

Summary
NLRP3 inflammasome-mediated immune responses are involved in the pathogenesis of multiple inflammatory diseases, but few clinical drugs are identified that directly target the NLRP3 inflammasome to treat these diseases to date. Here, we show that the anticancer agent tivantinib is a selective inhibitor of NLRP3 and has a strong therapeutic effect on inflammasome-driven disease. Tivantinib specifically inhibits canonical and non-canonical NLRP3 inflammasome activation without affecting AIM2 and NLRC4 inflammasome activation. Mechanistically, Tivantinib inhibits NLRP3 inflammasome by directly blocking NLRP3 ATPase activity and subsequent inflammasome complex assembly. In vivo, Tivantinib reduces IL-1β production in mouse models of lipopolysaccharide (LPS)-induced systemic inflammation, monosodium urate (MSU)-induced peritonitis and Con A-induced acute liver injury (ALI), and also has remarkable preventive and therapeutic effects on experimental autoimmune encephalomyelitis (EAE). In conclusion, our study identifies the anticancer drug tivantinib as a specific inhibitor of NLRP3 and provides a promising therapeutic agent for inflammasome-driven disease.
Subject areas: Health sciences, Biological sciences, Immunology
Graphical abstract
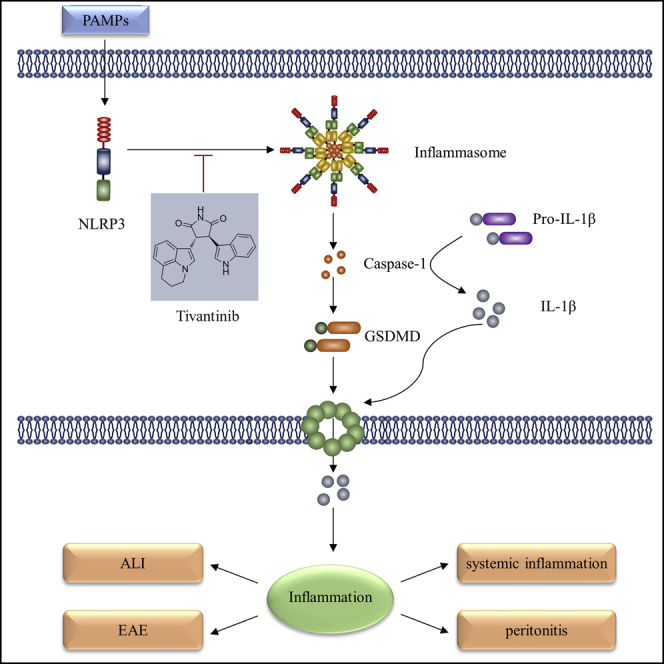
Highlights
-
•
Tivantinib inhibits NLRP3 inflammasome activation both in human and mouse cells
-
•
Tivantinib blocking NLRP3 ATPase activity and inflammasome complex assembly
-
•
Tivantinib is effective for the treatment of inflammatory diseases
Health sciences; Biological sciences; Immunology.
Introduction
The NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) is an important innate immune receptor in the cytoplasm that senses a wide range of structurally diverse factors, such as viral RNAs, microbial toxins, excess glucose, amyloids, uric acid crystals and ATP derived from pathogen, environment, and the host to initiate immune response.1,2,3,4,5 Upon activated by these pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), NLRP3 recruits and binds to apoptosis-associated speck-like protein containing a CARD (ASC) and cysteine protease caspase-1 to form a polymeric protein complex termed NLRP3 inflammasome.6 The primary function of NLRP3 inflammasome is to induce the production of pro-inflammatory cytokines IL-1β and IL-18 and mediate a form of inflammatory cell death called pyroptosis.7,8,9,10 Recent studies have reported that aberrant activation of NLRP3 inflammasome is also implicated in the pathogenesis of various acute and chronic inflammatory diseases in humans, such as sepsis, peritonitis, cryopyrin-associated periodic syndrome, morphine analgesic tolerance, gout, atherosclerosis, type 2 diabetes, multiple sclerosis, Alzheimer’s disease and COVID-19 disease.11,12,13,14,15,16,17 Therefore, targeting NLRP3 is a promising strategy for treating these inflammasome-driven diseases.
In recent years, several small molecules have been shown to alleviate inflammatory diseases by targeting NLRP3 inflammasome, including MCC950, CY-09, tranilast, oridonin, and 3,4-methylenedioxy-β-nitrostyrene (MNS).18,19,20,21,22 However, the clinical pharmacological effects and specificity of these inhibitors are still unknown, and their clinical safety also needs to be further evaluated. Current treatments for NLRP3 inflammasome-driven diseases in the clinic including canakinumab, anakinra, and rilonacept, three biological agents that target IL-1β.23,24,25 However, the NLRP3 inflammasome not only induces IL-1β secretion but also initiates the production of other pro-inflammatory cytokines, such as IL-18 and HMGB1.26 Furthermore, other inflammasomes, such as AIM2 and NLRC4, can also induce IL-1β secretion as well as NLRP3 inflammasome,27,28 so targeting IL-1β may be ineffective for some NLRP3 inflammasome-driven diseases and may have unexpected side effects. Therefore, it is urgent to develop new NLRP3 inhibitors with high safety and specificity that are closer to clinical application.
Tivantinib, also known as ARQ-197, is a selective non-ATP competitive inhibitor of c-Met with highly potent antitumor pharmacological effects, which is currently in phase III clinical trials for liver and lung cancers and phase II clinical trials for colon cancer.29,30,31 Importantly, tivantinib is a highly safe drug that is well tolerated by most patients, even at doses up to 360 mg twice daily continuously for 28 days. Tivantinib strongly inhibits the kinase activity of c-Met with a calculated inhibitory constant (Ki) of only 355 nmol/L by stabilizing its inactive nonphosphorylated configuration, causing the arrest of cell growth and inhibiting proliferation, invasion, metastasis.32,33,34 In addition to its antiproliferative and antitumor effects, recent studies have found that tivantinib can disrupt microtubule polymerization and inhibit mitosis in an c-Met independent manner,35 suggesting tivantinib may have therapeutic targets other than c-Met. Although tivantinib has shown favorable pharmacological effects against cancer, its roles and targets in inflammatory diseases are still unknown.
In this study, we showed that tivantinib is a highly specific inhibitor of NLRP3, which inhibits inflammasome activation by directly blocking NLRP3 ATPase activity, and preventing NLRP3 oligomerization. More importantly, tivantinib has remarkable pharmacological effects in mouse models of inflammasome-driven diseases, including sepsis, peritonitis, ALI, and EAE. Our study identified tivantinib as a specific inhibitor of NLRP3 and a potential therapeutic for inflammasome-driven diseases.
Results
Identification of tivantinib inhibiting NLRP3 inflammasome activation and pyroptosis
Although tivantinib (Figure S1A), a phase III clinical drug without cytotoxicity (Figures S1B and S1C), has shown favorable pharmacological effects against a variety of solid tumors, its role in inflammatory response remain unclear. To investigate its effect on the activation of NLRP3 inflammasome, we pretreated LPS-primed mouse primary bone marrow-derived macrophages (BMDMs) with various doses of tivantinib for 30 min and then stimulated the cells with the classic NLRP3 activator nigericin. We found that tivantinib dose-dependently inhibited caspase-1 cleavage, IL-1β, and IL-18 secretion without affecting the production TNF-α and IL-6 (Figures 1A–1E). Moreover, the cleavage of GSDMD and cell death can also be impaired by tivantinib (Figures 1F, 1G, S1D, and S1E), suggesting that tivantinib specifically inhibits nigericin-induced IL-1β secretion and pyroptosis. To further clarify the inhibitory effect of tivantinib on NLRP3 inflammasome activation, we treated PMA-predifferentiated human THP-1 macrophages with tivantinib and then stimulated the cells with nigericin, showing a significant reduction in the cleavage of caspase-1 and the production of IL-1β (Figures 1H and 1I). Taken together, these results indicate that tivantinib inhibits NLRP3 inflammasome activation in both human and mouse cells.
