

Abstract
To advance our ability to predict impacts of the protein scaffold on catalysis, robust classification schemes to define features of proteins that will influence reactivity are needed. One of these features is a protein's metal‐binding ability, as metals are critical to catalytic conversion by metalloenzymes. As a step toward realizing this goal, we used convolutional neural networks (CNNs) to enable the classification of a metal cofactor binding pocket within a protein scaffold. CNNs enable images to be classified based on multiple levels of detail in the image, from edges and corners to entire objects, and can provide rapid classification. First, six CNN models were fine‐tuned to classify the 20 standard amino acids to choose a performant model for amino acid classification. This model was then trained in two parallel efforts: to classify a 2D image of the environment within a given radius of the central metal binding site, either an Fe ion or a [2Fe‐2S] cofactor, with the metal visible (effort 1) or the metal hidden (effort 2). We further used two sub‐classifications of the [2Fe‐2S] cofactor: (1) a standard [2Fe‐2S] cofactor and (2) a Rieske [2Fe‐2S] cofactor. The accuracy for the model correctly identifying all three defined features was >95%, despite our perception of the increased challenge of the metalloenzyme identification. This demonstrates that machine learning methodology to classify and distinguish similar metal‐binding sites, even in the absence of a visible cofactor, is indeed possible and offers an additional tool for metal‐binding site identification in proteins.
Keywords: amino acids, convolutional neural network, image classification, iron–sulfur, metal‐binding sites, metalloenzyme, Rieske
1. INTRODUCTION
Biochemical reactions in nature are carried out by enzymes, which have precise control over the environment in which a specific chemical transformation will occur. Metalloenzymes are enzymes that contain at least one metal ion, and are found extensively in natural systems, with 40% of known enzyme structures containing metals (Andreini et al., 2008) and are an important aspect in many catalytic conversions (Fernandes et al., 2019; Holm et al., 1996; Warshel et al., 2006; Williams, 1971; Wolfenden and Snider, 2001). A significant amount of work has focused on creating artificial metalloenzymes because of the high specificity, selectivity, and ability of native metalloenzymes to function under mild conditions while employing non‐precious metals, as well as their potential to accelerate nonnatural reactions under mild conditions (Himiyama and Okamoto, 2020; Röthlisberger et al., 2008). Only a handful of design principles have been well established but, based on the often‐observed superior activity of enzymes over synthetic catalysts, more design principles have yet to be discovered. The ability to extract these design principles could advance catalysis in many different areas.
While there are many aspects to the design of artificial metalloenzymes (Roelfes, 2019; Schwizer et al., 2018; Yu et al., 2014; Thomas and Ward, 2005; Hosseinzadeh et al., 2016; Slater et al., 2018; Laureanti et al., 2019), predicting a protein's metal‐binding ability from amino acid sequence data is a key element. As a starting point, the structure of an artificial protein can be predicted with AlphaFold (Jumper et al., 2021). AlphaFold employs a 3D convolutional neural network (CNN), a deep learning architecture, to produce protein structures from protein sequences with accuracy similar to experimental structures (Jumper et al., 2021). However, AlphaFold does not predict metal binding sites.
Numerous methods to predict metal‐binding sites exist, yet all have inherent limitations. First, highly conserved amino acid sequences of protein metal cofactors alone have been used to predict metal binding for metalloenzymes. Metal‐binding sites often employ similar amino acid sequence motifs (Passerini and Frasconi, 2004). For example, in [2Fe‐2S] cluster containing proteins, multiple sequence motifs exist between the bacteria, plantae, and vertebrae with the most common bacterial motif of C‐X5‐C‐X2‐C‐X35‐C motif (Agar et al., 2003). As another example, c type cytochromes have a predominately C‐X2‐C‐H sequence motif where C is cysteine, X is any amino acid, and H is histidine (Andreeva, 2011). Databases built from conserved sequences alone are currently in use by systems such as MetalPredator. This system only predicts Fe‐S sites by comparing input sequences to the known binding sequence in a database for metal binding sites in the MetalPDB (Andreini et al., 2012). Importantly, MetalPredator was found to accurately predict 75.5% of known metal sites in the MetalPDB database (Valasatava et al., 2016).
In addition to amino acid sequence motifs, there are many other characteristics of a metal‐binding site that experts can use to both identify and classify possible sites. Residues involved in catalysis have been evaluated as one approach (Bartlett et al., 2002). An important characteristic is the immediate environment surrounding the Fe binding sites, which have been previously shown to be different for Fe, Cu, Mn, and Zn. The disparity leads to a binding site with features which can be defined, including the amino acid residues with the necessary functional groups, charge, orientation, and solvent accessibility, among other features (Karlin et al., 1997). While possible to identify manually, as demonstrated, this involves considerable time and the number of variables to consider can make it difficult to achieve high consistency or accuracy. In a recent example to try to accelerate identification of binding sites, only one feature, root‐mean‐square deviation of atomic positions (RMSD), was used to identify Zn and iron–sulfur ligand binding sites in AlphaFold structures. The RMSD value of ligands within proposed regions was calculated and the region was considered ligand binding if the RSMD values of a possible ligand orientation fell below a threshold (Wehrspan et al., 2022). The model from Wehrspan et al. was only moderately successful at locating [2Fe‐2S] binding sites, finding only 67% of the sites in the protein database UniProt (Consortium U, 2019), perhaps due the disulfide‐bonds between cysteines potentially being drawn incorrectly when using AlphaFold information. Improvement was observed for locating Zn binding sites while using experimentally derived data from UniProt as the input, with a recall of 84% (Wehrspan et al., 2022; Consortium U, 2019).
Recently, machine learning has been employed to classify metal‐binding sites, using expert‐defined features as inputs. Machine learning has also been used for related tasks, such as predicting enzyme mechanisms from sequence data (De Ferrari and Mitchell, 2014) and predicting catalytic residues based on the features of the environment (Bobadilla et al., 2007). These features might include physical descriptors of protein systems such as amino acid sequences and physicochemical properties such as polarity and surface tension (Bonetta and Valentino, 2020). These descriptors are ultimately features calculated from protein structural data. Although there are many advantages to using expert‐defined features, including the interpretability of the results, there are also disadvantages, as outlined in a recent paper by Torng and Altman (2017). First, creating features is a laborious and highly skilled process. Second, each biological question may require a different set of features. Finally, information is lost when protein structures are summarized with features, including the orientation information of the structures. For this reason, it can be useful to have a method that requires no feature input whatsoever (Torng and Altman, 2019). This is what image classification allows—classification without any information beyond the image.
In this work, for image classification, we will use a CNN, a deep learning architecture that has been used for more than 30 years for image classification. The input image can use pixels from either a 2D or 3D image (LeCun and Bengio, 1995). Some studies are successfully using CNNs on both 3D images, for functional site detection, including nitric oxide synthase and TRYPSIN‐like enzyme sites (Torng and Altman, 2019), and on 2D images of 3D structures, for protein structure classification (Nanni et al., 2020). Herein, we used a CNN model to classify 2D images of 3D metal‐binding sites in iron–sulfur proteins. The advantage of using 2D images rather than 3D images is that a 2D view of a 3D object has been found to be more accurate than 3D representations to classify 3D shapes with a neural network (Su et al., 2015). Nanni et al. (2020) also found that 2D representations of protein structures, using a limited number of 2D image rotations, can perform higher than the sequence/property analysis approach in classifying types of protein structure. They also compared the effects of various representations of the structures and found that the ball and stick representation was one of the top performing representations (Nanni et al., 2020). Other studies used mono‐colored 3D triangle mesh models from which the 2D images were obtained from different viewpoints (Qin et al., 2020).
