

Abstract
Atherosclerosis begins with accumulation of cholesterol-carrying lipoproteins on blood vessel walls and progresses to endothelial cell dysfunction, monocyte adhesion, and foam cell formation. Endothelialized tissue-engineered blood vessels (TEBVs) have previously been fabricated to recapitulate artery functionalities including vasoconstriction, vasodilation, and endothelium activation. Here the initiation of atherosclerosis is emulated by designing branched TEBVs (brTEBVs) of various geometries treated with enzyme-modified low-density-lipoprotein (eLDL) and TNF-α to induce endothelial cell dysfunction and adhesion of perfused human monocytes. Locations of monocyte adhesion under pulsatile flow are identified and the hemodynamics in the brTEBVs are characterized using particle image velocimetry (PIV) and computational fluid dynamics (CFD). Monocyte adhesion is greater at the side outlets than at the main outlets or inlets, and is greatest at larger side outlet branching angles (60° or 80° versus 45°). In PIV experiments, the branched side outlets are identified as atherosclerosis-prone areas where fluorescent particles show a transient swirling motion following flow pulses; in CFD simulations, side outlets with larger branching angles show higher vorticity magnitude and greater flow disturbance than other areas. These results suggest that the branched TEBVs with eLDL/TNF-α treatment provide a physiologically relevant model of early atherosclerosis for preclinical studies.
Keywords: atherosclerosis, bifurcated artery, microphysiological system, tissue-engineered blood vessel, pulsatile flow
1. Introduction
Atherosclerosis is an inflammatory disease in which plaque accumulates in arteries, and is the major precursor of cardiovascular disease, the cause of 31% of deaths worldwide in 2016.[1][2] Atherosclerosis is initiated by accumulation of low-density lipoproteins (LDLs) in the endothelial cell (EC) layer of arteries, and progresses to EC dysfunction with increased secretion of RANTES chemokine and overexpression of endothelial receptors including P- and E-selectin, vascular adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1).[3] These EC changes promote the adhesion and transmigration of monocytes (MCs), leading to formation of foam cell macrophages and plaque formation.
Atherosclerosis is site-specific and is associated with regions of low shear and oscillatory flow.[4]–[7] The outer walls of bifurcation areas and the inner walls of curvatures, which have low shear stress, show greater susceptibility to plaque and necrotic core formation.[8][9] Dai and coworkers defined “athero-prone” and “athero-protective” waveforms of blood-flow, based on flow patterns in coronary artery bifurcations in normal human patients captured using MRI and ultrasound and analyzed using three-dimensional (3D) computational fluid dynamics (CFD).[6] Further, EC dysfunction, a precedent event of MC adhesion, has been confirmed to be correlated with fluid dynamic characteristics in in vitro models.[6][10]
In vitro studies of the relationship between fluid dynamics and atherosclerosis have attempted to emulate physiological blood flow conditions in devices with a flat surface,[11][12] or molded into simple tubes or anatomically accurate models using silicone-elastomer.[13]–[15] A co-culture model[7] was developed to test the effect of hemodynamic forces on the interactions between smooth muscle cells (SMCs) and ECs, and identified athero-prone flow conditions in which the cells were more susceptible to inflammation. Recent studies reported more sophisticated in vitro microvascular structures[16][17] and were able to show disturbed flow on integrity of EC tight junctions or to recapitulate anatomy of arteries; however, it is often of difficulty representing in vivo vascular characteristics due to the choice of materials (i.e., PDMS- or Si-based) or inefficacy in connecting perfusion flow. In vitro models that more accurately capture the 3D architecture and mechanical properties of arteries are needed to better study early atherosclerosis.
We previously fabricated endothelialized tissue-engineered blood vessels (TEBVs)[18]–[20] using SMCs embedded in collagen; Ji and coworkers used induced SMCs (iSMCs) reprogrammed from endothelial progenitor cells (EPCs) or from patients’ fibroblasts[21] using human myocardin (MYOCD) viral vectors.[22] The iSMCs exhibited the contractile phenotype of primary SMCs,[23]–[25] and the resulting eTEBVs showed artery functionality including vasodilation and vasoconstriction.[18]–[20][26][27] The eTEBVs were cultured in a perfusion environment and were used as a preclinical model of rare diseases such as Marfan syndrome and Progeria syndrome.[21][28][29]
Here we further develop the TEBV technology to emulate early atherosclerosis by designing branched TEBVs (brTEBVs) with various branching angles to recapitulate bifurcation areas of coronary arterioles [30]–[32] and treating the brTEBVs with enzyme-modified LDL (eLDL) and TNF-α to induce EC dysfunction and MC adhesion. We describe (i) the design and fabrication of brTEBV chambers; (ii) the selective localization of MCs on the EC layers in the brTEBV lumen; and (iii) particle image velocimetry (PIV) experiments and CFD simulations used to characterize the hemodynamics in the brTEBV system. We found that the locations of MC focal adhesion in the brTEBVs coincided with regions of high vorticity in CFD simulations and high curl magnitude in PIV experiments and depended on the local bifurcation angle in the brTEBVs. The initiation and development of atherosclerosis as a focal phenomenon in arteries has drawn attention in clinics but largely been ignored in the field of microphysiological systems (MPS), where vascular networks are rarely incorporated as one of the components in tissue-chip construction. This unique, free-standing, bifurcated vascular platform highlights the potential utility of human cell-based vascular MPS as a preclinical model of early atherosclerosis and, further, a part of systemic inflammation model by integration with other multi-organ MPS.
2. Results
2.1. Design and operation of brTEBV chambers
The brTEBVs possess an inlet, a main outlet, and a side outlet (Figure 1a): three side outlet branching angles—45°, 60°, and 80°— to emulate coronary artery bifurcation angles.[30][31] The inner diameters (IDs) of the inlet and main outlet are 2 mm, the ID of the side outlet is 1.4 mm, and the vessel wall thickness is 2 mm. Each brTEBV chamber consists of a gel-molding chamber and a perfusion chamber (Figure 1b,c,e). In the gel-molding chamber, a hollow vessel is formed by placing a stainless steel mandrel across the middle of the chamber and adding a mixture of iSMCs and collagen which forms a gel. The vessel lumen has three perfusion ports (red arrows in Figure 1d), and the brTEBV chamber has two additional ports for refreshing chamber flow (green arrows in Figure 1d).
Figure 1. Design, assembly, and operation of brTEBVs.
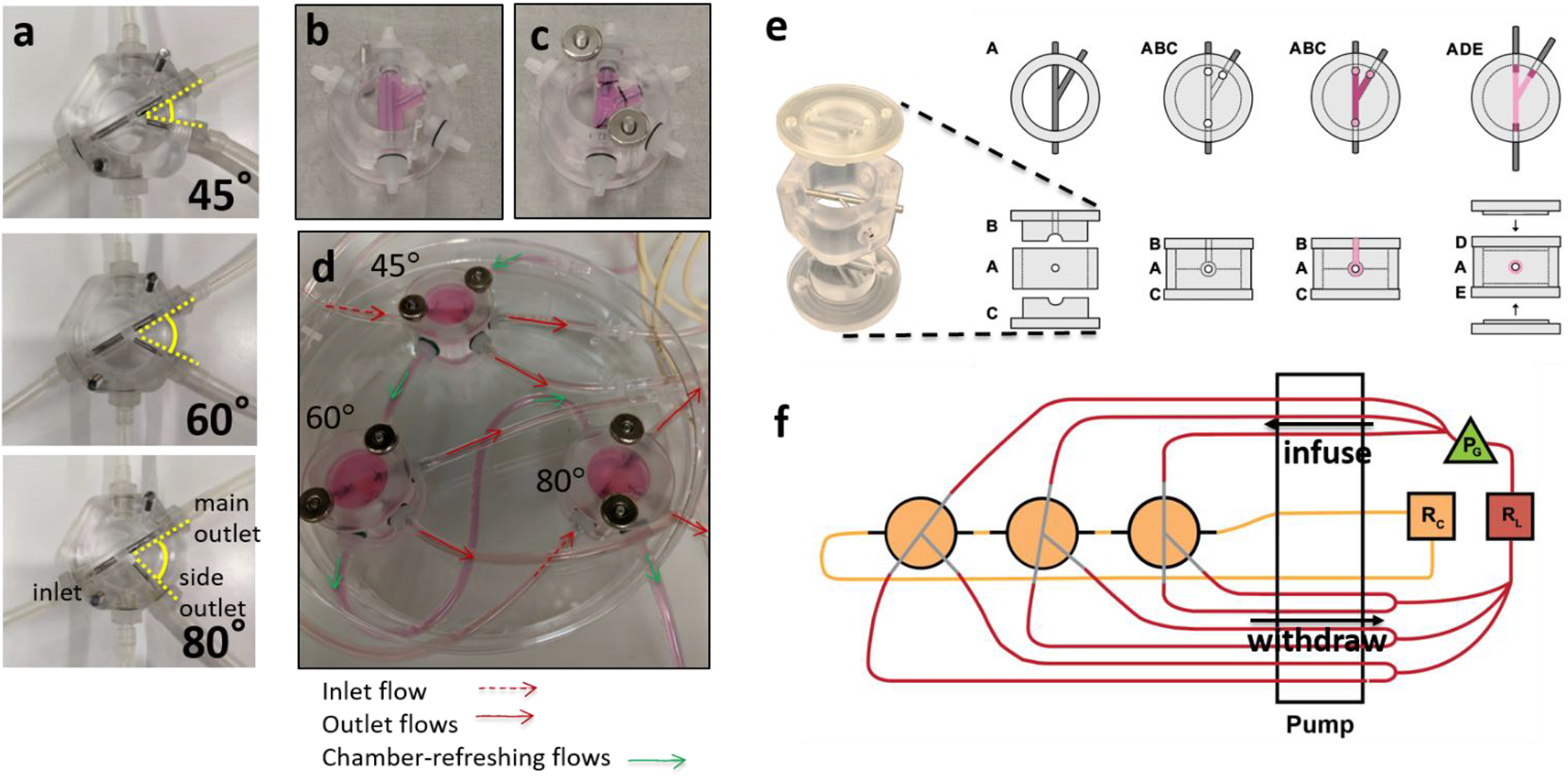
(a) Photographs of the three brTEBV chambers with side outlet branching angles of 45°, 60°, and 80°. (b) A collagen gel containing ECs is formed in the gel-forming chamber. (c) Mandrels are moved half-way out and the inlet and outlets of the gelated vessel structure are sutured onto them. (d) Each brTEBV flow chamber has five flow connections: one for inlet flow, two for outlet flow, and two for refreshing the chamber. (e) Schematic with top and side views of brTEBV chamber assembly. The top and bottom of the gel-forming chamber (Parts B and C) are assembled with Part A with mandrels tightly joined. The EC-collagen mixture is pipetted into the inlet hole on Part B (see top view) until the mixture exits the outlet holes in Part B. After gelation at 37 °C, Parts B and C are replaced with the top and bottom of the flow chamber (Parts D and E). (f) Diagram of brTEBV operation. A multi-channel peristaltic pump simultaneously infuses the inlets from the lumen reservoir (RL), withdraws flow from the outlets of multiple brTEBVs, and replaces media in the chamber from a separate reservoir (RC). A solenoid pinch valve acts as a pulse generator (PG) and is located immediately before the inlets to provide pulsatile flow into the vessel lumen.
A 24-channel peristaltic pump was used to infuse culture media into the inlets and withdraw fluid through the outlets of multiple brTEBV chambers simultaneously (Figure 1f). It was necessary to use both infusing and withdrawing mechanisms to obtain consistent flow distribution into both outlets and to maintain reproducible flow across multiple brTEBVs. The volumetric flow rates in the main and side outlets were 0.75× and 0.25× that of the inlet, respectively. The overall flow rate was 4 mL min−1, at which the wall shear stress (WSS) in the absence of branching or pulsing would be ~0.85 dyne cm−2. The flow into the brTEBVs was connected to a pulse generator (PG) to obtain a step pulse of 1 Hz (Figure 1f).