Figure 1.

Tivantinib inhibits nigericin-induced IL-1β release and pyroptosis
(A and B) ELISA analysis of IL-1β and IL-18 in supernatants (SN) of LPS-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min.
(C–F) Western blot analysis of IL-1β and caspase-1 (p20) in SN and pro-IL-1β, pro-caspase-1 in cell lysates (Input) of LPS-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min. (D, E) ELISA analysis of IL-6 (D) and TNF-α (E) in SN of LPS-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min. (F) LDH release in SN of LPS-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min.
(G) Western blot analysis of Gsdmd cleavage in Input of Flag-Gsdmd reconstituted Gsdmd−/− iBMDMs cells primed with LPS for 3 h in the presence of various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 1 h.
(H) ELISA analysis of IL-1β in SN of PMA-differentiated THP-1 cells treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 1 h.
(I) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of PMA-differentiated THP-1 cells treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 1 h. Data represent means ± SEM from four biological duplicates (A, B, D-F, and H). Statistical analysis was performed using one-way ANOVA of means test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Tivantinib is a specific inhibitor of NLRP3 but not of NLRC4 and AIM2 inflammasomes
Next, we explore the inhibitory effect of tivantinib on inflammasome activation induced by other NLRP3 stimuli, including monosodium urate crystals (MSU), ATP, and Alum. Consistent with nigericin, tivantinib inhibits these agonists-induced caspase-1 cleavages and IL-1β release (Figures 2A and 2B). In addition, intracellular LPS has been reported to induce caspase-11-dependent non-canonical NLRP3 inflammasome activation.8,36,37 Our results showed that tivantinib treatment also inhibited transfected LPS-induced caspase-1 cleavage and IL-1β release in a dose-dependent manner (Figures 2C and 2D). In conclusion, these results suggest that tivantinib is a common inhibitor for canonical and non-canonical NLPR3 inflammasome.
Figure 2.

Tivantinib specifically inhibits NLRP3 inflammasome activation
(A) ELISA analysis of IL-1β in SN of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with MSU (150 μg/mL), nigericin (5 μM), ATP (2.5 mM), Alum (300 μg/mL).
(B) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with MSU (150 μg/mL), nigericin (5 μM), ATP (2.5 mM), Alum (300 μg/mL).
(C) ELISA analysis of IL-1β in SN of Pam3CSK4-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 0.5 μg/mL cytosolic LPS (cLPS) for 16 h.
(D) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of Pam3CSK4-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 0.5 μg/mL cLPS for 16 h.
(E) ELISA analysis of IL-1β in SN of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin and 0.5 μg/mL poly (dA:dT).
(F) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin and 0.5 μg/mL Poly (dA:dT).
(G) ELISA analysis of IL-1β in SN of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin and Salmonella.
(H) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin and Salmonella. Data represent means ± SEM from four biological duplicates (A, C, E, and G). Statistical analysis was performed using one-way ANOVA of means test. ∗∗∗p < 0.001.
To test whether tivantinib has an inhibitory effect on other inflammasomes, we pretreated LPS-primed BMDMs with tivantinib and then stimulated the cells to induce AIM2 inflammasome activation by transfecting with the dsDNA analog Poly (dA:dT). Our results indicate that tivantinib does not attenuate IL-1β secretion, suggesting that it has no effect on AIM2 inflammasome activation (Figures 2E and 2F). Similarly, tivantinib also does not inhibit NLRC4 inflammasome activation induced by Salmonella typhimurium infection (Figures 2G and 2H). Thus, these results indicate that tivantinib is a specific inhibitor of NLRP3 inflammasome.
Tivantinib inhibits NLRP3 inflammasome activation independently of c-Met
We next investigated the underlying mechanism by which tivantinib inhibits NLRP3 inflammasome activation. Previous studies have shown that tivantinib is a selective inhibitor of c-Met with a wide range of antitumor pharmacological effects.32 To explore whether tivantinib blocks NLRP3 inflammasome activation by inhibiting c-Met, we pretreated LPS-primed BMDMs with three other inhibitors of c-Met, including NVP-BVU972, AMG-337, and SGX-523, and then stimulated the cells with nigericin. The results showed that these inhibitors attenuated the activation of c-MET signaling, but had no effect on NLRP3 inflammasome activation (Figures 3A, 3B, and S2A), suggesting that c-Met may not be involved in NLRP3 inflammasome activation. To further confirm these results, we knocked down c-Met with small interference RNA (siRNA) and found that it did not affect either NLRP3 inflammasome activation or the inhibitory effect of tivantinib (Figures 3C–3E). These results indicate that tivantinib inhibits NLRP3 inflammasome activation independently of c-Met.
Figure 3.

Tivantinib inhibits the NLRP3 inflammasome independently of c-Met
(A) ELISA analysis of IL-1β in SN of LPS-primed BMDMs treated with 5 μM Tivantinib, 1 μM NVP-BVU972, 1 μM AMG-337, 1 μM SGX-523, and then stimulated with 5 μM nigericin.
(B) Western blot analysis of IL-1β and p20 in SN and pro-IL-1β, pro-caspase-1 in Input of LPS-primed BMDMs treated with 5 μM Tivantinib, 1 μM NVP-BVU972, 1 μM AMG-337, 1 μM SGX-523, and then stimulated with 5 μM nigericin.
(C) The expression of c-Met in BMDMs transfected with siRNA.
(D) ELISA analysis of IL-1β in SN of LPS-primed BMDMs transfected with siRNA against c-Met and then stimulated with 5 μM nigericin for 30 min in the presence of various concentrations of Tivantinib (as labeled).
(E) Western blot analysis of IL-1β in SN and pro-IL-1β, pro-caspase-1 in Input of LPS-primed BMDMs transfected with siRNA against c-Met and then stimulated with 5 μM nigericin for 30 min in the presence of various concentrations of Tivantinib (as labeled). Data represent means ± SEM from three to four biological duplicates (A, C, and D). Statistical analysis was performed using one-way ANOVA of means test. ∗∗p < 0.01, ∗∗∗p < 0.001.
Tivantinib blocks NLRP3 inflammasome assembly by weakening NLRP3 ATPase activity
It has been reported that NLRP3 inflammasome activation requires two steps: priming and activation.38 We investigate which step is involved in the inhibition of tivantinib on NLRP3 inflammasome activation. Treating BMDMs with LPS and tivantinib, we found that tivantinib did not affect the expression of inflammasome components including NLRP3, pro-IL-1β, pro-caspase-1, NEK7, and ASC, nor did it affect the secretion of IL-6 and TNF-α (Figures S3A–S3C), indicating that it had no effect on the priming step.
We then studied the effect of tivantinib on the activation step of NLRP3 inflammasome, potassium efflux was reported to be a critical upstream event of NLRP3 inflammasome activation.39 Our results show that the NLRP3 agonists nigericin and ATP significantly induce potassium efflux, but tivantinib does not reverse this effect (Figures 4A and S4A), indicating that tivantinib has no effect on potassium efflux. Previous studies have shown that mitochondrial oxidation is another common event of NLRP3 inflammasome activation.40 By staining mitochondrial oxidation with mitoSOX, we found that tivantinib cannot block nigericin and ATP-induced mitochondrial ROS production (Figures 4B, 4C, S4B, and S4C). In addition, a requirement of chloride efflux in NLRP3 inflammasome activation was recently demonstrated.41,42 We examined the effect of tivantinib on intracellular chloride ions and found that it cannot prevent the chloride efflux induced by nigericin and ATP (Figures S4D and S4E). Collectively, these results suggest that tivantinib acts downstream of potassium and chloride efflux, and mitochondrial oxidation to inhibit NLRP3 inflammasome activation.
