

Abstract
Molecule–surface collisions are known to initiate dynamics that lead to products inaccessible by thermal chemistry. These collision dynamics, however, have mostly been examined on bulk surfaces, leaving vast opportunities unexplored for molecular collisions on nanostructures, especially on those that exhibit mechanical properties radically different from those of their bulk counterparts. Probing energy-dependent dynamics on nanostructures, particularly for large molecules, has been challenging due to their fast time scales and high structural complexity. Here, by examining the dynamics of a protein impinging on a freestanding, single-atom-thick membrane, we discover molecule-on-trampoline dynamics that disperse the collision impact away from the incident protein within a few picoseconds. As a result, our experiments and ab initio calculations show that cytochrome c retains its gas-phase folded structure when it collides onto freestanding single-layer graphene at low energies (∼20 meV/atom). The molecule-on-trampoline dynamics, expected to be operative on many freestanding atomic membranes, enable reliable means to transfer gas-phase macromolecular structures onto freestanding surfaces for their single-molecule imaging, complementing many bioanalytical techniques.
Short abstract
Efficient molecule-to-surface energy transfer in the landing of proteins on freestanding graphene preserves their gas-phase folding state on the surface, enabling them to be imaged one at a time.
Introduction
Molecular collision is one of the simplest ways for a molecule to obtain enough energy to initiate chemical reactions or conformational switches. Examining how energy flows in a molecule during its collision event is critical to understand—and eventually control—the outcome of any molecular collisions.
Molecule–surface collisions are especially important due to the ubiquity of molecule–surface interactions in many areas of chemistry such as heterogeneous catalysis,1−7 bottom-up material fabrication,8−10 mechanochemistry,1,11,12 astrochemistry,8 and macromolecular structure characterization.13−17 Intricate details of molecule–surface collisions have been revealed by molecule–surface scattering experiments in vacuo(1−7) that, for macromolecules, have led to rich applications enabled by the soft and reactive landing of molecules on surfaces.13 These studies highlight a unique feature of molecule–surface collision which promptly (sub-picoseconds) converts molecular translational energy toward the surface (T) to molecular vibrational energy associated with soft, flexible modes of the molecule (Vmol). Such a prompt energy transfer has been shown to lead to collision outcomes that are thermally inaccessible, such as conformational changes17,18 (small Vmol transferred), mechanochemical reactions,11 and molecular fragmentation14−16 (large Vmol transferred), thereby offering a means to manipulate molecular structures.
An important frontier in molecule–surface collision dynamics is the quest to minimize T → Vmol energy transfer in molecule–surface collision, which would preserve the nuclear and electronic state of gas-phase molecules when they are deposited on a surface in vacuo. Success in such undertaking will enable gas-phase molecular species to be preserved and immobilized on surfaces and structurally characterized one molecule at a time by single-molecule microscopy methods, such as electron microscopy19−21 or scanning probe microscopy techniques.22 These new capabilities will unlock new frontiers in both single-molecule microscopy and native mass spectroscopy, as it enables single-molecule visualization of any molecular species relevant to chemistry and biology that can be isolated from the solution phase by native electrospray. Such a combination has been shown for macromolecules by successful soft deposition and direct observation of folded proteins on a surface, ranging from small proteins (∼12 kDa) to large multiunit protein complexes (∼800 kDa).20,21,23−25
Realizing the full potential of this vision calls for strategies to divert the flow of molecular translational energy (T) away from the molecular internal coordinates in molecule–surface collision. One ideal destination for the energy is the surface, since it possesses a large number of soft vibrational/phonon modes (Vsurf), well suited to absorb the translational energy of the incident molecule. Collision dynamics dominated by T → Vsurf energy transfer would minimize the energy transferred to the internal coordinates of the molecule and preserve the gas-phase molecular structure on surface that can be subsequently characterized by many surface-based microscopy techniques.19−22 Given that the translational energy (T) would flow to the softest mode between the collision partners,26,27 be that mode residing in the molecule or the surface, replacing T → Vmol by T → Vsurf requires the surface to possess softer vibrational modes than the incident molecule. This requirement has been fulfilled in previous studies by noble-gas28,29 or organic adlayers30−32 on bulk surfaces, which cushion the impact of incident molecules on surfaces.28−34
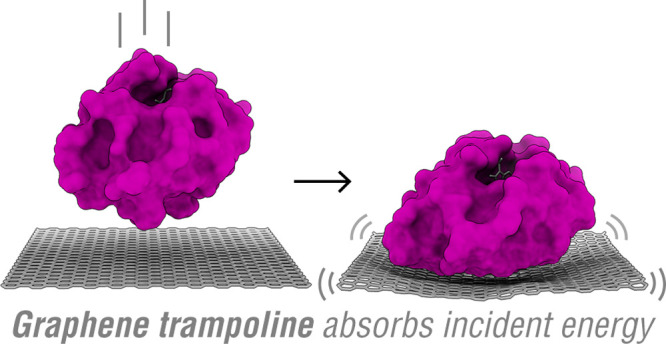
Here we point out that freestanding atomic membranes, such as single-layer graphene (SLG), possess a soft “trampoline” mode that fulfills the requirement for T → Vsurf dynamics. We demonstrate experimentally the use of freestanding graphene as the landing surface for cytochrome c (cyt c) proteins, which preserves the gas-phase folding state of cyt c on the surface. Ions of cyt c were generated by nanoelectrospray35,36 (nESI) and soft-landed on graphene in vacuo by an electrospray ion beam deposition (ESIBD) technique,13,37 whereupon the adsorbed cyt c structure was imaged one molecule at a time by low-energy electron holography20,21 (LEEH). We corroborate our experimental findings by simulating the collision dynamics of the cyt c on graphene using ab initio molecular dynamics (AIMD) implemented in density functional theory (DFT). Our simulation shows the dominance of T → Vsurf dynamics in the landing of molecules on the freestanding atomic membrane, where Vsurf is the soft acoustic vibrations of graphene. The efficient protein to graphene energy transfer preserves the gas-phase folding state of the protein on the surface, as observed in the experiment. The T → Vsurf dynamics on a freestanding atomic membrane, here termed molecule-on-trampoline dynamics, are expected to be operative in the encounter of rigid molecules with many freestanding 2D materials,38 such as graphene oxide, hexagonal boron nitride, metal chalcogenides (e.g. MoS2), etc. We focus our present work on graphene due to its widespread use across scientific disciplines that span from fundamental physics39 to structural biology.40 Our work shows the potential of the tandem combination of nESI + ESIBD + LEEH on graphene to reveal, at the single-molecule level, folded structures of molecular species generated from the native mass spectroscopy techniques.