In addition, when using 2D images, it is possible to take advantage of models pretrained on large amounts of image data found in datasets such as ImageNet, an image dataset with 15 million labeled images (Deng et al., 2009). It is often beneficial in image classification to use models that have been pretrained with large datasets to learn features that are generalizable to many tasks, and then to fine‐tune the model to the specific task, in our case, classifying amino acids and metal cofactor sites (Yosinski et al., 2014). Fine‐tuning involves training the pretrained model further on images pertaining to the specific task, and has been shown to increase the model's performance compared with starting with an untrained model (Yosinski et al., 2014).
We classified images of the environment around two iron‐based cofactors: a single iron (Fe) atom from a rubredoxin protein, and a [2Fe‐2S] cluster from ferredoxin proteins. The environments included amino acids within a 6.0 Å sphere of the metal cofactor. We chose to employ Fe cofactors as Fe is one of the top three most abundant metals found in enzymes (Andreini et al., 2008) and because the [2Fe‐2S] cofactor in particular is one of the most common but functionally diverse cofactor classes (Agar et al., 2003), providing an optimal training dataset. We performed three tasks that we believed would be progressively more difficult for the neural network model to classify. First, we classified 2D images of 3D amino acids. We employed each amino acid as a class, as a proof of principle of using a pretrained CNN architecture to classify protein structures, and to identify the most performant model. Then, this model was employed to classify images of the environment around a ligand in a metalloenzyme, including all amino acids within the 6.0 Å sphere of the following cofactors: Rieske [2Fe‐2S], standard [2Fe‐2S], and Fe atom. Two sets of images were prepared: with the cofactor present and with the cofactor hidden. The only items included in the image with the hidden cofactors were the ball and stick representation of the amino acids within the 6.0 Å radius for classification. Each task started with the original model, not the model after fine‐tuning the previous task.
2. RESULTS
2.1. Classification of all 20 standard amino acid residues
Our first task was to classify 2D images of random rotations of 20 standard amino acids, represented in a ball‐and‐stick model with color‐coded atoms and all backbone and side chains visible as shown in Figure 1. To find the best pretrained neural network model, we trained six different neural network models: ResNet (He et al., 2016), AlexNet (Krizhevsky et al., 2012), VGG (Simonyan and Zisserman, 2014), SqueezeNet (Iandola et al., 2016), DenseNet (Iandola et al., 2014), and Inception v3 (Szegedy et al., 2016). Training datasets consisted of 2D images of the 3D representation, generated in PyMol the molecular visualization software (Schrodinger, LLC, 2015), rotated at 36‐degree increments in the x, y, and z direction. After each model was trained, the best model was chosen by its performance with the validation set, and the performance metrics for that model were obtained with the test set. Both the validation dataset and the test dataset are composed of 250 independent 2D images of random rotations of the 3D PyMol representation of each amino acid. Of the six pre‐trained models we fine‐tuned to classify amino acids, ResNet proved to be the highest performing model for the full amino acid dataset, achieving 96% accuracy with the validation dataset, with a 96% average recall with 5% standard deviation and 95% average precision with a 4% standard deviation (Table 1a). The standard deviation represents differences between recall and precision of each class, the 20 amino acids.
FIGURE 1.
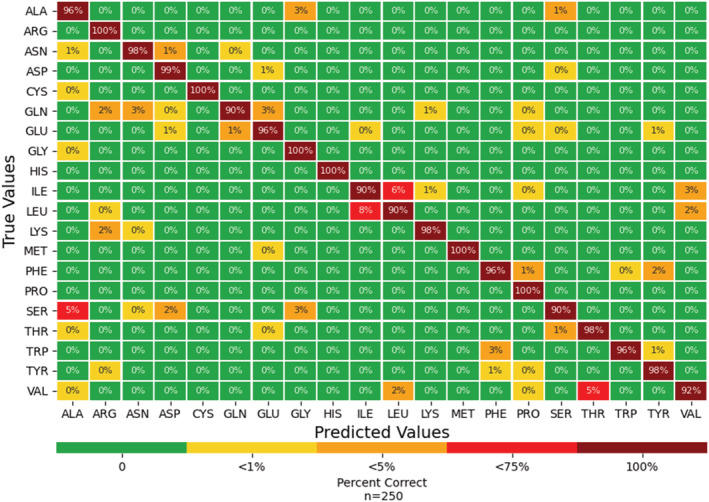
Confusion matrix from the ResNet model with the test dataset as input. Each amino acid employed 250 rotations.
TABLE 1a.
Recall (True Positive/[True Positive + False Negative]) for DenseNet, Inception, VGG, ResNet, AlexNet, and SqueezeNet for each of the 20 standard amino acids.
| Amino acid | Test set ResNet | ResNet | DenseNet | Inception | AlexNet | VGG | SqueezeNet |
|---|---|---|---|---|---|---|---|
| ALA | 96% | 96% | 97% | 94% | 93% | 98% | 92% |
| ARG | 100% | 100% | 99% | 100% | 98% | 99% | 98% |
| ASN | 98% | 98% | 98% | 96% | 98% | 98% | 90% |
| ASP | 99% | 99% | 91% | 85% | 97% | 96% | 100% |
| CYS | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| GLN | 90% | 85% | 88% | 80% | 92% | 84% | 94% |
| GLU | 96% | 97% | 98% | 97% | 96% | 86% | 98% |
| GLY | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| HIS | 100% | 100% | 100% | 100% | 99% | 100% | 97% |
| ILE | 90% | 85% | 91% | 73% | 70% | 61% | 57% |
| LEU | 90% | 87% | 85% | 81% | 78% | 69% | 65% |
| LYS | 98% | 99% | 100% | 100% | 97% | 98% | 100% |
| MET | 100% | 100% | 98% | 100% | 98% | 99% | 100% |
| PHE | 96% | 94% | 94% | 98% | 90% | 91% | 90% |
| PRO | 100% | 99% | 100% | 99% | 100% | 98% | 98% |
| SER | 90% | 95% | 98% | 99% | 96% | 95% | 92% |
| THR | 98% | 98% | 96% | 94% | 94% | 86% | 88% |
| TRP | 96% | 96% | 94% | 97% | 92% | 98% | 94% |
| TYR | 98% | 96% | 97% | 91% | 96% | 95% | 94% |
| VAL | 92% | 93% | 82% | 94% | 86% | 98% | 68% |
| Accuracy | 96% | 96% | 95% | 94% | 94% | 92% | 91% |
| Std. Dev. | 4% | 5% | 5% | 8% | 8% | 11% | 13% |
Note: Recall for ResNet was the highest of the neural networks investigated. Recall is shown from greatest to least from the left (ResNet) to right (SqueezeNet), with the first column showing recall for the test set. The remaining scores for the validation dataset are shown in bold. As the class sizes are equal, and the average recall is equal to the total accuracy. The standard deviation in the last row refers to the standard deviation of the recall scores per model.
Although we chose ResNet for further studies, other models performed with relative success, and none were statistically significantly different within error. Some of the models even surpassed that of ResNet for specific amino acid characterization, such as DenseNet or VGG for alanine predictions. Our continued use of ResNet for all future tasks was due to ResNet showing the highest accuracy.