Figure 4.

Tivantinib blocks NLRP3 inflammasome assembly by weakening NLRP3 ATPase activity
(A) Qualification of intracellular potassium level by ICP-OES in LPS-primed BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min.
(B) Confocal microscopy analysis in LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin, followed by staining with Mitosox and DAPI.
(C) The relative fluorescence intensity of mitoSOX in LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin.
(D) Western blot analysis of ASC oligomerization in cross-linked cytosolic pellets and input of BMDMs treated with 5 μM Tivantinib and then stimulated with various concentrations of nigericin (as labeled) for 30 min.
(E) Immunoprecipitation (IP) and western blot analysis of the interaction of endogenous ASC and NLRP3 in LPS-primed BMDMs treated with 5 μM Tivantinib and then stimulated with 5 μM nigericin for 30 min.
(F and G) Western blot analysis of NLRP3 oligomerization by SDD-AGE assay in BMDMs treated with various concentrations of Tivantinib (as labeled) and then stimulated with 5 μM nigericin for 30 min. (G) IP and western blot analysis of the interaction between Flag-NLRP3 and VSV-NLRP3 in the lysates of HEK-293T cells treated with 10 μM Tivantinib.
(H) ATPase activity assay for purified NLRP3 in the presence of different concentrations of Tivantinib (as labeled).
(I) Docking complex of NLRP3 with Tivantinib. Tivantinib is shown in sticks and colored light green, NLRP3 is shown in cartoon and colored light gray, key amino acid residues were shown as sticks.
(J) MST assay for the affinity between Tivantinib and purified NLRP3 protein. Data represent means ± SEM from four biological duplicates (A, C, H). Statistical analysis was performed using one-way ANOVA(A, H) or unpaired Student’s t test (C). ∗p < 0.05, ∗∗∗p < 0.001.
Since tivantinib has no effect on the common upstream events of NLRP3 inflammasome activation, we next assessed whether it affects the assembly of NLRP3 inflammasome. It has been reported that ASC oligomerization and NLRP3-ASC interaction are key events for NLRP3 inflammasome assembly.43 Consistent with previous studies, stimulation with nigericin-induced ASC oligomerization and the interaction between NLRP3 and ASC, which could be inhibited by tivantinib treatment (Figures 4D and 4E). These results suggest that tivantinib inhibits NLRP3 inflammasome activation by blocking inflammasome assembly. Recent studies have shown that NEK7 is another key component of inflammasome complex and maintains inflammasome activation by interacting with NLRP3.44,45,46 we examined the interaction between NEK7 and NLRP3 and found that tivantinib did not affect NLRP3-NEK7 interaction (Figures S5A and S5B), indicating that tivantinib acts downstream of NLRP3-NEK7 interaction to inhibit NLRP3 inflammasome assembly. The oligomerization of NLRP3 is a prerequisite for ASC oligomerization.47 By analyzing NLRP3 oligomerization with semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)20 and NLRP3-NLRP3 interaction by immunoprecipitation, we found that tivantinib treatment inhibited NLRP3 oligomerization and NLRP3-NLRP3 interaction (Figures 4F and 4G). Previous studies have shown that NLRP3 has ATPase activity, which is essential for NLRP3 oligomerization.48 We then tested the effect of tivantinib on the ATPase activity of NLRP3 and found that tivantinib significantly inhibited the ATPase activity of purified NLRP3 in a dose-dependent manner (Figures 4H and S5C). To further explore how tivantinib inhibited NLRP3 ATPase activity, we performed the template structure of human NLRP3 (PDB: 6NPY) and ADP bound docking calculations by AutoDock4. The results showed that tivantinib was readily docked into the ATP-binding pocket of NLRP3 (Figures 4I, S6A, and S6B), suggesting that tivantinib may interact with NLRP3. To determine that tivantinib directly targets NLRP3, we used microscale thermophoresis (MST) to analyze the interaction between tivantinib and purified NLRP3 and found that the equilibrium dissociation constant (KD) of tivantinib and NLRP3 was around 372.64 nM (Figure 4J). Thus, these results indicate that tivantinib binds to the ATP-binding pocket of NLRP3 and may competitively bind NLRP3 with ATP, thereby weakening NLRP3 ATPase activity and blocking NLRP3 oligomerization and inflammasome assembly.
Tivantinib inhibits NLRP3 activation in vivo
Since we have demonstrated that tivantinib attenuates NLRP3 inflammasome activation in vitro, we next investigated the inhibitory effects tivantinib in vivo. Intraperitoneal injection of LPS induces a NLRP3-dependent septic shock characterized by IL-1β production.49 We pretreated mice with vehicle or tivantinib 1 h before challenge with LPS, and found that tivantinib treatment significantly increased the survival rate of mice, and inhibited the production of IL-1β and IL-18 in serum without affecting TNF-α release (Figures 5A–5D). However, several other inhibitors of c-Met, including NVP-BVU972, AMG-337, and SGX-523, did not affect LPS-induced sepsis in mice (Figures S2B–S2E). The mouse model of peritonitis induced by intraperitoneal injection of MSU is closely related to the activation of NLRP3 inflammasome, which is pathologically characterized by IL-1β production and massive infiltration of neutrophil.3 Consistent with the therapeutic effect of tivantinib in LPS-induced septic shock, tivantinib treatment also suppressed MSU-induced IL-1β and IL-18 production and neutrophil infiltration (Figures 5E–5G). Con A-induced acute liver injury (ALI) is a well-known mouse model of hepatitis, and its pathologic development is closely related to NLRP3.50 To test whether tivantinib can alleviate ALI, we pretreated mice with vehicle or tivantinib 1 h before intraperitoneal injection of Con A. The results showed that tivantinib treatment significantly improved the survival rate of mice and alleviated liver injury (Figures 5H–5J). Consistent with this result, tivantinib also inhibited IL-1β and IL-18 expression in serum or liver tissue (Figures 5K and 5L). Taken together, these results suggest that tivantinib effectively inhibits NLRP3 inflammasome activation in vivo and alleviates NLRP3-dependent acute inflammatory diseases.
Figure 5.

Tivantinib inhibits inflammasome activation in vivo and has preventive effects in mouse models of NLRP3-driven inflammation
(A) Survival analysis of C57BL/6J mice intraperitoneally injected with LPS (20 mg/kg) and pretreated with vehicle or Tivantinib (20 mg/kg). n = 8 biologically independent mice.
(B–D) ELISA analysis of IL-1β (B), IL-18 (C), and TNF-α (D) in serum of C57BL/6J mice intraperitoneally injected with LPS and pretreated with vehicle or Tivantinib (20 mg/kg).
(E) FACS analysis of neutrophil numbers in the peritoneal cavity of C57BL/6J mice intraperitoneally injected with MSU and pretreated with vehicle or Tivantinib (20 mg/kg).
(F and G) ELISA analysis of IL-1β (F) and IL-18 (G) in the peritoneal cavity of C57BL/6J mice intraperitoneally injected with MSU and pretreated with vehicle or Tivantinib (20 mg/kg).
(H) Survival analysis of C57BL/6J mice tail vein injected with Con A (30 mg/kg) and pretreated with vehicle or Tivantinib (20 mg/kg). n = 10 biologically independent mice.
(I) Serum ALT levels of vehicle or Tivantinib-treated mice after Con A injection (15 mg/kg).
(J) Hematoxylin and eosin (H&E) staining in liver cross sections from vehicle or 20 mg/kg Tivantinib-treated mice after Con A injection (15 mg/kg). Scale bar, 100 μm.