Results and Discussion
Figure 1a shows a schematic of the experiment where cyt c ions ([M+7H]7+) were collided normal to a freestanding graphene held at room temperature. The proteins were collided at a selected energy of 35 eV collision energy (i.e. 5 eV/charge), previously shown to preserve the chemical structures of incident molecules on surfaces.13,41 The proteins adsorbed on graphene were subsequently imaged as single molecules using LEEH to reveal their structures, as inferred from their observed sizes and shapes.
Figure 1.

Landing folded proteins on freestanding single-layer graphene. (a) Cytochrome c (cyt c) [M+7H]7+ ions are landed normal to a freestanding single-layer graphene with a selected collision energy of 35 eV (i.e. 5 eV/charge, typical for soft-landing of molecules on a surface). (b) Folded cyt c on graphene imaged by low-energy electron holography (LEEH) showing agreement with folded cyt c computed from density functional theory (DFT). The shape of cyt c from the DFT calculations is given by the red outline obtained from the van der Waals representation of the protein. The deposition and imaging were done at room temperature.
The cyt c protein soft-landed on graphene was observed by LEEH as a folded protein (Figure 1b). The observed dimension of cyt c on graphene was found to agree well with the dimension of folded cyt c obtained by relaxing the final state of our AIMD calculations (Figure 1b). In addition, the measured size of cyt c on graphene with an apparent area of ∼13 nm2 was found to agree with the size of folded cyt c measured in the gas phase by ion-mobility experiments42 (∼14 nm2). Given the low energy involved in the landing event,13 the folding state of the adsorbed cyt c is expected to be similar to its gas-phase folded structure, consistent with the previous protein landing studies on graphene.20,21
We corroborate our findings by investigating the structural changes in the protein when it lands from the gas-phase onto the surface, using AIMD calculations at the level of DFT. We model the landing event by colliding a gas-phase cyt c7+ protein ion onto a freestanding graphene, whereby the protein possesses a 35 eV translational energy toward the graphene. We approximate the gas-phase structure of cyt c7+ ion by relaxing the crystal structure of cyt c (PDB ID 1HRC) in the gas phase with a net charge of +7. Our calculation gives a gas-phase cyt c structure that has a near-identical three-dimensional structure to its crystal structure, with the most significant changes being localized at the protein–vacuum interface. These structural changes are caused by the formation of salt bridges and hydrogen bonds among the side chains of the amino acids at the protein–vacuum interface, thereby confirming the “side chain collapse” in the literature43−46 at the ab initio level (Figure 2a, inset). These cohesive interactions at the protein–vacuum interface are understood to cause a surface-tension-like effect47 that maintains the folded structure of the protein in the gas phase. The gas-phase cyt c7+ structure from our calculations yields an ion-mobility cross section (∼12 nm2, from the IMPACT software48) that agrees well with its measured experimental value42 (∼14 nm2). The slight difference between theory and experimental cross sections may be attributed to the finite temperature effects, such as cyt c structural dynamics at ∼300 K, that have yet to be included in present DFT calculations.
Figure 2.

Nuclear and electronic structure of a protein on single-layer graphene. (a) cyt c adsorbed on graphene (red) is computed to be structurally similar to its crystal structure (blue, PDB ID 1HRC). The adsorbed cyt c structure when compared to its crystal structure possesses more “salt bridges” between the positively and negatively charged amino acid side chains. (b) Projected density of states (pDOS) calculations show strong overlap between cyt c and graphene electronic states, indicating an efficient path of molecule-to-surface electron/hole transfer. Comparatively, for hydrated cyt c, some unoccupied cyt c states do not overlap with water electronic states, indicating that electrons could be trapped in these states. (c) The positively charged cyt c attracts the graphene electrons into a pool underneath the protein (yellow density, isosurface 2 × 10–3 e Å–3) to establish charge–image charge interactions.
Our AIMD calculations, in agreement with the experiment, show that the principal features of the gas-phase cyt c are preserved when the protein lands on the surface. The AIMD calculation gives a final structure of cyt c on graphene whose size and shape agree well with the adsorbed cyt c observed by LEEH (Figure 1b). The computed structure of adsorbed cyt c has a near identical folding state to its crystal structure (PDB ID 1HRC) with the most significant changes located at the omega loops of the protein (residue 36 to 37, 44 to 46, 54 to 60, and 76 to 77) (Figure 2a). Our finding echoes the cyt c foldon hierarchy49 that regards the omega loops in cyt c as the most flexible part of the molecule and thus the structural motif that responds most to changes in the external environment of the protein.
