

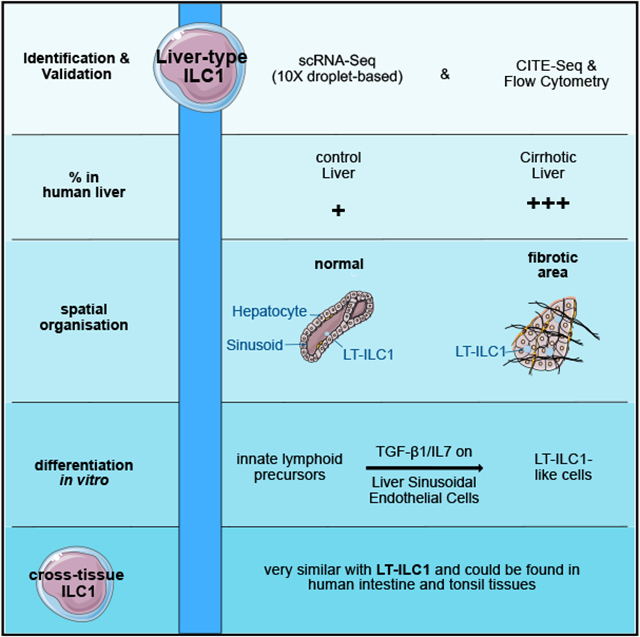
SUMMARY
Group 1 innate lymphoid cells (ILCs) comprise a heterogeneous family of cytotoxic natural killer (NK) cells and ILC1s. We identify a population of “liver-type” ILC1s with transcriptional, phenotypic, and functional features distinct from those of conventional and liver-resident NK cells as well as from other previously described human ILC1 subsets. LT-ILC1s are CD49a+CD94+CD200R1+, express the transcription factor T-BET, and do not express the activating receptor NKp80 or the transcription factor EOMES. Similar to NK cells, liver-type ILC1s produce IFN-γ, TNF-α, and GM-CSF; however, liver-type ILC1s also produce IL-2 and lack perforin and granzyme-B. Liver-type ILC1s are expanded in cirrhotic liver tissues, and they can be produced from blood-derived ILC precursors in vitro in the presence of TGF-β1 and liver sinusoidal endothelial cells. Cells with similar signature and function can also be found in tonsil and intestinal tissues. Collectively, our study identifies and classifies a population of human cross-tissue ILC1s.
Graphical Abstract
In brief
Krämer et al. report a population of human "liver-type" innate lymphocytes that differs from other previously characterized cells. They are expanded in cirrhotic livers and can be generated from precursors in co-culture with liver endothelial cells. Cells with similar properties can also be found in tonsil or intestinal tissue.
INTRODUCTION
The family of innate lymphoid cells (ILCs) consists of multiple non-T, non-B effector lymphocyte populations that regulate numerous physiologic and pathologic processes including lymphoid tissue organogenesis, mucosal homeostasis, pregnancy, infection, autoimmunity, obesity, and cancer.1 ILCs include cytotoxic natural killer (NK) cells and different populations of regulatory “helper”-ILCs. Overall, ILCs are classified into three broad groups based largely on their phenotypic and cytokine-producing profiles. Group 1 ILCs, including NK cells and ILC1s, produce T helper type 1 (Th1)-associated cytokines and express the transcription factor T-BET.2,3 Group 2 ILCs (ILC2s) produce Th2-associated cytokines and express high levels of the transcription factors GATA3, BCL11B, and RORα.4 Group 3 ILCs, including ILC3s and lymphoid tissue inducer cells, produce Th17-associated cytokines and express the transcription factor RORγt.5-8
Further research has revealed marked heterogeneity within each major group of ILCs. Group 1 ILCs are particularly diverse, and numerous tissue-specific and disease-associated subsets have been described.9,10 In mice, NK cells as well as all ILC1s can be identified by flow cytometry as lymphocytes that are “lineage antigen “ (Lin) negative (i.e., they lack surface markers that specifically denote other leukocyte populations) and express the natural cytotoxicity receptors (NCRs) NK1.1 and NKp46, as well as T-bet.11 Murine ILC1s also express CD49a (integrin α1) and were traditionally defined as lacking expression of the transcription factor EOMES, though recently described cytotoxic ILC1s can express low amounts of EOMES.12 In contrast, NK cells express high levels of EOMES and are mostly CD49a−. Thus, in some murinetissues, including the liver and adipose tissue, there exists a clear dichotomy (among Lin−NCR+ innate lymphocytes) between CD49a+Eomes− ILC1s and CD49a−Eomes+ NK cells.13,14 In other tissues, such as the salivary glands and uterus, or in specific pathologic settings of infection or malignancy, there are also dual positive CD49a+EOMES+ group 1 ILCs that may represent either activated NK cells with upregulated CD49a or an intermediate transitional group 1 ILC phenotype.15,16 Therefore, CD49a expression itself is not specific for either ILC1s or NK cells, and the distinction between these group 1 ILC populations is not always clear. Recently, it was shown that based on the expression of the inhibitory receptor CD200r1, murine ILC1s (which are CD200r1+) can be specifically distinguished from CD200r1− NK cells in liver and adipose tissue.14 Murine ILC1s and NK cells can thus be largely distinguished as Lin−NCR+CD49a+CD200r1+T-BET+EOMES− and Lin−NCR+CD49a−CD200r1−T-BET+EOMES+ cells, respectively.
In humans, “conventional” NK (cNK) cells, such as those found in the peripheral blood, were originally identified as Lin−CD56+ lymphocytes. However, it is now clear that the Lin−CD56+ phenotype is not specific to cNK cells but also encompasses non-conventional, tissue-resident NK (trNK) cells as well as other non-NK ILCs and ILC precursor cells (ILCPs) that can express CD56.6,17,18 Therefore, other markers, some of which are not expressed by murine ILCs, are now used to distinguish human ILC populations. These markers include NKp80, which is expressed by nearly all mature NK cells in healthy individuals19; CXCR6, which distinguishes human cNK cells (CXCR6−) from trNK cells (CXCR6+)20,21; and NKp44, which is non-specific, but in the absence of group 1 ILC-associated markers identifies human IL-22-producing group 3 ILCs.22
To date, two major human ILC1 populations have been described. Surface marker “nuN” ILC1s (or “classical ILC1s” [cILC1s] as we refer to here) were originally characterized as IFN-γ-producing Lin−CD127+ ILCs that lack expression of NK cell- (CD16, CD56, CD94), ILC2- (CD294), and ILC3-associated (CD117, NKp44) markers.7 These cILC1s were detected at low frequencies in healthy intestinal mucosal tissues yet shown to be expanded in patients with Crohn disease. A second population of human ILC1s, termed intraepithelial ILC1s (ieILC1s), was originally described in human tonsils to consist of Lin− CD56+CD103+CD127− NKp44+cells that produce IFN-γ and have features of IGF-β1 imprinting.2,3 Given that cILC1s are Lin−CD127+CD56−NKp44− and ieILC1s are Lin−CD127−CD56+ NKp44+, these two human ILC1 populations are mutually exclusive.
In this study, we provide single-cell transcriptional, phenotypic, and functional evidence for the existence of a distinct third population of human ILC1s, which we refer to as “liver-type” ILC1s given their original detection in human liver samples. We demonstrated that human liver-type ILC1s are not only distinct from cILC1s and tonsil-derived ieILC1s, but they can also be clearly distinguished from cNK cells, liver-derived trNK cells, ILC2s, and ILC3s. The human liver-type ILC1s expressed T-BET but not EOMES; they produced IFN-γ, TNF-α, GM-CSF, and IL-2, but not perforin or granzyme-B; and they could be derived in vitro from circulating ILCPs co-cultured in the presence of liver sinusoidal endothelial cells (LSECs), IL-7, and TGF-β1. Collectively, these findings support the identification of a population of human liver-type ILC1s.
RESULTS
Single-cell RNA sequencing identifies an ILC1-like population in the human liver
To the best of our knowledge, characterization of ILC1s in the human liver remains elusive. To evaluate human ILCs in the liver using an unbiased approach, we first incorporated single-cell RNA sequencing (scRNA-seq) data to classify the transcriptional profiles of liver-derived ILCs (experimental workflow; Figure S1A). We sorted total Lin−CD45+ lymphocytes from seven non-cirrhotic controls (two perfusate from healthy donors and five non-tumor tissue from liver cancer patients; Table S2; Figure S1B) and eight cirrhotic livers in two independent experiments (batch 1 and 2) and subsequently performed scRNA-seq analysis with compliance of quality standards and filtering of non-NK/ILC populations (Figures 1A, S1C, and S1D). As an additional comparison cohort, a publicly accessible scRNA-seq dataset from Heinrich et al.,23 including six patients with hepatocellular carcinoma, each with tumor margin and non-tumor liver tissue (12 samples; Table S2) was analyzed. Among the liver Lin−CD45+ lymphocytes, a total of 11 (batch 1) and 19 (batch 2 and Heinrich et al.) clusters, respectively, were identified and visualized using Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP). Visualization of cells by group identified a disease-specific distribution with clusters 1, 6, 9, 10 (batch 1) and clusters 7, 8, 14, 18, and 19 predominantly containing cells from cirrhotic livers (Figures S1E and S1F).
Figure 1. Single-cell RNA sequencing identified an ILC1-like population in the human liver.

(A) Cluster analysis of scRNA-seq data from ex vivo sorted hepatic Lin−CD45+ lymphocytes following quality control (clusters are indicated). Two experimentally independent scRNA-seq analyses (left: batch 1; middle: batch 2) and an external dataset (Heinrich et al.) are shown for comparison.
(B) Analysis of ex vivo sorted hepatic Lin−CD45+ lymphocytes with assignment of cell populations (trNK, tr/cNK, ILC3, cNK, ILC3/ILC2, CD49a+trNK, LT-ILC1) based on a match to known gene signatures and illustrated in UMAP. LT-ILC1s are marked with a circle. Two experimentally independent scRNA-seq analyses (left: batch 1; middle: batch 2) and an external dataset (Heinrich et al.) are shown for comparison.
(C) Heatmap showing a selection of the top 100 cells from each indicated cell type that has the highest expression for each gene. Two experimentally independent scRNA-seq analyses (left: batch 1; middle: batch 2) and an external dataset (Heinrich et al.) are shown for comparison.
(D) Dot plots depicting relative expression of selected genes from cell types defined in (B). Two experimentally independent scRNA-seq analyses (batches 1 and 2) and an external dataset (Heinrich et al.) are shown for comparison.
Next, we assigned each cell cluster to a biological ILC or NK cell subset (Figure 1B) based on known gene signatures of specific populations (Figures 1C, 1D, and S1F; cluster annotation in Table S1), which identified cNK cells, CD56brightcNK cells (only identified in batch 2), trNK cells, a population with shared cNK/trNK-features (batch 1 only), CD49a+trNK cells (batch 1 only), ILC3s, a population with shared ILC2/ILC3 features (batch 1 only), and ILC2s (Heinrich et al. external dataset).
Our analysis also revealed a population of cells within each of the three batches in cluster 10 (batch 1), cluster 18 (batch 2), and cluster 17 (Heinrich et al.) that did not specifically correspond to any of the aforementioned NK cell or ILC populations (marked with circle in Figure 1B). These cells expressed the NK cell-associated gene KLRD1 (CD94), indicating that they were distinct from cILC1s, ILC2s, and ILC3s, each of which are CD94− by definition. On the other hand, this population showed only weak or even undetectable expression of other genes that confer the transcriptional, phenotypic, and cytotoxic properties of NK cells, including EOMES, FCGR3A (CD16), KLRF1 (NKp80), GZMB (granzyme-B), PRF1 (perforin), NKG7, and GNLY (granulysin) (Figures 1C, 1D, and S1G). Differentially expressed genes (DEGs) for these cells include CD52, CD44, IL32, CXCR4, CXCR3, CD9, DUSP4, ZNF683 (HOBIT), and NCR3 (NKp30), with negligible expression found for the latter three genes in the other subsets of ILC/NK cells (Figures 1C and S1G). Of note, the cells in this population highly expressed CXCR6 and ITGA1 (CD49a), similar to hepatic ILC1s in mice.14 Further analysis indicated that additional genes that are differentially expressed by hepatic NK cells and ILC1s in mice, including LTB, IL7R, AHR, and ID3, were also highly expressed by these cells (data shown for analysis of batch 1) but not in human cNK/trNK cell clusters (Figure S1H). Using specific gene signatures for murine NK (mNK) and liver-ILC1s,14 module scores were calculated, which confirmed an overall similarity in the gene-expression profile between cluster 10 cells (data shown for analysis of batch 1) and hepatic ILC1s in mice (Figure S1I).
Based on these gene expression data, we concluded that the cells identified in clusters 10 (batch 1), 18 (batch 2), and 17 (Heinrich et al.) represented a population with ILC1-like features that were clearly distinct from other NK cell and ILC subsets. Importantly, the LT-ILC1s collectively included in the batch 2 and Heinrich et al. analyses were derived from a broad distribution of donors (Figure S1F). Based on the identification of these cells in human liver samples, we hereafter refer to this population as “liver-type” ILC1s (LT-ILC1s).