(K) Serum IL-1β and IL-18 levels of vehicle or Tivantinib-treated mice after Con A injection (15 mg/kg).
(L) Liver IL-1β mRNA expression of vehicle or 20 mg/kg Tivantinib-treated mice after Con A injection (15 mg/kg). Data represent means ± SEM (B-G, I, K, and L). Statistical analysis was performed using unpaired Student’s t test (B-G, I, K, and L) or generalized Wilcoxon test (A and H). ∗∗∗p < 0.001.
Tivantinib prevents the pathological development of EAE
EAE, a mouse model of human multiple sclerosis characterized by pro-inflammatory cytokine production and demyelination, has recently been demonstrated to be an NLRP3-driven inflammatory disease.51 We first investigated whether tivantinib was efficacious in preventing the pathological development of EAE. Treatment of mice with vehicle or tivantinib every two days from the day of EAE induction by immunization until the end of the experiment, we found that tivantinib treatment delayed the onset, ameliorated the severity of EAE, and reduced weight loss in EAE mice (Figures 6A and S7A). Hematoxylin-Eosin (H&E) and Luxol fast blue (LFB) staining of spinal cord sections revealed reduced inflammatory infiltration and demyelination in tivantinib-treated mice compared with vehicle-treated mice (Figure 6B). Flow cytometry analysis of immune cells in the CNS showed that treatment with tivantinib significantly decreased the frequencies and counts of infiltrating CD4+, CD11b+, and CD11b− cells (Figures 6C, 6D, and S7B). Furthermore, the expression of pro-inflammatory cytokines IL-1β and caspase-1 cleavage were also reduced in the spinal cord of tivantinib-treated mice (Figures 6E and 6F). Collectively, these results suggested that tivantinib alleviates neuroinflammation and prevents the pathological development of EAE.
Figure 6.

Tivantinib prevents the development of EAE
(A–E) Intraperitoneal injection of vehicle or Tivantinib (10 mg/kg) in C57BL/6J mice beginning at the induction of EAE every 2 days. (A) Clinical scores after EAE induction. (B) Sections of paraffin-embedded spinal cord tissues were stained with H&E and Luxol fast blue (LFB). (C) FACS analysis of live CD4+, CD8+, CD11b+, and CD11b− cells gate on CD45hi cells in the CNS on day 22 after EAE induction. (D) The numbers of live CD4+, CD8+, CD11b+, and CD11b− cells gate on CD45hi cells in the CNS on the 22 days after the induction of EAE. (E) The mRNA levels of IL-1β in CNS were evaluated by real-time PCR.
(F) Western blot analysis of NLRP3, ASC, and caspase-1 in the spinal cord of EAE mice. Data represent means ± SEM from five biological duplicates. Statistical analysis was performed using one-way ANOVA(A, D) or unpaired Student’s t test (E). ∗∗p < 0.01, ∗∗∗p < 0.001.
Tivantinib has a remarkable therapeutic effect on EAE
We further sought to investigate whether tivantinib was effective in reversing EAE severity after disease onset. We first induce EAE by immunization with a CNS auto-antigen and then treated with tivantinib from the day of disease onset (Figure 7A). Indeed, the treatment of mice with tivantinib alleviated the severity of EAE and significantly reduced the maximum and cumulative clinical score in EAE (Figures 7B and 7C). Consistent with previous results, we found that tivantinib had a protective effect on inflammatory infiltration and demyelination of the spinal cords by histological analysis of H&E and LFB staining (Figure 7D). Furthermore, the frequencies and counts of infiltrating CD4+, CD8+, CD11b+, and CD11b− cells in the CNS of tivantinib-treated mice was also reduced (Figures 7E and 7F). Thus, these results suggested that tivantinib has a remarkable therapeutic effect on EAE by inhibiting neuroinflammation.
Figure 7.

Tivantinib mediates therapeutic effects in EAE
(A–E) Intraperitoneal injection of vehicle or Tivantinib (10 mg/kg) in C57BL/6J mice beginning at the disease onset of EAE every 2 days. (A) Schematic diagram of Tivantinib administration in EAE. (B) Clinical scores after EAE induction. (C) The cumulative and maximum clinical score of EAE. (D) Sections of paraffin-embedded spinal cord tissues were stained with H&E and LFB. (E) FACS analysis of live CD4+, CD8+, CD11b+, and CD11b− cells gate on CD45hi cells in the CNS on the day 30 after EAE induction.
(F) The numbers of CD4+, CD8+, CD11b+, and CD11b− cells gate on CD45hi cells in the CNS on the 30 days after the induction of EAE. Data represent means ± SEM from six biological duplicates. Statistical analysis was performed using one-way ANOVA(B, F) or unpaired Student’s t test (C). ∗p < 0.05, ∗∗p < 0.01.
Discussion
In this study, we identify that tivantinib, a remarkable safe anticancer agent currently in phase III clinical trials evaluation for liver and lung cancers, acts as a potent specific NLRP3 inhibitor for the treatment of inflammasome-driven diseases, including LPS-induced systemic inflammation, MSU-induced peritonitis, Con A-induced ALI, and EAE. Tivantinib may serve as a target inhibitor of NLRP3 to regulate immune response and has the potential to be a new therapeutic agent for the treatment of inflammatory diseases.
Our results indicate that tivantinib inhibits canonical and non-canonical NLRP3 inflammasome activation in both mouse primary bone marrow-derived macrophage (BMDMs) and human THP-1 cells, but has no effects on AIM2 or NLRC4 inflammasome activation, suggesting it specifically targets NLRP3 inflammasome. Potassium efflux, reactive oxygen species (ROS) production, and chloride efflux have been reported as the common intermediate steps and upstream events for various stimuli-induced NLRP3 activation.39,40,52,53,54,55 Our results demonstrate that tivantinib blocks caspase-1 cleavage and IL-1β secretion independently of these signals, suggesting that it might directly target the complex of NLRP3 inflammasome. Further study revealed that tivantinib blocked ASC and NLRP3 oligomerization, indicating that it inhibits NLRP3 activation by preventing inflammasome complex assembly.
Although our results have demonstrated that tivantinib inhibited NLRP3 inflammasome assembly, its precise mechanism is still unclear. Previous results have shown that NEK7 is essential for NLRP3 activation and the interaction between NKE7 and NLRP3 is a prerequisite for inflammasome assembly.44,46 Moreover, several inhibitors, such as Licochalcone B, manoalide, RRx-001, and its analog, have been reported to inhibit inflammasome assembly by blocking NEK7-NLRP3 interaction,56,57,58,59 suggesting that tivantinib may inhibit inflammasome assembly by blocking NEK7-NLRP3 interaction. Unfortunately, our study found that tivantinib has no effect on NEK7-NLRP3 interaction. Furthermore, we found that tivantinib directly blocked NLRP3 ATPase activity and subsequent NLRP3 oligomerization. However, the precise mechanism by which tivantinib regulates the ATPase activity of NLRP3 is still unclear, one possible reason is that tivantinib binds directly to NLRP3 and competitively prevents ATP from binding to NLRP3.
