

Abstract
Though many folded proteins assume one stable structure that performs one function, a small‐but‐increasing number remodel their secondary and tertiary structures and change their functions in response to cellular stimuli. These fold‐switching proteins regulate biological processes and are associated with autoimmune dysfunction, severe acute respiratory syndrome coronavirus‐2 infection, and more. Despite their biological importance, it is difficult to computationally predict fold switching. With the aim of advancing computational prediction and experimental characterization of fold switchers, this review discusses several features that distinguish fold‐switching proteins from their single‐fold and intrinsically disordered counterparts. First, the isolated structures of fold switchers are less stable and more heterogeneous than single folders but more stable and less heterogeneous than intrinsically disordered proteins (IDPs). Second, the sequences of single fold, fold switching, and intrinsically disordered proteins can evolve at distinct rates. Third, proteins from these three classes are best predicted using different computational techniques. Finally, late‐breaking results suggest that single folders, fold switchers, and IDPs have distinct patterns of residue–residue coevolution. The review closes by discussing high‐throughput and medium‐throughput experimental approaches that might be used to identify new fold‐switching proteins.
Keywords: fold‐switching proteins, intrinsically disordered proteins, metamorphic proteins, protein folding, protein structure prediction
1. INTRODUCTION
Proteins have different levels of structural heterogeneity and stability, ranging from one stable fold to intrinsically disordered proteins (IDPs) with numerous unstable conformations. Between these two extremes lie a small but increasing number of experimentally characterized fold‐switching proteins (Kulkarni et al., 2018). Contrasting single‐fold proteins, which assume fixed secondary and tertiary structures in their folded states, fold switchers remodel their secondary and tertiary structures and change their functions in response to cellular stimuli (Porter & Looger, 2018). Contrasting IDPs, fold switchers spontaneously assume semi‐stable experimentally characterizable conformations in isolation. Exogeneous interactions, such as substrate binding, are required for IDPs—but not fold switchers—to fold. The structural transitions of fold‐switching proteins can be either irreversible or reversible (Kim & Porter, 2021); fold switchers that undergo reversible structural transitions are also called “metamorphic” (Murzin, 2008). The conformational transitions of fold switchers regulate biological processes (Kim & Porter, 2021) and perform diverse biological functions such as suppressing human innate immunity during severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) infection (Gao et al., 2021), regulating the expression of bacterial virulence genes (Kang et al., 2018), and controlling the periodicity of the cyanobacterial circadian clock (Chavan et al., 2021). Despite their biological importance, most fold switchers have been discovered by chance (Lopez‐Pelegrin et al., 2014), and their dual‐folding structures are usually missed by state‐of‐the‐art computational methods, such as AlphaFold2 (Chakravarty & Porter, 2022).
With the aim of advancing computational prediction and discovery of fold switchers, this review points out features that distinguish these shapeshifting proteins from their single‐fold and IDP counterparts. The first section discusses biophysical similarities and differences between fold switchers, single folders, and IDPs. All three sorts of proteins are dynamic and can interconvert between distinct conformations on similar timescales. Thus, timescale alone cannot be used to distinguish between the conformational changes that these three classes of proteins undergo. Nevertheless, the structures of isolated fold switchers tend to be more stable and less heterogeneous than IDPs but less stable and more heterogeneous than single folders. Second, the amino acid sequences of IDPs tend to evolve more rapidly than both fold switchers and single folders; in other words, the level of amino acid variation tends to be higher among homologous IDPs than among homologous fold switchers or single folders. In at least one case, the sequences of homologous fold switchers evolve more rapidly than single folders; in others, they evolve at similar rates. Thirdly, single folders, fold switchers, and IDPs are best predicted using different computational techniques. Deep‐learning‐based methods, such as AlphaFold2 (Jumper et al., 2021), often predict 3D structures of single folders with astonishingly high accuracy. In contrast, uncertainty scores in AlphaFold2 and RoseTTAfold (Baek et al., 2021) can be used to identify IDPs with good accuracy (He et al., 2022). Current deep‐learning‐based methods fail to predict fold switching in most cases (Chakravarty & Porter, 2022; Porter et al., 2022), but other approaches such as variable length secondary structure propensity comparison have been successful (Mishra et al., 2021; Porter et al., 2022). Emerging results also suggest that single folders, fold switchers, and IDPs have distinct patterns of coevolution. Finally, we discuss experimental approaches that hold promise for characterizing and screening fold‐switching proteins.
2. BIOPHYSICAL SIMILARITIES AND DIFFERENCES BETWEEN SINGLE FOLDERS, FOLD SWITCHERS, AND IDPS
Both in solution and in cells, proteins exist as dynamic ensembles of interconvertible conformations. The heterogeneity of these conformations tends to be inversely related to their stability, and dominant conformations often shift in response to environmental changes (Figure 1). Below we provide two illustrative examples of conformational changes from single‐fold, fold‐switching, and intrinsically disordered proteins. Timescales and reaction mechanisms are reported when known, though all three classes of proteins can exchange between conformations on similar timescales (Kulkarni et al., 2018) ranging from nanoseconds (e.g., domain reorientation) to tens or hundreds of seconds (e.g., proline isomerization; Guttman et al., 2020; Zosel et al., 2018). Nevertheless, the isolated—that is, unbound—structures of fold switchers tend to be less stable and more heterogeneous than single folders (Bryan & Orban, 2010) but more stable and less heterogeneous than IDPs.
FIGURE 1.
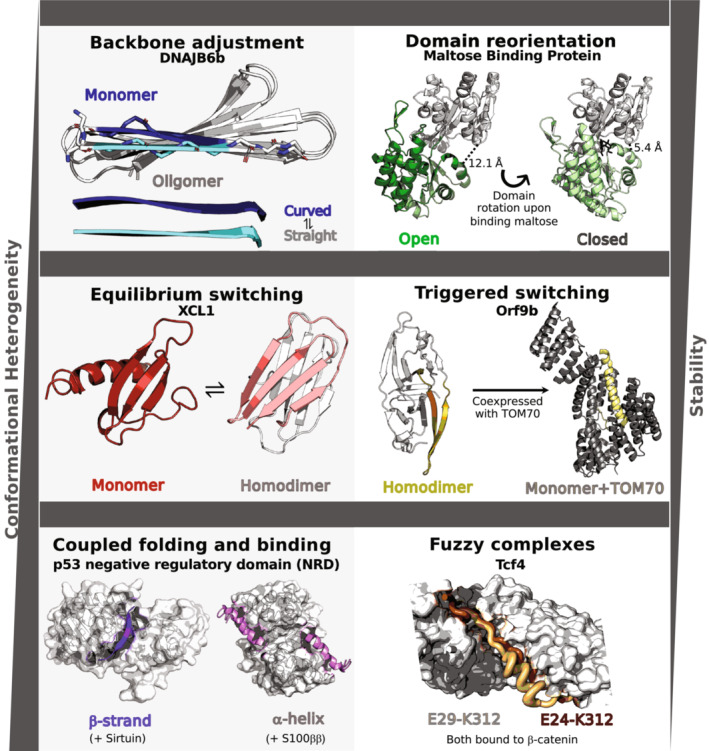
Increasing conformational heterogeneity and decreasing stability are generally observed among single‐fold, fold‐switching, and intrinsically disordered proteins, respectively. Examples of single‐fold proteins include backbone adjustment and domain reorientation. The former is illustrated by DNAJB6b, a chaperone whose N‐terminal β‐strand is twisted in its monomeric configuration (dark blue, PDB ID: 7JSQ, model 1) but straight in oligomers (cyan, PDB ID: 6U3R, Model 1); C‐terminal β‐sheets are light gray. The orientation of Maltose Binding Protein's N‐terminal (light gray) and C‐terminal domains differ in the open apo (dark green, PDB ID: 1JW4, Chain A) and close maltose‐bound (light green, 1ANF, Chain A) forms. Interdomain distances in both conformations are annotated. Structural heterogeneity in fold‐switching proteins can be categorized into equilibrium switching, in which both conformations are populated at detectable levels under certain conditions, and triggered switching, in which an external stimulus shifts the protein from one dominant fold to another. Equilibrium switching is exemplified by XCL1, a human chemokine that populates a monomeric α + β fold (red, PDB ID: 1J9O, Model 1) and a dimeric β‐sheet fold with a completely different hydrogen bond network (one unit salmon, the other light gray, PDB ID: 2 N54, Model 1) at about a 50:50 ratio under physiological conditions. Orf9b illustrates triggered switching; expressed in isolation, this severe acute respiratory syndrome coronavirus‐2 protein folds into a β‐sheet homodimer (olive, PDB ID: 6Z4U, Chains A and B), while a portion of it folds into an α‐helix (yellow, PDB ID: 7DHG, Chain B) when coexpressed with human TOM70 (light gray, PDB ID: 7DHG, Chain A), which it binds in situ. Intrinsically disordered proteins, which display the most structural heterogeneity, can fold into different secondary structures upon binding different partners (Coupled folding and binding) and upon binding the same partner (Fuzzy complex). Human tumor suppressor protein p53 demonstrates coupled folding and binding both by folding into a β‐sheet (purple, PDB ID: 1MA3, Chain B) when complexed with Sirtuin (gray surface, PDB ID: 1MA3, Chain A) and by folding into an α‐helix (pink, PDB ID: 1DT7, Chains X and Y) when complexed with S100ββ (gray surface, PDB ID: 1DT7, Chains A and B). Finally, Tcf4 forms a fuzzy complex with β‐catenin (gray surface, PDB ID: 1G3J, Chain A); two distinct bound conformations are shown in light orange (PDB ID: 1JDH, Chain B) and brown (PDB ID: 1G3J, Chain B). This brown conformation is a proposed model only as it shows the conformation of a highly similar homolog, Tcf3. Mutagenesis experiments demonstrate that Tcf4 can interact with β‐catenin in manners consistent with both structures.