We assessed the performance of ResNet with the test set. It is standard practice to use a separate dataset, a test dataset, to assess the performance of a model selected with the validation dataset due to the model having already been affected by the validation dataset. The test dataset had a 96% accuracy with the validation dataset, with a 96% average recall with a 4% standard deviation and 96% average precision with a 4% standard deviation (Table 1b). Of the 20 amino acids, 15 had a recall and precision above 95% of the time, and even the amino acid with the lowest recall, serine, had a recall of 90% and a precision of 98%. Example rotations that were misclassified are shown in Figure 2, and we believe these are due to the image blocking distinguishing atoms as discussed in more detail below.
TABLE 1b.
Precision (True Positive/[True Positive + False Positives]) for DenseNet, Inception, VGG, ResNet, AlexNet, and SqueezeNet for each of the 20 standard amino acids.
| Amino acid | Test set ResNet | ResNet | DenseNet | Inception | Alexnet | VGG | SqueezeNet |
|---|---|---|---|---|---|---|---|
| ALA | 95% | 91% | 98% | 100% | 89% | 98% | 95% |
| ARG | 97% | 100% | 98% | 98% | 99% | 100% | 97% |
| ASN | 95% | 92% | 87% | 98% | 87% | 99% | 95% |
| ASP | 99% | 98% | 98% | 92% | 89% | 82% | 99% |
| CYS | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| GLN | 100% | 99% | 100% | 97% | 93% | 94% | 100% |
| GLU | 92% | 98% | 97% | 88% | 96% | 89% | 92% |
| GLY | 97% | 98% | 97% | 98% | 97% | 94% | 97% |
| HIS | 100% | 99% | 100% | 100% | 99% | 98% | 100% |
| ILE | 87% | 87% | 94% | 81% | 81% | 58% | 87% |
| LEU | 88% | 90% | 89% | 81% | 89% | 70% | 88% |
| LYS | 96% | 94% | 97% | 95% | 97% | 96% | 96% |
| MET | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| PHE | 96% | 98% | 91% | 92% | 98% | 95% | 96% |
| PRO | 96% | 93% | 97% | 95% | 97% | 81% | 96% |
| SER | 98% | 87% | 79% | 92% | 91% | 95% | 98% |
| THR | 96% | 93% | 91% | 98% | 98% | 95% | 96% |
| TRP | 97% | 99% | 99% | 99% | 93% | 95% | 97% |
| TYR | 98% | 99% | 100% | 90% | 99% | 91% | 98% |
| VAL | 93% | 94% | 77% | 78% | 68% | 85% | 93% |
| Average precision | 96% | 95% | 94% | 94% | 93% | 91% | 96% |
| Std. Dev. | 4% | 4% | 7% | 7% | 8% | 11% | 4% |
Note: Precision for ResNet was the highest of the neural networks investigated. Precision is shown from greatest to least from the left (ResNet) to right (SqueezeNet), with the first column showing precision for the test set. The remaining scores for the validation dataset are shown in bold. The standard deviation in the last row refers to the standard deviation of the precision scores per model.
FIGURE 2.
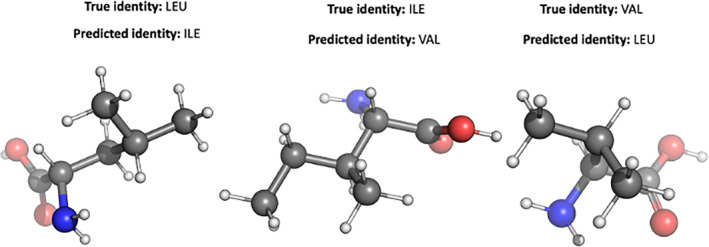
Improperly classified images from three structurally similar amino acids (valine, isoleucine, and leucine). The side chain branching patterns are masked at various rotations, leading to improper classification.
2.2. Classification of Fe and [2Fe‐2S] cofactors from metalloproteins
After finding that the pretrained ResNet model could be trained to distinguish amino acids, we fine‐tuned the original pretrained model to more difficult tasks: (1) classification of metal cofactors, and (2) specific folding/metal binding motifs within a class of metal cofactors. We employed ResNet as the base model since it performed well in the single amino acid investigation and fine‐tuned the pre‐trained original ResNet model with the new images. Using the Protein Data Bank (PDB), we found 120 unique proteins of Fe‐rubredoxin (FeRd) and 720 proteins of [2Fe‐2S] cofactor containing ferredoxins. We further split the [2Fe‐2S] ID's into the two sub‐categories: standard and Rieske cofactors with 474 and 253 unique proteins within each group, respectively. A small number of proteins contain multiple types of relevant cofactors and so the protein, but not the cofactors, are included in multiple groups.
Figure 3 allows a visual comparison of all metal cofactors employed and the connecting ligands, which consists of the: (1) Fe cofactor with four connecting cysteines; (2) standard [2Fe‐2S] with four connecting cysteines; and (3) Rieske [2Fe‐2S] cluster containing two cysteine ligands on one iron atom and two histidine ligands on the other iron atom. The bottom panel shows the cofactors as they appear in PyMol using the Crystallographic Information File (CIF) files from the PDB. The bonding information is often incorrect in these downloaded files. This could be corrected manually, but we wanted this workflow to be as automated as possible and we achieved a high accuracy despite not correcting the bonding information.
FIGURE 3.
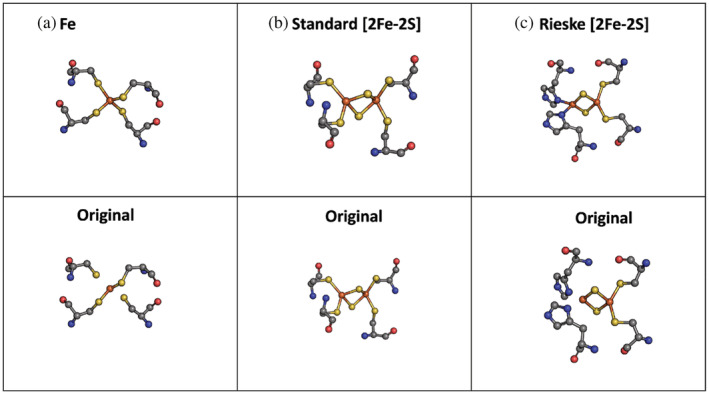
Metal cofactors employed in this study. (Panels a–c) A closeup view of the metal cofactors with the four coordinating protein residues with the correct bonds manually added (top) and as they appear without adjustments to the Crystallographic Information File (CIF) files in PyMol (bottom). Panel (a) displays an Fe cofactor from a rubredoxin metalloprotein (PDB 4D4O). Panels (b) and (c) show two [2Fe‐2S] metal cofactors: standard (PDB: 6TGA) and Rieske [2Fe‐2S] (PDB: 1BGY), respectively. Atoms were colored according to their respective element (C = gray, N = blue, O = red, H = white, S = yellow). PDB, Protein Data Bank.
For each metalloprotein type, the proteins were split into either a training or test dataset. The proteins within each class were used multiple times with different rotations if needed to create the 1000 training and 500 test images. The relevant metal cofactors and amino acids within 6.0 Å of the metal cofactors are shown and rotated to create 2D images. We initially planned to include a fourth group, type‐IV [2Fe‐2S] cluster. We found that our initial parameters within PyMol incorrectly included standard [2Fe‐2S] cofactors with the HIS amino acid within a sphere but not as ligand to the 2Fe‐2S. We discovered this after a spot‐check on the images revealed that the model misclassified this metal cluster. After this, we implemented a secondary check with a Python web‐scraper, MolQL Explorer (MolQL, n.d.; Rose et al., 2018) to verify the correct amino acids were bonded to the cofactor.
