

Abstract
Unlike familial Alzheimer’s disease, we have been unable to accurately predict symptom onset in presymptomatic familial frontotemporal dementia (f-FTD) mutation carriers, which is a major hurdle to designing disease prevention trials. We developed multimodal models for f-FTD disease progression and estimated clinical trial sample sizes in C9orf72, GRN, and MAPT mutation carriers. Models included longitudinal clinical and neuropsychological scores, regional brain volumes, and plasma neurofilament light chain (NfL) in 796 carriers and 412 non-carrier controls. We found that the temporal ordering of clinical and biomarker progression differed by genotype. In prevention-trial simulations employing model-based patient selection, atrophy and NfL were the best endpoints, whereas clinical measures were potential endpoints in early symptomatic trials. F-FTD prevention trials are feasible but will likely require global recruitment efforts. These disease progression models will facilitate the planning of f-FTD clinical trials, including the selection of optimal endpoints and enrollment criteria to maximize power to detect treatment effects.
Keywords: Frontotemporal lobar degeneration, C9orf72, GRN, MAPT, disease progression, neurofilament light chain, neuroimaging, neuropsychology, clinical trials
Editor summary:
Empirically-based models of disease progression in familial frontotemporal dementia reveal the relative ordering of clinical, neuroimaging, and fluid biomarker changes and facilitate novel clinical trial designs
Frontotemporal dementia (FTD), marked by impairments in behavior, language, and sometimes motor function, is a common form of early-onset dementia.1 Approximately 20–30% of FTD is caused by autosomal dominant mutations (familial, or f-FTD), usually in one of three genes: chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), or microtubule-associated protein tau (MAPT).2 FTD is uniformly fatal, and there are no approved therapies; however, a growing number of new treatments targeting C9orf72, GRN, and MAPT are moving into clinical trials.3,4 Experience from Alzheimer’s disease (AD), spinal muscular atrophy (SMA),5 and amyotrophic lateral sclerosis (ALS)6 suggests treating FTD will be most successful if treatment is initiated early in the disease course, ideally prior to the onset of symptoms. Such a disease prevention approach has been implemented in the Dominantly Inherited Alzheimer’s Network Trials Unit (DIAN-TU; https://dian.wustl.edu/our-research/clinical-trial/) platform clinical trial for dominantly inherited AD (DIAD) by including presymptomatic mutation carriers.7 Prevention trials in DIAD have also been facilitated by fluid and molecular PET imaging biomarkers that allow for the measurement of treatment-related changes in AD pathologies and neurodegeneration.8
There are many challenges to performing f-FTD clinical trials.9 Although the clinical manifestations of the f-FTD mutations are similar, the biology and neuropathology associated with C9orf72, GRN, and MAPT mutations are vastly different.2 Unlike AD,10 little is known about the ontogeny of biomarker and clinical changes in f-FTD that could be used to determine enrollment criteria or identify the best clinical trial endpoints at different disease stages. Also, the age at which symptoms present is variable even within a family (e.g., onset in the thirties vs. seventies in one family),11 making it difficult to identify the individuals in late presymptomatic stages most likely to benefit from therapies. For example, in GRN, familial age of onset only explains 14% of the variability in individual age at symptom onset.12
F-FTD is rare, with only hundreds of mutation carriers known to exist worldwide.12 Therefore, to prepare for f-FTD trials, the FTD Prevention Initiative (FPI, www.thefpi.org), an international collaboration focused on organizing f-FTD prevention trials, combined data from the two largest f-FTD natural history studies worldwide: ALLFTD in North America (www.allftd.org), and GENFI in Europe and Canada (www.genfi.org).13 In rare neurogenetic diseases such as f-FTD, the FDA has promoted the use of innovative approaches such as disease progression models (DPM) for selecting clinical trial endpoints, determining enrollment criteria, and analyzing the effects of novel interventions that might lead to deviations from expected disease progression,14 and such models have been employed successfully in DIAN-TU.7 We developed Bayesian DPMs that jointly model the best known measures of f-FTD global clinical status, neuropsychological performance, brain volume, and active neurodegeneration (plasma neurofilament light chain [NfL]) to model latent “Disease Age (DA),” which forecasts presymptomatic mutation carriers’ proximity to symptom onset and enables comprehensive quantification of disease progression. We then conducted simulations of prevention and early symptomatic treatment trials, exploring the use of DA, plasma NfL, and clinical measures as inclusion criteria to prioritize the recruitment of presymptomatic participants towards those most likely to exhibit measurable disease progression during a trial.
Results
Subject Characteristics
Demographic and clinical data are presented in Tables 1 and S1. Of the 796 mutation carriers, C9orf72 was the most common mutation (43.6%), followed by GRN (35.3%) and MAPT (21.1%). Across all three genetic groups, most participants were presymptomatic (CDR®+NACC-FTLD-Global=0, 54.4%). Most symptomatic participants presented with behavioral variant FTD (bvFTD, 68.6%), followed by primary progressive aphasia (PPA, 12.7%), which was driven largely by GRN (33.8% of symptomatic GRN). The average number of visits per mutation carrier was 2.1 (SD=1.1). The models incorporated 412 non-carrier family controls. A subset of participants had available NfL (n=981, 1,948 observations) and MRI data (n=882, 1,896 observations).
Table 1.
Characteristics of the study participants
| Characteristic | All Carriers | C9orf72+ | GRN+ | MAPT+ | Non-Carriers | p-value | Pairwise Comparisons |
|---|---|---|---|---|---|---|---|
| Sample Size | 796 | 347 | 281 | 168 | 412 | ||
| ALLFTD Sample Size | 275 | 127 | 68 | 80 | 161 | ||
| GENFI Sample Size | 521 | 220 | 213 | 88 | 251 | ||
| Age - yr (mean(SD)) | 50.2 (13.9) | 51.2 (13.7) | 52.2 (13.7) | 44.9 (13.3) | 45.9 (13.0) | <0.001 | (NC = MAPT) < (C9=GRN) |
| Female - n (%) | 447 (56.1%) | 188 (54.2%) | 167 (59.4%) | 92 (54.8%) | 239 (58.0%) | 0.51 | |
| Education - yr | 14.4 (3.2) | 14.5 (3.0) | 14.2 (3.4) | 14.7 (3.0) | 14.8 (2.9) | 0.07 | |
| Visits (total number) | 2.1 (1.1) | 2.0 (1.0) | 2.1 (1.1) | 2.5 (1.2) | 2.2 (1.1) | <0.001 | (C9=GRN,NC=GRN,C9<NC)< MAPT |
| N with 1 visit | 292 | 135 | 114 | 43 | 137 | ||
| N with 2 visits | 233 | 120 | 68 | 45 | 106 | ||
| N with 3 visits | 158 | 53 | 57 | 48 | 118 | ||
| N with ≥4 visits | 113 | 39 | 42 | 32 | 51 | ||
| Total number of observations | 1,695 | 690 | 592 | 413 | 910 | ||
| Follow-up Length (if > 1 visit) – yr | 2.0 (0.9) | 1.9 (0.9) | 2.1 (0.9) | 2.2 (0.9) | 2.2 (0.8) | <0.001 | C9< (GRN = MAPT = NC ) |
| Race - n (%) | |||||||
| White | 776 (97.5%) | 342 (98.6%) | 274 (97.5%) | 160 (95.2%) | 404 (98.0%) | 0.11 | |
| Non-White^ | 19 (2.4%) | 4 (1.2%) | 7 (2.5%) | 8 (4.8%) | 6 (1.5%) | ||
| Unknown | 1 (0.1%) | 1 (0.3%) | 0 | 0 | 2 (0.5%) | ||
| CDR®+NACC-FTLD Global- n (%) | |||||||
| 0 | 433 (54.4%) | 171 (49.3%) | 168 (59.8%) | 94 (56.0%) | 412 (100%) | 0.03^^ | C9<GRN, C9=MAPT, GRN=MAPT |
| 0.5 | 127 (16.0%) | 61 (17.6%) | 39 (13.9%) | 27 (16.1%) | NA | 0.45 | |
| ≥ 1 | 236 (29.7%) | 115 (33.1%) | 74 (26.3%) | 47 (28.0%) | NA | 0.16 | |
| Estimated Years Since Onset * | 4.4 (4.7) | 5 (4.7) | 2.7 (2.4) | 6 (7.8) | NA | <0.001 | GRN< C9, GRN < MAPT, C9 = MAPT |
| Symptomatic Diagnoses - n (%) | |||||||
| bvFTD | 162 (68.6%) | 85 (73.9%) | 38 (51.4%) | 39 (83.0%) | NA | <0.001 | GRN < (C9 = MAPT) |
| PPA | 30 (12.7%) | 4 (3.5%) | 25 (33.8%) | 1 (2.1%) | NA | <0.001 | (C9 = MAPT) < GRN |
| CBS | 2 (0.9%) | -- | 2 (2.7%) | -- | NA | 0.13 | |
| PSP | 3 (1.3%) | 1 (0.9%) | 1 (1.4%) | 1 (2.1%) | NA | 0.78 | |
| ALS | 4 (1.7%) | 4 (3.5%) | -- | -- | NA | 0.14 | |
| FTD-MND | 11 (4.7%) | 11 (9.6%) | -- | -- | NA | 0.002 | (GRN=MAPT) < C9 |
| MCI | 4 (1.7%) | 2 (1.7%) | 1 (1.4%) | 1 (2.1%) | NA | 1.0 | |
| AD Dementia | 5 (2.1%) | 1 (0.9%) | 3 (4.1%) | 1 (2.1%) | NA | 0.35 | |
| Other** | 4 (1.7%) | 3 (2.6%) | 1 (1.4%) | 1 (2.1%) | NA | NA | |
| Missing | 9 (3.8%) | 4 (3.5%) | 2 (2.7%) | 3 (6.4%) | NA | NA |
Note. Demographics were calculated using baseline values. Demographic variables and other participant characteristics were compared across genetic groups and controls using regression with pairwise group contrasts for most variables. Sex, race, CDR®+NACC-FTLD, and diagnostic categories were compared using chi-square with Bonferroni-adjusted pairwise comparisons when the omnibus test was significant. For chi-square tests in which any bins were < 10, the Fisher’s exact test was used. All tests were two-sided. Symptomatic clinical diagnoses were calculated in those with a CDR®+NACC FTLD Global ≥ 1
Due to the small number of non-White participants in this sample, a single bin was used to protect participants’ identities.
Controls not included in pairwise comparisons for CDR®+NACC FTLD
Median (IQR) of baseline values for symptomatic cases based on clinician-reported age of onset.
Other diagnoses include dementia not otherwise specified (n=2) or the clinician marked “other” without entering additional information.
Abbreviations: CDR®+NACC-FTLD: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration Module; bvFTD: Behavioral Variant Frontotemporal Dementia; PPA: Primary Progressive Aphasia; CBS: Corticobasal Syndrome; PSP: Progressive Supranuclear Palsy Syndrome; ALS: Amyotrophic Lateral Sclerosis; MND: Motor Neuron Disease; MCI: Mild Cognitive Impairment; AD: Alzheimer’s Disease
Disease Progression Models
Overview
When ALLFTD and GENFI participants were modeled separately, rates of progression were very similar between consortia on all measures (Figures 1, S1); subsequent models combined all participants. To understand the temporal ordering of biomarker and clinical changes, disease progression curves were graphed in relation to predicted DA (Figure 2).
Figure 1. Raw data points overlaid on model estimated fit.
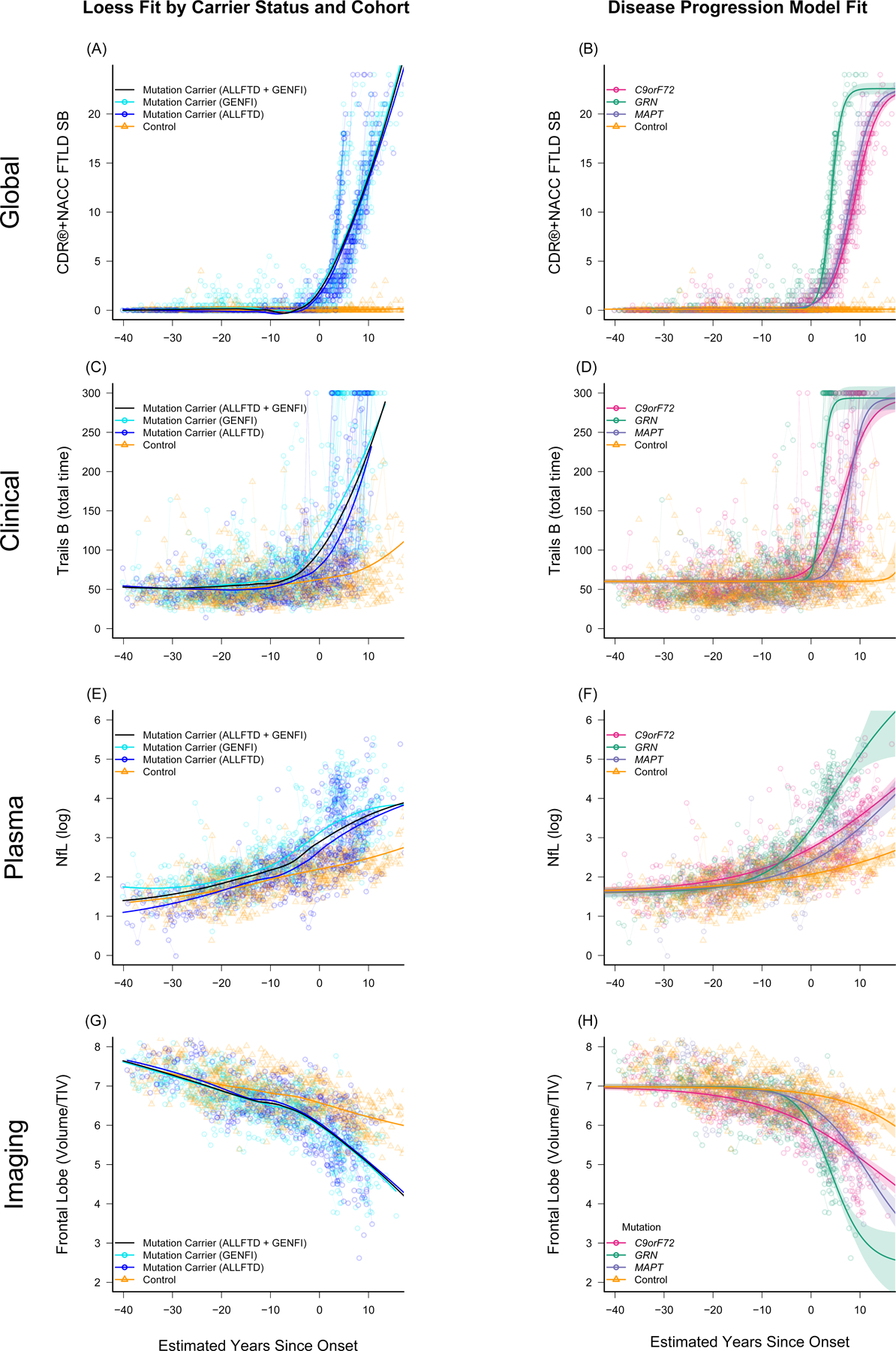
Panels A, C, E, and G display raw data points for mutation carriers (blue) and non-carrier controls (gold) for several representative measures as a function of model estimated Disease Age, with a loess fit to each group displayed using thick solid lines. In these panels, raw outcomes are plotted, and mutation carriers are color coded based on whether they were enrolled through ALLFTD or GENFI. These panels highlight the consistency in progression regardless of cohort. Panels B, D, F, and H display raw data points colored by mutation as a function of disease age. In these panels, the overall fit for each group was derived from the Bayesian disease progression model and is displayed using thick solid lines. Shaded areas indicate the 95% credible interval of the estimate. Age-related changes in controls are observed in panels C-H. Figures for all modeled measures are included in Supplemental Figure S1.
Abbreviations: CDR®+NACC FTLD SB: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center’s Frontotemporal Lobar Degeneration Module Sum of Boxes; Trails B: Trail Making Test, Part B (total time displayed in seconds); NfL (log): Log-transformed plasma neurofilament light chain; TIV: Total intracranial volume.
Figure 2. Temporal ordering of clinical and biomarker changes in F-FTD.