2.1. Single‐fold proteins maintain similar secondary and tertiary structures in the folded state
Backbone adjustments have been observed in the C‐terminal domain (CTD) of human DNAJB6b (Figure 1), an aggregation‐suppressing chaperone in the Heat Shock Protein 40 family. Its dominant monomeric conformation assumes a twisted β1‐strand conformation, while its sparsely populated homo‐oligomeric forms adopt flat β1‐strands (Karamanos et al., 2020). Occurring on the microsecond timescale (Cawood et al., 2022), this reversible structural interconversion affects the lengths and interactions of the neighboring β2‐strands and β3‐strands and likely impacts DNAJB6b's ability to assemble into functional homo‐oligomers (Karamanos et al., 2020). Homodimers—precursors to homo‐oligomers—form by β1‐strand association, likely facilitated by flat—but not twisted—β1‐strands. Indeed, a T142A mutation in the β1‐strand assumes the twisted conformation more frequently than wild‐type, increasing the dimer dissociation constant by a factor of 2.5 and lowering the population of oligomers by a factor of ~6 (Cawood et al., 2022). This mutationally triggered a decrease in homo‐oligomers likely inhibits Aβ42 amyloid fibril formation (Mansson et al., 2018).
Domain rearrangements, such as those observed in the bacterial periplasmic maltose‐binding protein (MBP; Figure 1), occur in many single‐fold proteins (Daily & Gray, 2007). Flexible loops and/or linkers interconnecting domains often function as hinges or pivot points for these rearrangements. Accordingly, amino acid substitutions within linkers can affect conformational rearrangement rates significantly (Aggarwal et al., 2022; Borden et al., 2020; Tian et al., 2009). The two domains of MBP's unbound (open) conformation are further apart than in its closed conformation, when it is bound to a substrate such as maltose (Figure 1). Interestingly, in the absence of ligand, unbound MBP undergoes conformational exchange on the nanosecond‐microsecond timescale between a dominant (~95%) open form and a minor (~5%) partially closed form (Tang et al., 2007). Thus, it may be tempting to hypothesize that MBP binds maltose only when it populates the partially closed form, a mechanism known as conformational selection. If so, the rate of MBP closing would limit its binding rate regardless of ligand concentration. The opposite was observed by single‐molecule Förster Resonance Energy Transfer experiments, which demonstrated that the closing rates of an MBP variant increase linearly with ligand concentration, strongly supporting an alternative induced‐fit mechanism whereby the ligand binds the open form of MBP and causes it to close (Kim et al., 2013). The sequence of the characterized MBP variant differed from wild‐type by one amino acid, and the authors proposed that wild‐type also functions by induced fit (Kim et al., 2013).
2.2. Fold‐switching proteins assume stable folds but remodel their secondary and tertiary structures in response to their environments
Contrasting the motions observed in the single‐fold proteins, which preserve all or most of the integrity of their folds, the fold‐switching transition of autoimmune disease associated (Lei & Takahama, 2012) human chemokine XCL1 involves completely repacking its hydrophobic core, reforming its hydrogen bonding network, and changing from an α/β to an all‐β fold (Dishman et al., 2021). This transition occurs slowly—on a timescale of seconds (Volkman et al., 2009). Under physiological conditions, XCL1 folds into two (approximately) equally populated conformations (Tuinstra et al., 2008): a monomeric α + β fold that activates the G‐protein coupled receptor XCR1 and dimeric fold that binds glycosaminoglycans (Figure 1) and may have antimicrobial activity (Dishman et al., 2021). Although the mechanistic details of XCL1's structural interconversion remain unsettled (Khatua et al., 2020; Tyler et al., 2011), a recent mutational study identified three important factors that contribute to its fold switching: (1) a stable dimer interface, (2) loop mutations that increase flexibility and/or strain, and (3) noncovalent amino acid contacts compatible with both folds (Dishman et al., 2021).
To date, few fold‐switching proteins have been found to populate two conformations simultaneously (Chakravarty & Porter, 2022) as XCL1 does. More have been observed to assume one dominant fold that shifts to an alternative conformation in response to environmental changes (Park et al., 2011; Stein et al., 1991; Xu et al., 2005; Zuber et al., 2019). For instance, when expressed in isolation, the SARS‐CoV‐2 protein Orf9b folds into a β‐sheet homodimer, but when coexpressed with its binding partner, human TOM70, the two proteins form a complex in which a ~35‐residue segment of Orf9b folds into a long α‐helix (Figure 1). Orf9b‐TOM70 interactions have been observed by x‐ray crystallography (Gao et al., 2021) and by cryogenic electron microscopy (cryo‐EM) along with coimmunoprecipitation and imaging in human cell lines (Gordon et al., 2020). These interactions are linked to suppression of type I interferon‐β (Jiang, Zhang, et al., 2020), a cytokine involved in antiviral immune responses (Murira & Lamarre, 2016). Immunoglobulin G (IgG) antibodies against Orf9b present in the sera of some convalescent SARS‐CoV‐2 patients (Jiang, Li, et al., 2020) further suggest an antigenic function. Interestingly, dimeric Orf9b does not bind to TOM70; instead, it requires coexpression (Gao et al., 2021) or possibly some other exogeneous factor in vivo. Other fold‐switching proteins can switch dominant states without changing their expression conditions. For instance, the C‐terminal domain of the bacterial transcription factor RfaH assumes a ground state α‐helical fold with no trace of its alternative β‐sheet fold, to which it switches when its N‐terminal domain binds both RNA polymerase and the operon polarity suppressor DNA sequence (Zuber et al., 2019). Interestingly, a recent biophysical study (Zuber et al., 2022) demonstrated that the unfolded state of RfaH's CTD may poise it to switch folds. Specifically, its folded β‐strand conformation exchanges with a minor (~5%) partially α‐helical conformation at a rate of 15.0 s−1. The thermodynamic stability of this fold‐switching domain is also less thermodynamically stable than its single‐fold counterparts (Zuber et al., 2022).