Human liver-type ILC1s were identified as Lin−CD45+CD49a+CD94+CD200R1+NKp80− cells
Having detected a putative LT-ILC1 population in human liver samples via unbiased scRNA-seq analyses (Figure 1B), we next sought to further characterize these cells based on their expression patterns of surface proteins. Using CITE-seq technique to analyze surface protein expression parallel to transcriptome data (Figures 2A and 2B), we were able to confirm robust expression of CD49a, CD94, CD56, CD200R1, CD103, CD69, CXCR6, KLRG1, and CD52 on LT-ILC1s, whereas NKp80 expression could not be observed. To further substantiate these findings, we next evaluated total Lin−CD45+ lymphocytes from human liver samples by flow cytometry for expression of CD94 (expressed by LT-ILC1s), NKp80 (expressed by cNK and trNK cells but not by LT-ILC1s), EOMES (expressed by cNK and trNK cells but not by LT-ILC1s), CD294 (expressed by ILC2s but not by LT-ILC1s), CD161 (expressed by all ILCs, including LT-ILC1s), CD117 (expressed by ILC3s and ILC2s but only minimally by LT-ILC1s), as well as CD49a, CD200R1, CXCR6, CD127, and CD52. Among liver Lin−CD45+ lymphocytes from two liver samples, nine clusters were identified by PhenoGraph and visualized using UMAP (Figures 2C and S2A). PhenoGraph cluster 6 cells were CD49a+CD200R1+CD94+CD56brighNKp80−CXCR6+CD127+CD52+CD294−CD117−/low and, thus, displayed an ILC1-like phenotype, which was further supported by low T-BET and lack of EOMES expression (Figure 2D). PhenoGraph clusters 1, 3, and 4 were assigned as trNK cells (CXCR6+CD56highTBETlowEOMEShighNKp80+CD94+); cluster 7 was formed by a mix of cNK/trNK cells (CXCR6lowCD56dimTBETlowEOMESlowNKp80lowCD94low); clusters 2, 5, and 8 comprised cNK cells (CXCR6−CD56dimTBEThighEOMESlowNKp80+CD94+); and cluster 9 was assigned as a mix of cells with ILC2- and ILC3-associated features (CD127+CD94−NKp80−CD117+CD294+CD161+). Next we evaluated whether the cells in PhenoGraph cluster 6 corresponded to a putative LT-ILC1 population based on a surface marker expression pattern associated with ILC1s (Figure 2E). Visualized in UMAP, CD49a+CD200R1+CD94+NKp80− cells showed a substantial match with cluster 6, supported by the identification of LT-ILC1-associated features (Figures 1C, 1D, and S1F) such as robust CXCR6, CD127, CD52, and CD161 expression, but only partial expression of CD103 and CD9 (Figures 2G and 2H). Among this population, lack of CD16 (FCGR3A) expression and positive expression of NKp30 (NCR3) also fit the transcriptional profile associated with LT-ILC1s (Figure S1). Even though KLRG1 was irregularly expressed in the batches (Table S1), it was clearly upregulated at the protein level on LT-ILC1 cells. Furthermore, high CD69 expression, as well as lack of CD49e expression, confirmed the tissue-resident characteristics of LT-ILC1s (Figures 2H and S2B). Of note, elevated transcript expression levels of CXCR3, CXCR4, CD44, and ZNF683 (HOBIT) observed via scRNA-seq analysis could not be confirmed at the protein level in LT-ILC1 (Figure S2C). ILC2s, ILC3s, trNK cells, and cNK cells were clearly identified based on their accepted definitions. However, cILC1s were not able to be assigned to any of the PhenoGraph clusters. Confirming recently published data,24 high CXCR6 expression among CD49a+EOMES+NKp80+CD94+ cells supported their assignment as trNK cells (Figures S2D and S2E). For this reason, we refer to CD49a+NK cells (as identified in Figures 1 and S1) as CD49a+ tissue-resident (tr) NK cells.
Figure 2. Identification and phenotypic characterization of human liver-type ILC1s.

(A) CITE-seq analysis (protein) of hepatic Lin−CD45+ lymphocytes showing relative expression of CD49a, CD94, and NKp80 in the LT-ILC1 cluster illustrated in UMAP (only batch 2).
(B) Relative expression levels of the indicated markers among each of the indicated populations as measured by CITE-seq analysis (only batch 2).
(C) PhenoGraph cluster analysis of flow cytometry data from hepatic Lin−CD45+ lymphocytes displayed in UMAP. Clustering and UMAP is based on following markers: CD94, CD45, CD56, CD117, CD52, CD200R1, CRTH2, T-BET, NKp80, CD161, CD49a, EOMES, CXCR6, and CD127.
(D) Heatmap showing the relative expression of the indicated markers from nine PhenoGraph clusters identified in (C) as measured by flow cytometry and calculated to a Z score for each marker.
(E) Representative flow cytometry plots showing the gating strategy from singlet Lin−CD45+ lymphocytes (from healthy livers) to identify liver-type (LT)-ILC1 (pink), cNK (turquoise), CD49a+trNK cells (violet), CD49a−trNK cells (red), classical (c) ILC1 (gray), ILC2 (blue), and ILC3 (green) populations.
(F) PhenoGraph cluster analysis of flow cytometry data from hepatic Lin−CD45+ lymphocytes with assignment of the indicated populations.
(G) Representative histograms showing the relative expression of the indicated markers in each identified population to distinguish ILCs from NK cell populations.
(H) Representative histograms showing the relative expression of the indicated markers in each identified population, each of which is elevated on LT-ILC1.
See Figure S2.
None of the aforementioned markers were exclusively expressed by LT-ILC1s. This included CD49a, which was expressed by small fractions of other ILCs as well as a minor proportion of Lin−CD56+CD94+NKp80+CXCR6+trNK cells (Figure 2G). Likewise, weak CD200R1 was also found on some of the trNK cells, as well as on ILC2s and ILC3s. Therefore, human LT-ILC1s appeared to be a population identified specifically by a distinct combination of markers, not by a single ILC1-associated “lineage” antigen. The simplified definition for LT-ILC1s used throughout the remainder of this study is Lin−CD45+CD49a+CD94+CD200R1+NKp80− lymphocytes.
Liver-type ILC1s are non-cytolytic but produce IFN-γ, TNF-α, IL-2, and GM-CSF
By convention, murine ILC1s have been defined as non-cytolytic, IFN-γ-producing ILCs, although they can also produce other cytokines including TNF-α, IL-2, and GM-CSF.2,3,25,26 To determine the functional profile of human liver-type ILC1s, we performed additional phenotypic and functional analyses of human liver-derived ILCs and NK cells. Although very low amounts of PRF1 and GZMB transcripts were detected among the cluster 10 cells (LT-ILC1s) found in the scRNA-seq analysis (Figure 1F), LT-ILC1s did not express detectable amounts of perforin or granzyme-B by intracellular flow cytometry (Figure 3A). Accordingly, we did not observe degranulation (as measured by induced surface CD107a expression) of LT-ILC1s following either stimulation by phorbol 12-myristate 13-acetate (PMA) and ionomycin or co-culture with K562 cells (Figure 3B), which was in sharp contrast to cNK and both CD49a− and CD49a+ trNK cell populations. Therefore, we concluded that liver-type ILC1s represent a non-cytolytic ILC population.
Figure 3. Liver-type ILC1s were non-cytolytic but produced IFN-γ, TNF-α, IL-2, and GM-CSF.

(A) Ex vivo intracellular flow cytometry showing perforin and granzyme-B expression patterns of human liver ILC and NK cell subsets as defined in Figure 2. Data are representative of at least three independent experiments.
(B) CD107a degranulation assay of the indicated populations following 5 h stimulation by either phorbol myristate acetate (PMA) and ionomycin or K562 target cells. All experiments were performed with healthy livers (n = 5).
(C) Analysis of cytokine production by NK cell and ILC populations from livers. For each cytokine, graphs depict intracellular flow cytometry analysis of hepatic lymphocytes following 5 h stimulation by PMA and ionomycin (IFN-γ, n ≥ 11, upper left panel; IL-2, n ≥ 4, upper right panel; TNF-α, n ≥ 5, lower left panel; GM-CSF, n ≥ 3, lower right panel).
(D) Representative histograms showing intracellular cytokine staining of the indicated hepatic ILC and NK cell subsets either unstimulated (control, semi-transparent histograms) or following 5 h stimulation with PMA and ionomycin (P/I, filled histograms).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, error bars represent SEM. See Figure S3.
In order to evaluate cytokine production from liver-derived ILC subsets, we performed intracellular flow cytometry of unfractionated Lin−CD45+ ILCs; or alternatively, we measured cytokine production using a cytokine bead array (CBA) using supernatants isolated from NK/ILC subsets purified with fluorescence-activated cell sorting. These experiments confirmed IFN-γ production by LT-ILC1s at amounts similar to those of trNK and cNK cells and significantly higher than observed for cILC1s, ILC2s, and ILC3s (Figures 3C, 3D, S3A, and S3B). Production of IL-2 was higher in LT-ILC1s compared with cILC1 and NK cell subsets, with significantly higher amounts of IL-2 produced by LT-ILC1s compared with NK cells (Figures 3C, 3D, and S3C). All ILC subsets produced TNF-α and GM-CSF. We did not detect production of the ILC3-associated cytokine IL-22 by LT-ILC1s (Figures 3C, 3D, S3C-S3E, and S3I). Furthermore, we used our scRNA datasets (Figures 1 and S1) to determine whether LT-ILC1s have a specific cytokine profile. However, no significant patterns could be detected that would distinguish them from the other cell-types (Figure S3F). Although we observed elevated expression of CXCR3, CXCR4, CXCR6, NCR3, and IL7R, no LT-ILC1-specific immune or chemokine receptors were observed (Figure S3G). CellPhone analysis was performed to identify potential interactions between LT-ILC1 and other subsets (Figure S3H). In particular, DLL1 (Notch ligand), FASLG (Fas ligand), TGFB1, and TNFSF10 (TRAIL) were highlighted.
Collectively, these functional data further strengthened our conclusion that LT-ILC1s represent a population that was phenotypically and functionally distinct from other liver-derived ILCs including cNK and CD49a+/−trNK cells.
Liver-type ILC1s are increased in cirrhotic liver tissues
Chronic disease causes changes to the tissue microenvironment, thereby altering the secretion of signaling molecules and expression of surface ligands. In the liver, this is frequently associated with ongoing inflammation and altered composition of the lymphocyte compartment.27-29 In cirrhotic liver samples, frequency of LT-ILC1s among total Lin−CD45+ lymphocytes was significantly higher compared with perfusate or tissue specimens obtained from control livers (Figure 4A). In cirrhotic livers, LT-ILC1s were present at amounts similar to those of other non-NK ILC subsets (Figures S4A and S4B). No association was observed between frequency of hepatic LT-ILC1s severity of liver disease, as assessed by MELD (Model of End Stage Liver Disease) score or serum liver enzyme levels (Figure 4B). TGF-β1 has been shown to critically affect ILC1 phenotypes among various tissues in both mice and humans.9,15,30 Thus, it was an interesting finding that we detected elevated levels of TGFB1 transcript by qPCR among cirrhotic livers in a reference cohort of liver samples (Figure S4B and Table S2). More importantly, we found intrahepatic LT-ILC1 frequencies to positively correlate with TGFB1 transcript levels in cirrhotic livers (Figures 4B and S4C), whereas no such association was found for IFNG, IL13, IL2, and IL7 mRNA levels (Figure 4B).
Figure 4. Liver-type ILC1s are increased in cirrhotic liver tissues.

(A) Comparison of LT-ILC1 (Lin−CD45+CD94+NKp80−CD200R1+CD49a+) frequencies calculated as percentages among total Lin− lymphocytes between control livers (perfusate n = 7), control livers (non-perfusate; n = 8), and cirrhotic livers (n = 29) (patient characteristics shown in Table S2).
(B) Correlation matrix estimated for LT-ILC1s (percent of Lin−), age, AST, ALT, gamma-GT, total bilirubin, MELD score (Model of End Stage Liver Disease), IFNG (mRNA), IL13 (mRNA), IL1B (mRNA), IL2 (mRNA), IL7 (mRNA), and TGFB1 (mRNA). LT-ILC1 frequencies were obtained from 25 cirrhotic livers. Spearman correlation coefficients (R) are given as color code. Statistically significant correlations are indicated with *p < 0.05, **p < 0.01, ***p < 0.001.
(C) Representative expression of the indicated markers by human LT-ILC1 (pink), cNK cells (turquoise), CD49a+trNK cells (violet), CD49a−trNK cells (red), cILC1s (gray), ILC2s (blue), and ILC3s (green), comparing populations derived from healthy and cirrhotic livers. Data are representative of at least three independent experiments.
