomeric stereoauxiliary aminocatalyst was efficiently expanded beyond the low molecular weight D-glucosamine to the biopolymer chitosan containing β-D-glucosamine as building blocks50 (Fig. 5a) (see Supplementary Method 6 and Supplementary Table 7). Interestingly, the use of chitosan demonstrates a recyclable aminocatalysis strategy and the reaction is highly efficient in H2O, while lithium salts are not required. As a result, various indolizine-aldehydes were obtained under the use of chitosan as sustainable aminocatalyst, such as products 4 (50%), 5 (41%), 6 (53%), 7 (72%), 8 (36%), 9 (45%), 10 (30%), 12 (24%), and 29 (32%). Even three cyclic α,β-unsaturated ketones efficiently delivered the corresponding products 35 (23%), 36 (60%), and 37 (52%). Although the use of chitosan for transforming halogenated aromatic α,β-unsaturated aldehydes led to lower yields compared to glucosamine (Fig. 5a), e.g., for 9 (81%), 10 (75%), and 12 (71%), chitosan as aminocatalyst resulted in higher yields for products 4-7 and 35-37.
Fig. 5. Chitosan as stereoauxiliary aminocatalyst for indolizine-2-carbaldehydes via [3+2] annulation.

a Scope of substrates. aGeneral procedure E: α,β-unsaturated aldehydes/ketones (0.2 mmol), heteroaryl ketones (2.5 equiv.), chitosan (20 mol%) and formic acid (4.0 equiv.) in H2O (1.0 mL) for 18 h at 120 °C under Ar atmosphere. bGeneral procedure F: α,β-unsaturated aldehydes/ketones (0.2 mmol), heteroaryl ketones (2.5 equiv.), and chitosan (20 mol%) in Formic acid : H2O (0.5 : 0.5 mL) for 36 h at 120 °C under Ar atmosphere. b Larger scale synthesis of indolizine-2-carbaldehyde. c Cycling catalytic experiments for the synthesis of indolizine-2-carbaldehyde.
Our strategy was compared with the state-of-the-art method28. For example, products with sensitive groups can be smoothly prepared with our protocol (7: 63%, 8: 46%, 14: 30%, 26: 32%, 27: 43% and 36: 60%), while only 2% NMR yield or even no products were obtained using the reaction condition as in the ref. 28 (7: not detected, 8: 2%, 14: not detected, 26: not detected, 27: 2% and 36: 8%). These results clearly demonstrated the robustness of our aminocatalysis protocol compared with ref. 28. Furthermore, product 4 can be successfully prepared by a one-pot method on a larger scale (2.0 mmol) with up to 62% yield (Fig. 5b) (see Supplementary Method 6). Chitosan can be used for multiple cycles as an aminocatalyst in the aqueous solution, and exhibited excellent catalytic performance even after 3 catalytic cycles under the standard conditions (see Supplementary Fig. 3). During the cycling catalytic reactions, product 4 can be easily isolated by organic solvent extraction, and the remaining aqueous phase can be directly used in the next catalytic cycle after adding 1a and 2 (Fig. 5c).
Mechanistic considerations
Under the standard condition, catalyst 3j with β-anomer smoothly achieved 97% yield of 4, while catalyst 3k with α-anomer only yielded 53% of 4 (Fig. 6a). This lower reactivity using 3k demonstrates the presence of a strong steric shielding from α-anomer that affects the efficient conversion to the desired product 4. To gain more insight into the reaction mechanism, imine intermedates of acetylated D-glucosamine, 3p as β-anomer and 3q as α-anomer, were synthesized, separated and tested under the standard conditions (Fig. 6b) (see Supplementary Method 7). Interestingly, product 4 with 51% yield was obtained using 3p (β-anomer), while 3q (α-anomer) could only deliver 16% yield of 4. Thus, the imine reaction pathway via aminocatalyst preferentially reacting with α,β-unsaturated aldehydes is verified by these control experiments. Besides, the lower yield of 4 with 3q (α-anomer) further provides a strong support for the existing steric hinderance from acetyl group at C1-position in 3q. In comparison, the stereoauxiliary effect from 3p (β-anomer) promoted the yield of 4. Therefore, a stereoauxiliary effect favored by β-anomer as well as a steric shielding effect from α-anomer were clearly verified by control experiments.
Fig. 6. Stereoauxiliary control experiments.