Aberrant activation of NLRP3 inflammasome has been reported to be implicated in the pathogenesis of various inflammatory diseases,1,2,3,5 suggesting that targeting NLRP3 is a promising strategy for treating these diseases. Although previous studies have shown that several small molecules, such as MCC950, CY-09, tranilast, oridonin, and 3,4-methylenedioxy-β-nitrostyrene (MNS), andrographolide and AI-44 inhibit NLRP3 and hold promise in treating inflammasome-driven diseases,18,19,20,21,22,60,61 most of these inhibitors are not available for clinical use. MNS, oridonin, and CY-09 has not been tested in clinical trials and its clinical safety has yet to be evaluated. IZD334, the derivative of MCC950, has only completed phase I clinical trials recently (NCT04086602). Furthermore, although IZD334 is modified from MCC950 and has a similar chemical structure, whether it also directly targets NLRP3 has yet to be demonstrated. Tranilast has reached phase II clinical trials to treat cryopyrin-associated periodic syndrome (CAPS) (NCT03923140), but the specific mechanism by which it targets NLRP3 and then blocks NLRP3 oligomeric remains unclear. Our study identified that tivantinib, a remarkably safe anticancer agent that has reached phase III clinical trials for the treatment of solid tumors, not only directly targets NLRP3 and inhibits NLRP3 oligomerization and inflammasome activation by blocking its ATPase activity, but also effectively alleviated LPS-induced systemic inflammation, MSU-induced peritonitis, and Con A-induced ALI. Furthermore, tivantinib also has remarkable preventive and therapeutic effects on the EAE mouse model at a dose of 10 mg/kg (equivalent to a dosage of 0.9 mg/kg/day in humans) every 2 days. Previous clinical trials have demonstrated that tivantinib is a highly safe drug that is well tolerated by most patients, even at doses up to 360 mg twice daily continuously in 28-day.32 Therefore, tivantinib is a promising clinical drug for the treatment of the above diseases. Importantly, whether tivantinib has an efficient pharmacological effect on other inflammasome-driven diseases, such as gout, atherosclerosis, type 2 diabetes, and Alzheimer’s disease, needs to be further studied.
Tivantinib is a selective non-ATP competitive inhibitor of c-Met, which blocks proliferation by causing the arrest of cell growth.35 In addition, tivantinib has been found to inhibit mitosis in an c-Met independent manner,33 suggesting tivantinib may have other therapeutic targets. Our data showed that tivantinib prevents inflammatory responses by inhibiting NLRP3 inflammasome, indicating that NLRP3 may be a new target for tivantinib. Moreover, although tivantinib as an antitumor agent has been shown to be highly effective against liver cancer, lung cancer, and colon cancer,29,30,31 its role in other diseases, especially inflammatory diseases, is rarely reported. Our study found that tivantinib can inhibit inflammatory response and alleviate systemic inflammation, peritonitis, ALI, and EAE in vivo. Considering that tivantinib has a significant and specific inhibitory effect on NLRP3 inflammasome activation both in vitro and in vivo, and has high safety in clinical trials, our studies suggest that tivantinib has excellent therapeutic potential in NLRP3 inflammasome-driven diseases.
Limitations of the study
The biggest limitation of this study is whether Tivantinib can alleviate inflammatory disease in the clinic. Although we demonstrated that Tivantinib has good safety as a clinical agent, and also revealed its anti-inflammatory effect in human cells and mouse models, its potential clinical application in inflammatory diseases remains to be further explored. Furthermore, NLRP3 inflammasome have been found to be closely related to the occurrence and development of a variety of inflammatory diseases. In addition to sepsis, peritonitis, liver injury, and EAE, whether Tivantinib, as an NLRP3 targeted inhibitor, can alleviate other inflammatory diseases is also worth studying.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse IL-1β | R&D Systems | Cat#AF-401-NA; AB_416684 |
| anti-NLRP3 | Adipogen | Cat#AG-20B-0014; AB_2885199 |
| anti-mouse caspase-1 (p20) | Adipogen | Cat#AG-20B-0042; AB_2490248 |
| Anti-β-actin | Abmart | Cat#P30002; AB_2222847 |
| Anti-human IL-1β | Proteintech | Cat#60136-1-Ig; AB_10597543 |
| Anti-human caspase-1 | Cell Signaling | Cat#2225; AB_2243894 |
| Anti-ASC | Santa Cruz | Cat#sc-22514-R; AB_2174874 |
| anti-NEK7 | Santa Cruz | Cat#SC-50756; AB_2235871 |
| Anti-Flag | Sigma | Cat#F2555; AB_796202 |
| anti-VSV | Sigma | Cat#V4888; AB_261872 |
| Chemicals, peptides, and recombinant proteins | ||
| Tivantinib | TargetMol | Cat#T6117 |
| NVP-BVU972 | TargetMol | Cat#T2680 |
| AMG-337 | TargetMol | Cat#T3209 |
| SGX-523 | TargetMol | Cat#T2293 |
| PMA (phorbol-12-myristate-13-acetate) | Sigma | Cat# P8139 |
| Poly (dA:dT) | Sigma | Cat#86828-69-5 |
| Nigericin | Sigma | Cat# N7143 |
| ATP | Sigma | Cat#34369-07-8 |
| MSU | Sigma | Cat#69-93-2 |
| protein G agarose | Sigma | Cat# P7700 |
| Pam3CSK4 | Invitrogen | Cat#112208-00-1 |
| ultrapure LPS | Invitrogen | Cat#297-473-0 |
| MitoSOX | Invitrogen | Cat#M36008 |
| Lipofectamine 2000 | Invitrogen | Cat#11668019 |
| DAPI | Invitrogen | Cat# D21490 |
| MQAE | Invitrogen | Cat#162558-52-3 |
| Critical commercial assays | ||
| LDH Cytotoxicity Assay Kit | Beyotime | Cat#C0017 |
| ADP-Glo Kinase Assay Kit | Promega | Cat#V6930 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | ATCC CRL-3216 |
| THP-1 | ATCC | ATCC TIB-202 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| Mouse IL-1β forward | Sangon | 5′TGCCACCTTTTGACAGTGATG-3′ |
| Mouse IL-1β reverse | Sangon | 5′AAGGTCCACGGGAAAGACAC-3′ |
| Mouse Gapdh forward | Sangon | 5′GGTGAAGGTCGGTGTGAACG-3′ |
| Mouse Gapdh reverse | Sangon | 5′CTCGCTCCTGGAAGATGGTG-3′ |
| Software and algorithms | ||
| ImageJ (v1.51) | ImageJ Software | ImageJ (nih.gov) |
| GraphPad Prism 8.0 | GraphPad Software | Prism - GraphPad |
| FlowJo software (v10.0.7) Article | FlowJo software | Download FlowJo | FlowJo, LLC |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wen Xu (xuwen6779@ustc.edu.cn).
Materials availability
This study did not generate new materials or reagents.
Experimental model and subject details
All studies were conducted using male C57BL/6J mice over 8 weeks of age and were purchased from Model Animal Research Center of Nanjing University. All mice were specific pathogen-free and housed under 12 h light/dark cycle at 22–24°C with unrestricted access to food and water. All animal experiment protocols and procedures were approved by the Ethics Committee of Jiangnan University.
Method details
Cell culture
Primary bone marrow-derived macrophages were derived from C57BL/6 mice aged 8-10 weeks and cultured for 4 days in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (P/S), 1 mM sodium pyruvate, 2 mM L-glutamine and 20 ng/mL M-CSF (Novoprotein). HEK293T cells were cultured in DMEM supplemented with 10% FBS and 1% P/S. THP-1 cells were cultured in RPMI 1640 medium containing 10% FBS and 1% P/S. THP-1 and HEK-293T cells were routinely tested for mycoplasma contamination.