2.3. IDPs lack stable secondary and tertiary structures in isolation; their binding partners select their conformations
Like Orf9b and RfaH, the intrinsically disordered C‐terminal negative regulatory domain (NRD) of tumor suppressor protein p53 (Rustandi et al., 2000) can fold into a β‐strand or an α‐helix (Oldfield et al., 2008), but its folded conformation is determined by its binding partner (Figure 1). Within a cleft of the deacetylating enzyme sirtuin (Sir2), the NRD peptide with acetylated K382 forms a β‐strand (Avalos et al., 2002) that links the two Sir2 domains together. Importantly, this posttranslational modification (PTM) alters DNA binding and likely occurs in response to DNA damage (Sakaguchi et al., 1998); human Sir2 can deacetylate this site in full‐length p53 (Avalos et al., 2002), regulating its activity (Borra et al., 2005). Contrastingly, in the presence of Ca2+, the p53 NRD binds to human S100ββ and folds into an α‐helix (Rustandi et al., 2000). This interaction blocks two phosphorylation and two acetylation sites, whose modifications are likely important for p53 activation (Rustandi et al., 2000). S100ββ binding rates from linewidth analysis of nuclear magnetic resonance (NMR) experiments suggest that, like MBP, the p53 NRD folds into a helix after binding, an induced‐folding (Berlow et al., 2015) or “fly‐casting” (Chen, 2009) mechanism that occurs on the sub‐microsecond timescale (Wafer et al., 2012). It is unclear how PTMs affect the NRD's secondary structure propensities, though they can affect the conformational propensities of other IDPs both in vitro (Bah et al., 2015) and in situ (Mylona et al., 2016), and multisite acetylation in full‐length p53 alters the chemical environment of some NRD residues (Krois et al., 2022). Various computational approaches suggest that the NRD peptide—unfolded in isolation—nevertheless has mixed secondary structure propensities (Kannan et al., 2016; Kumar et al., 2020), perhaps allowing it to assume different secondary structures with different partners.
While some IDPs assume a well‐defined fold when bound to their macromolecular partners, others remain unstructured or populate multiple stable conformations. The structural heterogeneity of these “fuzzy complexes” varies (Tompa & Fuxreiter, 2008). Interactions between intrinsically disordered transcription factor Tcf4 and transcriptional coactivator β‐catenin exemplify a polymorphic fuzzy complex in which Tcf4 assumes multiple distinct conformations. Cellular accumulation of β‐catenin is associated with many forms of cancer (Graham et al., 2001). Accordingly, interactions between β‐catenin and Tcf factors activate the transcription of several critical genes involved in cell proliferation, and Tcf4 is the major Tcf present in colon cancer cells (Graham et al., 2001). The crystal structure of the β‐catenin‐Tcf4 complex showed a well‐resolved structure of Tcf4 (Figure 1, orange), though it is intrinsically disordered in isolation. Interestingly, the conformation of Tcf3 (Graham et al., 2000), a highly similar homolog of Tcf4, complexed with β‐catenin differed from Tcf4 (Figure 1, brown). Importantly, regions of these proteins with identical sequences formed different functionally important salt bridges with K312 of β‐catenin: E24 in Tcf3 and E29 in Tcf4. Mutagenesis studies showed that E24 of Tcf4 can also form a salt bridge with K312, suggesting that Tcf4 can bind β‐catenin in multiple distinct conformations (Graham et al., 2001). More recent computational work suggests that both the homogeneity/heterogeneity of similarly charged residues and the charge patterns in IDPs can bias the thermodynamics of fuzzy complexes and may also contribute to binding specificity (Hazra & Levy, 2022). IDP–IDP interactions that occur in various protein condensates formed by liquid–liquid phase separation (Brangwynne et al., 2015) may also form fuzzy complexes. For instance, NMR and Raman spectroscopies did not provide any evidence showing that the low complexity domain of FUS, an RNA‐binding IDP, assumes traditional secondary structure in its liquid condensed phase; rather, it remains structurally heterogeneous, and nonspecific interactions between disordered subunits underlie its liquid–liquid phase separation (Murthy et al., 2019). A recent preprint reported similar findings: by combining single‐molecule FRET experiments with molecular dynamics simulations, Schuler and colleagues observed similar short‐range molecular environments in the dilute and condensed phases of the histone H1‐prothymosin α protein pair (Galvanetto et al., 2022).
3. EVOLUTION OF SINGLE FOLDERS, FOLD SWITCHERS, AND IDPS
Decades of research have established that single‐fold protein families have distinctive mutational patterns (Figures 2a,b). Increasingly sophisticated algorithms have leveraged these patterns to associate highly dissimilar sequences with similar folds and functions (Altschul et al., 1997; Bateman et al., 2004; Eddy, 2009; Remmert et al., 2011; Steinegger et al., 2019), and a recent deep learning approach has successfully used them to improve functional inference (Bileschi et al., 2022). In contrast, IDPs tend to evolve rapidly, and their sequences often lack obvious conservation patterns (Figures 2a,b). This absence of widespread conservation often confounds homology inference (Pancsa et al., 2018), though functionally important IDP/IDR subsequences known as molecular recognition features (Mohan et al., 2006) and short‐linear motifs (Van Roey et al., 2014) can be more highly conserved (van der Lee et al., 2014).
FIGURE 2.
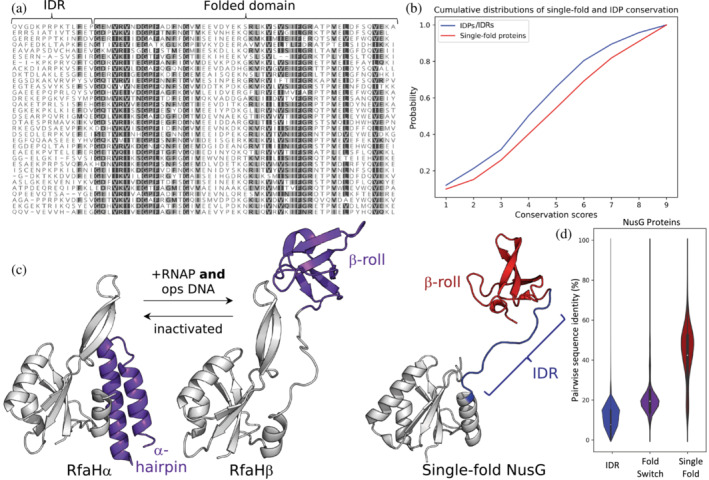
Sequence conservation patterns can differ among single‐fold, fold‐switching, and intrinsically disordered proteins. (a) The region of a NusG sequence alignment depicting its linker, an intrinsically disordered region (IDR), shows little sequence conservation while its single‐fold C‐terminal domain (folded domain) shows more. Conserved residues with dark gray/light gray backgrounds can be substituted with BLOSUM62 scores of 0 or higher and constitute 80%–99%/60%–80% of the amino acids in each column. Less conserved residues have white backgrounds. Alignment was generated by searching the Uniref30 database from February 2022 with the sequence of PDB 2JVV_1 using the HHblits online alignment tool (Steinegger et al., 2019; https://toolkit.tuebingen.mpg.de/tools/hhblits); alignment generated with Geneious Prime. (b) Consistent with the sequence alignment in (a), IDPs evolve more rapidly than single‐fold proteins. Cumulative distributions of conservation scores, calculated with Rate4Site in Chakravarty & Porter (2022), indicate that a sample of 100 randomly chosen IDPs evolves more quickly than a set of single‐fold proteins. Larger scores indicate stronger conservation and slower evolutionary rates. (c) RfaH, a member of the universally conserved NusG transcription factor family, has a C‐terminal domain (purple) that switches between completely α‐helical (PDB ID: 2OUG, Chain C) and β‐sheet folds (PDB ID: 6C6S, Chain D) in response to binding RNA polymerase and a specific DNA sequence known as ops. In contrast, the C‐terminal domains (CTDs) of all other NusGs with solved structures maintain the β‐sheet fold only (red). Their intrinsically disordered linker (blue) corresponds to the IDR region in (a); note that it lacks crystal density in the structure of RfaHα. N‐terminal domains of NusG and RfaH are colored gray. (d). Among NusG proteins, conformational heterogeneity and pairwise sequence identity appear inversely related. Median values (white dots) within the distributions of pairwise sequence identities between NusG IDRs (blue), fold‐switching CTDs (purple), and single‐fold CTDs (red) increase. Interquartile ranges are depicted within the distributions as bold black lines. Plots in (b) and (c) generated with matplotlib (Hunter, 2007) and seaborn (Waskom, 2021).
