

Abstract
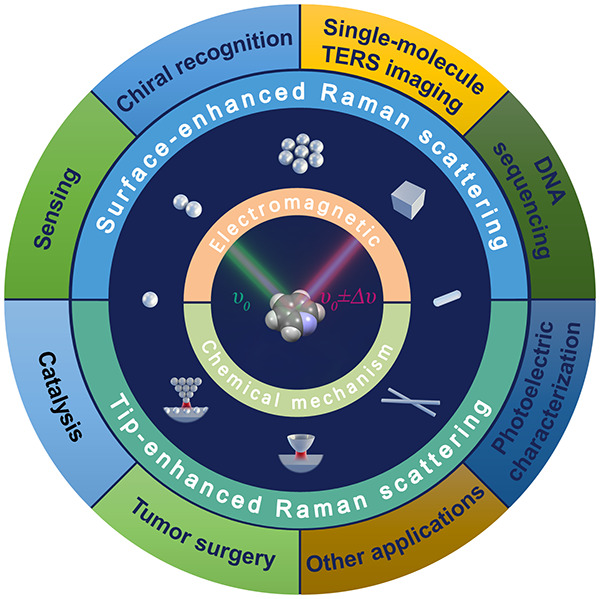
Surface-enhanced Raman scattering (SERS) and tip-enhanced Raman scattering (TERS) have opened a variety of exciting research fields. However, although a vast number of applications have been proposed since the two techniques were first reported, none has been applied to real practical use. This calls for an update in the recent fundamental and application studies of SERS and TERS. Thus, the goals and scope of this review are to report new directions and perspectives of SERS and TERS, mainly from the viewpoint of combining their mechanism and application studies. Regarding the recent progress in SERS and TERS, this review discusses four main topics: (1) nanometer to subnanometer plasmonic hotspots for SERS; (2) Ångström resolved TERS; (3) chemical mechanisms, i.e., charge-transfer mechanism of SERS and semiconductor-enhanced Raman scattering; and (4) the creation of a strong bridge between the mechanism studies and applications.
1. Introduction
The surface-enhanced Raman scattering (SERS) and tip-enhanced Raman scattering (TERS) have been expected to open up a new era from the time of their naming because they enable more efficient analysis of analytes by enhancing Raman scattering, which reflects vibrational information on analytes. SERS is the phenomena that molecules on specific metal surfaces exhibit large enhancement in Raman scattering cross-sections, and TERS is the controlled SERS phenomena generated on the specific metal needle tip of scanning microscopies. More than 45 and 20 years have passed since the discovery of SERS and the creation of TERS, respectively, as surveyed in Section 1.1.1−6 Thanks to the large efforts of their mechanistic studies, SERS and TERS have been understood as the results of electromagnetic (EM) and chemical enhancement as explained in Section 1.2.
Since then, SERS and TERS spectroscopies have created a variety of exciting research fields. In particular, numerous SERS applications have been proposed to various industries related to analytical chemistry, food safety, environmental monitoring, biochemistry, and other fields.7−14 The SERS and TERS communities have continuously expanded by connecting with various research fields including quantum electrodynamics (QED), photocatalysis, and medical diagnosis.15−23 These recent broadenings in the research fields of SERS and TERS raise a demand to re-examine the mechanisms of SERS and TERS in such new research fields, even though the EM and chemical mechanism has been investigated reasonably well.24−27 The quantum effects in the interactions between light and molecules within the subnanometer to piconanometer scale have revealed the necessity of modifying the EM mechanisms to apply them to SERS and TERS.15−17,25,27−29 In contrast to the EM mechanism, studies of chemical mechanisms are insufficient, even with the emergence of various remarkable semiconducting materials that exhibit significant chemical enhancement.30−34 In recent years, with the development of new SERS substrates, numerous applications of SERS and TERS have been proposed in various fields, including biomedicine, environmental monitoring, and chemical reaction monitoring;7−14,22,23,35−40 however, none of these proposals have reached real practical and common uses. This status indicates that the strategy for exploring the applications of SERS and TERS needs to be reconsidered.
The above situation calls for the need of a comprehensive review broadly covering recent progress in the fundamentals of SERS and TERS for researchers both inside and outside the SERS and TERS fields. Various review papers on SERS and TERS have been published over the last decades, as indicated above, many of which deal with their applications based on the EM and chemical mechanisms established by pioneers.41−43 However, these EM and chemical mechanisms have since been much improved, following the discovery of single-molecule SERS (SM-SERS).44,45 Thus, inclusive reviews based on these improved mechanisms are required.27 Notably, to date, reviews on recent progresses in chemical mechanisms and ones that discuss new directions and perspectives of SERS and TERS are very scarce.7,46 Thus, we consider that a comprehensive summary of these issues and recent progress in this field is highly desirable to pave the way for SERS and TERS applications.
The goals and scope of this review are to report new directions and perspectives of SERS and TERS mainly from the viewpoint of their recent mechanism studies. Thus, in this review, we discuss the breakdowns of approximations in the conventional EM mechanism of SERS and TERS in the subnanometer to pico-meter scale regions, namely plasmonic hotspots (HSs). Subsequently, to outline the chemical mechanism of SERS and TERS, we describe the mechanisms for SERS charge-transfer (CT) and semiconductor-enhanced Raman scattering. We also aim at overviewing the relationship between the mechanism studies and applications. Notably, it is important to clarify why SERS and TERS have not been used in practical applications. Thus, herein, we try to combine the SERS and TERS mechanism studies with application studies.
1.1. Brief History of SERS and TERS
1.1.1. Brief History of SERS
The SERS phenomenon was first measured by M. Fleischmann et al. in 1974,1 and this phenomenon was independently revealed as Raman scattering enhancement by a factor of ∼106 in 1977 by Van Duyne’s and Creighton’s groups.2,3 In all three studies,1−3 electrochemically roughened silver electrodes were used to generate enhanced Raman signals of pyridine molecules adsorbed on the electrodes. Therefore, the enhancement mechanism was first ascribed to an increase in the effective surface area due to roughness.41,47 However, this was subsequently denied because such a large enhancement could not be reproduced by the increase in the surface area alone, and the plasmon resonance contribution of the roughened metal surfaces was thought to be effective for the enhancement.41,48
In 1997, the groups of Nie and Kneipp discovered the huge Raman enhancement factor (EF) of ∼1012 in the SERS of dye molecules adsorbed on silver and gold nanoparticle (NP) aggregates. This value is sufficient to detect a single molecule (SM) with the support of the resonance Raman effect. These experiments were the origin of SM-SERS,44,45 and significantly impacted the study of the SERS mechanism. Theoretical and experimental investigations of the minimum aggregates, namely NP dimers, quantitatively demonstrated that SM-SERS signals are generated by a confined EM field across the dimer gaps, by directly observing the correlation between the SERS, plasmon resonance, and dimer structures.49−51 The success of SM-SERS indicates that SERS universally provides SM detection sensitivity under the resonance Raman condition. Organic molecules have commonly electronic resonance in a UV region. Thus, UV SERS using metals like aluminum showing plasmon resonance in the UV region is powerful for SM-SERS of various organic molecules.7 Yen et al. demonstrated rapid, label-free and ultrasensitive SERS detection of oligonucleotides in 12-mer ss-DNA using a deep UV excitation (266 nm).52 They used optimized periodic nanohole array formed on an epitaxial aluminum film as a SERS-active substrate providing EF of up to 106. They reported for the first time not only detection of all distinct nucleic bases but also their sequential single-base mutation by UV-SERS.
Two types of mechanisms have been discussed as the origin of SERS enhancement, namely the EM and chemical mechanisms.41,42 Of the two, EM enhancement, which is caused by the coupling of the incident and Raman EM fields with plasmon resonance on metallic surfaces, is thought to be the main contributor of the SERS effect.49−51 The chemical mechanism is characterized by the shifting of the Raman scattering from the nonresonance to the resonance region through the formation of CT complexes between the adsorbed molecules and metal surfaces.53,54 Although the existence of the chemical mechanism is experimentally confirmed by the appearance of CT resonance Raman properties in the SERS spectra, its contribution toward the total enhancement is thought to be relatively smaller than that of the EM mechanism.55
In 2016, Benz et al. demonstrated that the size of a HS reached atomic scale under cryogenic temperature conditions.56 In the atomic-scale regime, assumptions in classical spectroscopies, including long wavelength approximations, weak coupling approximations, and Kasha’s rule, are already broken down.27 Thus, SERS is now a tool for exploring the frontiers of light–matter interactions including exotic photochemical reactions.
1.1.2. Brief History of TERS
Ångström-resolved TERS is the latest topic in TERS, which will be discussed in Section 4, but here we begin with a brief history of TERS. In 1985, Wessel first proposed the concept and theoretical model of TERS based on a scanning tunneling microscopy (STM) configuration with a plasmonic NP probe tip.57 The NP plasmon resonance enhances and confines the excitation light, resulting in the sensitive Raman detection with high spatial resolution of the STM system. The mechanism of the enhancement in TERS is common to that of SERS, comprising both EM and CT enhancement mechanisms.41,42 However, the important factor in TERS compared to SERS is the control of the gap distance and gap positions of the HSs. Following the development of element technologies, such as TERS probes, in 2000 the groups of Zenobi, Kawata, Anderson, and Pettinger. independently published experimental setups and TERS results by combining Raman spectroscopy with atomic force microscopy (AFM) or STM.4−6,58 They reported an EF of <106 and a spatial resolution of ∼50 nm for TERS. The improvement of the EF and spatial resolution has been reported by many other groups using AFM, STM, or scanning force microscopy (SFM) under ambient or high vacuum with low temperature conditions.43,59,60 In 2013, Zhang et al. demonstrated SM-TERS mapping with a spatial resolution of 0.5 nm.61 Notably, in such a high atomic-scale spatial resolution, the assumptions made in classical spectroscopy are already broken down, similar to the atomic-scale HS of SERS.27,56 Thus, TERS has attracted attention for its potential application to in situ measurements on solid surfaces and as a promising tool for exploring the frontiers of light–matter interactions including exotic photochemical reactions.
The instrumentation for TERS has branched to several configurations although the instrumentation for SERS is basically common to that of conventional Raman spectroscopy. There are three typical configurations for TERS in terms of illumination modes: bottom-, top-, and side-illumination, whose details can be found in previous reviews.58,59 For the transparent sample substrate, the bottom illumination with backscattering collection of light is commonly used for TERS. The merit and demerit of this configuration are the use of high NA objective lens to obtain the high detection sensitivity and the impossibility for the opaque sample, respectively. For the opaque sample substrate, the top or side illumination with backscattering collection are commonly used for TERS. The merit and demerit of this configuration are the applicability to the opaque sample and the use of low NA objective lens due to long working distances, respectively. This demerit can be improved by using a parabolic mirror setup for the illumination and collection. In Section 4, we focus on the side-illumination configuration to describe field enhancement and confinement.
1.2. Outline of the SERS and TERS Mechanisms
1.2.1. Electromagnetic Mechanism
The EM mechanism in SERS describes the SERS enhancement as being induced by a strong EM field generated by plasmon resonance. Jeanmaire and Van Duyne were the first to discuss the electric field enhancement as a SERS mechanism.3 The EM mechanism by plasmon resonance was first developed by Moskovits et al.41,48 and then formulized by Kerker et al.62 Subsequently, Aravind, Inoue, and Xu et al. demonstrated that this mechanism could successfully explain the large Raman EF of ∼1010 of a molecule located in the gaps of a plasmonic NP dimer, i.e., the SERS HSs.50,63,64 There is not a rigorous definition of HS. If we assume that the definition is SM SERS detection, according to fluorescence cross-section ∼10–15 cm2 and resonance Raman cross-section ∼10–24 cm2, the EF of SERS is needed to be 109 to enable SM SERS detection. Thus, the gap distance is needed to be in the range between 1 to 2 nm.50 Therefore, the authors propose here to define the HS size of SM-SERS as 1–2 nm in terms of gap distance, based on the limitation of SERS enhancement by EM enhancement theory.
Here we begin to explain the basic theory of the EM
enhancement mechanism. The EM mechanism was confirmed to be dominant
for SERS by the quantitative reproduction of the experimentally obtained
SERS spectra using this mechanism.51 From
a physical viewpoint, enhancement of the optical response (e.g., Raman
enhancement), commonly requires “resonance” to enlarge
its cross-section. In the SERS EM mechanism, plasmon resonance is
a source of this “resonance”, which generates the Raman
enhancement. Plasmons are collective oscillation modes of conduction
electrons. The plasmon of a metal NP, which is usually called localized
plasmon or particle plasmon, induces a depolarization to cancel the
external electric fields.65 The explanation
is identical to that the negative real part of permittivity of metal
can enlarge the polarizability of NP 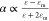 due to Re[ε + 2εm] = 0, where ε and εm are the permittivity
of metal and surrounding medium, respectively. This is particularly
evident for metals having a small value of Im[ε], such as gold
and silver. When a phase difference between the external electric
fields and dipole polarization of the NP reaches a value of π/2,
the two become resonant.65 We here call
this situation as plasmon resonance. At phase differences of zero
and π, which correspond to transmission and reflection, respectively,
the light resonating with plasmon cannot go through or back from the
NP, and is thus confined around the NP. If the electron-rich metal
NP forms a NP dimer, the light is further confined in the gap or junction
of the dimer, resulting in large EFs of ∼105 for
a single optical response.50,63,64 The spontaneous Raman process is composed of two processes, namely
excitation by light and de-excitation by vacuum fluctuation. Thus,
plasmon resonance contributes to the enhancement twice, by increasing
both the excitation rate and de-excitation rate, so that the SERS
EF increases significantly to (∼105)2.25,26 Note that the value of 1010 of
SERS HSs is much larger than those of standard SERS substrates. The
SERS substrates usually exhibit the EFs ∼106 because
of the averaging effect of whole substrate surfaces.
due to Re[ε + 2εm] = 0, where ε and εm are the permittivity
of metal and surrounding medium, respectively. This is particularly
evident for metals having a small value of Im[ε], such as gold
and silver. When a phase difference between the external electric
fields and dipole polarization of the NP reaches a value of π/2,
the two become resonant.65 We here call
this situation as plasmon resonance. At phase differences of zero
and π, which correspond to transmission and reflection, respectively,
the light resonating with plasmon cannot go through or back from the
NP, and is thus confined around the NP. If the electron-rich metal
NP forms a NP dimer, the light is further confined in the gap or junction
of the dimer, resulting in large EFs of ∼105 for
a single optical response.50,63,64 The spontaneous Raman process is composed of two processes, namely
excitation by light and de-excitation by vacuum fluctuation. Thus,
plasmon resonance contributes to the enhancement twice, by increasing
both the excitation rate and de-excitation rate, so that the SERS
EF increases significantly to (∼105)2.25,26 Note that the value of 1010 of
SERS HSs is much larger than those of standard SERS substrates. The
SERS substrates usually exhibit the EFs ∼106 because
of the averaging effect of whole substrate surfaces.
We here use cavity QED, which can treat the enhancement of light (or vacuum fluctuation) and a molecule in a resonator, for the introduction of EM mechanism. The EM mechanism has been explained using electromagnetism. However, Raman scattering includes a spontaneous emission process triggered by vacuum fluctuation, which does not explicitly appear in electromagnetism. To explicitly include the contribution of vacuum fluctuation to EM mechanism, cavity QED is useful. In cavity QED, plasmon resonance is treated as an optical resonator which enhances transitions of a molecule close to the resonator. In the EM mechanism, excitation and de-excitation are enhanced by the coupling between a molecular exciton and plasmon through photon exchange inside a HS.25 Under dipole approximation, which assumes that the wavelength of the incident light is sufficiently larger than the size of the molecule, the coupling energy ℏΩ, where ℏ and Ω are the Planck constant and coupling angular frequency, respectively, is determined by
| 1 |
where ⟨f|ex|i⟩ is the dipole moment of the transition
from the initial state |i⟩ to the final state
|f⟩, e is the elementary
charge of an electron (∼1.60 × 10–19 C), x is the position of the electron, and E is the amplitude of the electric field triggering the
transition.66 In the case of a spontaneous
scattering process, E becomes the amplitude of the
fluctuating vacuum electric field Evac. The zero-point energy 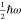 in the HS is equal to the EM field energy
integrated through the mode volume of the HS VHS. Thus,
in the HS is equal to the EM field energy
integrated through the mode volume of the HS VHS. Thus,
| 2 |
The VHS is defined as VHS = ∫2εrε0|E|2/max(εrε0|E|2)d3r, where Evac is the electric field amplitude inside the HS and εr and ε0 are the relative permittivity inside the HS and vacuum permittivity (∼8.854 × 10–12 F/m), respectively. Notably, the term 2εrε0Evac2 includes the energies of both the electric and magnetic fields.66 The oscillator strength of a molecule is described by the equation f = 2mωldip2/(e2ℏ), where ldip is the dipole length and m is the free electron mass (∼9.109 × 10–31 kg). Therefore, using eq 2, eq 1 can be rewritten as
| 3 |
where N is the number of dye molecules inside a HS.67 Notably, in eq 3, ℏΩvac is a function of VHS. The EF of the transition rate of a molecule inside a HS is expressed by the Purcell factor F = Wcav/Wfree at the resonant condition
| 4 |
where Wfree and Wcav are the transition rates of the molecule
in free space and a HS, respectively; λ is the resonant wavelength
of the resonator; n is the refractive index inside
the HS; and Q is the quality factor of the plasmon
resonance (assuming that the direction of the dipole in free space
is random, 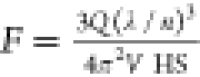 ).66 The EF of
a transition rate is a function of VHS. The term F in eq 4 includes both a radiative and nonradiative rate. Note
that the radiative and nonradiative rate are directly related to the
line width of a plasmon resonance spectrum. If the rate, which is
proportional to a line width Δω of the plasmon resonance
spectrum, is separated into the radiative rate ΔωR and nonradiative rate ΔωNR, then the
radiative portion of F is expressed as
).66 The EF of
a transition rate is a function of VHS. The term F in eq 4 includes both a radiative and nonradiative rate. Note
that the radiative and nonradiative rate are directly related to the
line width of a plasmon resonance spectrum. If the rate, which is
proportional to a line width Δω of the plasmon resonance
spectrum, is separated into the radiative rate ΔωR and nonradiative rate ΔωNR, then the
radiative portion of F is expressed as
| 5 |
where FR is the radiative portion of the Purcell factor, which is identical to the EM EF.25Equation 5 indicates that the radiative portion of the plasmon resonance can contribute toward the EM enhancement of Raman scattering. Note that the Raman process is composed of excitation and Raman scattering transitions. The EM enhancement contributes to both transitions. Thus, the SERS EM EF is composed of 2-fold enhancements of excitation and Raman scattering as described in eq 6
| 6 |
where EI and Eloc indicate the amplitudes of the incident and enhanced local electric fields, respectively; λex and λem denote the wavelengths of the incident and Raman-scattered light, respectively; and r is the position of a molecule in a HS. The large SERS EM EF is due to the small VHS value in eq 4. Notably, the values of ΔωR and ΔωNR in eq 5 are not constant but are functions of VHS.68 If VHS decreases, the contribution of the nonradiative plasmons (i.e., higher-order plasmon, CT plasmon due to an unscreened electron gas of a metal surface) to FR becomes dominant, resulting in the saturation or decrease in the EM enhancement.68
Yoshida et al. successfully reproduced SERS spectra by multiplying Raman spectra with plasmon resonance spectra as expressed by eq 7, revealing the correctness of the EM mechanism in SERS.51 In the reproduction, experimentally obtained coupled resonance between a plasmon and molecule exciton (molecule electronic transition dipole) is approximately treated as the plasmon resonance. Such treatment is called a “weak coupling approximation”. Specifically, eq 1 indicates that the coupled resonance is different from the original plasmon resonance (see Section 3.1 for the detailed discussion). The weak coupling assumes that Ωvac in eq 3 is sufficiently small compared to the dephasing rates of both the plasmon and molecular exciton resonances,25 so that it is unnecessary to distinguish between the plasmon and coupled resonances.
For the reproduction, the plasmon resonance and SERS spectra are experimentally obtained under dark- and bright-field excitation conditions, respectively, using a single dimer adsorbed by dye molecules at its HS.51 The point of measurement is the selection of a dipolar plasmon resonance showing an identical polarization dependence to the SERS light.51 This point is crucial for identifying the dipole plasmon inducing the FR in eq 5. Based on the shape of the dimer measured by scanning electron microscopy (SEM), the plasmon resonance spectra and FR are calculated by a finite-difference time-domain (FDTD) method.51 In the case of the dipole plasmon mode, FR commonly contributes to both Raman excitation and emission as a result of optical reciprocity.47 Thus, eq 6 indicates that the SERS cross-section σSERS(λex, λem) can be expressed as
| 7 |
where σRS(λex, λem) is the experimentally obtained Raman scattering cross-section of a molecule in free space. Figure 1g–j shows the excitation-laser wavelength dependence of the reproduced SERS spectra using eq 7, while the corresponding experimental data are presented in Figure 1b–e.51 The considerable spectral variations in SERS are well reproduced in the calculations. Specifically, the large combination and overtone modes (600–650 nm) in Figure 1c, which intensities are usually much smaller than those of the fundamental modes (550–600 nm), and anti-Stokes modes (600–625 nm) in Figure 1e, which intensities are usually much smaller than those of the Stokes modes, become comparable to the Stokes modes. These properties are quantitatively reproduced in the calculated spectra as overlapping between the combination and overtone modes or the anti-Stokes modes and the plasmon resonance expressed as σRS(λex, λem) and FR(λem, r), respectively, in eq 7. This reproduction indicates that the EM mechanism is dominant for SERS.
Figure 1.
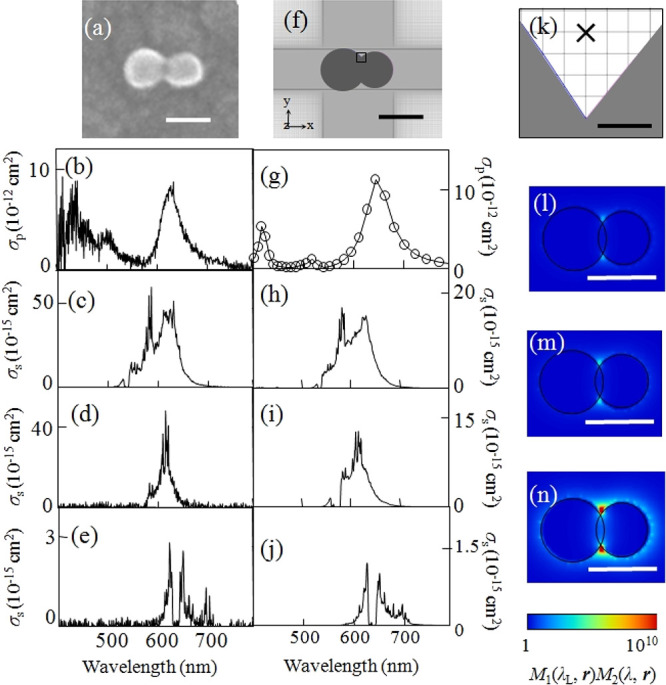
(a) SEM image of a silver NP dimer. (b) Experimental plasmon resonance spectrum and experimental resonant SERS spectra excited at (c) 532, (d) 561, and (e) 633 nm. (f) Modeled structure of a silver NP dimer in the FDTD calculation. (g) Calculated plasmon resonance spectrum and calculated SERRS spectra excited at (h) 532, (i) 561, and (j) 633 nm. (k) Magnified view in the vicinity of the crevice of the model structure in (f). Spatial distribution of the calculated SERRS-EM EF excited at (l) 532, (m) 561, and (n) 633 nm. Circularly polarized incident light (532, 561, and 633 nm), mesh size (0.2 nm), and refractive index (1.3) of the surrounding medium were selected for the FDTD calculation. The scale bars in panels a, f, and l–n are 50 nm, while that in panel k is 0.5 nm. Reproduced with permission from ref (51). Copyright 2010 American Physical Society.
The lower limit of VHS in eq 2 has been studied by van Savage et al., who reported a problem called the “plasmon quantum limit,” which is a breakdown of the classical model for the plasmon oscillations of conduction electrons.69 This limit indicates that the localized EM field volume cannot be smaller than the distribution volume of the unscreened metal surface electrons within the Debye radius. Zhu et al. experimentally confirmed through the SERS EFs that a quantum-based remodeling of the plasmons is necessary for a dimer gap smaller than 0.4 nm.70 The EM mechanism breaks down when the gap size gets sufficiently small so that electron tunneling can occur. Figure 2a shows that the SERS EF stagnates and even decreases once the gap size falls below 1 nm. Calculations using the classical EM mechanism are unable to reproduce this result, as shown in blue in Figure 2b. However, once a quantum mechanical correction that accounts for electron tunneling is introduced (red data in Figure 2b), the calculated results agree much better with the experimental data.70 This limitation is summarized as (1) the lowest limitation of the EM field confinement and (2) appearance of plasmon oscillations by tunneling electrons. These points cannot be derived from the classical EM mechanism assuming metal surfaces as dielectric surfaces. The tunneling-out of the conduction electrons, which loosens the field confinement, determines the lowest limit of the VHS, and thus, provides an estimation of the upper limitation of FR.70
Figure 2.