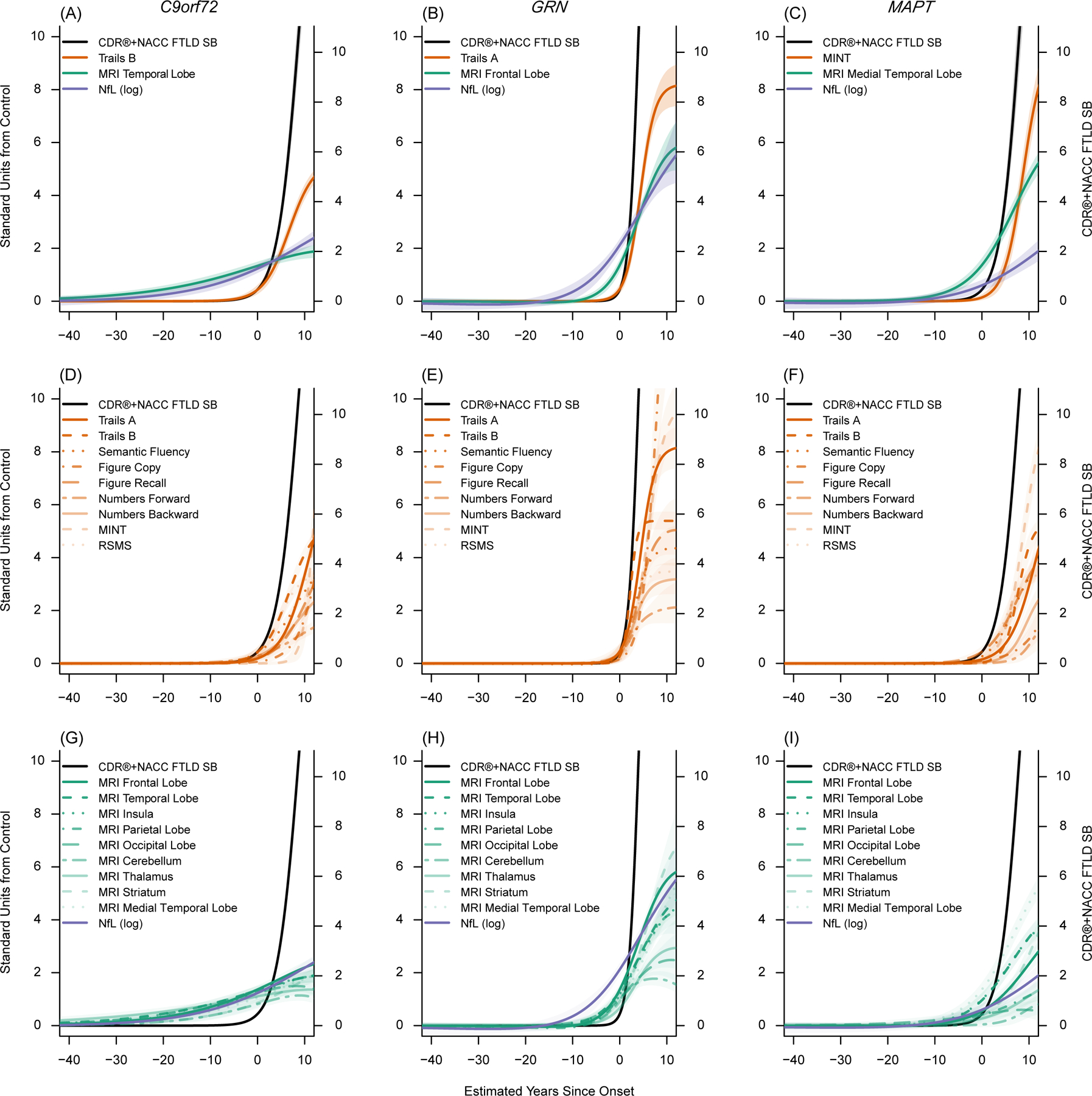
These figures display the empirically derived model-estimated curves in each genetic group. In all figures, model estimated time from onset (years) is on the x-axis. The left y-axis indicates the number of standard deviations (SD) of abnormality compared to controls. The right y-axis indicates CDR®+NACC FTLD Box Score units to provide a context for understanding the degree of clinical impairment associated with changes in the other biomarkers and to provide a raw estimate corresponding to the standardized CDR®+NACC FTLD Box Score (black line). Panels A-C display the mean curves for the CDR®+NACC FTLD Box Score, NfL, and a selected imaging and clinical measure for each genetic group, based on which measure is first elevated by one standard deviation from controls and the measure’s rate of longitudinal progression. All clinical, imaging, and fluid biomarkers are displayed in the remaining panels (D-I). The shaded areas indicate the 95% credible interval of the estimate. These figures suggest brain atrophy and elevations in neurofilament light chain levels are detectable prior to symptom onset, and that each mutation shows a unique cascade of biomarker changes.
Abbreviations: CDR®+NACC FTLD SB: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center’s Frontotemporal Lobar Degeneration Module Sum of Boxes; Trail B: Trail Making Test, Part B; MINT: Multilingual Naming Test; RSMS: Revised Self Monitoring Scale; MRI: magnetic resonance imaging; NfL (log): Log-transformed plasma neurofilament light chain; Stand: Standard
MR imaging and plasma NfL
In C9orf72, MRI was the first biomarker to change (Figures 2, 3, & Extended Data Figure 1; Tables 2, S2-S5), with visual inspection of the DPM curves suggesting that brain volumes deviate from controls up to 40 years before expected onset. Thalamic volume in C9orf72 was significantly lower than controls in the −40 to −10 epoch, with the largest effect size of all regions of interest (ROIs) (Extended Data Figure 1 & Table S5). Voxelwise quantification also underscored the early thalamic involvement (Extended Data Figure 2, Figure S2). In addition to the thalamus, most ROIs were smaller than controls (Extended data figure 2, Tables S5) and other mutation carriers (Table S6) in the −40 to −10 epoch. In the −10 to 0 epoch, the temporal lobe showed the largest effect size (Extended Data Figure 1), and it was the first ROI to deviate from controls (Table S4; deviated at DA = −6.1, 95%CI:−9.4,−3.2) by one standard deviation (SD), followed closely by parietal (DA = −6.1, 95%CI:−9.2,−3.2) and frontal (DA = −4.9, 95%CI:−7.5,−2.7) lobes. The largely overlapping credible intervals indicate these differences in temporal ordering are not statistically significant. The longitudinal rate of volume loss was relatively stable across the across epochs in C9orf72 compared to the other genetic groups (Table S4). Visual inspection of the DPM curves suggested mean NfL values in C9orf72 begin to deviate from controls approximately 30 years before estimated onset. NfL levels in C9orf72 were significantly higher than controls in all DA epochs and became elevated one SD above controls three years before estimated onset (95%CI:-0.7,-5.8).
Figure 3. Comparison of mutation carriers with controls at three epochs of disease age.
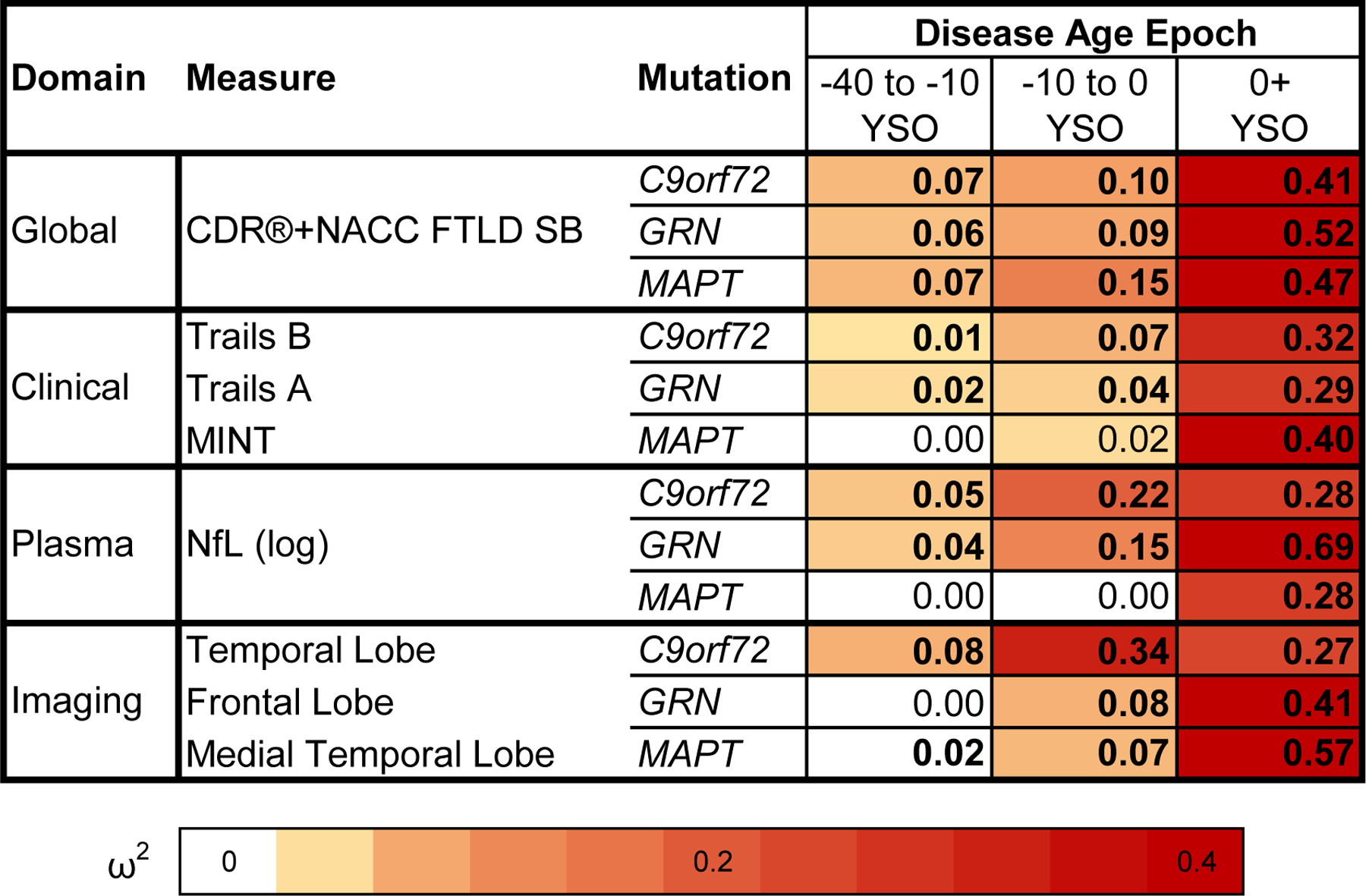
Cross-sectional baseline differences between mutation carriers and controls are presented as effect sizes (omega squared). Bolded cells indicate statistical significance (p < .05). Comparisons in which mutation carriers are more impaired than controls at an omega squared > 0.00 are colored, with darker shades illustrating larger effect sizes. CDR®+NACC FTLD SB scores and log-transformed NfL levels are presented for all genetic groups. Clinical and imaging measures were selected for each genetic group based on how early they deviated from controls in the disease progression model and rate of longitudinal progression. Note that statistical comparisons for the CDR®+NACC FTLD SB should be interpreted with caution given that controls were defined as having a baseline CDR®+NACC-FTLD=0 and thus have no variance due to this selection process. A similar figure including all modeled measures can be found in the extended data figures (Extended Data Fig 1). YSO = years since onset.
Table 2.
Baseline descriptive statistics of measures for each genetic group at three epochs.
| Mutation Status | Outcome Measure | Disease Age Epoch | |||
|---|---|---|---|---|---|
| −40 to −10 YSO | −10 to 0 YSO | 0+ YSO | |||
| Age-Matched Controls | N (prop) at baseline | 229 (0.56) | 85 (0.21) | 98 (0.24) | |
| Mean Age (SD) at baseline | 36.8 (7.7) | 52.6 (6.7) | 61.6 (7.7) | ||
| Mean Raw Score (SD; Range) | CDR®+ NACC FTLD SB | 0 (0; 0–0) | 0 (0; 0–0) | 0 (0; 0–0) | |
| Trails A (Total time in Seconds) | 22.76 (8.03; 8–78) | 26.36 (9.39; 12–61) | 31.07 (14.67; 12–89) | ||
| Trails B (Total time in Seconds) | 53.81 (21.93; 19–187) | 62.06 (29.48; 27–202) | 73.63 (30.43; 31–167) | ||
| MINT (Total Correct) | 29.92 (1.75; 24–32) | 29.94 (1.62; 26–32) | 29.95 (1.92; 25–32) | ||
| MRI Frontal/TIV | 7.07 (0.48; 5.39–8.21) | 6.68 (0.41; 5.83–7.55) | 6.33 (0.45; 5.27–7.28) | ||
| MRI Temporal/TIV | 4.76 (0.29; 3.76–5.62) | 4.54 (0.22; 4.07–5.03) | 4.24 (0.28; 3.46–4.79) | ||
| MRI Medial Temporal (MTL)/TIV | 1.03 (0.06; 0.81–1.22) | 1.02 (0.06; 0.89–1.19) | 0.97 (0.07; 0.8–1.13) | ||
| NfL (log) | 1.67 (0.43; 0.38–3.27) | 2.05 (0.38; 1.06–2.94) | 2.42 (0.43; 1.71–3.76) | ||
| C9orf72 | N (prop) at baseline | 135 (0.39) | 63 (0.18) | 149 (0.43) | |
| Mean Age (SD) at baseline | 38.3 (8.8) | 54.6 (8.2) | 61.5 (9) | ||
| Mean Raw Score (SD; Range) | CDR®+ NACC FTLD SB | 0.19 (0.57; 0–3) | 0.31 (0.69; 0–3.5) | 8.32 (6.23; 0–22) | |
| Trails B (Total time in Seconds) | 58.92 (21.85; 28–151) | 84 (45.61; 23–300) | 168.25 (88.4; 35–300) | ||
| MRI Temporal/TIV | 4.58 (0.29; 3.95–5.22) | 4.16 (0.32; 3.43–4.71) | 3.76 (0.46; 2.29–4.78) | ||
| NfL (log) | 1.89 (0.48; 0.94–3.89) | 2.58 (0.6; 1.72–4.76) | 3.31 (0.85; 1.54–5.54) | ||
| Mean Standardized Units from Control (SD; Range) | CDR®+ NACC FTLD SB | --- | --- | --- | |
| Trails B | 0.23 (1; −1.18–4.43) | 0.74 (1.55; −1.32–8.07) | 3.11 (2.91; −1.27–7.44) | ||
| MRI Temporal/TIV | −0.62 (1; −2.79–1.56) | −1.75 (1.43; −5.04–0.76) | −1.71 (1.65; −6.92–1.94) | ||
| NfL (log) | 0.51 (1.11; −1.68–5.1) | 1.37 (1.57; −0.85–7.06) | 2.07 (1.96; −2.01–7.18) | ||
| GRN | N (prop) at baseline | 125 (0.44) | 72 (0.26) | 84 (0.3) | |
| Mean Age (SD) at baseline | 41 (10.3) | 58.2 (7.5) | 63.7 (8.8) | ||
| Mean Raw Score (SD; Range) | CDR®+ NACC FTLD SB | 0.08 (0.26; 0–2) | 0.31 (0.71; 0–3) | 9.19 (6.53; 0–24) | |
| Trails A (Total time in Seconds) | 25.37 (9.2; 9–63) | 30.57 (10.73; 16–81) | 72.12 (46.48; 23–150) | ||
| MRI Frontal/TIV | 7.03 (0.52; 5.39–8.93) | 6.4 (0.52; 5.25–7.48) | 5.15 (0.92; 2.62–7.77) | ||
| NfL (log) | 1.87 (0.43; 0.82–3.34) | 2.45 (0.56; 1.57–4.27) | 4.04 (0.65; 2.14–5.35) | ||
| Mean Standardized Units from Control (SD; Range) | CDR®+ NACC FTLD SB | --- | --- | --- | |
| Trails A | 0.33 (1.15; −1.71–5.01) | 0.45 (1.14; −1.1–5.82) | 2.8 (3.17; −0.55–8.11) | ||
| MRI Frontal/TIV | −0.08 (1.09; −3.49–3.89) | −0.68 (1.26; −3.46–1.92) | −2.59 (2.02; −8.18–3.18) | ||
| NfL (log) | 0.46 (1; −1.95–3.84) | 1.04 (1.45; −1.25–5.79) | 3.74 (1.49; −0.62–6.75) | ||
| MAPT | N (prop) at baseline | 69 (0.41) | 37 (0.22) | 62 (0.37) | |
| Mean Age (SD) at baseline | 34.1 (9.2) | 46.3 (9.5) | 56.1 (8.6) | ||
| Mean Raw Score (SD; Range) | CDR®+ NACC FTLD SB | 0.15 (0.48; 0–2.5) | 0.39 (0.76; 0–3) | 7.9 (6.51; 0–24) | |
| MINT (Total Correct) | 29.88 (1.8; 25–32) | 29.16 (3; 17–32) | 21.22 (8.04; 1–32) | ||
| MRI MTL/TIV | 1.05 (0.06; 0.87–1.16) | 0.98 (0.07; 0.77–1.08) | 0.72 (0.14; 0.46–1.04) | ||
| NfL (log) | 1.69 (0.45; 0.39–2.53) | 1.98 (0.55; 0.93–3.44) | 3.04 (0.55; 1.93–5.1) | ||
| Mean Standardized Units from Control (SD; Range) | CDR®+ NACC FTLD SB | --- | --- | --- | |
| MINT | −0.02 (1.03; −2.82–1.19) | −0.48 (1.85; −7.98–1.27) | −4.56 (4.2; −15.12–1.07) | ||
| MRI MTL/TIV | 0.41 (1.04; −2.7–2.15) | −0.69 (1.29; −4.46–1.15) | −3.33 (1.86; −6.87–1.04) | ||
| NfL (log) | 0.04 (1.03; −2.95–1.98) | −0.19 (1.45; −2.91–3.63) | 1.45 (1.26; −1.11–6.17) | ||
Note. Baseline raw and standardized values for several measures are displayed for controls and mutation carriers at three Disease Age Epochs. Each participant was assigned to a Disease Age epoch based on the estimated Disease Age at their first visit. Clinical and imaging measures were selected by choosing the “best” measure for each genetic group based on when they became elevated compared to controls and the rate of longitudinal change (descriptive statistics for all modeled measures are displayed in Supplemental Table S2 and S3). Raw imaging measures are presented as percentage of total intracranial volume to account for head size. Mean standardized units from controls indicates the number of standard deviations from the control group.