(D) Comparison of IFN-γ production (healthy, n ≥ 7; cirrhotic, n ≥ 9), IL-2 production (healthy, n = 4; cirrhotic, n ≥ 4), CD107a degranulation (healthy, n = 3; cirrhotic, n = 5), TNF-α production (healthy, n ≥ 7; cirrhotic, n ≥ 5), and GM-CSF production (healthy, n ≥ 4; cirrhotic, n ≥ 3) between healthy and cirrhotic livers related to LT-ILC1 (pink), cNK cells (turquoise), CD49a+trNK cells (violet), CD49a−trNK cells (red), and ILC3s (green), from healthy and cirrhotic livers following 5 h PMA and ionomycin stimulation and measured by intracellular flow cytometry.
(E) Volcano plot showing DEG differences based on scRNA-seq data between LT-ILC1s comparing “control versus cirrhotic liver tissues” from batch 2.
*p < 0.05, ***p < 0.001, error bars represent SEM. See Figure S4.
We also compared the phenotypic and functional properties of LT-ILC1s isolated from healthy and cirrhotic liver tissues. No significant differences were observed regarding protein expression levels of EOMES, T-BET, CD49a, CD52, CD56, CD161, CD200R1, CXCR6, and KLRG1 between liver-type ILC1s from healthy versus cirrhotic liver samples (Figure 4C). Furthermore, we did not observe any significant differences in function between LT-ILC1s from healthy or cirrhotic livers (Figure 4D). Similarly, there was no difference in expression of these markers at the transcriptional level (based on scRNA-seq analysis) when comparing LT-ILC1s derived from control or cirrhotic livers (Figure 4E; batch 2). Collectively, these data demonstrated that while LT-ILC1s were expanded in liver cirrhosis, there were no appreciable differences in phenotype or function when comparing LT-ILC1s derived from healthy versus cirrhotic liver tissues.
Liver-type ILC1 cells accumulate in fibrotic areas within human liver tissues
Previous studies have localized ILCs in close proximity to LSECs in rodent livers.31 To our knowledge, there are no reports discussing ILC1 localization within the human liver or how the tissue distribution of ILCs may change during cirrhotic remodeling. Using the multicolor microscopy technique PhenoCycler (formerly Codex),32 we were able to detect and localize LT-ILC1s in human liver tissue sections. We hypothesized that human LT-ILC1s reside in close proximity to LSECs. Common LSEC-associated markers CD36 and CD32 identify two distinct subsets of LSECs: type 1 LSECs are CD36highCD32low and type 2 LSECs are CD36lowCD32high.33 To characterize the localization of LT-ILC1s within liver tissue, LT-ILC1s were defined as CD45+CD56+CD94+EOMES−KLRG1+CD3− lymphocytes and automatically recognized by HALO image analysis software (Figures 5 and S5A).
Figure 5. LT-ILC1 cells accumulate in fibrotic area of the human liver.

Representative images of three cirrhotic (A) and two control livers (C) generated with PhenoCycler (formerly Codex) with a selection of markers such as CD56, CD94, aSMA, CD36, and CD32, as well as DAPI for nucleus staining (A, C). An overview (left) and magnification (right) are shown side by side with scale bar and indicated diameters. Fibrotic regions were recognized and delineated with a white dashed line using aSMA-staining. The red arrows indicate LT-ILC1 cells automatically detected by the HALO image analysis software by CD45+CD3−CD56+CD94+EOMES−KLRG1+ definition (see staining in Figure S5). Images in (B) and (D) were artificially generated as cell contours using HALO image analysis and correspond to the respective section from (A) and (C). Only the identified LT-ILC1 cells were highlighted for enhanced visibility (surface is green, nucleus is blue and indicated with red arrows) with visualization of the non-fibrotic (NF) and fibrotic (F) regions.
LT-ILC1s accumulated in fibrotic areas within cirrhotic liver tissues characterized by abundant aSMA expression (Figures 5A and 5B; white dashed lines indicate the borders of fibrotic areas; red arrows indicate LT-ILC1s). LT-ILC1s were detected in the immediate vicinity of non-specific CD45−CD56+ non-leukocyte cells as well as CD32+CD36− (type 2) LSECs (Figures 5A, S5A, and S5B). As anticipated from our scRNA-seq and flow cytometry analyses, only a few LT-ILC1s could be detected in control (non-cirrhotic) livers; yet, these cells were similarly located close to either CD32+CD36− or CD32−CD36+ LSECs (Figures 5C and S5C). These data collectively show that LT-ILC1s, which are enriched within areas of fibrotic liver tissue, localize in close proximity to LSECs.
Liver sinusoidal endothelial cells supported the differentiation of liver-type ILC1s
We and others previously demonstrated that all major groups of human ILCs can differentiate from Lin−CD34−CD117+ ILC precursor cells (ILCPs) that reside within secondary lymphoid tissues or circulate in the blood.17,34 We hypothesized that human ILCPs would also be capable of giving rise to LT-ILC1s. To test this hypothesis, we purified human ILCPs from fresh healthy donor peripheral blood (PB) samples and cultured them for 2 weeks in the presence of recombinant human IL-7 and the murine bone marrow-derived OP9 stromal feeder cell line transduced to constitutively overexpress the human Notch ligand Delta-like 4 (OP9-DL4 cells). Consistent with previously published data, ILCPs cultured with OP9-DL4 stroma generated ILC2s, ILC3s, and Lin−CD94+ group 1 ILCs (Figure 6A).17,34 However, nearly all of the OP9-DL4-derived Lin−CD94+ cells expressed EOMES and perforin, and many expressed NKp80 consistent with an NK cell phenotype (Figures 6B and 6C), suggesting that other conditions might better support LT-ILC1 differentiation from ILCPs.
Figure 6. Liver sinusoidal endothelial cells supported the differentiation of liver-type ILC1s.

(A) Sorted peripheral blood ILCPs (Figure S5A; Lin−CD45+CD127+CD94−NKp80−CD45RA+CD117+CD294−KLRG1−NKP44−) from four healthy donors were cultured for 2 weeks on OP9-DL4 feeder cells or LSECs with IL-7 +/− TGF-β1 as shown to the left. The dot plots to the right show the expression patterns of CD56 and CD94 among Lin− lymphocytes derived in the indicated culture conditions.
(B) Frequencies of CD200R1+CD49a+EOMES− cells among total Lin−CD45+CD94+CD56+ cells derived in vitro following culture of ILCPs (n = 8) as in (A).
(C) Representative flow cytometry analyses of EOMES, T-BET, perforin, CXCR6, NKp80, CD200R1, and CD49a expression by Lin−CD45+CD94+CD56+ cells derived from ILCPs in the indicated culture conditions. Fluorescence minus one (FMO) control with the respective lacking specific antibody is shown as black dotted lines in each histogram.
(D) Intracellular flow cytometry analysis of IFN-γ, IL-2, GM-CSF, TNF-α, and IL-22 production by in vitro derived Lin−CD45+CD94+CD56+ cells generated from ILCPs as in (A) and following stimulation for 5 h with PMA and ionomycin. Unstimulated cells were set as controls (dotted black line).
(E) Representative flow cytometry analyses showing expression of the indicated markers by total Lin−CD45+ cells obtained after 2 weeks culture of primary sorted liver-type hepatic cNK, CD49a+trNK, or CD49a−trNK cells on OP9-DL4 feeder cells or LSECs with IL-7 +/− TGF-β1 as shown to the left. Data are representative of at least three independent experiments.
(F) Representative flow cytometry analyses showing expression of the indicated markers by total Lin−CD45+ cells obtained after 2-week culture of primary sorted liver-type ILC1s (LT-ILC1s) or hepatic NK cells (sorted as Lin−CD56+CD94+NKp80+) with OP9-DL4 cells and IL-7. Data are representative of at least three independent experiments.
(G) Frequencies of CD200R1+CD49a+EOMES−cells among total Lin−CD45+CD94+ lymphocytes obtained from cultures initiated with either total hepatic NK cells or liver-type ILC1s (F).
*p < 0.05, **p < 0.01, ***p < 0.001, error bars represent SEM. See Figure S6.
Based on our finding of human LT-ILC1 being located in close proximity to LSECs (Figures 5 and S5) and the observed association between intrahepatic TGF-β1 levels and LT-ILC1 frequencies, we hypothesized that primary LSECs (Figure S6I) and TGF-β1 may promote LT-ILC1 differentiation. To test this hypothesis, we cultured purified human PB-derived ILCPs (Figure S6A) in vitro with either primary human LSECs or OP9-DL4 cells together with recombinant human IL-7 in the presence or absence of recombinant human TGF-β1. Culturing ILCPs in the presence of LSECs induced the emergence of a prominent population of Lin−CD94+LT-ILC1-like cells that expressed CD49a, CD200R1, and T-BET but showed weak or undetectable expression of perforin, EOMES, and NKp80 (Figures 6B and 6C). This was particularly evident in cells cultured in the presence of TGF-β1. In contrast, only a small proportion of cells with a CD94+CD56+CD200R1+CD49a+EOMES− LT-ILC1-like phenotype were found in the OD9-DL4 co-cultures. Numbers of LT-ILC1-like cells increased in the presence of TGF-β1 but remained significantly lower than in the LSEC/TGF-β1 cultures. In addition, CD94+CD56+ cells arising in the presence of LSEC plus TGF-β1 were found to have robust CXCR6 expression, which was not observed in cells from the OP9-DL4 plus TGF-β1 cultures (Figure 6C). This altogether indicated that LSECs may promote the emergence of cells with an LT-ILC1-associated phenotype (Figure S6B). This hypothesis was further supported by functional analysis, where CD94+CD56+ cells from the OP9-DL4+TGF-β1 cultures were found to produce negligible cytokines, whereas CD94+CD56+ cells from the LSEC plus TGF-β1 cultures produced abundant cytokines (Figure 6D).
Given the observed effects of exogenous TGF-β1 on LT-ILC1 generation, we measured protein expression of TGF-β1 in the culture supernatants. In contrast to OP9-DL4, we detected in LSEC supernatant free active TGF-β1 (Figure S6C). This may suggest that the production of TGF-β1 by LSECs may be partly responsible for the observed effects of LSECs on LT-ILC1 differentiation in the absence of exogenous recombinant TGF-β1 (Figures 6A-6D). The amount of TGF-β1 produced by LSECs might be sufficient to cause an antiproliferative effect, because the number of Lin−CD45+ lymphocytes generated in the presence of LSECs was significantly lower compared with those generated in the presence of OP9-DL4 cells (Figure S6D). However, TGF-β1 blocking antibody had no significant effect on LT-ILC1 formation (Figure S6E). Moreover, no elevated LT-ILC1 formation was observed in the presence of human umbilical vein endothelial cells (HUVECs), another primary endothelial cell, suggesting LSEC-specific factors likely play a role in the generation of LT-ILC1s.
Given the previously published examples of human and murine NK cells differentiating into ILC1-like cells,13,15,30,35 we next tested whether human liver cNK, CD49a+trNK, CD49a− trNK, or PB cNK cells could differentiate into a population of cells with LT-ILC1-associated features. However, in none of the tested conditions did we observe any of the tested NK cell populations to acquire the phenotypic or functional characteristics of an LT-ILC1, suggesting that LT-ILC1s do not represent a transient NK cell activation state but rather a distinct ILC1 subset (Figures 6E and S6F).
Next, we sought to determine whether primary human LT-ILC1s could differentiate into other ILC populations or acquire cytotoxic properties. Purified LT-ILC1s were cultured for 2 weeks with IL-7 and OP9-DL4 cells, a condition that supports the generation of ILC2s, ILC3s, and EOMES+ NK cells from ILCPs (shown above in Figure 6A), or with LSECs in the presence of IL-7 and TGF-β1. Following co-culture in these conditions, LT-ILC1s stably maintained their non-cytotoxic phenotype and did not differentiate into other ILC subsets (Figures 6F, 6G, and S6G).
In summary, these in vitro cell culture data demonstrate that human PB-derived ILCPs differentiated into LT-ILC1s in the presence of LSECs and TGF-β1. In contrast, PB cNK or liver cNK/trNK cells did not differentiate into LT-ILC1-like cells under the same in vitro conditions. Moreover, LT-ILC1s appeared to represent a population of cells with a stable ILC1 phenotype distinct from other NK cell populations.
Populations of cells with LT-ILC1-associated features were identified in other tissues
To clarify whether cells with LT-ILC1-like phenotypic and functional properties may exist outside the liver, we examined Lin−CD45+ lymphocytes isolated from tonsils, duodenum, and colon by scRNA-seq (Figure 7A; batches 1 and 2). Here, in addition to the liver, a prominent cell population (cluster 8) with a signature compatible with LT-ILC1 was identified in all tonsil and duodenum samples, whereas only a few of such cells could be detected by scRNA-seq in the analyzed colon samples (Figures S7B, S7C, and S7E and Table S1). The highest proportion of these LT-ILC1-like cells was found in the duodenum (Figures 7A-7C). As could be expected, a number of DEGs were found among the cells of cluster 8 between the different tissues. However, expression of the signature genes characteristic of LT-ILC1 (Figures 1 and S1) remained very similar to the previous analysis of liver cells (Figures S7C and S7F).