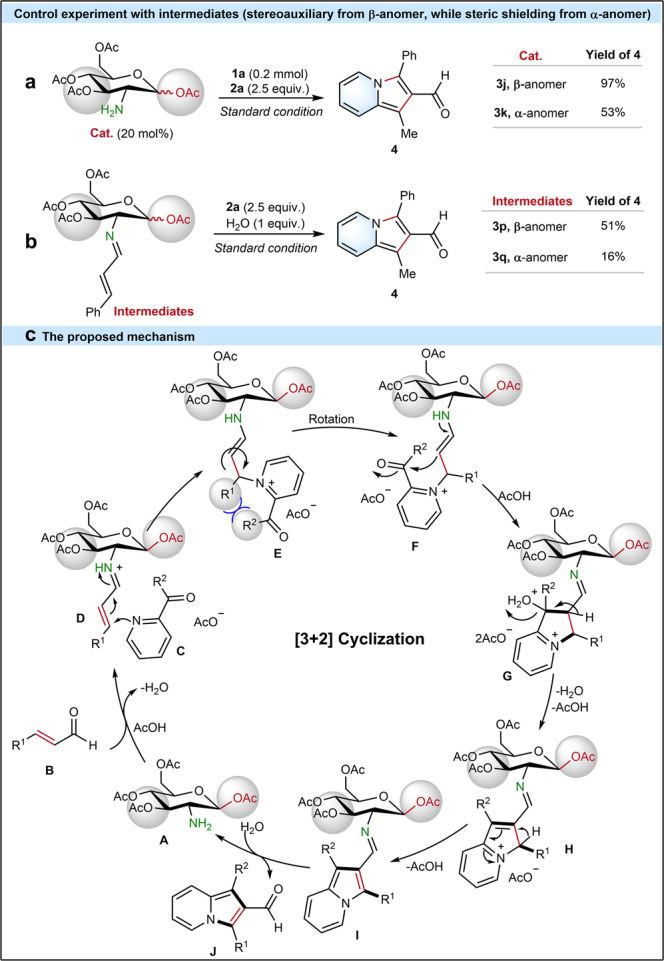
a Control experiment with 3j (β-anomer) and 3k (α-anomer). b Control experiment with intermediate 3p (β-anomer) and intermediate 3q (α-anomer). c Proposed mechanism.
Combining all results, a plausible mechanism is proposed (Fig. 6c). First, aminocatalyst A reacts with α,β-unsaturated aldehyde B to form iminium ion D32. Then, 2-acetylpyridine attacks the iminium ion D via Michael addition reaction to generate an enamine E32,51. Enamine F can be simply converted from E via the rotation, which will overcome the bulky steric hindrance between R1 and R2. Thereafter, an intermediate G forms via the intramolecular cyclization reaction in the enamine F. Then, an intermediate H generates from the intermediate G through a dehydration reaction, which leads to an intermediate I after deprotonation. Finally, the desired indolizine-2-aldehyde J forms via the hydrolysis reaction of intermedidate I and the catalyst A is regenerated (ESI-HRMS: m/z calcd. for C14H22NO9+ [M]: 348.1289, found 348.1297. see Supplementary Fig. 4; the conformation stability of catalyst 3j was proved with 1H NMR in Supplementary Fig. 5) for the next catalysis cycle. Computational investigations of the mechanistic and stereochemical aspects of this study are underway in the Houk lab at UCLA.
Conclusion
We have developed an unprecedented recyclable anomeric stereoauxiliary aminocatalytic approach using glucosamine/chitosan from biomass for the efficient one-pot preparation of versatilely decorated indolizine-2-carbaldehydes via [3+2] annulations of acyl pyridines and α,β-unsaturated aldehyde. This approach via an aminocatalysis pathway under mild conditions efficiently expands the scope of readily accessible trisubstituted indolizine-2-carbaldehydes relative to existing state-of-the-art methods. Mechanistic control studies provided strong support for the anomeric stereoauxiliary catalysis. Furthermore, a plethora of late-stage diversification and targeted modifications of bioactive molecules or drugs showcased the synthetic power of 1,2,3-trisubstituted indolizine-2-carbaldehydes that were assembled via this robust stereoaxuliary aminocatalysis approach. Moreover, biopolymer chitosan consisting of β-D-anhydroglucosamine units showed excellent catalytic performance in aqueous solution for various substrate diversifications, large-scale synthesis and recycling experiments. Overall, our anomeric stereoauxiliary catalytic approach provides a promising solution and an efficient green synthesis strategy towards addressing the challenges associated with the assembly of indolizine-2-aldehydes with versatile functional moieties, on which ongoing work is targeted to apply this strategy towards developing a wider range of catalytic applications.