Table 2a and Figure 4 show the tabulated data and the resulting confusion matrix, respectively, for the ResNet model when classifying individually between the three Fe systems shown in Figure 1, Fe ion, [2Fe‐2S], and Rieske [2Fe‐2S]. There was a 95% accuracy when classifying images as either a Rieske 2Fe‐2S, standard [2Fe‐2S], or a single Fe cofactor (Table 2b). The average recall was 95% with a 1% standard deviation and the average precision was 95% with a 2% standard deviation. Our success rate for successful image classification is high. We surpassed the results of Nanni et al. (2020) which achieved an accuracy of 90% for protein fold classification and 77% for protein class classification with 2D images of 3D proteins. We were able to make the prediction with fewer examples, using only one rotation per prediction instead of the 125 rotations used by Nanni et al., but with 95% accuracy. Situations in which the image is improperly classified often include rotations of the clusters that only show the histidine ring edge on. To demonstrate the improvement of using a pretrained model, Figure S1 shows the confusion table resulting from classifying without using a pretrained model. Figure 5 shows images of the Fe cofactor and the 6.0 Å environment around the cofactor, which the model incorrectly classified as [2Fe‐2S] cofactor types. The bottom panel shows the same metal cofactor as in the top panel, but rotated so the ligands are identifiable with the face‐on view of the histidine rings.
TABLE 2a.
Confusion table for the ResNet neural network to properly classify the image of the environment around the metal cofactor, amino acids within 6.0 Å.
| Metal cofactor name | Fe | [2Fe‐2S] standard | [2Fe‐2S] Rieske | Total images |
|---|---|---|---|---|
| Fe | 478 | 3 | 19 | 500 |
| [2Fe‐2S] standard | 10 | 482 | 8 | 500 |
| Rieske | 2 | 27 | 471 | 500 |
Note: Metal cofactors were included.
FIGURE 4.
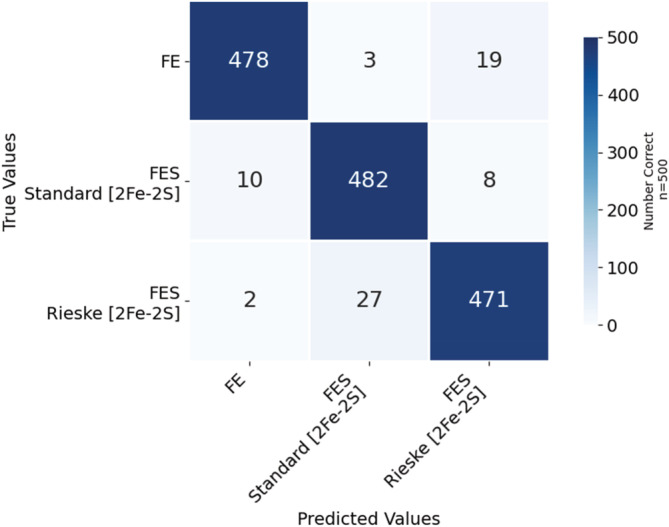
Confusion matrix for the ResNet neural network to properly classify the environment around the metal cofactor, amino acids within 6.0 Å, as belonging to the metal cofactor. Each metal center employed 500 images.
TABLE 2b.
Recall, precision, and accuracy for the ResNet neural network to properly classify the image of the environment around the metal cofactor, amino acids within 6.0 Å.
| Class | Recall | Precision | Total accuracy |
|---|---|---|---|
| Fe | 96% | 98% | 95% |
| [2Fe‐2S] standard | 96% | 94% | |
| [2Fe‐2S] Rieske | 94% | 95% | |
| [2Fe‐2S] average | 95% | 94% | |
| Average | 95% | 95% | |
| Std. Dev. | 1% | 2% |
Note: Metal cofactors were included.
FIGURE 5.
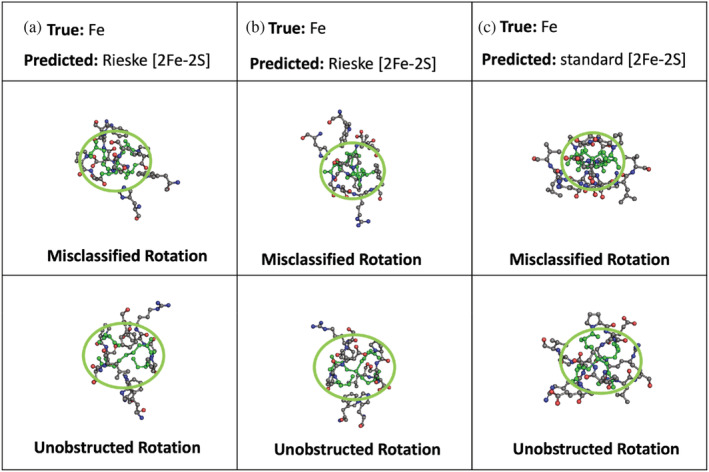
Incorrect classification of the image of the environment around the metal cofactor—the amino acids within 6.0 Å of a metal cofactor, and the metal cofactor. The model incorrectly classified the Fe cofactor as a [2Fe‐2S] cofactor at the rotation shown in the top panels. The bottom panels show the same cofactors but rotated so that the cysteine ligands are clearly visible (circled and colored in green). The green color is for illustrative propose—the image used by the model had the same color scheme used for all atoms. The metal residual identification of the images are: (a) PDB: FD4O, Chain: C, Residue: 501; (b) PDB id: 4X33, Chain: A, Residue: 101; (c) PDB id: 6J27, Chain: C, Residue: 401. PDB, Protein Data Bank.
2.3. Classification of Fe and [2Fe‐2S] cofactors from metalloproteins in the absence of metal cofactors
To further test the robustness and flexibility of our model, we also investigated the original neural network's ability to classify the metalloprotein images with the metal cofactor hidden. After fine‐tuning the originally downloaded ResNet model, ResNet did very well in classifying the 2D images as metal binding sites. The images included all amino acids within 6.0 Å of the Fe and [2Fe‐2S], with the metal cofactor hidden, and they were classified into the classes defined in the training data—a Rieske [2Fe‐2S], standard [2Fe‐2S], and Fe cofactor. Tables 3a and 3b and Figure 6 show the classification results when using the same residues and rotations as employed in Section 2.2 to build a library of images with the exception that the metal cofactor was omitted from this dataset of images. There was an 96% accuracy when classifying images as either a Rieske [2Fe‐2S], standard [2Fe‐2S], and single Fe ion cofactor. The average recall was 96% with a 3% standard deviation and the average precision was 96% with a 1% standard deviation.
TABLE 3a.
Confusion table for the ResNet neural network to properly classify the image of the environment around the metal cofactor, amino acids within 6.0 Å.
| Metal cofactor name | Fe | [2Fe‐2S] standard | [2Fe‐2S] Rieske | Total images |
|---|---|---|---|---|
| Fe | 468 | 18 | 14 | 500 |
| [2Fe‐2S] standard | 10 | 482 | 8 | 500 |
| Rieske | 0 | 6 | 494 | 500 |
Note: Metal cofactors were not included.