Impact of the quantum mechanical electron tunnelling on the SERS EF. (a) Maximum SERS EFs for 45 dimers measured by wavelength-scanned SERS (red circles). The horizontal error bars represent the errors in the gap-width determination, which is retrieved following the procedures described in the Supporting Information of ref (70). The vertical error bars represent the errors in the SERS EF measurements, which are calculated from the uncertainties in the reference Raman measurements for neat thiophenol (Supporting Information of ref (70)). The gap-widths range from (2.0 ± 0.6) Å to (9.1 ± 0.4) nm. The maximum SERS EF measured is (1.2 ± 0.2) × 109 for dimer III with a gap-width of (6.7 ± 1.1) Å. Two regions that show opposite trends for the SERS EF as a function of the gap width are observed. For the gap width ranging from 6.7 Å to 9.1 nm, the measured SERS EF generally follows the phenomenological linear fit (in a log–log scale) as log(EF) = 8.8–1.3log(gap width). The SERS EF generally increases as the gap width decreases in this region. As the gap width further decreases from 6.7 to 2.0 Å, the SERS EF does not increase but instead decreases significantly. (b) Simulated SERS EFs using both the quantum-corrected model (QCM) and classical EM model (CEM) for gap widths ranging from 1 Å to 10 nm. Only the EM SERS EF is considered (see Methods), and the simulated EFs are one to 2 orders of magnitude smaller than the measured values, mainly due to SERS chemical enhancement. The CEM simulations of Model 1 assume monolayer coverage of the thiophenol molecules on the gold surfaces. The CEM simulations of Model 2 consider the hypothetical case for which the thiophenol molecules cannot access the narrowest regions of the gap. Reproduced with permission from ref (70). Copyright 2014 Nature Research.
Furthermore, with the decrease in VHS, the contribution of the higher order plasmon resonances to FR increases, resulting in an increase in the nonradiative rate ΔωNR in eq 5. Thus, the value of FR2 exhibits a saturation tendency. Notably, the upper limitation of FR2 considerably depends on the quantum efficiency of the emission. In the case of the fluorescence and nonresonant Raman, the values are theoretically 102 and 1014, respectively, excluding the effect of the plasmon quantum limit.71
1.2.2. Chemical Mechanism
The chemical mechanism in SERS and TERS have been well discussed for several decades. Recent progress in the study of the chemical mechanism is presented in Section 5, so here we discuss the basics of the chemical mechanism. The chemical mechanism in SERS has been understood as another Raman enhancement caused by the formation of chemical bonds between the target molecule and SERS substrate, e.g., plasmonic NPs. The chemical mechanism was first proposed by Albrecht and Creighton to explain the Raman enhancement of pyridine molecules by combination with an electrochemically roughened silver surface.2 In fact, the chemical mechanism covers various effects including the resonance Raman effect.72 Jensen et al. grouped Raman enhancement in the chemical mechanism according to three effects: (A) resonance Raman enhancement due to a molecular electronic transition; (B) CT resonance Raman enhancement induced by substrate–molecule CT transitions; and (C) enhancement due to the ground-state chemical interactions between the molecule and NP.72 Mechanism (B) is commonly referred to as the chemical mechanism, and its description is closely related to that of mechanism (A). Thus, herein, we first explain (A), the resonance Raman, and then apply this explanation to (B), the CT resonance Raman, referring to the vibronic coupling regime. Recently, the chemical mechanism has been directly calculated using the density functional theory (DFT) method. However, here, we describe this mechanism through a quantum mechanical viewpoint to clarify the physics behind it.
The Raman polarization tensor ασρ is a product of an excitation and a scattering transition as given by the Kramers–Heisenberg–Dirac dispersion formula:73
| 8 |
where |i⟩, |f⟩, and |n⟩ are the wave functions of the initial state, final state, and an intermediate state of the molecular systems, respectively; Ei, Ef, and En are the respective energies of these states; ℏω0 is the excitation photon energy; Mσ and Mρ are the electronic dipole moments of the excitation and Raman scattering, respectively; and Γn is the dephasing energy of the intermediate state. The first term in eq 8 can resonate with light by the condition En – Ei – ℏω0 = 0, while the second term, which is a scattering transition, cannot because of the condition En – Ei + ℏω0; thus, the second term is usually neglected in a resonance Raman process. According to the Born–Oppenheimer approximation,74 |i⟩, |f⟩, and |n⟩ are described as products of electronic and vibrational states such as
| 9a |
| 9b |
| 9c |
where |g⟩|ν″⟩ is a wave function of the electronic ground state |g⟩ with vibrational states |ν″⟩, |g⟩|ν’⟩ is a wave function of |g⟩with vibrational states |ν″⟩, and |e⟩|ν⟩ is a wave function of the exited electronic state |e⟩ with vibrational states |ν⟩. These are called vibronic couplings, owing to the combination of electronic and vibrational states.
Figure 3 shows the
Raman processes classified into the off-, pre-, and rigorous-resonance.
Under the off-resonance Raman condition in Figure 3A, En – Ei ≫ℏω0, all intermediate states 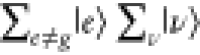 contribute to ασρ in eq 8. The contribution from
contribute to ασρ in eq 8. The contribution from 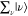 becomes
becomes 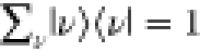 in eq 8 through eq 9c, owing to the closure relationship, and the contribution from
in eq 8 through eq 9c, owing to the closure relationship, and the contribution from 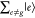 is expressed as the total molecular polarization,
as described by the Placzek approximation.75 Under the preresonance Raman condition in Figure 3B, En – Ei ∼
ℏω0,
is expressed as the total molecular polarization,
as described by the Placzek approximation.75 Under the preresonance Raman condition in Figure 3B, En – Ei ∼
ℏω0, 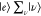 in
in 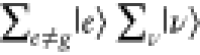 selectively contributes to ασρ. The contribution from
selectively contributes to ασρ. The contribution from 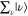 is rounded, owing to the closure relationship;
however, |e⟩ contributes exclusively to ασρ. Under the rigorous-resonance Raman condition
in Figure 3C, En – Ei = ℏω0, a specific
|e⟩|ν⟩ in
is rounded, owing to the closure relationship;
however, |e⟩ contributes exclusively to ασρ. Under the rigorous-resonance Raman condition
in Figure 3C, En – Ei = ℏω0, a specific
|e⟩|ν⟩ in 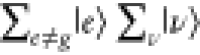 selectively contributes to ασρ.76
selectively contributes to ασρ.76
Figure 3.
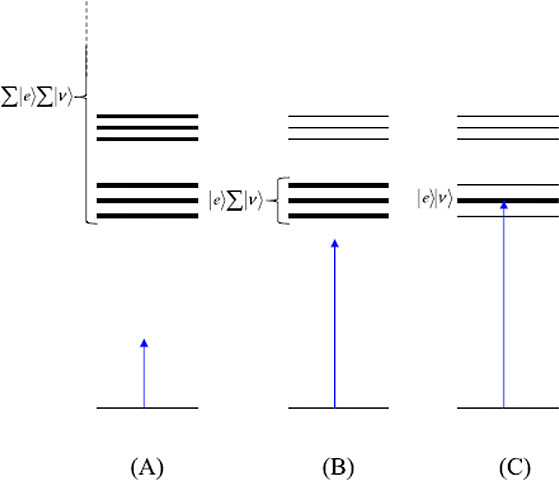
Illustration of the different
conditions of resonance Raman. (A)
Off-resonance Raman scattering condition, En – Ei ≫ ℏω0, in which many excited
electronic states 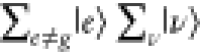 contribute (virtual intermediate states).
(B) Preresonance Raman scattering condition, where ℏω0 approaches En – Ei, and the
vibrational states of the lowest excited electronic state
contribute (virtual intermediate states).
(B) Preresonance Raman scattering condition, where ℏω0 approaches En – Ei, and the
vibrational states of the lowest excited electronic state 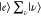 act as intermediate states. (C) Rigorous
resonance Raman scattering condition, where En – Ei ∼ ℏω0 and one particular
vibronic state, |e⟩|ν⟩, dominates
the scattering process. Reproduced with permission from ref (26). Copyright 2016 John Wiley
& Sons, Ltd.
act as intermediate states. (C) Rigorous
resonance Raman scattering condition, where En – Ei ∼ ℏω0 and one particular
vibronic state, |e⟩|ν⟩, dominates
the scattering process. Reproduced with permission from ref (26). Copyright 2016 John Wiley
& Sons, Ltd.
The first important property of the resonance Raman is the Raman enhancement. The EF of the resonant Raman effect can be roughly estimated using eq 8, in which Γn is of the order of 100 cm–1, En – Ei – ℏω0 is usually >104 cm–1 under the off-resonance Raman condition, and En – Ei – ℏω0 is ∼0 cm–1 under the pre- and rigorous-resonance Raman conditions. The value of ασρ for the resonance Raman is therefore ∼100 times larger than that for the off-resonance Raman. Thus, the resonance Raman cross-section ∝|ασρ|2 is ∼104 times larger than the off-resonance Raman cross-section. This enlargement in Raman cross-section is the origin of mechanism (B).
The second
important property of the resonance Raman is the change
in spectral lines. The Raman lines change depending on En – Ei – ℏω0, because a
specific state 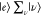 or |e⟩|ν⟩
selectively contributes to ασρ, as shown
in Figure 3b,c. Specifically,
ασρ is selectively generated by a certain
elementary Raman process, which is active for a resonant electronic
transition between |g⟩ and specific |e⟩|ν⟩.
or |e⟩|ν⟩
selectively contributes to ασρ, as shown
in Figure 3b,c. Specifically,
ασρ is selectively generated by a certain
elementary Raman process, which is active for a resonant electronic
transition between |g⟩ and specific |e⟩|ν⟩.
We here explain the changes of the Raman line positions as a result of the resonance effect based on a theory developed by Albrech.76 This theory comprises the coupling between the electronic states through nuclear motions, namely vibronic coupling.76 Under vibronic coupling, the “real” electronic potential becomes a function of the nuclear coordinate Qa by attractive interaction between the electrons and nuclei. Such a coupled electronic state can be expressed by the Herzberg–Teller (HT) expansion.76 Considering the first-order perturbation term from the HT expansion, |e⟩ and |g⟩ in eqs 9a–9c can be respectively rewritten as
| 10a |
and
| 10b |
In the above equations, |e0⟩ and Ee0, |s0⟩ and Es, and |t0⟩ and Et0 are electronic states and their energies, respectively, at the nuclear equilibrium position; V and Qa are the respective potential energy of the molecular system and nuclear coordinate; and (ha)es and (ha)gt are integrals showing the degree of vibronic coupling. By substituting both eqs 10a and 10b into eq 8, three terms are obtained as follows:
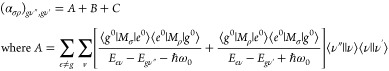 |
11a |
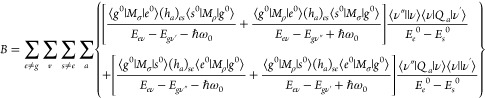 |
11b |
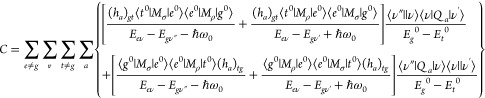 |
11c |
eqs 11a–11c are called the A (Franck–Condon; FC), B, and C (vibronic coupling) terms, respectively.76 The factor ⟨ν″||ν⟩⟨ν||ν’⟩ in the A term yields only totally symmetric vibrations, however, only when the minima of the ground and excited electronic states are at different Qa values.76 On the other hand, the factors ⟨ν″||ν⟩⟨ν|Qa|ν’⟩ and ⟨ν″|Qa|ν⟩⟨ν||ν’⟩ in the B and C terms yield both totally and nontotally symmetric vibrations. By assuming that the electronic energy gaps are much larger than the vibrational energy gaps, Ee0 – Eg ≫ (Eev – Ee0) – (Egv″ – Eg), the intensities of the A, B, and C terms (FA,FB, and FC, respectively) are expressed as
| 12a |
| 12b |
and
| 12c |
The denominators of eqs 12a–12c indicate that the intensity of these terms is resonantly enhanced in the rigorous-resonance Raman condition, indicating that the resonance Raman effect generates Raman lines of nontotally symmetric vibrations, which are forbidden in nonresonance Raman spectra.77
In the description of CT resonance Raman by Lombardi et al.,53,72 when a molecule is covalently bound to a metal surface, the molecule and metal surface can exchange an electron by a photoexcited transition through the new covalent bond formed between them; this transition is called a CT transition. Thus, Raman enhancement by CT transition resonance is expressed as the change from nonresonance to CT resonance Raman. The evidence of CT resonance is the emergence of resonance Raman lines due to vibronic coupling terms in eqs 12b and 12c in the SERS spectrum. The vibronic coupling between a molecule and metal surface through a covalent bond is assumed to be “weak” in CT resonance Raman.53 This assumption means that the molecular vibrational structure is not modified by vibronic coupling. Thus, the metal state only appears in the perturbation term of the HT expansion as (ha)es and (ha)gt in eqs 11b and 11c, respectively, where |s0⟩ and |t0⟩ are metal electronic states. Figure 4 shows the state diagram of possible CT transitions. These electronic states in the metal conduction band (CB) together with its energies are expressed as |F⟩ instead of |s0⟩ and |t0⟩with EF0 instead of Es and Et0, respectively. The modification in eqs 11b and 11c indicates that the intensity of the nontotally symmetric vibrations in the SERS spectrum resonantly increases with the inverse of (Ee – EF0)2-(ℏω0)2 and (Eg – EF0)2-(ℏω0)2. This means that there are two types of CT transitions, as illustrated in Figure 4: The molecule-to-metal electronic transition (Figure 4a) and the metal-to-molecule electronic transition (Figure 4b). In ref (53), Lombardi et al. assume that |e0⟩ in eq 11a or eq 12a may be altered by |F⟩. This alteration indicates that CT interaction changes the molecular electronic states, thereby resulting in enhancement of the totally symmetric vibrations of the A term by the CT resonance effect.
Figure 4.

State diagram of the molecule–metal system. Transitions
are allowed from the molecular ground state |g0⟩ to one or more of the excited states |e0⟩ through ⟨g0|Mσ|e0⟩, where Mσ is the dipole
moment of the electronic transition. (a) For the B term, a molecule-to-metal
CT transition is allowed through ⟨g0|Mσ|F⟩.
The states |F0⟩ and |e0⟩ are connected through the HT vibronic coupling
term 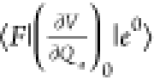 . For the C term, a metal-to-molecule CT
transition is allowed through ⟨F|Mσ|e0⟩. (b) States
|F⟩ and |g0⟩
are connected through the HT vibronic coupling term
. For the C term, a metal-to-molecule CT
transition is allowed through ⟨F|Mσ|e0⟩. (b) States
|F⟩ and |g0⟩
are connected through the HT vibronic coupling term 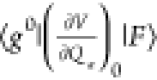 . Reprinted with permission from ref (26). Copyright 2016 John Wiley
& Sons, Ltd.
. Reprinted with permission from ref (26). Copyright 2016 John Wiley
& Sons, Ltd.
Two requirements on the molecular species need to be satisfied, to generate the CT resonant Raman effect. (1) The molecule must form a covalent bond with the metal surface and (2) the terms (Ee0 – EF)2 -(ℏω0)2 and (Eg0 – EF)2 -(ℏω0)2 in the denominators of eqs 12b and 12c must be small to obtain a large Raman polarizability. The first requirement imposes a condition for the electronic affinity between the molecule and a metal surface for the formation of CT complexes. Unpaired electrons of nitrogen or sulfur atoms in a molecule enhance such electronic affinity.78 The second requirement imposes the use of suitable Ee0 and Eg values for the π-electron orbital energy to attain large FB and FC values.79 Pyridine, pyrimidine, s-triazine, p-mercaptopyridine, and p-aminothiophenol, are known to satisfy this condition.80−82 The experimental evidence for the chemical mechanism is discussed in Section 5.1.
An excellent review by Lombardi et al. is highly recommended, which provides several examples that demonstrate the possible vibronic coupling in a metal–molecule system.83 The CT mechanism is a widely accepted explanation for semiconductor-enhanced Raman scattering, whereby the vibronic coupling mechanism in a semiconductor–molecule system is more complex than that in a metal–molecule system (see the detailed discussion in Section 5.4.1).
2. Nanometer to Subnanometer SERS HSs
2.1. Electromagnetic Enhancement of Various SERS HSs
The key to improving SM-SERS investigation is the effective development of HSs, which are existing gaps or junctions on plasmonic nanostructures.7,15,16,84 At these HSs, the EM field intensity is enhanced by up to 105, compared with its incident light, owing to coupling of the incident light and plasmon resonance of the plasmonic nanostructure. This results in a strong SERS signal by the EM mechanism. In this section, we introduce several types of basic HSs together with their plasmonic properties. HSs generate an FR2 in eq 6, of ∼1010, which enhances resonance Raman cross-sections of ∼10–24 cm2 to SERS cross-sections of ∼10–14 cm2, thereby enabling SM-SERS spectroscopy to a level of fluorescence cross-sections of ∼10–16 cm2.44,45 Various types of HSs have been developed by bottom-up and top-down methods to improve SM-SERS spectroscopy.84−86 The plasmonic properties of such HSs have been summarized in several review papers.7,15,16,84−86 Thus, we here briefly introduce the mechanistic studies of three basic HSs with respect to their configurations and plasmonic properties.
2.1.1. NP and Nanowire (NW) Dimers
We first introduce HSs at a gap or junction between a NP dimer. Such dimers have been confirmed to realize SM-SERS detection.49 SM-SERS was discovered by Kneipp and Nie using single NP aggregates or single NPs of silver or gold in 1997.44,45 By improving the spatial resolution of SEM, the minimum unit of such a NP was found to be a NP dimer by Xu et al. in 1999 (Figure 5A).49 The value of FR2 in eq 6, of ∼1010, required for SM-SERS detection, was theoretically predicted in a gap of a plasmonic NP dimer by Aravind, Inoue, and Xu using analytical calculations based on the EM mechanism (Figure 5B).49,63,64 The numerical calculations based on the EM mechanism have also predicted an FR2 value of ∼1010 for the HS of a NP dimer.50 Thus, the HS of a NP dimer has been widely accepted as a platform to realize SM-SERS research. Various types of dimers (gold, silver, gold and silver, and core@shell NP dimers; nanocube (NC) dimers; and their hybrid dimers) have been used for SM-SERS.25,87−94 These dimers have also been examined for other surface-enhanced spectroscopies, such as surface-enhanced fluorescence (SEF), surface-enhanced hyper Raman scattering (SEHRS), and other techniques.25,95
Figure 5.

(A) SEM images of immobilized silver NPs. (Aa) Overview of silver NP shapes and sizes, (Ab) Ag NP dimers observed after incubation with 10–11 M hemoglobin (Hb) for 3 h, and (Ac, Ad) SERS active dimers and corresponding single Hb molecule spectra. The double arrows in panels Ac and Ad indicate the polarization of the incident laser field. (B) Calculated EM EF for the midpoint between two silver spheres separated by d = 5.5 nm and for a point d/2 outside a single sphere. The solid and open circles indicate the position of the silver spheres and Hb molecule, respectively, in relation to the incident polarization vector (double arrows). Calculations were performed for spheres of diameters D = 60 (dashed curves), 90 (solid curves), and 120 (dotted curves) nm. Inset shows the plot of the enhancement versus D for an excitation light wavelength λI = 514.5 nm and a Stokes shift of 1500 cm–1 for configuration a. Reproduced with permission from ref (49). Copyright 1999 American Physical Society.
The origin of the high EFs at the HSs is the small value of VHS in eq 4.25 The small VHS of a dimer is due to the large coupling energy between the dipole plasmon resonances of two NPs. The value of FR attained from the dipole–dipole (DD)-coupled plasmon resonance is increased by decreasing the gap distance or increasing the degree of steepness in the junctions.25,49,63,64 The upper FR limit or minimum VHS value is determined by the overlapping between the unscreened electrons present on both metal surfaces as discussed in Section 1.2.1. The effective minimum gap distance was experimentally determined as ∼0.5 nm by the appearance of CT plasmon, which is a collective oscillation of the unscreened electron gas between the gaps or junctions, by closing or overlapping two TERS tips.69 The CT plasmon was theoretically evaluated by DFT calculation of a small metal cluster dimer.29 The EM mechanism was also modified by including dielectric properties of the unscreened electrons as a thin metal surface layer on a NP.29 Notably, the maximum FR value is reduced by the effect of the unscreened electron gas, when compared with the maximum FR value that does not consider this effect.
When the sizes of the NPs of a dimer differ significantly, the largest FR value is not generated by the DD-coupled plasmon but by dipole–quadrupole (DQ)-coupled plasmon resonance, which is a subradiant mode.96 In such a situation, the DD-coupled plasmon resonance contributes to transfer the excitation light energy to the DQ-coupled plasmon resonance, and emits SERS by receiving energy from the DQ-coupled plasmon resonance through near-field interactions.96,97 Briefly, SERS light generated by DQ-coupled plasmon resonance emits through DD-coupled plasmon resonance as discussed in Section 2.1.3. In this case, the plasmon resonance spectra obtained by dark-field spectroscopy are not identical to the FR spectra. Yoshida et al. excluded the contribution of DQ-coupled plasmon resonance to SERS spectra by selecting symmetric NP dimers that only induce DD-coupled plasmon.51,98
The volume of HS showing SM-SERS is only in the range of several nm3, indicating that the electric field inside a HS is a nanometer-scale structure.15,24,39,56 Thus, the fluctuation in the molecule positions induces SERS signal instability.71 Furthermore, the probability that a molecule can reach a HS is very low. This issue may be resolved by enlarging the VHS value. However, there is a trade-off between the VHS and FR for a HS of the dimer gap or junction as illustrated in eq 4. The NC dimer is expected to expand the HS area in two dimensions.24,39 However, the enhanced field experimentally showing SM-SERS sensitivity is only localized at the corners of the NC gap.24,39 Itoh et al. demonstrated that a gap or junction between NWs arranged in parallel, namely NW dimers, can be used for a SERS HS by DD-coupled plasmon resonance which direction is orthogonal to the NW long axis (Figure 6).99 The lengths of single HSs are one-dimensionally extended 104-fold to maintain a large FR value.99 SERS light propagates along this one-dimensional (1D) HS through coupling with the surface plasmons, wherein the coupled modes are formed from monopole, dipole, and quadrupole interactions.100 Such propagation phenomena of SERS light in one-dimensional HS may be useful for exploring the behavior of molecules that are strongly coupled with plasmon resonance inside the HSs.100 Another system comprises an NW with a NP attached on it, which enables remote excitation of the HS between the NP and NW through excitation of surface plasmon propagation.101,102
Figure 6.