Abbreviations: Prop: Proportion; CDR®+NACC FTLD SB: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center’s Frontotemporal Lobar Degeneration Module Sum of Boxes; Trail B: Trail Making Test, Part B; MINT: Multilingual Naming Test; MRI: magnetic resonance imaging; TIV: Total intracranial volume; NfL (log): Log-transformed plasma neurofilament light chain
In GRN, visual inspection suggested NfL begins to deviate from controls about 15 years prior to symptom onset, followed by MRI 5–10 years prior to onset (Figures 2, S1). Baseline plasma NfL concentrations in GRN were significantly elevated relative to controls in all DA epochs (Figure 3, Table S5) and elevated compared to all other genetic groups in the symptomatic phase (Table S6). NfL concentrations become elevated by one SD compared to controls 4.9 years prior to onset (95%CI:-3.4,-7). GRN also displayed the most rapid rates of NfL increase in the symptomatic epoch (Figures 1&2, Table S4). The frontal and temporal lobes were the first brain regions to differ from controls by one SD in the DPM (−1.1 and −1.2 years before estimated onset, respectively). The insula was significantly atrophied compared to controls in the −40 to −10 epoch (Extended Data Figure 1, Table S5), and all ROIs had smaller mean volumes than controls in the −10 to 0 epoch, except the striatum (p=0.057). In the symptomatic stage, volume loss in all ROIs was more rapid than the other genetic groups, with the frontal, temporal, medial temporal, insular, and striatal ROIs losing volume most rapidly (Figure S3, Table S4).
Medial temporal atrophy was the first observed biomarker change in MAPT, diverging from controls ~10 years before symptoms based on visual inspection (Figure 2), and reaching one SD below controls 1.8 years before onset (95%CI:-3.2,-0.5). The medial temporal lobe was the only region with significant volume loss compared to controls in the presymptomatic phase (Extended Data Figure 1, Table S5). The remaining temporal regions and insula were the next regions to become atrophied by one SD compared to controls (Table S4), with overlapping credible intervals. In the symptomatic stage, frontal, temporal and medial temporal, insular, and striatal regions showed the greatest degree of cross-sectional atrophy (Extended Data Figure 1, Figure S4, Table S5). Longitudinally, the medial temporal lobe (MTL), followed by the remainder of the temporal lobe, striatum, and insular regions were the regions to lose volume most rapidly in the symptomatic phase (Table S4). NfL levels began to diverge from controls closer to symptom onset in MAPT than C9orf72 or GRN, with mean values showing significant elevations during the symptomatic but not presymptomatic epochs (Extended Data Figure 1, Tables S5-6), and average values did not reach one SD above controls until 4.6 years after estimated symptom onset (95%CI:7.1,2.4).
We conducted a voxelwise sensitivity analysis in each DA epoch to complement the coarse-grained ROIs used in the DPMs and to illustrate the findings were not dependent on the DPMs. Results of this sensitivity analysis (Extended Data Figure 2, Figures S2-S4) supported the patterns observed using ROIs.
Global Ratings and Clinical Measures
Visual inspection of the curves revealed a rapid CDR®+NACC-FTLD-SB increase after symptom onset, and all genetic groups had cross-sectional elevations in CDR®+NACC-FTLD-SB prior to symptom onset (Figure 3, Table S5); note that statistical comparisons of this measure should be interpreted with caution given that controls were defined as having a baseline CDR®+NACC-FTLD=0 (as is typical in most clinical dementia research studies) and thus have no variance due to this selection process. Similar to the MRI results, GRN exhibited the most rapid CDR®+NACC-FTLD-SB changes following symptom onset (Figure 2, Table S4). Visual inspections of the curves indicated that neuropsychological and Revised Self-Monitoring Scale (RSMS) impairments relative to controls were generally observed only after symptom onset for all genetic groups (Figure 2 & Table S5), and no measure reached one SD worse than controls until after symptom onset (Table S4). In direct statistical comparison, C9orf72 expansion carriers performed worse than controls on Trails A and B at all DA epochs (Table S5). GRN performed worse than controls on Trails A at all epochs, and worse than controls on Trails B in the −10 to 0 epoch. MAPT mutation carriers exhibited impairments in the Figure Copy in the −10 to 0 epoch, with a trend towards impairment on the Multilingual Naming Test (MINT) in this epoch. Longitudinally, the most rapid change in the symptomatic stage relative to controls was observed for Trails A & B in C9orf72, Trails A, MINT, and Benson Copy in GRN, and the MINT and Trails B in MAPT (Table S4).
Raw values were modeled. The same pattern of findings was observed (Figure S5) in a sensitivity analysis adjusting for nuisance covariates (details in online methods).
Patient-level Estimates
DA estimates at baseline ranged from −40 to 21. The precision of individual DA estimates depends on the proximity to symptom onset and follow-up duration. In mutation carriers with at least one post-baseline visit who were >10 years from expected onset, the average uncertainty of the DA estimate (95%CI) was +/−14.6 years. For those −10 to 0 years from onset, this uncertainty decreased to +/−5.5 years, and after onset, the average uncertainty of the estimate was +/−0.9 years. To better understand the impact of level of impairment, rate of progression, and model priors (i.e., years since onset) on estimated DA, individual patient-level data were examined (Extended Data Figure 3). With increasing DA, performance is increasingly impaired across multiple measures, and there is a greater tendency for progressive impairment from baseline to final observations. In those furthest from onset, when most scores tend to be within normal limits, prior information about their age has a large influence on estimated DA. Examining cases that the model estimated to be presymptomatic (DA<0) despite a CDR®+NACC-FTLD-SB>0, these participants tend to perform in the average range across other measures and stay stable or show improvements over time.
Application to Clinical Trials
The DPM curves suggest that clinical trial endpoint selection might differ by genetic group and disease stage (Figure 2). To explore this further, simulation studies based on the natural history data were conducted to estimate the sample sizes required to measure a 50% reduction in various potential endpoints for two- and four-year presymptomatic prevention trials and 1.5 and two-year early symptomatic treatment trials (Table 3; 1:1 randomized parallel design; details in online methods). Prevention trial designs included only participants with a CDR®+NACC-FTLD-Global = 0 at baseline. Simulations explored the use of baseline NfL and DA as additional inclusion criteria to define a high-risk population most likely to show clinical change over the course of the trial, thereby increasing power. Sample size estimates for prevention trials were generally lowest when using biomarkers (NfL or MRI) as the outcome. For example, a two-year prevention trial requiring a DA within five years of onset would require sample sizes of 52 total participants for GRN (MRI Frontal), 108 for MAPT (MRI MTL), and 424 for C9orf72 (MRI Temporal) if MRI is used as an endpoint. Based on the estimated number of eligible participants from the FPI dataset (assuming no additional recruitment efforts), two-year trials appear to be feasible for GRN if MRI is used as the outcome, whereas a four-year trial would be required for MAPT. Additional recruitment would be required for a C9orf72 prevention trial to be sufficiently powered to detect a 50% treatment effect.
Table 3.
Clinical trial sample size estimates
|
Pre-symptomatic prevention trial (CDR®+NACC-FTLD Global = 0)
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Primary endpoint - Sample Size Estimates (50% Treatment Effect) | ||||||||||
|
| ||||||||||
| Genetic Group | Estimated number of eligible participants | Inclusion Criteria | CDR®+NACC-FTLD-SB | Neuropsychological Tests | NfL (log) | MRI Volume | ||||
|
| ||||||||||
| 2 Yrs | 4 Yrs | 2 Yrs | 4 Yrs | 2 Yrs | 4 Yrs | 2 Yrs | 4 Yrs | |||
|
C9orf72 MRI=Temporal NP = Trails B |
171 | All CDR 0 |
>10000 | 4994 | >10000 | 6784 | 3397 | 699 | 1639 | 394 |
| 13 | CDR 0 & NfL (log) > 3 |
582 | 334 | 1113 | 386 | >10000 | 638 | 537 | 173 | |
| 38 | CDR 0 & DA > −5 |
508 | 224 | 657 | 184 | 527 | 153 | 424 | 119 | |
| 20 | CDR 0 & DA > −2.5 | 266 | 111 | 364 | 96 | 439 | 123 | 402 | 102 | |
|
|
||||||||||
|
GRN MRI=Frontal NP=Trails A |
168 | All CDR 0 |
3144 | 1526 | 3844 | 1576 | 684 | 271 | 826 | 459 |
| 7 | CDR 0 & NfL (log) > 3 |
250 | 179 | 250 | 140 | 158 | 51 | 71 | 46 | |
| 26 | CDR 0 & DA −5 |
297 | 182 | 267 | 130 | 99 | 30 | 52 | 27 | |
| 10 | CDR 0 & DA −2.5 | 182 | 104 | 159 | 79 | 84 | 26 | 37 | 24 | |
|
|
||||||||||
|
MAPT
MRI=MTL NP=MINT |
94 | All CDR 0 |
7073 | 2733 | >10000 | 3741 | 3059 | 802 | 1492 | 526 |
| 4 | CDR 0 & NfL (log) > 3 |
283 | 188 | 373 | 220 | >10000 | 501 | 147 | 72 | |
| 19 | CDR 0 & DA −5 |
362 | 190 | 641 | 265 | 595 | 149 | 108 | 39 | |
| 14 | CDR 0 & DA −2.5 | 191 | 97 | 311 | 134 | 438 | 117 | 72 | 24 | |
|
Early symptomatic treatment trial (All CDR®+NACC-FTLD Global = 1 enriched with 0 and 0.5 participants)
| ||||||||||
| Primary endpoint - Sample Size Estimates (50% Treatment Effect) | ||||||||||
|
| ||||||||||
| Genetic Group | Estimated number of eligible participants | Inclusion Criteria | CDR®+NACC-FTLD-SB | Neuropsychological Tests | NfL (log) | MRI Volume | ||||
|
|
||||||||||
| 1.5 Yrs | 2 Yrs | 1.5 Yrs | 2 Yrs | 1.5 Yrs | 2 Yrs | 1.5 Yrs | 2 Yrs | |||
|
C9orf72 MRI=Temporal NP = Trails B |
94 | ALL CDR 0.5 and 1 |
188 | 129 | 340 | 203 | 811 | 483 | 639 | 367 |
| 37 | All CDR 1 & (CDR 0 & 0.5 if NfL > 3) |
161 | 115 | 370 | 222 | 1806 | 782 | 645 | 358 | |
| 83 | All CDR 1 & (CDR 0 & 0.5 if DA > −2.5) |
176 | 124 | 400 | 207 | 740 | 423 | 678 | 360 | |
| 67 | All CDR 1 & (CDR 0 & 0.5 if DA > 0) | 117 | 79 | 275 | 161 | 628 | 384 | 669 | 359 | |
|
|
||||||||||
|
GRN MRI=Frontal NP=Trails A |
67 | ALL CDR 0.5 and 1 |
76 | 66 | 115 | 79 | 133 | 76 | 44 | 30 |
| 33 | All CDR 1 & (CDR 0 & 0.5 if NfL > 3) |
97 | 84 | 124 | 92 | 182 | 110 | 49 | 36 | |
| 48 | All CDR 1 & (CDR 0 & 0.5 if DA > −2.5) |
79 | 68 | 105 | 74 | 127 | 75 | 36 | 26 | |
| 38 | All CDR 1 & (CDR 0 & 0.5 if DA > 0) | 39 | 32 | 62 | 41 | 124 | 72 | 32 | 22 | |
|
|
||||||||||
|
MAPT
MRI=MTL NP=MINT |
43 | ALL CDR 0.5 and 1 |
175 | 136 | 300 | 196 | 845 | 437 | 124 | 74 |
| 11 | All CDR 1 & (CDR 0 & 0.5 if NfL > 3) |
89 | 66 | 138 | 91 | 1719 | 769 | 95 | 59 | |
| 43 | All CDR 1 & (CDR 0 & 0.5 if DA > −2.5) |
164 | 120 | 244 | 163 | 779 | 419 | 109 | 63 | |
| 31 | All CDR 1 & (CDR 0 & 0.5 if DA > 0) | 96 | 66 | 150 | 104 | 627 | 359 | 83 | 48 | |
Note. Sample size estimates (total n for a trial) are first presented for pre-symptomatic prevention trials in which all enrolled participants are presymptomatic based on CDR®+NACC-FTLD = 0. Within each genetic group, sample sizes are estimated for trials enrolling all presymptomatic participants as well as three additional scenarios in which NfL or Disease Age are used to enroll high-risk participants likely to be proximal to symptom onset. In the bottom half of the table, estimates are presented for an early symptomatic trial in which all participants with a CDR®+NACC-FTLD Global = 1 are eligible, and those with CDR®+NACC-FTLD < 1 are included based on different inclusion criteria. The estimated number of eligible participants refers to the number of participants in the current dataset that meet the specified inclusion criteria. For each genetic group, we select a representative MRI and neuropsychological measures (displayed in the first column). Bolded cells indicate that the sample size estimates are less than or within 15 participants of the number eligible. All trial designs assume 1:1 randomization treatment vs. control, 10% attrition per year, and have a primary analysis of a change from baseline in the primary endpoint. Additional details of the assumptions underlying these simulations can be found in Table S9.
Abbreviations: CDR®+NACC-FTLD-SB/CDR: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center’s Frontotemporal Lobar Degeneration Module Sum of Boxes; NfL(log): Log-transformed plasma neurofilament light chain; MRI: magnetic resonance imaging; NP: Group-specific neuropsychological measure; Trails A/B: Trail Making Test, Parts A & B; MTL: Medial Temporal Lobe; MINT: Multilingual Naming Test; DA = Disease Age
Symptomatic trial simulations included all participants with a CDR®+NACC-FTLD-Global=1 and subsets of high-risk participants with a CDR®+NACC-FTLD-Global of 0 or 0.5 defined based on elevated NfL (log(NfL) > 3.0) or an estimated DA within 2.5 years of onset (Table 3). Based on these simulations, it would be feasible to power trials for all three genetic groups using the CDR®+NACC-FTLD-SB and neuropsychological tests, measures most likely to be approvable by regulatory bodies as clinically meaningful endpoints.15 For example, within a population having a CDR®+NACC-FTLD-Global=1 or a DA within 2.5 years of onset in those with a CDR®+NACC-FTLD-Global<1, the estimated sample sizes using CDR®+NACC-FTLD-SB as the primary endpoint for a two-year trial were 68 total participants for GRN, 120 for MAPT, and 124 for C9orf72.
Discussion
We present the efforts of the international FTD Prevention Initiative (FPI) to establish the largest known cohort of f-FTD cases worldwide, gathered from North American (ALLFTD) and European/Canadian (GENFI) natural history studies. We harmonized clinical, neuropsychological, biofluid, and neuroimaging measurements to build DPMs that allow direct comparisons of effect sizes for mean values and rates of change between the best available measures for characterizing FTD. The DPMs revealed important insights about the earliest manifestations of f-FTD and the temporal ordering of biomarker and clinical changes. Across all three FTD mutation carrier groups, regional brain atrophy and plasma NfL elevations were the first measurable manifestations of disease, potentially developing 10 to 40 years before the earliest clinical features. Neuropsychological changes typically occurred later, contemporaneous with the emergence of informant-reported symptoms (CDR®+NACC-FTLD-SB). The genetic groups displayed differences in patterns of disease progression that are relevant for clinical care and clinical trial planning. The striking concordance in disease progression between the two independent North American and European cohorts supports the validity of the models, suggesting that the natural history of the disease is strongly determined by pathogenic mutations and that global clinical trials of f-FTD therapies are feasible. Finally, we leveraged the DPMs and natural history data to simulate prevention and treatment clinical trial scenarios, including candidate participant selection criteria and primary endpoints, to provide evidence for the feasibility of running presymptomatic prevention trials and symptomatic treatment trials in f-FTD.