Methods
Preparation of indolizine-2-carbaldehydes derivatives
General procedure A
A mixture of α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketone, catalyst 3j (0.04 mmol), AcOH (2.0 equiv.) and LiSO3CF3 (3.0 equiv.) in the CF3CH2OH (0.9 mL) were stirred at 80 °C under Ar atmosphere for 18 h.
General procedure B
A mixture of α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketone, catalyst 3j (0.04 mmol) and LiSO3CF3 (3.0 equiv.) in the CF3CH2OH : AcOH (0.5 : 0.4 mL) were stirred at 80 °C under Ar atmosphere for 36 h.
General procedure C
A mixture of α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketone, catalyst 3j (0.04 mmol), AcOH (4.0 equiv.) and LiSO3CF3 (3.0 equiv.) in the CF3CH2OH (0.9 mL) were stirred at room temperature under Ar atmosphere for 42 h.
General procedure D
A mixture of α,β-unsaturated aldehyde (0.2 mmol), heteroaryl ketone, catalyst 3j (0.04 mmol), AcOH (2.0 equiv.) and LiSO3CF3 (3.0 equiv.) in the CF3CH2OH (0.9 mL) were stirred at 80 °C under Ar atmosphere for 36 h.
Workup General procedure A–D
The reaction temperature was directly read from temperature detector of IKA apparatus that was calibrated with thermometer. After cooling to room temperature, the reaction mixture was basified up to pH 7 via stad. Na2CO3 aqueous solution, then extracted by diether (3 × 3 mL) and dried over anhydrous Na2SO4. After filtration and concentration on rotary evaporator, the crude product was purified with flash chromatography on silica gel (ethyl acetate : n-hexane) to give products.
General procedure E
A mixture of α,β-unsaturated aldehyde/α,β-unsaturated ketone (0.2 mmol), heteroaryl ketone (2.5 equiv.), catalyst chitosan (0.04 mmol), formic acid (4.0 equiv.) in H2O (1.0 mL) were stirred at 120 °C under Ar atmosphere for 18 h.
General procedure F
A mixture of α,β-unsaturated aldehyde/α,β-unsaturated ketone (0.2 mmol), heteroaryl ketone (2.5 equiv.), catalyst chitosan (0.04 mmol) in formic acid : H2O (0.5 : 0.5 mL) were stirred at 120 °C under Ar atmosphere for 36 h.
Workup for General procedure E–F
The reactions were conducted in a sealed Schlenk tube and heated by an IKA magnetic heating agitator with heating block. The reaction temperature was directly read from temperature detector of IKA apparatus that was calibrated with thermometer. After cooling to room temperature, the reaction mixture was extracted by diether (3 × 3 mL) and dried over anhydrous Na2SO4. After filtration and concentration on rotary evaporator, the crude product was purified with flash chromatography on silica gel (ethyl acetate : n-hexane) to give products.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Kai Zhang thanks German Research Foundation (DFG) for the financial support for the project with the grant number ZH546/3-1 and University of Göttingen for the Department Start-up funding. Kui Zeng thanks China Scholarship Council for the PhD grant. We sincerely thank Prof. Dr. Kendall N. Houk and Jonathan J. Wong from the Department of Chemistry and Biochemistry, University of California, Los Angeles for the helpful discussions.
Author contributions
K.Zh. conceived the concept and supervised the project. K.Z. and R.M. undertook all of the experimental work, analytical characterization, and spectroscopic analysis. S.D. performed X-ray crystallography. K.Z., L.A., and K.Zh. analyzed the data and wrote the manuscript.
Peer review
Peer review information
Communications Chemistry thanks Daniel Gryko and the other, anonymous, reviewers for their contribution to the peer review of this work.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
The data that support the findings of this study are available in the Supplementary Information (experimental procedures and characterization data). The NMR spectra of all compounds are available in Supplementary Data 1. The X-ray crystallographic coordinates for structures 19, reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under CCDC 2079110 (19, Supplementary Data 2). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-023-00828-2.