Inflammasome activation assays
BMDMs and THP-1 cells (pretreated with 100 nM PMA for 4 h) were seeded at 5 × 105 cells/ml in 12-well plates. The overnight medium was replaced with Opti-MEM supplemented with 1% FBS and ultrapure LPS (50 ng/mL) for 3 h or Pam3CSK4 (400 ng/mL) for 4 h. After priming, cells were treated with DMSO or inhibitors for 30 min and then stimulated with MSU (150 μg/mL), Alum (300 μg/mL), S. typhimurium infection and poly (dA:dT) (0.5 μg/mL) transfection for 4 h or with ATP (2.5 mM) and nigericin (3 μM) for 30 min. Cells were transfected with LPS (500 ng/mL) for 16 h through the use of Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). Supernatants were analyzed by ELISA kits according to the manufacturer’s instructions (R&D Systems) or by immunoblotting. LDH release was measured using the LDH Cytotoxicity Assay Kit (Beyotime).
siRNA interference
BMDMs were seeded at 3 × 105 cells/ml in 12-well plates. The overnight medium was replaced with Opti-MEM and then were transfected with 50 nM siRNA by Lipofectamine 2000 according to the manufacturer’s guidelines (Invitrogen). The siRNA sequences were chemically synthesized by GenePharma and their sequences were as follows: cMET siRNA-1 (GCCAATCTTGCTAAGCAAA), cMET siRNA-2 (GCTACTTATGTGAATGTAA).
Confocal microscopy
BMDMs were seeded at 3 × 105 cells/ml on coverslips (Thermo Fisher Scientific) in 12-well plates. The overnight medium was replaced with Opti-MEM supplemented with 1% FBS and ultrapure LPS (50 ng/mL) for 3 h. After priming, cells were treated with inhibitors for 30 min, and then stimulated with nigericin and stained with MitoSOX (5 μM). Removing the medium and washing the cells with PBS for three times. Fixing the cells with 4% PFA in PBS for 15 min and then washing three times with PBST. Confocal microscopy analyses were carried out using a Zeiss LSM700.
Intracellular potassium and chloride concentrations assay
For accurate measurement of the intracellular potassium, BMDMs were seeded at 5 × 105 cells/ml in 6-well plates. The overnight medium was replaced with Opti-MEM supplemented with 1% FBS and ultrapure LPS (50 ng/mL) for 3 h. After priming, cells were treated with DMSO or different concentrations of tivantinib for 30 min and then stimulated with nigericin (3 μM) for 30 min. The culture supernatant was removed and the cells were lysed with 3% ultrapure HNO3. Intracellular K+ measurements were performed by inductively coupled plasma optical emission spectrometry (ICP-OES) with a PerkinElmer Optima 2000 DV spectrometer using yttrium as the internal standard.
For accurate measurement of the intracellular chloride, BMDMs were seeded at 5 × 105 cells/ml in 12-well plates. The overnight medium was replaced with Opti-MEM supplemented with 1% FBS and ultrapure LPS (50 ng/mL) for 3 h. After priming, cells were treated with DMSO or different concentrations of tivantinib for 30 min and then stimulated with nigericin (3 μM) for 20 min. The culture supernatant was removed and the cells were lysed with ddH2O. Then, cell lysate was collected and centrifuged at 10,000 × g for 5 min 50 μL supernatants was then transferred the to a 1.5 mL EP tube and mixed with 50 μL MQAE (10 μM). The absorbance was tested by BioTek Multi-Mode Microplate Readers (Synergy2).
Immunoprecipitation
For the endogenous interaction assay, BMDMs were seeded in 6-well plates and stimulated with nigericin. Then, the BMDMs lysed by NP-40 lysis buffer with complete protease inhibitor. The cell lysates were incubated overnight at 4°C with anti-ASC or anti-NEK7 antibodies and Protein G Mag Sepharose (GE Healthcare). The antibody-bound proteins were precipitated by protein G beads and subjected to western blotting analysis.
For the exogenous interaction assay, HEK-293T cells were seeded in 6-well plates and transfected with plasmids via Lipofectamine 2000. After 24 h, cells were collected and lysed by NP-40 lysis buffer with complete protease inhibitor. Extracts were immunoprecipitated with anti-Flag antibody and beads and then were assessed by western blotting.
Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)
NLRP3 oligomerization was analyzed as described.20 Briefly, BMDMs were lysed with triton X-100 lysis buffer (0.5% Triton X-100, 50 mM Tris–Hcl, 10% glycerol, 150 mM NaCl, 1 mM PMSF and protease inhibitor cocktail). Cell lysate was collected and then resuspended in sample buffer (0.5× TBE, 2% SDS, 10% glycerol and 0.0025% bromophenol blue). Loading the sample onto a vertical 1.5% agarose gel and electrophoresis in the running buffer (1× TBE and 0.1% SDS) for 1 h with a constant voltage of 80 V at 4°C. and then the proteins were transferred to Immobilon membrane (Millipore) for immunoblotting.
Quantitative real-time PCR
Total RNA was isolated from BMDMs or cervical spinal cords by extraction with TRIzol reagent (Takara). RNA (800 ng) of each sample was used for reverse transcription with Thermo Scientific RevertAid MM (Thermo Fisher SCIENTIFIC) according to the manufacturer’s instructions. qPCR was performed using SYBR Green premix (Takara Bio) in a Roche LightCycler 96. GAPDH was used as the reference gene. The sequences of the gene-specific primers used were as follows: Mouse IL-1β forward, TGCCACCTTTTGACAGTGATG; Mouse IL-1β reverse, AAGGTCCACGGGAAAGACAC; Mouse Gapdh forward, GGTGAAGGTCGGTGTGAACG, Mouse Gapdh reverse, CTCGCTCCTGGAAGATGGTG.
ASC oligomerization assay
BMDMs were seeded at 5 × 105 cells/ml in 6-well plates. The overnight medium was replaced with Opti-MEM supplemented with 1% FBS and ultrapure LPS (50 ng/mL) for 3 h. After priming, cells were treated with DMSO or tivantinib for 30 min and then stimulated with nigericin (3 μM) for 30 min. Removing the supernatants and rinsing the cells with ice-cold PBS, and then lysing the cells with NP-40 for 30 min. Cell lysate was collected and centrifuged at 330 × g for 10 min at 4°C. The pellets were washed in 1 mL ice-cold PBS and resuspended in 500 μL PBS. 2 mM disuccinimydylsuberate (DSS) was added to the resuspended pellets and co-incubated with rotation at room temperature for 30 min. Samples were then centrifuged at 330 × g for 10 min at 4°C. The cross-linked pellets were resuspended in 30 μL sample buffer and then were boiled and analyzed by western blotting.
NLRP3 ATPase activity assay
Purified recombinant human NLRP3 was co-incubation with different concentrations of tivantinib at 37°C for 15 min in the reaction buffer. Add ultra-pure ATP (250 μM) to the mixture and incubate at 37°C for another 40 min. The amount of ATP converted into adenosine diphosphate (ADP) was determined by luminescent ADP detection with ADP-Glo Kinase Assay Kit (Promega) according to the manufacturer’s protocol. The results were expressed as percentage of residual enzyme activity to the vehicle-treated enzyme.
Microscale thermophoresis assay
KD values were measured using Monolith NT.115 instrument (NanoTemper Technologies). Tivantinib (concentrations from 5 mM to 3.1361 nM) were incubated with 200 nM purified His-GFP-NLRP3 protein for 30 min in assay buffer (50 mM HEPES, 10 mM MgCl2, 100 mM NaCl (pH 7.5), and 0.05% Tween 20). The samples were loaded into Nano Temper glass capillaries, and MST was performed using an LED power of 100% and an MST power of 80%. KD values were calculated using the mass action equation with Nano Temper software from duplicate reads of an experiment.
Protein expression and purification
For NLRP3 protein purification, HEK-293T cells were transfected with plasmids encoding Flag-NLRP3 for 48 h. The culture medium was removed and cells were collected, and then incubated with Flag-M2 monoclonal antibody–agarose beads for 4 h at 4°C on rotation. The beads were then incubated in elution buffer (50 mM HEPES, pH 7.4, 500 mM NaCl, 0.1% CHAPS and 100 mg/mL Flag peptide) for 2 h at 4°C on rotation. The eluted fractions were concentrated by centrifugation and then filtered using an ultrafiltration device (Merck Millipore) to remove proteins of less than 100 kDa.