How conserved are fold‐switching sequences compared with those of single folders and IDPs? One might expect robust conservation since fold‐switching sequences must specify two stable conformations. Nevertheless, a recent study contradicts that expectation (Porter et al., 2022). It focused on bacterial NusG transcription factors, which share a two‐domain architecture: a highly conserved N‐terminal NGN domain that binds RNA polymerase and a less conserved C‐terminal domain (CTD) that interacts with proteins involved in various biochemical pathways (Kang et al., 2018), such as the integral ribosomal subunit, S10 (Burmann et al., 2012). While the CTDs of most structurally characterized NusGs share a conserved β‐roll fold (Figure 2c), the CTD of a specialized member of the family, known as RfaH, has been shown to switch reversibly from a completely α‐helical ground state fold into an excited β‐roll conformation (Figure 2c) upon binding both RNA‐polymerase and a specific DNA sequence, ops (Zuber et al., 2019). Computational predictions, based on inconsistencies between inferred secondary structures (Kim et al., 2021; Mishra et al., 2019, 2021), suggested that 24% of NusG proteins switch folds. These predictions were confirmed by circular dichroism and NMR experiments characterizing 10/10 dissimilar NusG variants (Porter et al., 2022). Pairwise sequence identity distributions (Figure 2d) indicated that the diversity of fold‐switching NusG sequences exceeded that of their single‐fold counterparts. By comparison, pairwise sequence identities of the disordered linker connecting N‐terminal and C‐terminal NusG domains are the lowest, indicating the most diversity (Figure 2D). The sequence diversity of fold switchers may depend on the protein family, however. Phylogenetic analysis suggests that ancestors with <60% sequence identity to fold‐switching protein XCL1 (Figure 1) lose their ability to switch folds (Dishman et al., 2021), and an overall comparison of the evolutionary rates between 98‐fold switching and single‐fold found no statistically significant difference (Chakravarty & Porter, 2022).
4. SEQUENCE‐BASED PREDICTIONS OF STRUCTURAL HETEROGENEITY IN PROTEINS
Deep‐learning‐based methods have revolutionized protein structure prediction. Before 2020, the median heavy‐atom root mean square deviations between well predicted and experimentally determined single‐domain protein structures had stagnated around 6 Å (AlQuraishi, 2021). This accuracy metric dropped precipitously to ~2 Å in 2020 after the development of AlphaFold2 (AlQuraishi, 2021). Since then, other deep‐learning‐based methods have been developed to accurately predict conformations of interacting proteins (Baek et al., 2021; Evans et al., 2022), conformations of orphan protein sequences (Chowdhury et al., 2022), and the folds of ~617 million metagenomic sequences (Lin et al., 2022). AlphaFold2 may have also learned some physical principles of protein folding (Roney & Ovchinnikov, 2022), though not enough to reproduce experimentally observed folding pathways (Outeiral et al., 2022). This is not surprising since pattern recognition—rather than biophysics—underlies deep learning algorithms (Chen et al., 2023; Rose, 2021).
The limitations of deep‐learning‐based methods are evidenced by their predictions of fold switchers and somewhat for IDPs. For instance, AlphaFold2 consistently predicts one conformation of fold‐switching proteins and favors the excited conformation 30% of the time (Chakravarty & Porter, 2022), indicating that it cannot always decipher the lowest energy states of fold switchers. Additionally, it often does not distinguish between the sequences of fold‐switching and single‐fold proteins, both of which are predicted with high confidence (Chakravarty & Porter, 2022). Furthermore, AlphaFold2, Robetta, EVCouplings, and PHYRE2 all failed to predict the ground‐state α‐helical conformation of a sequence‐dissimilar homolog of RfaH whose secondary structure was determined by NMR (Porter et al., 2022). Instead, they predicted ground‐state β‐sheet folds, as do RoseTTAfold, ESM‐fold, and RGN2 (Figure 3a). Relatedly, AlphaFold2 does not accurately model the conformational heterogeneity of IDPs (Ruff & Pappu, 2021). Nevertheless, AlphaFold2's uncertainty metric, known as its per‐residue local distance difference test (pLDDT) score, can be used to infer intrinsic disorder with considerable accuracy because AlphaFold2 is not confident in its predictions of IDPs (He et al., 2022). Conditionally folding IDPs can sometimes be inferred from AlphaFold2 predictions also: ~60% of a set of 350 IDPs experimentally observed to fold conditionally have high pLDDT scores (Alderson et al., 2022). Other bioinformatic methods can also infer intrinsic disorder and conditional folding with reasonable accuracy (Disfani et al., 2012; Erdos et al., 2021; Xue et al., 2010).
FIGURE 3.