Our ab initio calculations on the entire protein shed light into the electronic structure of cyt c on graphene, revealing its density of states and its binding mechanism to the surface. Figure 2b shows the projected density of states (pDOS) of the adsorbed cyt c and the underlying graphene, revealing a strong overlap between the cyt c and graphene electronic states. Such a strong overlap provides an explanation for the remarkable stability of the proteins on graphene from the continuous ∼100 eV electron irradiation in the LEEH imaging (i.e. no event with the probability higher than ∼10–12 per incident electron was observed; see Methods for details). Given that low-energy electrons (50–150 eV) are known to efficiently ionize molecules (M + e → M+ + 2e) and cause molecular dissociations,50−52 the apparent immunity of proteins on graphene toward electron-induced dissociations indicates an efficient hole removal from the protein. This efficient hole transfer mechanism is understood to originate from the large electronic transition probability between the protein and graphene, enabled by the strong electronic state overlap between them—a common feature for molecules adsorbed on conductive surfaces.53 For molecules adsorbed on metal surfaces,54,55 the lifetime of transient charges trapped in the molecules was measured to be below 5 fs—a time scale that is too short for any molecular bonds to significantly stretch to cause any chemical reactions, which typically requires tens or hundreds of femtoseconds.56 This insight highlights the importance of fast electronic relaxation in suppressing electron-induced reactive events in adsorbed molecules.
We contrast the cyt c-graphene pDOS to that for hydrated cyt c (Figure S1), which shows the nonoverlap between unoccupied cyt c states and the water electronic states between the Fermi level to ∼3 eV above the Fermi level (Figure 2b, lower panel). These “unprotected” cyt c states consist of the π-states in the heme, the nitrogen base, and the peptide bonds of the protein (Figure S2). The nonoverlap implies that electrons can be trapped on these “unprotected” states to cause molecular dissociations, since these trapped charges can only be removed by slow nuclear dynamics (e.g. a Grothuss-like mechanism57) as opposed to fast electronic dynamics. Our comparison thereby highlights the importance of protein contact to a conductive medium (e.g. graphene) in providing fast electronic de-excitation pathways for the protein.
The binding of proteins to graphene is driven by noncovalent (electrostatic and van der Waals) interactions, as revealed by our calculation that shows that every C atom on the graphene retains its C-sp2 geometry. Our calculations estimate the cyt c–graphene binding energy to be ∼33 eV, originating primarily from electrostatic interactions (77%) and secondarily from van der Waals interactions (23%). The dominant electrostatic cyt c–graphene interaction is understood to arise from the net positive cyt c (+3.2e) accumulating a pool of electrons on the underlying graphene (−3.2e) that leads to a charge–image charge interaction between cyt c and graphene (Figure 2c). The positive charge in the adsorbed cyt c originates from all the retained protons that are initially attached to the protein in the gas phase. These protons are likely to remain attached to the protein due to their high detachment barriers, computed to be above +0.9 eV if detached from histidine and above +1.4 eV from lysine. Given that our relaxation calculations show that all protons initially attached to the protein in the gas phase remain attached to the adsorbed protein, we note that the computed +3.2e net charge on the adsorbed protein is lower than the +7e charge in the gas-phase protein. This charge difference thereby indicates a significant graphene-to-protein electron injection during the landing of the protein on the surface.
We now detail the mechanism that connects the initial, gas-phase protein structure to the final, adsorbed protein structure. Figure 3a shows the soft-landing dynamics of cyt c on graphene, as per our experiment, at 35 eV collision energy (i.e. 5 eV/charge) and at an approach angle normal to the surface. The calculation shows that landing a protein at such energy on graphene preserves its primary structure and most of its secondary and tertiary structure from the gas phase to the surface, consistent with the conclusions from the experiment. The computed dynamics show that the protein–surface collision creates a soliton-like collective oscillation in the protein, which propagates as fast as ∼2.5 nm/ps away from the surface (Figure S3a), similar to the previously reported “proteinquake” collective oscillation in a protein.58,59 This oscillation subsequently is followed by a slight compression (∼10%) and decompression of the entire protein prior to its stable adsorption on graphene (Figure 3A and Figure S4a).
Figure 3.

Ab initio molecular dynamics of protein landing on single-layer graphene. Folded cyt c is landed on graphene with an energy of 35 eV (i.e. 5 eV/charge—typical for soft-landing), shown in (a), and 350 eV (i.e. 50 eV/charge—typical for reactive-landing), shown in (b). The collision compresses the incident protein, initiating the protein unfolding in the latter case. The graphene has traveled downward by 3.1 nm at the 9.4 ps mark in (a) and by 4.7 nm at the 3.8 ps mark in (b); these distances are the greatest distance traveled by graphene in their respective trajectories.
The protein compression evidences the conversion of its molecular translational energy toward its molecular vibrational energy. Specifically, the most flexible, collective vibrational modes of the protein are excited upon landing, whose magnitude dictates the extent of structural change in the protein upon its landing on surface. We verify this claim by comparing AIMD calculations of cyt c at soft landing (35 eV, 5 eV/charge, Figure 3a and Figures S3a and 4a) and reactive landing regimes (350 eV, 50 eV/charge, Figure 3b and Figures S3b and 4b). In the low-energy regime, the gained vibrational energy is insufficient to cause major conformational changes, as shown by the similar protein folding between the final adsorbed structure and the crystal structure (Figure 2a). In the high-energy regime, the gained vibrational energy is sufficient to alter the tertiary structure of cyt c to initiate the unfolding of the protein while some secondary structures (i.e. the helices) remain intact (Figure 3b). The surface-induced protein compression causes a prompt (within 1–2 ps) and specific structural perturbation on the protein that could lead to thermally inaccessible products/outcomes, such as, in the case for small molecules, bond-selective mechanochemical reactions,11 or in the case of macromolecules, conformation changes27 and fragmentations.14−16,31,32 We thereby argue that the surface-collision-induced compression is the key to access unique fragmentation pathways observed in the surface-induced dissociation (SID) technique15,16 that uses a high-energy collision of macromolecules on surfaces to induce their fragmentations for macromolecular structure characterization.