2.2. Confirmation of brTEBV function and effect of eLDL treatment
To obtain iSMCs, human EPCs were transduced with m2rtTA and MYOCD vectors at different multiplicity of infections (MOIs), followed by treatment with doxycycline to induce MYOCD expression. Using both vectors at an MOI of 20, the EPCs were successfully reprogrammed to iSMCs as indicated by expression of calponin (CNN) and α-smooth muscle actin (α-SMA, Figure 2a). At an MOI of 10, the EPCs were only partially reprogrammed; at an MOI of 30, a significant fraction of the cells died (data not shown). Figure 2b shows EC activation and MC adhesion following eLDL and TNF-α treatment, indicated by expression of ICAM-1 on ECs and CD80 on MCs.
Figure 2. SMC and activated EC markers and MC adhesion in 2D culture.
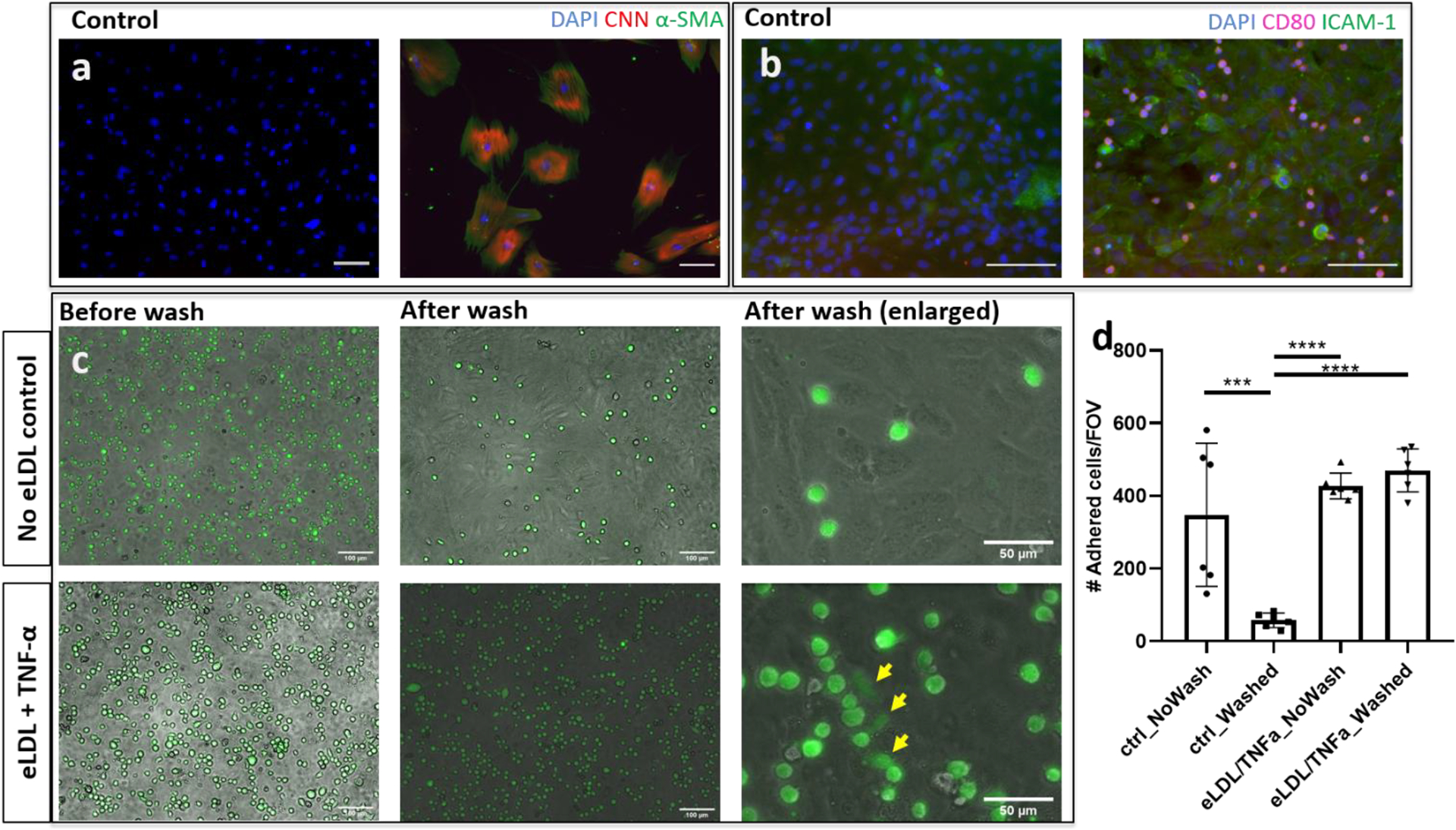
(a) SMC markers calponin (CNN, red) and α-SMA (green) are observed in iSMCs reprogrammed from human EPCs using MYOCD expression. The control shows EPCs without MYOCD viral transduction. (b) After eLDL and TNF-α treatment, ECs express ICAM-1 (green) and THP-1 cells expressing CD80 (magenta) adhere to the ECs. The control shows ECs without eLDL/TNF-α treatment. (c) THP-1 cells remained adhered to the surface of the EC layer, and some THP-1 cells changed morphology (yellow arrows) after eLDL/TNF-α treatment. Without treatment, the majority of THP-1 cells were washed away. (d) Quantitation of THP-1 adhesion; n = 6, representative data from ≥ 5 independent experiments; one-way ANOVA Tukey test. *** P<0.001, **** P<0.0001. Scale bars: 100 μm (a-c), 50 μm (c, after wash (enlarged)). All panels represent 2D experiment results.
Adhesion of MCs was significantly greater following eLDL and TNF-α treatment than without treatment in 2D well plates (Figure 2c). In the absence of eLDL and TNF-α treatment, MCs were washed away; following eLDL and TNF-α treatment, the majority of MCs remained adhered to the activated ECs after washing (Figure 2d). The MCs not only adhered but also changed morphology (yellow arrows in Figure 2c), implicating potential transvasation into the vessel wall to form the foam cells in vivo.[33]
Foam cell formation following MC adhesion was also investigated. The brTEBVs were perfused for at least 7 days for maturation before treatment with eLDL for 3 days and with TNF-α for 12 h, followed by perfusion with THP-1 MCs for 3 days. In brTEBVs with eLDL/TNF-α treatment, many MCs adhered and the majority showed internalized lipids (Figure 3b,c). Without eLDL/TNF-α treatment, few MCs adhered to the vessel lumen (Figure 3a) in the perfusion environment, likely due to the inactivated ECs. Foam cell formation was also confirmed by using Oil-red-O staining of frozen sections of the brTEBVs (Figure 3d–f). Well-developed EC layers are visualized in the brTEBV at various locations after eLDL and TNF-α treatment (Figure 3g–i). Main and side outlet areas showed more aligned EC layer in contrast to the center area where the ECs were randomly oriented. Phenotypic markers of iSMCs in the brTEBV tissue sections were also examined (Figure 3j,k). Colocalization of CNN and α-SMA is indicated by yellow fluorescence in Figure 3j; colocalization of TAGLN and MYH11 is indicated by yellow fluorescence in Figure 3k. ICAM-1 expression in brTEBV ECs indicates EC activation by the inflammatory eLDL/TNF-α treatment (Figure 3g–i).
Figure 3. TEBV function and effect of eLDL/TNF-α treatment.
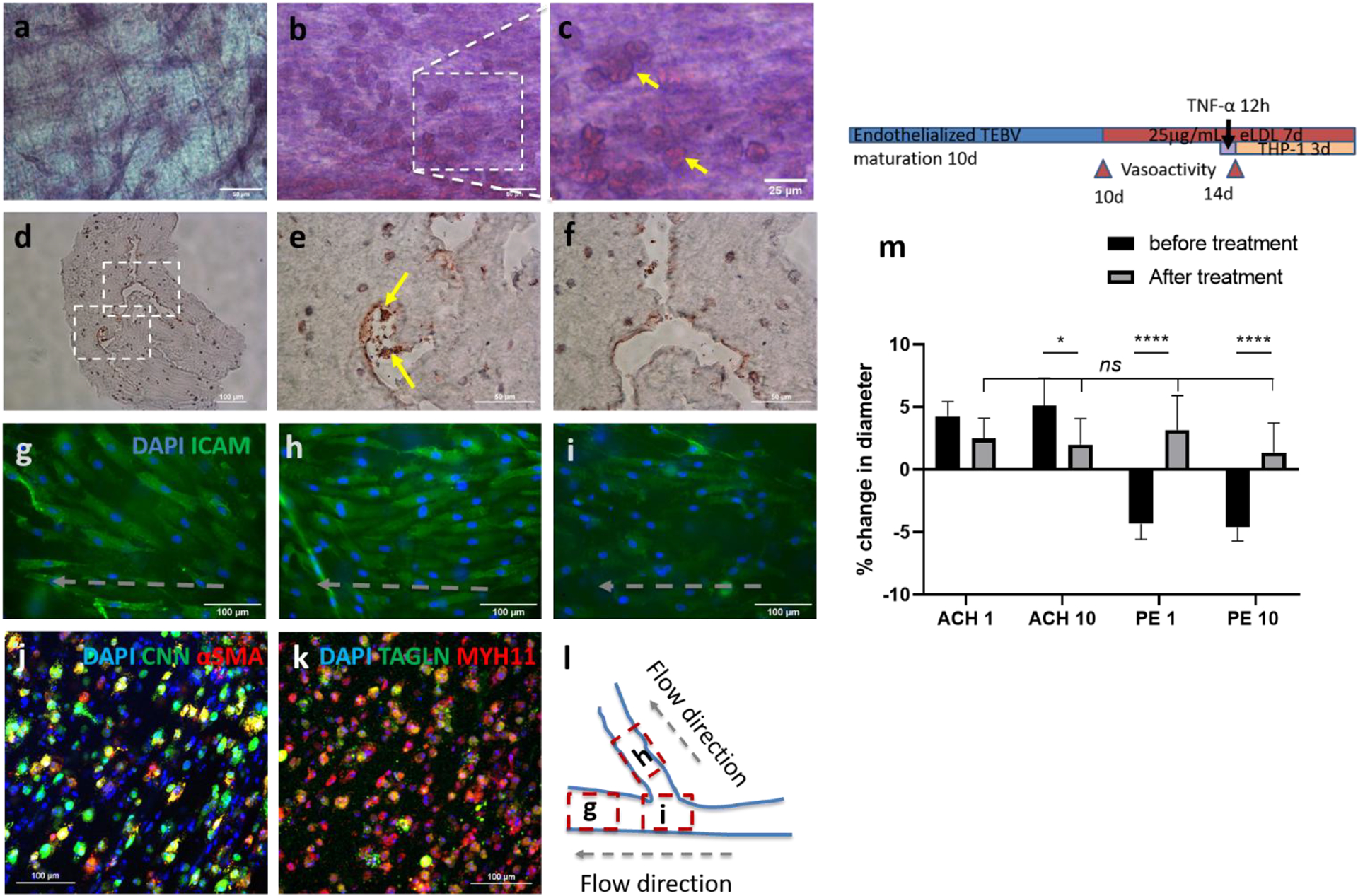
Oil-red-O staining of lipid-laden macrophages (foam cells) in brTEBVs without (a) and with (b-f) eLDL and TNF-α treatment: (a-c) tissue staining, (d-f) staining of frozen sections. (g-i) Immunofluorescence images of endothelial layer in brTEBV tissue, with ICAM-1 (green) and DAPI (blue), after 5 days of culture and 72 h treatment of 10 μg mL−1 eLDL and 8 h TNF-α at main (g), side (h), and center (i.e., branching area) (1) Schematic diagram indicating the location of each image and direction of flow in (g-i). (j-k) Confocal immunofluorescence images of TEBV tissues after the treatment; (j) Calponin (CNN, green) and α-SMA (red); (k) TAGLN (green) and MYH11 (red). Colocalization of CNN and α-SMA, and of TAGLN and MYH11 is indicated by yellow fluorescence. (m) The change in vasoconstriction and dilation capabilities in linear TEBVs induced by 1 or 10 μm acetylcholine (Ach) and phenylephrine (PE), before and after 96 h-treatment with eLDL and TNF-α treatment; n = 6 (3 locations/ vessel, 2 vessels/ condition), two-way ANOVA multiple comparisons. * P<0.05, **** P <0.0001. Scale bars: 100 μm (d,g-k), 50 μm (a,b,e,f), 25 μm (c). Panels (a-k) are from brTEBVs and panel (m) is from linear TEBV.