(A): (Aa1) SEM image of an NW dimer. Insets: (Aa2, Aa3) enlarged SEM images of the edges of the dimer crevice. (Ab, Ac) Dark field and SERS images of the dimer. (d) Superposition of the SEM and dark-field image. (Ae) Superposition of the SEM and SERS images. Scale bars in (Aa–Ac) are 5 μm. (B): (Ba) Spectrum of the elastic light scattering for the NW dimer and (inset) monomer perpendicular (red line) and parallel (black line) to the dimer length. (Bb) Spectrum of the SERS intensity perpendicular (red line) and parallel (black line) to the dimer length. (Bc) Polarization dependence (circles and solid line) of the plasmon resonance maxima at 520 (red) and 720 (black) nm. (Bd) Polarization dependence of the SERS intensity at 560 nm (red circles and red solid line). Reproduced with permission from ref (99). Copyright 2017 American Physical Society.
2.1.2. NPs on a Mirror
Second, we introduce the HS of a gap or junction between a NP and a metal plane substrate. The coupling between the dipole plasmon of a NP and its mirror dipole on the plane also realizes high EM field enhancement, resulting in SM-SERS detection.15,56,69 This type of HS is suitable for measuring SM-SERS phenomena owing to its considerable stability. Specifically, the field enhancement in this NP–mirror system is stable against a few nanometer changes in the position of the NPs, because the HS does not disappear by such small position changes of the NP on the substrates.24 In contrast, the field enhancement of a HS at a NP dimer gap decreases significantly following such position changes of each NP of a NP dimer.103 The other benefit of this configuration is the use of surface plasmon propagation through the plane to excite the HS. Li et al. demonstrated remote excitation of the HSs on a gold substrate by surface plasmon propagation from a photoexcited NW on the gold substrate.104
The mechanism realizing a large field enhancement and small mode volume comprises coupling between the dipole plasmon of a NP and its mirror dipole on the plane substrates at the gaps or junctions between the NP and plane substrate.105,106 Thus, in this configuration, the VHS and FR in eq 4 become smaller and larger, respectively, with decreasing gap distance, as is observed for the NP dimer. The field enhancement in this configuration was formulated by Aravind et al. using analytical calculations based on the EM mechanism.105 Nordlander et al. developed the intuitive description of the substrate thickness dependence of the coupling as the interaction between the NP plasmons and the continuum of the surface plasmon of the plane substrate based on the Anderson–Fano model.107
The limitation of the EFs of the HS in in NP–mirror system is determined by the overlapping of the unscreened electrons between the gaps, as is observed for a NP dimer HS.69,70,108 This effect is called the nonlocal effect, and is defined as the quantum repulsion of an electron gas that resists the compression induced by an applied EM field.69,70,108 Ciraci et al. described the nonlocal effects by a hydrodynamic model of the collective motion of the electrons inside a metal.108 The model well predicts the plasmon resonance blueshifts, which do not appear in the model that only assumes local effects, and the saturation of the field EFs (Figure 7).108 The blueshifts have been experimentally confirmed by Ciraci et al.108
Figure 7.
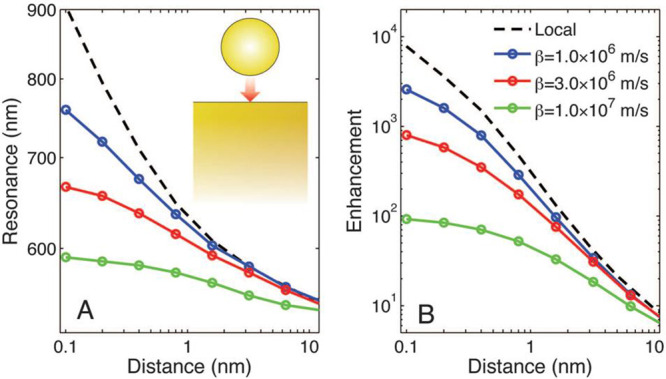
Behavior of the film-coupled nanosphere, assuming a local model and a nonlocal model with various values of β (nonlocal parameter), as a function of the separation distance. Calculations refer to a gold nanosphere of radius r = 30 nm on a 300 nm-thick film. (A) Position of the peak scattering intensity as a function of gap size. (B) Corresponding field enhancement ratio. Notably, in the absence of nonlocal effects, the peak scattering wavelength is extreme, and the field enhancement grows to enormous values; thus, nonlocality places a limit on the ultimate enhancement. Reproduced with permission from ref (108). Copyright 2012 American Association for the Advancement of Science.
This type of HS is suitable for SERS spectroscopy of the molecular physical and chemical fluctuations inside a HS.15,56,109−113 Furthermore, an atomic-scale subnanometer structure inside a HS generates a “picocavity,” for which VHS is smaller than 1 nm3 under cryogenic temperatures.56,109−111 The interaction between a molecule and confined electric field on the subnanometer structure inside a HS breakdowns the conventional selection rules of photoinduced transitions, resulting in the observation of both Raman- and IR-active vibrational modes.56 The large coupling energy from the ultrasmall VHS enables strong coupling between a plasmon and single molecular excitation.15,29 This has led to a new research field related to cavity QED, called “polariton chemistry,” which makes use of HSs.15,17 Thus, in polariton chemistry, the photochemical properties of molecules in a HS are modified by strong coupling and monitored by SERS and other surface-enhanced spectroscopies such as surface-enhanced absorption (SEA) and SEF, as discussed in Section 3.1.2.
2.1.3. Higher-Order NP Clusters
Third we introduce another HS located in the gaps or junctions inside higher-order plasmonic nanostructures, e.g., NP clusters. This type of HS uses higher-order coupled plasmon resonance such as Fano resonance.114 In fact, the plasmon resonance of SERS HSs, which is described in Section 2.1.2, is bright (superradiant) resonance (dipole plasmon resonance), which resonance generates a weaker enhanced field than the enhanced field of higher-order coupled plasmon resonance. This is because excitation light from a free space should resonate with plasmon, and scattering light should be sent to the free space from the plasmon.25 However, dark plasmon (subradiant) resonance (i.e., quadrupole plasmon) has an advantage for SERS HSs because the intensity of the enhanced field generated by dark plasmon is much higher than that generated by bright plasmon.96,97 Therefore, the higher enhancement is due to the lack or suppression of radiation loss in dark plasmon.96,97 The EM enhancement process of SERS by Fano resonance is indirect as those: the light energy which is received by bright plasmon resonance transfer to Fano resonance which is enhancing SERS signals and bright plasmon emits SERS light received from Fano resonance through near-field interactions.97 Experimentally, Fano resonance appears in the scattering spectrum as a dip-like structure, because bright plasmon resonance loses light energy by transferring it to the Fano resonance through near-field interactions.96,97,114 Here we introduce two experimental studies of SERS using Fano resonance.
Ye et al. applied the Fano resonance of gold NP heptamers to SERS and observed that a maximum SERS intensity could be obtained by tuning the excitation laser line at the dip in the scattering spectrum (Figure 8).97 This property is inverse to the relationship between the DD-coupled resonance scattering spectrum and SERS intensity.25 The property of the Fano resonance of heptamers was consistently reproduced by numerical simulation using the FDTD method.97 Zhang et al. applied the Fano resonance of a gold NP quadrumer to surface-enhanced coherent anti-Stokes Raman scattering (SECARS) with a high EF of ∼1.5 × 1010, achieving SM SECARS demonstration.115 The SECARS EFs were consistently reproduced by the FDTD method based on the EM mechanism.115
Figure 8.

Far-field scattering spectra, near-field enhancement spectra, and SERS properties of individual heptamers with varying sizes: (i) 85-, (ii) 130-, and (iii) 170 nm-diameter constituent disks. In all structures, the height of the disks is 30 nm and the gaps between the disks are ∼15 nm. (A) SEM images, (B) experimentally obtained dark-field scattering spectra, (C) FDTD-calculated scattering spectra, and (D) SERS spectra of a monolayer of para-mercaptoaniline (p-MA) molecules for the individual heptamers (i–iii) obtained with horizontal polarization. In panels B and C, the scattering spectra of the individual heptamers exhibit a significant red shift with chemisorption of a monolayer of p-MA molecules. Pristine structure (red) after p-MA binding (black). (E) Total near-field enhancement intensity (|E/E0|2) spectra calculated by integrating over all surfaces of the heptamer. (F) Spatial distribution of the SERS enhancement (=|Eex/E0|2 · |Estokes/E0|2) for the p-MA Stokes line at 1080 cm–1 for each of the individual heptamers (i–iii). Enhancement evaluated at half the height (left) and 2 nm above the top surface (right) of the cluster. (G, H) Calculated near-field enhancement (|E/E0|) spectra at HSs (G) 1 and (H) 2, indicated on the left side of the spectra in the accompanying SERS maps. Dashed lines in panels B, C, E, G, and H denote the excitation laser at 785 nm (black) and the Raman Stokes lines of p-MA at 1080 (blue) and 1590 (pink) cm–1. These modes are indicated by the blue and pink arrows in Dii, respectively. Reproduced with permission from ref (97). Copyright 2012 American Chemical Society.
Kleinman et al. measured the relationship between the SERS FR and plasmon resonance in scattering spectra using gold NP aggregates116 and observed that the two were not correlated. This uncorrelation was well reproduced by their numerical simulation. In their paper, they discuss the contribution of dark plasmon to FR and the discrepancy between the near-field effects, SERS, far-field effects, and dark-field scattering.116 Here we consider that their discussion is identical to the contribution of the Fano resonance to SERS.
Many theoretical studies have been carried out to clarify the relationship between the Fano resonance and field enhancement.117−119 However, the discussion in many papers reporting on this relationship remains experimentally insufficient with respect to SERS. This may be because of the difficulty in fully identifying the Fano resonance in the scattering spectra. Absorption spectroscopy of the HSs is required to directly measure the Fano resonance.96 Thus, many opportunities remain for exploiting the Fano resonance for SERS.
2.2. Monitoring of the Molecular Fluctuations at the SERS HS
Studies of the intensity and spectral fluctuations in SERS are useful for in situ probing dynamics in the ground and excited states of a molecule, such as movement, desorption, structural rearrangement, chemical reaction, and decomposition.44,45,49,120−124 From 1997, the SERS spectral fluctuation itself has been thought to be indirect evidence of SM observation.44,45,49 To obtain direct evidence, the bianalyte SERS technique has been employed for direct SM observation, in which a mixture of two chemically similar but Raman-distinguishable molecules has been used.125 The direct evidence of SM observation was achieved by the lack of simultaneous detection of isotopically differentiated dye SMs at the HSs in a concentration of ∼10–9 M.126 Based on the above background, we next introduce several recent mechanistic studies of physical and chemical fluctuations of a molecule inside a HS by SERS spectroscopy.
2.2.1. Physical Fluctuations at the SERS HS
We here introduce several SERS studies of the physical fluctuations inside HSs. These studies mainly extract positional information on the molecules at a resolution ranging from several nanometers to a few Ångströms (i.e., super-resolution SERS imaging),127 SERS and SEF intensity analysis,71 statistical analysis of on–off SERS blinking,128 spectral analysis broadening of SERS lines,129 and control of fluctuation using host–guest complexes.130
Super-resolution SERS imaging of the molecular fluctuation inside HSs has been first reported by Willets et al.127 In conventional optical microscopy, the separation distance should be larger than the diffraction limit. Super-resolution SERS imaging is the method used to determine the centroid position of an emitter within a resolution below the diffraction limit by two-dimensional Gaussian fitting or more complex dipole-based fitting for emitters separated from each other.127,131 The estimated position of the emitters by such fitting is a convolution between the EM field distribution and the position of the molecule emitting SERS. Interestingly, Willets clarifies that the centroid position of multiple molecules (MMs) in a HS is more stable than that of an SM, because of the averaging effect of the SERS signals.131 He also demonstrated that the centroid position becomes more unstable in an SM than in MMs by reflecting the fluctuation of the SM (Figure 9).131 In another study, Willet and his group demonstrated the SM motions by monitoring the SERS centroid position with a spatial resolution of several nanometers and a temporal resolution of 0.4 s.132 In this study, they also successfully discriminated isotopically differentiated dye molecules in a HS by super-resolution imaging of the SERS spectra.132 Recently, super-resolution imaging of SERS HSs has been used to differentiate chemical signatures from biological cells and shows great potential for application to other biological structures of interest.133,134 Super-resolution imaging of SERS has now been applied to analysis inside biological cells with a precision of up to 6 nm by using HSs as SERS nanotags.135
Figure 9.
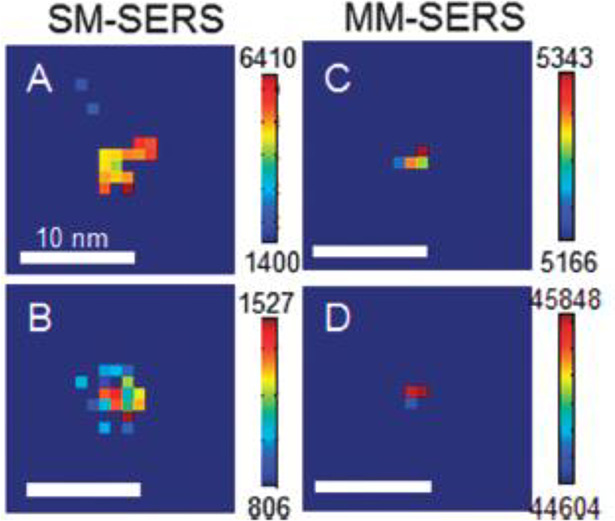
Spatial intensity maps for SERS active nanoparticle dimers in the (A, B) single-molecule and (C, D) multiple-molecule limits. Reprinted with permission from ref (131). Copyright 2014 Royal Society of Chemistry.
The fluctuations in the distance between a dye molecule and metal surface inside a HS were discussed in the scale of several Ångström through the analysis of the SERS and SEF intensity ratios.71 EM enhancement of SERS and SEF are identical because both are induced by a common dipole plasmon.136−138 However, the quenching of the two differs because SEF is much more sensitive for quenching by higher-order plasmon than SERS.136−138 Johansson et al. formulated the EM enhancement and quenching for the SERS and SEF intensities as a function of the distance between a molecule and metal surface d, and showed a difference in d that is dependent on SERS and SEF, as discussed in eq 4.138 Galloway et al. estimated d by comparing the SERS and SEF intensities and showed that d is in the scale of several Ångströms.136 Based on these studies, Itoh et al. evaluated the temporal fluctuations in d inside the HSs.71 They observed that the temporal fluctuation in SERS and SEF corresponds to the fluctuations in d in the region from 0.4 to 1.0 nm.71 This method shows great application potential as an analysis method for the molecular positions on plasmonic nanostructures with subnanometer scale resolution.103
The optical potential inside a HS, which restricts the several-nanometer fluctuation of a molecule, was investigated by Kitahama et al. They employed SERS fluctuation analysis based on a diffusion-controlled electron transfer model, which was developed for fluorescence blinking of a single quantum dot.128 Restriction of SM motion by the optical fields enhanced by plasmon resonance was theoretically predicted in the literature. However, it was difficult to directly observe this phenomenon because the energy of the optical potential is much smaller than the potential energy of Brownian motion.139 Kitahama et al. demonstrated such restriction by the analysis of the probability distribution of bright and dark states using on–off SERS fluctuation.128 Further, a simple exponential kinetics model has led to power-law statistics in the probability distribution of bright and dark states.140 However, a potential barrier between the bright and dark states breaks the power-law statistics, resulting in a bending or truncating tail in the probability distribution.140 Kitahama et al. reported the truncation in the probability distribution of dark states.128 Regarding such optical potential, a molecule inside a HS (in the bright state) is exposed to a deeper potential than a molecule outside a HS (in the dark state). Specifically, Kitahama et al. analyzed this truncation as an effect of optical potentials on the molecular motion.128 They also applied the method to simultaneously determine the truncation of the SERS of a dye molecule and fluorescence of Ag clusters inside a HS.141 Notably, if this method is combined with super-resolution imaging, the potential distribution inside a HS can be analyzed.
SERS spectral fluctuation results in inhomogeneous broadening of the SERS spectral lines, i.e., interactions with the substrate, isotopic effects, and instrumental contributions.129 Improvement of the CCD camera performance has led to a decrease in both the detection rates of the SERS intensity and detection duration of the SERS spectra to values of only 1 MHz and 50 ms, respectively.142,143 Thus, such inhomogeneous broadening in the SERS spectra can be directly discussed as the molecular dynamic of SM on a metal surface by excluding the random noise, impurity, and time-average effects under ambient conditions.142,143 Zong et al. also successfully demonstrated the neutral state of the R6G dye—stabilized by a cationic state in the SERS HSs—as rare events, by separating them from popular and impurity events during SERS fluctuation (Figure 10).143 Such a high-speed SERS measurement also shows great potential to separately provide both molecular vibrational and electronical information to allow the better understanding of surface catalysis and biochemical processes in living cells.
Figure 10.
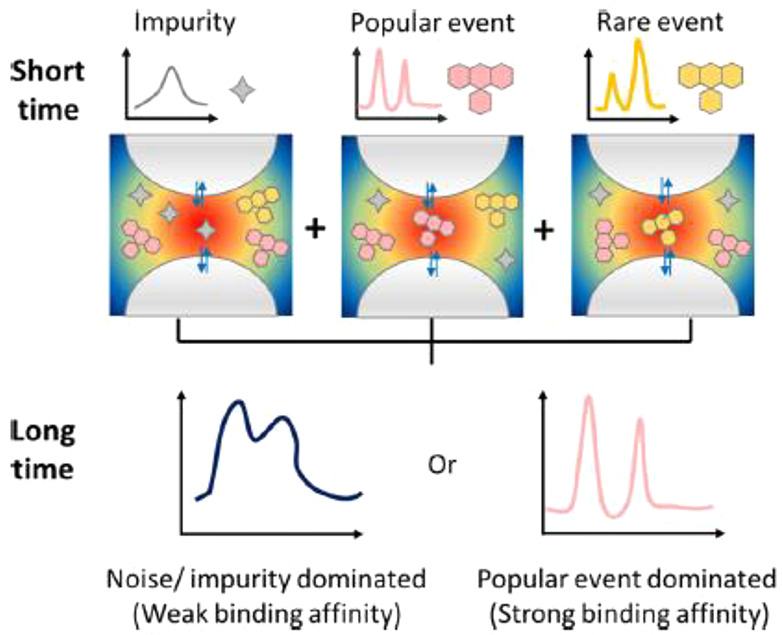
Schematic diagram of dynamic SERS detection. In the traditional SERS measurement (long time exposure), either the popular events or the noise-dominated SERS spectrum is obtained. In the dynamic SERS measurement (short time exposure), the signals from the impurity and popular and rare events of the target are well-distinguished. Reproduced with permission from ref (143). Copyright 2020 American Chemical Society.
The SERS spectral fluctuations of a host–guest complex inside a HS have been used to monitor the guest molecule fluctuation inside the host molecule.130 In this study, the HS was composed of a silver NP and silver substrate bridged by a host molecule as previously discussed.130 Cucurbit[n]urils (CB[n]), macrocyclic host molecules with 0.9 nm dimensions, have been used as host molecules.144 The host molecule works as a stable HS inside the gap of the NPs with a distance of ∼0.9 nm. Thus, one can detect the process of a single guest molecule entering inside the host molecule by spectral changes in the SERS. This insertion of the guest molecule into the host molecule was possible because of the intrinsic affinity between the guest and host molecules.130 The electrochemical (EC) or photochemical control of the guest molecule inside a HS has also been reported.145 Furthermore, such a HS has been used for the simultaneous detection of spectral changes in SERS and plasmon resonance, indicating strong coupling between a plasmon and guest exciton as discussed in Section 3.1.15 The combinations of host–guest complexes are versatile and the HS using a CB has high stability and reproductivity for analysis of SERS spectral fluctuation,146 resulting in various applications including the identification of guest molecules and monitoring of photochemical reactions inside the CB.147,148
2.2.2. Chemical Fluctuations at the SERS HS
In this section, we focus on the mechanistic studies of the SERS spectral fluctuation, reflecting photochemical reactions inside the HSs. Their applications to EC and electrocatalytic processes will be discussed later in Section 6.2.2. Analysis of the SERS spectral fluctuation has provided various information on the chemical interactions between two molecules or a molecule and metal surface inside a HS.7,18,149 The fluctuations in SM-SERS from a HS have been investigated to clarify the chemical interactions, excluding inhomogeneity, wherein the relationship between the reactions and plasmon-induced hot carriers was also been discussed.150 Research of the chemical fluctuations in SM-SERS has been mainly carried out using model reactions such as photocatalytic reduction from 4-nitrobenzenethiol (NBT) to 4-aminobenzenethiol (ABT) or 4,4′-dimercaptoazobenzene (DMAB) (Figure 11).151 Photocatalytic oxidation from 4-aminothiophenol (4-ATP) to 4-nitrothiophenol (4-NTP) has also been carried out.152 Dye SMs were also used to analyze the chemical fluctuations of SM-SERS. Lombardi et al. measured the SERS spectral fluctuation of R6G SMs and observed on–off cycling of the vibronic coupling modes, which are evidence of the resonance Raman effect, as discussed in Section 1.2.2.153 They discussed the on–off cycling in terms of the appearance and disappearance of the CT resonance between an R6G SM and a metal surface.153 Habuchi et al. measured the SERS spectral fluctuation of the exocyclic C=C mode of a green fluorescent protein SM, and discussed the fluctuation in terms of molecular protonation and deprotonation.123 Moreover, Itoh et al. measured the SERS spectral fluctuation of the C–N mode of a tetraphenylporphine SM and discussed Ag–N bond formation between tetraphenylporphine and a metal surface.154
Figure 11.

SERS trajectory showing a spectral feature (peak at νHABT ≈ 1360 cm–1) of a reaction intermediate. Discrete blinking of this peak is also temporally anticorrelated to those of DMAB (ν1, ν2, and ν3). The blinking in this case corresponds to the reversible interconversion of a single DMAB and single intermediate molecule (see inset). The intermediate is assigned to be hydroxylaminobenzenethiol (HABT). The right panel shows selected SERS spectra at the number position. Reprinted with permission from ref (151). Copyright 2019 American Chemical Society.
Over this past decade, several important chemical reactions have been achieved on plasmonic metal surfaces under conditions that deviate from their conventional reaction conditions, such as H2 dissociation, CO2-to-fuel conversion, water splitting, ammonia synthesis, and catalytic oxidation reactions (e.g., ethylene epoxidation, CO oxidation, and NH3 oxidation).155−159 These reactions are called plasmon-enhanced chemical reactions, and both SERS and TERS have been important tools for the analysis of their mechanisms.160 The insight obtained from the analysis of these chemical reactions is the contribution of hot carriers, tentatively generated after the dephasing of plasmon (coherent oscillation of conduction electrons), within a few picoseconds, induced by plasmon resonance.161 There are several reviews of these plasmon-induced chemical reactions and their analysis methods.19,160,161
There are still many unclear points regarding the optimization of the plasmon-enhanced photochemical reaction. Cai et al. demonstrated the importance of steric hindrance and the energetic barrier effect on the plasmon-enhanced photochemical coupling reaction using TERS imaging, STM, and DFT simulations.162 They investigated the chemical conversion of 4-NTP to DMAB on single-crystal Au(111) and polycrystalline TS-Au surfaces by TERS spectral changes and clearly showed a higher reaction efficiency in the polycrystalline TS-Au phase compared to that observed with the thermodynamically single-crystal phase. Furthermore, they compared the reaction efficiency of a 4-NTP adlayer on Au(111) between the disordered phases (the low coverage ∼7.2%) and ordered phases (the high coverage ∼72.3%) and showed a higher reaction efficiency in the disordered phase. These results were simulated by a DFT method and confirmed that a combination of a steric hindrance effect and energetic barrier clearly favors a higher reaction efficiency in the disordered phase, as compared to that of a well-packed ordered phase.162 These fundamental insights are important to establish suitable environmental conditions for photocatalytic coupling reactions.
Clarification of the relationship between plasmon-induced hot electron and plasmon-enhanced chemical reaction is important to fundamentally understand the reaction mechanism. Thus, to understand the mechanism, Huang et al. developed a method for the simultaneous measurement of the photoinduced reaction and photoinduced current using SERS and an ultramicroelectrode.163 They observed that the conversion of 4-ATP to form DMAB simultaneously occurs with the generation of a photoinduced current (Figure 12). They linked this reaction to the activated oxygenated species by hot-electron transfer. Moreover, they compared the photocurrent with the conventional EC current of the oxygen reduction reaction, indicating, with the support of DFT calculation, that the activated oxygenated species are oxidative in transforming 4-ATP to DMAB.
Figure 12.