An energy analysis of protein landing on graphene reveals the dominance of T → Vsurf energy transfer as its most striking feature (Figure 4 and Figure S5). Figure 4a shows that the molecular translational energy (T) is converted largely to the surface vibrational energy (Vsurf) (77%) and, to a lesser extent, to the molecular vibrational energy (Vmol) (19%). These results are in stark contrast to that for molecular landing on bulk surfaces,11,18 whereby T is converted primarily to Vmol (34%), and secondarily to Vsurf (23%). The different energy transfer dynamics on hard, bulk surfaces and soft, freestanding graphene echo the previously reported experiments31,32 and calculations33 of peptides scattering on hard, fluorinated self-assembled monolayers (SAMs) and soft, hydrocarbon SAMs. We additionally note that these dynamics also minimize the T → Rmol energy transfer (Rmol = molecular rotational energy), which suggests that the observed molecular orientation on the surface would be largely similar to the orientation of the gas-phase molecule prior to its landing. Protein rotations only become significant in the collision dynamics at very low collision energy, such as at 3.5 eV (Figure S6), because the slowly moving protein has ample time to experience the attractive forces from the surfaces that reorients the protein throughout its surface collision (also known as “the dynamic steering effect”60).
Figure 4.

Quantitative analysis of protein landing dynamics on single-layer graphene at 35 eV collision energy. (a) A protein–graphene collision principally converts the protein translational energy (Etrans,mol) to the graphene vibrational energy (Evib,surf and Etrans,surf). The graphene translational energy (Etrans,surf) would approximate the energy received by the “trampoline” mode of the graphene. The sum of Evib,surf and Etrans,surf would give the total vibrational energy of graphene (termed Vsurf in the main text). (b) A protein–graphene collision creates a soliton in the graphene that serves as a mechanism to promptly transport energy away from the landing site, as fast as ∼3–4 nm/ps (black atoms denote C atoms moving away from the protein).
The dominance of T → Vsurf in a protein–graphene collision evidences a coupling between incident protein translation and graphene vibration. Specifically, our calculations show the collision exciting the out-of-plane, soft “trampoline” vibrational mode in graphene, whose fundamental is measured to be in megahertz61,62 due to the low mass and the freestanding state of graphene. The protein–graphene collision generates a soliton-like, traveling transverse wavepacket in the graphene (Figure 4b) (also called a stress wave63 or an elastic deformation wave64), understood to be the superposition of various out-of-plane modes of freestanding graphene. At short time scales (approximately picoseconds) covered by our AIMD calculations, our result shows that the soliton travels as fast as 3–4 nm/ps, in agreement with previously computed values.65 At long time scales (approximately nanoseconds to approximately milliseconds), we expect the soliton to gradually disperse into various out-of-plane modes of the graphene,66 as its kinetic energy equipartitions to all degrees of freedom in the graphene. The soliton dynamics highlight the fast protein-to-surface energy dissipation channel for the protein translational energy, which enables the preservation of protein folded states when they land on freestanding surfaces. These molecule-on-trampoline dynamics are expected to be general in the collision of rigid molecules with many freestanding 2D materials, which opens up new research opportunities at the intersection of reaction dynamics, macromolecular chemistry, nanoelectromechanical (NEMS) resonators,67 and 2D materials research. One interesting avenue is to examine the scaling behavior of these dynamics across different projectile sizes, starting from a small molecule (e.g. amino acid) to a large macromolecular complex (e.g. membrane proteins in a micelle), and to examine whether their surface encounters could be discerned by transient electrical transport measurements.68
Conclusions
This work provides important physical descriptions of protein nuclear and electronic structures when they are isolated in vacuo and adsorbed on graphene, critical to assess new opportunities afforded by the tandem combination of native electrospray ionization, soft-landing technology, and single-molecule microscopy techniques. Our experiments and ab initio calculations reveal a fast protein-to-surface energy transfer mechanism in a protein–graphene collision that allows the gas-phase protein structures to be preserved at a freestanding surface. We point out that surface collision on an atomic membrane could provide a means to access the ground and excited conformational states of macromolecules via their compressions. For molecular collisions on organic adlayers,30 we anticipate that a similarly effective energy dissipation mechanism involving different surface modes33,34 may be operative to preserve the gas-phase protein structures on these surfaces. Observing these gas-phase structures on surface one at a time by single-molecule microscopy techniques would complement the structural studies conducted by ion-mobility and native mass spectroscopy techniques. This single-molecule approach could prove particularly valuable for small, flexible proteins, glycans, or glycoproteins, which are difficult to observe by ensemble-averaged approaches.