References
- 1.Majumdar, K. C. & Chattopadhyay, S. K. Heterocycles in Natural Product Synthesis (John Wiley & Sons, 2011).
- 2.Vemula VR, Vurukonda S, Bairi CK. Indolizine derivatives: recent advances and potential pharmacological activities. Int. J. Pharm. Sci. Rev. Res. 2011;11:159–163. [Google Scholar]
- 3.Singh GS, Mmatli EE. Recent progress in synthesis and bioactivity studies of indolizines. Eur. J. Med. Chem. 2011;46:5237–5257. doi: 10.1016/j.ejmech.2011.08.042. [DOI] [PubMed] [Google Scholar]
- 4.Sharma V, Kumar V. Indolizine: a biologically active moiety. Med. Chem. Res. 2014;23:3593–3606. doi: 10.1007/s00044-014-0940-1. [DOI] [Google Scholar]
- 5.Kim E, Lee Y, Lee S, Park SB. Discovery, understanding, and bioapplication of organic fluorophore: a case study with an indolizine-based novel fluorophore, Seoul-Fluor. Acc. Chem. Res. 2015;48:538–547. doi: 10.1021/ar500370v. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Song H, Qin Y. Total synthesis of indoline alkaloids: a cyclopropanation strategy. Acc. Chem. Res. 2011;44:447–457. doi: 10.1021/ar200004w. [DOI] [PubMed] [Google Scholar]
- 7.Čerņaks D. Recent methods for the synthesis of indolizines (microreview) Chem. Heterocycl. Compd. 2016;52:524–526. doi: 10.1007/s10593-016-1921-8. [DOI] [Google Scholar]
- 8.Wang LX, Tang YL. Cycloisomerization of pyridine‐substituted propargylic alcohols or esters to construct indolizines and indolizinones. Eur. J. Org. Chem. 2017;2017:2207–2213. doi: 10.1002/ejoc.201601530. [DOI] [Google Scholar]
- 9.de Souza CR, Gonçalves AC, Amaral M, Dos Santos A, Clososki G. Recent synthetic developments and reactivity of aromatic indolizines. Targets Heterocycl. Syst. 2016;20:365–392. [Google Scholar]
- 10.Hui J, Ma Y, Zhao J, Cao H. Recent advances in synthesis of indolizine and its derivatives by radical cyclization/cross-coupling. Org. Biomol. Chem. 2021;19:10245–10258. doi: 10.1039/D1OB01431E. [DOI] [PubMed] [Google Scholar]
- 11.Sadowski B, Klajn J, Gryko DT. Recent advances in the synthesis of indolizines and their π-expanded analogues. Org. Biomol. Chem. 2016;14:7804–7828. doi: 10.1039/C6OB00985A. [DOI] [PubMed] [Google Scholar]
- 12.Boekelheide V, Windgassen R., Jr Syntheses of pyrrocolines unsubstituted in the five-membered ring. J. Am. Chem. Soc. 1959;81:1456–1459. doi: 10.1021/ja01515a044. [DOI] [Google Scholar]
- 13.Scholtz MDie. Einwirkung von Essigsäureanhydrid auf α-Picolin. Ber. Dtsch. Chem. Ges. 1912;45:734–746. doi: 10.1002/cber.191204501108. [DOI] [Google Scholar]
- 14.Tschitschibabin A. Tautomerie in der Pyridin-Reihe. Ber. Dtsch. Chem. Ges. (A B Ser.) 1927;60:1607–1617. doi: 10.1002/cber.19270600724. [DOI] [Google Scholar]
- 15.Bora U, Saikia A, Boruah RC. A novel microwave-mediated one-pot synthesis of indolizines via a three-component reaction. Org. Lett. 2003;5:435–438. doi: 10.1021/ol020238n. [DOI] [PubMed] [Google Scholar]
- 16.Poissonnet G, Théret-Bettiol M-H, Dodd RH. Preparation and 1, 3-dipolar cycloaddition reactions of β-carboline azomethine ylides: a direct entry into C-1-and/or C-2-functionalized indolizino [8, 7-b] indole derivatives. J. Org. Chem. 1996;61:2273–2282. doi: 10.1021/jo951520t. [DOI] [Google Scholar]
- 17.Katritzky AR, Qiu G, Yang B, He H-Y. Novel syntheses of indolizines and pyrrolo [2, 1-a] isoquinolines via benzotriazole methodology. J. Org. Chem. 1999;64:7618–7621. doi: 10.1021/jo9906936. [DOI] [Google Scholar]