TABLE 3b.
Recall, precision, and accuracy for the ResNet neural network to properly classify the image of the environment around the metal cofactor, amino acids within 6.0 Å.
| Class | Recall | Precision | Accuracy |
|---|---|---|---|
| Fe | 94% | 98% | 96% |
| [2Fe‐2S] standard | 96% | 95% | |
| [2Fe‐2S] Rieske | 99% | 96% | |
| [2Fe‐2S] average | 98% | 95% | |
| Average | 96% | 96% | |
| Std. Dev. | 3% | 1% |
Note: Metal cofactors were not included.
FIGURE 6.
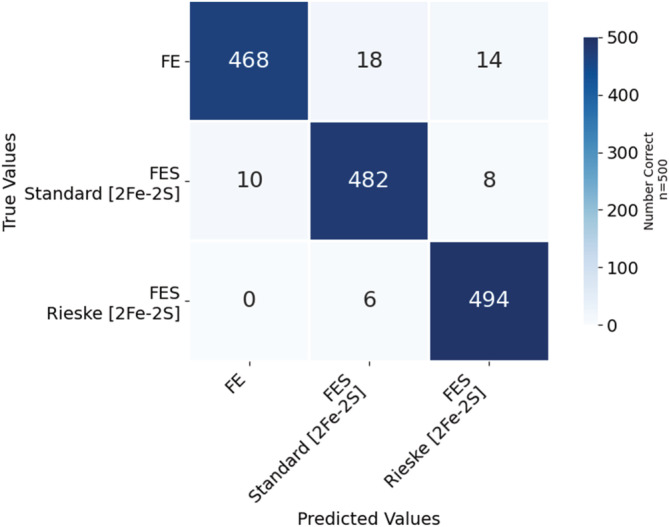
Confusion matrix for the ResNet neural network to properly classify the environment around the metal cofactor—the amino acids within 6.0 Å of a metal cofactor—as belonging to the metal cofactor in the absence of the native metal cofactors. The images used the same database of PDB IDs and same rotations as in Figure 4 with the exception that the atoms of the Fe containing metal cofactors were removed.
2.4. Classification of a predicted AlphaFold structure
To demonstrate the model's ability to classify the metal binding site for a predicted protein structure, we searched the PDB (Berman et al., 2003) for the PetF protein from Phaelodactylum tricornutum, which is a standard [2Fe‐2S] containing ferredoxin. Although 14 structures from P. tricornutum were located in the PDB, PetF was not one of the deposited structures. However, we did find the protein associated with the petF gene in the AlphaFold Protein Structure Database (Jumper et al., 2021; Berman et al., 2003). As this protein is a predicted protein structure rather than a structure produced from experimental efforts and deposited in the PDB, it is an ideal candidate to test the model.
Using our trained model, we classified images of rotations of the PetF protein structure, predicted using the AlphaFold Protein Structure Database. These images included all amino acids within 6.0 Å or 6.5 Å of the center of four cysteine residues, where the cysteine residues were identified by an expert. Residues with at least one atom located within the 6.0 Å sphere from any atom in the metal cofactor were previously used to create the training image, rather than 6.0 Å from the center of the metal cofactor. Therefore, we increased the radius to 6.5 Å to compensate for measuring from the center rather than the nearest atom of the metal cofactor. As we only had one structure, we classified incremental rotations of the cluster and found that the model correctly identified the images as standard [2Fe‐2S] in 117 out of 125 rotations (Table 4).
TABLE 4.
Confusion table for the ResNet neural network to properly classify the image of the environment around the center of four cysteine residues in a protein structure predicted from AlphaFold, Phaelodactylum tricornutum (PetF), which includes the cysteine residues and additional amino acids within 6.0 Å and 6.5 Å.
| Metal cofactor name | Fe | [2Fe‐2S] standard | [2Fe‐2S] Reiske | |
|---|---|---|---|---|
| 6.0 Å | [2Fe‐2S] standard | 29 | 96 | 3 |
| 6.5 Å | [2Fe‐2S] standard | 7 | 117 | 1 |
Note: The correct cofactor, [2Fe‐2S], was identified for 117 of the 125 rotations.
3. DISCUSSION
Providing alternate methods to analyze metal binding sites beyond the analysis of the primary protein structure could provide significant advances in the development of artificial enzymes that mimic features of nature. In this work, we used CNNs to determine if image classification can be used as a tool to identify metal binding sites both with and without the metal in the pocket. Similar to previous studies where they were studying protein classes or protein structure classes, we also found success in classifying structures based on 2D images (Qin et al., 2020) and specifically using ball and stick representations (Nanni et al., 2020) when classifying metal‐binding sites.
The best fine‐tuned models identified single amino acids and metal cofactors with a >95% accuracy, demonstrating the ability of CNNs to distinguish these 3D protein structures from 2D images. Most interesting was the ability of the model to properly classify a protein scaffold that binds a single Fe atom from a protein scaffold that binds a [2Fe‐2S] using only the protein microenvironment that directly encapsulates the coordinated metallocenter.
We found that ResNet did very well in identifying amino acids. It had a 100% recall for arginine, histidine, and proline, and above 95% recall of 15 out of 20 amino acids, and it had a 100% precision for cysteine, glutamine, and methionine, and above 95% precision of 16 out of 20 amino acids. One difficulty of using neural networks to classify is that only input and outputs are observable, thus it can be difficult to diagnose the reasons the model does or does not classify correctly. We can only infer patterns in the images that were correctly and incorrectly classified. These images are 2D snapshots of the 3D cluster taken at a random angle, with color‐coded atoms and connections drawn by PyMol.
Likewise, classifying confusion within amino acids of similar physical properties is sensible as a terminal functional group may be shared, but the carbon chain connecting the functional group to the amino acid backbone may differ by only one carbon atom. For instance, isoleucine images were confused as leucine 6% of the time, and leucine was confused as isoleucine 8% of the time (Figure 2). Figure 3 shows some of the images of these amino acids that the model confused. Each residue side chain contains saturated sp3 carbon atoms which branch and terminate in methyl groups. Given the similarities of the physical composition, ResNet may have trouble distinguishing different configurations of carbon branching patterns. Perhaps the model more often confused isoleucine and leucine with each other rather than with valine because valine is slightly more structurally dissimilar to isoleucine and leucine, as valine has a shorter chain length than both isoleucine and leucine. It is also possible that the improperly classified images simply indicate that any given random rotation can be an inherently poor descriptor when the amino acid side chains are not fully visible, and thus are not ideal for identifying residues via image analysis.
In some cases, it is difficult to find a reason why the model may have confused amino acids as some misclassification was between groups that would likely be readily distinguished by human classifiers. For example, serine was confused with alanine 5% of the time and valine was confused with threonine 5% of the time despite the presence or absence of an oxygen atom in the confused amino acid. When using random rotations to define a structure, there will be inherently non‐opportunistic poses that place important side chain atoms at an angle that is obstructed for the identification of features in the image. The result is an image that shows only the edge of an amino acid or an accessory amino acid that is obstructing a metal‐bonding amino acid from view.