An unbranched, linear TEBV was used to confirm iSMC vasodilation and vasoconstriction and to observe the effect of inflammatory signals on these capabilities. Measurement of changes in vessel diameters by vasoconstrictor or vasodilator was more reliable in linear TEBVs than in brTEBVs. The linear TEBV vessel diameter increased ~4% following treatment with acetylcholine (Ach) at 1 or 10 μM, and decreased ~4% following treatment with phenylephrine (PE) at 1 or 10 μM (Figure 3m). The iSMC vasodilation and vasoconstriction capabilities were lost following 4 days of treatment with eLDL and TNF-α, which have affected not only EC dysfunction but also iSMCs underlying medial layer.
2.3. Localization of human monocytes in brTEBVs
We hypothesized that the selective localization of MCs in the lumen of brTEBVs would be affected by the side outlet branching angles. To monitor MC adhesion, CellTracker Green-labeled THP-1 cells were perfused at 4 mL min−1 with a 1 Hz pulse into endothelialized brTEBVs treated with eLDL and TNF-α. Based on previous pathological findings,[8][34] the brTEBV was divided into three areas (dotted circles in Figure 4a): inlet, outer wall of the main outlet, and outer wall of the side outlet. These low shear stress areas of the main and side outlets were areas of particular interest as indicated in many reports.[34][35] For all three branching angles of brTEBVs, MC adhesion was greater on the outer walls of the side outlet areas than on the inlet or main outlet areas (Figure 4b–c); MC adhesion was greatest in the side outlet areas at branching angles of 60° and 80°. Interestingly, in the 45° brTEBV, MC adhesion in the side outlet was significantly greater than that in the main outlet, but not significantly different from that in the inlet. MC adhesion in inlets and main outlets were similar across all branching angles. These results demonstrate that the brTEBV model recapitulates the focal nature of initiation of atherosclerotic lesions, and suggest that lesion formation is affected by the bifurcation angle; i.e., a greater change in the direction of flow results in greater susceptibility to MC adhesion and lesion formation. In the absence of eLDL/TNF-α treatment, minimal MC adhesion was observed (Figure 4d). The large error bars in Figure 4c,d resulted from experiment-to-experiment variation, possibly due to the use of different passages of THP-1 cells, or variation in stimulation of ECs by the different batches of eLDL.
Figure 4. Localization of MC adhesion in brTEBVs at various branching angles.
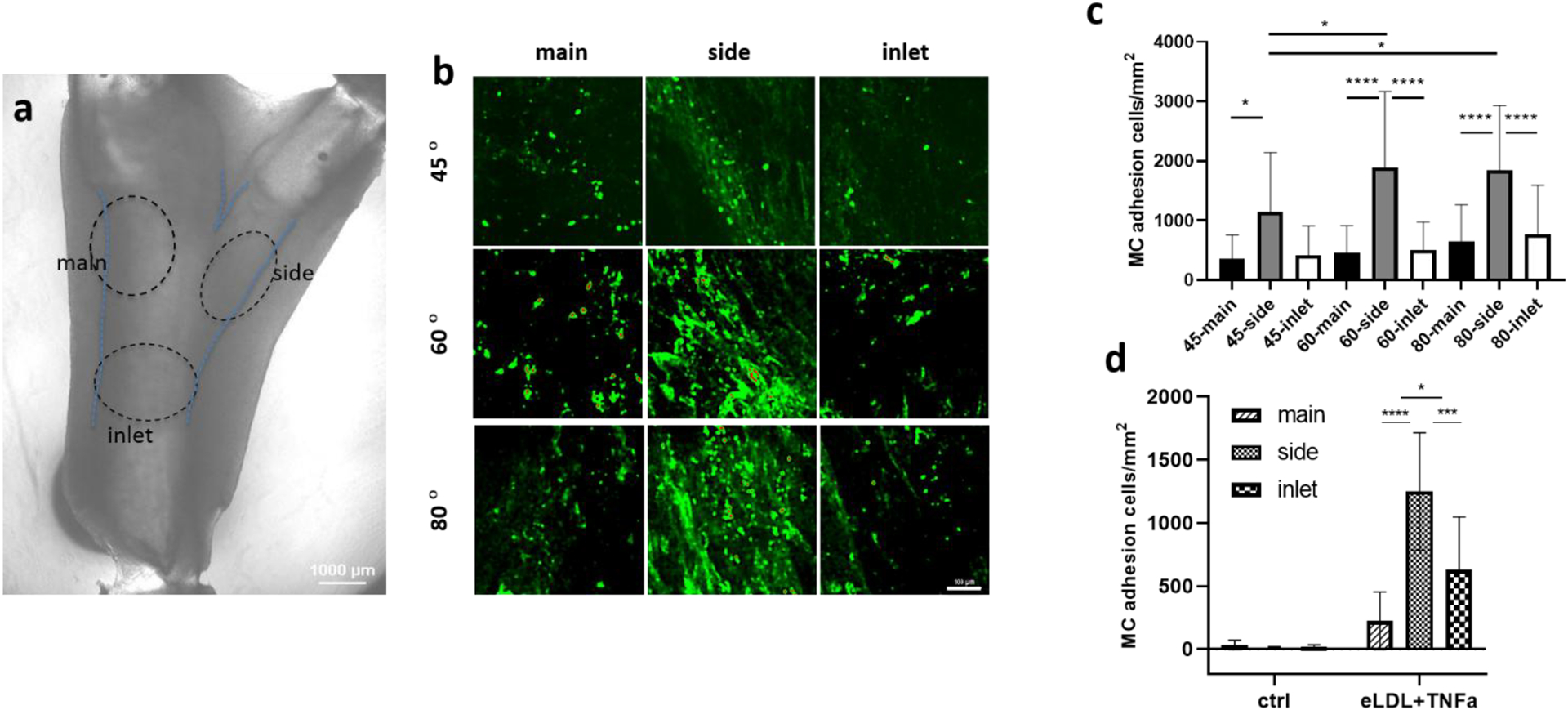
(a) Brightfield image of brTEBV with branching angle of 45° is shown with categorized area of inlet, main and side outlets (dotted circles), where MC adhesion was considered. (b) Fluorescence images of CellTracker Green-labeled MCs in each brTEBV area. Scale bar: 100 μm (c) Quantitative analysis of effect of branching angle on MC adhesion in each area with 10 μg mL−1 eLDL and 50 ng mL−1 TNF-α treatment; n ≥ 4 vessels from 5 independent experiments; Kruskal–Wallis two-way ANOVA test multiple comparisons. (d) Effect of eLDL and TNF-α on MC adhesion; n ≥ 3 vessels from 2 independent experiments; one-way ANOVA Tukey test. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001.
2.4. Particle image velocimetry reveals brTEBV flow profile
To understand the behavior of MCs at different locations in brTEBVs, we used PIV with three branched PDMS vessels and captured the motion of 10-μm fluorescent beads at 500 fps for 10 s. Movie S1 shows the fluorescent beads moving in an 80° branched PDMS vessel at a flow rate of 4 mL min−1 with 1 Hz pulse. There was significant circulation of flow near the side outlet after flow was established, and a chaotic relaxation of flow at the branching area when flow was stopped by closing the valve. In contrast, when the flow rate was reduced to 1 mL min−1 (Movie S3) while keeping all other conditions the same (including pulsatile flow), no circulation of flow at the side outlet or chaotic relaxation was observed.
We analyzed the PIV results to examine and quantify the particle movements. In our brTEBV design, the flow appeared laminar without pulses; the Reynolds number was ~50 in the circular inlet tubes, much lower than that of traditional threshold for turbulent flow. To quantify the flow separation and strong recirculation with oscillatory inflow in the branched tubes, we first considered the vorticity field (ω), defined as the curl of velocity field ν(vx, vy) as shown in equation (1):
| (1) |
and the spatial and temporal correlations of velocities, c(R, T), as shown in equation (2).
| (2) |
These correlations reflect the variation rates in the spatial and temporal domains. With pulsatile flow, the changes in velocity magnitude and direction became irregular and more stochastic, and therefore, the combined spatial and temporal correlation was lower than that for typical laminar flow. Figure 5 compares the flow profiles in a 60° brTEBV at a flow rate of 4 mL min−1 with and without pulses. Velocity magnitude maps showed the average velocities in the brTEBV. Without pulses, the flow appeared laminar; with pulses, the flow was disturbed in the side outlet area (see arrows indicating velocity in Figure 5a,b). Curl magnitude maps showed significant vorticity near the side outlets with pulsatile flow (yellow dashed circle in Figure 5d). Flow was more disturbed near the side outlet than near the inlet or main outlet as indicated by lower spatial and temporal correlation in the flow pattern at the side outlet areas (yellow dotted circle, Figure 5f).
Figure 5. Quantitative analysis of PIV results.
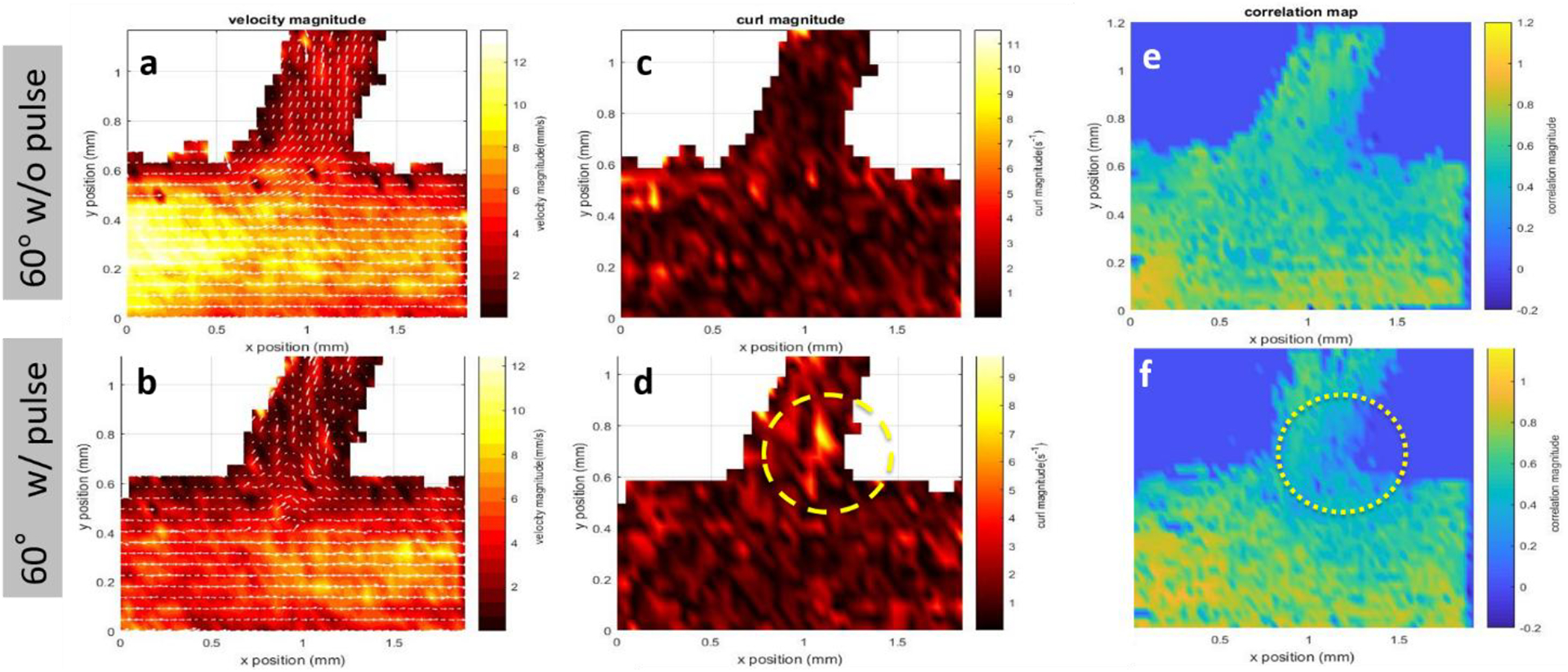
PIV results for a brTEBV with branching angle of 60° at 4 mL min−1 with 1 Hz pulsing (bottom row: b,d,f), and without pulsing (top row: a,c,e). (a-b) Velocity magnitude profiles. (c-d) Curl magnitude maps. (e-f) Spatial and temporal correlation maps.