Schematic comparison between conventional EC-SERS on a macroelectrode (a) and the microphotoEC (μPEC)-SERS on an ultramicroelectrode (UME) (b). (c) Schematic illustration of the correlation of the plasmon-mediated chemical reactions (PMCRs) with hot electron transfer in μPEC-SERS, (d) optical microscopy image of the UME in the μPEC-SERS measurement, and (e) SEM image of the SERS-active silver UME. Reproduced with permission from ref (163). Copyright 2020 American Chemical Society.
Hot carriers are well-known to play important roles in plasmon-enhanced photocatalytic reaction and light harvesting.161 However, there is difficulty in directly measuring hot charge carrier generation, resulting in most measurements being performed on bulk scales or under ultrahigh vacuum. de Nijs et al. investigated the behavior of a hot electron under ambient conditions using a stable HS between a NP and a mirror containing a CB molecule at the HS.164 Using SERS spectral fluctuations they demonstrated the changes in efficiency of the coherent tunneling of hot electrons through a gap made by a NP and a mirror, which HS comprised several types of SMs (biphenyl-4,4′-dithiol, 4,4′-bis(mercaptomethyl)biphenyl, 5,5′-bis(mercaptomethyl)-2,2′-bipyridine, ether bipyridine, 1-methyl-4-phenylpyridinium iodide, and methyl viologen dichloride), which exhibit different degrees of preventing coherent tunneling. In this study, SERS spectral fluctuation analysis clarified that hot-electron-induced redox processes are largely enhanced by the increase in the prevention of coherent tunneling. Briefly, they demonstrated that the transition from coherent to hopping electron transport occurs by an increase in the tunneling barrier height and dephasing strength.164 This result signifies that the observation of redox processes in real time at the SM level was achieved.
Apart from the well-investigated plasmon-enhanced photochemical reactions, such as the conversion of 4-ATP to DMAB, there are many other unclear photochemical reactions that occur inside HSs. Szczerbinski et al. demonstrated a TERS study to clarify the mechanism of these unclear reactions inside a single HS.165 They measured the TERS spectral changes in the reaction products using self-assembled monolayers of 1-hexadecanethiol, biphenyl-4-thiol, and 1H,1H,2H,2H-perfluorodecane-1-thiol molecules. Interestingly, they observed that for these products, the plasmon-enhanced photochemical reactions are similar to the ones already known from X-ray or e-beam-induced surface chemical reactions, despite the >100-fold energy difference between visible and X-ray photons. In the X-ray or e-beam-induced surface chemical reactions, such products are generated by secondary electrons. Therefore, formation of such similar products indicate that hot electrons play a similar role to that of secondary electrons. Moreover, in both cases, the formation of side products (amorphous carbon) occurs upon desorption of molecular fragments from the surface in the presence of secondary/hot electrons that drive the carbonization.165
Plasmon-induced carbonization is one of the interesting topics in plasmon-enhanced photochemical reactions. Raman spectroscopy of sp2 carbon systems has been extensively investigated in terms of strain and stress, disorder, oxidation, hydrogenation, and nitridization.166 Thus, plasmon-induced carbonization can be evaluated from the correlation between the SERS fluctuation and these previous studies on sp2 carbon systems. Itoh et al. reported the SERS analysis of a single sp2 carbon cluster generated from near-single dye molecules in single HSs of silver NP dimers.167 They observed that the number of SERS lines of the dye decreased after the SERS spectrum fluctuation, with two lines finally remaining around the G and D modes of the sp2 carbon cluster. They estimated the crystallite size of this cluster to be less than 2.0 nm from the G line width. They also showed that temporal changes in the ratios of the G and D lines can be attributed to the changes in the crystallite size in the order of several Ångströms. Thus, they concluded that the temporal SERS spectral changes can be attributed to the generation and annihilation of topological defects inside the sp2 carbon systems, showing the usefulness of the SERS fluctuation for structural fluctuations of SMs with an order of several Ångströms.167
3. Unique Phenomena Occurring at the Subnanometer SERS HSs
The unique interactions that occur between the electric field confined by plasmon resonance and a molecule inside a HS have been extensively studied by SERS, TERS, and other SEF spectroscopies. This is because such interactions can be applied to control the molecular properties,15−17,27 and thus, the photochemical reactions.28,168−172 There are several reviews related to phenomena induced by these interactions, including modified emission, changes in the excited-state quantum yields, effects on photochemical reactions, vibrational strong coupling, effects on the ground state, intermolecular communications, and energy transfer.28−31
However, the mechanisms of such phenomena remain unclear, resulting in missing or omission of many important results. This lack of clarity of the mechanism is increased by the classical assumptions used in spectroscopy. Indeed, the coupling energy, amplitude of vacuum fluctuation, and size of the electric field inside HSs are beyond these assumptions.28−31 Here we point out four types of the phenomena, labeled I to IV below, induced by the breakdown of the assumptions inside HSs.
-
(I)
Strong coupling between plasmon and exciton resonances. In eq 1, the starting point for the theory of SERS, we naturally assume a weakly coupled regime. However, if the coupling rate Ωvac, as in eq 1, between the plasmon and molecular exciton resonances becomes comparable to or larger than their dephasing rates, both resonances coherently exchange excitation energy. This results in hybridization of both resonances, a phenomenon called strong coupling.17,25,27,173 Thus, the weak coupling approximation cannot be applicable to the hybridized system because |i⟩ and |f⟩ in eq 1 are not equal to the ones observed in a weak coupling system.
-
(II)
Ultrafast SEF. Another assumption to be considered exists in eq 5. If the FR in eq 5 enhances the radiative rate so that it becomes greater than the molecular vibrational decay rate of the electronic excited state, the molecule emits SEF from the vibrational excited state in the electronic excited state.25,27,174 This SEF indicates the breakdown of Kasha’s rule, in which a molecule is supposed to emit fluorescence from the vibrational ground state, and has been investigated as blue-shifted SEF or excitation-energy dependent SEF.25,27,174
-
(III)
Vibrational pumping effect on anti-Stokes SERS. If the FR in eq 5 enhances the excitation rate so that it becomes greater than the molecular vibrational decay rate in the electronic ground state, the molecule emits unexpectedly strong anti-Stokes Raman light from the vibrational excited state in the electronic ground state.175,176 This anti-Stokes Raman indicates the breakdown of the assumption that anti-Stokes-to-Stokes ratios can be expected from a Boltzmann distribution, and has been investigated as the excitation power dependence of the anti-Stokes Raman intensity.175,176
-
(IV)
Field-gradient effect on SERS. If the local electric field-gradient inside the HSs becomes comparable to the size of the electronic structure of a molecule, the long wavelength approximation, in which the size of the electronic structure is much smaller than that of the electric structure of light, breaks down.27,56 This breakdown signifies that ⟨f|ex|i⟩ and E in eq 1 cannot be expressed by a simple product. Hence, the excitation of multipoles, such as a quadrupole, which probability is negligible in most cases of excitation by light in free space, becomes non-negligible. The selection rule for the molecular electronic excitation therefore needs to be modified. This modification has been investigated through the observation of forbidden transition modes.56
3.1. Strong Coupling between a Plasmon and Molecular Exciton at the SERS HS
This phenomenon is induced by an enlarged coupling energy ℏΩvac in eq 1 between a plasmon and molecule exciton inside a HS. The breakdown of the weak coupling approximation results in hybridized resonance between the plasmon and molecule exciton resonances.17,25,27,173 Here we review recent studies related to both SERS and hybridized resonance. The hybridized resonance systems, as discussed in I in Section 3 exhibit different optical and chemical properties from the original systems.28,168−172 One can optimize these properties by controlling the hybridized resonance. Such studies have also been performed for QED-like low-threshold lasing and the fabrication of ultrafast optical switching devices, among others.28,168 SERS is a fundamental tool to investigate these phenomena.17,24,25,27,47 A comprehensive review treating this strong coupling has been published recently;177 thus, here we focus on strong coupling realized by SERS HSs.
3.1.1. Theoretical Approaches to Strong Coupling at the SERS HS
We introduce several models to evaluate the strong coupling of a dimer with SM inside the HS. Strong coupling properties are evaluated by analyzing the anticrossing properties, observed by tuning plasmon resonance peaks across the exciton resonance.173 We first explain the coupled-oscillator model composed of plasmon and molecular exciton resonances.178,179 In this model, the molecular exciton is assumed to be a two-level system (Figure 13A). This model illustrates the relationship between the spectral changes of the hybridized resonance and coupling energy (Figure 13B). We proceed to explain a modified coupled-oscillator model, in which the molecular exciton is a multilevel system reflecting the FC mechanism.68 This model provides more realistic spectral changes in the hybridized resonance that reflect the multilevel properties. Both of these models explicitly include the coupling energy. Finally, we describe an analytical model based on electromagnetism.180 In this model, the coupling energy is implicitly included in the dielectric parameter and structures of the dimer and molecule.
Figure 13.
(A) Schematic image of the strong coupling between the plasmon and molecular resonances. Here, |g⟩ and |e⟩ are the ground and excited states of the two-level system, respectively; |0⟩ and |1⟩ are the zero- and one-photon states of the plasmon resonance; 2ℏg indicates vacuum Rabi splitting (VRS), which is the energy split of the excited state of the strong coupling system formed by the two-level system and plasmonic resonance. The red and blue arrows indicate the two coupled resonance energies. (B) Calculated anticrossing property of the strong coupling between a plasmon and an exciton of a two-level system with a coupling energy of 400 meV. Inset: Absorption spectrum of the aqueous solution of R6G (green curve). Reproduced with permission from ref (17). Copyright 2021 Royal Society of Chemistry.
We next show a numerical model based on electromagnetism using the FDTD method.178,181 This numerical calculation model is applicable to arbitral dielectric parameters and structures, correctly reproducing the experimental results. We proceed to explain a model based on QED and a time-dependent density-functional theory (TDDFT) model.182,183 In this model, a time-dependent Schrödinger equation of a general quantum system is directly solved to reproduce the strong coupling. This model includes quantum effects, i.e., electron-tunneling CT plasmon and metal–molecule CT effects.
The coupled-oscillator model has been frequently used to evaluate the coupling energy of hybridized resonance.173,178,179 In this model, two oscillators represent a dipole plasmon resonance and the dipole electronic transition resonance of a SM, respectively. Both oscillators are coupled through the electric near-field of the vacuum fluctuation (not external fields) with a coupling energy of ℏΩvac in eq 1. The equations of motion for the two oscillators are composed of oscillation, dissipation, and restoring force terms, a term for coupling with another oscillator, and an external force term, as follows:
| 13 |
| 14 |
where xp and xm are the coordinates of the plasmon and molecular exciton oscillations, respectively; γp and γm are the dephasing rates of the plasmon and molecular exciton resonances, respectively; ωp and ωm are the resonance frequencies of the plasmon and molecular electronic transitions, respectively; and Fp and Fm are the driving forces of the two oscillators, respectively. The driving forces represent the external electric field of light. Fm(t) is usually set to zero because the molecular extinction cross-section is negligible compared to that of the silver NP dimer: Fp(t) ≪ Fm(t).178,179 By assuming the driving force as Fp(t) = Fpe–iωt, where ω is the incident light frequency, xp(t) and xm(t) can be derived as solutions of eqs 13 and 14. In the quasi-static limit, the scattering cross-section is described as Csca = (8π/3k)|α|2,65 where k = ωn/c is the wavevector of light and α = Fpxp is the polarizability of the dimer. By substituting xp(t) into this expression, Csca can be described as
| 15 |
The elastic light-scattering spectra are evaluated by changing the value of Ωvac in eq 15. The evaluation is carried out by tuning the plasmon resonance energy against the exciton resonance energy. The split gap in the hybridized resonance (vacuum Rabi splitting) corresponds to 2ℏΩvac, as illustrated in Figure 13B. Wu et al. compared the results of the coupled-oscillator model with those from the FDTD method.178 Both sets of calculations indicate common optical properties in the elastic light-scattering spectra of the silver NP dimers which HSs contain quantum dots, e.g., the vacuum Rabi splitting and Fano interference induced by the strong coupling.178 These results show that the classical coupled-oscillator model can be applied to evaluate the strong coupling raised inside HSs.
Itoh et al. proposed a modified coupled-oscillator model.68 The coupled-oscillator model assumes a molecule as a two-level system.178 However, in real situations, the FC mechanism, which provides electron–vibration coupling, can reproduce multilevel properties using phonon replicas of the exciton line.137,138 The molecular–exciton oscillator of eq 14 is therefore modified into multiple oscillators representing this multilevel property. Thus, the equations of motion become
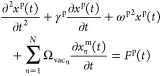 |
16 |
| 17 |
where xnm indicates the coordinates of the plasmon or the nth exciton oscillation (n = 1 indicates an exciton and n > 1 indicates its phonon replicas), respectively; γn is the corresponding line width; ωnm is the corresponding frequency; and ℏΩvacn is the coupling rate between the plasmon and nth exciton.68 The oscillator strengths, ωn, and γnm, of the nth exciton resonance can be derived from the absorption spectra, as shown in Figure 14a. In the quasi-static limit, Csca is obtained from the equation
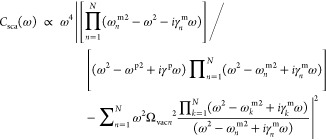 |
18 |
Figure 14.

(a) Absorption spectrum of an aqueous solution of R6G (red curves) fitted with four Lorentzian curves representing the exciton and its three phonon replicas (dashed curves); ωnm and γn are indicated in the panel. The black curve is the sum of the Lorentzian curves and values of fn are indicated in the panel. Inset: Photoinduced transitions from the electronic excited state S1 to the electronic ground state S0, including the vibrational energy levels corresponding to the four Lorentzian peaks. (b) Experimental plasmon resonance spectra before (red curve) and after (black curve) the loss of SERRS activity. (c) Spectra calculated by the coupled oscillator model (eq 18) with ℏg1 = 500 meV using a single exciton (dashed red curve), an exciton with three phonon replicas (red curve), and no exciton coupled with the plasmon (black curve). The hybridized resonances in the higher energy region are indicated by a dashed arrow (strong coupling between the plasmon and a single exciton) and solid arrows (strong coupling between the plasmon and a single exciton with its phonon replicas), respectively. (d) Anticrossing properties appearing in the plasmon resonance spectra calculated using eq 18 under the conditions with ℏΩvac 2 values of (d1) 0, (d2) 50, (d3) 100, (d4) 150, (d5) 200, and (d6) 250 meV. Reproduced with permission from ref (68) for panels a–c. Copyright 2018 American Institute of Physics. Reproduced with permission from ref (180) for panel d. Copyright 2020 American Institute of Physics.
Itoh et al. compared the results from the model with the experimental results, as shown in Figure 14b,c.68 The experimental spectral changes of the silver NP dimers, which HSs contain near-SM, were reproduced as the multiple strong coupling between a plasmon and an exciton with a coupling energy of approximately 200 meV. However, if such spectral changes are evaluated without considering the multilevel properties, one may largely overestimate the coupling energy, as shown in Figure 14d.180
We next discuss an analytical model based on classical electromagnetism. The previous two models explicitly include coupling energy. In the electromagnetism model, the coupling energy is implicitly included in the dielectric properties of the materials and the structures of the system, i.e., the imaginary part of the dielectric function is connected to de-excitation by vacuum fluctuation.180 In the model, the dimer including a molecule is represented as a dimer coated with a very thin molecule film. The polarizability of a coated NP dimer was formulized by Ruppin.184 Thus, the anticrossing can be evaluated using the elastic scattering spectrum of the coated dimer by changing the parameters, such as the molecule-film thicknesses, gap distances, and refractive indexes of its surrounding media.180 Itoh et al. demonstrated that the anticrossing characteristics of the coated dimers are most effectively observed by changing the refractive indexes of the surrounding media because other parameters do not need to be changed. Anticrossing appears even when the coated film thickness is only 0.3 nm.180 Considering that the size of the dye molecule is ∼0.5 nm, this result indicates that anticrossing is induced by near-SM conditions.
The FDTD method has been used to evaluate the strong coupling of various SERS HSs.181,185−187 In the FDTD method, Maxwell’s equation is discretized with Yee’s mesh.51 The accuracy of the calculations depends on the mesh size because the EM fields are averaged within the mesh.51 To correctly reproduce the spatial structures of the EM fields inside the SM-SERS HSs, the mesh size should be approximately 0.2 nm.51 Chikkaraddy et al. calculated the coupled resonance spectra with a mesh size of 0.3 nm, under the condition that the dye molecules are located inside an SERS HS between the gold NPs and a gold substrate.187 They demonstrated a high coupling energy of approximately 150 meV in the hybridized resonance spectra by placing 10 dye molecules inside a HS with mode volumes of less than 50 nm3, as shown in Figure 15. The calculations well reproduced the experimental results, indicating that FDTD is a powerful tool for analyzing the strong HS couplings of arbitral structures.
Figure 15.

(a) FDTD simulated scattering spectrum from nanoparticle-on-mirror
(NPoM) geometry with a Lorentz absorbing material with 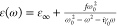 , where ε∞, oscillator
strength f, resonant energy ℏω0, and dephasing energy ℏγ0 are 1.96, 0.27,
1.86, and 85 meV, respectively. (b) Experimentally obtained scattering
spectrum from NPoM. (c) FDTD-simulated near-electric field maps at
ω+ (595 nm), ω0 (665 nm), and ω– (756 nm) [blue dashes in panel a]. Reproduced with
permission from ref (187). Copyright 2016 Nature Research.
, where ε∞, oscillator
strength f, resonant energy ℏω0, and dephasing energy ℏγ0 are 1.96, 0.27,
1.86, and 85 meV, respectively. (b) Experimentally obtained scattering
spectrum from NPoM. (c) FDTD-simulated near-electric field maps at
ω+ (595 nm), ω0 (665 nm), and ω– (756 nm) [blue dashes in panel a]. Reproduced with
permission from ref (187). Copyright 2016 Nature Research.
The TDDFT, which directly solves the time-dependent Schrödinger equation of a general quantum system, is a potential method for automatically including quantum (electron-tunneling CT and EM) effects in the calculation.183 Neuman et al. developed the analytical expression of the TDDFT model, which can quantum mechanically treat the QED of both the molecule and dimer.182 In the electromagnetism-based models, the bulk dielectric functions are used for the dielectric properties of the dimer.180,181,185−187 However, this use is not suitable for HSs because of the presence of an electron tunneling effect when the NP separation distance is below 0.5 nm, as discussed in Section 1.2.1.69,70 In addition, an electromagnetism-based method does not account for electronic interactions between the molecules and metal surfaces, such as the CT effect.183 The TDDFT model automatically includes such effects. However, the TDDFT method has difficulties describing long-range collective effects. Thus, the TDDFT method has been applied to relatively small metal clusters composed of <2,000 atoms adsorbed by a small molecule such as pyridine. Rossi et al. calculated the coupled resonance spectra of a benzene molecule bonded to the surface of an aluminum cluster (Figure 16).183 They demonstrated that the anticrossing in hybridized resonance corresponds to a coupling energy of approximately 200 meV with a VHS of 3.3 nm3. Such an approach can be applied to the SERS HSs of NP dimers to reveal the modification of molecular electronic structures and their dynamics by strong coupling. We expect that this approach will unify the EM and chemical mechanisms because the low branch of hybridized resonance may change a nonresonant SERS condition to one that is resonant. This expectation is further supported by the appearance of vibronic coupling terms in eqs 11b and 11c in the SERS spectra.
Figure 16.

Strong plasmon coupling
in Al nanoparticles (NPs) and excitations
in benzene molecules. The calculated photoabsorption spectra of NP–molecule
systems composed of (a) Al201, (b) Al586, and
(c) Al1289 NPs coupled with N benzene
molecules at 3 Å separation (solid lines) clearly show separated
lower and upper polaritons, the splitting of which increases with
increasing N. Spectra of the bare Al particles and
benzene molecule are shown for reference (dotted lines). (d) Coupling
strengths of the NP–molecule systems. Coupling strengths g for different numbers N of molecules
placed 3 Å above the NP. The symbols denote the fitted g values from the data in Figure 2 of ref (183). The dotted lines mark
the ideal theoretical coupling strengths 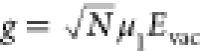 , where μ1 and Evac are the transition dipole moment and amplitude
of vacuum fluctuation, respectively. The dashed lines mark the ideal
dependence multiplied by the efficiency factors between the exciton
and cavity η for η201 = 0.47, η586 = 0.61, and η1289 = 0.78 (see Figure 4a and its in-text description
for details of ref (183)). Reproduced with permission from ref (183). Copyright 2019 Nature Research.
, where μ1 and Evac are the transition dipole moment and amplitude
of vacuum fluctuation, respectively. The dashed lines mark the ideal
dependence multiplied by the efficiency factors between the exciton
and cavity η for η201 = 0.47, η586 = 0.61, and η1289 = 0.78 (see Figure 4a and its in-text description
for details of ref (183)). Reproduced with permission from ref (183). Copyright 2019 Nature Research.
3.1.2. Experimental Approach to Strong Coupling at the SERS HS
Comprehensive reviews of strong coupling plasmonic systems have been recently published.177,188 Thus, here we mainly review the experimental studies of the relationships between SERS and strong coupling using HSs. The evidence of strong coupling, which is the anticrossing appearing in the dispersion curve of the hybridized resonance (Figure 13B), has been mainly investigated using the surface plasmon of metal surfaces. This is because its resonance energy can be tuned against the exciton resonance energy by changing the excitation angles.173 Such tuning techniques are not applicable to dimer HSs or NPs on mirrors. Notably, because the number of dimers should be measured to examine the anticrossing properties,177,188 studies of the relationships between SM-SERS and strong coupling using HSs are rather limited. However, these relationships are fundamentally important for its application, i.e., polariton chemistry.28,168−172 This is because the excited state dynamics, such as the energy transfer from the hybridized to the dark exciton states, which properties are the same as those of the original exciton states, becomes dominant as N (the number of molecules) inside the HS increases.17,168 In Section 3.1.2, we first review the strong coupling with SERS using single NPs,181,189−194 followed by collective HSs,195,196 the single HS of a NP dimer,197−202 and finally, the single HS between a NP on a mirror surface containing a small number of molecules.187,203−207
The strong coupling between the plasmon of a single isolated NP and molecular exciton has been investigated by several groups,181,189−194 all of which reported strong coupling systems composed of an isolated NP (including a spherical NP, nanorod (NR), nanoprism, and nanoring) with molecules on it.181,189−194 Two different methods were applied to tune the plasmon resonance energy against the exciton resonance energy for the isolated NPs. The first is the collection of multiple NPs with different plasmon resonance energies.181,189−192 The key to this method is a uniform environment surrounding the NPs. The second method involves changing the refractive index surrounding the NPs.192−194 The coupling energies around several hundred meV were reported by covering or attaching J-aggregate molecules on the NP. Shegai’s group investigated the relationships between the hybridized resonance and SERS with SEF, including spectral changes in the SEF by hybridized resonance.189−191 Zhang et al. demonstrated the strong coupling of plasmon and two types of excitons.193 Specifically, they showed that hybridized resonance is further hybridized by other excitons, resulting in a triplet-like spectral shape (Figure 17). They also discussed the possibility of the ultrafast control of two excitons via strong coupling. These hybridized resonance spectra are reasonably reproduced by the coupled oscillator model (Section 3.1.1).181,189−192 Shegai’s group reported the application of strong coupling to photochemistry, as suppression of the photo-oxidation of organic chromophores by the coupling energy.191
Figure 17.