Figure 7. Population of cells with liver-type ILC1-associated features are identified in other tissues.

(A) scRNA-seq analysis of Lin−CD45+ lymphocytes from control livers, cirrhotic livers, tonsils, duodenum, and colon from batches 1 and 2 with assignment of cell populations trNK, cNK, ILC3/ILC2, and LT-ILC1-like cells based on a match to known gene signatures and illustrated in UMAP with 16,187 cells (assignment in Figures S7A-S7C; G). LT-ILC1-like cells are marked with a black circle.
(B) scRNA-seq analysis showing the relative proportion of cNK, ILC2/ILC3, LT-ILC1-like, and trNK populations among control liver, cirrhotic liver, tonsil, duodenum, and colon tissues (n = 3 per compartment).
(C) Dot plot showing CD45+Lin− lymphocytes with the relative distribution of the indicated populations identified in Figures 1B and S7I among peripheral blood, tonsil, liver, duodenum, and colon tissues of each three subjects displayed in UMAP (based on markers indicated in Table S3). The left black dashed circle includes LT-ILC1-like cells, and the right dashed circle includes intraepithelial ILC1 cells.
(D) Representative histograms showing the relative expression of the indicated markers among the populations identified in Figures 1B and S7I in peripheral blood, tonsil, colon, and duodenum tissues in addition to control liver.
(E) Representative histogram of intracellular flow cytometry analysis of IFN-γ, TNF-α, and IL-2 produced by LT-ILC1-like cells (same gating as for Figures 1C and 1D) following ex vivo stimulation for 5 h with PMA and ionomycin. Unstimulated cells were set as controls.
(F) Relative frequency of LT-ILC1-like cells calculated as the percentage of total CD45+ lymphocytes in the duodenum (n = 6), liver (n = 29), colon (n = 7), tonsil (n = 6), and peripheral blood (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, error bars represent SEM. See Figure S7.
These results were also confirmed via CITE-seq analysis that the tonsil- and duodenum-derived cells in cluster 8 displayed a CD49a+CD200R1+CD94+NKp80− phenotype (Figure S7G). To corroborate these findings further, multiparametric flow cytometry analyses were performed, which confirmed the similarity of these cells identified in the different tissues (Figure 7C). In particular, the expression of signature markers such as CD49a, CD200R1, CD52, and CD127, as well as an EOMES−T-BET+ transcription factor profile, was confirmed here in all tissues examined and in peripheral blood. In agreement with the gene-expression analyses, certain tissue-specific differences between the respective LT-ILC1-like populations were also found at the protein level. In particular, CXCR6 was expressed exclusively on hepatic cells, whereas CD103 was detectable only on cells from duodenum or liver. Functional assays confirmed the production of ILC1-associated cytokines, including IFN-γ, TNF, and IL-2, by LT-ILC1-like cells in all tissues examined (Figure 7E). Flow cytometry analysis confirmed the highest percentage of these cells in the duodenum, followed by the liver, colon, and tonsil; they were only minimally detected in circulating blood relative to total lymphocytes (Figure 7F).
These results showed that cells with LT-ILC1-like features could be identified in tissues outside the liver, including the tonsil. Given the previously published definition of intraepithelial ILC1s (ieILC1s) found in the tonsils,3 we next checked for a potential overlap with LT-ILC1-like cells. In our integrated scRNA-seq dataset, no distinct cluster with ieILC1-associated features could be identified, as cells with a significant CD103 expression (CITE-seq) were only detected in the trNK cell cluster without forming an independent cluster (Figure S7G). Thus, we next analyzed tonsil ieILC1s for their expression of EOMES, CD200R1, and NKp80 by flow cytometry. Among Lin−CD56+NKp44+ cells in the tonsil, the population of CD103+IFN-γ+ cells (i.e., functional ieILC1s) expressed EOMES and NKp80, but it was mostly negative for CD200R1. Therefore, we concluded that the population of tonsillar ieILC1s is distinct from LT-ILC1-like cells, which are EOMES−NKp80−CD200R1+ (Figure S7H). This could be further confirmed in multiparametric flow cytometric analysis by distinguishing ieILC1s (ex vivo in Figure S7I) from LT-ILC1s in UMAP analysis (Figure 7C).
Taken together, although LT-ILC1s appear to be a transcriptionally, phenotypically, and functionally distinct ILC subset in the liver, we also identified cells with similar properties in other tissues such as the tonsil and duodenum, suggesting that these “cross-tissue ILC1s” are not liver-restricted.
DISCUSSION
The field of ILC biology is rapidly progressing and evolving, and the classification of human ILC1s is not yet complete. Previous groups have identified human tissue-resident ILC populations with an ILC1-associated phenotype,2,3,28,36 but the identification of a global human ILC1 phenotype that spans all tissues remains elusive. Indeed, there is accumulating evidence for the existence of multiple ILC1 phenotypes in different tissues.2,3,7,37 However, to the best of our knowledge, human hepatic ILC1s have not yet been specifically characterized. Here we identified a population of ILC1s in the human liver, which we refer to as “liver-type”-ILC1s (LT-ILC1s) given their original characterization in the liver and their similarity to murine liver ILC1s. However, we could also find a population with an identical signature in other tissues, such as tonsils and duodenum. This cross-tissue population showed conventional ILC1 properties including IFN-γ production, T-BET expression, and lack of cytolytic degranulation. However, based on extensive ex vivo phenotypic and functional characterization, these cells were distinct from cILC1s and tonsil-derived ieILC1s as well as from cNK cells, CD49a+/− trNK cells, ILC2s, and ILC3s. Thus, we propose that this characterized cross-tissue population represents a distinct ILC1 subset in humans.
Using a combination of CITE-seq and scRNA-seq data, LT-ILC1s were identified based on distinct gene-expression patterns that clearly differed from that observed in other ILC populations. Comparing the relative expression levels of the respective proteins using flow cytometry, LT-ILC1s were also distinguished from other previously established human ILC subsets by PhenoGraph clustering. In contrast to cNK and CD49a+/−trNK cells, LT-ILC1s expressed neither EOMES nor NKp80, and they lacked perforin and granzyme-B expression. However, they did produce large amounts of IL-2. Regarding their differences compared with previously described CD127+ helper ILC populations, LT-ILC1s showed a higher capacity for IFN-γ production compared with cILC1s. LT-ILC1s also expressed CD94, which is not expressed by cILC1s, ILC2s, or ILC3s. Moreover, LT-ILC1s expressed high amounts of surface CD200R1 and were therefore distinct from ieILC1s, which we observed were mostly CD200R1lowEOMES+NKp80+.3,37 Based on these observations, we utilized the Lin−CD45+CD49a+CD94+CD200R1+NKp80− immunophenotype for prospective purification and analysis of LT-ILC1s. Importantly, this surface antigen profile was derived from an unbiased approach using single-cell-multiomics data, which also revealed numerous parallels with murine hepatic ILC1s. Given the many similarities between murine hepatic ILC1s and the human LT-ILC1s we describe here, we propose that LT-ILC1s most closely represent the human counterpart to murine hepatic ILC1s.
In light of the evidence that LT-ILC1s represent a distinct ILC subset in humans, we note that the overall frequencies of LT-ILC1s were very low in the liver, though their frequencies were significantly increased in the presence of liver cirrhosis and were overall similar to proportions seen for other non-NK ILCs. This raises the question as to what extent LT-ILC1s can significantly influence inflammatory processes in the liver. While further investigation is clearly needed, we do anticipate that LT-ILC1s likely play various roles in healthy and cirrhotic liver, given the published evidence that other low-frequency ILC populations are involved in immune responses in liver diseases.38-40 In addition, it is now well established that in mice, hepatic ILC1s are involved in the initial immune response to viral infection via rapid secretion of IFN-γ.14 Human LT-ILC1s had a high capacity for IFN-γ production and thus may have a similar role in the early immune response. Recently, murine ILC3-derived IL-2 was described as a crucial factor in the homeostasis of regulatory T cells in the intestine.41 Since we observed that human liver-type ILC1s produced IL-2 ex vivo, it is possible that they maintain a regulatory role in the liver as well. A previously unknown function of ILC1s was recently demonstrated, in which ILC1s patrol the basal layers of the oral epithelium under steady-state conditions, thereby priming uninfected tissue to limit viral replication.42 Even though the frequency of LT-ILC1s is very low in healthy liver, these cells may have a similar function.
In addition to the liver, we also found lymphocytes with LT-ILC1-like features in tonsils and especially in the duodenum, whereas frequencies of these cells in colon tissue were rather low. This may also explain why previous scRNA-seq analysis of intestinal tissue, primarily examining colon tissue, did not identify this cross-tissue ILC1 population.43
Previous reports demonstrated that hepatic CD49a+ NK cells are enriched in cases of obesity or liver cell carcinoma and that this likely occurs at least in part via signaling through TGF-β1.30,44,45 TGF-β1 imprinting in the murine system, specifically in the tumor microenvironment, was also shown to support the differentiation of ILC1-like cells.15 We observed that LT-ILC1s were selectively enriched in fibrotic areas within cirrhotic liver tissues. In particular, we observed that LT-ILC1s resided in close proximity to LSECs. Given that TGF-β1 is a primary mediator of extracellular matrix production by fibroblasts,46 we hypothesized that TGF-β1 signaling may be an important factor in the differentiation of human LT-ILC1s. However, our results indicate that TGF-β1 alone may not be sufficient to generate functional LT-ILC1s, as our in vitro cultures with PB-derived ILCPs in the presence of TGF-β1 and OP9-DL4 stroma only generated cells with negligible ability to produce LT-ILC1-associated cytokines. In contrast, ILCPs co-cultured with human LSECs gave rise to cells capable of producing IFN-γ, IL-2, TNF-α, and GM-CSF, consistent with an LT-ILC1-associated functional phenotype. This effect was further augmented in the presence of TGF-β1. Indeed, while TGF-β1 in most cases inhibits NK cell functions, the influence on LT-ILC1 is opposite and could possibly be related to the small differences between healthy and cirrhotic LT-ILC1 in function and phenotype. Although we observed LSECs to produce TGF-β1 in vitro, confirming previous reports by others,47 we found that blocking TGF-β1 did not significantly affect LT-ILC1 formation in LSEC co-cultures. These data indicated that while TGF-β1 may be at least partially involved, synergism with other liver-specific factors may play an important role in the mechanism of LT-ILC1 differentiation and homeostasis. Recently, it has been shown that human ILCPs can interact with endothelial cells in a feedback loop to promote intercellular adhesion, speculating that these interactions may also have an impact on ILCP differentiation.48 Thus, we co-cultured ILCPs with HUVECs as an endothelial cell substitute for LSECs, yet we did not observe a rise in cells with LT-ILC1 phenotype. However, the possibility of such a mechanism still remains, and it could play a role in the interaction with LSECs. Further evidence that LT-ILC1s originate from ILCPs and not from mature NK cells is supported by the inability to completely reduce EOMES expression in human NK cells via a TGF-β1-mediated mechanism.49 Moreover, we were not able to generate functional LT-ILC1-like cells from co-culture of mature liver-derived NK cells with LSECs and TGF-β1. This is in contrast to a similar mechanism described in mice for TGF-β1-mediated conversion of NK cells to ILC1s, suggesting that if such a pathway exists for human NK cells and ILC1s, additional factors may be involved.9,15
In summary, we have identified a population of human cross-tissue ILC1s defined as Lin−CD45+CD49a+CD94+CD200R1+NKp80−. This population displayed the hallmark features of ILC1s in that they expressed T-BET and were capable of producing ILC1-associated cytokines (IFN-γ, IL-2, TNF-α, and GM-CSF). Yet they were nonetheless distinct from previously described human cILC1s and ieILC1 subsets and also differed from CD49a+/−trNK and cNK cells due to their lack of EOMES, NKp80, and cytolytic granule expression. Although quite rare in healthy livers, LT-ILC1s were expanded in the tissues of patients with liver cirrhosis. We also demonstrated that LT-ILC1-like cells could be derived in vitro from PB-derived ILCPs, although not from cNK or trNK cells, in the presence of LSECs + TGF-β1. Our results provide further insight into the definition and properties of this ILC1 subset in humans as well as establish a basis for further research into their role in human diseases.
Limitations of the study
Due to low cell numbers, we only determined the cytotoxic activity of LT-ILC1 via CD107a degranulation assay but not the direct lysis of target cells, which may be mediated via FASL, TRAIL, or TNF. For the scRNA-seq analysis, we could only analyze cells from healthy liver perfusates as controls, and we did not have tissue from healthy livers available that we could enzymatically digest in the same way as the cirrhotic livers. Instead, we have used normal liver tissue samples as controls that were maximally distant from a liver tumor. Proteomics via CITE-seq analysis with the critical markers such as CD200R1, CD49a, NKp80, CD94, and CD56 was only available to us for “batch 2” and not for the other two datasets "batch 1" and "Heinrich et al.".