Methods
Experiment
Cytochrome c (cyt c) (Sigma-Aldrich, >95%, from Equine Heart, Catalog Number C7752) was dissolved in a 200 mM ammonium acetate solution and desalted twice (Bio-Rad BioSpin P6 column) to give a cyt c spray solution (1.5 mg/mL). The solution was loaded to a metal-coated nanoelectrospray glass emitter and sprayed at 1.0–1.5 kV to a 80 °C capillary inlet to prevent denaturation of the protein. No unexpected or unusually high safety hazards were encountered. Using an electrospray ion beam deposition (ESIBD) setup, described in detail elsewhere,21,37 the ions were mass-selected to yield an ion beam that consisted of +7 (major, m/z = 1764) and +8 (minor, m/z = 1543) cyt c ions and subsequently deposited on a freestanding single-layer graphene (SLG) held at room temperature under ultrahigh vacuum (P < 10–10 mbar). The protein–graphene collision energy was controlled by applying a selected voltage to decelerate the incident protein ion. After the deposition, the graphene sample was transferred in vacuo to a low-energy electron holography (LEEH) instrument, operated at room temperature under ultrahigh vacuum (P < 10–10 mbar). Low-energy electrons between 50 and 150 eV were used in LEEH imaging with a dose estimated at ∼10 nA per 50 nm2. Illuminating a protein for ∼1 s was sufficient to record a high-quality hologram, which was subsequently reconstructed using the previously described method21 to reveal the real-space image of the adsorbed protein.
The cyt c protein shown in Figure 1b was observed continuously for 23 s using an electron energy of 147 eV. The protein structure was observed to remain unchanged throughout the illumination time, during which a total of ∼4 × 1011 electrons had been scattered on the protein. Given that no inelastic event (e.g. diffusion, rotation, or dissociation) was observed, our data thereby set an upper limit for any inelastic event probability to be ∼3 × 10–12 event per electron.
Theory
Ab initio calculations at the level of density functional theory (DFT) as implemented in the code OpenMX69−71 (version 3.9.2) was used to model the experiment. The code employed norm-conserving pseudopotentials, pseudoatomic localized basis functions, periodic boundary conditions, and van der Waals correction using Grimme’s DFT-D3 method.72 Our calculations employed a cutoff energy of 300 Ry, an electronic temperature of 300 K, and a supercell with dimensions of 51.33 Å (X axis), 49.39 Å (Y axis), and 50.00 Å (Z axis). All calculations sampled only the gamma point of the k-mesh and used an electronic convergence criterion of 4 × 10–8 hartree, employing the divide-conquer with localized natural orbitals (DC-LNO) method.73 The graphene was modeled by 960 C atoms, and the cyt c was modeled with a total of 1744 atoms as C558H878O155N148FeS4. The relaxation calculations were performed until the forces in all atoms were below 4 × 10–4 hartree/bohr. Charge analyses of the relaxed structures were performed using a Bader analysis.74 Visualization of the molecular structures was performed using the ChimeraX software.75,76
Born–Oppenheimer MD calculations were performed as a microcanonical ensemble that preserved the total number of atoms (N), volume (V), and energy (E). The MD calculations were performed with a 0.5 fs time step, which gave a negligible total energy drift of as low as ∼2 meV/ps per atom. The initial state of the MD placed a gas-phase relaxed cyt c ∼7 Å above the graphene. All atoms were subsequently initialized with random velocities sampled from the Boltzmann distribution at room temperature (298 K). The thermalization of these velocities is evident in the first few femtoseconds at the start of the trajectory, shown in the Evib,mol and Evib,surf of Figure 4a and Figure S5a. On top of these velocities, all atoms in cyt c receive a constant velocity toward the graphene that corresponded to the translational energy of the protein toward the graphene of either 35 or 350 eV. In the MD calculations, all of the C atoms of the graphene were free to move, causing the act of restoring force in the graphene “trampoline” onto the protein to be unaccounted for in the MD calculations. However, these forces, estimated to be ∼0.3–0.6 nN from ref (61), were minor in comparison to the forces experienced by the protein during the collision with the graphene at ∼10 nN for 5 eV/c landing and ∼50 nN for 50 eV/c landing (Figure S4) and thereby were not expected to be the major factor in the landing dynamics of the protein on graphene.
Acknowledgments
K.A. thanks Alexander von Humboldt Foundation for financial support and Dr. Tim Esser for discussions. The calculations were performed at the Max-Planck Computing and Data Facility in Garching. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. The authors declare no competing financial interests.
Data Availability Statement
All data required to evaluate the conclusions of the paper is present in the main text or the Supporting Information. Supporting Information contains analysis of hydrated cyt c7+ and cyt c7+ colliding with graphene. The AIMD trajectories for cyt c landing on graphene, as well as the atomic coordinates for the gas-phase cyt c7+, hydrated cyt c7+, and the adsorbed cyt c on graphene are available at the Data Repository of the Max Planck Society (10.17617/3.VJ9ZIM).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.2c00815.