2.5. Computational fluid dynamics
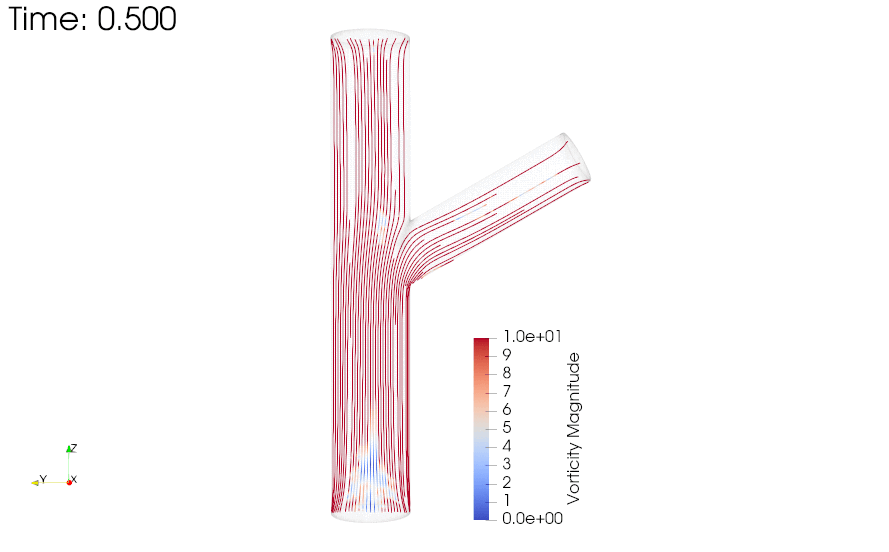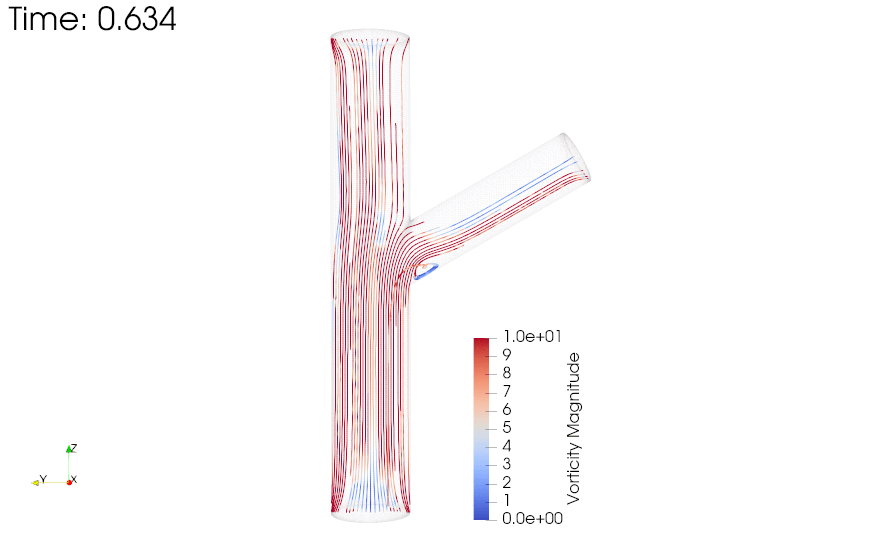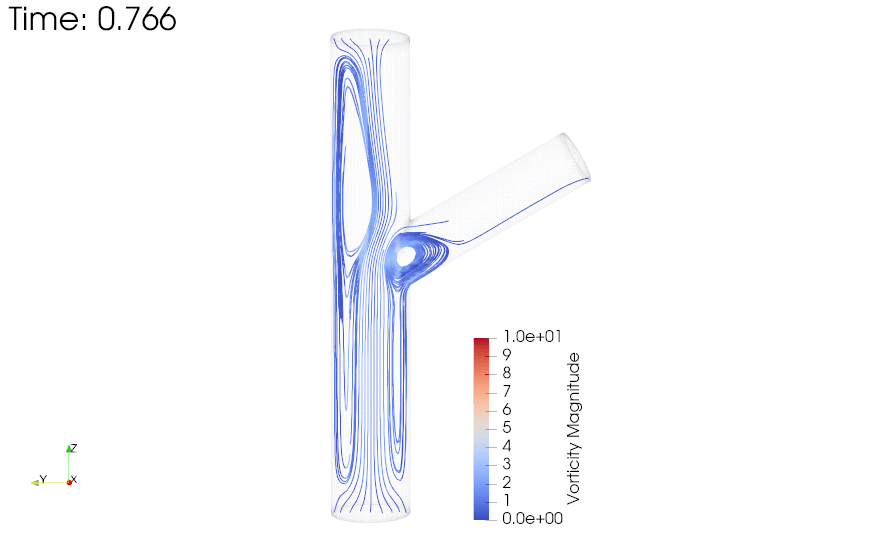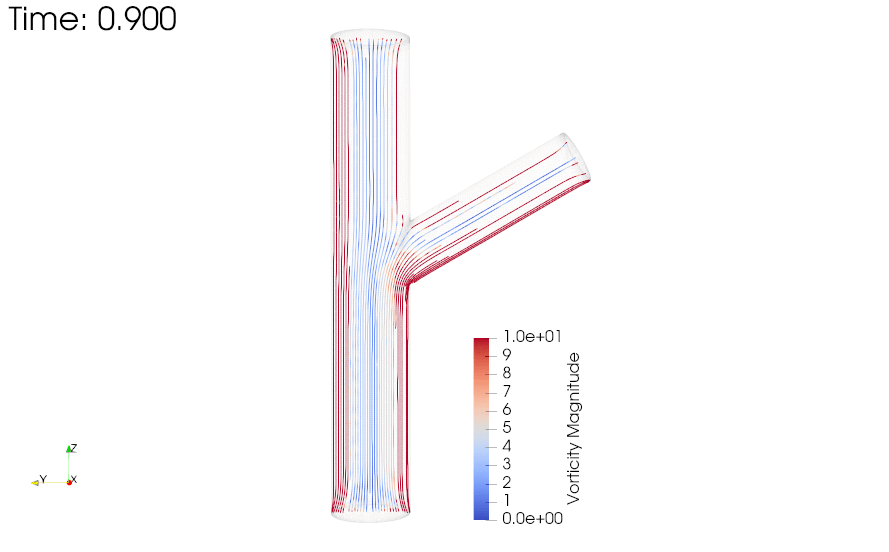
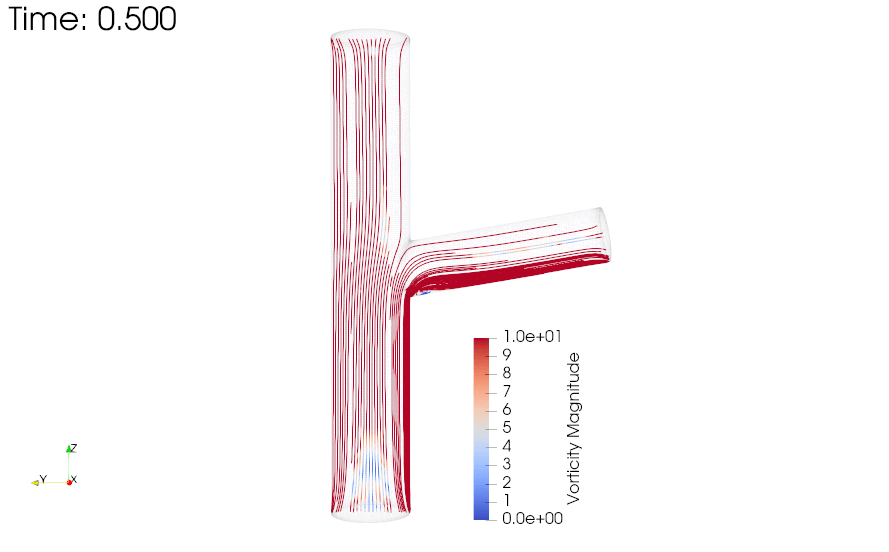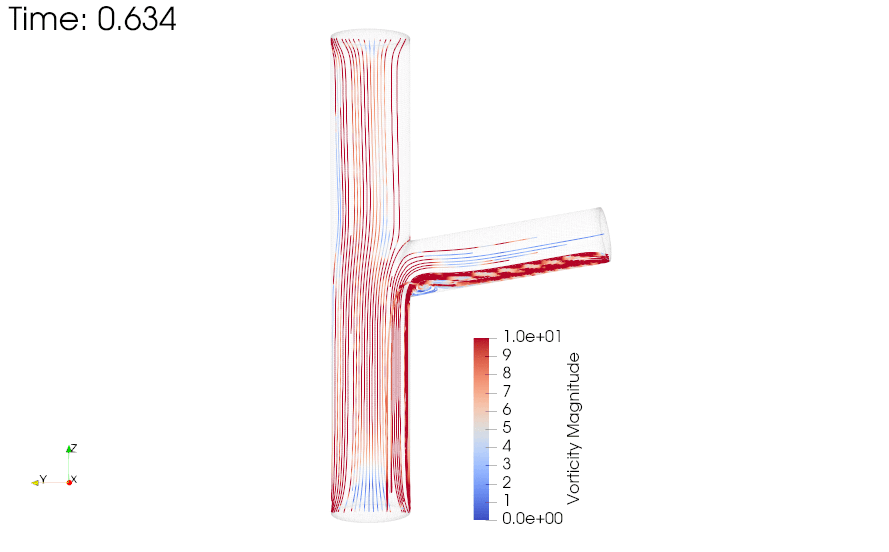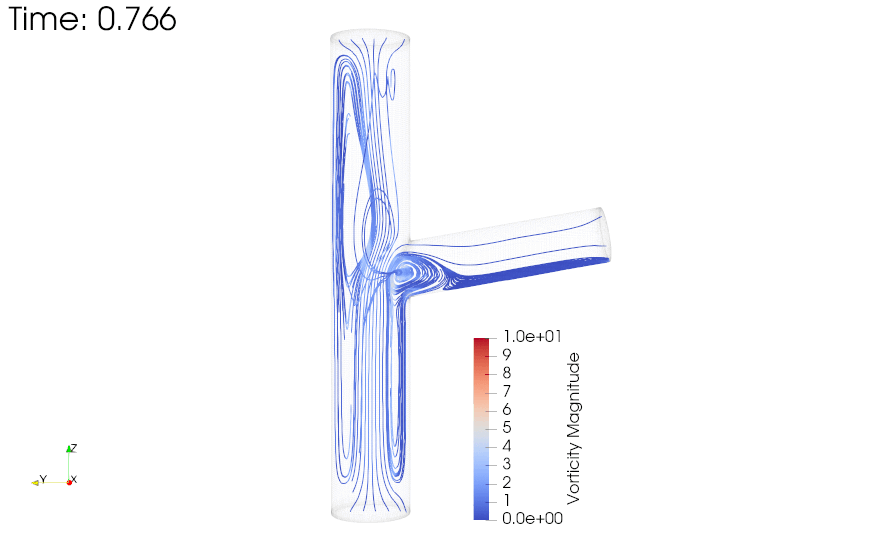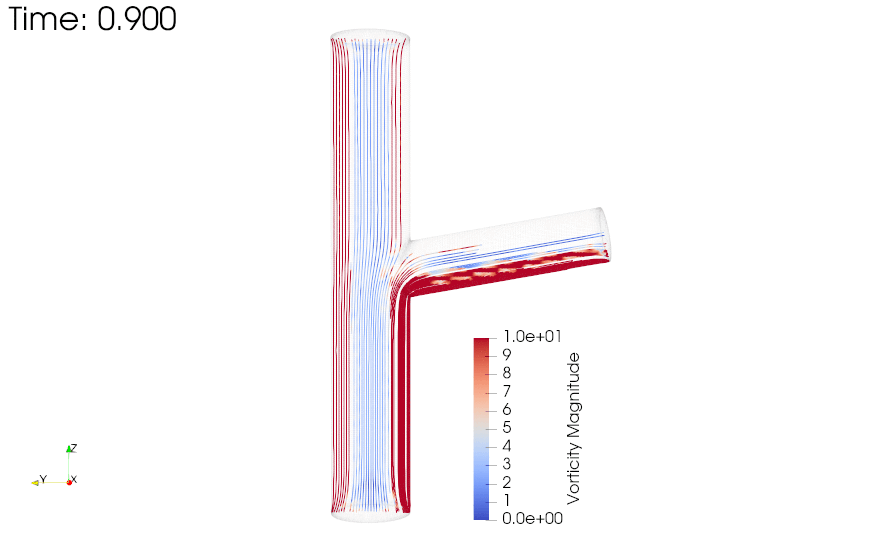
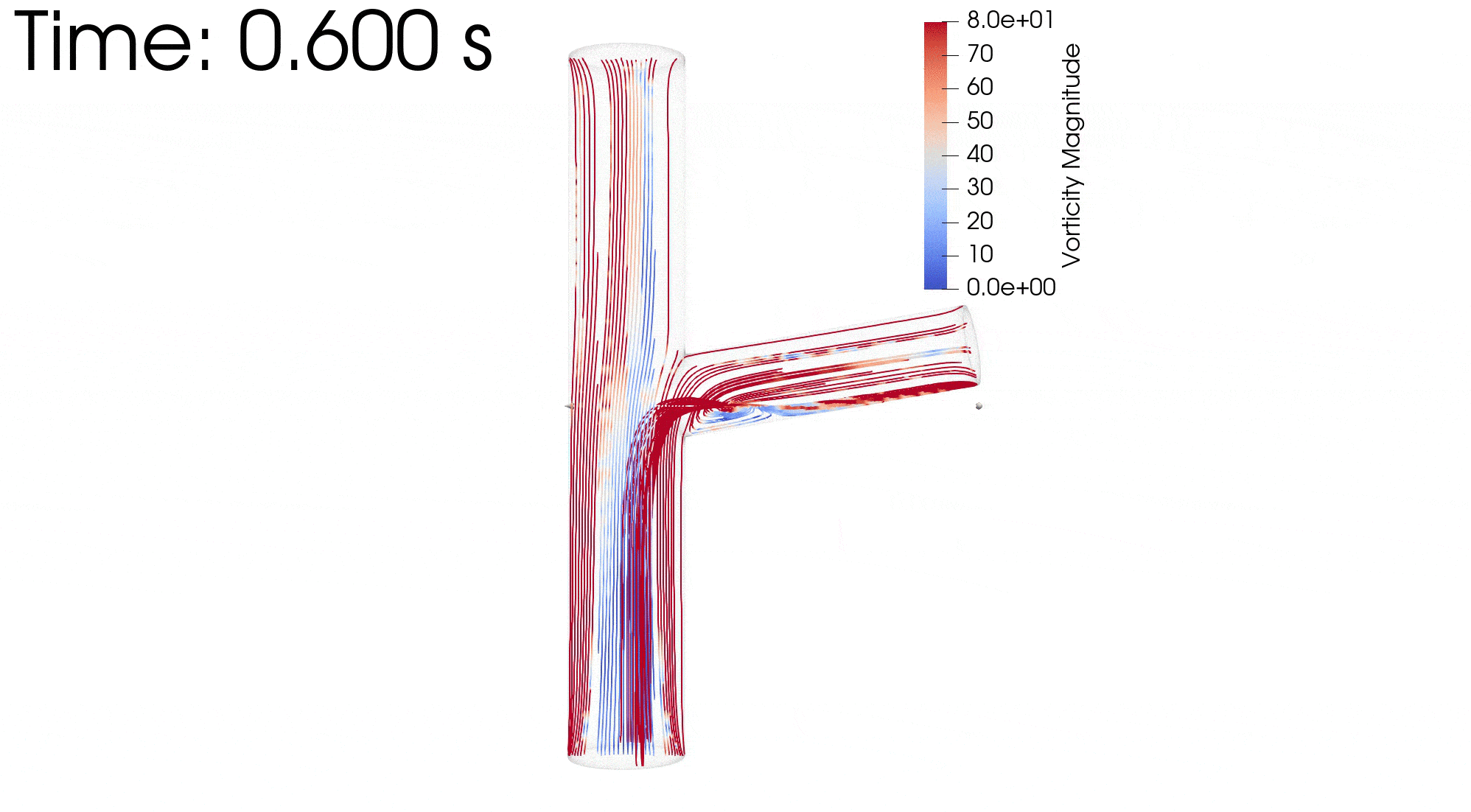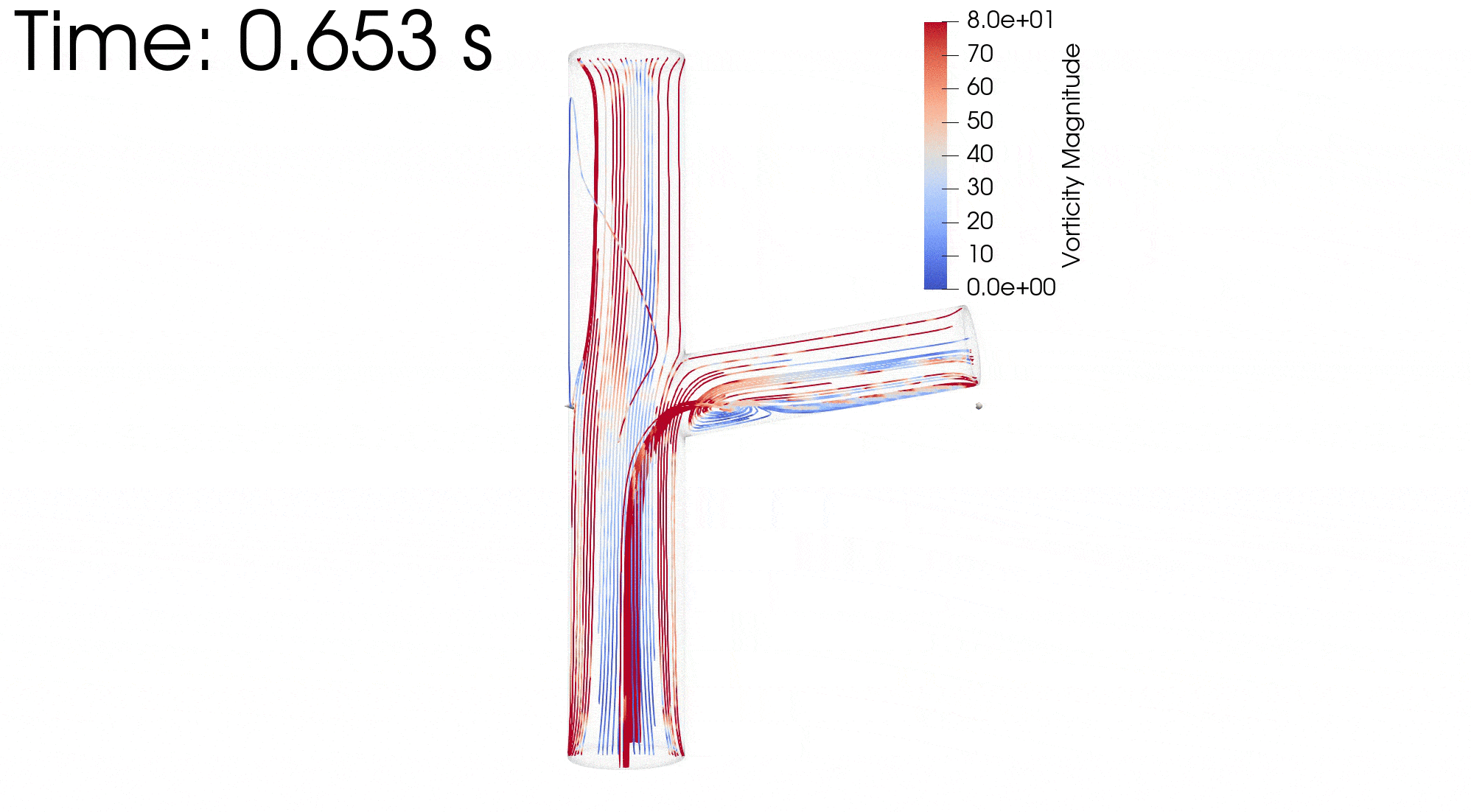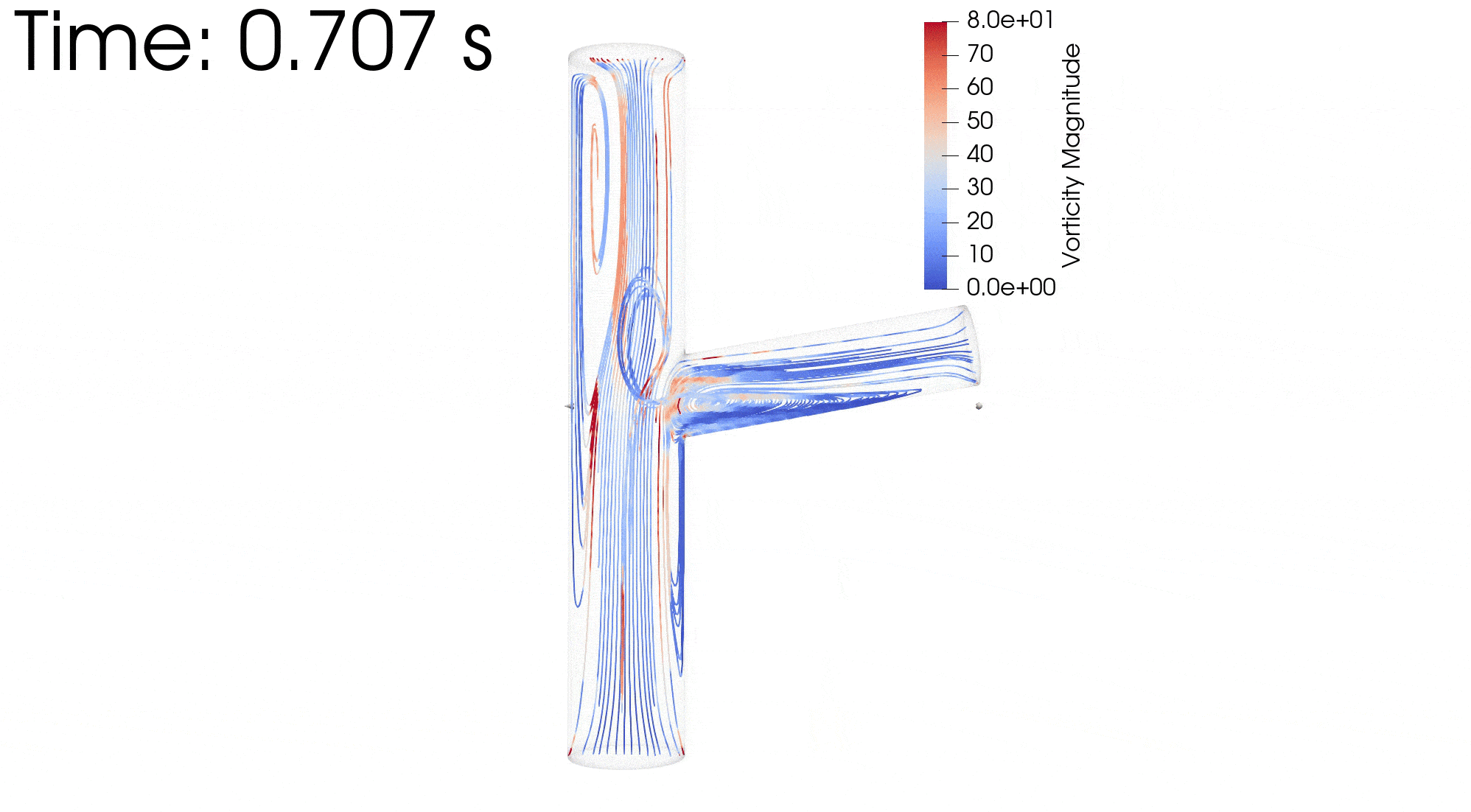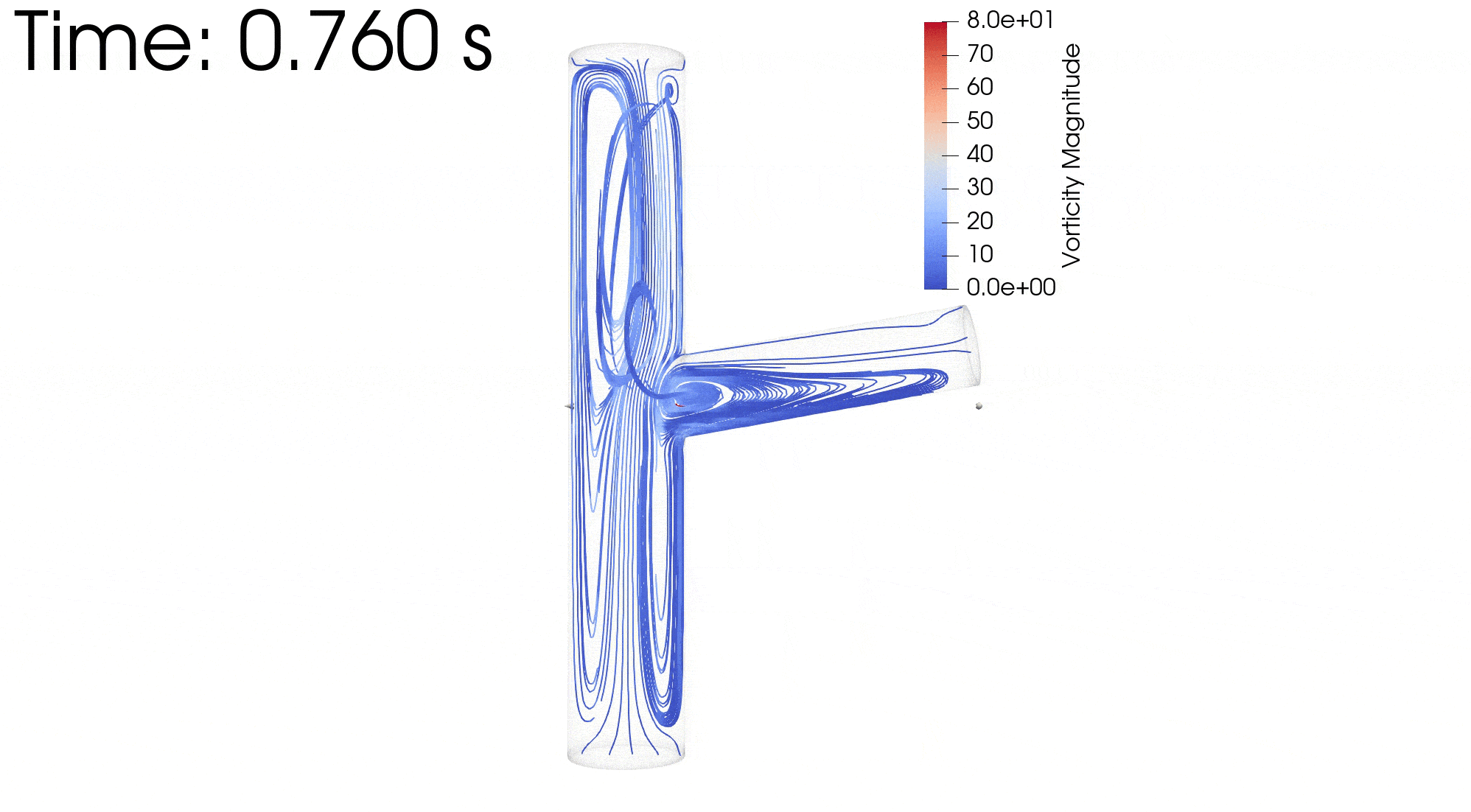
CFD simulations were performed to compare the flow profiles in brTEBVs with different branching angles. The simulations solved the unsteady Navier-Stokes equation for the flow pattern in the brTEBVs. To mimic pulsatile inflow driven by the pulse-generating solenoid valve, a sinuous function was used for the inlet flow rate. The volumetric flow rates at the main and side outlets were 0.75× and 0.25× that at the inlet, respectively, as in brTEBV experiments. Figure S2 shows simulation results when the outlet flow partitioning was 0.5:0.5 instead of 0.75:0.25. Figure 6b shows the flow velocity at the inlet and two outlets over one period; red arrows indicate the time points at which the flow velocity was at its maximum (0.250 s) and minimum (0.755 s). Figure 6c–h show the post-processed flow profiles of brTEBVs at these two time points with 3D-streamlines colored according to the velocity magnitude. For all three branching angles, irregular and curled flow streamlines were observed extensively at t = 0.755 s; in contrast, normal and straight streamlines were observed at t = 0.250 s. During the relaxation period (at around t = 0.755 s), there were few lines passing through the side outlet; instead, recirculation of fluid was mainly observed (Figure 6f–h). At this time point, for all three branching angles, the flow velocity from the inlet to the main outlet was greater than that to the side outlet. As marked by the black arrows in Figure 6i, areas with relatively lower speed were observed, indicating secondary flow such as swirl or curl motions. Figure 6j compares vorticity streamlines in a brTEBV under steady and pulsatile flow at t = 0.696 s (i.e., immediately before complete relaxation); it is confirmed that swirl or recirculating motion of flow appears only in the pulsatile flow condition, as shown in PIV. The vorticity magnitude itself was lower with pulsatile flow during this relaxation period. Figure 6k shows the vorticity streamlines near the branching area of each brTEBV in detail. A small and strong recirculation