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin Krämer(Benjamin.kraemer@ukbonn.de).
Materials availability
Materials generated from this study are available on request from the lead contact.
Data and code availability
The scRNASeq data analyzed here are accessible with corresponding documentation on EGA(European Genome-Phenome Archive) with the accession GEO: EGAS00001006847. The Heinrich et al. dataset23 is available from GEO with the accession GEO: GSE179795. Other primary data, as raw FCS files, raw PCR data, CBA data, or microscopy images, are available from the authors upon reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics statement
All subjects gave written informed consent in accordance with the Declaration of Helsinki. Experiments performed in Bonn were approved by the local ethics committee of the Medical Faculty, University of Bonn(#275/13, #040/16, #035/14, and #417/17). Experiments performed in Berlin were approved by the ethics committee of the Charité-Universitätsmedizin Berlin(No. EA1/353/16) and carried out in compliance with its guidelines. Experiments performed in the Freud Lab were performed in accordance with protocols approved by The Ohio State University Institutional Review Board(OSU IRB). De-identified human pediatric tonsil specimens were obtained fresh via the NCI-approved Cooperative Human Tissue Network(CHTN) from Nationwide Children’s Hospital(Columbus, OH USA).
Patients
A total of 40 cirrhotic(mean age: 53.8, 60% male) as well as 19 healthy livers(age and gender only for 11 donors available; see Table S2), 8 non-cirrhotic(mean age: 71.8, 75% male) control livers from non-tumor tissue, 8 peripheral blood samples(mean age: 40.8, 60% male) collected from healthy donors, 7 colon(mean age: 30.2, 50% male) and 6(mean age: 46.2, 66% male) duodenum biopsies obtained from patients that underwent cancer screening, and 8 tonsils obtained from patients undergoing elective tonsillectomy were enrolled into this study. In a reference cohort, we used for scRNA-Seq-analysis data from hepatic NK cells/ILCs studied by Heinrich et al., in which 6 hepatocellular carcinoma patients with non-tumor and tumor margin tissue were enrolled.23 For measurement of transcriptional TGFB1 expression a reference cohort with 20 cirrhotic and 12 healthy livers was examined(age and gender not available). The patient characteristics are shown in Table S2.
METHOD DETAILS
Human lymphocyte isolation
Isolation of lymphocytes from liver perfusate was performed as follows: shortly before implantation of the donor liver, the UW(University of Wisconsin)-solution was rinsed out and collected in a plastic bag(Southard and Belzer, 1993). This solution was filtered through a 70 μm mesh and centrifuged at 500g for 10 min. Single lymphoid cells were isolated using Ficoll-Paque gradient centrifugation, washed with DPBS and directly cryopreserved until further usage.
Explant livers, liver resections, and donor livers were washed in DPBS(Biochrome, Germany)-Heparine(Sigma, USA) and then tissue pieces with an edge length of 2 cm were cut and placed in a sterile plastic bag to be washed twice with cold heparin(+) DPBS. The pieces were again freed from all liquid and the bag containing a maximum of 20 pieces was filled with 10 mL of digestion medium [RPMI 1640 medium(Gibco Life, USA), 2% FCS(Sigma, USA), 1% penicillin/streptomycin, 1 mg/mL Collagenase IV(Worthington, USA), 25 U/ml Endonuclease(MoBiTec, Germany), and 0.5 mg/mL Soybean-Trypsin-Inhibitor(Thermo-Fisher, Germany)]. The bag was clamped in a laboratory homogenizer(Stomacher, UK) and processed for 10 min at 230 rpm. The bag was filled with 100 mL cold 1%-HSA(PAN-Biotech, Germany)-DPBS, and the liquid was filtered through a 100 μm gauze sieve and stored at 4°C. The liver pieces were put back into the bag and the whole process was repeated at least 4 times. The liquid was always collected and kept at 4°C. The remaining liver pieces were again cut with a scalpel, then intensively crushed with a spatula on a metal sieve and washed with DPBS. The liquid was merged with that collected so far and centrifuged, resuspended in 40.5% Percoll-solution(General Electric, USA), filtered through a 40 –m sieve, layered on a 58.5% Percoll-solution, and centrifuged again. The interphase was collected, washed and the cells were directly cryopreserved.
For isolation of tonsil-derived lymphocytes, tissue specimens were cut into small pieces and squeezed through a stainless steel mesh. Single lymphoid cells were isolated using Ficoll(Biochrome, Germany)-Paque gradient centrifugation(Biochrom AG, Berlin, Germany), washed with DPBS and directly cryopreserved. For some experiments, total NK cells and ILCs were enriched fresh from tonsil single cell suspensions according to previously published methods.50
Isolation of lymphocytes from endoscopic duodenum and colon biopsies was performed based on recently published protocols.28 In brief, biopsy samples were cut into small pieces and incubated in Ca2+/Mg++-free Hanks Balanced Salt Solution(GIBCO, Germany) containing 154 μg/mL DTT(Dithiothreitol; Sigma Aldrich, Germany), 5 mM EDTA(AppliChem, Germany), 0.25% NAC(N-acetylcysteine; Sigma, USA) and 1% penicillin/streptomycin(PAN Biotec, Germany) for 45 min at 37°C to dissolve epithelial tight junctions and eliminate mucus. Following centrifugation(50g for 3 min) supernatant was collected and tissue specimens were digested for 45 min at 37°C in RPMI 1640 medium supplemented with 10% FCS and 1% penicillin/streptomycin(complete RPMI medium) containing 1 mg/mL Collagenase II(Worthington, USA) and 25 U/ml Endonuclease(MoBiTec, Germany). Cell suspensions were filtered through a 70μm Nylon cell-strainer(BD, Germany) and washed with PBS.
Peripheral blood mononuclear cells(PBMC) were isolated using Ficoll-Paque gradient centrifugation(Biochrom AG, Berlin, Germany), washed with DPBS, and directly cryopreserved.
FACS-analysis and cell sorting
Phenotypic-analysis of cells was performed using either an LSR-II Flow Cytometer, an LSR-Fortessa Cytometer, or a BD Aria Fusion(BD Biosciences, USA). In brief, all antibodies were titrated, the panels were tested using FMO controls, and constant conditions were ensured by plate staining to guarantee an optimal staining result. The antibodies used in these studies are listed in Table S3. The following antibodies were selected for the Lineage cocktail: BDCA-2(dendritic cell), CD1a(dendritic cell), CD3(T cell), CD4(T cell), CD5(T cell), CD14(monocyte), CD19(B cell), CD20(B cell), CD34(progenitor cell), CD123(dendritic cell), FcεR1(mast cell), TCRαβ(T cell), and TCRγδ(T cell). Of note, due to the current uncertainty in the field as to whether CD4 and CD5 T cell-associated antigens can also be expressed by a subset of human ILCs,37,51,52 we purified human liver-derived Lin−CD4+CD5+CD117−CD127+CD294−CD94−NKp80−lymphocytes and then cultured these in vitro with OP9-DL4 stroma and IL-7 plus FL (Figure S5F). After 2 weeks, only T-lineage cells were formed; thus for our study we concluded that such cells were likely T lineage cells, and so we included CD4 and CD5 in the lineage antigen cocktail.
For intracellular analyses of transcription factors the Foxp3 Transcription Factor Staining Kit(eBioscience, Germany) was used for permeabilization, fixation, and washing. For sorting by flow cytometry an ARIA Fusion(BD Biosciences, USA) was used. Data were analyzed with FlowJo software V10.6.1(BD Biosciences, USA).
CD107a degranulation assay
Liver lymphocytes were coincubated at a ratio of 1:40 with major histocompatibility complex–deficient K562 cells or stimulated for 5 h with phorbol-12-myristat-13-acetat(PMA, 50 ng/mL; Cell Signaling Technology Europe, Netherlands) and Ionomycin(1000 ng/ml; Cell Signaling Technology Europe) in complete RPMI 1640 medium in the presence of CD107a specific antibody. After 1 h stimulation addition of Golgi-Stop(BD Biosciences, USA) was added for the remainder of the incubation. After staining with the viability dye Zombie Aqua(Biolegend, USA) and surface antibodies, the samples were analyzed by flow cytometry.
Intracellular cytokine staining
Lymphocytes were stimulated for 5 h with phorbol 12-myristate 13-acetate(PMA, 50 ng/mL; Cell Signaling Technology Europe, Netherlands) and Ionomycin(1000 ng/ml; Cell Signaling Technology Europe) or IL-12(10 ng/ml)/IL-15(50 ng/ml) or K562/IL-12/IL-15 combined in complete RPMI 1640 medium in the presence of brefeldin A(BFA, 5 μg/mL; Enzo, Germany) for the final 4 h. Cells were washed, stained, and permeabilized using the Cytofix/Cytoperm Kit(BD Biosciences, USA). IFN-γ, IL-2, TNF-α, GM-CSF, and IL-22 were detected with specific antibodies by intracellular staining. Data were acquired with an LSR-Fortessa Cytometer(BD Biosciences, USA) and analyzed with FlowJo software V10.6.1(BD Bioscience, USA). A complete list of the antibodies used in this study is given in Table S3.
Cytokine bead array
For the detection of IFN-γ, IL-2, TNF-α, and GM-CSF in in vitro-derived supernatants, cell-free liquid volumes were collected following stimulation of 1000 FACS-purified cells with PMA/Ionomycin(see above), and a LegendPlex TH cytokine kit(Biolegend, USA) was applied according to the manufacturer’s instructions. For detection of free active TGF-β1, supernatant from 5 days culture of LSEC, OP9-DL4 or medium control was performed using LEGENDplex HU Essential Immune Response Panel. The beads were measured with a BD Canto II flow cytometer(BD Biosciences, USA) and the data were analyzed with Legendplex 8.0 software(Biolegend, USA).
Single cell RNA sequencing
Lin−CD45+lymphocytes from 2 explant livers, 2 liver perfusates, and 2 tonsils were sorted using an Aria Fusion(BD Biosciences, USA). Cell concentrations were adjusted to 12,000 cells per sample and the 2 samples of each tissue were each combined into one sample. Droplet based single cell RNA sequencing datasets were generated(Chromium platform, 10x Genomics). The standard protocol for the 10x single cell kit(V3 chemistry) was used for all steps including cell loading, library preparation, and quality control. The concentrations of the respective cDNA were adjusted to the required conditions of the S2 flow cell and sequenced using Illumina NextSeq.
Single cell data analysis
After sequencing, fastq files were generated using Cellranger mkfastq(version 3.0.2). The raw reads were mapped to the human reference genome(refdata-cellranger-GRCh38–3.0.0) using cellranger count. Digital expression matrix was extracted from the filtered_feature_bc_matrix folder outputted by the cell ranger count pipeline. To identify different cell-types and find signature genes for each cell-type, the R package Seurat(version 3.1.1) was used to analyze the digital expression matrix. Cells with less than 500 unique molecular identifiers(UMIs), less than 100 genes, or greater than 50% mitochondrial expression were removed. The Seurat function NormalizeData was used to normalize the raw counts. Variable genes were identified using the FindVariableFeatures function. The ScaleData function was used to scale and center expression values in the dataset, the number of unique molecular identifiers(UMI) was regressed against each gene. Principal component analysis(PCA) and uniform manifold approximation and projection(UMAP) were used to reduce the dimensions of the data, and the first 2 dimensions were used in the plots. The FindClusters function was used to cluster the cells. Marker genes were found using the FindAllMarkers function for each cluster. Cell-types were annotated based on the marker genes and their match to canonical markers. For the analysis with samples from different batches, Seurat integration was used. The FindIntegrationAnchors and IntegrateData were used to create an integrated Seurat object, then the same normalizing, scaling and clustering methods were applied to the object. To match the flow cytometry data and focus the analysis on ILCs and NK cells, we removed the B cell, T cell, myeloid and low-quality clusters, and re-clustered on the rest of the cells. The barcode, origin, cluster and cell-type information are provided in Table S1. Differential expression analysis between any two groups of cells were carried out using the FindMarkers function.
Ligand receptor interaction analysis
CellphoneDB(v2.0.0) was applied for ligand-receptor analysis. Each cell-type was separated by their disease classifications, and a separate run was performed for each disease classification. If a subtype contains fewer than 10 cells for a disease classification, it was not considered in the ligand-receptor analysis for this disease classification. Pairs with p value >0.05 were filtered out from further analysis. To compare between the two disease conditions, each pair was assigned to the condition in which it showed the higher interaction score. The circos plots were generated using the R package circlize.
Murine cell bulk RNA sequencing
The murine cell bulk RNA-Seq data of hepatic ILC1 and mature NK cells are published and available in a public database.14 The module scores (Figure S1I) were calculated using the Seurat function AddModuleScore with default parameters, which measure the average expression levels of a set of genes, subtracted by the average expression of randomly selected control genes(signature genes are listed in Table S1).