Author Contributions
K.A., L.M., S.R., and K.K. conceived and supervised the project. H.O. and S.S. performed the ESIBD deposition and LEEH measurements. K.A. performed the DFT calculations and their analyses. K.A. wrote the paper with inputs from all the coauthors. All authors contributed to the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ceyer S. T. New Mechanisms for Chemistry at Surfaces. Science. 1990, 249, 133–139. 10.1126/science.249.4965.133. [DOI] [PubMed] [Google Scholar]
- Hou H.; Gulding S. J.; Rettner C. T.; Wodtke A. M.; Auerbach D. J. The Stereodynamics of a Gas-Surface Reaction. Science. 1997, 277, 80–82. 10.1126/science.277.5322.80. [DOI] [Google Scholar]
- Beck R. D.; et al. Vibrational Mode-Specific Reaction of Methane on a Nickel Surface. Science. 2003, 302, 98–100. 10.1126/science.1088996. [DOI] [PubMed] [Google Scholar]
- Smith R. R.; Killelea D. R.; DelSesto D. F.; Utz A. L. Preference for Vibrational over Translational Energy in a Gas-Surface Reaction. Science. 2004, 304, 992–995. 10.1126/science.1096309. [DOI] [PubMed] [Google Scholar]
- Killelea D. R.; Campbell V. L.; Shuman N. S.; Utz A. L. Bond-Selective Control of a Heterogeneously Catalyzed Reaction. Science. 2008, 319, 790–793. 10.1126/science.1152819. [DOI] [PubMed] [Google Scholar]
- Yoder B. L.; Bisson R.; Beck R. D. Steric Effects in the Chemisorption of Vibrationally Excited Methane on Ni(100). Science. 2010, 329, 553–556. 10.1126/science.1191751. [DOI] [PubMed] [Google Scholar]
- Jiang B.; Yang M.; Xie D.; Guo H. Quantum dynamics of polyatomic dissociative chemisorption on transition metal surfaces: mode specificity and bond selectivity. Chem. Soc. Rev. 2016, 45, 3621–3640. 10.1039/C5CS00360A. [DOI] [PubMed] [Google Scholar]
- Jacobs D. C. Reactive Collisions Of Hyperthermal Energy Molecular Ions With Solid Surfaces. Annu. Rev. Phys. Chem. 2002, 53, 379–407. 10.1146/annurev.physchem.53.100301.131622. [DOI] [PubMed] [Google Scholar]
- Barth J. V.; Costantini G.; Kern K. Engineering atomic and molecular nanostructures at surfaces. Nature 2005, 437, 671. 10.1038/nature04166. [DOI] [PubMed] [Google Scholar]
- Dubey G.; et al. Chemical Modification of Graphene via Hyperthermal Molecular Reaction. J. Am. Chem. Soc. 2014, 136, 13482–13485. 10.1021/ja5046499. [DOI] [PubMed] [Google Scholar]
- Krumbein L.; et al. Fast Molecular Compression by a Hyperthermal Collision Gives Bond-Selective Mechanochemistry. Phys. Rev. Lett. 2021, 126, 056001. 10.1103/PhysRevLett.126.056001. [DOI] [PubMed] [Google Scholar]
- O’Neill R. T.; Boulatov R. The many flavours of mechanochemistry and its plausible conceptual underpinnings. Nat. Rev. Chem. 2021, 5, 148–167. 10.1038/s41570-020-00249-y. [DOI] [PubMed] [Google Scholar]
- Grill V.; Shen J.; Evans C.; Cooks R. G. Collisions of ions with surfaces at chemically relevant energies: Instrumentation and phenomena. Rev. Sci. Instrum. 2001, 72, 3149–3179. 10.1063/1.1382641. [DOI] [Google Scholar]
- Laskin J.; Bailey T. H.; Futrell J. H. Shattering of Peptide Ions on Self-Assembled Monolayer Surfaces. J. Am. Chem. Soc. 2003, 125, 1625–1632. 10.1021/ja027915t. [DOI] [PubMed] [Google Scholar]
- Harvey S. R.; et al. Relative interfacial cleavage energetics of protein complexes revealed by surface collisions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8143–8148. 10.1073/pnas.1817632116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D. T.; Harvey S. R.; Wysocki V. H. Surface-induced Dissociation Mass Spectrometry as a Structural Biology Tool. Chem. Rev. 2022, 122, 7442–7487. 10.1021/acs.chemrev.1c00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donor M. T.; Mroz A. M.; Prell J. S. Experimental and theoretical investigation of overall energy deposition in surface-induced unfolding of protein ions. Chem. Sci. 2019, 10, 4097–4106. 10.1039/C9SC00644C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggara K.; et al. Exploring the Molecular Conformation Space by Soft Molecule-Surface Collision. J. Am. Chem. Soc. 2020, 142, 21420–21427. 10.1021/jacs.0c09933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X.; McMullan G.; Scheres S. H. W. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Longchamp J.-N.; et al. Imaging proteins at the single-molecule level. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 1474–1479. 10.1073/pnas.1614519114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochner H.; et al. Low-energy electron holography imaging of conformational variability of single-antibody molecules from electrospray ion beam deposition. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2112651118 10.1073/pnas.2112651118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian K.; et al. Scanning probe microscopy. Nat. Rev. Methods Prim. 2021, 1, 36. 10.1038/s43586-021-00033-2. [DOI] [Google Scholar]
- Mikhailov V. A.; Mize T. H.; Benesch J. L. P.; Robinson C. V. Mass-Selective Soft-Landing of Protein Assemblies with Controlled Landing Energies. Anal. Chem. 2014, 86, 8321–8328. 10.1021/ac5018327. [DOI] [PubMed] [Google Scholar]