of flow was observed at the outer wall of the side outlet and a wide and long recirculation near the outer wall of the main outlet in each brTEBV. Changes in vorticity during the relaxation period of each brTEBV are shown in Movies (Movies S6–8). The swirl motion appeared more complicated and irregular with larger side branching angles. It is notable that clinical observations of atherosclerotic locations are consistent with the two regions showing swirl motion, indicated by flow velocity and vorticity fields (in Figure 6i and 6k). The WSSs along the outer walls of each brTEBV from inlet to main outlet or from center to side outlet are plotted in Figure 6l (walls depicted as A to B or G to F). The low WSS areas along the walls of A to B and G to F were consistent with areas of recirculation during both infusing and relaxation periods (t = 0.25 s and 0.696 s, respectively).
Figure 6. CFD simulations reveal brTEBV flow profiles.
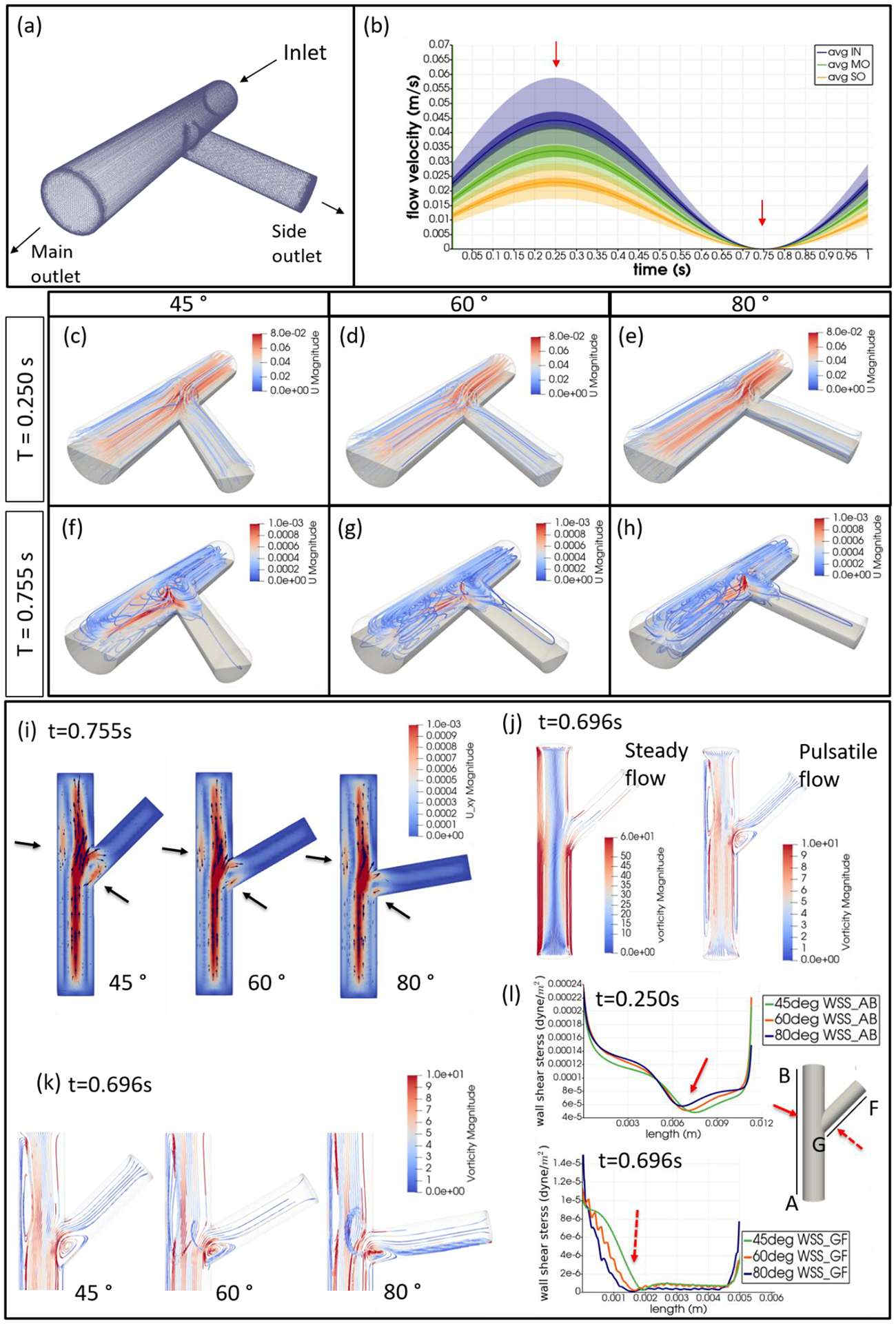
(a) Mesh diagram for the 80° brTEBV. (b) Sinuous inflow at the inlet (purple lines), and outflow at two outlets over one time period (green lines for the main outlet, yellow lines for the side outlet). (c-h) 3D velocity streamlines in the three brTEBVs at t = 0.250 s and t = 0.755 s. (i) Velocity profiles in the longitudinal direction in three brTEBVs at t = 0.755 s, with black arrows denoting secondary flow at the outer walls of main and side outlets. (j) Vorticity field in brTEBV in steady and pulsatile flow at t = 0.696 s. (k) Detailed views of vorticity streamlines near the branching area at t = 0.696 s. (l) Wall shear stress (WSS) along the outer walls of inlet to main outlet (AB) and branched area to side outlet (GF).
Overall, the brTEBV experiments, PIV experiments, and CFD simulations showed consistent results. PIV experiments showed particle swirling motion near the side outlet branch after each pulse; similarly, in CFD simulations, the side outlet area showed greater curl magnitude than at the inlet or main outlet, and lower temporal and spatial correlation indicating greater flow disturbance. Together, the CFD and PIV results suggested a greater chance of collision of MCs with the EC wall at blood vessel branches, consistent with the observation of greater MC focal adhesion and foam cell formation at branches in the brTEBV model.
3. Discussion