In vitro cell cultures
OP9-DL4 cells were obtained from Andreas Diefenbach(Charite, Berlin) and maintained in DMEM(GIBCO LIFE, USA, Germany) supplemented with 1% penicillin/streptomycin and 10% FCS. Primary human liver sinusoidal epithelial cells(LSECs), primary hepatic stellate cells(HSC), and primary hepatocytes were purchased53,54(Sciencell, USA) and expanded with endothelial cell medium, stellate cell medium or hepatocyte medium(Sciencell, USA), respectively, to passage 3 as instructed by the supplier. Primary Human umbilical vein endothelial cells(HUVECs) were purchased from PromoCell(USA) and cultured with endothelial cell medium(Sciencell, USA). LSEC specific markers from the literature55 were tested by qPCR to verify the phenotype of primary LSECs used in long-term culture and compared with HSCs as well as hepatocytes (Figure S6I; see qPCR below).
Before OP9-DL4 cells, LSECs or HUVECs became 90% confluent, the cells were detached and irradiated in suspension at 20 Gy(Faxitron MultiRad MR 160-225, USA). Then 8000 OP9-DL4 cells or LSECs were plated in a 96 well flat bottom plate, left in culture in their specific media for 3 days, and washed twice before co-culture experiments were started.
Differentiation assays used media containing DMEM(Gibco Life, USA) and F12(2:1), 1% antibiotic and antimycotic(Gibco Life, USA), 20 mg/mL ascorbic acid(Sigma, USA), 24 mM 2-mercaptoethanol(Gibco Life, USA), 0.05 mg/mL sodium selenite(Sigma, USA), and 10% heat-inactivated human AB serum(Sigma, USA) based on previous protocols(Cichocki and Miller, 2010).
For the isolation of peripheral blood ILCPs(ILC precursor cells), PBMCs from healthy subjects were first depleted of Lin+ cells and NK cells using negative magnet-based separation(biotin-CD3, -CD14, -CD19, -CD20, -CD94, -NKp80; Miltenyi Biotec) and then sorted according to a known definition(Lin−CD94−CD34−NKp80−CD127+CD117+CD294−CD45RA+NKp44−IL1R1+)34 using a BD Aria Fusion(BD, Biosciences, USA). ILC/NK cell populations from the liver or PBMC were sorted directly without prior enrichment steps with the definitions described in Figure 2E.
For in vitro co-cultures, 1000 cells per well were added to wells containing the adherent LSECs, HUVECs or OP9-DL4 cells. Media was supplemented with human IL-7(10 ng/mL; Miltenyi Biotec) with or without TGF-β1(10 ng/ml; Miltenyi Biotec) and with or without TGF-β1 blocking antibody(1 μg/ml, Biolegend). For testing the lineage differentiation potentials of liver-derived Lin−CD4+CD5+CD117−CD127+CD294− cells, the latter were sorted and cultured on OP9-DL4 cells with the addition of FL(FLT3LG, 10 ng/mL; Immunotools, Germany) and IL-7(10 ng/mL; Miltenyi Biotec) for 2 weeks (Figure S6H). Fresh media and cytokines were replenished every 3 days. Feeder cells and LSECs were monitored under the microscope throughout the experiments and samples were discarded where no intact monolayer existed. Following in vitro culture, lymphocytes were harvested and analyzed by flow cytometry. For functional studies, half of the cells were stimulated with PMA/Ionomycin(see above) for 5 h following by analysis via intracellular flow cytometry.
PCR-analysis
Tissue specimens or primary cells were placed in NAP-buffer56 to stabilize mRNA, subsequently frozen at −80°C, and stored until analysis. Before mRNA isolation, the tissue was thawed, excess buffer was removed, and the tissue was homogenized by bead-beating and lysed(Fast Prep 24, M.P. Biomedicals, USA). From the lysate, messenger RNA(mRNA) was extracted using the GeneJet RNA purification kit(Thermo Scientific, Germany) and cDNA was transcribed using the QuantiTect reverse transcription kit(Qiagen, Germany) in accordance with the manufacturer’s protocol. qPCR was performed on a LightCycler(Roche, Germany) with the SYBR Green Master Mix(Biorad, Germany) using the primer sets depicted in Table S4. Relative expression of the respective target genes is given in relation to housekeeper gene(Geomean of Cq from RPL19 and EEF1A1) expression as 2−ΔCq. Since the mRNA samples of the healthy liver were not available for comparison between healthy and cirrhotic liver, a reference cohort was examined, the clinical characteristics of which are shown in Table S2.
Flow cytometry clustering analysis
For the clustering of flow cytometric data in Figures 2C and 2F, gated CD45+Lin−lymphocytes in 2 livers were initially downsampled to 9800 cells in Flowjo(10.7.1; plugins: www.flowjo.com/exchange/#/) using the DownSample plugin(v.3.3). For Figure 7C, gated CD45+Lin−lymphocytes from circulating blood, tonsils, liver, duodenum, and liver of each 3 subjects were used. Subsequently, the samples to be compared were concatenated and for Figures 2C and 2F a cluster-analysis was performed using the FlowJo plugin Phenograph(v. 3.0) and clusters were visualized using the UMAP(v. 3.1) or t-SNE plugin. For UMAP visualization in Figures 2C and 2F the following markers were included: CD161, CD49a, CD127, CD117, CD52, CD200R1, CD294, CD45, TBET, NKp80, CD56, CD94, EOMES, and CXCR6. BATCH effects were checked by displaying individual specimens in UMAP(for Figures 2C and 2F in Figure S2A in the same panel). The heatmap with the protein expressions of the respective clusters was generated in the software Biovinci(v.3.0). The expressions are given as Z score [z = (x - μ)/σ)] and were calculated from the gMFI (geometric mean fluorescence intensity) optained from FlowJo (v. 10.7.1). For t-SNE visualization in Figure S6B we included these markers: CD49a, CD52, CD117, CD200R1, CD294, CD45, TBET, NKp80, CD56, CD94, EOMES, KLRG1, and CXCR6. For UMAP visualization in Figure 7C we included these markers: CD103, CD49a, CD127, CD9, CD52, CD117, CD200R1, CD294, CD45, TBET, NKp80, CD56, CD94, EOMES, KLRG1, and CXCR6.
Immunohistostaining of fresh-frozen human liver for PhenoCycler(formerly CODEX)
Antibody conjugation, tissue preparation and staining were performed according to the Nolan Lab protocol.32 Notably, blocking buffer contained 0.05 mM naked oligonucleotides(Biomers) and dissolving of maleimide-modified oligonucleotides was performed in 1x DPBS in the presence of the vapor phase of LN2.
Purified, carrier free(no BSA) antibodies were added to pre-blocked 50k MWCO filter columns(ThermoFisher, UFC505096) with 0.1% PBS-Tween. After spinning, reduction of antibodies was applied in TCEP solution(2.5 mM EDTA and 2.5 mM TCEP in 1x DPBS pH 7.0) followed by a washing step in Buffer C(Tris-buffer containing 1 mM Tris(pH 7.0), 1 mM Tris(pH 7.5), 150 mM NaCl, 1 mM EDTA(pH 8.0) and 0.02%(w/v) NaN3). Afterward, reduced antibodies were incubated with maleimide-modified oligonucleotides(Biomers) in a 2:1(w/w) ratio. The lyophilized maleimide-modified oligonucleotides were dissolved directly before use in 1x DPBS in the presence of vapor phase of LN2. After incubation, antibody-oligonucleotide solution was washed three times in high-salt PBS(1 M NaCl and 0.02%(w/v) NaN3 in 1x DPBS). Finally, antibodies were dissolved in a concentration of 0.5 mg/mL in Antibody stabilizer PBS solution(Candor) containing 0.2%(w/v) NaN3, 0.5 M NaCl and 5 mM EDTA. The fresh-frozen liver samples were sectioned at 5 μm and directly adhered onto poly-L-lysine- coated coverslips and stored under vacuum until staining was performed. Sections were dehydrated in acetone followed by tissue rehydration in 1x DPBS-Tween 0.1%(v/v) and by a pre-fixation in 1.6% PFA containing 1X DPBS-Tween 0.1%(v/v). Afterward, samples were incubated at room temperature with blocking buffer containing 1 mg/mL mouse IgG(Sigma, I5381) in S2(61 mM Na2HPO4, 39 mM NaH2PO4 and 250 mM NaCl in a 1:0.7 v/v S1(1X DPBS containing 5 mM EDTA, 0.5% w/v BSA(BSA) and 0.02% w/v NaN3)), 1 mg/mL rat IgG(Sigma, I4131) in S2, 10 mg/mL sheared salmon sperm DNA(Thermo Fisher, AM9680) and 0.05 mM naked oligonucleotides(Biomers) diluted in S2. Antibody staining solution was prepared in blocking buffer(1:200 dilution of surface antibodies and 1:100 dilution of transcription factor antibodies from 0.5 mg/mL stock solution of conjugated antibodies, respectively). Before usage, antibody staining solution was once filtered through 50 kDa Amicon Ultra Filters and resolved in blocking buffer. Following, tissue was incubated with the antibody staining solution in a humidity chamber overnight at 4°C. After antibody staining, tissues were washed twice in S2 followed by a three step fixation. First tissues were fixed with 1.6% PFA in S4 buffer(1x DPBS containing 5 mM EDTA, 0.5% w/v BSA, 0.02% w/v NaN3 and 0.5 M NaCl), followed by a fixation in ice-cold methanol and a final fixation in 8 mM Bis(sulfosuccinimidyl)suberate(ThermoFisher, 21,580). After each fixation step, tissue was washed three times in 1x DPBS. Before storing the tissue in S4 buffer, tissue was incubated with TrueBlack Lipofuscin Autofluorescence Quencher(Biotium, 23,007). Tissue sections were acquired using multicycle CO-Detection by IndEXing(CODEX) technology combined with a Zeiss Axio Observer 7 inverted microscope with the Colibri 7 LED Light source(Carl Zeiss) and a Prime BSI PCIe camera(Teledyne Photometrics). Complementary fluorescent oligonucleotides(Biomers) with a final concentration of 400 nM were aliquoted in Corning black 96-well plates in 250 μL of plate buffer(150 mM NaCl, 10 mM Tris pH 7.5, 10 mM MgCl2 · 6 H2O, 0.1% w/v TritonTM X-100 and 0.02% w/v NaN3 in ddH2O), 1:300 DAPI and 0.5 mg/mL sheared salmon sperm DNA.