The validity of our DPM models is supported by the results of previous studies focusing on individual biomarkers or clinical measures in f-FTD. Because the models incorporate both new and some previously analyzed historical data, we were able to replicate and extend the results of previous studies. We also directly compared the relative utility of different assessments at different stages of disease. Consistent with previous MRI studies demonstrating brain atrophy can be detected in presymptomatic f-FTD,16–19 MRI-measured brain atrophy was the first biomarker to change in C9orf72 and MAPT, but our models revealed that NfL elevations preceded atrophy by a few years in GRN. In C9orf72, the thalamus and most other brain regions were smaller than controls 10 to 40 years prior to onset, supporting the hypothesis that C9orf72 repeat expansions may affect early brain development.19,20 Also consistent with prior work, the most rapid rates of atrophy occurred in GRN with widespread brain involvement within 10 years of onset.21,22 Despite differences in analytic methods, and the inclusion of a much larger dataset, the DPMs developed in this study allowed us to replicate the findings of earlier, smaller analyses. In an earlier MRI study, Rohrer and colleagues17 defined expected disease onset based on each genetic group’s mean age of onset rather than using model derived DA employed here. Similar to the previous study, we detected medial temporal atrophy in MAPT 15 years prior to onset followed by atrophy of the insula. Temporal lobe atrophy in presymptomatic MAPT has been consistently reported,16,18,23 and the insula may be a common region of early atrophy in MAPT.24
We and others have previously shown that NfL concentrations are elevated in the plasma25–27 and CSF28,29 of symptomatic FTD patients compared to other neurological conditions. In the current study, we verified that the genotype-related patterns of plasma NfL elevation that were measured in two different laboratories, in two independent f-FTD cohorts, were very similar and for the purposes of DPM, could be combined. In C9orf72, NfL levels began to deviate from controls approximately 30 years prior to onset and remained significantly elevated compared to controls in all presymptomatic epochs. In GRN, NfL levels begin to increase 15 years prior to onset and were elevated compared to controls in the late presymptomatic stages. In contrast, NfL levels begin to increase just proximal to symptom onset in MAPT, and presymptomatic MAPT mutation carriers did not show increased levels compared to controls. In the symptomatic stage, NfL levels rose more than twice as fast in GRN than the other genetic groups. These results extend previous fluid biomarker studies showing NfL concentrations become elevated early in f-FTD, are harbingers of symptom onset, and rise most rapidly in GRN.25,27,30–32
Paralleling the biomarker findings, global disease severity (CDR®+NACC-FTLD-SB) and neuropsychological measures declined more rapidly in GRN than C9orf72 or MAPT mutation carriers. Although GRN was previously shown to have the longest disease course in an international f-FTD cohort,12 disease duration in that study was determined based on clinical interview rather than the data-driven approach taken in the current study; moreover, the C9orf72 sample in the prior study had a higher proportion of participants with ALS or FTD with motor neuron disease than the current study (30.3% v 13.1%), and these diagnoses were associated with more rapid disease progression.12,33 Neuropsychological impairments relative to age-matched controls were typically observed after symptom onset in all groups, although abnormalities on a few measures were detected in the presymptomatic stages. These findings add to prior studies suggesting that cognitive changes can be detected in the presymptomatic phases of f-FTD and that there are genotype-specific cognitive profiles.34–37 Future work should continue to explore the development and validation of novel neuropsychological measures for early detection and monitoring, including digital cognitive tests and cognitive composite scores (e.g. GENFI-COG) that may improve early detection and reduce sample size estimates.37
An overarching aim of this study was to develop models that inform the design of f-FTD clinical trials. Simulation studies were conducted to estimate the sample sizes necessary to power prevention and early symptomatic treatment trials. These studies also explored the use of NfL and DA estimates as inclusion criteria to enroll presymptomatic mutation carriers at heightened risk for clinical progression during a trial. The simulations revealed important information that will be directly applicable to clinical trial design. First, using NfL and MRI biomarkers as surrogate endpoints for prevention trials would allow trials to be conducted with many fewer participants than clinical measures. Second, prevention trials appear most feasible for MAPT and GRN relative to the estimated number of eligible participants based on our dataset, however, given that C9orf72 is the most common mutation causing FTD and ALS, recruiting the estimated sample sizes may be feasible. Third, using estimated DA to select high-risk presymptomatic participants for trial enrollment leads to a sizeable reduction in sample sizes. This reduction in sample size must be balanced against the reduction in number of eligible participants (of that DA), but these simulations show that GRN and MAPT trials enrolling presymptomatic participants within five years of estimated onset would be feasible based on the estimated number of eligible participants from our current dataset. Fourth, clinical measures perform very well in the early symptomatic trial simulations, and sample sizes for trials using the CDR®+NACC-FTLD-SB as a primary outcome are feasible for all three genetic groups. Not only was this measure statistically powerful for measuring change, but given that it reflects informant-reported clinical status, it could also be considered a clinically meaningful outcome and approvable endpoint from a regulatory perspective.15
The clinical trial simulations included in this study used standard, two-arm, parallel-group clinical trial designs. Future work to explore innovative trial designs and analysis methods may enable trials with smaller samples sizes and/or increased power for smaller (but clinically meaningful) treatment effects. With the incorporation of a treatment effect parameter, the DPM-predicted versus post-treatment progression could potentially be used as a primary endpoint in clinical trials to estimate the slowing in disease progression across multiple endpoints.7,38 In rare diseases such as f-FTD analysis methods may also simulate data from natural history participants to generate “synthetic” participants to decrease sample sizes and reduce allocation to placebo as has been encouraged in recent FDA guidance.14 Additionally, platform trials based on DPMs allow multiple therapies to be tested simultaneously with comparisons made to a shared placebo group further improving trial efficiency in rare populations.39
There are important limitations to this work. Known genetic modifiers of f-FTD disease progression were not included, such as specific mutations (for MAPT) and TMEM106B, a modifier of penetrance in GRN.2,40 We were also limited in the clinical measures that we could include in the analysis to those that were readily harmonizable between ALLFTD and GENFI, excluding a variety of promising novel measures that were not available in both cohorts.34,35 Future models will likely be improved by including a more exhaustive collection of measures and biomarkers41 and approaches accounting for heterogeneity in f-FTD features.42 Because disease onset was defined as CDR®+NACC-FTLD-SB=0.5, non-carrier controls by definition had CDR®+NACC-FTLD-SB=0 at baseline, which reduced the variance in this measure, thereby potentially overestimating the effect size relative to other measures where there was more variance in the controls. Because abnormal global status may reflect other brain pathologies in the controls that could potentially obscure important findings, we believe that the requirement for CDR®+NACC-FTLD-SB=0 in controls was appropriate.
The DPMs produced for the current study have additional limitations related to less informative clinical data at early stages of disease and missing data at late stages of disease. In subjects estimated to be within 10 years of symptom onset, the accuracy is +/- 5.5 years, which approaches the accuracy of familial age of onset-based estimates which are useful in DIAD,43 but not possible in most f-FTD syndromes.12 However, individuals furthest from onset are typically within normal limits on all contributing measures forcing the model to rely heavily on prior information about participants’ chronological age to estimate DA. This results in considerable uncertainty around exact DA in those furthest from expected onset (e.g., +/-14 years in the −40 to −10 epoch). To visually assess how the weight of evidence (number of measures that changed over the range of visits) related to each subject’s DA, we color coded measurements in each individual mutation carrier in Extended Data Figure 3. This revealed that in more severely impaired mutation carriers at later DA, there was more missing data, particularly MRI. This suggests an important limitation to the use of MRI as an outcome in symptomatic mutation carriers: data may be missing because scans are harder to acquire in advanced patients, possibly because they either cannot travel to research centers or they cannot lie still in a MRI scanner. Such informative missing data also impacts the DPMs, potentially biasing the models towards a smaller standard deviation from normal; this is a limitation and a direction for future research. Finally, the current study is limited by the lack of racial and ethnic diversity of the sample. Improving the diversity of participants in FTD research is an urgent priority,44 however, it should be noted that in genetic f-FTD there are known founder effects for C9orf7245 and GRN mutations46 with European ancestry, leading to strong associations with particular racial and ethnic groups.
In conclusion, these DPMs will facilitate the planning of f-FTD clinical trials, including selection of optimal endpoints and enrollment criteria to maximize power to detect treatment effects.14 Brain atrophy and plasma NfL elevations are measurable years prior to symptom onset, paving the way for using these biomarkers in clinical trials of agents that could prevent or delay the clinical manifestations of f-FTD. The models also highlight the challenges of conducting adequately powered trials in rare f-FTD populations and provide a roadmap for development of new biomarkers and clinical endpoints that may improve power to detect effects in presymptomatic stages of disease and create a renewed sense of urgency to identify eligible trial participants outside of Europe and North America.
Online Methods
Participants
Participants included 796 carriers of pathogenic mutations in the C9orf72, GRN, or MAPT genes and 412 non-carrier controls from families with a known mutation in one of these genes. Participants were enrolled through Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL; NCT02365922) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS; NCT02372773),47 which recently combined into the ARTFL/LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD; NCT04363684) study. These studies enrolled participants through a consortium of 18 centers across the US and Canada between 2015 and 2020. Participants were also enrolled through the Genetic Frontotemporal Initiative (GENFI), which involves 25 research centers across Europe and Canada. GENFI 2 participants from the 5th Data Freeze (2015–2019) were included. All participants were required to have completed the Clinical Dementia Rating Scale (CDR®) plus Behavioral and Language Domains from the National Alzheimer’s Coordinating Center (NACC) FTLD module (CDR®+NACC-FTLD). GENFI 1 (2012 – 2015) participants were excluded because the CDR®+NACC-FTLD was not collected during that study period. Some participants in GENFI 2 and ALLFTD cohorts underwent longitudinal evaluations, and all available data were included. ALLFTD participants received travel compensation and remuneration up to $100 based on the study they participated in. For GENFI, Travel, accommodations, or other reasonable expenses are offered to the participants to cover any costs they incur in order to attend the research visits. The ALLFTD study was approved through the Trial Innovation Network (TIN) at Johns Hopkins University. Local ethics committees at each of the sites approved the study, and all participants provided written informed consent or assent with proxy consent.
ALLFTD Inclusion/Exclusion Criteria Relevant to this study:
Inclusion criteria:
Participants must be a member of a family with a known pathogenic mutation in the GRN or MAPT genes, or with a pathogenic expansion in the C9orf72 gene. The participant does not have to know their own genetic status but must be at least 18 years of age. The predominant phenotype in most families is cognitive or behavioral. However, families may present with motor-dominant syndromes without exclusion. Participants must have a reliable informant who personally speaks with or sees that subject at least weekly. Participants are sufficiently fluent in English to complete all measures. Participants must be willing and able to consent to the protocol and undergo yearly evaluations over three years. Participants must be willing and able to undergo neuropsychological testing (at least at baseline visit) and have no contraindication to MRI imaging. Non-carrier family controls were included in the current study if they were clinically normal at baseline, defined by a CDR®+NACC-FTLD Global Score = 0.
Exclusion criteria:
Known presence of a structural brain lesion (e.g., tumor, cortical infarct). Presence of another neurologic disorder that could impact findings (e.g., multiple sclerosis).
GENFI Inclusion/Exclusion Criteria Relevant to this study:
Inclusion Criteria:
Participants are at least 18 years old. Participants must be a member of a family with a known pathogenic mutation in the GRN or MAPT genes, or with a pathogenic expansion in the C9orf72 gene. If the participant is cognitively impaired, there must be an available caregiver that can escort them. The participant must have an identified informant. The participant must be fluent in the language of their country of assessment. Non-carrier family controls were included in the current study if they were clinically normal at baseline, defined by a CDR®+NACC-FTLD Global Score = 0.
Exclusion criteria:
Participant has another medical or psychiatric illness that would interfere in completing assessments. Participant is pregnant. Local MRI and lumbar puncture contraindications. The predominant phenotype in most families is cognitive or behavioral. However, families may present with motor-dominant syndromes without exclusion.
Genetic Testing
ALLFTD participants had genetic testing at the University of California, Los Angeles using published methods.48 GENFI participants were genotyped at their local sites according to previous methods.17 Briefly, in ALLFTD and GENFI, DNA samples were screened using targeted sequencing of a custom panel of genes previously implicated in neurodegenerative diseases, including GRN and MAPT. The presence of hexanucleotide repeat expansions in C9orf72 was detected in ALLFTD using both fluorescent and repeat-primed PC and in GENFI using repeat-primed PCR.
Clinical Assessment
The ALLFTD and GENFI multidisciplinary assessments includes neurological history and examination and collateral interview.17 Documented years since onset, which was entered as prior in the model, was based on clinical interview.
The CDR®+NACC-FTLD module is an eight-domain rating scale based on informant report.49–51 A Global Score was calculated to categorize disease severity as presymptomatic (0), questionable or mild symptoms of neurodegenerative disease (0.5), or clear symptoms of dementia (1, 2, or 3).49 A sum of the eight box scores (CDR®+NACC-FTLD-SB) was also calculated; this score ranges from 0 –24, with higher scores indicating greater functional impairment.49
A subset of neuropsychological tests from the Uniform Data Set (UDS) Neuropsychological Battery, version 3.0 was available for both consortia: Trail Making Test Parts A & B, the Multilingual Naming Test (Boston Naming Test in GENFI), Number Span Forward and Backward (Digit Span in GENFI), Benson Figure Copy and Delayed Recall, and Animal Fluency. Conversion tables from the UDS Crosswalk study were used to harmonize Number Span/Digit Span and the MINT/BNT.52 Upon review of neuropsychological test scores in the controls, one outlier score was removed. As a sensitivity analysis to consider the impact of additional demographic covariates (i.e., sex, education, language), statistical harmonization of the neuropsychological data was conducted using a W-score approach,42,53 which is a standardized score controlled for nuisance covariates. Regression models were built using baseline neuropsychological test scores in the non-carrier controls, with separate models in each consortium. All regressions included sex and education. In the GENFI cohort, primary language was included as an additional categorical covariate. Next, in all participants at every time point, the difference between their actual score and predicted score (based on regression conducted in controls) was divided by the standard deviation of the control group to derive a standardized estimate compared to controls with the same demographic background.
Neuroimaging
Image Acquisition
Details of image acquisition, processing, and harmonization can be found below and have been published elsewhere.54 ALLFTD participants were scanned at 3T on MRI scanners (scanner types are displayed in Supplemental Table S7). T1-weighted images from ALLFTD were acquired as Magnetization Prepared Rapid Gradient Echo (MP-RAGE) images using the following parameters: 240x256x256 matrix; about 170 slices; voxel size = 1.05x1.05x1.25 mm3; flip angle, TE and TR varied by vendor. A standard imaging protocol was used across all centers, managed, and reviewed for quality by a core group at the Mayo Clinic, Rochester.
GENFI participants underwent volumetric T1-weighted MRI using the standard GENFI protocol.17,55 A variety of 1.5T and 3T scanners were used across the sites: Siemens Trio, Siemens Skyra, Siemens Prisma, Philips, and General Electric. The scan protocols were designed at the start of the GENFI study to ensure that there was adequate matching between the scanners and the quality of the images. T1-weighted images from GENFI were acquired using the following parameters: 256x256x208 matrix; 208 slices; voxel size = 1.1 mm isotropic, flip angle = 8°, TE and TR varied by vendor. All scans were quality checked and those with movements or artifacts were removed. Furthermore, if any participants displayed moderate to severe vascular disease or any other brain lesions, they were also excluded from the analysis.
Image Processing
The same image processing steps were performed on ALLFTD and GENFI data. Before any prepossessing of the images, all T1-weighted images were visually inspected for quality control. Images with excessive motion or image artifact were excluded. T1-weighted images underwent bias field correction using N3 algorithm.56 The segmentation was performed using SPM12 (Wellcome Trust Center for Neuroimaging, London, UK, http://www.fil.ion.ucl.ac.uk/spm) unified segmentation.57 A customized group template was generated from the segmented gray and white matter tissues and cerebrospinal fluid by non-linear registration template generation using the Large Deformation Diffeomorphic Metric Mapping framework.58 Subjects’ native space gray and white matter were geometrically normalized to the group template, modulated, and then smoothed in the group template. The applied smoothing used a Gaussian kernel with 8~mm full width half maximum. Every step of the transformation was carefully inspected from the native space to the group template.