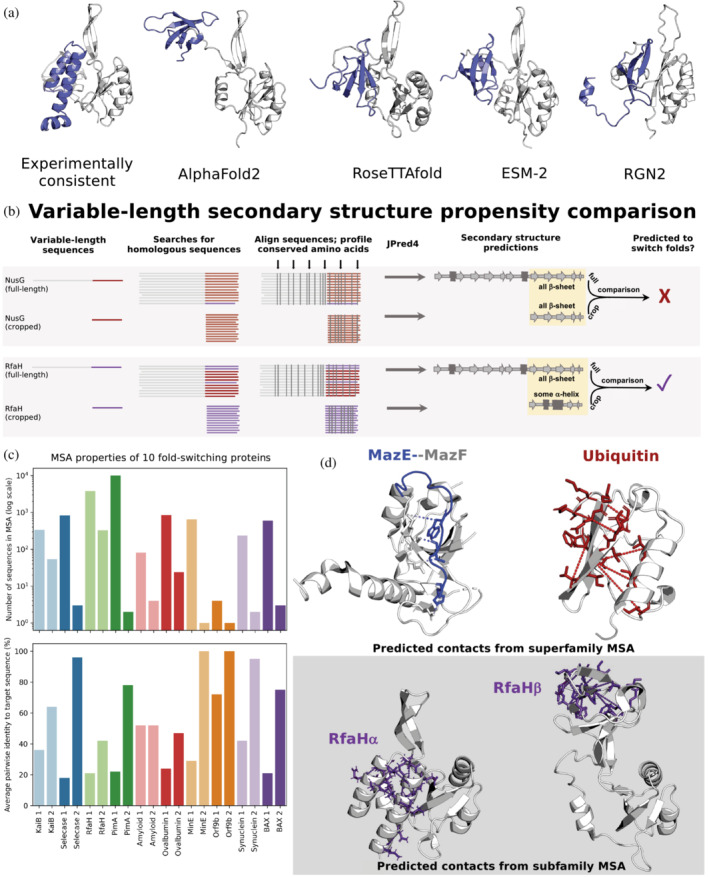
Predictive features of fold‐switching proteins. (a) All state‐of‐the‐art deep‐learning‐based methods fail to predict the experimentally characterized α‐helical ground state of Variant 5, a member of the NusG family with ≤29% sequence identity to its homologs with experimentally determined structures (Porter et al., 2022). The 3D helical model of Variant 5 (left), generated using Rosetta‐CM (Song et al., 2013) with RfaH (PDB 5OND, Chain A) as a template, is consistent with its chemical‐shift‐derived secondary structures (Porter et al., 2022). In contrast, all state‐of‐the‐art methods, including trRosetta (Du et al., 2021), EVCouplings (Hopf et al., 2019), and PHYRE2 (Kelley et al., 2015; shown in Porter et al., 2022), predict the activated β‐roll fold. In all cases, Variant 5's fold‐switching CTD is slate while its single‐fold NTD is gray. (b) Contrastingly, Variant 5's ground state α‐helical fold was successfully inferred from variable‐length secondary structure propensity comparison. While both full‐length and cropped NusG sequences have similar amino acid conservation patterns (gray vertical lines, top gray panel), conservation patterns differ for full‐length and cropped RfaH (gray vertical lines, bottom gray panel). Similar/different full‐length and cropped conservation patterns lead to similar/different secondary structure predictions, suggesting that NusG does not switch folds (top) while RfaH does (bottom). These different patterns likely result from different multiple sequence alignment (MSA) homogeneities. The sequence distributions depicted are for illustrative purposes only since true sequence distributions are unknown. (c) For 10 fold‐switching proteins successfully predicted by JPred4 (Mishra et al., 2021), full‐length alignments yielding secondary structure prediction 1 are deeper (plot above) and more diverse (plot below), indicating the presence of both fold‐switching and single‐fold sequences. In contrast, cropped sequence MSAs, yielding secondary structure prediction 2, are shallower (plot above) and more similar to the target sequence (plot below), reflecting fold‐switching subfamily properties. (d). Some single‐fold, fold‐switching, and intrinsically disordered proteins coevolve differently. The bacterial IDP MazE (blue, PDB ID: 5CQX, Chain C) coevolves with its folded binding partner, MazF (gray, PDB ID: 5CQX, Chain A); predicted contacts taken from Pancsa et al. (2018). Stablizing intrachain contacts coevolve in single‐folding ubiquitin (red, PDB ID: 1UBQ, Chain A); predicted contacts generated using GREMLIN (Balakrishnan et al., 2011; Kamisetty et al., 2013). In contrast, GREMLIN can successfully predict inter‐residue contacts unique to both folds of RfaH when using an MSA composed of sequences that yield similar partially helical JPred4 predictions (Porter et al., 2022).
Although sophisticated deep learning methods are currently unable to consistently predict both conformations of fold‐switching proteins, recent work has shown that inconsistent secondary structure predictions can be used to infer fold switching (Figure 3b; Chen et al., 2020; Kim et al., 2021; Mishra et al., 2019, 2021; Porter & Looger, 2018). This approach has been applied successfully to RfaH (Porter et al., 2022) and nine other fold‐switching proteins that undergo α‐helix ↔ β‐strand transitions (Mishra et al., 2021), including Orf9b (Porter, 2021).
JPred4 (Drozdetskiy et al., 2015), the secondary structure predictor that best identifies fold switchers overall (Mishra et al., 2019), requires a multiple sequence alignment (MSA) as input. MSAs with different depths and similarities to their sequence targets yield different secondary structure predictions that can be leveraged to infer fold switching (Figure 3b; Porter et al., 2022). Though initially recognized in the RfaH family, this observation applies to the other nine JPred4‐predicted fold‐switching proteins (Mishra et al., 2021) as well (Figure 3c), indicating that MSAs of varying depths and diversity might provide more information relevant to fold switching than can be obtained from a deep MSA alone (Schafer & Porter, 2023). It should be noted, however, that sequence lengths also vary between JPred4 predictions, and their effects on inference have not been examined systematically.
AlphaFold2 also relies on MSAs to make reliable predictions (Jumper et al., 2021). Several studies suggest that inputting shallow MSAs (Del Alamo et al., 2021; Del Alamo et al., 2022) or masking coevolutionary patterns (Stein & McHaourab, 2022) can steer AlphaFold2 to predict alternative protein conformations accurately. Could this approach work for fold switchers? A recent preprint reports that AlphaFold2 was able to accurately predict the alternative conformations of 3/6‐fold switchers when shallow, subfamily‐specific MSAs were inputted (Wayment‐Steele et al., 2022). Furthermore, AlphaFold2 predicted α‐helix ↔ β‐sheet switching in reconstructed ancestors of bacterial response regulator proteins with high pairwise sequence identity (Chakravarty et al., 2022).
Interestingly, emerging work suggests that fold‐switching proteins may have distinctive coevolutionary patterns (Figure 3d), which play a central role in deep‐learning‐based protein structure prediction (Jumper et al., 2021; Lin et al., 2022; Rao et al., 2021). Coevolution describes when the occurrence of an amino acid at a site in a protein's sequence depends upon the identities of amino acids at other positions (Ivankov et al., 2014). In folded proteins, coevolving amino acid pairs usually contact one another directly (Anishchenko et al., 2017), typically stabilizing protein structure or promoting function. Coevolved contacts can greatly limit the number of possible conformations that computational methods must sample to predict a protein's fold (Yang et al., 2020), driving the development of increasingly sensitive methods that infer amino acid coevolution from MSAs (Lockless & Ranganathan, 1999; Morcos et al., 2011; Rao et al., 2021). Single‐fold proteins with deep MSAs tend to display robust coevolution between many diverse amino acid pairs proximal in experimentally determined structures of single‐fold proteins (e.g. Figure 3d). In contrast, because IDPs often evolve quickly, it can be difficult to obtain MSAs with high enough quality to perform robust coevolutionary analysis. Interestingly, a recent coevolutionary study indicates that amino acids in some IDPs coevolve with those of their binding partners (Pancsa et al., 2018; Figure 3d), suggesting that exogenous factors may influence the evolution of some IDPs. To our knowledge, only one systematic study has analyzed the intrachain IDP coevolution (Basu & Bahadur, 2022). It suggested that residues in IDPs may coevolve—not to form residue‐residue contacts—but to maintain high intrachain entropy. By comparison, recent work indicates that residue–residue contacts unique to both conformations of RfaH (Porter et al., 2022) and KaiB (Wayment‐Steele et al., 2022) coevolve, suggesting that their structural interconversions may confer evolutionary advantage (Schafer & Porter, 2023). If the coevolution of fold‐switching proteins is a general phenomenon, it may provide the key to accurately predicting two 3D structures from one amino acid sequence.
5. EXPERIMENTALLY CHARACTERIZING AND SCREENING FOLD SWITCHERS
Computational methods that predict which proteins switch folds require subsequent experimental testing. Solution NMR has been a method of choice because it can recognize multiple conformations exchanging on a slow timescale (Dishman et al., 2021; Tuinstra et al., 2008) and sparsely populated conformations that may facilitate fold‐switch transitions (Cai et al., 2019; Zuber et al., 2022). Recently, 19F NMR experiments have also probed fold switching of the tuberculosis‐associated protein PimA (Liebau et al., 2020), demonstrating another avenue for probing the dynamics fold switching. These experiments could potentially monitor fold switching in situ as well (Thole et al., 2021). Additionally, single‐molecule experiments that have successfully characterized protein misfolding pathways (Yu et al., 2012) could potentially be used to characterize intermediates that occur during fold‐switch transitions. Despite their utility and promise, neither NMR nor single‐molecule experiments can be used for high‐throughput screening of fold‐switching candidates because they tend to be slow; NMR experiments can also be expensive, and they require large amounts of pure isotopically labeled protein.