- 18.Chuprakov S, Hwang FW, Gevorgyan V. Rh‐catalyzed transannulation of pyridotriazoles with alkynes and nitriles. Angew. Chem. 2007;119:4841–4843. doi: 10.1002/ange.200700804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kel’in AV, Sromek AW, Gevorgyan V. A novel Cu-assisted cycloisomerization of alkynyl imines: efficient synthesis of pyrroles and pyrrole-containing heterocycles. J. Am. Chem. Soc. 2001;123:2074–2075. doi: 10.1021/ja0058684. [DOI] [PubMed] [Google Scholar]
- 20.Seregin IV, Gevorgyan V. Gold-catalyzed 1, 2-migration of silicon, tin, and germanium en route to C-2 substituted fused pyrrole-containing heterocycles. J. Am. Chem. Soc. 2006;128:12050–12051. doi: 10.1021/ja063278l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antón-Cánovas T, Alonso F. The Eschenmoser’s salt as a formylation agent for the synthesis of indolizinecarbaldehydes and their use for colorimetric nitrite detection. Angew. Chem. 2023;62:e202215916. doi: 10.1002/anie.202215916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biagetti, M., Accetta, A., Capelli, A. M., Guala, M. & Retini, M. Indolizine derivatives as phosphoinositide 3-kinases inhibitors. US patent No. US 2015/0361100 A1. (17-12-2015).
- 23.Burnett, D. A & Vacca, J. P. Compounds, compositions and methods of use. International patent No. WO 2018/195075 A1. (25-10-2018).
- 24.Singh PK, Singh VK. Highly enantioselective Friedel−Crafts reaction of indoles with 2-enoylpyridine 1-oxides catalyzed by chiral pyridine 2, 6-bis (5′, 5′-diphenyloxazoline)−Cu (II) complexes. Org. Lett. 2008;10:4121–4124. doi: 10.1021/ol8016929. [DOI] [PubMed] [Google Scholar]
- 25.Meazza M, et al. Synergistic catalysis: highly enantioselective acetyl aza-arene addition to enals. Chem. Eur. J. 2019;24:13306–13310. doi: 10.1002/chem.201802303. [DOI] [PubMed] [Google Scholar]
- 26.Basavaiah D, Veeraraghavaiah G, Badsara SS. Ketones as electrophiles in two component Baylis–Hillman reaction: a facile one-pot synthesis of substituted indolizines. Org. Biomol. Chem. 2014;12:1551–1555. doi: 10.1039/c3ob42064g. [DOI] [PubMed] [Google Scholar]
- 27.Basavaiah, D. & Rao, A. J. First example of electrophile induced Baylis–Hillman reaction: a novel facile one-pot synthesis of indolizine derivatives. Chem. Commun. 604–605 (2003). [PubMed]
- 28.Yuan Y-C, Liu T-Z, Zhao B-X. Metal-free catalyzed synthesis of fluorescent indolizine derivatives. J. Org. Chem. 2021;86:12737–12744. doi: 10.1021/acs.joc.1c01292. [DOI] [PubMed] [Google Scholar]
- 29.Kroehnke F. The specific synthesis of pyridines and oligopyridines. Synthesis. 1976;1976:1–24. doi: 10.1055/s-1976-23941. [DOI] [Google Scholar]
- 30.List B, Lerner RA, Barbas CF. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000;122:2395–2396. doi: 10.1021/ja994280y. [DOI] [Google Scholar]
- 31.Ahrendt KA, Borths CJ, MacMillan DW. New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels−Alder reaction. J. Am. Chem. Soc. 2000;122:4243–4244. doi: 10.1021/ja000092s. [DOI] [Google Scholar]
- 32.Erkkilä A, Majander I, Pihko PM. Iminium catalysis. Chem. Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- 33.Lelais G, MacMillan DW. Modern strategies in organic catalysis: the advent and development of iminium activation. Aldrichim. Acta. 2006;39:79–87. [Google Scholar]
- 34.Bertelsen S, Jørgensen KA. Organocatalysis—after the gold rush. Chem. Soc. Rev. 2009;38:2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 35.van Der Helm MP, Klemm B, Eelkema R. Organocatalysis in aqueous media. Nat. Rev. Chem. 2019;3:491–508. doi: 10.1038/s41570-019-0116-0. [DOI] [Google Scholar]
- 36.Chauhan P, Mahajan S, Enders D. Achieving molecular complexity via stereoselective multiple domino reactions promoted by a secondary amine organocatalyst. Acc. Chem. Res. 2017;50:2809–2821. doi: 10.1021/acs.accounts.7b00406. [DOI] [PubMed] [Google Scholar]