Regional volume estimates were calculated from individual subjects’ smoothed, modulated grey matter in template space, by taking the mean of all voxels in several a priori regions of interest (ROIs)59 by taking the mean of all voxels within the following regions: Frontal, Temporal, Medial Temporal (consisting of amygdala, hippocampus, entorhinal cortex, and parahippocampal gyrus ROIs), Parietal, and Occipital Lobes, Striatum, Insula, Thalamus, and Cerebellum. Volume estimates were then represented as percentage of total intracranial volume. To understand the effects of scanner and to present voxelwise maps, a W-score was created at each voxel to represent volume relative to controls after adjusting for covariates. First, a multivariable linear model was fit for each voxel in a reference group that consisted of the first available scan for non-carrier family controls. Predictors in this model were total intracranial volume (TIV) and scanner platform.42,53 Next, for each voxel of every available MRI in the study, the same model was fit, entering TIV and scanner, using the coefficients from the reference group to extract a residual. This residual was then divided by the standard deviation of the residuals in the reference group. Therefore, the W-score represents the gray matter content at that voxel as the number of standard deviations away from the expected mean for a reference group, accounting for TIV and scanner platform. We then created a mean W-score value for each ROI and entered it into the model as a sensitivity analysis. Mean W-scores at each voxel in mutation carriers are also presented in supplemental figures.
Plasma Neurofilament Light Chain (NfL)
ALLFTD Methods
Plasma NfL light concentrations were measured at the Mayo Clinic in Jacksonville using the Quanterix single-molecule array technology (Simoa) @ NF-Light Advantage Kit (Cat#103186, Lot 501992) and the HD-X instrument according to the instructions provided. Samples were tested in duplicate using kits from the same lot. In addition to the two quality control samples provided with the kit, all assays included five inter-assay controls. Prior to each assay, plasma samples were thawed, mixed thoroughly by low-speed vortexing, centrifuged at 10,000 g for five minutes, and transferred to 96-well plates that were then sealed to minimize sample evaporation. Samples were diluted four times by the instrument. If levels of NfL in a sample exceeded the upper limit of the calibration curve, the sample was retested at a higher dilution. Across all assays, the percent coefficient of variations of the mean NfL concentration for the inter-assay controls were below 10%.
GENFI Methods
Plasma NfL concentrations were measured at baseline with Simoa, using the commercially available Simoa Neurology 4-Plex A kit (Quanterix, Lexington, MA, Cat# 102153, Lot#: 50216). Plasma samples were thawed at room temperature (one cycle), mixed thoroughly, and centrifuged at 14,000g for 3 minutes. The supernatant was loaded onto a Quanterix HD-1 Analyzer with a 1:4 specified dilution. Measures were completed in duplicate over a total of six batches, each with an eight-point calibration curve tested in triplicate and two controls tested in duplicate. Plasma concentrations were interpolated from the calibration curve within the same batch and corrected for the dilution. All samples were quantifiable within the dynamic range of 0.69 to 2,000 pg/mL and with an average coefficient of variation below 10%. Instrument operators were blinded to clinical and genetic information.
Prior publications
Prior publications have included some of the data included in these models, including publications describing MRI,16,18,23,42,54,60–62 NfL,25,27,30 and clinical data.12,31,35,37,41,51,54 For full lists of publications from these consortia see https://www.allftd.org/publications and https://www.genfi.org/publications/. This study is the first comprehensive effort to combine clinical, imaging, and plasma biomarker data across consortia.
Statistical Analyses
All available data were included in the statistical analyses. Complete cases were not required, and no imputation was conducted. Statistical tests were two-sided.
Participant characteristics
Demographic variables and other participant characteristics (Table 1) were compared across genetic groups and controls using regression with pairwise group contrasts for most variables. Sex, race, CDR®+NACC-FTLD, and diagnostic categories were compared using chi-square with Bonferroni-adjusted pairwise comparisons when the omnibus test was significant. For chi-square tests in which any bins were < 10, the Fisher’s exact test was used.
Disease Progression Model
Disease progression models were built using a Bayesian mixed effects framework, with the goal of estimating a single latent disease stage parameter for each person, which we refer to as Disease Age. The disease progression model is a joint model of all 20 measures listed in Supplemental Table S8. Disease Age is the estimated difference between an individual’s chronological age and the age of symptom onset (defined for this study as a CDR®+NACC-FTLD-SB = 0.5). This estimate is positive for symptomatic cases and negative for presymptomatic cases. The model included priors based on an individual’s time from expected symptom onset. In symptomatic cases, we used the clinician’s estimate of time from symptom onset, sampled from a normal distribution with a small amount of error (SD=4) to acknowledge the imperfection of this estimate. For presymptomatic cases and non-carrier controls, we used the mean age of the mutation group as a prior, sampled from a normal distribution with more noise (SD=10). The prior standard deviations of 4 and 10 were chosen to be relatively non-informative. For a subject with an observed clinician’s estimate of time since symptom onset, there is a 95% prior probability that the true age of onset was within +/- 8 years of the clinician’s estimate. For a subject whose onset has not yet been observed, there is a 95% prior probability that the true age of onset was within +/- 20 years of the mean estimated age of onset from the same mutation group.
Disease age was then estimated from a joint analysis of all available clinical, neuropsychological, imaging, and NfL data. Simultaneously, overall disease progression of each endpoint was modeled as a function of latent disease age with several parameters, including expected value at “normal,” total decline, endpoint and mutation- specific rate and timing of progression. To account for variability in values of each endpoint at healthy across subjects, we included subject-specific random effects that were correlated across similar endpoints (see Grouping variable in Table S8).
First, models were built separately in each cohort. Visual inspection suggested sufficient alignment between disease progression of all endpoints across the two consortia and subsequent models combined both cohorts within a single analysis. A detailed description of the approach follows:
Latent Disease Stage Disease Progression Model
Model each endpoint, , for each subject, , for each visit, , as a function of latent disease stage. Where is the value of the endpoint for subject at visit , and is the age for subject at visit .
Disease age was defined as years since onset (YSO): age at visit minus age at onset, . Age at onset is a latent variable that is estimated for each subject.
- The observed value was assumed to be distributed normally with a subject and endpoint-specific mean and endpoint-specific variance that is a function of the mean.
- The subject and endpoint-specific mean decay function, , followed an exponential decay as a function of disease age with location and scale parameters that are mutation specific. Mutations are denoted for C9orf72, GRN, MAPT, and non-carriers respectively, is an indicator of the mutation (m=1:4) for subject .
Model parameters and prior distributions for each parameter are described below.
Model Components and prior distributions
: Value of the endpoint at normal/healthy state. Normal prior distribution with mean fixed based on expected value of endpoint at a normal state (see Table S8) and SD of 10.
: Worst value for the endpoint (floor). Normal prior distribution with mean fixed based on expected worst value of the endpoint (see Table S8) and SD of 10.
-
: Subject and endpoint-specific random effects in value of the endpoint at normal state that are correlated across similar endpoints (see Table S8 for groupings). Random effects are standardized based on the estimated endpoint-specific variability across subjects at normal, , and have a hierarchical prior distribution with subject-specific standardized mean for each group, , of endpoints, , and group-specific variability across endpoints within a subject, .
Hyper-prior distributions for the endpoint-specific variability across subjects at normal and the group-specific variability across endpoints in that group within a subject have a mean value of 1 on the precision and a SD of 10.
-
: Endpoint and mutation-specific overall location of mean decay function. Location parameter was set for endpoint, , that corresponds to CDR®+NACC-FTLD-SB so that the model is anchored to assume that a disease age of 0 corresponds to a value on CDR®+NACC-FTLD-SB of 0.5. For all other endpoints, we placed a non-informative prior distribution on the location parameter.
In particular, , provides the percentage of the total decline of the endpoint at “onset” (DA =0). A value of 1 implies the endpoint is fully declined, a value of 0.5 implies 50% of the total decline. Under the above non-informative prior, 95% of the distribution of is between 0 (<.00001) and 1 (>.99999) with a median value of 0.50.
- : Endpoint and mutation-specific overall slope of mean decay function. For all endpoints and mutations, we placed a non-informative prior distribution on the scale parameter.
-
: Age at onset per subject
- If value was observed within the dataset, we assumed that the prior distribution of a subjects age of onset was normal with a mean of the observed value and a SD of 4.
- If value was not observed within the dataset, we assumed that the prior distribution of a subjects age of onset was normal with a mean of the imputed value (imputed as the mutation and study-specific mean from all observed ages of onset) and a SD of 10.
: Imputed or observed age of onset per subject
: 4 if observed 10 if imputed
- : Endpoint-specific measurement error.
Computation
The Bayesian model was computed in R version 4.1.2, using the rjags package. This package uses Markov Chain Monte Carlo (MCMC) to generate a sequence of dependent samples from the posterior distribution of the parameters. The MCMC had a burnin of 10,000 samples, followed by 100,000 samples.
Secondary analyses using estimated Disease Age
After building the DPMs, we extracted estimates of disease age for each observation. We then further explored the data in two different ways. For each endpoint, we first plotted raw values for mutation carriers and non-carriers as a function of disease age. For each measure, we provide mutation-specific estimates for the age at which that measure deviates from controls by one SD. Second, we binned mutation carriers and controls based on their disease age at baseline (i.e., Epoch 1: Disease Age = −40 to −10; Epoch 2: Disease Age = −10 to 0; Epoch 3: >0). Epochs were chosen for illustrative purposes and to allow for a frequentist statistical analysis. For the cross-sectional data, we first compared the three genetic groups within an epoch by fitting a linear regression with the clinical measure or biomarker as the outcome, and genetic group as a three-level categorical variable. Multiple comparisons were controlled for using the Tukey method. Within each epoch, we also compared carriers to controls. Using the first available MRI scan for each participant, voxelwise mean W-scores for each bin were displayed for illustrative purposes. We also provide estimates of rates of change within each epoch based on the Bayesian DPM. Each Disease Age estimate is associated with a 95% credible interval. The mean of these credible intervals is presented for each epoch to provide an estimate of how the model accuracy varies as a function of Disease Age; we hypothesized greater uncertainty further away from onset as most measures will be in the normal range at this stage and thus the model is more reliant on prior knowledge (i.e., baseline age for presymptomatic cases).
Clinical Trial Simulation
Virtual clinical trial simulations are used to understand operating characteristics of proposed clinical trial designs. We simulated virtual patient outcomes under different assumptions for key design parameters to create simulated example trials. Within clinical trial simulation, generally, thousands of example trials are simulated under different sets of assumptions (scenarios) including trial sample size, randomization ratio, length of follow-up, targeted population, control progression rates and variability, and treatment effects. Overall average operating characteristics may then be summarized to quantify important characteristics of the proposed design (e.g. type I error, power, treatment effect estimates).
Clinical trial simulation requires assumptions to be made about the underlying data. Results from the disease progression model can be used to create evidence-based assumptions about rates of progression and variability of progression of each endpoint for a target population.
To create a single simulated clinical trial dataset of participant-level endpoint values over time we used the following approach for subject i at visits for endpoints
Simulate CDR®+NACC-FTLD global score at baseline given the mutation of the subject and distribution specified in Supplemental Table S9 (informed by natural history data).
Simulate the disease age at baseline given the CDR®+NACC-FTLD global score and the mutation type from the distribution specified in Supplemental Table S9 (informed by natural history data).
Simulate a subject-level random effect at normal for each endpoint k by first simulating the overall subject-level standard units from normal for each group of endpoints,
and then simulating the subject and endpoint-specific effect using sampled subject-level standard units from above for each group, , and posterior estimates from the DPM
Simulate observed value of endpoint , at visit , from a normal distribution with a subject and endpoint-specific mean and endpoint-specific variance based on the posterior mean results DPM, the subject-level DA at each visit, , and the subject-level random effect at normal, simulated above:
Subject may additionally be accepted / rejected on enrollment into the simulated clinical trial based on inclusion/exclusion criteria for CDR®+NACC FTLD-global score, Disease Age at baseline, and/or NfL at baseline.
The expected change from baseline (mean and SD) over different timepoints for each endpoint for a placebo participant given a set of enrollment criteria are calculated using the above simulation strategy across 10,000 simulated datasets. The expected mean and SD of the change from baseline for a placebo participant is then used to calculate the sample size needed (N) to achieve 80% power for a 50% slowing in progression assuming 10% attrition rate per year and 1:1 randomization.
Enrollment criteria was defined based on baseline values of CDR®+NACC-FTLD Global, log(NfL), and estimated Disease Age. Presymptomatic trial designs consider only participants with a baseline CDR®+NACC-FTLD Global = 0 and explored inclusion criteria to define a subpopulation at heightened risk for symptom onset based on elevated NfL (log(NfL) > 3.0) or an estimated disease age within 5 years or 2.5 years of onset. The hypothesis was that enrolling those presymptomatic cases close to onset would reduce the sample size needed to detect an effect by increasing the likelihood that the participants change on the endpoints during the trial period. Early symptomatic trial designs (CDR®+NACC-FTLD = 0, 0.5, and 1) included all participants with a baseline Global score = 1. These simulations explored additional inclusion criteria for presymptomatic participants (Global score of 0 or 0.5) to define a high risk subpopulation based on NfL or an estimated Disease Age cutoff (-2.5 or 0).
Extended Data
Extended Data Fig. 1. Baseline comparisons between mutation carriers and controls by disease age epoch.
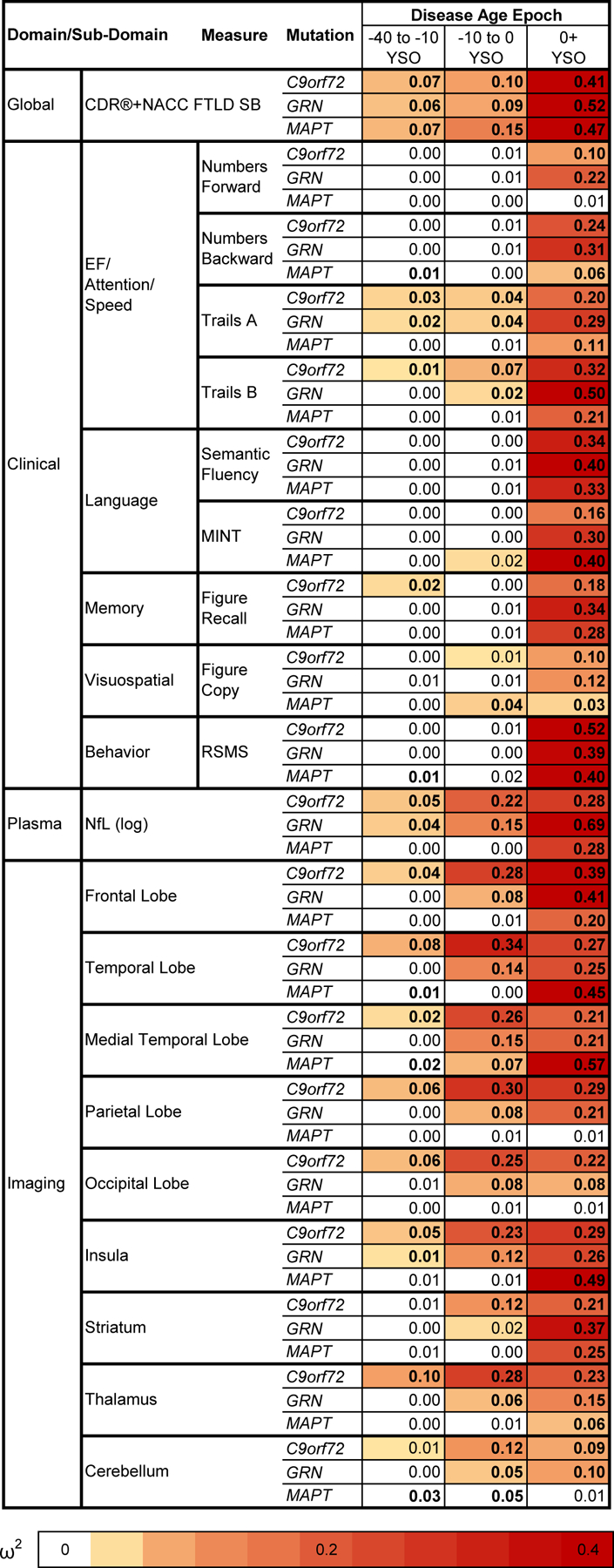
Cross-sectional baseline differences between mutation carriers and controls are presented as effect sizes (omega squared). Bolded cells indicate statistical significance (p < .05) using two-sided tests without multiple comparison correction. Comparisons in which mutation carriers are more impaired than controls are colored, with darker shades illustrating larger effect sizes. Note that statistical comparisons for the CDR®+NACC FTLD SB should be interpreted with caution given that controls were defined as having a baseline CDR®+NACC-FTLD=0 and thus have no variance due to this selection process.
Abbreviations: EF: Executive Functioning; NfL: Plasma neurofilament light chain levels; RSMS: Revised Self Monitoring Scale. CDR®+NACC FTLD SB: Clinical Dementia Rating Scale plus National Alzheimer’s Coordinating Center’s Frontotemporal Lobar Degeneration Module Sum of Boxes
Extended Data Fig. 2. Voxelwise atrophy by estimated disease stage in familial frontotemporal dementia.