Imaging cycles were performed using an Akoya CODEX instrument and CODEX instrument manager software(Akoya Biosciences). Automated images were performed using the the Plan-Apochromat 20X/0,8 M27(a = 0.55 mm) objective(Zeiss) and the imaging pipeline was controlled by a Focus strategy with autofocus for each support point created, creating a three z stack image with a distance of 1.5 μm. DAPI nuclear stain was imaged in each cycle. The images were processed with CODEX Processor(Akoya Biosciences) and analyzed with HALO Image Analysis software(Indica Labs). Segmentation and identification of LT-ILC1 cells of liver sections were performed using implemented software tools(Nuclei Seg, Membrane and Nuclear Detection). LT-ILC1 cells were defined as CD45+, CD3−, CD94+, CD56+, Eomes−(only DAPI overlay positively identified due to transcription factor) and KLRG1+.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and visualization
Descriptive statistics were calculated for each parameter in Microsoft Excel. Data variance was determined by controlling the False Discovery Rate(Benjamini and Yekutieli, 2001) for multiple comparisons following one-way ANOVA in GraphPad PRISM 9.1(Graphpad software, Inc., La Jolla, CA). From a case number above 8 a normal distribution test according to D’Àgostino-Pearson was applied, below 8 individuals non-parametric tests were performed. Therefore, for unpaired/normally distributed data ordinary ANOVA was performed; for unpaired/non-normally distributed data Kruskal-Walis test was performed; for paired/normally distributed data ANOVA with Greenhaus-Geiser correction was performed; and for paired/non-normally distributed data Friedman test was performed. For a comparison between 2 groups, a t test was performed for normally distributed data and a Mann-Whitney test for non-normally distributed data. A p value below the limit of 0.05 was considered significant, and figures were produced using GraphPad Prism. All figures were created with the graphic program "Affinity Designer"(v.1.9.1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Biotin A0436 | Biolegend | RRID: AB_2801086; Cat#409008 |
| BDCA-2 FITC (AC144) | Miltenyi | RRID: AB_244167; Cat# 130-090-510 |
| CD103 A0145 | Biolegend | RRID: AB_2749996; Cat#350231 |
| CD103 AF700 (Ber-ACT8) | NOVUS | RRID: AB_11189940; Cat# NBP1-97564AF700 |
| CD107a PE (H4A3) | Biolegend | RRID: AB_1186062; Cat#328607 |
| CD117 APC-Fire750 (104D2) | Biolegend | RRID: AB_2632949; Cat#313240 |
| CD117 BUV395 (YB5.B8) | BD Biosciences | RRID: AB_2743211; Cat#745740 |
| CD117 BV650 (104D2) | Biolegend | RRID: AB_2721550; Cat#313222 |
| CD117 PE-Cy7 (104D2 | Biolegend | RRID: AB_893222; Cat#313212 |
| CD127 BV605 (A019D5) | Biolegend | RRID: AB_2562022; Cat#351334 |
| CD14 FITC (M5E2) | Biolegend | RRID: AB_314186; Cat#301804 |
| CD16 APC-H7 (B73.1) | BD Biosciences | RRID: AB_10643005; Cat#561306 |
| CD161 APC Cy7 (HP-3G10) | Biolegend | RRID: AB_2563967; Cat#339928 |
| CD19 FITC (HIB19) | Biolegend | RRID: AB_314236; Cat#302206 |
| CD1a FITC (HI149) | Biolegend | RRID: AB_314018; Cat#300104 |
| CD20 FITC (2H7) | Biolegend | RRID: AB_314252; Cat#302304 |
| CD200R APC (OX-108) | BioLegend | RRID: AB_2564351; Cat#329308 |
| CD200R Biotin (REA725) | Miltenyi | RRID: AB_2656117; Cat#130-111-289 |
| CD200R PE/Dazzle 594 (OX-108) | Biolegend | RRID: AB_2565526; Cat#329310 |
| CD200R1 BV421 (OX-108) | Biolegend | RRID: AB_2800856; Cat#329314 |
| CD200R1 PE (OX-108) | Biolegend | RRID: AB_2074200; Cat#329306 |
| CD3 FITC (UCHT1) | Biolegend | RRID: AB_314060; Cat#300406 |
| CD34 FITC (581) | Biolegend | RRID: AB_1731852; Cat#343504 |
| CD4 APC-Cy7 (RPA-T4) | Biolegend | RRID: AB_314086; Cat#300518 |
| CD44 Pe-Cy7 (BJ18) | Biolegend | RRID: AB_2716000; Cat#338815 |
| CD45 BUV395 (HI 30) | BD Biosciences | RRID: AB_2869519; Cat#563792 |
| CD45 BUV805 (HI30) | Biolegend | RRID: AB_2174123; Cat#304029 |
| CD45RA APC-Vio770 | Miltenyi | RRID: AB_2658317; Cat#130-108-717 |
| CD49a A0575 | Biolegend | RRID: AB_2783195; Cat#328315 |
| CD49a PerCP-e710 (TS2/7) | eBiosciences | RRID: AB_2573891; Cat#46-9490-42 |
| CD49e APC (NKI-SAM-1) | Biolegend | RRID: AB_2128193; Cat#328012 |
| CD5 PE/Dazzle 594 (UCHT2) | Biolegend | RRID: AB_2632673; Cat#300634 |
| CD52 A0033 | Biolegend | RRID: AB_2734292; Cat#316017 |
| CD52 APC (QA19A22) | Biolegend | RRID: AB_830793; Cat# 318906 |
| CD56 A0047 | Biolegend | RRID: AB_2749970; Cat#362557 |
| CD56 BUV563 (NCAM16.2) | BD Biosciences | RRID: AB_2744431; Cat#565704 |
| CD69 A0146 | Biolegend | RRID: AB_2749997; Cat#310947 |
| CD69 BV785 (FN50) | BD Biosciences | RRID: AB_2738441; Cat#563834 |
| CD9 APC Vio770 (REA1071) | Miltenyi | RRID: AB_2733145; Cat#130-118-871 |
| CD94 A0867 | Biolegend | RRID: AB_2814142; Cat#305521 |
| CD94 AF700 (DX22) | Novus | RRID: AB_962803; Cat#NB100-64104AF700 |
| CD94 BUV737 (HP-3D9) | BD Biosciences | RRID: AB_2873190; Cat#748787 |
| CD94 FITC (DX22) | Biolegend | RRID: AB_314534; Cat#305504 |
| ckit PE-Cy7 (104D2 | Biolegend | RRID: AB_893222; Cat#313212 |
| CRTH2 AF647 (BM16) | Biolegend | RRID: AB_10642025; Cat#350104 |
| CXCR3 BV605 (G025H7) | Biolegend | RRID: AB_2562200; Cat#353727 |
| CXCR4 BV421 (12G5) | Biolegend | RRID: AB_11146018; Cat#306518 |
| CXCR6 A0804 | Biolegend | RRID: AB_2800961; Cat#356021 |
| CXCR6 BV421 (K041E5) | BioLegend | RRID: AB_2563873; Cat#356014 |
| EOMES PE/eFluor610 (WD1928) | eBiosciences | RRID:; Cat#61-4877-42 |
| FC Blocking Reagent | Miltenyi | RRID: AB_2892112; Cat#130-059-901 |
| FcERJa FITC (AER-37) | Biolegend | RRID: AB_1227653; Cat#334608 |
| GM-CSF PE (BVD2-21C11) | BioLegend | RRID: AB_2085532; Cat#502306 |
| Grz B PE (GB11) | BD Biosciences | RRID: AB_10561690; Cat#561142 |
| Hobit (ZNF683) AF647 (Sanquin-Hobit/1) | BD Biosciences | RRID: AB_2739629; Cat#566250 |
| IFNg BV421 (4S.B3) | Biolegend | RRID: AB_2561398; Cat#502532 |
| IL1R1 PE (polyclonal) | Biotechne | RRID: AB_2124912; Cat#FAB269P-100 |
| IL2 (MQ1-17H12) | Biolegend | RRID: AB_2563878; Cat#500334 |
| IL22 APC (REA466) | Miltenyi | RRID: AB_2652429; Cat#130-106-959 |
| Ki-67 BV421 (Ki-67) | Biolegend | RRID: AB_2563860; Cat#350506 |
| KLRG1 A0153 | Biolegend | RRID: AB_2750373; Cat#367721 |
| KLRG1 BV785 (2F1/KLRG1) | Biolegend | RRID: AB_2629749; Cat#138429 |
| KLRG1 PE/Dazzle 594 (368,608) | Biolegend | RRID: AB_2572135; Cat#368608 |
| NKp30 711 (p30-15) | BD Biosciences | RRID: AB_2738169; Cat#563383 |
| Nkp30 PE (P30-15) | Biolegend | RRID: AB_756111; Cat#325207 |
| NKp44 BV786 (P44-8) | BD Biosciences | RRID: AB_2742134; Cat#744304 |
| Nkp80 A0923 | Biolegend | RRID: AB_2814274; Cat#346709 |
| NKp80 APCVio770 (REA845) | Miltenyi | RRID: AB_2653031; Cat#130-112-593 |
| NKp80 FITC (4A4.D10) | Miltenyi | RRID: AB_10829948; Cat#130-094-843 |
| Nkp80 PE (REA845) | Miltenyi | RRID: AB_2653022; Cat#130-112-590 |
| Perforin BV421 (dG9) | Biolegend | RRID: AB_2566204; Cat#308122 |
| Perforin Pe-Cy7 (B-D48) | Biolegend | RRID: AB_2571973; Cat#353316 |
| TBET 711 (O4-46) | BD Biosciences | RRID: AB_2738136; Cat#563320 |
| TCRa/b FITC (IP26) | Biolegend | RRID: AB_314644; Cat#306706 |
| TCRy/d FITC (B1) | Biolegend | RRID: AB_1575108; Cat#331208 |
| TNF-a BV785 (FN50) | BD Biosciences | RRID: AB_2738441; Cat#563834 |
| anti-human TGF-b1 | Biolegend | RRID: AB_2810654; Cat#521708 |
| Eomes | R&D Systems | RRID: AB_10569705; Cat#AF6166 |
| CD56 | R&D Systems | RRID: AB_442152; Cat#AF2408 |
| αSMA | ThermoFisher | RRID: AB_2572996; Cat#14-9760-82 |
| CD45 | NovusBio | RRID: AB_2864384; Cat#NBP2-34528 |
| CD3e | Abcam | RRID: AB_1191514; Cat#ab271850 |
| CD94 | biorbyt | RRID: AB_2922808; Cat#orb308626 |
| CD32 | BioLegend | RRID: AB_314333; Cat#303201 |
| CD36 | Miltenyi | RRID: AB_2819615; Cat#130-124-322 |
| KLRG1 | Miltenyi | RRID: AB_2889702; Cat#130-126-458 |
| mouse IgG | Sigma | RRID: AB_1163670 I; Cat#5381 |
| Chemicals, peptides, and recombinant proteins | ||
| IL2(IS) | Miltenyi | Cat#130-097-748 |
| IGF-β1 | Miltenyi | Cat#130-095-066 |
| IL-7 | Miltenyi | Cat#130-095-363 |
| IL-15 | Immunotools | Cat#11340155 |
| IL-12 | Immunotools | Cat#11349125 |
| Viability Zombie Green | Biolegend | Cat#423112 |
| viability Zombie-Aqua | Biolegend | Cat#423102 |
| FLT3L | Immunotools | Cat#11343303 |
| Medium F-12 Nut Mix (+) L-Glutamin | Gibco Life | Cat#21765-029 |
| Antibiotic-Antimyotic | Gibco Life | Cat#15240-062 |
| Human Serum AB Plasma | Sigma | Cat#H3667-100mL |
| Sodium Selenite | Sigma (Merck) | Cat#S5261 |
| 2- Mercaptoethanol | Gibco Life | Cat#31350-010 |
| Ascorbic Acid | European Pharmacopoeia Reference | Cat#A1300000 |
| DMEM | Gibco Life | Cat#41965 |
| Penicillin/Streptomycin | PAN | Cat#P06-07100 |
| Fetal bovine serum low in endotoxin A.H. | Sigma | Cat#F7524-500mL |
| Deposited data | ||
| scRNASeq data from Heinrich et al.23 | re-analyzing external dataset | GEO: GSE179795 |
| scRNASeq data batch 1 | This paper | GEO: EGAS00001006847 |
| scRNASeq data batch 2 | This paper | GEO: EGAS00001006847 |
| scRNASeq data tissue batch 1&2 | This paper | GEO: EGAS00001006847 |
| Experimental models: Cell lines | ||
| Human LSECs | ScienCell | Cat#5000 |
| Human Hepatocytes | ScienCell | Cat#5200 |
| Human Hepatic Stellate Cells | ScienCell | Cat#5300 |
| Human Umbilical Vein Endothelial Cells | ScienCell | Cat#8000 |
| Oligonucleotides | ||
| Costumized primer Oligos for qPCR are listed in Table S4 and are all tested for product length by gel electrophoresis | IDT | N/A |
| TrueBlack® Lipofuscin Autofluorescence Quencher | Biotium | Cat#23007 |
| Ultra Filters | Amicon | N/A |
| salmon sperm DNA | Thermo Fisher | Cat#AM9680 |
| MWCO filter columns | ThermoFisher | Cat#UFC505096 |
| Software and algorithms | ||
| Prism 9 | Graphpad | N/A |
| Excel 2011 | Microsoft | N/A |
| Powerpoint 2001 | Microsoft | N/A |
| Cellranger mkfastq 3.0.2 | 10X Genomics | N/A |
| R package Seurat 3.1.1 | cran.r-project.org | N/A |
| NormalizeData | Function R package | N/A |
| FindVariableFeatures | Function R package | N/A |
| ScaleData | Function R package | N/A |
| FindClusters | Function R package | N/A |
| FindAllMarkers | Function R package | N/A |
| AddModuleScore | Function R package | N/A |
| CellphoneDB 2.0.0 | Function R package | N/A |
| FindMarkers | Function R package | N/A |
| FlowJo 10.7.1 | BD Bioscience | N/A |
| Affinity Designer 2.0 | Affinity tools | N/A |
| Affinity Publisher 2.0 | Affinity tools | N/A |
| HALO® Image Analysis software ( | Indica Labs | N/A |
| CODEX® instrument manager software (Akoya Biosciences | Akoya | N/A |
| Other | ||
| Axio Observer 7 inverted microscope | Zeiss | N/A |
| CODEX® instrument | Akoya | N/A |
| multicycle CO-Detection by IndEXing | Akoya | N/A |
Highlights.