LPS-induced systemic inflammation
Ten-week-old male C57BL/6 mice were intraperitoneally injected with vehicle or tivantinib (20 mg/kg). One hour later, intraperitoneally injected with LPS (Sigma Aldrich) to induce systemic inflammation. Serum samples were collected 4 h later, and serum levels of IL-1β and TNF-α were measured by ELISA according to the manufacturer’s protocol.
MSU-induced peritonitis
Ten-week-old male C57BL/6 mice were intraperitoneally injected with vehicle or tivantinib (20 mg/kg) 1 h before intraperitoneally with MSU (Sigma Aldrich). After 6 h, mice were sacrificed and peritoneal cavities underwent lavage with 5 mL PBS. Peritoneal lavage fluid was assessed by flow cytometry (BD) with the neutrophil markers Ly6G and CD11b for analysis of the recruitment of neutrophils and determined IL-1β production by ELISA.
ConA-induced acute liver injury mice models
Ten-week-old male C57BL/6 mice were intraperitoneally injected with vehicle or tivantinib (20 mg/kg) 1 h before tail vein with Con A (Sigma Aldrich). For survival analysis, mice were monitored for 72 h after Con A (30 mg/kg) injection. For serum alanine aminotransferase (ALT) and IL-1β assay, serum was collected via mouse eyeball 4 h after Con A (15 mg/kg) injection. For H&E staining analysis, liver specimens were collected 24 h after Con A (15 mg/kg) injection.
Induction and assessment of EAE
8-week-old male C57BL/6 mice were injected subcutaneously in the dorsal flanks with 3 mg/ml MOG35-55 peptide (300 μg per mouse) in CFA containing 5 mg/ml heat-killed Mycobacterium tuberculosis (500 μg per mouse) on day 0. Mice were injected with Pertussis toxin (150 ng per mouse, list labs) on days 0 and day 2 through the retroorbital vein. In Tivantinib prevention trails, Tivantinib was intraperitoneally injected into mice (10 mg/kg) beginning at day 0 to day 22 every two days. In Tivantinib treatment trails, Tivantinib was intraperitoneally injected into mice (10 mg/kg) beginning at disease onset (day 10) to day 30 every two days. Control group mice were injected with vehicle containing 95% PBS and 5% DMSO at the same time points. Disease severity was scored on a scale of 0–5 as follows: 0, no abnormalities; 1, limp tail or waddling gait with tail tonicity; 2, wobbly gait; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, death. To analyze CNS infiltrates, both the brain and spinal cord were harvested from mice perfused with PBS, and mononuclear cells were isolated by 30% Percoll separation.
Histological analysis
The cervical spinal cords were fixed in 4% paraformaldehyde (PFA) overnight and sliced after embedding in paraffin. Sections were prepared and stained with hematoxylin &eosin (H&E) and Luxol Fast Blue (LFB). Slides were examined under a Nikon ECL IPSE Ci biological microscope, and images were acquired with a Nikon DS-U3 color digital camera.
Quantification and statistical analysis
Data are presented as the means ± SEMs. Statistical analysis was carried out with the unpaired t-test for two groups or one-way ANOVA(GraphPad Software) using for multiple groups with all data points showing a normal distribution. No data points were excluded. Sample sizes were selected on the basis of preliminary results to ensure adequate power. Data were considered significant when the p value was <0.05.
Acknowledgments
We thank Dr. Rongbin Zhou (University of Science and Technology of China) for his helpful and insightful comments on the article, and Lingyun Xia (Westlake University) for providing the computing resources for docking analysis. This research was supported by the National Natural Science Foundation of China (82202038, 32201159) and the Natural Science Foundation of Jiangsu Province (BK20221085).
Author contributions
Wen Xu, Yun Guo, and Yan Zhou performed the experiments of this work; Wen Xu, Yun Guo, Yingli Luo, and Yi Huang designed the research. Yingli Luo and Yi Huang wrote the article. Yi Huang supervised the project.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 1, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106062.
Contributor Information
Yi Huang, Email: hy527@jiangnan.edu.cn.
Yingli Luo, Email: luoyingl@mail.ustc.edu.cn.
Wen Xu, Email: xuwen6779@ustc.edu.cn.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon reasonable request.
References
- 1.Heneka M.T., Kummer M.P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.C., et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 4.Swanson K.V., Deng M., Ting J.P.Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 6.Martinon F., Mayor A., Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 7.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 8.Kayagaki N., Stowe I.B., Lee B.L., O'Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T., et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y., Xu W., Zhou R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021;18:2114–2127. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu W., Huang Y. Regulation of inflammatory cell death by phosphorylation. Front. Immunol. 2022;13:851169. doi: 10.3389/fimmu.2022.851169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masters S.L., Dunne A., Subramanian S.L., Hull R.L., Tannahill G.M., Sharp F.A., Becker C., Franchi L., Yoshihara E., Chen Z., et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue M., Williams K.L., Gunn M.D., Shinohara M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halle A., Hornung V., Petzold G.C., Stewart C.R., Monks B.G., Reinheckel T., Fitzgerald K.A., Latz E., Moore K.J., Golenbock D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duewell P., Kono H., Rayner K.J., Sirois C.M., Vladimer G., Bauernfeind F.G., Abela G.S., Franchi L., Nuñez G., Schnurr M., et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z., Zhang S., Xiao Y., Zhang W., Wu S., Qin T., Yue Y., Qian W., Li L. NLRP3 inflammasome and inflammatory diseases. Oxid. Med. Cell. Longev. 2020;2020:4063562. doi: 10.1155/2020/4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng J., Xie X., Feng X.L., Xu L., Han J.B., Yu D., Zou Q.C., Liu Q., Li X., Ma G., et al. Specific inhibition of the NLRP3 inflammasome suppresses immune overactivation and alleviates COVID-19 like pathology in mice. EBioMedicine. 2022;75:103803. doi: 10.1016/j.ebiom.2021.103803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q., Su L.Y., Sun C., Jiao L., Miao Y., Xu M., Luo R., Zuo X., Zhou R., Zheng P., et al. Melatonin alleviates morphine analgesic tolerance in mice by decreasing NLRP3 inflammasome activation. Redox Biol. 2020;34:101560. doi: 10.1016/j.redox.2020.101560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coll R.C., Robertson A.A.B., Chae J.J., Higgins S.C., Muñoz-Planillo R., Inserra M.C., Vetter I., Dungan L.S., Monks B.G., Stutz A., et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H., He H., Chen Y., Huang W., Cheng J., Ye J., Wang A., Tao J., Wang C., Liu Q., et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017;214:3219–3238. doi: 10.1084/jem.20171419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y., Jiang H., Chen Y., Wang X., Yang Y., Tao J., Deng X., Liang G., Zhang H., Jiang W., Zhou R. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018;10:e8689. doi: 10.15252/emmm.201708689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He H., Jiang H., Chen Y., Ye J., Wang A., Wang C., Liu Q., Liang G., Deng X., Jiang W., Zhou R. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018;9:2550. doi: 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Y., Varadarajan S., Muñoz-Planillo R., Burberry A., Nakamura Y., Núñez G. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014;289:1142–1150. doi: 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lachmann H.J., Kone-Paut I., Kuemmerle-Deschner J.B., Leslie K.S., Hachulla E., Quartier P., Gitton X., Widmer A., Patel N., Hawkins P.N., Canakinumab in CAPS Study Group Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009;360:2416–2425. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 24.Brogan P.A., Hofer M., Kuemmerle-Deschner J.B., Koné-Paut I., Roesler J., Kallinich T., Horneff G., Calvo Penadés I., Sevilla-Perez B., Goffin L., et al. Rapid and sustained long-term efficacy and safety of canakinumab in patients with cryopyrin-associated periodic syndrome ages five years and younger. Arthritis Rheumatol. 2019;71:1955–1963. doi: 10.1002/art.41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jesus A.A., Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu. Rev. Med. 2014;65:223–244. doi: 10.1146/annurev-med-061512-150641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willingham S.B., Allen I.C., Bergstralh D.T., Brickey W.J., Huang M.T.H., Taxman D.J., Duncan J.A., Ting J.P.Y. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y., Yang J., Shi J., Gong Y.N., Lu Q., Xu H., Liu L., Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 29.Rimassa L., Assenat E., Peck-Radosavljevic M., Pracht M., Zagonel V., Mathurin P., Rota Caremoli E., Porta C., Daniele B., Bolondi L., et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018;19:682–693. doi: 10.1016/S1470-2045(18)30146-3. [DOI] [PubMed] [Google Scholar]