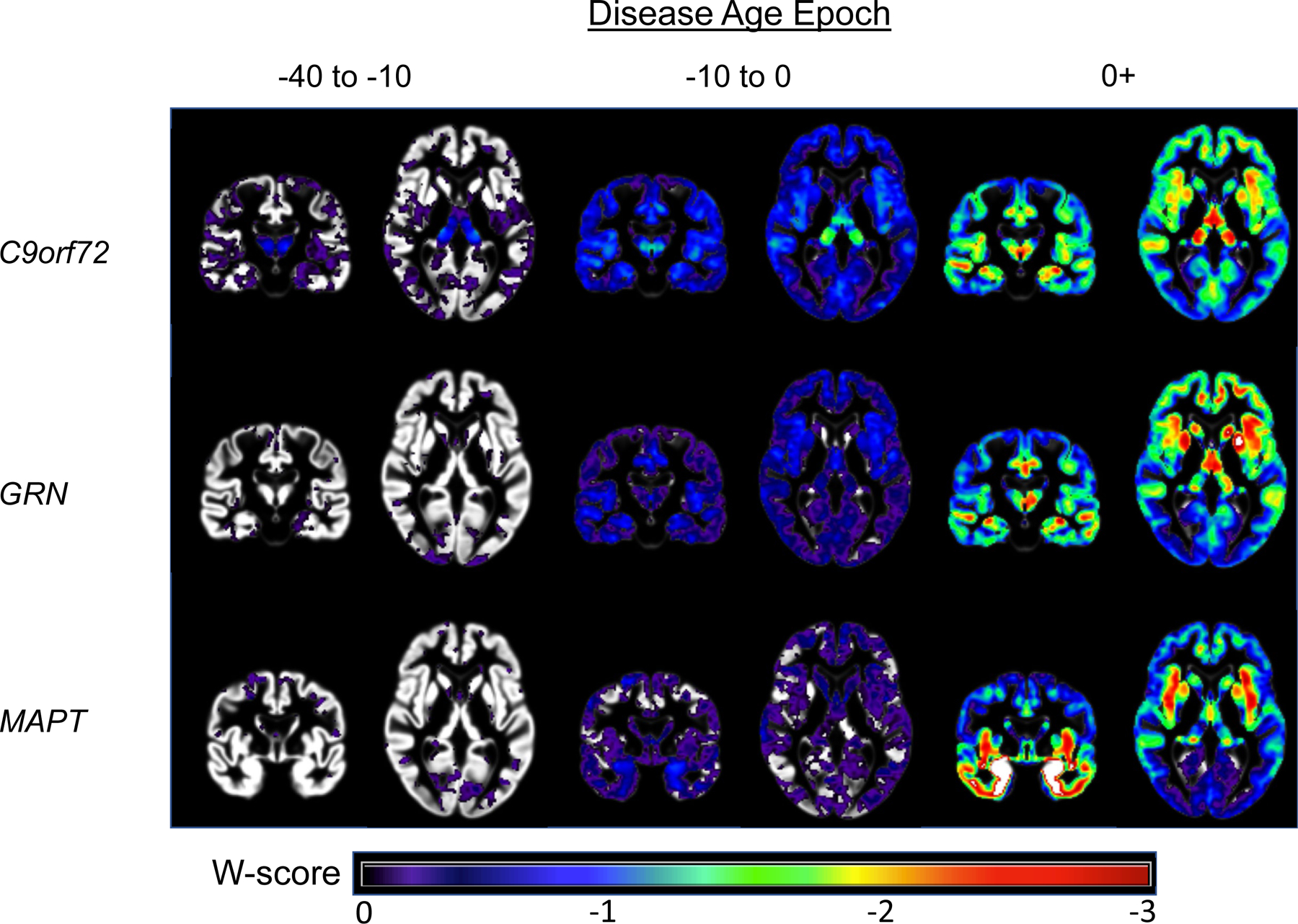
Voxelwise maps display brain atrophy as the number of standardized units from controls (W-scores), controlling for head size and scanner. Images are shown in radiological orientation (i.e., right is left). Voxelwise results are presented with a greater number of axial slices in Figures S2-S4. Results were generally consistent with the region of interest findings, supporting the validity of the DPM approach. In C9orf72, thalamic atrophy, particularly in the pulvinar, was the primary region of atrophy in the −40 to −10 epoch and continued to be a region of prominent atrophy throughout the disease course. Medial temporal lobe volume loss became prominent in the −10 to −0 epoch. Frontoinsular, medial parietal, and medial temporal atrophy became prominent in the symptomatic phase (see also Figure S2). In GRN, subtle early cerebellar atrophy was observed (−40 to −10), along with atrophy in frontotemporal, subcortical, and insular structures in the −10 to 0 epoch. In the symptomatic stage, atrophy extended into the temporal lobe, frontoparietal regions, and striatum (see also Figure S3). Atrophy in MAPT appeared to begin in the medial temporal lobe and temporal pole (−10 to 0), and symptomatic mutation carriers showed temporal, insular, ventral and medial frontal, and striatal atrophy (see also Figure S4).
Extended Data Fig. 3. Patient-level data contributing to the disease progression models.
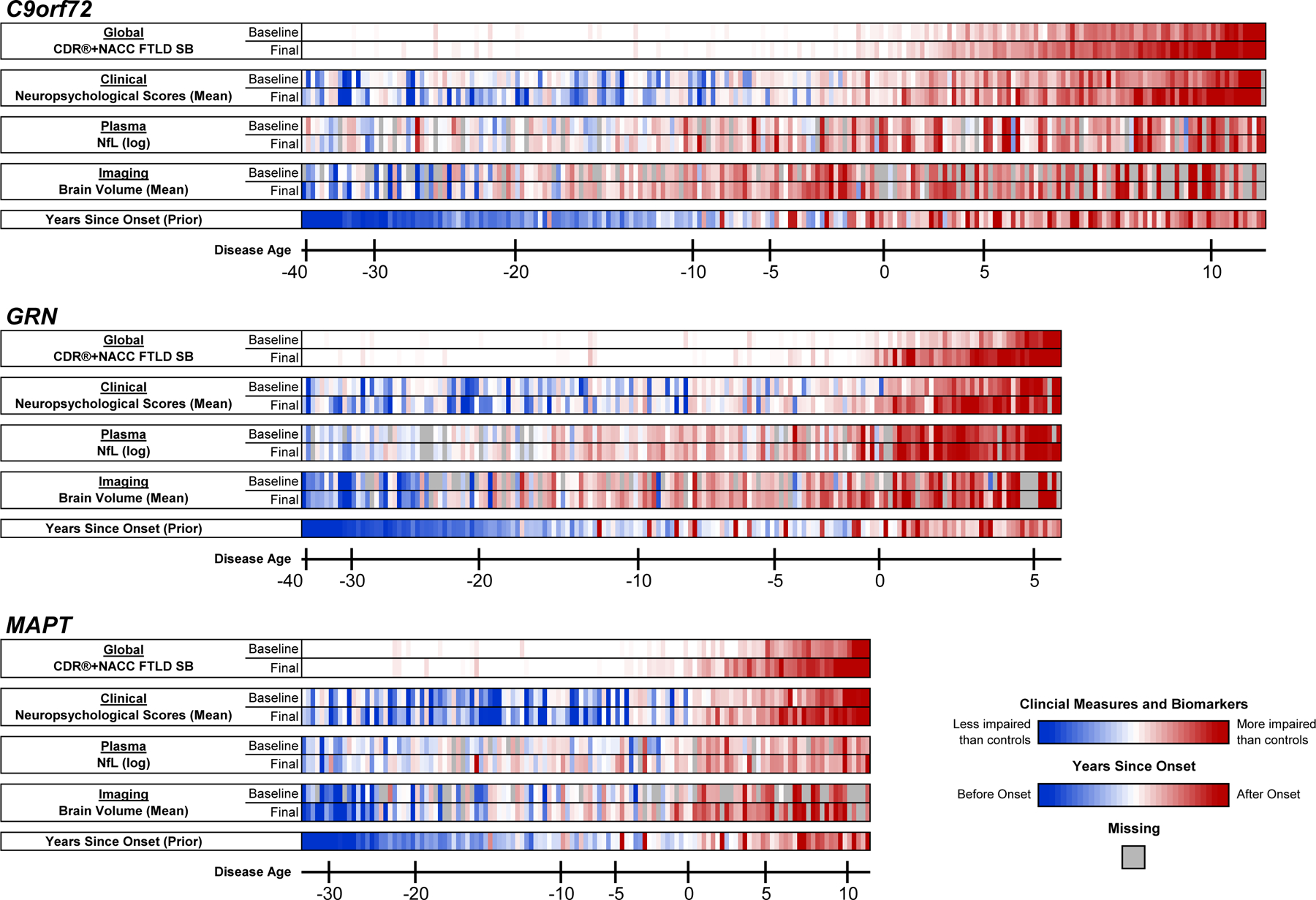
For each genetic group, each mutation carrier with longitudinal data is displayed in a single column, organized on the x-axis by their model estimated Disease Age at baseline. Participants’ baseline and last available (Final) observation for each outcome are presented. For the CDR®+NACC-FTLD-SB, white cells indicate a score of 0, and increasingly dark red tones denote higher scores (i.e., more severe impairments or greater atrophy). Log transformed plasma NfL concentrations and the mean of all available neuropsychological scores and regional gray matter volume estimates are also presented, with the color scale indicating their scores relative to controls of the same Disease Age. Lastly, the model’s prior estimate of Years Since Onset is displayed. For participants with documented onset, we display the difference between their chronological age and the clinician estimated age of onset. For those participants in whom clinical onset has not yet occurred (or this data was unavailable), we display the difference between their chronological age and the mean age of onset for their mutation.
Supplementary Material
Acknowledgements
Data collection and dissemination of the data presented in this manuscript was supported by the ALLFTD Consortium (U19: AG063911, funded by the National Institute on Aging and the National Institute of Neurological Diseases and Stroke) and the former ARTFL & LEFFTDS Consortia (ARTFL: U54 NS092089, funded by the National Institute of Neurological Diseases and Stroke and National Center for Advancing Translational Sciences; LEFFTDS: U01 AG045390, funded by the National Institute on Aging and the National Institute of Neurological Diseases and Stroke). The manuscript has been reviewed by the ALLFTD Executive Committee for scientific content. The authors acknowledge the invaluable contributions of the study participants and families as well as the assistance of the support staffs at each of the participating sites. This work is also supported by the Association for Frontotemporal Degeneration (including the FTD Biomarkers Initiative), the Bluefield Project to Cure FTD, Larry L. Hillblom Foundation [2018-A-025-FEL (AMS)], the National Institutes of Health [AG038791 (ALB), AG032306 (HJR), AG016976 (WAK), AG062677 (RCP), AG019724 (BLM), AG058233 (SEL), AG072122 (WAK), P30 AG062422 (BLM), K12 HD001459 (NG), K23AG061253 (AMS), AG062422 (RCP), K24AG045333 (HJR)], and the Rainwater Charitable Foundation. Samples from the National Centralized Repository for Alzheimer Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant [U24 AG021886 (TF)] awarded by the National Institute on Aging (NIA), were used in this study.
This work was also supported by the Medical Research Council UK GENFI grant [MR/M023664/1 (JDR)], the Bluefield Project, the National Institute for Health Research including awards to Cambridge and UCL Biomedical Research Centres, and the JPND GENFI-PROX grant (2019–02248). Several authors of this publication are members of the European Reference Network for Rare Neurologic Diseases, project No. 739510. JDR and LLR are also supported by the NIHR UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. JDR is also supported by the Miriam Marks Brain Research UK Senior Fellowship and has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). MB is supported by a Fellowship award from the Alzheimer’s Society, UK (AS-JF-19a-004–517). RC/CG are supported by a Frontotemporal Dementia Research Studentships in Memory of David Blechner funded through The National Brain Appeal (RCN 290173). J.B.R. is supported by NIHR Cambridge Biomedical Research Centre (BRC-1215–20014; The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care); The Wellcome Trust (220258) The Cambridge Centre for Parkinson-plus; The Medical Research Council (SUAG/092 G116768); ILB is supported by ANR-PRTS PREV-DemAls, PHRC PREDICT-PGRN and several authors of this publication are members of the European Reference Network for Rare Neurological Diseases - Project ID No 739510. JL is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198). RS-V was funded at the Hospital Clinic de Barcelona by Instituto de Salud Carlos III, Spain (grant code PI20/00448 to RSV) and Fundació Marató TV3, Spain (grant code 20143810 to RSV). MLS has been supported by the German Federal Ministry of Education and Research (BMBF) by a grant given to the German FTLD Consortium (FKZ O1GI1007A), and by the German Research Foundation DFG (SCHR 774/5–1). MM was, in part, funded by the UK Medical Research Council, the Italian Ministry of Health and the Canadian Institutes of Health Research as part of a Centres of Excellence in Neurodegeneration grant, and also Canadian Institutes of Health Research operating grants (Grant #s: MOP- 371851 and PJT-175242) and funding from the Weston Brain Institute to Mario Masellis. R.L. is sup-ported by the Canadian Institutes of Health Research and the Chaire de Recherche sur les Aphasies Primaires Progressives Fondation Famille Lemaire. C.G. is supported by the Swedish Frontotemporal Dementia Initiative Schörling Foundation, Swedish Research Council, JPND Prefrontals, 2015–02926,2018–02754, Swedish Alzheimer Foundation, Swedish Brain Foundation, Karolinska Institutet Doctoral Funding, KI Strat-Neuro, Swedish Dementia Foundation, and Stockholm County Council ALF/Region Stockholm. J.L. is supported by Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (German Research Foundation, EXC 2145 Synergy 390857198). The Dementia Research Centre is supported by Alzheimer’s Research UK, Alzheimer’s Society, Brain Research UK, and The Wolfson Foundation. This work was supported by the National Institute for Health Research UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK.