Schemes of strong coupling plexcitonic nanosystems with resonant (upper) and detuned (lower) emitters. (a, d) Energy level diagrams showing the couplings between the plasmon and emitters: (a) Resonant emitter ensemble (e.g., single type of excitons) collectively represented by a single two-level system and (d) detuned emitter ensemble (e.g., diexcitons) treated as two different two-level systems. (b, e) Representative constructions of the single-excitonic and diexcitonic strong coupling nanosystems: (b) AuNC coated with a layer of J-aggregates (AuNC@J-agg) and (e) AuNC@J-agg hybrid nanoparticle (HNP) sitting on the WS2 monolayer. (c, f) Optical characteristics of the respective nanosystems: (c) Experimentally measured scattering spectra showing Rabi splitting (top) and anticrossing (bottom) and (f) three-peak spectrum (top) and the temporal dynamics (bottom) for illustration purposes. Reproduced with permission from ref (193). Copyright 2021 American Chemical Society.
The strong coupling between the plasmon of collective HSs and a molecular exciton has also been investigated by several groups.195,196,208 Cade et al. reported the relationship between SERS, the strong coupling between the plasmon resonances of nanostructured silver films having HSs on them, and the molecular excitons of a monolayer of R6G dye adsorbed on the films.208 They observed anticrossing in the dispersion curve of the hybridized resonance, yielding a coupling energy of 200 meV. The strength of this coupling is proportional to the square root of the molecular density. The SERS spectra exhibit a maximum intensity when the hybridized resonance lies in the middle of the Stokes Raman lines.208 Nagasawa et al. reported a coupling energy of ∼200 meV by investigating the hybridized resonance between the plasmon and exciton resonances of dye molecules (1,1′,3,3,3′,3′-hexamethylin-dotricarbocyanine perchlorate) using HSs in a NP dimer array, which they prepared by angle-resolved nanosphere lithography.195 They tuned the plasmon resonance by varying the distance between the silver dimers, and observed that the SERS spectra show maximum intensity by overlapping the hybridized resonance and excitation laser line.195 They also applied the array to control the photoEC reactions.209 Huang et al. reported a coupling energy of 200 meV by investigating the hybridized resonance between the plasmon and excitons of a fluorescent dye included in a polymer film, using HSs between silver NCs on a silver substrate.196 The plasmon resonance was dynamically tuned by varying the polymer film thickness (gap distance), which decreases with increasing temperature. They estimated the VHS to approximate 0.002(λ/n), where λ and n are the plasmon resonance wavelength and refractive index of the gaps, respectively.196
The single HS of a single NP dimer is a suitable place to directly evaluate the strong coupling between the plasmon and single molecular exciton resonances because the HS realizes SM-SERS, as discussed in Section 2.1.1. There are several evaluations for this strong coupling using elastic scattering and extinction spectra of the single dimers.197−202 Schlather et al. investigated the strong coupling using the J-aggregates in HSs at the single-(gold)-dimer level, tuning the plasmon resonance energy by varying the size of the dimers.197 They derived a coupling energy in the range of 115–200 meV from the anticrossing in the hybridized resonance using the coupled oscillator model, as discussed in Section 3.1.1. Itoh et al. simultaneously measured the scattering and absorption spectral changes in the plasmon resonance and SERS during SERS quenching processes, using a single silver dimer (Figure 18a–d).210 The modified coupled oscillator model, described in Section 3.1.1, consistently reproduced the changes with decreasing coupling energy from 200 to 0 meV.210 They also derived the enhancement and quenching factors from the coupling energy and quantitatively reproduced the SERS and SEF spectra, taking into consideration the ultrafast electronic dynamics (Figure 18e,f).68 Santhosh et al. demonstrated the strong coupling of the single silver dimer and single QD inside the gap;198 such a single exciton system is important to eliminate the dark exciton states. They derived a coupling energy of 120 meV by evaluating the scattering spectra using the coupled oscillator model.198 Bitton et al. demonstrated strong coupling between the dark plasmon resonance of a single silver dimer and exciton of a single QD by electron energy loss (EEL) spectroscopy.202 The strong coupling between the dark or subradiant plasmon resonance, which exhibits high Q factors, and the exciton resonance is important for the application of QED. Luo et al. also measured the strong coupling of a single gold dimer and single-to-few QDs inside the gap,201 and showed that the SEF spectra differ from the scattering spectra. The numerical analysis clarified that the QD excitons are strongly coupled with the octupolar plasmon resonance.201 Such strong coupling involving subradiant plasmon may improve the degree of freedom in the application.
Figure 18.

(a, b) Images of the temporal changes in the plasmon resonance and resonant SERS in the SEF spectra for the NP dimer, respectively. The white bar indicates the loss by the laser notch filter. (c, d) Plasmon resonance spectra and resonant SERS with SEF spectra extracted from panels a and b, respectively, at detection times indicated in the panels. The maxima of the envelopes are indicated by arrows. (e) Combined spectra of the resonant SERS and SEF calculated from the product of eq 18 and the sum of σR (Raman cross-section) and σF (fluorescence cross-section). Values of ℏg1(coupling energy) and ηRf (quantum efficiency) for the SEF are indicated in the panels, while the value of ηRR (quantum efficiency) for the resonant SERS is fixed at 0.001; maxima of the envelopes are indicated by arrows. (f) Schematic image of the supposed changes from ultrafast to conventional SEF with decreasing ℏg1. Reproduced with permission from ref (68). Copyright 2018 American Institute of Physics.
Recently, the strong coupling at a HS between a single gold NP and substrate has attracted considerable attention, as discussed in Section 2.1.2. This is because of the strong coupling (∼100 meV) realized using a single dye molecule in the HS reported by Baumberg’s group.187 In their strong coupling systems, the single dye molecule is captured in a CB molecule inside the HS. They observed a step-like increase in the coupling energies, indicating the number of dye molecules captured by the CB molecules.187 They also observed fluctuation of the hybridized resonance spectra, indicating the fluctuation of the SM inside a CB molecule.187 Recently, Baumberg’s experimentally demonstrated classical Rabi oscillations derived by the light field (not by vacuum fluctuation), which was theoretically predicted by Xu et al.137 inside the HS, using the excitation power dependence of fluorescence intensity (Figure 19).205 Leng et al. investigated the Fano and hybridized resonances in a strong coupling system using a single QD in HSs between a single gold NP and silver substrate.203 The QD was linked to gold NP through their capping molecule, and the coupling energy was estimated at ∼100 meV using the coupled oscillator mode. They also clearly showed that the decay rate of the single QD was increased by being strongly coupled to the plasmon at the HS.203
Figure 19.

Time-resolved and power-dependent measurements. (a, b) Time-resolved emission under 120 fs pulsed excitation of a 10 μM concentration ensemble of Atto647 in solution (orange circles) and (b) single Atto647 in NPoM (red circles), together with the instrument response (gray circles). Lines are single exponential fits. (c, d) Measured intensity vs optical field on the sample for ensemble Atto647 in water and SMs in NPoM, respectively. Curves are fits to the experimental data, as described in the text. Error bars are the fluctuations of the measured counts on the detector. Reproduced with permission from ref (205). Copyright 2019 Nature Research.
3.2. Ultrafast SEF at the HS
The ultrafast SEF, which appears as the background of SERS, is induced by an enhanced radiative de-excitation rate by a factor of FR, at the HS, in eq 5. The ultrafast SEF provides information on the electronic dynamics of the SERS active molecules. Indeed, the electronic dynamics during the de-excitation of electronically excited molecules determines their subsequent photochemical reaction.168−171,191 Spontaneous fluorescence spectroscopy is a powerful tool to evaluate the molecular electronic dynamics. The electronic dynamics of an SM inside a HS are expected to be largely deviated from that of the molecule in the free space, owing to the largely enhanced vacuum fluctuation caused by plasmon resonance.25,27,136,174 A molecule usually emits fluorescence from the bottom of a vibrational state in an electronically excited state (S1), a phenomenon known as Kasha’s rule. However, the molecule inside a HS may emit fluorescence from the S1 vibrationally excited state before returning to the ground state.25,27,174 Such fluorescence, called ultrafast SEF and observed in the background of the SERS spectrum, must be investigated to elucidate the modified electronic dynamics on the molecule inside a HS.
We provide a more quantitative picture of the ultrafast SEF based on a Jablonski diagram (Figure 20).174 The total radiative decay rate of a molecule in the free space is expressed as Kr0 (=∫0∞kr0(ωem)dωem), where kr0(ωem)dωem is the decay rate at ωem.25,174 The vacuum fluctuation yields the decay rates of fluorescence. The total radiative decay rates enhanced by plasmon resonance are calculated by the formula KRad = ∫0kr0(ωem)FR(ωem)dωem, using FR from eq 4.25,174 For a molecule, the total internal relaxation rate Kint (∼1012 s–1) is much larger than the total radiative decay rate Kr0 (∼108,9 s–1). Thus, the fluorescence spectrum of a molecule in the free space is a radiative transition from the S1 vibrational ground state (Figure 20a). For the normal SEF Kint ≫ KRad, the excitation and fluorescence transitions of a molecule are identical to those of the molecule in the free space (Figure 20b). However, because of the large value of FR ∼ 105 inside the HSs, Kint (1012 s–1) < KRad (105 × 109 s–1).25,174 Thus, the SEF has a component emitting from the S1 vibrational excited state before relaxing to the S1 vibrational ground state (Figure 20c). The highest photon energy of the ultrafast SEF is therefore blue-shifted from the fluorescence photon of the molecule in the free space, and the value of the highest photon energy depends on the excitation laser energy.25,174
Figure 20.
Simplified Jablonski diagrams depicting the processes involved for a fluorophore in the three possible situations discussed in the text: (a) Standard fluorescence; (b) SEF in the slow-dynamics regime, in which the vibrational decay rate is faster than the enhanced emission rate of slow-dynamics SEF; and (c) SEF in the fast-dynamics regime, in which the vibrational decay rate is slower than the enhanced emission rate of fast-dynamics SEF. Reprinted with permission from ref (174). Copyright 2007 American Chemical Society.
There are a reasonable number of studies reporting on the ultrafast decay of SEF using various species, such as dye molecules and QDs, located inside HSs.177 The large Purcell factors of ultrafast decay include both the radiative and nonradiative decay components of the de-excitation processes.25,27,136,174 Thus, the direct evidence of ultrafast SEF is the observation of blue-shifted SEF spectra emitting from the S1 vibrational excited state.174,211 The number of papers reporting such blue-shifted SEF are still limited in the SERS field. Moreover, the hot electroluminescence process observed from an STM tip is identical to the de-excitation processes of ultrafast SEF, because the gap between the STM tip and metallic substrate functions as a HS.212,213 Notably, the electroluminescence is excited by a tunneling electron and not by a photon. Thus, we next review the studies related to both ultrafast SEF and hot electroluminescence.
We first discuss studies on ultrafast SEF using plasmonic NPs.68,174,211,214−218 Le Ru et al. first developed a theoretical model for ultrafast SEF as the photoemission from the vibrational excited states prior to electron relaxation to the bottom of S1. They treated the ultrafast SEF spectra as a deviation from the conventional SEF spectra reproduced by the product of a fluorescence spectrum and a plasmon resonance spectrum, which is similar to the description in eq 7.174 Itoh et al. experimentally averaged out the spectral variations in the SEF of dye molecules induced by plasmon resonance by measuring a huge number of HSs.211 Notably, the spectral modulation by FR(ωem), as defined in eq 7, makes separation of the ultrafast SEF spectra from the total SEF spectra in Figure 20 difficult. They successfully observed that the SEF spectra exhibit excitation laser energy dependence, blue shifts, and superbroadening, predicted as evidence of ultrafast SEF.211 Itoh et al. also evaluated changes from ultrafast to normal SEF by decreasing the coupling energy of eq 1 using a single dimer.68 Cho et al. demonstrated ultrafast SEF using resonant whispering-gallery plasmon nanocavities comprising CdS-SiO2/silver core/shell NWs.214,215 They tuned the cavity size to match the whispering-gallery resonances, and achieved the transition from the thermalized exciton to ultrafast SEF by observing blue-shifted SEF. They also estimated the radiative rate enhancement to be >103 and predicted ultrafast lifetimes at the subpicosecond scale, indicating that they correctly observed ultrafast SEF.214,215 Bingi et al. demonstrated the SEF of dye molecules adsorbed inside HSs on plasmonic random media fabricated from silver nanostructures with different aspect ratios for varying plasmon resonances.216 They reported that the SEF from the dye molecules were blue-shifted compared to the free-space emissions, thereby evidencing ultrafast SEF.216 Aspetti et al. observed SEF from silicon NWs coupled with silver nanocavities supported by high-order hybrid nanocavity-surface plasmon modes.217 Excitation energy dependence of the SEF spectra reveals that the SEF includes the phonons of an ultrafast SEF process (Figure 21).217 They also reported the temperature dependence of the measured intensity that is indicative of ultrafast SEF and rules out the resonant Raman process.217 Gokbulut et al. reported spectral changes in the SEF profiles of perovskite NWs by surrounding them with gold NP clusters in random polymer media.218 Blue-shifted SEF profiles for the perovskite NWs were reported. They also discussed that plasmon-mediated optical pumping generates higher vibrational excited energy state levels, resulting in ultrafast SEF because of the strong plasmon and exciton interactions of the perovskite NWs inside the HSs.218
Figure 21.

Photoluminescence spectrum of plasmonically coupled silicon nanowire (diameter = 150 nm) plotted versus the (a) absolute emission energy and (b) energy shift from the laser line for various excitation energies in the range 1.959–2.708 eV. Spectra in panel b are plotted with a constant offset of 2 × 103 counts for clarity. The dashed boxes labeled A and B denote high-intensity emission bands. (c) Schematic of the electronic dispersion of silicon featuring carrier excitation (magenta arrows), intraband relaxation and hot luminescence (green arrows), and radiative recombination from thermalized carriers (blue arrows). This process is examined for relaxation toward the selected conduction band minima at both the X- (1.12 eV) and L- (∼2.1 eV) points. Reproduced with permission from ref (217). Copyright 2014 American Chemical Society.
We next review studies of hot electroluminescence using STM.212,219−222 Dong et al. demonstrated hot electroluminescence, of which the de-excitation process is identical to ultrafast SEF, directly from the S1 vibrational excited states of porphyrin molecules located inside a HS between an STM tip and metal substrate, along with the upconversion electroluminescence, by spectrally tuning the frequency of plasmons.219 Chen et al. developed the model to evaluate the hot electroluminescence by combining the classical generalized Mie theory with the quantum master equation.220 They discovered that the radiative decay rate of electroluminescence can be enhanced by 3 to 5 orders of magnitude by plasmon resonance, so that the radiative process occurs on the lifetime scale of picoseconds and becomes competitive with the vibrational relaxation.220 Chong et al. demonstrated SM hot electroluminescence by placing a porphyrin molecule inside the HS.221 They clearly observed hot electroluminescence, which photon energy is higher than the energy gap between S1 and S0 owing to their vibrational energies (Figure 22). Bergfield developed a quantum master equation approach to investigate the electroluminescence of the molecules inside a HS. They calculated the STM-induced spontaneous emission of a single porphyrin molecule coupled to a plasmon,212 and observed excellent agreement with the experimental results in the weak coupling regime.212 In the strong coupling regime, they predicted the spectral features indicative of the formation of hybridization states.212
Figure 22.
Vibronic features and emission mechanism. (a) Light emission spectra of fused-5,15-(dinaphthalene)-10,20-(dibromo)-porphyrin (DPP), -5-(phenyl)-10,20-(dibromo)-porphyrin (MPP), and -5,15-(dinaphthalene)-10,20-(dibromo)-porphyrin (DNP) plotted as a function of the energy shift from the 0–0 lines. The raw (smoothed) data appear in gray (red). (b) Schematic representation of the emission mechanism. The energy lost by an inelastic tunneling electron (1) is transferred to the molecular emitter that is excited from S0 to S1 (2). Eventually, the emitter relaxes to its electronic ground state by emitting a photon (3). (c) Details of the excitation and de-excitation processes. Three possible paths are described: The green arrows characterize the most efficient path leading to the 0–0 emission line, which corresponds to a molecule excited to the S1 vibrational ground level and then relaxing to the S0 vibrational ground level. The red and blue arrows characterize much less efficient paths. In path II (red) the molecule is also excited to the S1 vibrational ground level but decays to the S0 vibrational excited level (R-band). Path III (blue) corresponds to a molecule excited to the S1 vibrational excited levels which relaxes to the S0 vibrational ground level (B-band). Reproduced with permission from ref (221). Copyright 2016 American Chemical Society.
3.3. Vibrational Pumping at the HS
Vibrational pumping is induced by an enhanced excitation rate at a HS by a factor of FR in eq 5. The vibrational state in the electronic ground state S0 affects the pathway of the subsequent chemical reaction.168−170,223,224 Thus, vibrational pumping is expected to be applicable to control the chemical reaction. The vibrational decay rate is much faster, on the order of approximately 1012 s–1, than the conventional photo excitation rates. Thus, the Boltzmann distribution after reaching thermal equilibrium dominates the vibrational state in the molecule. The photoexcitation rate of a molecule inside a HS is much larger than that in free space, by FR in eq 5. Thus, one can expect vibrational pumping, which is the population inversion of the molecular vibrations, for controlling its chemical reaction pathway.168−170 Vibrational pumping was first discovered by Kneipp in 1996, as a quadratic increase in the anti-Stokes SERS intensity against incident light power.175 The subsequent quantitative evaluation, attained by comparing their enhanced excitation and vibrational decay rates, indicated that vibrational pumping may occur by plasmon-enhanced resonance absorption and de-excitation processes but not by plasmon-enhanced Raman processes.137,138,176,225−231 Recently, the possibility of controlling vibrational pumping by coupling between the plasmon and vibrations has been reported.232
Vibrational pumping based on a Jablonski diagram is illustrated in Figure 23.176 Several research groups have provided a simple evaluation method for understanding this phenomenon.137,138,225 The population of the molecules that are in vibrationally excited states determines the probability of vibrational pumping. Xu et al. described that this probability is expressed as CFR2σA(ωex)ϕph/γvib, where C ≤ 1 is a numerical factor, σA ∼ 10–16 cm2 is the absorption cross-section at ωex, ϕph is the photon flux, and γvib ∼ 1012 s–1 is the vibrational decay rate.137 In the description, the excited vibrational state is mainly pumped by repeated absorption and de-excitation. If ϕph= 1023 photons cm–2 s–1, corresponding to 5 × 104 W/cm2, then CFR2σA(ωex)ϕph/γvib is ∼1 for FR2 ∼ 105. This is a reasonable value in HSs, indicating that vibrational pumping likely occurs. If one uses the typical value of the resonant Raman cross-section σRS∼ 10–24 cm2 instead of σA, then CFR2σRS(ωex)ϕph/γvib ∼ 10–8, indicating that vibrational pumping may not occur.137,138,225 Thus, the reported vibrational pumping may be induced by molecule exciton resonance. We consider vibrational pumping under nonresonant conditions observed by Kneipp et al.,175 may be related to strong coupling, which induces a resonant effect between the incident light and lower branch of the hybridized resonance (Section 3.1).
Figure 23.

Schematic (Jablonski) diagrams (top) showing the basic elements of the electronic structure of a molecule. In quantum mechanical terms, a (nonresonant) Stokes process (right) consists of a transition to a virtual state, followed by a re-emission, leaving the molecule in the first (v = 1) vibrational excited state. This corresponds to positive Raman shifts in the spectrum. The anti-Stokes process (negative Raman shifts) on the top left follows the opposite path, thus producing a photon with a higher energy than that of the incoming one. The anti-Stokes process depends on the population of the excited level, while the Stokes process is (at low intensities where Raman stimulation is negligible) independent of this population. The processes depicted in the top-right (top-left) diagram contribute to the Stokes (anti-Stokes) part of the spectrum at lower (higher) energies with respect to the laser, as depicted in the bottom figure. Reproduced with permission from ref (176). Copyright 2008 Royal Society of Chemistry.
As described above, the vibrational pumping phenomenon in the SERS spectra was first discovered by Kneipp et al. in 1996,175 the mechanism of which was discussed as Stokes Raman pumping.175 However, Haslett et al. showed that the cross-section of the Raman scattering is too small to induce the observed vibrational pumping and suggested the contribution of resonance or preresonance to the pumping.225 Etchegoin’s group comprehensively examined the origin of the vibrational pumping with respect to various processes, including Raman pumping, fluorescence pumping, and resonance effects.176,227−229 Furthermore, they demonstrated SM vibrational pumping of the resonant SERS of a dye molecule.230 Kozich et al. separated the thermal and vibrational pumping effects with subpicosecond time-resolved SERS spectra of R6G molecules.231 They observed an increase in the anti-Stoke SERS intensity that is delayed by 1.0 ps after pulse excitation, indicating that the vibrational pumping originates from electronic excitation followed by fluorescence and ultrafast internal conversion and not from Stokes SERS.231 Pozzi et al. examined the vibrational pumping in the SERS spectra of R6G molecules under low extraction power <2.0 W/cm2 using HSs in silver NP aggregates.233 They did not show a quadratic increase in the anti-Stokes SERS intensity against the incident light power and found that the intensity varies as a result of the variations in the EFs with changing local temperatures.233 Their results are reasonable when considering a required laser power >5 × 104 W/cm2 for the vibrational pumping.
Recently, vibrational pumping has been investigated in the field of cavity QED, where the vibrational pumping is described as the optical response of a coupled system between plasmon and molecular excitons or vibrations. Benz et al. demonstrated the vibrational pumping in the SERS spectra of biphenyl-4-thiol molecules using a picocavity (Section 2.1.2) inside HSs between a gold substrate and gold NP.56 They observed a quadratic increase in the anti-Stokes SERS intensity from a rather low incident light power of 6 × 103 W/cm2. This increase was explained by a QED model and demonstrated the relationship between the vibrational pumping and coupling energy between a Raman dipole and vacuum fluctuation for several Raman lines, indicating optomechanical effects on the vibrational pumping.56
Several groups developed a QED model for SERS, to calculate the vibrational pumping under the effect of coupling between the plasmon and SM vibrations, using experimentally realistic parameters of a HS.232,234 Schmidt et al. showed that the anti-Stokes SERS intensity mainly depends on the temperature for an excitation power of <107 W/cm2 and on the vibrational pumping for values >108 W/cm2 under nonresonant conditions.235 Roelli et al. showed that the coupling between the plasmon and vibrations induces sharpening of the SERS excitation profile by considering the dynamical vibrational pumping effect.232 Neuman et al. developed a QED model of resonant SERS to investigate the vibrational pumping under weak and strong coupling between the plasmon and exciton resonances.182 Their calculation, using a coupling energy of 100 meV, showed that the vibrational pumping appears with much weaker excitation power (<105 W/cm2) than that observed under nonresonant conditions (>108 W/cm2). Interestingly, the maximum of the anti-Stokes line appears at an excitation power of 5 × 106 W/cm2, while the Stokes emission peaks are maximized at an excitation power of 2 × 106 W/cm2. Zhang et al. developed a QED model of resonant SERS that includes the higher-order plasmon modes and optomechanical effects. They reported that the vibrational pumping can be controlled by tuning the coupling energy.236 We predict that these results will be experimentally demonstrated by using ultrafast laser pulse excitation to avoid photodecomposition of the molecules.
Several groups have reported on electrically driven vibrational pumping based on a two-temperature model of the metal.237,238 Crampton et al. examined the vibrational pumping under nonresonant SERS conditions with picosecond laser pulse excitation.237 They measured the SERS of 1,2-di(4-pyridyl) ethylene (BPE) molecules inside a HS of gold NP dimers and estimated the temperature using the SERS of the BPE molecules and electronic Raman scattering (ERS) of the dimer with continuous wave and picosecond pulsed excitation (Figure 24).237 Unusual temperatures were observed for the BPE molecules (e.g., −3000 K), and even the pulsed excitation temperature of the dimer electrons was ∼3000 K.237 From the large deference in the temperature, they proposed that the vibrational pumping is induced by the coupling between the excited electrons in a nonequilibrium state of the dimer and the vibrations of the BPE molecules during the electron–phonon scattering process.237
Figure 24.