Several experimental alternatives could potentially be used to screen for fold switching. Hydrogen‐Deuterium eXchange Mass Spectroscopy (HDX‐MS) has successfully distinguished between two different prefusion conformations of the SARS‐CoV‐2 spike protein trimer (Costello et al., 2022), suggesting that it may be sensitive enough to distinguish between two alternatively folded populations of proteins exchanging on a slow timescale. Since fold switching often occurs on the order of seconds or slower (Liebau et al., 2020; Luo et al., 2004; Solomon et al., 2023; Volkman et al., 2009; Zuber et al., 2022), HDX‐MS is a promising screen. Additionally, FRET experiments can be used to probe protein dynamics in vitro (Feng et al., 2019), suggesting another promising screen if the predicted conformations of fold switchers have different end‐to‐end distances. Finally, circular dichroism experiments were recently used to screen for fold switching in the NusG protein family (Porter et al., 2022). These experiments were consistent with subsequent NMR analyses and likely succeeded because the ground state structures of fold‐switching RfaHs have considerably more helical content than single‐folding NusGs.
6. CONCLUSIONS
Several features of proteins can reveal where they might fall on the structural heterogeneity and stability scales (Figure 1). First, in general, the isolated structures of fold switchers are less stable and more heterogeneous than single folders but more stable and less heterogeneous than IDPs. Second, the rates of amino acid variation differ between homologous sequences of single folders, fold switchers, and IDPs. Third, single folders, fold switchers, and IDPs are best predicted using different computational techniques. Finally, emerging results suggest that these three protein classes may have distinct patterns of coevolution. Many discoveries related to fold‐switching proteins have occurred in the past 5 years. By combining recent advances in deep learning with state‐of‐the‐art experimental techniques, it may become possible to predict two stable conformations from one sequence accurately and consistently in the next 5 years.
AUTHOR CONTRIBUTIONS
Devlina Chakravarty: Conceptualization (equal), visualization (supporting), and writing – original draft (lead). Joseph W. Schafer: Visualization (supporting) and writing – review and editing (supporting). Lauren L. Porter: Conceptualization (equal), formal analysis (lead), funding acquisition (lead), supervision (lead), visualization (lead), and writing – review and editing (lead).
ACKNOWLEDGMENTS
This work was carried out by staff of the National Library of Medicine (NLM), National Institutes of Health, with support from NLM (ZIA 202011‐04).
Chakravarty D, Schafer JW, Porter LL. Distinguishing features of fold‐switching proteins. Protein Science. 2023;32(3):e4596. 10.1002/pro.4596
Review Editor: John Kuriyan
Funding information U.S. National Library of Medicine, Grant/Award Number: ZIA 202011‐04; National Institutes of Health
REFERENCES
- Aggarwal A, Liu R, Chen Y, Ralowicz AJ, Bergerson SJ, Tomaska F, Hanson TL, Hasseman JP, Reep D, Tsegaye G. 2022. Glutamate indicators with improved activation kinetics and localization for imaging synaptic transmission. bioRxiv. 2022; 10.1101/2022.02.13.480251 [DOI] [PMC free article] [PubMed]
- Alderson TR, Pritišanac I, Moses AM, Forman‐Kay JD. 2022. Systematic identification of conditionally folded intrinsically disordered regions by Alphafold2. bioRxiv. 2022; 10.1101/2023.01.18.524637 [DOI] [PMC free article] [PubMed]
- AlQuraishi M. Machine learning in protein structure prediction. Curr Opin Chem Biol. 2021;65:1–8. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishchenko I, Ovchinnikov S, Kamisetty H, Baker D. Origins of coevolution between residues distant in protein 3d structures. Proc Natl Acad Sci U S A. 2017;114(34):9122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10(3):523–35. [DOI] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373(6557):871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519(7541):106–9. [DOI] [PubMed] [Google Scholar]
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79(4):1061–78. [DOI] [PubMed] [Google Scholar]
- Basu S, Bahadur RP. Conservation and coevolution determine evolvability of different classes of disordered residues in human intrinsically disordered proteins. Proteins. 2022;90(3):632–44. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths‐Jones S, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32(Database issue):D138–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow RB, Dyson HJ, Wright PE. Functional advantages of dynamic protein disorder. FEBS Lett. 2015;589(19 Pt A):2433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bileschi ML, Belanger D, Bryant DH, Sanderson T, Carter B, Sculley D, et al. Using deep learning to annotate the protein universe. Nat Biotechnol. 2022;40(6):932–7. [DOI] [PubMed] [Google Scholar]
- Borden PM, Zhang P, Shivange AV, Marvin JS, Cichon J, Dan C, Podgorski K, Figueiredo A, Novak O, Tanimoto M. A fast genetically encoded fluorescent sensor for faithful in vivo acetylcholine detection in mice, fish, worms and flies. bioRxiv. 2020; 10.1101/2020.02.07.939504 [DOI]
- Borra MT, Smith BC, Denu JM. Mechanism of human sirt1 activation by resveratrol. J Biol Chem. 2005;280(17):17187–95. [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11(11):899–904. [Google Scholar]
- Bryan PN, Orban J. Proteins that switch folds. Curr Opin Struct Biol. 2010;20(4):482–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann BM, Knauer SH, Sevostyanova A, Schweimer K, Mooney RA, Landick R, et al. An alpha helix to beta barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell. 2012;150(2):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Huang Y, Shen Y, Li M, Mizuuchi M, Ghirlando R, et al. Probing transient excited states of the bacterial cell division regulator MinE by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci U S A. 2019;116(51):25446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawood EE, Clore GM, Karamanos TK. Microsecond backbone motions modulate the oligomerization of the dnajb6 chaperone. Angew Chem Int Ed Engl. 2022;61(20):e202116403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Porter LL. Alphafold2 fails to predict protein fold switching. Protein Sci. 2022;31(6):e4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Sreenivasan S, Swint‐Kruse L, Porter L. 2022. Identification of a covert evolutionary pathway between two protein folds. bioRxiv. 2022; 10.1101/2022.12.08.519646 [DOI] [PMC free article] [PubMed]
- Chavan AG, Swan JA, Heisler J, Sancar C, Ernst DC, Fang M, et al. Reconstitution of an intact clock reveals mechanisms of circadian timekeeping. Science. 2021;374(6564):eabd4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Intrinsically disordered p53 extreme c‐terminus binds to S100B(betabeta) through "fly‐casting". J Am Chem Soc. 2009;131(6):2088–9. [DOI] [PubMed] [Google Scholar]
- Chen N, Das M, LiWang A, Wang LP. Sequence‐based prediction of metamorphic behavior in proteins. Biophys J. 2020;119(7):1380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Hassan M, Jernigan RL, Jia K, Kihara D, Kloczkowski A, et al. Opinion: protein folds vs. protein folding: differing questions, different challenges. Proc Natl Acad Sci U S A. 2023;120(1):e2214423119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R, Bouatta N, Biswas S, Floristean C, Kharkare A, Roye K, et al. Single‐sequence protein structure prediction using a language model and deep learning. Nat Biotechnol. 2022;41(11):1617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello SM, Shoemaker SR, Hobbs HT, Nguyen AW, Hsieh CL, Maynard JA, et al. The SARS‐CoV‐2 spike reversibly samples an open‐trimer conformation exposing novel epitopes. Nat Struct Mol Biol. 2022;29(3):229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily MD, Gray JJ. Local motions in a benchmark of allosteric proteins. Proteins. 2007;67(2):385–99. [DOI] [PubMed] [Google Scholar]
- Del Alamo D, Govaerts C, McHaourab HS. Alphafold2 predicts the inward‐facing conformation of the multidrug transporter LMRP. Proteins. 2021;89(9):1226–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Alamo D, Sala D, McHaourab HS, Meiler J. Sampling alternative conformational states of transporters and receptors with Alphafold2. Elife. 2022;11:e75751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, et al. MorfPred, a computational tool for sequence‐based prediction and characterization of short disorder‐to‐order transitioning binding regions in proteins. Bioinformatics. 2012;28(12):i75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishman AF, Tyler RC, Fox JC, Kleist AB, Prehoda KE, Babu MM, et al. Evolution of fold switching in a metamorphic protein. Science. 2021;371(6524):86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, Barton GJ. Jpred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43(W1):W389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Su H, Wang W, Ye L, Wei H, Peng Z, et al. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc. 2021;16(12):5634–51. [DOI] [PubMed] [Google Scholar]
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23(1):205–11. [PubMed] [Google Scholar]
- Erdos G, Pajkos M, Dosztanyi Z. Iupred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021;49(W1):W297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, O'Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J. 2022. Protein complex prediction with Alphafold‐multimer. BioRxiv.2021.2010. 2004.463034.