We developed a vascular microphysiological system to emulate early atherosclerosis at bifurcated areas of blood vessels. The brTEBVs treated with eLDL and TNF-α were employed to study the localization of perfused human MCs and to test the effect of bifurcation angle on preferential MC focal adhesion at certain areas. MC localization was significantly greater near branched side outlets than near the inlets or main outlets. It was particularly interesting that the focal adhesion of MCs at side outlets corresponded to the location where fluorescent particles were swirling in PIV and furthermore, was consistent with clinical observations of atherosclerotic-prone locations. In consideration of Murray’s law of flow partitioning,[35] the volumetric flow rates at the main outlet and the side outlet were controlled to be 0.75× and 0.25× that of the inlet, respectively. Without application of pulsatile flow, neither swirl motion nor curl formation was observed in PIV or CFD (shown in Movie S2, top row of Figure 5, or Figure 6j). Interestingly, evidence of flow disturbance was not found at a low flow rate (~1 mL min−1), even with pulses (Movie S3). These results were similar to previous observations[36] of swirl motion disappearing at relatively lower Re with flow partitioning following Murray’s law. A significant backflow from the main outlet to the side outlet was observed during the relaxation periods when a flow partitioning ratio of 0.5:0.5 (deviating from Murray’s law) was applied (Movie S5, Figure S2). We applied flow in both an infusion mode through inlets and a withdrawal mode through the main and side outlets to precisely control flow conditions. When infusion flow was applied without withdrawal, the flow partitioning ratio could not be controlled due to the difference in pressure drop in the outlet flow connections. MC localization at branched side outlets was greater at the larger branching angles of 60° and 80° than at 45°. CFD results highlighted the secondary recirculation flow shown near the outer walls of the main and side outlets (Figure 6i), where WSS was relatively lower than other areas in both infusing and relaxation periods under pulsatile flow conditions (Figure 6l). During relaxation, the outer wall of the side outlet of the 80° brTEBV showed not only perturbed, recirculating flow, but also denser streamlines with low WSS (Figure 6k and 6l); this may have contributed to the preferential focal adhesion of MCs at the side outlets with larger angles. While the effect of hemodynamics was clear on the focal phenomena of early atherosclerosis, it is possible that the specific hemodynamics near the bifurcated area caused EC dysfunction to a different degree,[10] which resulted in the MC localization observed in this study.