(A) SERS spectra recorded on single nantennas via (i) continuous wave excitation (633 nm, 30 μW/μm2) and (ii–iv) pulsed excitation (785 nm, 7 ps, 76 MHz, 100 μW/μm2). In all cases, the polarization of the incident field is aligned with the long axis of the dimer. Micrographs of each dimer, acquired after the SERS measurements, are shown in the insets. (B) Anti-Stokes branch of the electronic Raman scattering (ERS) continuum for continuous wave (cw) (open circles) and pulsed (inset) excitation after removing the molecular lines. The fittings into eq 1 of ref (237) in the text (continuous lines) yield metal electronic temperatures of 385, 3400, 3100, and 2400 K for the spectra shown in panels i–iv, respectively. (C) S-to-AS ratios extracted from the five prominent vibrations of 1,2-di(4-pyridyl)ethylene (BPE) (open circles) and the predicted ratios, assuming equilibration between metal and molecule and using the measured electronic temperature of 385 K (blue trace; see eq 2 of ref (237)). Reproduced with permission from ref (237). Copyright 2018 American Chemical Society.
3.4. Modification of the Excitation Selection Rule at the SERS HS
This phenomenon is induced by the extremely small value of VHS in eq 3. The interaction Hamiltonian between the electric field and a molecule is described as Ĥ = μ·Ê(r̂, t), where μ is the dipole moment, r̂ is the electron position operator, and Ê is the operator for the electric field.66 The light wavelength (several hundred nanometers) is much larger than the molecular size (below one nanometer) in free space. Thus, the molecule cannot feel the inhomogeneous electric field of light, and the transition matrix element between the electronic ground state |ψg⟩ and electronic excited state |ψe⟩ is ⟨ψg|r̂|ψe⟩·E.66 Such an assumption is called the long-wavelength or dipole approximation.66 However, the size of the electric field structure inside the HSs is comparable to the size of the electronic structure of a molecule. Thus, the transition matrix element is given by ⟨ψg|r̂·Ê(r, t)|ψe⟩, indicating that the selection rule of the transition is dependent on the position inside the HS. This modification of the selection rule has been observed as forbidden Raman transition modes in SERS.239−243 The relationship between the picocavity and modification of the selection rule is discussed in detail in Section 4.
The modification of the selection rule by the confined electric field, i.e., that inside the HSs, has been examined using the Raman inactive lines appear in the SERS spectra.239−243 We next introduce the modification as the appearance of Raman inactive lines from an analytical viewpoint.239 Considering the higher-order terms, μ is expressed as
| 19 |
where α, β, γ correspond to x, y, z, respectively, and the tensor convention is assumed for the repeated subscripts.239−243 Here μαp is the permanent dipole moment, and ααβ and Aαβγ are the electric dipole–dipole and electric dipole–quadrupole polarizabilities, respectively.239−243 The effect of the dipole–quadrupole term was first proposed by Moskovits et al. as the activation of inactive Raman modes on a silver film,240−243 while Ayars et al. reported the lowest order of the field-gradient effect.239 Using partial differentiation, eq 19 is expanded by the coordinate of the vibration Q as
| 20 |
The terms related to Q are collected as
| 21 |
The first and second terms in eq 21 correspond to infrared (IR) absorption and Raman scattering, respectively. The third term is called the gradient-field Raman (GFR) term. In free space, the ratio of the GFR to the Raman terms is on the order of 2πa/λ, where a and λ represent an atomic size of ∼0.2 nm and wavelength of ∼500 nm, respectively. Thus, this ratio is ∼10–3, and the GFR contribution is insignificant. The situation differs at a SERS HS, where the ratio of the GFR term to the Raman term approaches unity, indicating the modified selection rule in SERS by the field-gradient effect at the HSs.239
The modification of the selection rule by the confined fields inside the HSs has been investigated using TERS, which is the main topic discussed in Section 4. The mechanism behind the appearance of Raman forbidden modes in spectra is common for SERS and TERS. Here we introduce several studies treating the modification by SERS.
Takase et al. demonstrated that the field-gradient effect can induce forbidden electronic transitions in the SERS signals from carbon nanotubes, and observed a forbidden Raman transition using carbon nanotubes inside HSs (Figure 25).244 This breakdown of the selection rules was reproduced using the transition dipole moment of the excitons without approximating the transition matrix element. They successfully derived the transition dipole moment of the forbidden modes in the SERS spectra. This work shows that a HS not only enhances the EM-field intensity but also increases the optical excitation channels, including the excitation of forbidden Raman modes beyond the selection rules. The demonstration of the importance of the field-gradient effect by the advances of the field-gradient effect are summarized by Aikens et al.245
Figure 25.
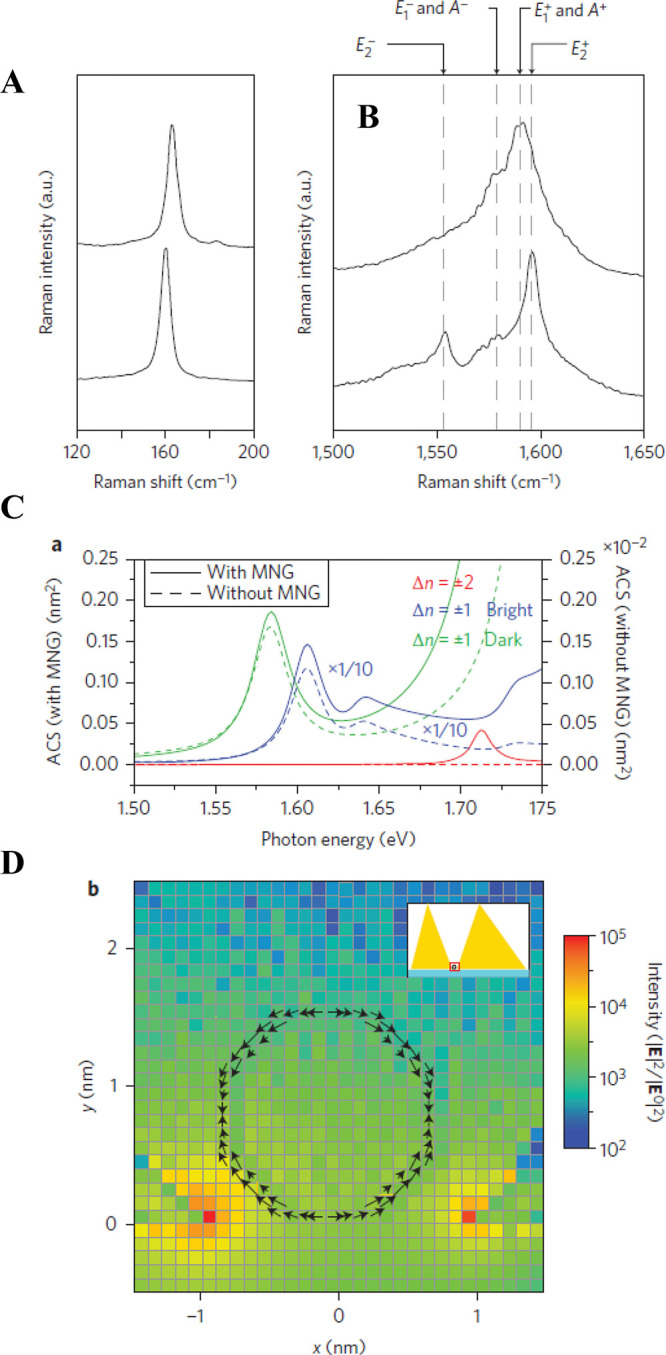
(A) Characteristic SERS spectra at a radial-breathing mode (RBM) of ∼160 cm–1. (B) Upper and lower SERS spectra are obtained via the E11M exciton (see Figure 1 of ref (244)) of a chiral vector (15, 6). Single-walled carbon nanotube (SWNT) with an RBM of ∼163 cm–1 and the E14 (E41S) exciton (see Figure 1 of ref (244)) of a chiral vector (13, 9) SWNT with an RBM of ∼161 cm–1, respectively. (C) Selection-rule breakdown as indicated by the numerical calculation. Absorption cross-section with (solid lines, left-hand scale) and without (dotted lines, right-hand scale) a metallic nanogap, as given by the numerical calculation. Results indicated by the solid and dotted blue lines are reduced by a factor of 10. (D) Cross-sectional view of the calculated intensity of the EM field, including both the longitudinal and transverse components, under resonant excitation conditions of the E14 (E41S) exciton (see Figure 1 of ref (244)). The calculated quadrupole polarization of the E14 exciton (see Figure 1 of ref (244)) is indicated by black arrows. The grids show the cell size in the present numerical calculation. Reproduced with permission from ref (244). Copyright 2013 Nature Publishing Group.
Benz et al. demonstrated the modification of the selection rule in the SERS of a single biphenyl-4-thiol molecule inside a picocavity, which is an atomic-scale subnanometer structure inside a HS under cryogenic temperature conditions.56 The low temperature and field confinement at subnanometer scales enable the stable observation of the modification of the selection rule. They reproduced spectral variations in SERS, i.e., the appearance of an IR active mode via a position-dependent picocavity on a SM, using DFT calculation (Figure 26). Picocavities have been widely used for investigating the relationships between internal structures and the modified Raman selection rules. Theoretical models have been developed to treat the results of picocavities based on analytical and numerical methods.240,246−252 Zhang et al. have proposed a quantum-mechanical model for describing Benz’s SERS results.252 They showed the importance of the full spatial properties of both the excitonic transition charge density and the photonic fields to correctly describe the plasmon–exciton coupling. They also successfully described the optical-selection-rule breaking of molecular excitations inside picocavities from the plasmon–exciton coupling.252
Figure 26.

Raman spectra reveal changes of the SERS selection rules. (A) Anti-Stokes/Stokes spectra at a time when additional picocavity-induced lines are present. Colors correspond to the same lines on the Stokes and anti-Stokes sides, with vibrational eigenmodes and effective temperatures shown. (B) Time-series anti-Stokes (aS) and Stokes (S) SERS. Dashed lines (aS) mark the expected aS line positions corresponding to the ever present (persistent) S lines from the many molecules in the larger nanocavity. (C, D) Comparison of two experimental spectra (bottom) with DFT simulated spectra (top) under the assumption of atomic-scale field confinement (inset shows geometries; red sphere denotes field localization for the simulations). Reprinted with permission from ref (56). Copyright 2016 American Association for the Advancement of Science.
4. Ångström-Resolved TERS: State-of-the-Art Advances
Characterization of the chemical structure of a SM adsorbed on surfaces with chemical-bond sensitivity and Ångström-level spatial resolution can provide important insights into the understanding of chemical reactions, catalytic processes, surface science, and molecular electronics/optoelectronics. However, as we have discussed in Section 1, the cross-section of ordinary Raman scattering is usually very low: every 106 to 1010 incident photons can only produce one Raman photon.253 Thus, plasmon-enhanced Raman spectroscopy was developed to enhance the Raman signals based on the strong and highly localized electric fields near the surface of the nanostructures.253 Based on the different situations of plasmonic enhancement and confinement, the enhanced Raman technique can be classified into SERS1−3,7 and TERS,4−6,58 which have been thoroughly discussed in Sections 1 and 2. Briefly, in SERS, metallic nanostructures, such as small NPs and structured surfaces with a typical roughness at the nanoscale, are employed to confine the strongly enhanced local plasmonic fields near the metal surface.254 On the other hand, in the TERS technique, the local fields are provided by a metallic probing tip and/or the nanogap formed with a metallic substrate.49,255 For the molecules inside such local fields, enhanced Raman signals that are stronger than those of the molecules in the free space can be obtained, even enabling the detection of a SM.61,256−258
Although SERS and TERS share the same enhancement mechanisms, as explained in Section 1.2, the introduction of a probing tip in TERS provides advantages to circumvent some of the disadvantages encountered in SERS: (1) Controlling the HS properties. In the SERS configuration, the spatial distributions of the EFs in the HSs are usually unknown in advance, and it is difficult to precisely control the HSs for the nanostructures fabricated. In contrast, TERS is generally operated through the combination of a scanning probe microscopy (SPM) instrument and a Raman microscope with synchronized control. The probing tip not only provides the controllability of a single HS by tuning the nanogap distance and tip morphology,219,259 but also enables real-space imaging through the tip scanning process with high spatial resolutions.61 Specifically, TERS provides a mobile HS with control in both vertical field enhancement and the lateral HS position on the surface. (2) Controlling the single molecular responses. In the SERS measurements, the molecular responses can only be induced through macroscopic methods, such as the introduction of external light or electric fields across the whole samples.260 In the TERS technique, the position of the probing tip can be localized with atomistic precision, and the field confinement can also approach atomic scale through the use of atomistic protrusions at the tip apex. In this case, the Raman signals can be mainly contributed from the local chemical groups or bonds inside a SM, enabling the possibility of probing the intramolecular structure and its changes at the single-chemical-bond level.261,262
Because of these advantages in combining both the sensitive chemical specificity and high spatial resolution, scientists had been attempting to develop the TERS technique since the concept of TERS was proposed in 1985.57 The first experimental demonstrations of TERS were achieved in 2000 by coupling Raman spectrometry with AFM4−6 or STM,58 as introduced in Section 1.1.2. Later developments have enabled the TERS technique to allow the visualization of individual molecules with chemical identification,61,258 even achieving Ångström-resolved Raman imaging with single-chemical-bond resolution.256,257 Because of the strong spatial confinement and enhancement of near-field EM fields at the apex of the metallic probing tip, the Abbe diffraction limit in the far-field measurement is no longer valid, providing another way to fulfill the super-resolution microscopy circumventing the fluorescent labeling techniques.263−265 Such a super resolution in real space, together with the high sensitivity in chemical specification, makes the TERS technique a versatile tool to either investigate the molecular structures or induce surface reactions at the subnanometer scale in a controlled manner, which is very important in chemistry,261,266 catalysis,267,268 materials science,269,270 and biology.271−273
In this section, we begin with an introduction of the theoretical background for ultrahigh spatial resolution, including the plasmonic response and molecular response under light excitation. In particular, attention is paid to the concept of the plasmonic “picocavity” in TERS (see also Section 2.1.2 for “picocavity” in SERS) and the Raman scattering process of a SM under the “illumination” of such an atomically confined light. The spatial resolution limit will also be discussed briefly from the theoretical aspect. We will then move on to describe in detail recent experimental advances in subnanometer resolved TERS imaging, especially the latest Ångström-resolved Raman spectromicroscopy for chemical structural reconstruction, namely scanning Raman picoscopy (SRP). The future outlook of the field, particularly regarding Ångström-resolved TERS, will be provided in Section 7 from both the technical and application aspects.
4.1. Theoretical Considerations on Ångström-Resolved TERS
Since Ernst Abbe first discovered the fundamental “diffraction limit” of optical imaging in 1873,274 many efforts have been devoted toward improving the optical spatial resolution. To circumvent this limit and apply the optical methods to material systems down to the SM scale, different schemes have been adopted to achieve subdiffraction imaging, which can be generally classified as far-field and near-field techniques. For the far-field approaches, the most well-known technique is super-resolution fluorescence microscopy, based on fluorescent labeling, which was awarded the 2014 Nobel Prize in Chemistry.263−265 Another far-field technique used to break the diffraction limit is based on metamaterials, in which the optical resolution beyond the diffraction limit is realized by conveying both the propagating and evanescent waves from the object to the image.275 The diffraction limit can also be circumvented using the near-field approach. This is because at very close distances to the surface, the nonpropagating evanescent waves would dominate, and thus, imaging resolution would no longer need to abide by the far-field diffraction rule.276 Such a method is usually known as scanning near-field optical microscopy (SNOM) or near-field scanning optical microscopy (NSOM). There are generally two ways to realize such near-field imaging: The first follows the concept proposed by Edward Synge in 1928, where a subwavelength light source, generated through a subwavelength hole on a thin film, is introduced to illuminate a sample at very close distances.277 Technically, such a light source was realized in the early 1980s through a tapered fiber with a subwavelength aperture at its end, thus called aperture-SNOM.277,278 For the aperture-SNOM based nano Raman techniques, because both the incident light and the emitted photons are manipulated in the near field, the far-field scattering background can be eliminated, with the spatial resolution reaching about 50 nm. However, the spatial resolution is hard to be further improved because of the limit in the aperture diameter. The power transmission efficiency for an aperture diameter below 50 nm would be too low for the detection of weak photon signals in practice.279 The second method follows the concept proposed by John Wessel in 1985, where a nanoscale scatter or antenna is introduced as a probe either to convert the incident light as the local field or to convey the local scattered signals to the far-field region.57 The most commonly used probe comprises a metal (or metal-coated) tip without an aperture at the tip apex, and the TERS technique belongs to this so-called apertureless-SNOM.280 Being free from the requirement of an aperture to deliver light, the spatial resolution of TERS is only limited by the size of the tip apex, which can even go down to an atomic scale. As described previously, the first experimental demonstrations of TERS were realized in 2000 by four independent groups, namely, the groups of Zenobi,4 Pettinger,58 Kawata,6 and Anderson.5 After nearly two decades of development, the spatial resolution of TERS has been improved from tens of nanometers to the Ångström level.61,256−258,281−286
In the subsequent text, we first focus on the physical mechanisms of the high spatial resolution in TERS measurements. Two aspects are believed to be responsible for the resolution: The confinement of the local plasmonic field and the molecular response to this local field. For the plasmonic response, the local field can be analyzed using both classical and quantum theory, while for the molecular response, both linear and nonlinear processes can affect the spatial resolution and generally require quantum chemical calculations.
4.1.1. Plasmonic Response: Confinement of Light at the Atomic Scale
One strategy to obtain deep-subwavelength spatial resolution in optical microscopy is to use the spatially confined light fields induced by a local probe. As shown in Figure 27a, for the probing tip used in the TERS measurement, the incident light is first scattered by the shaft of the tip, converting from far-field to near-field SPP propagation along the tip surface. The tip apex behaves like a NP to further convert the propagating surface plasmon polariton (SPP) to the ultraconfined localized surface plasmon (LSP). As shown in Figure 27b, the local fields near the tip apex are strongly influenced by the morphology of the tip, especially the geometry of the apex.287,288 For the tip apex with an atomistic protrusion attached to it, the electric fields are first localized and enhanced near the surface, producing a background nanocavity plasmon (NCP) field associated with the tip shaft. The NCP field can be strongly enhanced especially under resonance conditions. Moreover, the “atomic-scale lightning rod effect” associated with the atomistic protrusion would then produce an additional enhancement over this background, resulting in an atomic-scale HS, as demonstrated in Figure 27c. A comparison with the quantum results calculated by the TDDFT method289 reveals that the main features of the field localization and enhancement in atomistic protrusions can be correctly addressed by classical and analytical simulations, through the consideration of the sharp curvature with an atomistic profile, giving rise to similar near-field distributions (Figure 27c). Therefore, the spatial extension of the enhanced local fields can go down to the atomistic scale, which is believed to be primarily responsible for the high spatial resolution of TERS mapping images down to the Ångström level.
Figure 27.
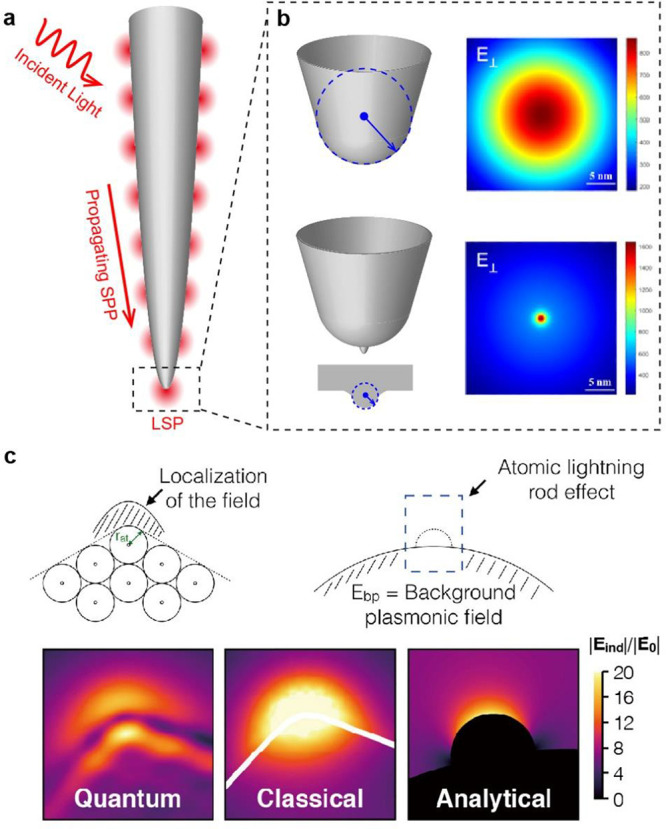
Confinement of light at the atomic scale through a metallic tip in TERS. (a) Conversion from the far-field light to the near-field SPP propagating along the tip surface and further to the LSP confined at the tip apex. (b) Spatial distribution of the local electric field near the tip apex with and without atomistic protrusion. Adapted with permission from ref (287). Copyright 2020 Springer Nature. (c) Comparisons between the quantum, classical, and analytical simulation results of local electric fields, exhibiting similar features in both field intensities and spatial distributions. Adapted with permission from ref (289). Copyright 2018 American Chemical Society.
The enhancement and spatial resolution of TERS
can be further improved
through the formation of a nanogap, by approaching the tip to a metal
surface. The electric field in the direction perpendicular to the
substrate is determined by the gap distance, which can be decreased
to subnanometer scale, until the quantum tunneling effect sets in
and limits the field confinement and enhancement, as explained in Section 1.2.1. On the
other hand, the field confinement in the direction parallel to the
substrate follows approximately 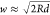 ,290,291 which suggests that
the local field confinement can be improved through reducing the gap
distance (d) and tip radius (R).
However, since the scattering cross-section of a NP is proportional
to R,6 a very small isolated
NP alone is not an effective setup for the field enhancement, even
when the NP is within subnanometer proximity to the metal surface.288
,290,291 which suggests that
the local field confinement can be improved through reducing the gap
distance (d) and tip radius (R).
However, since the scattering cross-section of a NP is proportional
to R,6 a very small isolated
NP alone is not an effective setup for the field enhancement, even
when the NP is within subnanometer proximity to the metal surface.288
The gap plasmons confined between two metal surfaces can be sensitive to single-atom placement.56,292 Therefore, a further level of optical confinement can be achieved via the localization of EM fields in close proximity to the atomistic structures within the plasmonic nanocavities.15,56,111 Because the effective mode volume induced by the atomistic structures can be less than a cubic nanometer, such structures are sometimes termed as picocavities. Picocavities can be formed as one or a few metal atoms protrude out of a metal surface; however, the exact configuration is still difficult to control experimentally. One experimental method employed to form picocavities is to deliver intense laser power to plasmonic nanostructures, so that near the plasmonic HSs, the energy delivered by the laser can activate surface atoms to become mobile and protrude out of the metal surface.56 Picocavities can also be created by controlled gentle indentations between the metal tip and substrate, which can generate an atomistic protrusion at the tip apex.287 On the other hand, to stabilize the picocavity, the system must be cooled down to cryogenic temperatures and the incident laser power must be kept low to avoid thermal diffusion. The plasmonic picocavity features cascaded enhancement from both the EM field enhancement of a plasmonic nanostructure, such as the tip shaft, and the atomic-scale field enhancement associated with the atomistic protrusion, which offers optical resolutions down to the subnanometer or even Ångstrom scale. Such an ultrahigh spatial resolution enables direct access to the internal structure of a SM, as demonstrated in recent TERS256,257 and tip-enhanced photoluminescence (TEPL)287 experiments. In this situation, the SM cannot be considered as a simple point dipole, instead, the internal electronic structure of the molecule must be taken into account, as explained previously in the modification of excitation selection rules in Section 3.4.
The cascaded enhancement mentioned above implies that the formation of an effective picocavity for high-resolution spectroscopy demands not only the formation of appropriate atomistic protrusions at the tip apex, but also effective coupling between the protrusion and gap modes induced by the tip shaft.289 Because the direct coupling between single atoms and the free-space photons is very weak, intermediate stages are necessary to deliver the energy of the free-space photons to atomic-scale volumes successively and efficiently. As shown in Figure 28, owing to the good impedance matching between the free-space photons and plasmonic antennas with typical sizes comparable to photon wavelengths, the free-space propagating EM wave is first efficiently coupled to the plasmonic antenna and converted into the gap mode. In a second step, the localized gap plasmons are further enhanced and concentrated by the atomic-scale protrusion, thus successively focusing the free-space photons to the picocavities.
Figure 28.

Schematic of local plasmonic field confinement from the isolated tip to a nanocavity, and further, to an atomistic picocavity.
Therefore, the enhancement and concentration of local EM fields by atomistic structures in nanogaps can be attributed to two factors: The first is the nonresonant atomic-scale lightning rod effect, owing to the sharp curvatures of the atomistic protrusion.288,289,293 The second is the plasmonic hybridization between the gap modes and protrusion-induced EM modes, which generates strong and ultraconfined EM modes in proximity to the protrusion.56,111 Although the nonresonant lightning rod effect alone, without the plasmonic substrate, could also account for the main functionality of the picocavity, it was reported that when the incident photon is resonant with the hybridized protrusion modes, the field intensity and spatial confinement are greatly enhanced due to the resonant accumulation of conduction electrons around the atomistic protrusion.288,294
However, the spatial resolution is unlikely to be improved down to the sub-Ångström level because of the quantum effects involved. As shown in Figure 29, within a local EM response model, the induced electron charges are assumed to distribute on a layer of infinitesimal thickness on the metal surface. However, in real metals, the electron densities would smear outside the ideal metal surface slightly. Such an electron spill-out effect would restrict the further improvement of spatial resolution.29,69 Due to such a plasmonic nolocal effect, the spatial resolution is said to be limited by a Thomas-Fermi screening length of approximately 0.1 nm.295,296 On the other hand, the quantum tunneling effect in the subnanometer gap could significantly change the optical response and even quench the electric field enhancements,29 as described in Section 1.2.1. Moreover, when the size of a NP becomes small, the electron scattering at the surface could reduce the surface plasmon mean free path, thus increasing the decay of plasmons into electron–hole pairs, a process known as the Landau damping.297−300 Owing to the extremely small size of an atomistic protrusion, Landau damping could be a significant factor that may quench the optical fields around the protrusion and thus limit the field intensity in the picocavities.299,301,302 Fortunately, a recent theoretical work by Li et al.294 has shown that the coupling of the protrusion modes to the host modes will accelerate the radiation rate of the former so that it can compete with the intrinsic Landau damping.
Figure 29.