Frontotemporal Prevention Initiative (FPI) Investigators
ALLFTD Investigators
Liana Apostolova,70 Brian Appleby MD,7 Sami Barmada,71 Bradley Boeve MD,5 Yvette Bordelon MD PhD,8 Hugo Botha MD,5 Adam L. Boxer MD PhD,1 Andrea Bozoki,19 Danielle Brushaber BS,6 David Clark,70 Giovanni Coppola,8 Ryan Darby,72 Bradford C. Dickerson MD,9 Dennis Dickson,4 Kimiko Domoto-Reilly MD,10 Kelley Faber,12 Anne Fagan,14 Julie A. Fields PhD,11 Tatiana Foroud PhD,12 Leah Forsberg PhD,5 Daniel Geschwind MD PhD,8,13 Nupur Ghoshal MD PhD,14 Jill Goldman MS MPhil,15 Douglas R. Galasko,20 Ralitza Gavrilova MD,5 Tania F. Gendron PhD,4 Jonathon Graff-Radford MD,5 Neill Graff-Radford MD,16 Ian M. Grant,27 Murray Grossman MD EdD,17 Matthew GH Hall MS,1 Eric Huang,1 Hilary W. Heuer PhD,1 Ging-Yuek Hsiung MD MHSc,18 Edward D. Huey MD,15 David Irwin MD,17 Kejal Kantarci MD,5 Daniel Kaufer MD,19 Diana Kerwin,73 David Knopman MD,5John Kornak PhD,69 Joel Kramer PsyD,1 Walter Kremers PhD,6 Maria Lapid,5 Argentina Lario Lago PhD,1 Suzee Lee,1 Gabriel Leger,20 Peter Ljubenkov MD,1 Irene Litvan MD,20 Diane Lucente MS,9 Ian R. Mackenzie MD,21 Joseph C. Masdeux,74 Scott McGinnis,9 Mario Mendez,8 Carly Mester BA,6 Bruce L. Miller MD,1 Chiadi Onyike MD,22 M. Belen Pascual,74 Leonard Petrucelli PhD,4 Peter Pressman,75 Rosa Rademakers PhD,4 Vijay Ramanan MD PhD,5 Eliana Marisa Ramos PhD,8 Meghana Rao MPH,5 Katya Rascovsky PhD,17 Katherine P. Rankin PhD,1 Aaron Ritter,76 Julio C. Rojas MD PhD,1 Howard J. Rosen MD,1 Rodolfo Savica MD PhD,5 William W. Seeley,1 Jeremy Syrjanen,5 Adam M. Staffaroni PhD,1 M. Carmela Tartaglia MD,26 Jack C. Taylor,1 Lawren VandeVrede MD PhD,1 Sandra Weintraub PhD,24 Bonnie Wong PhD 9
1 University of California, San Francisco, Weill Institute for Neurosciences, Department of Neurology, Memory and Aging Center, San Francisco, CA
4 Mayo Clinic, Department of Neuroscience, Jacksonville, FL, USA
5 Mayo Clinic, Department of Neurology, Rochester, MN, USA
6 Mayo Clinic, Department of Quantitative Health Sciences, Rochester, MN, USA
7 Case Western Reserve University, Department of Neurology, Cleveland, OH, USA
8 University of California, Los Angeles, Department of Neurology, Los Angeles, CA, USA
9 Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston MA
10 University of Washington, Department of Neurology, Seattle, WA, USA
11 Mayo Clinic, Department of Psychiatry and Psychology, Rochester, MN, USA
12 Indiana University School of Medicine, National Centralized Repository for Alzheimer’s, IN, USA
13 Institute for Precision Health, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA
15 Columbia University, Department of Neurology, New York, NY, USA
16 Mayo Clinic, Department of Neurology, Jacksonville, FL, USA
17 University of Pennsylvania, Department of Neurology, Philadelphia, PA, USA
18 University of British Columbia, Division of Neurology, Vancouver, BC, CA
19 University of North Carolina, Department of Neurology, Chapel Hill, NC, USA
20 University of California, San Diego, Department of Neurosciences, La Jolla, CA, USA
21 Department of Pathology, University of British Columbia. Vancouver, BC, CA
22 Johns Hopkins University, Department of Psychiatry and Behavioral Sciences, Baltimore, MD, USA
25 University of Alabama at Birmingham, Department of Neurology, Birmingham, AL
26 Tanz Centre for Research in Neurodegenerative Diseases, Division of Neurology, University of Toronto, Toronto, Ontario, Canada
27 Northwestern University, Department of Neurology, Chicago, IL, USA
69 University of California, San Francisco, Department of Epidemiology and Biostatistics, San Francisco, CA
70 Indiana University, Department of Neurology, Indianapolis, IN, USA
71 University of Michigan, Department of Neurology, Ann Arbor, MI, USA
72 Vanderbilt University, Department of Neurology, Nashville, TN, USA
73 UT Southwestern, Department of Neurology, Dallas, TX, USA
74 Houston Methodist, Department of Neurology, Houston, TX, USA
75 University of Colorado, Department of Neurology, Aurora, CO, USA
76 Lou Ruvo Center for Brain Health, Cleveland Clinic Las Vegas, Las Vegas, NV, USA
GENFI Investigators
Aitana Sogorb Esteve,3,77 Annabel Nelson,3 Arabella Bouzigues,3 Carolin Heller,3 Caroline V Greaves,3 David Cash,3 David L Thomas,78 Emily Todd,3 Hanya Benotmane,77 Henrik Zetterberg, 77,79 Imogen J Swift,3 Jennifer Nicholas,80 Kiran Samra,3 Martina Bocchetta,3 Rachelle Shafei,3 Rhian S Convery,3 Carolyn Timberlake,28 Thomas Cope,28 Timothy Rittman,28 Alberto Benussi,29 Enrico Premi,81 Roberto Gasparotti,82 Silvana Archetti,83 Stefano Gazzina,84 Valentina Cantoni,29 Andrea Arighi,31 Chiara Fenoglio,31 Elio Scarpini,31 Giorgio Fumagalli,31 Vittoria Borracci,32 Giacomina Rossi,32 Giorgio Giaccone,32 Giuseppe Di Fede,32 Paola Caroppo,32 Pietro Tiraboschi,32 Sara Prioni,32 Veronica Redaelli,32 David Tang-Wai,85 Ekaterina Rogaeva,26 Miguel Castelo-Branco,86 Morris Freedman,87 Ron Keren,88 Sandra Black,33 Sara Mitchell,33 Christen Shoesmith,34 Robart Bartha,89,90 Rosa Rademakers,23 Jackie Poos,35 Janne M. Papma,35 Lucia Giannini,35 Rick van Minkelen,35 Yolande Pijnenburg,91 Benedetta Nacmias,92 Camilla Ferrari,92 Cristina Polito,93 Gemma Lombardi,92 Valentina Bessi,92 Michele Veldsman,38 Christin Andersson,94 Hakan Thonberg,40 Linn Öijerstedt,40 Vesna Jelic,95 Paul Thompson,42 Tobias Langheinrich,42 Albert Lladó,46 Anna Antonell,46 Jaume Olives,46 Mircea Balasa,46 Nuria Bargalló,96 Sergi Borrego-Ecija,46 Ana Verdelho,97 Carolina Maruta,98 Catarina B. Ferreira,99 Gabriel Miltenberger,47 Frederico Simões do Couto,100 Alazne Gabilondo,48 Ana Gorostidi,49 Jorge Villanua,99 Marta Cañada,102 Mikel Tainta,49 Miren Zulaica,49 Myriam Barandiaran,48,49 Patricia Alves,48,49 Benjamin Bender,103 Carlo Wilke,51 Lisa Graf,50 Annick Vogels,104 Mathieu Vandenbulcke,105 Philip Van Damme,53 Rose Bruffaerts,24 Koen Poesen,106 Pedro Rosa-Neto,107 Serge Gauthier,108 Agnès Camuzat,57 Alexis Brice,57 Anne Bertrand,57 Aurélie Funkiewiez,109 Daisy Rinaldi,109 Dario Saracino,57 Olivier Colliot,57 Sabrina Sayah,57 Catharina Prix,60 Elisabeth Wlasich,60 Olivia Wagemann,60 Sandra Loosli,60 Sonja Schönecker,60 Tobias Hoegen,60 Jolina Lombardi,63 Sarah Anderl-Straub,63 Adeline Rollin,66 Gregory Kuchcinski,64,66 Maxime Bertoux,66 Thibaud Lebouvier,64,66 Vincent Deramecourt,64,66 Beatriz Santiago,110 Diana Duro,111 Maria João Leitão,68 Maria Rosario Almeida,67 Miguel Tábuas-Pereira,110 Sónia Afonso, 112 Annerose Engel,113 Maryna Polyakova,113,114
3 Dementia Research Centre, Department of Neurodegenerative Disease, UCL Queen Square London
23 Applied and Translational Neurogenomics Group, VIB Center for Molecular Neurology, VIB, Antwerp, Belgium
24 Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium
26 Tanz Centre for Research in Neurodegenerative Diseases, Division of Neurology, University of Toronto, Toronto, Ontario, Canada
28 Department of Clinical Neurosciences and Cambridge University Hospitals NHS Trust and Medical Research Council Cognition and Brain Sciences Unit, University of Cambridge, Cambridge, UK
29 Centre for Neurodegenerative Disorders, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
31 Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy
32 Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy
33 Division of Neurology, Department of Medicine, Sunnybrook Health Sciences Centre; Hurvitz Brain Sciences Program, Sunnybrook Research Institute; University of Toronto, Toronto, Canada
34 Department of Clinical Neurological Sciences, University of Western Ontario, London, Ontario Canada
35 Department of Neurology, Erasmus Medical Centre, Rotterdam, Netherlands
38 Nuffield Department of Clinical Neurosciences, Medical Sciences Division, University of Oxford, Oxford, UK
40 Center for Alzheimer Research, Division of Neurogeriatrics, Department of Neurobiology, Care Sciences and Society, Bioclinicum, Karolinska Institutet, Solna, Sweden
42 Division of Neuroscience and Experimental Psychology, Wolfson Molecular Imaging Centre, University of Manchester, Manchester, UK.
46 Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Institut d’Investigacións Biomèdiques August Pi I Sunyer, University of Barcelona, Barcelona, Spain
47 Faculty of Medicine, University of Lisbon, Lisbon, Portugal
48 Cognitive Disorders Unit, Department of Neurology, Donostia University Hospital, San Sebastian, Gipuzkoa, Spain
49 Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Gipuzkoa, Spain
50 Department of Neurodegenerative Diseases, Hertie-Institute for Clinical Brain Research and Center of Neurology, University of Tübingen, Tübingen, Germany
51 Center for Neurodegenerative Diseases (DZNE), Tübingen, Germany
53 Neurology Service, University Hospitals Leuven, Belgium
57 Sorbonne Université, Paris Brain Institute – Institut du Cerveau – ICM, Inserm U1127, CNRS UMR 7225, AP-HP - Hôpital Pitié-Salpêtrière, Paris, France
60 Neurologische Klinik und Poliklinik, Ludwig-Maximilians-Universität, Munich, Germany
63 Department of Neurology, University of Ulm, Ulm, Germany
64 Univ Lille, France
66 CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France
67 University Hospital of Coimbra (HUC), Neurology Service, Faculty of Medicine, University of Coimbra, Portugal
68 Center for Neuroscience and Cell Biology, Faculty of Medicine, University of Coimbra, Coimbra, Portugal
77 UK Dementia Research Institute at University College London, UCL Queen Square Institute of Neurology, London, UK
78 Neuroimaging Analysis Centre, Department of Brain Repair and Rehabilitation, UCL Institute of Neurology, Queen Square, London, UK
79 Department of Psychiatry and Neurochemistry, the Sahlgrenska Academy at the University of Gothenburg, Mölndal, Sweden
80 Department of Medical Statistics, London School of Hygiene and Tropical Medicine, London, UK
81 Stroke Unit, ASST Brescia Hospital, Brescia, Italy
82 Neuroradiology Unit, University of Brescia, Brescia, Italy
83 Biotechnology Laboratory, Department of Diagnostics, ASST Brescia Hospital, Brescia, Italy
84 Neurology, ASST Brescia Hospital, Brescia, Italy
85 The University Health Network, Krembil Research Institute, Toronto, Canada
86 Faculty of Medicine, University of Coimbra, Coimbra, Portugal
87 Baycrest Health Sciences, Rotman Research Institute, University of Toronto, Toronto, Canada
88 The University Health Network, Toronto Rehabilitation Institute, Toronto, Canada
89 Department of Medical Biophysics, The University of Western Ontario, London, Ontario, Canada
90 Centre for Functional and Metabolic Mapping, Robarts Research Institute, The University of Western Ontario, London, Ontario, Canada
91 Amsterdam University Medical Centre, Amsterdam VUmc, Amsterdam, Netherlands
92 Department of Neuroscience, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy
93 Department of Biomedical, Experimental and Clinical Sciences “Mario Serio”, Nuclear Medicine Unit, University of Florence, Florence, Italy
94 Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden
95 Division of Clinical Geriatrics, Karolinska Institutet, Stockholm, Sweden
96 Imaging Diagnostic Center, Hospital Clínic, Barcelona, Spain
97 Department of Neurosciences and Mental Health, Centro Hospitalar Lisboa Norte - Hospital de Santa Maria & Faculty of Medicine, University of Lisbon, Lisbon, Portugal
98 Laboratory of Language Research, Centro de Estudos Egas Moniz, Faculty of Medicine, University of Lisbon, Lisbon, Portugal
99 Laboratory of Neurosciences, Faculty of Medicine, University of Lisbon, Lisbon, Portugal
100 Faculdade de Medicina, Universidade Católica Portuguesa
101 OSATEK, University of Donostia, San Sebastian, Gipuzkoa, Spain
102 CITA Alzheimer, San Sebastian, Gipuzkoa, Spain
103 Department of Diagnostic and Interventional Neuroradiology, University of Tübingen, Tübingen, Germany
104 Department of Human Genetics, KU Leuven, Leuven, Belgium
105 Geriatric Psychiatry Service, University Hospitals Leuven, Belgium; Neuropsychiatry, Department of Neurosciences, KU Leuven, Leuven, Belgium
106 Laboratory for Molecular Neurobiomarker Research, KU Leuven, Leuven, Belgium
107 Translational Neuroimaging Laboratory, McGill Centre for Studies in Aging, McGill University, Montreal, Québec, Canada
108 Alzheimer Disease Research Unit, McGill Centre for Studies in Aging, Department of Neurology & Neurosurgery, McGill University, Montreal, Québec, Canada
109 Centre de référence des démences rares ou précoces, IM2A, Département de Neurologie, AP-HP - Hôpital Pitié-Salpêtrière, Paris, France; Sorbonne Université, Paris Brain Institute – Institut du Cerveau – ICM, Inserm U1127, CNRS UMR 7225, AP-HP - Hôpital Pitié-Salpêtrière, Paris, France
110 Neurology Department, Centro Hospitalar e Universitario de Coimbra, Coimbra, Portugal
111 Faculty of Medicine, University of Coimbra, Coimbra, Portugal
112 Instituto Ciencias Nucleares Aplicadas a Saude, Universidade de Coimbra, Coimbra, Portugal
113 Clinic for Cognitive Neurology, University Hospital Leipzig, Leipzig, Germany
114 Department for Neurology, Max Planck Institute for Human Cognitive and Brain Sciences, University Hospital Leipzig, Leipzig, Germany
Competing Interest Statement
Appleby B - receives research support from CDC, NIH, Ionis, Alector, and the CJD Foundation. He has provided consultation to Acadia, Ionis, and Sangamo.
Boccheta M – nothing to disclose
Boeve B – has served as an investigator for clinical trials sponsored by EIP Pharma, Alector and Biogen. He receives royalties from the publication of a book entitled Behavioral Neurology of Dementia (Cambridge Medicine, 2009, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from NIH, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program and the Little Family Foundation.
Bordelon Y – nothing to disclose
Botha H – receives research support from NIH.
Boxer A – receives research support from NIH, the Tau Research Consortium, the Association for Frontotemporal Degeneration, Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, the Alzheimer’s Drug Discovery Foundation and the Alzheimer’s Association. He has served as a consultant for Aeovian, AGTC, Alector, Arkuda, Arvinas, Boehringer Ingelheim, Denali, GSK, Life Edit, Humana, Oligomerix, Oscotec, Roche, TrueBinding and Wave, and received research support from Biogen, Eisai and Regeneron.
Brushaber D – nothing to disclose
Cobigo Y – Nothing to disclose
Dickerson B – Dr. Dickerson is a consultant for Acadia, Alector, Arkuda, Biogen, Denali, Eisai, Genentech, Lilly, Merck, Novartis, Takeda, Wave Lifesciences. Dr. Dickerson receives royalties from Cambridge University Press, Elsevier, Oxford University Press. Dr. Dickerson receives grant funding from the NIA, NINDS, NIMH, and the Bluefield Foundation.
Domoto-Reilly K – receives research support from NIH, and serves as an investigator for a clinical trial sponsored by Lawson Health Research Institute.
Ducharme S - has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by the following companies: Biogen, Ionis Pharmaceuticals, Wave Life Sciences, and Janssen. He has received speaking honorarium/advisory fees from the following companies: Eisai, Biogen, Innodem Neurosciences, NeuroCatch. Dr. Ducharme receives salary support from the Fond de Recherche du Québec – Santé.
Faber K – receives research support from NIH
Fields J – receives research support from NIH
Foroud T – receives research support from NIH
Forsberg L – receives research support from NIH.
Gavrilova R – receives research support from NIH
Gendron T – receives research support from NIH
Ghoshal N - has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by the following companies: Bristol Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen Immunotherapy, Novartis, Pfizer, Wyeth, SNIFF (The Study of Nasal Insulin to Fight Forgetfulness) study, and A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial. She receives research support from Tau Consortium and Association for Frontotemporal Dementia and is funded by the NIH.
Goh SYM – Nothing to disclose
Goldman, JS – is serving as a consultant to the Novartis Alzheimer’s Prevention Advisory Board. She receives research support from NIH, HDSA, New York State Department of Health (RFA # 1510130358)
Graff-Radford J – receives research support from the NIH and is on the editorial board of Neurology.
Graff-Radford N – receives royalties from UpToDate, has participated in multicenter therapy studies by sponsored by Biogen, TauRx, AbbVie, Novartis and Lilly. He receives research support from NIH.
Grossman M - receives grant support from NIH, Avid and Piramal; participates in clinical trials sponsored by Biogen, TauRx, and Alector; serves as a consultant to Bracco and UCB; and serves on the Editorial Board of Neurology.
Hall MGH – nothing to disclose
Heuer H – nothing to disclose
Hsiung G-Y – has served as an investigator for clinical trials sponsored by AstraZeneca, Eli Lilly, and Roche / Genentech. He receives research support from Canadian Institutes of Health Research and the Alzheimer Society of British Columbia.
Huey E – receives research support from NIH
Irwin D – receives support from NIH, Brightfocus Foundation and Penn Institute on Aging.
Jones D- – receives research support from NIH and the Minnesota Partnership for Biotechnology and Medical Genomics
Kantarci K - served on the Data Safety Monitoring Board for Takeda Global Research & Development Center, Inc.; data monitoring boards of Pfizer and Janssen Alzheimer Immunotherapy; research support from the Avid Radiopharmaceuticals, Eli Lilly, the Alzheimer’s Drug Discovery Foundation and NIH
Kerwin, D has served on an Advisory Board for AbbVie and as site PI for studies funded by Roche/Genentech, AbbVie, Avid, Novartis, Eisai, Eli Lilly and UCSF.
Knopman D - serves on the DSMB of the DIAN-TU study, is a site PI for clinical trials sponsored by Biogen, Lilly and the University of Southern California, and is funded by NIH.