Based on our success with amino acids, we chose ResNet as the preferred model for the classification of the metal cofactors. This task is unrelated to the task of classifying amino acids, and the original pretrained neural network was fine‐tuned for the task. In this case, the classification model is not required to identify amino acids within the images, which would be object detection. Instead, the model is classifying images as associated with a metal cofactor based on the entire image. After fine‐tuning, ResNet had higher average recall and precision within the [2Fe‐2S] cofactor class at 98% and 95%, respectively (Table 2b) compared to the Fe cofactor class at 95% and 94%, respectively (Table 2a). One possible reason behind the increase in recall and precision for [2Fe‐2S] and Rieske cofactors as opposed to the Fe cofactor was that the Fe cofactor classes were created from a dataset containing 6 times more unique proteins than the [2Fe‐2S] clusters due to the lower availability of experimentally derived protein structures.
Figure 5 shows improperly classified images of an Fe cofactor class that the model classified as one of the two types of [2Fe‐2S] clusters. Not only do unfavorable poses place atoms in positions that make them difficult to assess with this tool, but atomic information can also be hidden when rotated in the direction orthogonal to the 2D view, leading to a loss of both connectivity and color value information to be used as structural reference material. The bottom panel in Figure 5 shows the same metal cluster that was misclassified but rotated so that the cysteine ligands are clearly identifiable. In addition, the model used color images, and the backbone nitrogen atoms may impede correct classification as unfavorable poses will place blue nitrogen atoms where chelating histidine residues could possibly reside. While this highlights some of the limitations of this technique, the overall predictive performance was very high, with an accuracy of 96%. It is expected that using 600–800 images would allow us to approach 99%, however, the level of accuracy achieved with this approach positions us to use this technique to identify binding pockets with a high level of confidence.
In the absence of metals, the average recall is 96 with a standard deviation of 3% and the average precision is 96 with a standard deviation of 1% when distinguishing three possible cofactor classes: Rieske [2Fe‐2S], standard [2Fe‐2S], and a single Fe ion, which is slightly higher than with metals (average recall of 95% with a standard deviation of 1% and average precision is 95% with a standard deviation of 2%). This is interesting as these results suggest that the metal cofactor is introducing increased error in some image rotations. Symmetrical operations may produce identical images in the single Fe binding site and the [2Fe‐2S] cofactor binding site when the metal atoms overlap. Removal of the metal cofactors likely lowers the number of disadvantageous viewing angles in the created images by allowing atoms that would normally be hidden behind the Fe atom to be visible, and thus included in metal binding site predictions.
The high level of classification accuracy even in the absence of the metals implies that an aspect of each cofactor must be consistent within each class. Considering that both the Fe ions and standard [2Fe‐2S] cofactors are both connected by sulfur atoms from four cysteine residues, yet still can be distinguished from each other, this hints at the conservation of traits independent of each cofactor type. Further, recognition of a metal binding site must involve more than the atoms of ligands directly bound to the metal, that is, the first coordination sphere. These additional features may include the influence of the exogenous molecule bound to those ligands, and thus a second coordination sphere and the intermittent orientations or locations of any of the amino acids within the 6.0 Å sphere may inhabit. The ability of the model to correctly identify images of a predicted protein structure as standard [2Fe‐2S] in 117 out of 125 rotations when using 6.5 Å from the center of the expert‐identified cysteine residues shows this methodology can be applied to predicted structures within the AlphaFold database.
Counterintuitively, ResNet had similar accuracy for the three classification tasks: (1) single amino acids, (2) protein structural images containing atomic information for amino acid residues and metal cofactors, and (3) protein structural images containing atomic information for only amino acid residues and no atomic information for the metal cofactors. Since each task had the same starting model and did not build sequentially upon each other, our initial expectation was to observe a decrease in classification performance as our perception of the complexity of the task increased from 1 to 2 to 3. However, incorrectly classified images may have been the result of poor bonding information, unfavorable rotational space, or a lack of structural examples. From our analysis, it appears that evolutionary pressure conserves important structural information, such as metal‐binding environments, and thus allows for reasonable classification of metal‐binding proteins from information derived purely from the images of the environment around the metal cofactors.
Evolutionary conservation is also observed via amino acid sequence motifs, which are contained within the original PDB file and can guide the elucidation of known metal binding sites. However, a portion of the same motif employed by all [2Fe‐2S] clusters, C‐X2‐C, has been shown to be used by protein scaffolds to coordinate all metal ions in the PDB except for Mg2+ (Belmonte and Mansy, 2017). This is an example where our CNN for image classification of metal binding sites can enable another layer of quality assurance when classifying novel protein structures.
Ultimately, we would like to accelerate our ability to predict metal binding pockets to create artificial metalloenzymes. Deep learning models like AlphaFold, which has been shown to predict protein folding with a very high level of accuracy, have accelerated our ability to predict protein structure. However, posttranslational modifications such as the incorporation of metal cofactors are not currently included, and additional techniques are needed to identify binding pockets that can host metals. Methods such as AlphaFill have become available recently that allow the prediction of small molecules and metal ions (Hekkelman et al., 2022). Our methods described in this paper, of using 2D images of 3D structures have advantages over using expert‐defined features in classification. First, the process of categorizing an enzyme into a list of features may result in the loss of important structural or other information. For example, the exact orientation of each amino acid relative to the others might be lost in an individual image, while the global set of images retains all required information. Second, the model requires only the images of the cluster, but no prior knowledge of what is visible in the image. The result is that expert input will not be needed beyond defining the parameters for the training data. In addition, a lower need for prior knowledge means that this method can easily be adjusted for new questions and classification tasks even if features are not defined or understood. The strength of this method then is that it requires low cost and effort compared with using expert‐defined features, while rapidly delivering high predictive performance.
4. CONCLUSION
We have successfully updated the pretrained CNN ResNet to classify all 20 standard amino acid residues with an overall accuracy >96%. Resnet showed the highest accuracy among six evaluated CNN architectures and was employed for more detailed studies of metal cofactors and their metal binding pockets. The original ResNet was trained to successfully classify the amino acids within a 6.0 Å sphere around two metal cofactors, Fe ion and [2Fe‐2S] cluster, with and without the metal cofactor visible. We further showed the ability to differentiate among the coordination spheres of two types of [2Fe‐2S] cofactor containing ferredoxins.
Classifying known metal‐binding sites is a necessary aspect of identifying metal‐binding sites in nonmetal binding proteins, that is, artificial metalloenzyme studies. Our work illustrates a method to distinguish similar clusters from each other with minimal expert input and time. Our method can be run quickly with PyMol scripts and requires limited time from experts beyond spot checks and the initial definition of the sites used to create the training images. It is the first case, in our knowledge, of using 2D images of 3D structures to classify metal cofactor sites and opens an opportunity for the rapid classification of metal binding sites in proteins, with potential to extend to other binding sites in proteins.
5. METHODS
5.1. Image database of amino acid rotations
The molecular graphics program, PyMol, was employed to produce all images in the training/validation/testing datasets. Each amino acid was downloaded from PDB within PyMol. Training was completed using 2D snapshots of the 3D representation for each amino acid. The amino acids were represented as ball and sticks, where atoms are represented as spheres and bonds as cylinders. Atoms were colored according to their respective element: C = gray, N = blue, O = red, H = white, S = yellow. Depth and shadow were included to emphasize the foreground and background of the amino acid. Training data were created from amino acids rotated in each possible combination of 36‐degree increments around the x, y, and z‐axis. At each rotation a 2D snapshot was saved, resulting in 1000 images for each amino acid. Model validation and testing datasets employed amino acid CIF files that were rotated by random degrees on the x, y, and z‐axis and 2D snapshots were taken at each rotation, resulting in 250 snapshots of each amino acid in the validation dataset, used to select the optimal neural network model, and 250 snapshots in the test dataset.