- 37.Diéguez M, Pamies O, Claver C. Ligands derived from carbohydrates for asymmetric catalysis. Chem. Rev. 2004;104:3189–3216. doi: 10.1021/cr0306889. [DOI] [PubMed] [Google Scholar]
- 38.Castillón S, Claver C, Díaz Y. C 1 and C 2-symmetric carbohydrate phosphorus ligands in asymmetric catalysis. Chem. Soc. Rev. 2005;34:702–713. doi: 10.1039/b400361f. [DOI] [PubMed] [Google Scholar]
- 39.Woodward S, Diéguez M, Pàmies O. Use of sugar-based ligands in selective catalysis: Recent developments. Coord. Chem. Rev. 2010;254:2007–2030. doi: 10.1016/j.ccr.2010.03.005. [DOI] [Google Scholar]
- 40.Kong S, Fan W, Wu G, Miao Z. Enantioselective synthesis of tertiary α‐hydroxy phosphonates catalyzed by carbohydrate/cinchona alkaloid thiourea organocatalysts. Angew. Chem. Int. Ed. 2012;51:8864–8867. doi: 10.1002/anie.201204287. [DOI] [PubMed] [Google Scholar]
- 41.Kolcsár VJ, Szőllősi G. Chitosan as a chiral ligand and organocatalyst: preparation conditions–property–catalytic performance relationships. Catal. Sci. Technol. 2021;11:7652–7666. doi: 10.1039/D1CY01674A. [DOI] [Google Scholar]
- 42.Zeng K, et al. Anomeric stereoauxiliary cleavage of the C‐N bond of D‐glucosamine for the preparation of imidazo [1, 5‐α] pyridines. Chem. Eur. J. 2022;28:e202200648. doi: 10.1002/chem.202200648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim, S.-K. Chitin, Chitosan, Oligosaccharides and their Derivatives: Biological Activities and Applications (CRC Press, 2010).
- 44.Seebach D. Structure and reactivity of lithium enolates. from pinacolone to selective C‐alkylations of peptides. difficulties and opportunities afforded by complex structures. Angew. Chem. Int. Ed. 1988;27:1624–1654. doi: 10.1002/anie.198816241. [DOI] [Google Scholar]
- 45.Klier L, Tur F, Poulsen PH, Jørgensen KA. Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers. Chem. Soc. Rev. 2017;46:1080–1102. doi: 10.1039/C6CS00713A. [DOI] [PubMed] [Google Scholar]
- 46.Chan KS, et al. Ligand-enabled cross-coupling of C (sp3)–H bonds with arylboron reagents via Pd (II)/Pd (0) catalysis. Nat. Chem. 2014;6:146–150. doi: 10.1038/nchem.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q. Functionalization of C (sp3)–H bonds using a transient directing group. Science. 2016;351:252–256. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong L, Sun W, Yang D, Li G, Wang R. Additive effects on asymmetric catalysis. Chem. Rev. 2016;116:4006–4123. doi: 10.1021/acs.chemrev.5b00676. [DOI] [PubMed] [Google Scholar]
- 49.Glavnik V, Simonovska B, Vovk I. Densitometric determination of (+)-catechin and (−)-epicatechin by 4-dimethylaminocinnamaldehyde reagent. J. Chromatogr. A. 2009;1216:4485–4491. doi: 10.1016/j.chroma.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 50.Zeng K, Mei R, Zhang XC, Andreas LB, Zhang K. Direct nitrogen interception from chitin/chitosan for imidazo [1, 5-a] pyridines. Chem. Commun. 2022;58:6068–6071. doi: 10.1039/D2CC01060G. [DOI] [PubMed] [Google Scholar]
- 51.Moyano A, Rios R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011;111:4703–4832. doi: 10.1021/cr100348t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support the findings of this study are available in the Supplementary Information (experimental procedures and characterization data). The NMR spectra of all compounds are available in Supplementary Data 1. The X-ray crystallographic coordinates for structures 19, reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under CCDC 2079110 (19, Supplementary Data 2). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.