Kornak J - – has provided expert witness testimony for Teva Pharmaceuticals in Forest Laboratories Inc. et al. v. Teva Pharmaceuticals USA, Inc., Case Nos. 1:14-cv-00121 and 1:14-cv-00686 (D. Del. filed Jan. 31, 2014 and May 30, 2014) regarding the drug Memantine; for Apotex/HEC/Ezra in Novartis AG et al. v. Apotex Inc., No. 1:15-cv-975 (D. Del. filed Oct. 26, 2015, regarding the drug Fingolimod. He has also given testimony on behalf of Puma Biotechnology in Hsingching Hsu et al, vs. Puma Biotechnology, INC., et al. 2018 regarding the drug Neratinib. He receives research support from the NIH.
Kramer J – receives royalties from Pearson Inc.
Kremers W - receives research funding from AstraZeneca, Biogen, Roche, DOD and NIH.
Lapid M – receives research support from the NIH.
Levin J - reports speaker fees from Bayer Vital, Biogen and Roche, consulting fees from Axon Neuroscience and Biogen, author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers, non-financial support from Abbvie and compensation for duty as part-time CMO from MODAG, outside the submitted work.
Litvan I– receives research support from the National Institutes of Health grants: 2R01AG038791–06A, U01NS100610, U01NS80818, R25NS098999; U19 AG063911–1 and 1R21NS114764–01A1; the Michael J Fox Foundation, Parkinson Foundation, Lewy Body Association, CurePSP, Roche, Abbvie, Biogen, Centogene. EIP-Pharma, Biohaven Pharmaceuticals, Novartis, Brain Neurotherapy Bio and United Biopharma SRL - UCB. She was a member of the Scientific Advisory Board of Lundbeck and is a Scientific advisor for Amydis and Rossy Center for Progressive Supranuclear Palsy University of Toronto. She receives her salary from the University of California San Diego and as Chief Editor of Frontiers in Neurology.
Ljubenkov P - is a site primary investigator for clinical trials by Alector, AbbVie, and Woolsey. He serves as an advisor for Retrotrope. He receives research and salary support from the NIH-NIA and the Alzheimer’s Association- Part the Cloud partnership
Lucente D - receives research support from NIH
Mackenzie I – receives research funding from Canadian Institutes of Health Research, Alzheimer’s Association US, NIH, Weston Brain Institute.
Masellis - MM reports grant funding from the Canadian Institutes of Health Research relating to this work. MM reports grants from the Canadian Institutes of Health Research, Woman’s Brain Health Initiative, Brain Canada, Ontario Brain Institute, Weston Brain Institute and Washington University, outside of this submitted work. MM has received personal fees for serving on a Scientific Advisory Committee for Ionis Pharmaceuticals, Alector Pharmaceuticals, Wave Life Sciences, and Biogen Canada, outside of this submitted work. MM has received royalties from Henry Stewart Talks, outside of this submitted work. MM is a clinical trial site investigator for Roche, and Alector Pharmaceuticals, outside of this submitted work.
McGinnis S – has served as an investigator for clinical trials sponsored by AbbVie, Allon Therapeutics, Biogen, Bristol-Myers Squibb, C2N Diagnostics, Eisai Inc., Eli Lilly and Co., Genentech, Janssen Pharmaceuticals, Medivation, Merck, Navidea Biopharmaceuticals, Novartis, Pfizer, and TauRx Therapeutics. He receives research support from NIH.
Mendez M - receives research support from NIH.
Miller B – Dr. Miller is Director and Internal Advisor of The Bluefield Project to Cure FTD, Co-Director and Scientific Advisor Tau Consortium, Co-Director of the Global Brain Health Institute (GBHI) – Co-Director, Medical Advisor for The John Douglas French Foundation, Scientific advisor for The Larry L. Hillblom Foundation, Scientific Advisor for Association for Frontotemporal Degeneration, Scientific Advisor for National Institute for Health Research Cambridge Biomedical Research Centre and its subunit, the Biomedical Research Unit in Dementia, External Advisor to University of Washington ADRC, Stanford University ADRC, Arizona Alzheimer’s Disease Center (ADC). Massachusetts Alzheimer Disease Research Center, and scientific advisor to The Buck Institute for Research on Aging. Serves as Editor-in-Chief for Neurocase, on the editorial board of ALS/FTD Journal (Taylor & Francis), Section Editor for Frontiers, and editor for PLOS Medicine. He receives royalties from Cambridge University Press, Guilford Publications, Inc., Johns Hopkins Press, Oxford University Press, Taylor & Francis Group, Elsevier, Inc., and Up-to-Date.
Ong E – reports no disclosures.
Onyike CU - receives research funding from the NIH, Lawton Health Research Institute, National Ataxia Foundation, Alector Inc., and Transposon, Inc. He is also supported by the Robert and Nancy Hall Brain Research Fund, the Jane Tanger Black Fund for Young-Onset Dementias, and the gift from Joseph Trovato. He is a consultant with Alector, Inc. and Acadia Pharmaceuticals.
Petrucelli L – receives research support from NIH.
Quintana M – is an employee of Berry Consultants, LLC where she serves as a consultant to numerous pharmaceutical and device companies
Rademakers R – receives research funding from NIH and the Bluefield Project to Cure Frontotemporal Dementia. RR is on the Scientific Advisory Board of Arkuda Therapeutics and receives royalties from progranulin-related patent. She is also on the Scientific Advisory Board of the Fondation Alzheimer.
Ramanan V – reports no disclosures.
Ramos E – receives research support from NIH.
Rankin K – receives research support from NIH and NSF, and serves on a Medical Advisory Board for Eli Lilly.
Rascovsky K – receives research support from NIH.
Roberson ED – receives research support from NIH, Bluefield Project to Cure Frontotemporal Dementia, Alzheimer’s Association, BrightFocus Foundation, Biogen, and Alector, has served as a consultant for AGTC and on a DSMB for Lilly, and owns intellectual property related to tau.
Rohrer JD - has served on a Medical Advisory Board and had a consultancy agreement with Alector, Arkuda Therapeutics, Wave Life Sciences, and Prevail Therapeutics, and had a consultancy agreement also with UCB, AC Immune, Astex Pharmaceuticals, Biogen, Takeda and Eisai.
Rojas JC – receives research support from NIH and is a site PI for clinical trials sponsored by Eli-Lilly and Eisai.
Rosen HJ – has received research support from Biogen Pharmaceuticals, has consulting agreements with Wave Neuroscience and Ionis Pharmaceuticals, and receives research support from NIH.
Rowe J - has research grants unrelated to the current work from AstraZeneca, Janssen, Lilly, GSK via the Dementias Platform UK; and has provided consultancy unrelated to the current work, to Asceneuron, Astex, Curasen, UCB, SV Health, WAVE and Alzheimer Research UK.
Russell LR – nothing to disclose
Sanchez-Valle R - receives personal fees from Wave pharmaceuticals for attending Advisory board meetings, personal fees from Roche diagnostics, Janssen and Neuraxpharm for educational activities, and research grants to her institution from Biogen and Sage Therapeutics outside the submitted work.
Savica R- receives support from the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, the Parkinson’s Disease Foundation, and Acadia Pharmaceuticals
Staffaroni AM – received research support from the NIA/NIH, the Bluefield Project to Cure FTD, and the Larry L. Hillblom Foundation. He has provided consultation to Passage Bio and Takeda.
Tartaglia M.C. has served as an investigator for clinical trials sponsored by Biogen, Avanex, Green Valley, and Roche / Genentech, Bristol Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen. She receives research support from Canadian Institutes of Health Research.
Tatton N – was employed by the Association for Frontotemporal Degeneration and is now employed by Alector.
Taylor J – nothing to disclose
VandeVrede L – receives research support from the Alzheimer’s Association, American Academy of Neurology, American Brain Foundation, and the NIH, and has provided consultation for Retrotope.
Weintraub S – receives research support from the NIH
Wendelberger B – is an employee of Berry Consultants, LLC where she serves as a consultant to numerous pharmaceutical and device companies
Wong B – receives research support from the NIH
Footnotes
Code Availability Statement:
Custom R code is available at 10.5281/zenodo.6687486.
Peer review information:
Primary Handling editor: Jerome Staal, in collaboration with the Nature Medicine team
Peer review information:
Nature Medicine thanks Sebastian Palmqvist and Jorge Llibre-Guerra for their contribution to the peer review of this work.
Data Availability Statement:
The datasets analyzed for the current study reflect collaborative efforts of two research consortia: ALLFTD and GENFI. Each consortium provides clinical data access based on established policies for data use: processes for request are available for review at allftd.org/data for ALLFTD data and by emailing genfi@ucl.ac.uk. Certain data elements from both consortia (e.g. raw MRI images) may be restricted due to the potential for identifiability in the context of the sensitive nature of the genetic data. The deidentified combined dataset will be available for request through the FTD Prevention Initiative in 2023 (https://www.thefpi.org/).
References
- 1.Knopman DS & Roberts RO Estimating the number of persons with frontotemporal lobar degeneration in the US population. J. Mol. Neurosci 45, 330–335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greaves CV & Rohrer JD An update on genetic frontotemporal dementia. J. Neurol 266, 2075–2086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai RM & Boxer AL Therapy and clinical trials in frontotemporal dementia: past, present, and future. J. Neurochem 138, 211–221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boeve BF, Boxer AL, Kumfor F, Pijnenburg Y & Rohrer JD Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet. Neurol 21, 258–272 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Mercuri E et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med 378, 625–635 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Miller T et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med 383, 109–119 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Bateman RJ et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers. Dement 13, 8–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salloway S et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med 27, 1187–1196 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boxer AL et al. New directions in clinical trials for frontotemporal lobar degeneration: Methods and outcome measures. Alzheimer’s Dement 16, 131–143 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman RJ et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leverenz JB et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain 130, 1360–1374 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Moore KM et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol 19, 145–156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohrer JD & Boxer AL The Frontotemporal Dementia Prevention Initiative: Linking Together Genetic Frontotemporal Dementia Cohort Studies. Adv. Exp. Med. Biol 1281, 113–121 (2021). [DOI] [PubMed] [Google Scholar]
- 14.U.S. Food & Drug Administration. Human Gene Therapy for Neurodegenerative Diseases. Draft Guidance for Industry. FDA-2020-D-2101
- 15.Rentz DM et al. Building clinically relevant outcomes across the Alzheimer’s disease spectrum. Alzheimer’s Dement. Transl. Res. Clin. Interv 7, e12181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staffaroni AM et al. Rates of Brain Atrophy across Disease Stages in Familial Frontotemporal Dementia Associated with MAPT, GRN, and C9orf72 Pathogenic Variants. JAMA Netw. Open 3, e2022847–e2022847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohrer JD et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 14, 253–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Q et al. Rates of lobar atrophy in asymptomatic MAPT mutation carriers. Alzheimer’s Dement. Transl. Res. Clin. Interv 5, 338–346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SE et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. NeuroImage Clin 14, 286–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caverzasi E et al. Gyrification abnormalities in presymptomatic c9orf72 expansion carriers. J. Neurol. Neurosurg. Psychiatry 90, 1005–1010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitwell JL et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur. J. Neurol 22, 745–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohrer JD et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53, 1070–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu SA et al. Brain volumetric deficits in MAPT mutation carriers: a multisite study. Ann. Clin. Transl. Neurol 8, 95–110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young AL et al. Characterizing the Clinical Features and Atrophy Patterns of MAPT-Related Frontotemporal Dementia With Disease Progression Modeling. Neurology 97, e941–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Ende EL et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet. Neurol 18, 1103–1111 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Illán-Gala I et al. Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease. Neurology 96, e671–e683 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gendron TF et al. Comprehensive cross-sectional and longitudinal analyses of plasma neurofilament light across FTD spectrum disorders. Cell Reports Med 3, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bridel C et al. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis. JAMA Neurol 76, 1035–1048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scherling CS et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann. Neurol 75, 116–126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rojas JC et al. Plasma Neurofilament Light for Prediction of Disease Progression in Familial Frontotemporal Lobar Degeneration. Neurology 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panman JL et al. Modelling the cascade of biomarker changes in GRN-related frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry (2021). doi: 10.1136/jnnp-2020-323541 [DOI] [PMC free article] [PubMed]
- 32.Saracino D et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J. Neurol. Neurosurg. Psychiatry (2021). doi: 10.1136/jnnp-2021-326914 [DOI] [PMC free article] [PubMed]
- 33.Glasmacher SA, Wong C, Pearson IE & Pal S Survival and Prognostic Factors in C9orf72 Repeat Expansion Carriers: A Systematic Review and Meta-analysis. JAMA Neurol 77, 367–376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore K et al. A modified Camel and Cactus Test detects presymptomatic semantic impairment in genetic frontotemporal dementia within the GENFI cohort. Appl. Neuropsychol. Adult 1–8 (2020). doi: 10.1080/23279095.2020.1716357 [DOI] [PubMed]
- 35.Staffaroni AM et al. Assessment of executive function declines in presymptomatic and mildly symptomatic familial frontotemporal dementia: NIH-EXAMINER as a potential clinical trial endpoint. Alzheimers. Dement 16, 11–21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barker MS et al. Recognition memory and divergent cognitive profiles in prodromal genetic frontotemporal dementia. Cortex 139, 99–115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poos JM et al. Cognitive composites for genetic frontotemporal dementia: GENFI-Cog. Alzheimers. Res. Ther 14, 10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintana M et al. Bayesian model of disease progression in GNE myopathy. Stat. Med 38, 1459–1474 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Paganoni S et al. Adaptive Platform Trials to Transform Amyotrophic Lateral Sclerosis Therapy Development. Ann. Neurol 91, 165–175 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Pottier C et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol 17, 548–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Ende EL et al. A data-driven disease progression model of fluid biomarkers in genetic frontotemporal dementia. Brain 145, 1805–1817 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staffaroni AM et al. Individualized atrophy scores predict dementia onset in familial frontotemporal lobar degeneration. Alzheimer’s Dement 16, 37–48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oxtoby NP et al. Data-driven models of dominantly-inherited Alzheimer’s disease progression. Brain 141, 1529–1544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onyike CU, Shinagawa S & Ellajosyula R Frontotemporal Dementia: A Cross-Cultural Perspective. Adv. Exp. Med. Biol 1281, 141–150 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Mok K et al. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol. Aging 33, 209.e3–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Zee J et al. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain 129, 841–852 (2006). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 47.Boeve B et al. The longitudinal evaluation of familial frontotemporal dementia subjects protocol: Framework and methodology. in Alzheimer’s and Dementia 16, 22–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos EM et al. Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimers. Dement 16, 118–130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyagawa T et al. Utility of the global CDR® plus NACC FTLD rating and development of scoring rules: Data from the ARTFL/LEFFTDS Consortium. Alzheimers. Dement 16, 106–117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staffaroni AM et al. Longitudinal multimodal imaging and clinical endpoints for frontotemporal dementia clinical trials. Brain 142, 443–459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyagawa T et al. Use of the CDR® plus NACC FTLD in mild FTLD: Data from the ARTFL/LEFFTDS consortium. Alzheimer’s Dement 16, 79–90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monsell SE et al. Results From the NACC Uniform Data Set Neuropsychological Battery Crosswalk Study. Alzheimer Dis. Assoc. Disord 30, 134–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jack CR et al. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 49, 786–94 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olney NT et al. Clinical and volumetric changes with increasing functional impairment in familial frontotemporal lobar degeneration. Alzheimer’s Dement 16, 49–59 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russell LL et al. Social cognition impairment in genetic frontotemporal dementia within the GENFI cohort. Cortex 133, 384–398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sled JG, Zijdenbos AP & Evans AC A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans. Med. Imaging 17, 87–97 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Ashburner J & Friston KJ Unified segmentation. Neuroimage 26, 839–51 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Ashburner J & Friston KJ Diffeomorphic registration using geodesic shooting and Gauss-Newton optimisation. Neuroimage 55, 954–67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desikan RS et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–80 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Bocchetta M et al. Thalamic atrophy in frontotemporal dementia - Not just a C9orf72 problem. NeuroImage. Clin 18, 675–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Young AL et al. Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat. Commun 9, 4273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bocchetta M et al. Differential early subcortical involvement in genetic FTD within the GENFI cohort. NeuroImage. Clin 30, 102646 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed for the current study reflect collaborative efforts of two research consortia: ALLFTD and GENFI. Each consortium provides clinical data access based on established policies for data use: processes for request are available for review at allftd.org/data for ALLFTD data and by emailing genfi@ucl.ac.uk. Certain data elements from both consortia (e.g. raw MRI images) may be restricted due to the potential for identifiability in the context of the sensitive nature of the genetic data. The deidentified combined dataset will be available for request through the FTD Prevention Initiative in 2023 (https://www.thefpi.org/).