Schematic illustrating processes that could limit the spatial confinement of the nanocavity plasmon field. Middle: Illustration of the NCP field in the junction between the tip and substrate. Left: In the subnanometer gaps, the AC quantum tunneling current Jω across the junction significantly changes the optical response and quenches the electric field enhancement. Upper right: Considering the nonlocality of the surface response, the equilibrium electron density n0(r) exponentially decays across the metal surface and the induced charges n1(r) are smeared both inside and outside the metal–air interface, a phenomenon known as the Thomas-Fermi screening effect. The vertical dashed line represents the ideal metal surface disregarding the electron spill-out effect. Lower right: The NCP field is quenched by directly exciting electron–hole pairs in the metal via Landau damping. The momentum mismatch between the NCP and electrons can be compensated by scattering with phonons, defects, or surfaces.
4.1.2. Molecular Responses: Raman Scattering of a Single Molecule in an Atomically Confined Light Field
As described in Section 3, such a highly confined plasmonic field can interact with the molecule and results in many unique phenomena, ranging from the strong coupling to the field-gradient effect, and the selection rule can also be modified. In fact, the spatial resolution of TERS could be influenced as well because of this atomically confined field, and many theoretical models have been proposed by taking into account such additional effects, including the local field response,250,303 nonlinear optical process,304 field gradient (see the discussion in Section 3.4),305,306 self-interaction,307 optomechanical process,232,235 and chemical effects (see discussion in Section 1.2.2).308 Among all these models, the local response model of a SM in the confined plasmonic field can provide detailed analyses at the single-chemical-bond level, and we will focus on such a model in the ensuing discussion.
The optical response of a molecule under the illumination of incident light can be described as the polarized dipole p = αmolEloc(rmol), where αmol is the polarizability of the molecule defined in eq 8 and Eloc(rmol) is the local electric field at the molecular position rmol. Conventionally, the electric field distribution is much broader than the size of a molecule, so it is reasonable to regard the polarized molecule as a point dipole. However, as we discussed in the previous section, the local electric field inside a nanocavity can be extremely confined, even down to the atomistic scale, with a size comparable to or even smaller than the molecule, as shown in Figure 30. In this situation featuring an inhomogeneous field distribution, the point-dipole approximation would not be valid, as discussed in Section 3.4. Thus, the expression of polarizability αmol or the polarized molecular dipole p = αmolEloc(rmol) should be modified by taking into account the spatial distribution of the local electric field:
| 22 |
where |Ψi(r,f)⟩ (corresponding to |i⟩, |n⟩, |f⟩ in eq 8) is the molecular wave function of the initial (intermediate, final) state; ωri(rf) (corresponding to En – Ei (En – Ef) in eq 8) is the transition energy between state r and state i(f); and ω is the frequency of the electric field. By expanding the wave functions in the normal coordinates of the vibrations under the Born–Oppenheimer approximation, the polarized dipole can be finally expressed as three terms in analogy to Albrecht’s theory:
| 23 |
in which the first term pA is related to the FC term, and the second (pB) and third (pC) terms arise from the HT coupling, which derivations are described in eqs 8–11b. For the nonresonance Raman scattering, the contribution from the A term can be neglected without any loss of accuracy,309,310 and the Raman signals are mainly contributed by the B and C terms, which can be simplified as
| 24 |
where ρ(σ) refers to the x-, y-, or z-component of the transition dipole moment operator; |ψi(r)⟩ is the molecular electronic wave function of the initial (intermediate) state; |vi(vr′)⟩ is the molecular vibrational wave function of the initial (intermediate) state; and Qk is the normal coordinate of the kth vibrational mode. By transforming the coordinates from the normal-mode coordinates (Qk) to the atomic Cartesian coordinates (ξ(n)), and assuming that only the local electronic wave function near the nth atom plays a dominant role in the polarized dipole moment within the region of local electric field E(rn) at the atomic position, we can write
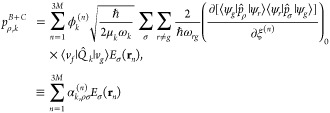 |
25 |
which defines the atomic Raman polarizability αk,ρσ(n) to account for the contributions from each atom in the molecule. Thus, the total polarized dipole moment corresponding to the Raman scattering process can be written as
| 26 |
where M is the number of atoms in the molecule. The emission of a collection of such polarized atomic dipoles would be further scattered by the nanocavity acting as a nanoantenna through Green’s function G⃡k(r∞,rn). Thus, the Raman scattering cross-section can be written as
| 27 |
Therefore, we can conclude that the TERS selection rule for a SM in an atomically confined light field can be stated as follows: only the contributions from the vibrating atoms within the local field region would dominantly contribute to the Raman signals of a SM.
Figure 30.
Schematic showing different Raman responses of a SM in a homogeneous light field versus an atomically confined light field.
4.2. Recent Experimental Advances in Subnanometer Resolved TERS Imaging
Owing to the atomically confined light field within a nanocavity and the atomistic contribution from single-molecule Raman scattering, the spatial resolution of TERS for a SM could be dramatically improved from the 15 nm reported in 2005281 to the subnanometer scale achieved in recent years.61,256,257 In 2013, Dong’s group first pushed the spatial resolution of TERS mapping down to the subnanometer scale (∼0.5 nm) under ultrahigh vacuum and liquid nitrogen temperature, and optically resolved the intramolecular structure of a single porphyrin (H2TBPP) molecule on Ag(111), as shown in Figure 31a.61 Such a high spatial resolution of TERS imaging broke the nanometer barrier and somehow reset the prevailing beliefs of the scientific community on the limits of both optical imaging and field confinement at that time. The ultrahigh spatial resolution also allowed to distinguish different molecular species and surface structures at the SM level, such as distinguishing between adjacent H2TBPP and ZnTPP molecules on the Ag(111) surface (Figure 31b–d)311,312 or identifying the conformational switch between two metastable surface-mediated isomers of the H2TBPP molecule on the Cu(111) surface at room temperature (Figure 31e–g).313 Recently, Kim’s group reported the resonant Raman effect of a single CuNc molecule on a three-monolayer-thick NaCl film supported by a Ag(111) substrate, wherein the SM-TERS image pattern was associated with symmetries of different vibrational modes (Figure 31h).258
Figure 31.

Single-molecule TERS imaging with subnanometer resolution. (a) TERS spectra on the representative positions of a single H2TBPP molecule adsorbed on Ag(111) and TERS mappings for different Raman peaks. Adapted with permission from ref (61). Copyright 2013 Springer Nature. (b) STM image for molecular chains of ZnTPP and H2TBPP adsorbed at the silver step edges. (c) TERS-VCA image acquired at the area marked by the dashed square in panel b. (d) Corresponding endmember spectra for four endmembers used in panel c. Adapted with permission from ref (312). Copyright 2017 Springer Nature. (e) STM image of the H2TBPP molecular island on Cu(111). (f) TERS line-profile obtained along the green dotted line in panel e. (g) STM height profile and a TERS intensity line-profile of 1502 cm–1 for the same line trace as that in panel e. Adapted with permission from ref (313). Copyright 2016 American Chemical Society. (h) TERS mappings of a single CuNc molecule adsorbed on the NaCl surface. The Raman mappings show different patterns depending on the symmetry of the corresponding vibrational mode. Adapted with permission from ref (258). Copyright 2020 Springer Nature.
The subnanometer resolved TERS technique not only allows to study molecular systems with the ability of chemical identification at the submolecular level, it can also be applied to other systems such as 2D-materials. For example, Jiang’s group reported the interfacial interaction of the DBP/borophene vertical heterostructrue by Ångström-resolved TERS spectra and revealed subtle ripples and compressive strains of the borophene lattice underneath the tetraphenyldibenzoperiflanthene (DBP) layer (Figure 32a,b).314 Wu’s group achieved a TERS enhancement of 109 and a spatial resolution of 0.5 nm using monolayer silicene on Ag(111), and identified the vibrational properties of the different silicene phases and the defects and domain boundaries in silicone (Figure 32c–e).270 Ren’s group demonstrated the edge-related vibrational properties of MoS2 on a Au film via AFM-TERS, and directly visualized the defect-induced band-bending region and electronic transition region at the edge in real space (Figure 32f–i).315 By using a specially fabricated tip, namely the plasmon-tunable tip pyramid, Jorio’s group realized the TERS spectroscopic imaging of the crystal superlattice in reconstructed twisted bilayer graphene with a lateral spatial resolution of 20 nm and revealed the subtle influence of electron–phonon coupling on the Raman intensity and peak line width (Figure 32j–l).269 Moreover, subnanometer resolved TERS can be used to specify the catalytic sites on surfaces. For example, Ren’s group reported the site-specific catalytic and plasmonic properties of a Pd/Au bimetallic system with 3 nm resolution, using phenyl isocyanide (PIC) as a probe molecule.267 Notably, the signal sensitivity of high-resolution TERS can be enhanced through the formation of an atomic point contact between the tip and sample surface, as demonstrated by Kumagai’s group.316,317
Figure 32.

Applications of TERS
for charactering two-dimensional materials.
(a) TERS spectra obtained on two types of borophene (red and blue
dots) and the Ag(111) surface (black dot), as shown in the inset STM
image. (b) TERS line profile across the edge between two types of
borophene. The TERS intensity profiles for the 189 and 180 cm–1 Raman modes show a spatial resolution of 5 Å.
Adapted with permission from ref (314). Copyright 2021 American Chemical Society.
(c) Gap-distance dependence of the TERS spectra for the silicone 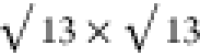 phase. (d) STM image of the silicene
phase. (d) STM image of the silicene 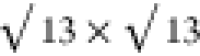 phase on Ag(111). (e) TERS intensity profile
for the A1 Raman mode across the edge of silicene marked
in panel d. Adapted with permission from ref (270). Copyright 2017 American
Physical Society. (f) AFM image of a MoS2 layer on Au substrate.
(g) Raman spectra on the representative positions of a MoS2 layer under tip approach and retraction conditions. (h) AFM height
and TERS line profiles across the edge of a bilayer MoS2. (i) Normalized TERS intensity profiles for the 396 and 406 cm–1 Raman modes across a bilayer edge. The inset shows
the intensity profile of the 406 cm–1 peak after
deconvolution of the EM field. Adapted with permission from ref (315). Copyright 2019 Springer
Nature. (j) TERS mapping of reconstructed twisted bilayer graphene
for the G′-band mode. (k) TERS line width
mapping of the G-band mode for the area marked with a dashed square
in panel j. (l) TERS line width profile for the G-band mode along
the high-symmetry directions shown by the orange and red lines in
panel k. Adapted with permission from ref (269). Copyright 2021 Springer Nature.
phase on Ag(111). (e) TERS intensity profile
for the A1 Raman mode across the edge of silicene marked
in panel d. Adapted with permission from ref (270). Copyright 2017 American
Physical Society. (f) AFM image of a MoS2 layer on Au substrate.
(g) Raman spectra on the representative positions of a MoS2 layer under tip approach and retraction conditions. (h) AFM height
and TERS line profiles across the edge of a bilayer MoS2. (i) Normalized TERS intensity profiles for the 396 and 406 cm–1 Raman modes across a bilayer edge. The inset shows
the intensity profile of the 406 cm–1 peak after
deconvolution of the EM field. Adapted with permission from ref (315). Copyright 2019 Springer
Nature. (j) TERS mapping of reconstructed twisted bilayer graphene
for the G′-band mode. (k) TERS line width
mapping of the G-band mode for the area marked with a dashed square
in panel j. (l) TERS line width profile for the G-band mode along
the high-symmetry directions shown by the orange and red lines in
panel k. Adapted with permission from ref (269). Copyright 2021 Springer Nature.
Furthermore, considering that the TERS signals of a SM are very sensitive to the local EM environment, one can even use a SM as a probe to explore the local electric field in the nanocavity. As shown in Figure 33a–e, a single 4,4′-bipyridine molecule was adopted as a local probe to reflect the spatial distributions of the local electric field at the tip apex.318 A carbon monoxide (CO) molecule at the tip apex can serve as a scanning electrometer to “feel” the change in the electrostatic fields from a SM through the shift of the C–O stretching vibrational mode, enabling the measurement of the electrostatic fields with submolecular resolution (Figure 33f–h).319 By rationally designing the structure of the molecular probe, such as using a set of viologen-based self-assembly monolayers, the vertical distribution of the plasmonic field within a nanocavity can be precisely measured with a longitudinal resolution down to 2 Å, as shown in Figure 33i–k.320 Therefore, the evidence from both the highly resolved chemical structures and the precisely measured local electric fields indicate that the subnanometer resolved TERS signals are mainly originated from the local chemical bonds or groups within the local plasmonic fields, which inspires the further development of Ångström-resolved Raman spectromicroscopy.
Figure 33.

Probing the field distributions of nanocavity plasmon and SMs by Ångström-resolved TERS. (a) STM image of a single 44BPY molecule on Ag(111) and the corresponding TERS mappings for the 1342 and 1607 cm–1 vibrational modes. (b) STM height line profile and (c) TERS intensity line profile of the 1342 cm–1 vibrational mode for a single 44BPY molecule along the line trace from the STM and TERS image in panel a, respectively. (d) Calculated local field distribution for the Ag tip shown in panel e. (e) Atomistic model of the closely packed Ag tip with the top two-layer atoms removed. Panels a–e are adapted with permission from ref (318). Copyright 2019 Wiley-VCH GmbH. (f) Simultaneously obtained STM and TERS images on the intensity, frequency shift, and line width for a single ZnEtio molecule under constant-current and -height modes. (g) Stark shift of the CO vibration corresponding to the dashed line from the constant-height frequency-shift map in panel f. (h) Electrostatic field surface obtained by color-coding the Stark shift on the STM constant-height topography. Panels f–h are adapted with permission from ref (319). Copyright 2018 American Association for the Advancement of Science. (i) Schematic of the molecular conformations for a series of radical cation viologens in the nanocavity of a SERS system. (j) Normalized experimental (solid symbols) and simulated (open symbols) Raman peak intensities for V2+ viologens in the nanocavity formed by a Au nanoparticle (NP) and Au(111) surface. (k) Normalized electric field distribution of the nanocavity formed by a Au NP and Au(111) surface. Panels i–k are adapted with permission from ref (320). Copyright 2020 Springer Nature.
4.3. Scanning Raman Picoscopy: Ångström-Resolved Raman Spectromicroscopy for Chemical Structural Reconstruction
The recent advances in subnanometer resolved TERS imaging described above have stimulated the exploration of Ångström-resolved Raman spectromicroscopy to resolve the chemical bonds or groups within a SM. However, the development of such a technique would still require an exquisite control over the tip structure and stability, especially the atomistic control of the tip apex to form a picocavity for the local field confinement at the subnanometer scale. Therefore, the tip quality is crucial for ultrahigh resolution TERS experiments. In this regard, ultrahigh-vacuum (UHV) and low-temperature conditions are highly desirable for fabricating and maintaining a stable plasmonic picocavity with an atomistic protrusion at the tip apex, along with other advantages such as contamination-free SM sample preparation and good thermal stability.321 Given the crucial role of the tip quality for TERS experiments, here we present a brief description of the typical tip fabrication procedure adopted in our work.287 Usually, the TERS tip is prepared using an EC etching process,322 followed by sputtering and annealing under UHV to ensure a clean and smooth tip with a radius at the tens-of-nanometer scale. Next, it is essential to decorate the tip apex with an additional atomistic structure to produce the “picocavity”. Such a requirement is fulfilled by gently indenting the tip to the metal substrate repeatedly, until some metal atoms or clusters are formed at the tip apex to yield TERS-active scattering signals. Notably, the stabilization of a picocavity structure requires cryogenic conditions, and liquid helium temperature (<10 K) is highly desirable in this regard. Otherwise, at higher temperatures such as those under ambient conditions, the local structure formed by metal atoms at the apex could be easily destroyed due to atom diffusion. In this case, the picocavity is reconstructed dynamically, resulting in the fluctuation of TERS signals, as discussed in Section 2.2. Nevertheless, it is worth pointing out that, although signal fluctuation is often viewed as a registry of SM experiments, it is definitely not an intrinsic feature of such experiments. Indeed, the signal can remain very stable over a long time as long as the molecule and local environment, such as the picocavity, are stable without changes, as demonstrated below for Ångström-resolved TERS imaging at liquid helium temperatures.
Using a tip fabrication method similar to that described above, in 2019, Apkarian’s group reported mode-resolved spatial mapping for a single Co(II)-tetraphenyl porphyrin (CoTPP) molecule on Cu(100).257 As shown in Figure 34a, each Raman peak shows its own characteristic image with rich details corresponding to different vibrational modes, and a spatial resolution down to ∼1.67 Å was obtained through the TERS line-profile measurement over individual C–H bonds.257 A similar spatial resolution of ∼1.5 Å was also reported in the same year for a single Mg-porphine (MgP) molecule on an Ag(100) surface, as shown in Figure 34b.256 The full vibrational-mode imaging of the MgP molecule for all the resolved Raman peaks could reveal molecular structural details with distinct characteristics, indicating distinctive contributions from different chemical groups or bonds corresponding to different vibrational modes.256 Therefore, a new methodology called scanning Raman picoscopy (SRP) was proposed for visually constructing the chemical structure of a SM through a Lego-like building process.
Figure 34.

Ångström-resolved Raman spectromicroscopy for a SM. (a) TERS mapping images for representative vibrational normal modes of CoTPP on Cu(100). Adapted with permission from ref (257). Copyright 2019 Springer Nature. (b) Spatial mapping images for various Raman peaks of a single MgP molecule on Ag(100). Adapted with permission from ref (256). Copyright 2019 Oxford University Press on behalf of China Science Publishing & Media Ltd.
As demonstrated in Figure 35, the assembling process of the “unknown” MgP molecular chemical structure (here, by “unknown” we assume its structure is not known in advance) can be realized stepwise by identifying different chemical units (groups or bonds) in space, with the help of a Raman fingerprint database and an interference effect discovered for pairs of symmetric and antisymmetric vibrational mapping images.256 For example, the highly localized C–H stretching vibration at 3072 cm–1 can be identified as the sp2 C–H stretching mode, and the out-of-phase destructive interference associated with an antisymmetric vibration of two neighboring C–H bonds would result in a well-resolved “eight bright dot” pattern, with these dots marking the positions of eight sp2 C–H bonds and their connections. Following similar procedures, a conjugated five-membered pyrrole ring structure can be determined for each lobe in the mapping images corresponding to the Raman peak at 1463 cm–1, and the connecting piece between the neighboring pyrrole rings can be determined as C–H bonds through a relevant out-of-plane bending vibration at 841 cm–1. The central atom can be identified as the Mg metal atom from the images at 211 cm–1, corresponding to the out-of-plane vibration of the central atom. With all these “Lego” pieces identified, the chemical structure of the target MgP molecule can be fully determined in real space.256
Figure 35.

Reconstruction of the chemical structure of a SM from TERS mapping. (a) STM topograph of the target molecule only shows the outline of the molecular shape without any structural details. (b) Reconstruction of the full molecular structure from the TERS mapping images of representative vibrational modes by assembling constituent chemical units together, including pyrrole C–H bonds, pyrrole rings, bridging C–H bonds, and a central Mg atom. Adapted with permission from ref (256). Copyright 2019 Oxford University Press on behalf of China Science Publishing & Media Ltd.
By exploiting the capability of the SRP technique in resolving the single chemical bonds inside a molecule, one can not only reconstruct chemical structures, but also in situ monitor the bond breaking and making processes at the single-bond level. Figure 36a,b shows three pentacene derivatives (α, β, γ) on the Ag(110) surface, where the C–H bonds at the central benzene ring are broken in sequence from α to γ, and the pentacene skeleton is also deformed with atomic rearrangements at the center. Through the Raman mapping of these molecules for the C–H stretching mode in the wavenumber range of 2800–2900 cm–1, the breaking of two C–H bonds at the center can be clearly observed and coincides with the skeleton features in the AFM images.262 The SRP technique can also be applied to investigate the adsorption behavior of up-standing molecules on surfaces. As shown in Figure 36c–e, through the analysis of the TERS signals related to the N–H stretching vibrations, one can not only specify two N–H bonds at the top of a single up-standing melamine molecule and other two N–H bonds at the bottom becoming dehydrogenated upon chemisorption on Cu(100), but also demonstrate the vertical resolving ability of TERS with a detection depth of ∼4 Å, albeit relatively limited. Furthermore, by monitoring the spectral evolution, one can even track the breaking of one top N–H bond and the formation of a new N–H bond on the cyanuric ring, resulting in the transformation from an adsorbed melamine to its tautomer through photon-induced hydrogen transfer.261 The SRP technique can also be applied to more complex molecular systems, e.g., for the H2TBPP porphyrin molecule on Ag(100), the orientation of the N–H bonds around the porphyrin core can be directly identified through the spatial mapping.323 For the [12] cycloparaphenylene nanohoop molecule, its adsorption configuration and structural deformation on different metal substrates can be directly identified through different features in the spatial mapping images, revealing the local structural deformation associated with different tilting of the benzene units in real space caused by inhomogeneous molecule–substrate interactions.324
Figure 36.

Monitoring the bond breaking and making processes at the single-chemical-bond level. (a) STM topograph of three types of “pentacene” molecules (α, β, γ) and the simultaneously acquired SRP image for the C–H stretching vibration and theoretically simulated Raman maps. (b) Optimized geometries of α-, β-, and γ-pentacene and the corresponding Q-plus AFM images. Adapted with permission from ref (262). Copyright 2021 American Association for the Advancement of Science. (c) Line-trace TERS measurements over a single melamine molecule on Cu(100) for the symmetric and antisymmetric N–H stretching vibration modes and corresponding TERS mapping images. (d) Schematic showing the vertical resolving ability of TERS for the N–H stretching vibrations at the bottom. (e) Sketch for the dehydrogenation and hydrogen tautomerization processes. Adapted with permission from ref (261). Copyright 2021 Oxford University Press on behalf of China Science Publishing & Media Ltd.
As a brief summary to the SRP methodology, the various experimental studies described above have demonstrated its strong structural characterization ability, regardless of whether a molecule is planar or three-dimensional. Such a Lego-like building process can be generalized with the aid of artificial intelligence (AI), including imaging recognition and machine learning, and could become even more powerful when combined with noncontact AFM or inelastic tunneling probe techniques that can reveal molecular skeletons. Ångström-resolved TERS is believed to serve as a versatile tool for probing and identifying chemical structures in various systems at the single-bond level, and will have a wide range of applications including surface science, surface catalysis and chemical reactions, 2D materials, and biomolecular imaging. More details on the future outlook of TERS are included in Section 7.
5. Chemical Mechanism of SERS and Semiconductor-Enhanced Raman Scattering
5.1. Chemical Mechanisms of SERS
Although the EM mechanism provides a reasonable theory to clarify the enhancement performance of a SERS-active substrate, it does not predict the observed selective enhancement of different vibrational modes under particular conditions.325 A chemical mechanism based on the CT theory has been proposed to unravel this problem.53,326 This mechanism can be easily understood by acknowledging that the chemical interaction of the adsorbed molecules with the substrate surface could directly affect the electron density distribution of the molecules; thus, the polarizability of the adsorbed molecules is increased, thereby leading to the enhancement of Raman scattering.26 The CT mechanism can be divided into three categories based on the nature of the CT process:
-
(I)
Ground-state CT model (also called a static chemical enhancement), which can be attributed to the bonding action between the molecule and substrate and is not involved with any excitations of the molecule–substrate system.327
-
(II)
Excited-state CT model (also called photoinduced CT), in which the existence of a photoinduced CT transition is assumed, where an electron is excited from the substrate to the adsorbed molecule or vice versa.
-
(III)
Electronic resonant transition within the molecule, which is the same as the resonance Raman scattering effect. However, the energy level of the substrate may also be involved in the electron transition under specific conditions.
The ground-state CT model is based on specific interactions, such as electronic coupling between the molecule and substrate and the formation of an adsorbate–surface complex. These interactions increase the polarizability of the adsorbed molecule in the complex compared to that of a free molecule in a normal Raman measurement.82,328 Assuredly, the ground-state CT model relies on the fact that even in the ground state there is a CT interaction between the substrate and adsorbed molecule. This CT interaction does not need the aid of light radiation. Specifically, such a CT contribution always lies in a chemical-binding-based SERS system, and its contribution is an increasing function of the electronic coupling strength. The ground-state CT contribution to the EFs is of the order of 10–102.82,327−329
Among the three CT models, the photoinduced CT model is considered the most important for SERS enhancement.53,83,330 The quantum mechanical description of the photoinduced CT model is explained in Section 1.2.2. When the Fermi level of the metal lies between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of the molecule, the ground-state electrons can be excited from the Fermi level of the metal and transferred to the LUMO of the molecule. Alternatively, the ground state electrons can be excited from the HOMO of the molecule and transferred to the Fermi level of the metal. After a short relaxation period, the photoexcited electrons immediately revert to their initial states and subsequently release a Raman photon with the adsorbed molecule in a vibrational state. Indeed, the photoinduced CT model enables the electron transition, which is an allowed transition in the molecular electronic states; however, the excitation energy is not sufficient to directly excite such a transition. The transition becomes a high probability process through the auxiliary of the metal energy level, and the polarizability of the system is significantly improved to obtain high Raman intensity.
5.1.1. Surface Selection Rules
The HT surface selection rules are widely applicable and reasonably precise in predicting the relative intensities of the Raman lines in a SERS spectrum for a molecule–metal system.83 The simplest expression for the HT surface selection rules can be represented by
| 28 |
where Γ(Qk) is the irreducible representation of the allowed vibrational mode (Qk). Γ(μCT)⊥ and Γ(μmol) are the irreducible representations to which CT belongs, perpendicular to the surface, and the allowed optical transition that belongs to the excited state of the molecule, respectively. Only the vibrational modes ascribed to Γ(Qk) will be active (strongly enhanced) by borrowing the intensity from the CT transitions, which indicates that specific Raman modes (Qk) will be selectively enhanced by the HT contribution via CT. This effect is highly dependent on the excitation frequency and/or voltage that modulate/s the CT process.
For example, 3,4,9,10-perylene tetracarboxylic acid (PTCA) was adsorbed on an Ag NP surface via weak Ag–O bonds, and the PTCA molecular plane was inclined several degrees from the vertical direction of the Ag surface (Figure 37).331 Following the deprotonation of the carboxyl group, the symmetry of PTCA should be lowered from the D2h to the C2v point group with no inversion symmetry element. According to the energy level diagrams, CT occurs from the Fermi level of the Ag NPs to the LUMO level of PTCA. The CT dipole moment operator is associated with A1 (θ = 0°) and the vertical component A2 (θ > 0°). If we assign transition symmetries to the C2v point group, only one type of transition (π–π* transition) between the occupied and unoccupied molecular orbitals is allowed for PTCA. The lowest transition corresponds to the singlet state with B1 symmetry. Applying the HT surface selection rules, the Raman bands belonging to b1 (A1 × B1) and b2 (A2 × B1) symmetry are expected to be selectively enhanced via the CT contribution. As expected, the b1 and b2 vibrational modes of PTCA initiate the dominance of the SERS spectrum, owing to the CT resonance (785 nm excitation).
Figure 37.