- Westphall M. S.; et al. Three-dimensional structure determination of protein complexes using matrix-landing mass spectrometry. Nat. Commun. 2022, 13, 2276. 10.1038/s41467-022-29964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser T. K.; et al. Mass-selective and ice-free electron cryomicroscopy protein sample preparation via native electrospray ion-beam deposition. PNAS Nexus 2022, 1, pgac153. 10.1093/pnasnexus/pgac153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck R. D.; Rockenberger J.; Weis P.; Kappes M. M. Fragmentation of C60+ and higher fullerenes by surface impact. J. Chem. Phys. 1996, 104, 3638–3650. 10.1063/1.471066. [DOI] [Google Scholar]
- Mowrey R. C.; Brenner D. W.; Dunlap B. I.; Mintmire J. W.; White C. T. Simulations of buckminsterfullerene (C60) collisions with a hydrogen-terminated diamond {111} surface. J. Phys. Chem. 1991, 95, 7138–7142. 10.1021/j100172a011. [DOI] [Google Scholar]
- Bromann K.; et al. Controlled Deposition of Size-Selected Silver Nanoclusters. Science. 1996, 274, 956–958. 10.1126/science.274.5289.956. [DOI] [PubMed] [Google Scholar]
- Cheng H.-P.; Landman U. Controlled Deposition, Soft Landing, and Glass Formation in Nanocluster-Surface Collisions. Science. 1993, 260, 1304–1307. 10.1126/science.260.5112.1304. [DOI] [PubMed] [Google Scholar]
- Gologan B.; Green J. R.; Alvarez J.; Laskin J.; Graham Cooks R. Ion/surface reactions and ion soft-landing. Phys. Chem. Chem. Phys. 2005, 7, 1490–1500. 10.1039/b418056a. [DOI] [PubMed] [Google Scholar]
- Laskin J.; Futrell J. H. Energy transfer in collisions of peptide ions with surfaces. J. Chem. Phys. 2003, 119, 3413–3420. 10.1063/1.1589739. [DOI] [Google Scholar]
- Fernández F. M.; Smith L. L.; Kuppannan K.; Yang X.; Wysocki V. H. Peptide sequencing using a patchwork approach and surface-induced dissociation in sector-TOF and dual quadrupole mass spectrometers. J. Am. Soc. Mass Spectrom. 2003, 14, 1387–1401. 10.1016/j.jasms.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Meroueh O.; Hase W. L. Dynamics of Energy Transfer in Peptide-Surface Collisions. J. Am. Chem. Soc. 2002, 124, 1524–1531. 10.1021/ja011987n. [DOI] [PubMed] [Google Scholar]
- Pratihar S.; Barnes G. L.; Laskin J.; Hase W. L. Dynamics of Protonated Peptide Ion Collisions with Organic Surfaces: Consonance of Simulation and Experiment. J. Phys. Chem. Lett. 2016, 7, 3142–3150. 10.1021/acs.jpclett.6b00978. [DOI] [PubMed] [Google Scholar]
- Wilm M.; Mann M. Analytical Properties of the Nanoelectrospray Ion Source. Anal. Chem. 1996, 68, 1–8. 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- Juraschek R.; Dülcks T.; Karas M. Nanoelectrospray—more than just a minimized-flow electrospray ionization source. J. Am. Soc. Mass Spectrom. 1999, 10, 300–308. 10.1016/S1044-0305(98)00157-3. [DOI] [PubMed] [Google Scholar]
- Rauschenbach S.; Ternes M.; Harnau L.; Kern K. Mass Spectrometry as a Preparative Tool for the Surface Science of Large Molecules. Annu. Rev. Anal. Chem. 2016, 9, 473–498. 10.1146/annurev-anchem-071015-041633. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Beyer A. Mechanics of free-standing inorganic and molecular 2D materials. Nanoscale 2021, 13, 1443–1484. 10.1039/D0NR07606F. [DOI] [PubMed] [Google Scholar]
- Cao Y.; et al. Unconventional superconductivity in magic-angle graphene superlattices. Nature 2018, 556, 43–50. 10.1038/nature26160. [DOI] [PubMed] [Google Scholar]
- Naydenova K.; Peet M. J.; Russo C. J. Multifunctional graphene supports for electron cryomicroscopy. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 11718–11724. 10.1073/pnas.1904766116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portz A.; et al. Chemical Analysis of Complex Surface-Adsorbed Molecules and Their Reactions by Means of Cluster-Induced Desorption/Ionization Mass Spectrometry. Anal. Chem. 2018, 90, 3328–3334. 10.1021/acs.analchem.7b04876. [DOI] [PubMed] [Google Scholar]
- Clemmer D. E.; Hudgins R. R.; Jarrold M. F. Naked Protein Conformations: Cytochrome c in the Gas Phase. J. Am. Chem. Soc. 1995, 117, 10141–10142. 10.1021/ja00145a037. [DOI] [Google Scholar]
- Steinberg M. Z.; Elber R.; McLafferty F. W.; Gerber R. B.; Breuker K. Early Structural Evolution of Native Cytochrome c after Solvent Removal. ChemBioChem. 2008, 9, 2417–2423. 10.1002/cbic.200800167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18145–18152. 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo R. R. O.; Loo J. A. Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J. Am. Soc. Mass Spectrom. 2016, 27, 975–990. 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtiari M.; Konermann L. Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. J. Phys. Chem. B 2019, 123, 1784–1796. 10.1021/acs.jpcb.8b12173. [DOI] [PubMed] [Google Scholar]
- Dadarlat V. M.; Post C. B. Adhesive-cohesive model for protein compressibility: An alternative perspective on stability. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14778–14783. 10.1073/pnas.2434157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund E. G.; Degiacomi M. T.; Robinson C. V.; Baldwin A. J.; Benesch J. L. P. Collision Cross Sections for Structural Proteomics. Structure 2015, 23, 791–799. 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Mayne L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15873–15880. 10.1073/pnas.1411798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhler E.; Warneke J.; Swiderek P. Control of chemical reactions and synthesis by low-energy electrons. Chem. Soc. Rev. 2013, 42, 9219–9231. 10.1039/c3cs60180c. [DOI] [PubMed] [Google Scholar]