The brTEBV structure was intended to mimic bifurcation in the coronary arterioles, especially branching from the left main (LM), left anterior descending (LAD), and left circumflex (LCx) coronary arteries.[36][37] The brTEBV lumen diameters were designed to match L3 of LAD for the main outlet and C3 of LCx for the side outlet.[30] To avoid enlargement of the platform, the length of the brTEBVs does not match the physiology of the coronary artery. The brTEBV platform utilized physiologically meaningful biological materials. It consisted of collagen-embedded, contractile iSMCs with ECs lined in the lumen. Three SMC phenotypes are commonly observed in vivo and in vitro: proliferative, synthetic, and contractile.[25] The iSMCs, transdifferentiated from human EPCs using lentiviral gene delivery of MYOCD, showed the contractile phenotype based on SMC markers and gene expression,[19] and their alignments were sensitive to cyclic stretch caused by pulsatile flow as shown in Figure S3. In addition, the contractile iSMCs clearly contributed the vasodilation and vasoconstriction capabilities demonstrated in Figure 3m. The WSS of the brTEBV was estimated to be ~0.85 dyne cm−2 under steady flow, and as high as 1.8 dyne cm−2 under pulsatile flow, both of which are within the range of WSS in the contractile SMC phenotype.[38] We modeled slightly smaller vessels towards proximal locations of LM, LAD or LCx; the WSS in these areas are lower than the typical WSS in a human coronary artery, which is ~6.8 dyne cm−2 by mean value.[39] The flow rate we chose (4 mL min−1 with pulses) resulted in a lower WSS than the physiologically relevant values summarized in Table S1. At high flow rates such as 32 mL min−1, there would be leaks caused by the pressurized perfusion chamber. In order to meet the physiological WSS, reducing vessel diameter instead of increasing flow rate would be a viable strategy to resolve the issue. A CFD simulation at inlet flow rate ~32 mL min−1 (Figure S5 and Movie S9), which produced a mean WSS of 6.8 dyne cm−2, revealed that velocity streamlines and vorticity fields exhibited similar patterns with the ones with the inlet flow rate of 4 mL min−1. PIV experiments and CFD simulations in this study did not consider the elastic properties of blood vessels. For PIV, optically transparent PDMS was chosen to replace the semi-opaque collagen for high speed capture of moving fluorescent particles.
To induce THP-1 MC adhesion, differentiation to macrophages, and foam cell formation, we treated the ECs with eLDL (to model cholesterol) and TNF-α (an inflammatory cytokine secreted by activated ECs). eLDL has been reported to be more effective in inducing foam cell formation than LDL or oxidized LDL.[40] EC dysfunction by eLDL and TNF-α was shown by ICAM-1 expression on well-developed EC layer in brTEBVs (Figure 3g–i). However, eLDL alone did not significantly affect THP-1 adhesion (Figure S4); an addition of TNF-α, even at a later time point, caused THP-1 cell adhesion, expression of CD80, and a change in cell morphology (Figure 2). These results suggest that a longer maturation and perfusion culture period would be necessary to cause inflammation by eLDL-only treatment than by treatment with both eLDL and TNF-α. The loss of vasodilation and vasoconstriction in linear TEBVs after eLDL and TNF-α treatment is likely due to the inflamed SMC/EC at the medial layer, although the possibility of phenotype switching in iSMC cannot not be excluded.[19]
The results from this study indicate that the brTEBV platform may present the utility for testing patient-iPSC-derived SMCs or ECs to understand the effects of systemic inflammation on initiation of atherosclerosis in branched blood vessel areas. Patients with chronic inflammatory diseases such as rheumatoid arthritis are our next target to test their iPSC-derived SMCs and ECs for susceptibility to atherosclerosis as well as patient-specific drug screening. Further, testing the effect of PSCK9 genes, either by gain-of-function or loss-of-function mutation, on such systemic inflammatory diseases using this vascular MPS may hold a potential to develop patient-specific drugs without addressing liver function or other associated genes.[41][42]
4. Conclusions
We developed a tissue-engineered vascular MPS that recapitulates key features of bifurcated coronary arterioles and, when treated with eLDL and TNF-α, emulates the focal phenomenon of MC adhesion and foam cell formation in early atherosclerosis. Selective localization of MCs was observed at the vessel branch; a larger bifurcation angle (60° or 80°) caused greater focal MC adhesion than a smaller angle (45°). We used PIV experiments and CFD simulations to characterize the hemodynamics in the branched TEBV system and observed significant similarities between the in vitro brTEBV experiments and the PIV and CFD results. These results warrant further investigation of this vascular MPS as a physiologically relevant platform for in vitro preclinical studies of early atherosclerosis.
5. Experimental Section
Cell culture, transfection, and transduction:
Human EPCs were derived from human umbilical cord blood[19][20] and were cultured in T75 cell culture flasks in complete EBM-2 medium with EGM-2 supplement, 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin (P/S), unless otherwise indicated. Doxycycline (2 μg mL−1; Sigma) was added to induce transgenic expression of human MYOCD. When EPCs were cultured a couple of passages in culture flasks with above EGM-2 supplemented medium, EPCs expressed EC markers, such as CD31, CD105, and von Willebrand Factor (vWF) and were used for endothelialization of the TEBVs.[19][20]
MYOCD and M2rtTA plasmid vectors and their viral particles were prepared by replacing OCT4 with MYOCD in FU.tet.on.OCT4.[18][19][22] To produce viral particles, we used the psPAX2 packaging vector (Addgene plasmid 12660) and the pMD2.G envelope vector (Addgene plasmid 12559). Briefly, HEK293T cells were maintained in standard DMEM supplemented with 10% FBS, 1% P/S, 1× non-essential amino acids (NEAA), 1× sodium pyruvate, and β-mercaptoethanol, and were expanded in T75 flasks precoated with 0.1% porcine gelatin (Sigma). HEK293T cells at 90% confluence were transfected in serum-free medium with 12.94 μg of the three lentiviral vectors (6.47 μg of expression vector, 4.31 μg of psPAX2, and 2.16 μg of pMD2.G) using Calfectin (Signagen, SL100478). The medium was exchanged at 24 h after addition of vectors, and 20 mL of supernatant containing the viral particles was collected at 48 h and 96 h. The supernatant was concentrated using Amicon Ultra-15 centrifugal filter units (Millipore) and stored at 4 °C for short-term use or in aliquots at −80 °C for long-term use. Viral particle concentration was determined by qPCR using a lentivirus titration kit (Abmgood, LV900).
To transduce human EPCs, the EPCs were plated at ~15,000 cells cm−2 in a T75 flask. The culture medium was removed on the following day and replaced with 20 mL of medium containing 10–20 virus particles per cell (FUW.M2rtTA, FU.tet.on.MYOCD); 10 μg mL−1 polybrene (hexadimethrine bromide; Sigma) was added. The virus-containing culture medium was replaced with fresh medium on the third day, and doxycycline (20 μg mL−1; Sigma) was added.
Enzyme-modified LDL preparation:
Human plasma LDL (Lee Biosolutions, 360–10) was enyzme-modified as described previously.[43] Briefly, LDL was dissolved in saline solution and the LDL concentration was determined using a Modified Lowry Protein Assay (Thermo Fisher, 23240). Trypsin (7 μg per mg LDL cholesterol; Life Technologies, 25200–056) was added and the solution was incubated for 6 h at 37 °C. Cholesterol esterase (12 μg per mg LDL cholesterol; Sigma, C9281) was then added and the solution was incubated for an additional 10 h. These steps were followed by the sequential addition of 24 μg of trypsin and 29 μg of cholesterol esterase per mg LDL cholesterol and incubation at 37 °C for 6 h and 48 h, respectively. The resulting eLDL preparation appeared cloudy and was extensively dialyzed against phosphate-buffered saline (PBS), filter-sterilized twice, and stored at 4 °C for up to 3 months. eLDL size was ~200 nm based on DLS measurements (Malvern Zetasizer ZS90). The toxicity of eLDL on EPCs was assessed by adding eLDL at various concentrations to EPCs and measuring cell viability using a live/dead cell probe (Molecular Probes, L3224). An eLDL concentration of 10 μg mL−1 was selected for subsequent experiments.
Human monocyte adhesion on EPCs (2D):
THP-1 human MCs (ATCC) were maintained in RPMI 1640 medium supplemented with 10% FBS, 1% P/S, and β-mercaptoethanol, and were passaged every 3–4 days to maintain a cell density of 2–8×105 cells mL−1. THP-1 cells were labeled with CellTracker Green CMFDA or CellTracker Red CMTPX (Thermo Fisher) before perfusion into eTEBVs to monitor adhesion to the TEBV lumen.
EPCs were seeded at 75,000 cells/well and maintained on 12-well plates for two days, followed by addition of 10 μg mL−1 eLDL for 24 h and 50 ng mL−1 TNF-α for 4 h. THP-1 cells were then labeled with CellTracker Green CMFDA at 1 μM and were added into a 12-well plate at 300,000 cells per well. After 1 h, bright-field and fluorescence images were acquired before and after washes with growth medium.
Design, fabrication, and operation of brTEBV chambers:
The brTEBV chambers were composed of a collagen gel-molding chamber and a perfusion chamber (Figure 1). The brTEBV vessels featured an inlet and main outlet with 2-mm inner diameter (ID), a side outlet with 1.4-mm ID, and a wall thickness of 2 mm. A hollow vessel was formed by mandrels that cross in the middle of the gel-molding chamber. The diameter of a vessel lumen was determined by the outer diameter of the mandrels, and the vessel wall thickness was determined by the mandrels and the gel mold. Fabrication of molding and perfusion chambers (acrylic) and mandrels (stainless steel) was performed using milling techniques. The mandrel pieces were assembled, forming a brTEBV chamber with a branching angle of 45°, 60°, or 80°. Mandrels were fit tight for molding steps (Figure 1b,e) and were released for suturing and connection to perfusion flow (Figure 1c,e).
The brTEBV flow chambers had five flow connections (Figure 1d,f): three for flow through the vessel lumen (red in Figure 1d), and two for refreshing media (green in Figure 1d). A 24-channel peristaltic pump (Ismatec ISM394) was used to simultaneously infuse into inlets and withdraw from outlets. The volumetric flow rates at the main and side outlets were 0.75× and 0.25× that at the inlet, respectively, controlled by using different tubing diameters (Cole-Parmer EW-96460–36 for inlet, EW-95692–32 for main outlet, and EW-95723–28 for side outlet). The overall flow rate was maintained at 4 mL min−1. Two culture media reservoirs were used: one for perfusing the lumen (RL), another for refreshing the chamber (RC). EGM-2 media (Lonza CC3156 and CC4176) for ECs was used for perfusing the lumen; M231 media (Gibco M231) for SMCs was used for refreshing the chamber. The flow from RL was connected to a pulse generator (PG) and was split into the inlet ports of multiple chambers; the two outlets (main and side) from each chamber were combined after withdrawal and were returned to RL; chamber-refreshing flow was also returned to RC.
brTEBVs were subject to pulsatile flow using a pulse generator: a two-way, normally-closed, solenoid pinch valve (EW-98302–02, Cole Parmer) connected between RL and the chamber inlets. The valve was actuated at 1 Hz with a 50% duty cycle (0.5 s open, 0.5 s closed) using a custom-built external circuit with slight modification from a previous work.[44] Figure S1 shows schematics of the oscillator circuit for valve actuation and the driver circuit for the solenoid pinch valve.