- 30.Scagliotti G.V., Shuster D., Orlov S., von Pawel J., Shepherd F.A., Ross J.S., Wang Q., Schwartz B., Akerley W. Tivantinib in combination with erlotinib versus erlotinib alone for EGFR-mutant NSCLC: an exploratory analysis of the phase 3 MARQUEE study. J. Thorac. Oncol. 2018;13:849–854. doi: 10.1016/j.jtho.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Rimassa L., Bozzarelli S., Pietrantonio F., Cordio S., Lonardi S., Toppo L., Zaniboni A., Bordonaro R., Di Bartolomeo M., Tomasello G., et al. Phase II study of tivantinib and cetuximab in patients with KRAS wild-type metastatic colorectal cancer with acquired resistance to EGFR inhibitors and emergence of MET overexpression: lesson learned for future trials with EGFR/MET dual inhibition. Clin. Colorectal Cancer. 2019;18:125–132.e2. doi: 10.1016/j.clcc.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Bagai R., Fan W., Ma P.C. ARQ-197, an oral small-molecule inhibitor of c-Met for the treatment of solid tumors. Idrugs. 2010;13:404–414. [PubMed] [Google Scholar]
- 33.Porta C., Giglione P., Ferrari A., Reversi F., Liguigli W., Imarisio I., Ganini C. Tivantinib (ARQ197) in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2015;15:615–622. doi: 10.1586/14737140.2015.1050383. [DOI] [PubMed] [Google Scholar]
- 34.Puzanov I., Sosman J., Santoro A., Saif M.W., Goff L., Dy G.K., Zucali P., Means-Powell J.A., Ma W.W., Simonelli M., et al. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Invest. New Drugs. 2015;33:159–168. doi: 10.1007/s10637-014-0167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katayama R., Aoyama A., Yamori T., Qi J., Oh-hara T., Song Y., Engelman J.A., Fujita N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73:3087–3096. doi: 10.1158/0008-5472.CAN-12-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rühl S., Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015;45:2927–2936. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 37.Moretti J., Jia B., Hutchins Z., Roy S., Yip H., Wu J., Shan M., Jaffrey S.R., Coers J., Blander J.M. Caspase-11 interaction with NLRP3 potentiates the noncanonical activation of the NLRP3 inflammasome. Nat. Immunol. 2022;23:705–717. doi: 10.1038/s41590-022-01192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin X., Zhou R., Huang Y. Role of inflammasomes in HIV-1 infection and treatment. Trends Mol. Med. 2022;28:421–434. doi: 10.1016/j.molmed.2022.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B.L., Rajendiran T.M., Núñez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 41.Domingo-Fernández R., Coll R.C., Kearney J., Breit S., O'Neill L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017;292:12077–12087. doi: 10.1074/jbc.M117.797126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang T., Lang X., Xu C., Wang X., Gong T., Yang Y., Cui J., Bai L., Wang J., Jiang W., Zhou R. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017;8:202. doi: 10.1038/s41467-017-00227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stutz A., Horvath G.L., Monks B.G., Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- 44.He Y., Zeng M.Y., Yang D., Motro B., Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmid-Burgk J.L., Chauhan D., Schmidt T., Ebert T.S., Reinhardt J., Endl E., Hornung V. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2016;291:103–109. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi H., Wang Y., Li X., Zhan X., Tang M., Fina M., Su L., Pratt D., Bu C.H., Hildebrand S., et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagar A., Rahman T., Harton J.A. The ASC speck and NLRP3 inflammasome function are spatially and temporally distinct. Front. Immunol. 2021;12:752482. doi: 10.3389/fimmu.2021.752482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duncan J.A., Bergstralh D.T., Wang Y., Willingham S.B., Ye Z., Zimmermann A.G., Ting J.P.Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA. 2007;104:8041–8046. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y., Franchi L., Núñez G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J. Immunol. 2013;190:334–339. doi: 10.4049/jimmunol.1202737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong T., Wang X., Yang Y., Yan Y., Yu C., Zhou R., Jiang W. Plant lectins activate the NLRP3 inflammasome to promote inflammatory disorders. J. Immunol. 2017;198:2082–2092. doi: 10.4049/jimmunol.1600145. [DOI] [PubMed] [Google Scholar]
- 51.Gris D., Ye Z., Iocca H.A., Wen H., Craven R.R., Gris P., Huang M., Schneider M., Miller S.D., Ting J.P.Y. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010;185:974–981. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green J.P., Yu S., Martín-Sánchez F., Pelegrin P., Lopez-Castejon G., Lawrence C.B., Brough D. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. USA. 2018;115:E9371–E9380. doi: 10.1073/pnas.1812744115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo W., Liu W., Chen Z., Gu Y., Peng S., Shen L., Shen Y., Wang X., Feng G.S., Sun Y., Xu Q. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat. Commun. 2017;8:2168. doi: 10.1038/s41467-017-02351-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jo E.K., Kim J.K., Shin D.M., Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016;13:148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y., Wang H., Hao Y., Lin H., Dong M., Ye J., Song L., Wang Y., Li Q., Shan B., et al. Myeloid PTEN promotes chemotherapy-induced NLRP3-inflammasome activation and antitumour immunity. Nat. Cell Biol. 2020;22:716–727. doi: 10.1038/s41556-020-0510-3. [DOI] [PubMed] [Google Scholar]
- 56.Li Q., Feng H., Wang H., Wang Y., Mou W., Xu G., Zhang P., Li R., Shi W., Wang Z., et al. Licochalcone B specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep. 2022;23:e53499. doi: 10.15252/embr.202153499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li C., Lin H., He H., Ma M., Jiang W., Zhou R. Inhibition of the NLRP3 inflammasome activation by manoalide ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Cell Dev. Biol. 2022;10:822236. doi: 10.3389/fcell.2022.822236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin H., Yang M., Li C., Lin B., Deng X., He H., Zhou R. An RRx-001 analogue with potent anti-NLRP3 inflammasome activity but without high-energy nitro functional groups. Front. Pharmacol. 2022;13:822833. doi: 10.3389/fphar.2022.822833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y., He H., Lin B., Chen Y., Deng X., Jiang W., Zhou R. RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell. Mol. Immunol. 2021;18:1425–1436. doi: 10.1038/s41423-021-00683-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo W., Sun Y., Liu W., Wu X., Guo L., Cai P., Wu X., Wu X., Shen Y., Shu Y., et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10:972–985. doi: 10.4161/auto.28374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu W., Guo W., Zhu Y., Peng S., Zheng W., Zhang C., Shao F., Zhu Y., Hang N., Kong L., et al. Targeting peroxiredoxin 1 by a curcumin analogue, AI-44, inhibits NLRP3 inflammasome activation and attenuates lipopolysaccharide-induced sepsis in mice. J. Immunol. 2018;201:2403–2413. doi: 10.4049/jimmunol.1700796. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon reasonable request.