- Feng R, Gruebele M, Davis CM. Quantifying protein dynamics and stability in a living organism. Nat Commun. 2019;10(1):1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvanetto N, Ivanović MT, Chowdhury A, Sottini A, Nüesch M, Nettels D, Best R, Schuler B. 2022. Ultrafast molecular dynamics observed within a dense protein condensate. bioRxiv. 2022; 10.1101/2022.12.12.520135 [DOI]
- Gao X, Zhu K, Qin B, Olieric V, Wang M, Cui S. Crystal structure of SARS‐CoV‐2 Orf9b in complex with human Tom70 suggests unusual virus‐host interactions. Nat Commun. 2021;12(1):2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, et al. Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science. 2020;370(6521):eabe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TA, Ferkey DM, Mao F, Kimelman D, Xu W. Tcf4 can specifically recognize beta‐catenin using alternative conformations. Nat Struct Biol. 2001;8(12):1048–52. [DOI] [PubMed] [Google Scholar]
- Graham TA, Weaver C, Mao F, Kimelman D, Xu W. Crystal structure of a beta‐catenin/Tcf complex. Cell. 2000;103(6):885–96. [DOI] [PubMed] [Google Scholar]
- Guttman M, Padte NN, Huang Y, Yu J, Rocklin GJ, Weitzner BD, et al. The influence of proline isomerization on potency and stability of anti‐HIV antibody 108. Sci Rep. 2020;10(1):14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra MK, Levy Y. Affinity of disordered protein complexes is modulated by entropy‐energy reinforcement. Proc Natl Acad Sci U S A. 2022;119(26):e2120456119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Turzo SBA, Seffernick JT, Kim SS, Lindert S. Prediction of intrinsic disorder using Rosetta residuedisorder and Alphafold2. J Phys Chem B. 2022;126(42):8439–46. [DOI] [PubMed] [Google Scholar]
- Hopf TA, Green AG, Schubert B, Mersmann S, Scharfe CPI, Ingraham JB, et al. The EVcouplings python framework for coevolutionary sequence analysis. Bioinformatics. 2019;35(9):1582–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng. 2007;9(3):90–5. [Google Scholar]
- Ivankov DN, Finkelstein AV, Kondrashov FA. A structural perspective of compensatory evolution. Curr Opin Struct Biol. 2014;26(100):104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HW, Li Y, Zhang HN, Wang W, Yang X, Qi H, et al. SARS‐CoV‐2 proteome microarray for global profiling of COVID‐19 specific IgG and IgM responses. Nat Commun. 2020;11(1):3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, et al. SARS‐CoV‐2 Orf9b suppresses type I interferon responses by targeting Tom70. Cell Mol Immunol. 2020;17(9):998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with Alphafold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution‐based residue‐residue contact predictions in a sequence‐ and structure‐rich era. Proc Natl Acad Sci U S A. 2013;110(39):15674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Mooney RA, Nedialkov Y, Saba J, Mishanina TV, Artsimovitch I, et al. Structural basis for transcript elongation control by NusG family universal regulators. Cell. 2018;173(7):1650–1662 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S, Lane DP, Verma CS. Long range recognition and selection in idps: the interactions of the C‐terminus of p53. Sci Rep. 2016;6:23750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanos TK, Tugarinov V, Clore GM. An S/T motif controls reversible oligomerization of the HSP40 chaperone DNAJB6b through subtle reorganization of a beta sheet backbone. Proc Natl Acad Sci U S A. 2020;117(48):30441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatua P, Ray AJ, Hansmann UHE. Bifurcated hydrogen bonds and the fold switching of lymphotactin. J Phys Chem B. 2020;124(30):6555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AK, Looger LL, Porter LL. A high‐throughput predictive method for sequence‐similar fold switchers. Biopolymers. 2021;112(10):e23416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AK, Porter LL. Functional and regulatory roles of fold‐switching proteins. Structure. 2021;29(1):6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Lee S, Jeon A, Choi JM, Lee HS, Hohng S, et al. A single‐molecule dissection of ligand binding to a protein with intrinsic dynamics. Nat Chem Biol. 2013;9(5):313–8. [DOI] [PubMed] [Google Scholar]
- Krois AS, Park S, Martinez‐Yamout MA, Dyson HJ, Wright PE. Mapping interactions of the intrinsically disordered C‐terminal regions of tetrameric p53 by segmental isotope labeling and NMR. Biochemistry. 2022;61(23):2709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni P, Solomon TL, He Y, Chen Y, Bryan PN, Orban J. Structural metamorphism and polymorphism in proteins on the brink of thermodynamic stability. Protein Sci. 2018;27(9):1557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Kumar P, Kumari S, Uversky VN, Giri R. Folding and structural polymorphism of p53 C‐terminal domain: one peptide with many conformations. Arch Biochem Biophys. 2020;684:108342. [DOI] [PubMed] [Google Scholar]
- Lei Y, Takahama Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012;14(3):262–7. [DOI] [PubMed] [Google Scholar]
- Liebau J, Tersa M, Trastoy B, Patrick J, Rodrigo‐Unzueta A, Corzana F, et al. Unveiling the activation dynamics of a fold‐switch bacterial glycosyltransferase by (19)F NMR. J Biol Chem. 2020;295(29):9868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, dos Santos Costa A, Fazel‐Zarandi M, Sercu T, Candido S. 2022. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv.