SMC-embedded vessel formation and brTEBV assembly:
Collagen I (high concentration, rat tail; Corning, 354249) was used to produce dense vessel constructs as described previously, with slight modification.[19][20] Briefly, the gel-forming chamber was prepared by connecting the mandrels and securely closing the top to the bottom (Figure 1e). For two of the brTEBV chambers, 1.86 mL of collagen at ~9 mg mL−1 was mixed with 0.235 mL of 10× DMEM in a 50 mL conical tube while swirling, followed by addition of 1 M NaOH to neutralize the pH. 3×106 iSMCs in 0.25 mL of EGM-2 media were added to the collagen solution and swirled to mix homogeneously, producing a total volume of ~2.5 mL of cell and gel mixture. 0.93 mL of the cell-collagen solution was pipetted into the inlet hole at the top of the gel-formation chamber until solution exited from the two outlet holes on top of the chamber (Figure 1e). This procedure was repeated for another set of two chambers. Avoiding air bubbles while pipetting the collagen and cell mixture was crucial, as bubbles would result in holes in the vessels and cause leakages. The collagen-filled chambers were incubated at 37 °C in a humidified incubator for 30 min until a gel formed. After gelation, the branched construct was removed from the gel-forming chamber and was reassembled in the flow chamber (Figure 1c,e). M231 media was added to hydrate the gel construct for 30 min, followed by dehydration using autoclaved Kimwipes for 1–2 s per side; this dehydration step was critical to achieve dense collagen gel. The three stainless steel mandrel pieces were pulled half-way from each inlet and outlet port to function as flow connection ports. Each end of the gel construct was attached to the end of the three mandrels with surgical sutures (Figure 1c). The flow chamber was then assembled completely and filled with M231 media.
ECs were seeded into the sutured construct at 6×106 cells mL−1 in EGM-2 media. 0.1 mL of ECs were seeded into each inlet and outlet and were incubated for 2 h on a rotation device at ~10 rph. The chambers were then removed from the rotator and flow was connected as described above (Figure 1d,f).
Addition of eLDL, TNF-α, and human monocytes:
4 mL min−1 of perfusion flow with a 1 Hz pulse was maintained for 3–5 days to mature ECs and iSMCs before addition of eLDL (10 μg mL−1) into EGM-2 media on Day 5. On Day 6, cell-tracker labeled THP-1 cells at 0.5×106 cells mL−1 were added to the perfusion flow for another 3 days. 50 ng mL−1 TNF-α was added to the media for the last 8 h of THP-1 perfusion. TEBV samples were fixed using 4% paraformaldehyde (PFA) for 24 h before paraffin-embedding or further immunofluorescent sample preparation.
Immunofluorescence and confocal microscopy:
Fluorescence imaging was performed using a Nikon Eclipse TE2000-U microscope with a Roper Scientific CoolSnap HQ camera. Confocal z-stack images of the entire TEBVs were acquired using a Nikon A1RMP laser scanning system on an Eclipse Ti stand with a 10× air objective; 3D images were reconstructed using NIS Elements software.
Primary antibodies used for immunofluorescence experiments were the following: anti-αSMA (Abcam, ab7817), anti-calponin (Abcam, ab46794), anti-TAGLN (Abcam, ab14106), anti-MYH11 (Abcam, ab683), anti-vWF (Abcam, ab6994), anti-CD31 (Abcam, ab32457), anti-ICAM1 (Santa Cruz, sc-107), and anti-CD80 (Abcam, ab225674). Secondary antibodies were purchased from Life Technologies and conjugated to Alexa Fluor 555, Alexa Fluor 488, or Alexa Fluor 647. Cell nuclei were stained using DAPI (Life Technologies, D1306). The actin cytoskeleton was stained using phalloidin conjugated to Alexa Fluor 488 or 647 (Life Technologies). Cells were fixed using 4% paraformaldehyde (Electron Microscopy Sciences) in PBS, and were permeabilized using 0.1% Triton X-100 (Sigma) in PBS. Nonspecific adhesion was blocked using 5% bovine serum albumin or 10% goat serum. MCs were labeled with CellTracker Green CMFDA or CellTracker Red CMTPX (Life Technologies) before perfusion into the brTEBV lumen.
Statistical Analysis
The number of samples for analysis is indicated in each figure caption. Quantitative data are presented at mean ± SD. Group differences were accessed by one-way or two-way ANOVA with multiple comparisons using GraghPad Prism 8. Statistical significance was set at p > 0.05 (ns), P < 0.05(*), P < 0.005 (**), P < 0.0005 (***), and P < 0.0001 (****).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was funded by NIH grant UH3TR002142. MC adhesion images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grants #P30 CA013696 and #S10 RR025686 (National Cancer Institute). PIV experiments were performed with a Nikon microscope equipped with a high-speed camera (Phantom V 630) kindly provided by Dr. Kyle Bishop’s lab at Columbia University.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library.
Contributor Information
Jounghyun H. Lee, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Zaozao Chen, School of Biological Sciences and Medical Engineering, Southeast University, Nanjing, 210096, China.
Siyu He, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA.
Joyce K. Zhou, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Alexander Tsai, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA.
George A. Truskey, Department of Biomedical Engineering, Duke University, Durham, NC 27708, USA
Kam W. Leong, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
References
- [1].World Health Organization, Cardiovascular diseases fact sheet (2017), https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds), accessed: Jan. 2021.
- [2].Pahwa R, Jialal I, Atherosclerosis, StatPearls Publishing, Treasure Island, FL, USA: 2019. [Google Scholar]
- [3].Ghattas A, Griffiths HR, Devitt A, Lip GYH, Shantsila E, J. Am. Coll. Cardiol 2013, 62, 1541. [DOI] [PubMed] [Google Scholar]
- [4].McMillan DE, Stroke 1985, 16, 582. [DOI] [PubMed] [Google Scholar]
- [5].Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA, Circ. Res 2007, 101, 723. [DOI] [PubMed] [Google Scholar]
- [6].Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, García-Cardeña G, Gimbrone MA, Proc. Natl. Acad. Sci. U. S. A 2004, 101, 14871 LP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR, Am. J. Physiol. - Cell Physiol 2007, 293, 1824. [DOI] [PubMed] [Google Scholar]
- [8].Nakazawa G, Yazdani SK, Finn AV, Vorpahl M, Kolodgie FD, Virmani R, J. Am. Coll. Cardiol 2010, 55, 1679. [DOI] [PubMed] [Google Scholar]
- [9].Lu M, Cui Y, Peng P, Qiao H, Cai J, Zhao X, J. Atheroscler. Thromb 2019, 26, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Martorell J, Santomá P, Kolandaivelu K, Kolachalama VB, Melgar-Lesmes P, Molins JJ, Garcia L, Edelman ER, Balcells M, Cardiovasc. Res 2014, 103, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chiu J-J, Chen C-N, Lee P-L, Yang CT, Chuang HS, Chien S, Usami S, J. Biomech 2003, 36, 1883. [DOI] [PubMed] [Google Scholar]
- [12].Blackman BR, García-Cardeña G, A. GM Jr., J. Biomech. Eng 2002, 124, 397. [DOI] [PubMed] [Google Scholar]
- [13].Farcas MA, Rouleau L, Fraser R, Leask RL, Biomed. Eng. OnLine 2009, 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Menon NV, Su C, Pang KT, Phua ZJ, Tay HM, Dalan R, Wang X, Li KHH, Hou HW, Biofabrication 2020, 12, 045009. [DOI] [PubMed] [Google Scholar]
- [15].Rouleau L, Copland IB, Tardif J-C, Mongrain R, Leask RL, Ann. Biomed. Eng 2010, 38, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jin Q, Bhatta A, Pagaduan JV, Chen X, West-Foyle H, Liu J, Hou A, Berkowitz D, Kuo SC, Askin FB, Nguyen TD, Gracias DH, Romer LH, Sci. Adv 2020, 6, eaaz2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bouhrira N, DeOre BJ, Sazer DW, Chiaradia Z, Miller JS, Galie PA, Biofabrication 2020, 12, 25020. [DOI] [PubMed] [Google Scholar]
- [18].Jung Y, Ji H, Chen Z, Fai Chan H, Atchison L, Klitzman B, Truskey G, Leong KW, Sci. Rep 2015, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ji HY, Atchison L, Chen Z, Chakraborty S, Jung Y, Truskey GA, Christoforou N, Leong KW, Biomaterials 2016, 85, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fernandez CE, Yen RW, Perez SM, Bedell HW, Povsic TJ, Reichert WM, Truskey GA, Sci. Rep 2016, 6, 21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ji H, Ph.D. Thesis, Columbia University, 2018. [Google Scholar]
- [22].Christoforou N, Chellappan M, Adler AF, Kirkton RD, Wu T, Addis RC, Bursac N, Leong KW, PLoS ONE 2013, 8, e63577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Owens GK, Physiol. Rev 1995, 75, 487. [DOI] [PubMed] [Google Scholar]
- [24].Owens GK, Kumar MS, Wamhoff BR, Physiol. Rev 2004, 84, 767. [DOI] [PubMed] [Google Scholar]
- [25].Truskey GA, Int. J. High Throughput Screen 2010, 1, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z, Nat. Rev. Cardiol 2019, 16, 727. [DOI] [PubMed] [Google Scholar]
- [27].Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM, Cardiovasc. Res 2018, 114, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Atchison L, Zhang H, Cao K, Truskey GA, Sci. Rep 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Atchison L, Abutaleb NO, Snyder-Mounts E, Gete Y, Ladha A, Ribar T, Cao K, Truskey GA, Stem Cell Rep. 2020, 14, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dodge JTJ, Brown BG, Bolson EL, Dodge HT, Circulation 1992, 86, 232. [DOI] [PubMed] [Google Scholar]
- [31].Pflederer T, Ludwig J, Ropers D, Daniel WG, Achenbach S, Invest. Radiol 2006, 41, 793. [DOI] [PubMed] [Google Scholar]
- [32].Sakellarios A, Bourantas CV, Papadopoulou SL, Kitslaar PH, Girasis C, Stone GW, Reiber JHC, Michalis LK, Serruys PW, De Feyter PJ, Garcia-Garcia HM, Fotiadis DI, EuroIntervention 2017, 13, e1084. [DOI] [PubMed] [Google Scholar]
- [33].Chen Z, Tang M, Huang D, Jiang W, Li M, Ji H, Park J, Xu B, Atchison LJ, Truskey GA, Leong KW, Lab. Chip 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Doutel E, Carneiro J, Campos JBLM, Miranda JM, Eur. J. Mech. BFluids 2018, 67, 341. [Google Scholar]
- [35].Zheng X, Shen G, Wang C, Li Y, Dunphy D, Hasan T, Brinker CJ, Su BL, Nat. Commun 2017, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kang S-J, Mintz GS, Kim W-J, Lee J-Y, Oh J-H, Park D-W, Lee S-W, Kim Y-H, Lee CW, Park S-W, Park S-J, Circ. Cardiovasc. Interv 2011, 4, 355. [DOI] [PubMed] [Google Scholar]
- [37].Cui Y, Zeng W, Yu J, Lu J, Hu Y, Diao N, Liang B, Han P, Shi H, PloS One 2017, 12, e0174352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tada S, Tarbell JM, Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1589. [DOI] [PubMed] [Google Scholar]
- [39].Doriot PA, Dorsaz PA, Dorsaz L, De Benedetti E, Chatelain P, Delafontaine P, Coron. Artery Dis 2000, 11, 495. [DOI] [PubMed] [Google Scholar]
- [40].Chellan B, Rojas E, Zhang C, Hofmann Bowman MA, Sci. Rep 2018, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, Wang Z, Wei DH, Liu LS, Zheng XL, Jiang ZS, Atherosclerosis 2017, 262, 113. [DOI] [PubMed] [Google Scholar]
- [42].Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, Reis RJS, Varughese KI, Sawamura T, Mehta JL, Cardiovasc. Res 2015, 107, 556. [DOI] [PubMed] [Google Scholar]
- [43].Chellan B, Reardon CA, Getz GS, Bowman MAH, Arterioscler. Thromb. Vasc. Biol 2016, 36, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brown MA, Iyer RK, Radisic M, Biotechnol Prog 2008, 24, 907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.