5.2. Image database of metal cofactor rotations
Clusters containing Fe and [2Fe‐2S] were found by downloading a list of all proteins containing (1) Fe (III) and Fe (II) ion and (2) FeS, respectively, from the Protein Data. A PyMol script identified the cofactors that contained one iron atom and four cysteine residues within a 3.0 Å sphere of the Fe residue. The PyMol script filtered the [2Fe‐2S] cofactors into three classes: standard, Rieske, and other. Standard [2Fe‐2S] was chosen if the residue FeS contained at least four cysteine residues with 3.0 Å. Rieske [2Fe‐2S] cofactors were chosen if the cysteine residues were equal or greater than two and the histidine residues were equal to two. All FeS residues that did not fit into either of these two categories were discarded. The classification was confirmed by using a Python web scraper to download a list of residues bonded to the metals from the MolQL Explorer website (MolQL, n.d.), with residues that did not meet the bonding requirement discarded. Seventeen proteins were not downloadable from MolQL, so the classification of their residues was confirmed with a visual inspection of the orientation of the amino acids to the metal cofactor in PyMol.
For each class of cofactors, the CIF files were downloaded within PyMol. A 6.0 Å sphere around each metal cofactor, Fe or FeS, in the protein, was created using PyMol commands to show only the metal cofactor of interest and the residues with at least one atom located within the 6.0 Å sphere from the metal cofactor. Training was completed using 2D snapshots of the 3D representation for each of the spheres centered by, and including the metal cofactors. A 1000 images were created for each cofactor (Fe, Rieske 2Fe‐2S, and standard 2Fe‐2S) in the training and 500 in the test dataset. Separate proteins were used in training and test sets. Tables S2–S4 identify the metal cofactor residues used as the center of each cofactor.
A ball and stick visualization method within PyMol was used with the same atom coloring scheme as the single amino acid representations, but without depth or shadow as the overlapping amino acids within the cluster already provide information on which structures are in the foreground and background. To create the training data, a list was created of each possible combination of 36‐degree increments about the x, y, and z‐axis to create a rotation list with a 1000 items. For each rotation combination and for each possible cofactor (Fe only, standard 2Fe‐2S, Rieske 2Fe‐2S) in the training dataset, a random unique protein ID was fetched and rotated. If multiple metal cofactor residues existed in one protein, the duplicate residue numbers on different chains were not used. The script used a new metal cofactor residue ID associated with the protein each time the protein is selected (restarting with the initial residues once all residues are used). To create the test data, 500 random cofactors from the test dataset were rotated by random degrees on the x, y, and z‐axis, and a 2D snapshot was taken at each rotation. As only ResNet was used for classifying metal cofactors, there was no need for model validation sets. The same residues and rotations were employed to create the images without metals, with the PyMol script hiding the metal cofactors.
This method was modified slightly to test classification on an AlphaFold protein (entity A0A6B9XN65). As the metal cofactor is not included in an AlphaFold structure, the image was centered instead by the four cysteine residues identified by an expert. We used a radius of 6.0 Å or 6.5 Å to create the images. The image was rotated at 72‐degree increments across the x, y, and z‐axis. These images were only used to test the previously trained models—they were not used in the training set.
5.3. Machine learning models
We fine‐tuned and evaluated the performance of the models ResNet (He et al., 2016), AlexNet (Krizhevsky et al., 2012), VGG (Simonyan and Zisserman, 2014), SqueezeNet (Iandola et al., 2016), DenseNet (Iandola et al., 2014), and Inception v3 (Szegedy et al., 2016). A Python library used for computer vision, PyTorch (Paszke et al., 2019), was employed to execute the models. All data preprocessing, model fine‐tuning, and inference was performed on NVIDIA P100‐based GPU nodes. The script used was modified from the webpage, Finetuning Torchvision Models Tutorial (Inkawhich, 2017). Test images were resized and normalized to match the required input of the neural network, and output of the last layer was fed into a classification layer, for example, for amino acids the output layer was modified for 20 classes. We used pretrained models, which allow the parameters of the neural network to be preloaded based on 15 million images from an image dataset, ImageNet. The models were tuned using 50 epochs each, a batch size of 16, and updating weights of the entire model as opposed to only updating the final layer. We then selected the model with the highest accuracy score in the validation set to assess performance in the test set for amino acid classification. We also used the original pretrained model to both train and test the metal cofactor classification. The same machine learning methods were used for the metal cofactor classification.
AUTHOR CONTRIBUTIONS
Marjolein Oostrom: Formal analysis (equal); investigation (equal); methodology (equal); software (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Sarah Akers: Investigation (supporting); software (supporting); writing – review and editing (supporting). Noah Garrett: Investigation (supporting). Emma Hanson: Investigation (supporting). Wendy Shaw: Funding acquisition (equal); supervision (equal); writing – review and editing (lead). Joseph A. Laureanti: Funding acquisition (equal); supervision (equal); investigation (equal); methodology (equal); writing – original draft (equal); writing – review and editing (equal).
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
This work is supported by the PNNL Laboratory Directed Research and Development (LDRD) program and in part by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), Division of Chemical Sciences, Geosciences & Biosciences, Catalysis Science program, FWP 47319 and in part by the U.S. DOE, Office of Science, Office of Workforce Development for Teachers and Scientists (WDTS) under the Science Undergraduate Laboratory Internships Program (SULI).
Oostrom M, Akers S, Garrett N, Hanson E, Shaw W, Laureanti JA. Classifying metal‐binding sites with neural networks. Protein Science. 2023;32(3):e4591. 10.1002/pro.4591
Review Editor: Nir Ben‐Tal
Funding information Pacific Northwest National Lab; U.S. Department of Energy
Contributor Information
Wendy Shaw, Email: wendy.shaw@pnnl.gov.
Joseph A. Laureanti, Email: laureanti.joseph@gmail.com.