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295–9. [DOI] [PubMed] [Google Scholar]
- Lopez‐Pelegrin M, Cerda‐Costa N, Cintas‐Pedrola A, Herranz‐Trillo F, Bernado P, Peinado JR, et al. Multiple stable conformations account for reversible concentration‐dependent oligomerization and autoinhibition of a metamorphic metallopeptidase. Angew Chem Int Ed Engl. 2014;53(40):10624–30. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, et al. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11(4):338–45. [DOI] [PubMed] [Google Scholar]
- Mansson C, van Cruchten RTP, Weininger U, Yang X, Cukalevski R, Arosio P, et al. Conserved S/T residues of the human chaperone DNAJB6 are required for effective inhibition of Abeta42 amyloid fibril formation. Biochemistry. 2018;57(32):4891–902. [DOI] [PubMed] [Google Scholar]
- Mishra S, Looger LL, Porter LL. Inaccurate secondary structure predictions often indicate protein fold switching. Protein Sci. 2019;28(8):1487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Looger LL, Porter LL. A sequence‐based method for predicting extant fold switchers that undergo alpha‐helix ↔ beta‐strand transitions. Biopolymers. 2021;112(10):e23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, et al. Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006;362(5):1043–59. [DOI] [PubMed] [Google Scholar]
- Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, et al. Direct‐coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A. 2011;108(49):E1293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murira A, Lamarre A. Type‐I interferon responses: from friend to foe in the battle against chronic viral infection. Front Immunol. 2016;7:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy AC, Dignon GL, Kan Y, Zerze GH, Parekh SH, Mittal J, et al. Molecular interactions underlying liquid‐liquid phase separation of the fus low‐complexity domain. Nat Struct Mol Biol. 2019;26(7):637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG. Biochemistry. Metamorphic proteins. Science. 2008;320(5884):1725–6. [DOI] [PubMed] [Google Scholar]
- Mylona A, Theillet FX, Foster C, Cheng TM, Miralles F, Bates PA, et al. Opposing effects of Elk‐1 multisite phosphorylation shape its response to erk activation. Science. 2016;354(6309):233–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14‐3‐3 with their partners. BMC Genomics. 2008;9(Suppl 1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiral C, Nissley DA, Deane CM. Current structure predictors are not learning the physics of protein folding. Bioinformatics. 2022;38:1881–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancsa R, Zsolyomi F, Tompa P. Co‐evolution of intrinsically disordered proteins with folded partners witnessed by evolutionary couplings. Int J Mol Sci. 2018;19(11):3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KT, Wu W, Battaile KP, Lovell S, Holyoak T, Lutkenhaus J. The Min oscillator uses MinD‐dependent conformational changes in mine to spatially regulate cytokinesis. Cell. 2011;146(3):396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LL. Predictable fold switching by the SARS‐CoV‐2 protein Orf9b. Protein Sci. 2021;30:1723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LL, Kim AK, Rimal S, Looger LL, Majumdar A, Mensh BD, et al. Many dissimilar NusG protein domains switch between alpha‐helix and beta‐sheet folds. Nat Commun. 2022;13(1):3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LL, Looger LL. Extant fold‐switching proteins are widespread. Proc Natl Acad Sci U S A. 2018;115(23):5968–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RM, Liu J, Verkuil R, Meier J, Canny J, Abbeel P, Sercu T, Rives A. MSA transformer. International Conference on Machine Learning, PMLR; 2021.
- Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning‐fast iterative protein sequence searching by HMM‐HMM alignment. Nat Methods. 2011;9(2):173–5. [DOI] [PubMed] [Google Scholar]
- Roney JP, Ovchinnikov S. State‐of‐the‐art estimation of protein model accuracy using alphafold. Phys Rev Lett. 2022;129(23):238101. [DOI] [PubMed] [Google Scholar]
- Rose GD. Reframing the protein folding problem: entropy as organizer. Biochemistry. 2021;60(49):3753–61. [DOI] [PubMed] [Google Scholar]
- Ruff KM, Pappu RV. Alphafold and implications for intrinsically disordered proteins. J Mol Biol. 2021;433(20):167208. [DOI] [PubMed] [Google Scholar]
- Rustandi RR, Baldisseri DM, Weber DJ. Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat Struct Biol. 2000;7(7):570–4. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, et al. DNA damage activates p53 through a phosphorylation‐acetylation cascade. Genes Dev. 1998;12(18):2831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer JW, Porter L. 2023. Evolutionary selection of proteins with two folds. bioRxiv. 2023; 10.1101/2023.01.18.524637 [DOI] [PMC free article] [PubMed]
- Solomon TL, He Y, Sari N, Chen Y, Gallagher DT, Bryan PN, et al. Reversible switching between two common protein folds in a designed system using only temperature. Proc Natl Acad Sci U S A. 2023;120(4):e2215418120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, et al. High‐resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PE, Leslie AG, Finch JT, Carrell RW. Crystal structure of uncleaved ovalbumin at 1.95 a resolution. J Mol Biol. 1991;221(3):941–59. [DOI] [PubMed] [Google Scholar]
- Stein RA, McHaourab HS. Speach_af: sampling protein ensembles and conformational heterogeneity with Alphafold2. PLoS Comput Biol. 2022;18(8):e1010483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, Meier M, Mirdita M, Vohringer H, Haunsberger SJ, Soding J. HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Schwieters CD, Clore GM. Open‐to‐closed transition in apo maltose‐binding protein observed by paramagnetic NMR. Nature. 2007;449(7165):1078–82. [DOI] [PubMed] [Google Scholar]
- Thole JF, Fadero TC, Bonin JP, Stadmiller SS, Giudice JA, Pielak GJ. Danio rerio oocytes for eukaryotic in‐cell NMR. Biochemistry. 2021;60(6):451–9. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, et al. Imaging neural activity in worms, flies and mice with improved gcamp calcium indicators. Nat Methods. 2009;6(12):875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein‐protein interactions. Trends Biochem Sci. 2008;33(1):2–8. [DOI] [PubMed] [Google Scholar]
- Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci U S A. 2008;105(13):5057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler RC, Murray NJ, Peterson FC, Volkman BF. Native‐state interconversion of a metamorphic protein requires global unfolding. Biochemistry. 2011;50(33):7077–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114(13):6589–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roey K, Uyar B, Weatheritt RJ, Dinkel H, Seiler M, Budd A, et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev. 2014;114(13):6733–78. [DOI] [PubMed] [Google Scholar]
- Volkman BF, Liu TY, Peterson FC. Chapter 3. Lymphotactin structural dynamics. Methods Enzymol. 2009;461:51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wafer LN, Streicher WW, McCallum SA, Makhatadze GI. Thermodynamic and kinetic analysis of peptides derived from CapZ, NDR, p53, HDM2, and HDM4 binding to human S100B. Biochemistry. 2012;51(36):7189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskom ML. Seaborn: Statistical data visualization. Journal of Open Source Software. 2021;6(60):3021. [Google Scholar]
- Wayment‐Steele HK, Ovchinnikov S, Colwell L, Kern D. Prediction of multiple conformational states by combining sequence clustering with Alphafold2. bioRxiv. 2022; 10.1101/2022.10.17.512570 [DOI]
- Xu M, Arulandu A, Struck DK, Swanson S, Sacchettini JC, Young R. Disulfide isomerization after membrane release of its Sar domain activates p1 lysozyme. Science. 2005;307(5706):113–7. [DOI] [PubMed] [Google Scholar]
- Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. Pondr‐fit: a meta‐predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804(4):996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci U S A. 2020;117(3):1496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Liu X, Neupane K, Gupta AN, Brigley AM, Solanki A, et al. Direct observation of multiple misfolding pathways in a single prion protein molecule. Proc Natl Acad Sci U S A. 2012;109(14):5283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zosel F, Mercadante D, Nettels D, Schuler B. A proline switch explains kinetic heterogeneity in a coupled folding and binding reaction. Nat Commun. 2018;9(1):3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber PK, Daviter T, Heissmann R, Persau U, Schweimer K, Knauer SH. Structural and thermodynamic analyses of the beta‐to‐alpha transformation in RfaH reveal principles of fold‐switching proteins. Elife. 2022;11:e76630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber PK, Schweimer K, Rosch P, Artsimovitch I, Knauer SH. Reversible fold‐switching controls the functional cycle of the antitermination factor RfaH. Nat Commun. 2019;10(1):702. [DOI] [PMC free article] [PubMed] [Google Scholar]