- Burns A. R.; Stechel E. B.; Jennison D. R. Desorption by electronically stimulated adsorbate rotation. Phys. Rev. Lett. 1987, 58, 250–253. 10.1103/PhysRevLett.58.250. [DOI] [PubMed] [Google Scholar]
- Burns A. R.; Jennison D. R.; Stechel E. B. Charge-transfer screening and the dynamics of electronically stimulated adsorbate dissociation and desorption. J. Vac. Sci. Technol. A 1990, 8, 2705–2709. 10.1116/1.576654. [DOI] [Google Scholar]
- Nilsson A.; Pettersson L. G. M. In Chemical Bonding at Surfaces and Interfaces; Nilsson A., Pettersson L. G. M., Nørskov J. K., Eds.; Elsevier: 2008; pp 57–142. 10.1016/B978-044452837-7.50003-4. [DOI] [Google Scholar]
- Bartels L.; et al. Dynamics of Electron-Induced Manipulation of Individual CO Molecules on Cu(111). Phys. Rev. Lett. 1998, 80, 2004–2007. 10.1103/PhysRevLett.80.2004. [DOI] [Google Scholar]
- Garg M.; et al. Real-space subfemtosecond imaging of quantum electronic coherences in molecules. Nat. Photonics 2022, 16, 196–202. 10.1038/s41566-021-00929-1. [DOI] [Google Scholar]
- Eisenstein A.; Leung L.; Lim T.; Ning Z.; Polanyi J. C. Reaction dynamics at a metal surface; halogenation of Cu(110). Faraday Discuss. 2012, 157, 337–353. 10.1039/c2fd20023f. [DOI] [PubMed] [Google Scholar]
- Cukierman S. Et tu, Grotthuss! and other unfinished stories. Biochim. Biophys. Acta - Bioenerg. 2006, 1757, 876–885. 10.1016/j.bbabio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Ansari A.; et al. Protein states and proteinquakes. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 5000–5004. 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barends T. R. M.; et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science. 2015, 350, 445–450. 10.1126/science.aac5492. [DOI] [PubMed] [Google Scholar]
- Gross A.; Wilke S.; Scheffler M. Six-Dimensional Quantum Dynamics of Adsorption and Desorption of H2 at Pd(100): Steering and Steric Effects. Phys. Rev. Lett. 1995, 75, 2718–2721. 10.1103/PhysRevLett.75.2718. [DOI] [PubMed] [Google Scholar]
- Bunch J. S.; et al. Electromechanical Resonators from Graphene Sheets. Science. 2007, 315, 490–493. 10.1126/science.1136836. [DOI] [PubMed] [Google Scholar]
- Blaikie A.; Miller D.; Alemán B. J. A fast and sensitive room-temperature graphene nanomechanical bolometer. Nat. Commun. 2019, 10, 4726. 10.1038/s41467-019-12562-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K.; Zhan H.; Hu D.; Gu Y. Failure mechanism of monolayer graphene under hypervelocity impact of spherical projectile. Sci. Rep. 2016, 6, 33139. 10.1038/srep33139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizao R. A.; Machado L. D.; de Sousa J. M.; Pugno N. M.; Galvao D. S. Scale Effects on the Ballistic Penetration of Graphene Sheets. Sci. Rep. 2018, 8, 6750. 10.1038/s41598-018-25050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang F.; Wu H. Anisotropic propagation and upper frequency limitation of terahertz waves in graphene. Appl. Phys. Lett. 2013, 103, 071904. 10.1063/1.4818683. [DOI] [Google Scholar]
- Ackerman M. L.; et al. Anomalous Dynamical Behavior of Freestanding Graphene Membranes. Phys. Rev. Lett. 2016, 117, 126801. 10.1103/PhysRevLett.117.126801. [DOI] [PubMed] [Google Scholar]
- Rosłoń I. E.; Japaridze A.; Steeneken P. G.; Dekker C.; Alijani F. Probing nanomotion of single bacteria with graphene drums. Nat. Nanotechnol. 2022, 17, 637. 10.1038/s41565-022-01111-6. [DOI] [PubMed] [Google Scholar]
- Hu J.; Vanacore G. M.; Cepellotti A.; Marzari N.; Zewail A. H. Rippling ultrafast dynamics of suspended 2D monolayers, graphene. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E6555–E6561. 10.1073/pnas.1613818113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 2003, 67, 155108. 10.1103/PhysRevB.67.155108. [DOI] [Google Scholar]
- Ozaki T.; Kino H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B 2004, 69, 195113. 10.1103/PhysRevB.69.195113. [DOI] [Google Scholar]
- Ozaki T.; Kino H. Efficient projector expansion for the ab initio LCAO method. Phys. Rev. B 2005, 72, 45121. 10.1103/PhysRevB.72.045121. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Ozaki T.; Fukuda M.; Jiang G. Efficient O(N) divide-conquer method with localized single-particle natural orbitals. Phys. Rev. B 2018, 98, 245137. 10.1103/PhysRevB.98.245137. [DOI] [Google Scholar]
- Bader R. F. W.Atoms in Molecules: A Quantum Theory; Clarendon Press: 1990. [Google Scholar]
- Goddard T. D.; et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data required to evaluate the conclusions of the paper is present in the main text or the Supporting Information. Supporting Information contains analysis of hydrated cyt c7+ and cyt c7+ colliding with graphene. The AIMD trajectories for cyt c landing on graphene, as well as the atomic coordinates for the gas-phase cyt c7+, hydrated cyt c7+, and the adsorbed cyt c on graphene are available at the Data Repository of the Max Planck Society (10.17617/3.VJ9ZIM).