DATA AVAILABILITY STATEMENT
The datasets used in the paper are available here: https://data.pnnl.gov/group/nodes/dataset/33238. The code used in the paper is available here: https://github.com/pnnl/mlprotein
REFERENCES
- Agar JN, Dean DR, Johnson MK. Iron‐sulfur cluster biosynthesis. Biochemistry and physiology of anaerobic bacteria. Berlin, Germany: Springer; 2003. p. 46–66. [Google Scholar]
- Andreeva A. Classification of proteins: available structural space for molecular modeling. Homology modeling. Berlin, Germany: Springer; 2011. p. 1–31. [DOI] [PubMed] [Google Scholar]
- Andreini C, Bertini I, Cavallaro G, Holliday GL, Thornton JM. Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem. 2008;13(8):1205–18. [DOI] [PubMed] [Google Scholar]
- Andreini C, Cavallaro G, Lorenzini S, Rosato A. MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2012;41(D1):D312–D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324(1):105–21. [DOI] [PubMed] [Google Scholar]
- Belmonte L, Mansy SS. Patterns of ligands coordinated to metallocofactors extracted from the protein data bank. J Chem Inf Model. 2017;57(12):3162–71. [DOI] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H. Announcing the worldwide protein data bank. Nat Struct Mol Biol. 2003;10(12):980. [DOI] [PubMed] [Google Scholar]
- Bobadilla L, Nino F, Cepeda E, Patarroyo MA. Characterizing and predicting catalytic residues in enzyme active sites based on local properties: a machine learning approach. 2007 IEEE 7th International Symposium on BioInformatics and BioEngineering. 2007; IEEE, pp. 938–945. [Google Scholar]
- Bonetta R, Valentino G. Machine learning techniques for protein function prediction. Proteins Struct Funct Bioinform. 2020;88(3):397–413. [DOI] [PubMed] [Google Scholar]
- Consortium U . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari L, Mitchell JB. From sequence to enzyme mechanism using multi‐label machine learning. BMC Bioinform. 2014;15(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Dong W, Socher R, Li L‐J, Li K, Fei‐Fei L. Imagenet: A large‐scale hierarchical image database. 2009 IEEE conference on computer vision and pattern recognition. 2009; IEEE, pp. 248‐255. [Google Scholar]
- Fernandes HS, Teixeira CSS, Sousa SF, Cerqueira NM. Formation of unstable and very reactive chemical species catalyzed by metalloenzymes: a mechanistic overview. Molecules. 2019;24(13):2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016;770–778. [Google Scholar]
- Hekkelman ML, de Vries I, Joosten RP, Perrakis A. AlphaFill: enriching AlphaFold models with ligands and cofactors. Nat Methods. 2022;20:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himiyama T, Okamoto Y. Artificial metalloenzymes: from selective chemical transformations to biochemical applications. Molecules. 2020;25(13):2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm RH, Kennepohl P, Solomon EI. Structural and functional aspects of metal sites in biology. Chem Rev. 1996;96(7):2239–314. [DOI] [PubMed] [Google Scholar]
- Hosseinzadeh P, Marshall NM, Chacón KN, Yu Y, Nilges MJ, New SY, et al. Design of a single protein that spans the entire 2‐V range of physiological redox potentials. Proc Natl Acad Sci. 2016;113(2):262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iandola F, Moskewicz M, Karayev S, Girshick R, Darrell T, Keutzer K. Densenet: implementing efficient convnet descriptor pyramids. arXiv preprint. 2014; arXiv:14041869. [Google Scholar]
- Iandola FN, Han S, Moskewicz MW, Ashraf K, Dally WJ, Keutzer K. SqueezeNet: AlexNet‐level accuracy with 50× fewer parameters and <0.5 MB model size. arXiv preprint. 2016; arXiv:160207360. [Google Scholar]
- Inkawhich N.; 2017. Finetuning Torchvision Models PyTorch.org: PyTorch. Available from https://pytorch.org/tutorials/beginner/finetuning_torchvision_models_tutorial.html.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Zhu Z‐Y, Karlin KD. The extended environment of mononuclear metal centers in protein structures. Proc Natl Acad Sci. 1997;94(26):14225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. Adv Neural Inf Proces Syst. 2012;25:84–90. [Google Scholar]
- Laureanti JA, O'Hagan M, Shaw WJ. Chicken fat for catalysis: a scaffold is as important for molecular complexes for energy transformations as it is for enzymes in catalytic function. Sustain Energy Fuels. 2019;3(12):3260–78. [Google Scholar]
- LeCun Y, Bengio Y. Convolutional networks for images, speech, and time series. The Handbook of Brain Theory and Neural Networks. 1995;3361(10):1995. [Google Scholar]
- MolQL . Available from: http://molql.org/explorer.html.
- Nanni L, Lumini A, Pasquali F, Brahnam S. iProStruct2D: identifying protein structural classes by deep learning via 2D representations. Expert Syst Appl. 2020;142:113019. [Google Scholar]
- Passerini A, Frasconi P. Learning to discriminate between ligand‐bound and disulfide‐bound cysteines. Protein Eng Des Sel. 2004;17(4):367–73. [DOI] [PubMed] [Google Scholar]
- Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, et al. Pytorch: an imperative style, high‐performance deep learning library. Adv Neural Inf Proces Syst. 2019;32:8026–8037. [Google Scholar]
- Qin S, Li Z, He L, Lin W. Similarity analysis of 3D structures of proteins based tile‐CNN. IEEE Access. 2020;8:44622–31. [Google Scholar]
- Roelfes G. LmrR: a privileged scaffold for artificial metalloenzymes. Acc Chem Res. 2019;52(3):545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AS, Sehnal D, Bliven S, Burley SK, Velankar S. Molql: towards a common general purpose molecular query language. Biophys J. 2018;114(3):342a. [Google Scholar]
- Röthlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453(7192):190–5. [DOI] [PubMed] [Google Scholar]
- Schrodinger, LLC (2015) The PyMOL molecular graphics system, version 2.0. New York, NY. [Google Scholar]
- Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, et al. Artificial metalloenzymes: reaction scope and optimization strategies. Chem Rev. 2018;118(1):142–231. [DOI] [PubMed] [Google Scholar]
- Simonyan K, Zisserman A. Very deep convolutional networks for large‐scale image recognition. arXiv preprint. 2014; arXiv:14091556. [Google Scholar]
- Slater JW, Marguet SC, Monaco HA, Shafaat HS. Going beyond structure: nickel‐substituted rubredoxin as a mechanistic model for the [NiFe] hydrogenases. J Am Chem Soc. 2018;140(32):10250–62. [DOI] [PubMed] [Google Scholar]
- Su H, Maji S, Kalogerakis E, Learned‐Miller E. Multi‐view convolutional neural networks for 3D shape recognition. Proceedings of the IEEE international conference on computer vision; 2015;945–953. [Google Scholar]
- Szegedy C, Vanhoucke V, Ioffe S, Shlens J, Wojna Z. Rethinking the inception architecture for computer vision. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016;2818–2826. [Google Scholar]
- Thomas CM, Ward TR. Artificial metalloenzymes: proteins as hosts for enantioselective catalysis. Chem Soc Rev. 2005;34(4):337–46. [DOI] [PubMed] [Google Scholar]
- Torng W, Altman RB. 3D deep convolutional neural networks for amino acid environment similarity analysis. BMC Bioinform. 2017;18(1):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torng W, Altman RB. High precision protein functional site detection using 3D convolutional neural networks. Bioinformatics. 2019;35(9):1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasatava Y, Rosato A, Banci L, Andreini C. MetalPredator: a web server to predict iron–sulfur cluster binding proteomes. Bioinformatics. 2016;32(18):2850–2. [DOI] [PubMed] [Google Scholar]
- Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106(8):3210–35. [DOI] [PubMed] [Google Scholar]
- Wehrspan ZJ, McDonnell RT, Elcock AH. Identification of iron‐sulfur (Fe‐S) cluster and zinc (Zn) binding sites within proteomes predicted by DeepMind's AlphaFold2 program dramatically expands the metalloproteome. J Mol Biol. 2022;434(2):167377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. Catalysis by metallo‐enzymes: the entatic state. Inorg Chim Acta Rev. 1971;5:137–55. [Google Scholar]
- Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34(12):938–45. [DOI] [PubMed] [Google Scholar]
- Yosinski J, Clune J, Bengio Y, Lipson H. How transferable are features in deep neural networks? Adv Neural Inf Proces Syst. 2014;27:3320–3328. [Google Scholar]
- Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, et al. Protein design: toward functional metalloenzymes. Chem Rev. 2014;114(7):3495–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
The datasets used in the paper are available here: https://data.pnnl.gov/group/nodes/dataset/33238. The code used in the paper is available here: https://github.com/pnnl/mlprotein