scRNA-seq and proteomics identify a “liver-type” (LT) ILC1 subset in human livers
LT-ILC1s accumulate in fibrotic areas of cirrhotic livers
LT-ILC1-like cells can be derived from progenitors in co-culture with TGF-β1/LSECs
Similar cross-tissue ILC1 cells could also be found in tonsil and intestinal tissues
ACKNOWLEDGMENTS
This work was supported by grants from the DFG (KR 4521/1-1 to B.K., SPP1937 to J.N., SFB/TR 57 to U.S. and J.N.), H.W.& J. Hector Foundation (M69 to J.N.), DZIF (TTU 04.912&TTU 04.810 to J.N.), National Institutes of Health/National Cancer Institute (CA199447&CA208353 to A.G.F. and CA236063 to A.P.N.), AACR 17-20-46-MUND to B.L.M., and the Pelotonia-Organization to A.P.N. and to M.L. J.N., M.H., and T.B. are supported by DFG under Germany’s-Excellence-Strategy – EXC2151– 390873048. We thank the Cooperative Human Tissue Network of Nationwide Children’s Hospital, Columbus, Ohio, USA, for providing us with human pediatric tonsil samples. We would like to acknowledge the assistance of the Flow Cytometry-Core Facility at the Institute of Experimental Immunology, Medical Faculty at the University of Bonn. We want to also thank the Genomics Core Facility of the University Hospital Bonn under the direction of Dr. Andre Heimbach. S.E. and S.L. are supported by post-doc research fellowships within the Mildred-Scheel School of Oncology-Cologne-Bonn supported by the German Cancer Aid – Project ID 70113307 (Deutsche Krebshilfe). This work was also supported by the Genomics Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111937.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate lymphoid cells: 10 Years on. Cell 174, 1054–1066. 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, et al. (2013). Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol 14, 221–229. 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-Producing cells. Immunity 38, 769–781. 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, and Spits H (2011). Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol 12, 1055–1062. 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 5.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JKM, Doherty JM, Mills JC, and Colonna M (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725. 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, and Spits H (2009). Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol 10, 66–74. 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- 7.Hazenberg MD, and Spits H (2014). Human innate lymphoid cells. Blood 124, 700–709. 10.1182/blood-2013-11-427781. [DOI] [PubMed] [Google Scholar]
- 8.Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, Mao C, Giovenzana C, Nuovo G, Wei L, et al. (2010). Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 32, 803–814. 10.1016/j.immuni.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, Gilfillan S, Fuchs A, Vivier E, Sun JC, et al. (2016). Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity 44, 1127–1139. 10.1016/j.immuni.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riggan L, Freud AG, and O’Sullivan TE (2019).True detective: unraveling group 1 innate lymphocyte heterogeneity. Trends Immunol. 40, 909–921. 10.1016/j.it.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Sullivan TE (2019). Dazed and confused: NK cells. Front. Immunol 10, 2235. 10.3389/fimmu.2019.02235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nixon BG, Chou C, Krishna C, Dadi S, Michel AO, Cornish AE, Kansler ER, Do MH, Wang X, Capistrano KJ, et al. (2022). Cytotoxic granzyme C-expressing ILC1s contribute to antitumor immunity and neonatal autoimmunity. Sci. Immunol 7, eabi8642. 10.1126/sciimmunol.abi8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park E, Patel S, Wang Q, Andhey P, Zaitsev K, Porter S, Hershey M, Bern M, Plougastel-Douglas B, Collins P, et al. (2019). Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife 8, e47605. 10.7554/eLife.47605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weizman O-E, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE (2017). ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808.e12. 10.1016/j.cell.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, et al. (2017). Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol 18, 1004–1015. 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 16.Montaldo E, Vacca P, Chiossone L, Croxatto D, Loiacono F, Martini S, Ferrero S, Walzer T, Moretta L, and Mingari MC (2015). Unique Eomes(+) NK cell subsets are present in uterus and decidua during early pregnancy. Front. Immunol 6, 646. 10.3389/fimmu.2015.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Youssef Y, Robinson C, Ernst GF, Carson MY, Young KA, Scoville SD, Zhang X, Harris R, Sekhri P, et al. (2018). CD56 expression marks human group 2 innate lymphoid cell divergence from a shared NK cell and group 3 innate lymphoid cell developmental pathway. Immunity 49, 464–476.e4. 10.1016/j.immuni.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cichocki F, Grzywacz B, and Miller JS (2019). Human NK cell development: one road or many? Front. Immunol 10, 2078. 10.3389/fimmu.2019.02078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freud AG, Keller KA, Scoville SD, Mundy-Bosse BL, Cheng S, Youssef Y, Hughes T, Zhang X, Mo X, Porcu P, et al. (2016). NKp80 defines a critical step during human natural killer cell development. Cell Rep. 16, 379–391. 10.1016/j.celrep.2016.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lugthart G, Melsen JE, Vervat C, van Ostaijen-Ten Dam MM, Corver WE, Roelen DL, van Bergen J, van Tol MJD, Lankester AC, and Schilham MW (2016). Human lymphoid tissues harbor a distinct CD69+CXCR6+ NK cell population. J. Immunol 197, 78–84. 10.4049/jimmunol.1502603. [DOI] [PubMed] [Google Scholar]
- 21.Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, Christophides T, Pallett LJ, Peppa D, Dunn C, Fusai G, et al. (2016). CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci. Rep 6, 26157. 10.1038/srep26157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoorweg K, Peters CP, Cornelissen F, Aparicio-Domingo P, Papazian N, Kazemier G, Mjösberg JM, Spits H, and Cupedo T (2012). Functional differences between human NKp44− and NKp44+ RORC+ innate lymphoid cells. Front. Immunol 3, 72. 10.3389/fimmu.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinrich B, Gertz EM, Schäffer AA, Craig A, Ruf B, Subramanyam V, McVey JC, Diggs LP, Heinrich S, Rosato U, et al. (2022). The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut 71, 1161–1175. 10.1136/gutjnl-2021-325288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stary V, Pandey RV, Strobl J, Kleissl L, Starlinger P, Pereyra D, Weninger W, Fischer GF, Bock C, Farlik M, and Stary G (2020). A discrete subset of epigenetically primed human NK cells mediates antigen-specific immune responses. Sci. Immunol 5, eaba6232. 10.1126/sciimmunol.aba6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356. 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, et al. (2014). Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife 3, e01659. 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter S, Willcox CR, Davey MS, Kasatskaya SA, Jeffery HC, Chudakov DM, Oo YH, and Willcox BE (2018). Human liver infiltrating γδ T cells are composed of clonally expanded circulating and tissue-resident populations. J. Hepatol 69, 654–665. 10.1016/j.jhep.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krämer B, Goeser F, Lutz P, Glässner A, Boesecke C, Schwarze-Zander C, Kaczmarek D, Nischalke HD, Branchi V, Manekeller S, et al. (2017). Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 13, e1006373. 10.1371/journal.ppat.1006373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, Lim TKH, Yeong J, Toh HC, Lee SY, et al. (2019). Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 68, 916–927. 10.1136/gutjnl-2018-316510. [DOI] [PubMed] [Google Scholar]
- 30.Cuff AO, Sillito F, Dertschnig S, Hall A, Luong TV, Chakraverty R, and Male V (2019). The obese liver environment mediates conversion of NK cells to a less cytotoxic ILC1-like phenotype. Front. Immunol 10, 2180. 10.3389/fimmu.2019.02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikulak J, Bruni E, Oriolo F, Di Vito C, and Mavilio D (2019). Hepatic natural killer cells: organ-specific sentinels of liver immune homeostasis and physiopathology. Front. Immunol 10, 946. 10.3389/fimmu.2019.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CODEX multiplexed tissue imaging with DNA-conjugated antibodies ∣ Nat. Protoc https://www.nature.com/articles/s41596-021-00556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauss O, Phillips A, Ruggiero K, Bartlett A, and Dunbar PR (2017). Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep 7, 44356. 10.1038/srep44356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, et al. (2017). Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell 168, 1086–1100.e10. 10.1016/j.cell.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 35.Salomé B, Gomez-Cadena A, Loyon R, Suffiotti M, Salvestrini V, Wyss T, Vanoni G, Ruan DF, Rossi M, Tozzo A, et al. (2019). CD56 as a marker of an ILC1-like population with NK cell properties that is functionally impaired in AML. Blood Adv. 3, 3674–3687. 10.1182/bloodadvances.2018030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yudanin NA, Schmitz F, Flamar A-L, Thome JJC, Tait Wojno E, Moeller JB, Schirmer M, Latorre IJ, Xavier RJ, Farber DL, et al. (2019). Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity 50, 505–519.e4. 10.1016/j.immuni.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo S-L, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang C-L, et al. (2017). Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity 46, 148–161. 10.1016/j.immuni.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ducimetière L, Lucchiari G, Litscher G, Nater M, Heeb L, Nuñez NG, Wyss L, Burri D, Vermeer M, Gschwend J, et al. (2021). Conventional NK cells and tissue-resident ILC1s join forces to control liver metastasis. Proc. Natl. Acad. Sci. USA 118, e2026271118. 10.1073/pnas.2026271118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forkel M, Berglin L, Kekäläinen E, Carlsson A, Svedin E, Michaëlsson J, Nagasawa M, Erjefält JS, Mori M, Flodstrom-Tullberg M, et al. (2017). Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur. J. Immunol 47, 1280–1294. 10.1002/eji.201646890. [DOI] [PubMed] [Google Scholar]
- 40.Jeffery HC, McDowell P, Lutz P, Wawman RE, Roberts S, Bagnall C, Birtwistle J, Adams DH, and Oo YH (2017). Human intrahepatic ILC2 are IL-13positive amphiregulinpositive and their frequency correlates with model of end stage liver disease score. PLoS One 12, e0188649. 10.1371/journal.pone.0188649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, et al. (2019). Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409. 10.1038/s41586-019-1082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shannon JP, Vrba SM, Reynoso GV, Wynne-Jones E, Kamenyeva O, Malo CS, Cherry CR, McManus DT, and Hickman HD (2021). Group 1 innate lymphoid-cell-derived interferon-γ maintains anti-viral vigilance in the mucosal epithelium. Immunity 54, 276–290.e5. 10.1016/j.immuni.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krabbendam L, Heesters BA, Kradolfer CMA, Haverkate NJE, Becker M.a.J., Buskens CJ, Bemelman WA, Bernink JH, and Spits H (2021). CD127+ CD94+ innate lymphoid cells expressing granulysin and perforin are expanded in patients with Crohn’s disease. Nat. Commun 12, 5841. 10.1038/s41467-021-26187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stiglund N, Strand K, Cornillet M, Stål P, Thorell A, Zimmer CL, Näslund E, Karlgren S, Nilsson H, Mellgren G, et al. (2019). Retained NK cell phenotype and functionality in non-alcoholic fatty liver disease. Front. Immunol 10, 1255. 10.3389/fimmu.2019.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J, Wen H, Lin R, Qu K, Li K, et al. (2019). Accumulation of tumor-infiltrating CD49a+ NK cells correlates with poor prognosis for human hepatocellular carcinoma. Cancer Immunol. Res 7, 1535–1546. 10.1158/2326-6066.CIR-18-0757. [DOI] [PubMed] [Google Scholar]
- 46.Bauer M, and Schuppan D (2001). TGFbeta1 in liver fibrosis: time to change paradigms? FEBS Lett. 502, 1–3. 10.1016/s0014-5793(01)02655-2. [DOI] [PubMed] [Google Scholar]
- 47.Karrar A, Broomé U, Uzunel M, Qureshi AR, and Sumitran-Holgersson S (2007). Human liver sinusoidal endothelial cells induce apoptosis in activated T cells: a role in tolerance induction. Gut 56, 243–252. 10.1136/gut.2006.093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanoni G, Ercolano G, Candiani S, Rutigliani M, Lanata M, Derré L, Marcenaro E, Schneider P, Romero P, Jandus C, and Trabanelli S (2021). Human primed ILCPs support endothelial activation through NF-κB signaling. Elife 10, e58838. 10.7554/eLife.58838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harmon C, Jameson G, Almuaili D, Houlihan DD, Hoti E, Geoghegan J, Robinson MW, and O’Farrelly C (2019). Liver-derived TGF-β maintains the EomeshiTbetlo phenotype of liver resident natural killer cells. Front. Immunol 10, 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scoville SD, Keller KA, Cheng S, Zhang M, Zhang X, Caligiuri MA, and Freud AG (2015). Rapid column-free enrichment of mononuclear cells from solid tissues. Sci. Rep 5, 12490. 10.1038/srep12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernink JH, Mjösberg J, and Spits H (2017). Human ILC1: to Be or not to Be. Immunity 46, 756–757. 10.1016/j.immuni.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roan F, and Ziegler SF (2017). Human group 1 innate lymphocytes (ILC1) are negative for surface CD3ε but express CD5. Immunity 46, 758–759. 10.1016/j.immuni.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McMahan RH, Porsche CE, Edwards MG, and Rosen HR (2016). Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: insights into non-alcoholic fatty liver disease. PLoS One 11, e0159217. 10.1371/journal.pone.0159217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shido K, Chavez D, Cao Z, Ko J, Rafii S, and Ding B-S (2017). Platelets prime hematopoietic–vascular niche to drive angiocrine-mediated liver regeneration. Signal Transduct. Target. Ther 2, 16044. 10.1038/sigtrans.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, et al. (2018). Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun 9, 4383. 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camacho-Sanchez M, Burraco P, Gomez-Mestre I, and Leonard JA (2013). Preservation of RNA and DNA from mammal samples under field conditions. Mol. Ecol. Resour 13, 663–673. 10.1111/1755-0998.12108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNASeq data analyzed here are accessible with corresponding documentation on EGA(European Genome-Phenome Archive) with the accession GEO: EGAS00001006847. The Heinrich et al. dataset23 is available from GEO with the accession GEO: GSE179795. Other primary data, as raw FCS files, raw PCR data, CBA data, or microscopy images, are available from the authors upon reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.