Wavelength–dependent SERS spectra of the Ag/PTCA system and a schematic illustration of the CT mechanism in the Ag/PTCA system. Adapted with permission from ref (331). Copyright 2017 American Chemical Society.
5.1.2. Degree of CT
A method for the quantitative evaluation of the CT contribution to the overall SERS enhancement was proposed by Lombardi and Birke.83,332 The degree of CT (ρCT) for a vibrational mode k can be defined as
| 29 |
where Ik(CT) is the intensity of a k-band in which the CT resonance as an additional contributes to its total SERS intensity. SPR means surface plasmon resonance. Two reference bands should be selected in the spectral region, I0(SPR) is the intensity of a totally symmetric band where only the SPR contributes to its intensity. If the k-band is totally symmetric, the main contribution to its intensity stems from SPR, wherein Ik(SPR) = I0(SPR). If it is not totally symmetric, i.e., it displays nontotal symmetry, the CT contribution dominates its intensity. At this moment, Ik(SPR) is negligible and can be neglected in many cases. Accordingly, the value of ρCT is zero when the CT does not contribute to the intensity of the k-band. However, this value approaches one when the CT contribution plays a leading role in the intensity of the k-band. The value of ρCT is 0.5, while the SPR and CT contributions are approximately equal.
The degree of the CT enables one to separate the CT contribution from the other contributions to the SERS signal; however, it requires that the Raman spectra be obtained using different scanning parameters, such as the excitation wavelength, applied potential, particle size, and interparticle distance.332 These scanning parameters change the resonance conditions for a CT between the Fermi level of the substrate and the specific energy level of the molecule. Thus, the CT contribution can be compared from the values of the degree of CT under resonant and nonresonant excitation. However, the CT resonance cannot always be observed by the potential modulated method, owing to the mismatching between the excitation energy and CT transition energy.
A CT complex system, comprising a continuously adjustable energy level structure, was designed by Ji et al. to investigate the CT contribution through a layer-by-layer assembly of TiO2 over a 4-mercaptophenol (4-MPH)-modified Ag NP film (Figure 38A).333−335 This approach enables control of the TiO2 thickness with monolayer precision, and the extent of the CT contribution to SERS can be modulated in the Ag/4-MPH/n-TiO2 system. Assuming that 4-MPH is vertically adsorbed on the Ag surface, the μCT has A1 symmetry (in the C2v point group). A strongly allowed singlet state of the π–π* transition in this system is the B2 symmetry.336 According to the HT surface selection rules, strong CT contributions will be anticipated from the b2 (A1 × B2) symmetry modes. As expected, the bands associated with the b2 modes were selectively enhanced in the SERS spectra of the Ag/4-MPH/n-TiO2 system, compared to those of the Ag/4-MPH system (Figure 38B).334 Considering the relative isolation from interference of the neighboring bands, the intensity of the band at 1163 cm–1 (C–H bend, b2 mode) was selected as Ik(CT), and the intensity of the band at 1072 cm–1 (C–S stretch, a1 mode) was defined as I0(SPR). Subsequently, the values of ρCT were calculated for the Ag/4-MPH/n-TiO2 system using the different excitation wavelengths. Based on the ρCT results, the absorption threshold of the Ag/4-MPH/1-TiO2 system was estimated to be approximately 1.95 eV, which is in accordance with the DFT results. Figure 38C illustrates the plots of the degree of CT against the number of layers of TiO2. Here, ρCT represents a resonance-shaped profile and reaches a maximum value at different layer numbers for the different excitation wavelengths. These results indicate that the resonant absorption band of the Ag/4-MPH/n-TiO2 system is blue-shifted as the number of layers increases because of the increase in the size of TiO2.337 Evidently, the layer-number-dependent SERS spectrum can be studied by analyzing ρCT.
Figure 38.

(A) Schematic illustration of the Ag/MPH/n-TiO2 system. (B) SERS intensity ratio of I1163/I1072 as a function of the excitation wavelengths. Inset shows the SERS spectra of 4-MPH in the Ag/MPH/1-TiO2 system. Adapted with permission from ref (333). Copyright 2011 Royal Society of Chemistry. (C) Correlation between the degree of CT (ρCT) in the sample Ag/MPH/n-TiO2 system and the layer number of TiO2 under three different excitation wavelengths. Panels A and C are adapted with permission from ref (334). Copyright 2012 American Chemical Society.
5.2. Applications of CT-Mechanism-Based SERS (CT-SERS)
For the convenience of the subsequent discussion, we here refer to the CT-mechanism-based SERS as CT-SERS. As mentioned above, the chemical mechanism of SERS is very sensitive to the electronic structure of the molecule. The noncovalent interaction of the molecule with another molecule or atom will affect the electronic structure of the molecule. Based on this, CT-SERS can be utilized to study the noncovalent interaction between a molecular probe and target species (including inorganic and organic species) and subsequently identifies the target species. In addition, both CT-SERS and photovoltaic devices involve electron transfer and separation. This intrinsic relevance enables CT-SERS to become a potential technique for studying the CT process in photovoltaic devices. In this section, we will introduce the typical applications of metal-based CT-SERS for inorganic species sensing, enantiomeric discrimination, and photovoltaic characterization. The semiconductor-based CT-SERS applications will be introduced in Section 5.5.
5.2.1. Inorganic Species Sensing
Inorganic species either have a small Raman scattering cross-section or they do not exhibit any vibrational modes in atomic systems. Thus, the highly sensitive SERS sensing of inorganic species cannot be realized by the direct detection of the Raman signals of inorganic species.338 By monitoring the vibrational changes of a ligand probe (inorganic species–sensitive molecule) induced by the introduction of an inorganic species, several research groups have progressed in the development of SERS-based sensing methods for inorganic species.339,340 The structure of the ligand probe usually comprises a conjugated π-system with a high Raman cross-section and a receptor unit with a specific coordination ability for the target inorganic species. The receptor units are the common functional groups and elements contained within organic ligands, such as carboxy, amino, hydroxy, N, and S groups. Generally, the binding of inorganic ions with the ligand probe induces the electronic perturbation of a metal–molecule system, which leads to the alteration of the Raman polarizability and eventually yields characteristic changes in the SERS spectra.341 According to the CT enhancement mechanism, the changes are mainly reflected in the frequency shift and/or relative intensity changes of the spectral bands that can be qualitatively and/or quantitatively correlated with the presence of inorganic species. Therefore, most SERS-based sensing principles have been proposed based on the CT mechanism.342,343 Notably, the molecular orientation on the substrate surface may vary after binding of the inorganic species to the ligands. In most cases, the CT mechanism and surface selection rule (associated with the EM mechanism) collaborate to detect inorganic ions.
The spectral alterations in the SERS spectrum focus on the vibrational modes associated with the binding groups and/or π-conjugated structures. The C–C skeleton vibration mode is the most sensitive to the coordination of metal species. For example, a 4-mercaptophenylboric acid (MPBA)-modified Ag NP-decorated silicon wafer (Si@Ag NPs chip) was used for the SERS analysis of F– ions.343 When the MPBA-modified Si@Ag NPs chip was exposed to a solution containing F– ions, a dehydration reaction occurred between the F– ions and boron center of MPBA. In the SERS spectra, the intensities of a pair of intense bands associated with the C–C stretching modes at 1589 and 1576 cm–1 exhibit a reverse change trend upon the bonding of fluoride ions to MPBA. The DFT results reveal that this change is due to the breaking of the structural symmetry by the F– ions and the charge redistribution of phenylboronic acid. A ratiometric signal of the area ratio (A1576/A1589) between the F–-bond MPBA molecules and unoccupied MPBA molecules was utilized as a spectral marker for the quantitative detection of F– ions.
The electronic distribution of a conjugated π-system in a molecule is highly dependent on the nature of the metal ion. According to the CT mechanism, a slight difference in the electronic structure is reflected in the SERS spectrum, owing to the coupling of the electronic and vibration states via the HT coupling constant. The multiplex detection of inorganic ions can also be achieved through the evaluation of specific changes induced by different metal ions.341,344 For example, the simultaneous SERS detection of Cu2+ and Co2+ ions was reported by using the 4′-(N-piperazinyl)terpyridine dithiocarbamate salt (TPY-DTC) as a Raman reporter.341 TPY ligands have a high affinity toward first-row transition metal ions, such as Cu2+ and Co2+ ions. The UV–vis spectra of the Co-TPY and Cu-TPY complexes indicate that these two complexes have different electronic states. In the SERS spectra, a marked shift in the ring-breathing mode of TPY at ∼1000 cm–1 was observed before and after the coordination of these two metal ions to the TPY ligands. Specifically, for the Co-TPY and Cu-TPY complexes, this peak shifted to 1033 and 1040 cm–1, respectively. This can be ascribed to the coupling of the different vibronic states of the Co-TPY and Cu-TPY complexes with the metal substrates. The simultaneous identification of Cu2+ and Co2+ ions can be achieved by monitoring the relative Raman intensities of the metal-ion-coordinated and uncoordinated TPY molecules. Furthermore, the SERS spectra of several metal-4-mercaptobenzoic acid (metal-4-MBA) complexes (metal: Cd2+, Pb2+, Pd2+, Hg2+, Zn2+, Fe2+, Cu2+, and Ag+) were studied by Moskovits et al.345 Based on this finding, a multiplex detection system was constructed by assembling MBA-modified Au NPs on the interior of a siloxylated glass capillary.
5.2.2. Enantiomeric Discrimination
Enantiomeric discrimination is crucial in the synthesis of chiral drugs and the study of their physiological mechanisms. Multiple chiroptical spectroscopic techniques, such as optical rotation and circular dichroism, have been proposed for the detection and quantification of enantiomers. Typically, enantiomeric discrimination requires the use of circularly polarized light and/or chiral molecules to achieve selective responses to different chiral enantiomers. Liu et al. reported chiral nanostructured Au films that exhibit local chiral EM fields.346 The local fields enhance Raman scattering for both enantiomers; however, the scattering due to one of the enantiomers is substantially stronger because of the matching of the structural handedness between the chiral nanostructured Au films and molecular chirality. The SERS-chiral anisotropy effect in this work relies on the chiral nanostructured Au films, which are synthesized using the chiral ligands. Recently, a novel label-free chiral SERS method based on the CT mechanism was proposed for enantiomeric discrimination, which excluded the use of chiral agents.
Wang et al. first demonstrated the application of CT-SERS for the enantiomeric discrimination of various chiral alcohols. The “chiral probe” in this work is the N atom arranged in self-assembled monolayers of 4-MPy on Ag NPs. This is because 4-MPy can easily form O–H···N hydrogen bonds with various alcohols (Figure 39A).347 The S and R enantiomer-4-MPy complexes may differ in their energy states, owing to the difference in their stereochemistry, which influences the CT transitions between 4-MPy and the Ag NPs. This slight difference in the CT transitions will increase with the resonant excitation of the enantiomer complexes, leading to the selective enhancement of the nontotally symmetric mode in the SERS spectra of 4-MPy, which has C2v symmetry. The relative intensity between 1202 and 1220 cm–1 was selected as an indicator to discriminate chiral alcohols, such as (R/S)-1,1,1-trifluoro-2-propanol (TFIP), (R/S)-2-butanol, and (R/S)-1-methoxy-2-propanol. The enantiomeric excess (ee) value (%) is positively associated with the relative intensities of 1202/1220 cm–1, indicating that the current CT-SERS method can be used to determine the enantiomeric purity.
Figure 39.

(A) SERS spectra of 4-MPy for the enantioselective discrimination of TFIP and (R)-TFIP with various ee values; correlation between the relative intensity ratio of I1202/I1220 and the ee values (%). Adapted with permission from ref (347). Copyright 2014 Wiley-VCH Verlag. (B) SERS spectra of MPBA in the presence of different chiral aromatic molecules; schematic illustration of the chiral discrimination mechanism. Adapted with permission from ref (348). Copyright 2020 Wiley-VCH Verlag.
More importantly, enantiospecific molecular fingerprints could be achieved simultaneously in the SERS spectrum. For instance, MPBA and n-hexanethiol comodified Au NR arrays have been ingeniously designed for the enantiomeric discrimination of small aromatic molecules (Figure 39B).348 In this study, the function of n-hexanethiol is to block the nonspecific binding of impurities on the surface of the Au NRs surface, and to capture chiral molecules through hydrophobic interaction. In this way, the molecular structure information on each chiral target could be clearly identified. However, the interaction of the chiral group with the boric acid group of MPBA has implications for the CT process between the Fermi level of the Au NRs and MPBA molecular orbital level. The symmetry of MPBA can be ascribed to the C2v point group, and the Raman bands associated with the b2 mode are more sensitive to the CT variation based on the surface selection rules. As expected, the b2 mode intensity of MPBA increased significantly when the system was incubated with D (or R)-type enantiomers; however, its intensity decreased markedly when the system involved L- (or S-) type enantiomers. Similar to earlier work, the variation in the relative intensity of b2/a1 can also be utilized as an indicator to differentiate between enantiomeric molecules. Because semiconductor materials have energy band structures, the anchoring of Ag NPs on the TiO2 surface can improve the sensitivity of nontotally symmetric modes to the CT process, thereby realizing efficient enantiomeric discrimination even at low concentrations.349
5.2.3. Photovoltaic Characterization
CT is a fundamental process that determines the energy conversion efficiency of photovoltaic devices; thus, exploring the CT process is beneficial for the rational design of photovoltaic devices.350−352 The interfacial electron transfer process is generally closely associated with the CT process from the Fermi level of the metal to the LUMO level of the adsorbed dye molecules with suitable laser excitation, or vice versa. This results in distinct changes in the relative intensity of the SERS spectra of the dye molecules. CT-SERS provides an in situ/operando characterization technology for studying the dynamics and interfacial processes of the photovoltaic devices. A series of proof-of-concept demonstrations was performed with one part of a dye-sensitized solar cell (DSSC), such as the independent interfaces or separate electrodes.
Wang et al. constructed a DSSC-like system to demonstrate the potential application of SERS for the characterization of electron transfer pathways.350 They compared the electron transfer pathways in three organic/inorganic interfaces, including Ag/4-MPy, Ag/4-MPy-iron phthalocyanine (FePc), and a solar cell-like TiO2/Ag/MPy-FePc system. Their results revealed that the interfacial electron transfer direction could be changed using mesoporous TiO2 NPs. The relative intensity between the nontotally symmetric mode (b2 mode) of 4-MPy at 1577 cm–1 and the totally symmetric mode (a1 mode) at 1612 cm–1 was used as an index to evaluate the CT process at the organic/inorganic interface. In the Ag-4-MPy system, the electron can be excited from the Fermi level of the Ag NPs to the LUMO of 4-MPy under 633 nm excitation, resulting in the selective enhancement of the b2 mode of 4-MPy due to the CT contribution. The CT process was inhibited after anchoring the FePc molecule with the N atom of 4-MPy. This effect is reflected in the reduction of the b2 mode of 4-MPy in the SERS spectrum. However, with the introduction of TiO2 NPs, the interfacial electron transition from the LUMO level of the FePc molecule to the CB level of the TiO2 NPs is more likely to occur. This transition is followed by transfer to the Fermi level of the Ag NPs, leading to the promotion of the CT process and a further increase in the intensity of the b2 mode. This study demonstrated that SERS can be used as an efficient method for estimating the electron transfer behavior in photovoltaic devices.
The interfacial CT properties of DSSCs can also be revealed in detail by analyzing the CT degree of the functional dye molecules in the SERS spectrum. For example, Wang et al. constructed a series of cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II) (N3) dye-sensitized DSSC photoanodes on SERS-active substrate surfaces.351 The effect of the Ag NPs on the CT process for the TiO2/N3/Ag system was first studied. Compared to the TiO2/N3 system, the Raman band at 1270 cm–1, which is associated with the coupling mode of the C–N and C–C stretching in the pyridine ring was found to be more sensitive to the CT in the TiO2/N3/Ag system. However, the Raman band at 1024 cm–1, that is assigned to ring breathing was insensitive to the CT.351 The Raman bands at 1270 and 1024 cm–1 were selected as Ik(CT) and I0(SPR), respectively, to calculate the degree of CT (ρCT) for the further study of the interfacial CT. The increase in ρCT in the TiO2/N3/Ag system indicated that the absorption capacity of this DSSC system improved significantly. This is because the SPR effect of the Ag NPs can broaden the optical response range and enhance the optical absorption of this system and the Fermi level of the Ag NPs provides a new CT pathway. This study provides a reasonable understanding of the effects of the Ag NPs on the DSSC performance of TiO2/N3 systems.
Based on this demonstration, the effect of the crystal structure on the CT performance can be further discussed by comparing the degree of CT in a TiO2/N3/Ag system that consists of TiO2 NPs sintered at different temperatures.351 Furthermore, the synergistic effect of the semiconductor on the DSSC performance can be revealed by the design of the ZnO-TiO2/N3/Ag structure.352 The semiconductor heterojunctions are good candidates for improving the efficiency of DSSCs, because their band structures favor electron–hole separation. This effect is evidenced by the significant increase in the degree of CT in the SERS spectrum of N3 in the ZnO-TiO2/N3/Ag system.
5.3. Semiconductor-Enhanced Raman Scattering: Another Possibility of SERS Application
5.3.1. Brief Development History
SERS requires the use of a suitable substrate with nanoscale roughness to achieve the enhancement of the Raman signal of a surface-adsorbed molecule. Since the discovery of SERS in 1974, coinage metal materials (Au, Ag, and Cu) are the first choice to realize a substantial SERS effect, because of their strong EM contribution due to the LSP resonance.1 Except for the attenuated total-reflection (ATR) and interference-enhanced Raman scattering, the experimental observation of SERS on semiconducting materials can be traced back to the 1980s.353−356 For instance, the first application of the CT theory to semiconductor materials was reported in the study by Yamada et al. as early as 1983.355 They observed SERS from pyridine adsorbed on the surface of single-crystal NiO(110). The enhancement was suggested to be from the CT transition from the crystal substrate to pyridine. However, the reported EF was only 10 orders of magnitude. Hayashi et al. first observed that the enhanced Raman scattering originated from the EM mechanism of copper Pc molecules on GaP NPs in 1988.353 The EF was estimated to be 700. The appearance of semiconductor-enhanced Raman scattering is extremely important for the better understanding of the SERS phenomenon itself; however, the reported relatively weak enhancement performance was not noteworthy for practical application. Indeed, the development of semiconductor SERS was stagnant for a long time in the 1990s.
Renewed interest in semiconductor SERS was stimulated by the requirement of in situ characterization techniques in semiconductor-material-involved fields.357 Since 2000, a growing number of semiconductor materials have been reported with SERS activity, further prompting the development of semiconductor-enhanced Raman scattering techniques.358−370 For example, Zhao cooperated with Lombardi et al. to demonstrate a series of SERS-active semiconductor materials, including metal oxides, metal tellurides, and metal sulfides.358−363 They discovered that the defect state in semiconductor materials introduced by self-doping and transition metal-doping can significantly increase the sensitivity of semiconductor SERS.365−370 Based on these works, they proposed a clear CT model to explain the enhanced Raman observed on semiconductor materials. Another representative work was reported by Zhang et al., in which Raman enhancement was observed on a graphene surface.364 Graphene is an ideal semiconductor substrate to further verify the semiconductor-enhanced Raman scattering effect and investigate the CT mechanism, owing to its flat surface and strong chemical interaction with organic molecules.342,343,371−375 Following these works, other researchers have proposed more methods to prepare and optimize the semiconductor substrates (see the detailed discussion in Section 5.4). The semiconductor-enhanced Raman scattering effect breaks the long limitation of SERS in which only metals can exhibit huge Raman enhancement, and provides a new understanding of the intrinsic nature of SERS. More importantly, semiconductor SERS injects new vitality into traditional SERS, possibly leading to promising functions based on their unique optical, chemical, electrical, and catalytic properties (see the detailed discussion in Section 5.5).
5.3.2. Advantages of Semiconductor SERS
SERS-active semiconductor materials have been thoroughly reviewed elsewhere, and thus, they are excluded from further discussion in this chapter.357,376−378 Instead, this chapter focuses on the mechanism and applications of SERS-active semiconductor materials. Before starting, the main advantages of semiconductor SERS, compared to those of metal-based SERS, may be summarized as follows:
-
(1)
High chemical stability. Semiconductor materials have excellent thermodynamic stability compared to conventional metals, which can effectively avoid the problem of signal reproducibility caused by substrate oxidation. In addition, semiconductor substrates widen the applicable media of SERS technology, such as strong oxidation, strong acid, and high-pressure media.
-
(2)
Good biocompatibility. Semiconductor materials contain abundant hydroxyl groups on their surface, which can combine with proteins through amino and carboxyl groups. Such binding modes do not damage the spatial conformation and biological activity of proteins, thereby solving the biocompatibility problem of metal SERS technology.
-
(3)
Surface modifiability. Metal materials can only be used for efficient adsorption of analytes containing sulfhydryl groups. In contrast, most common functional groups can be directly adsorbed on semiconductor material surfaces, with the modification strategies of semiconductor materials being more flexible than those of metal materials. This advantage facilitates the determination of analytes without functional group limitation.
-
(4)
Abundant species, low cost, and mature preparation process. The energy level structure of semiconductor materials can be regulated by size, shape, material, doping, and other methods. According to the test condition of the analyte, a suitable SERS-active semiconductor substrate can be selected to meet different SERS detection requirements. The synthesis of semiconductor substrates is cost-effective and diverse synthetic techniques may be used, thus making the application of semiconductor substrates in SERS feasible.
-
(5)
Wide application range. Semiconducting materials are widely used as active components in practical devices related to electronics, optoelectronics, energy storage and conversion, catalysis, biological sensors, and other fields. The wide applications of semiconductor materials provide more possibilities for expanding the application range of SERS technology.
5.4. Mechanism of Semiconductor-Enhanced Raman Scattering
EM and CT enhancements are the two primary mechanisms operating in metal-based SERS,25,83 with CT being a widely accepted explanation of the mechanism of semiconductor SERS. The CT process depends on the vibronic coupling between the CB and valence band (VB), with the molecular excited and ground states.379 It has recently been revealed that the Mie resonance plays an important role in semiconductor SERS.353,380,381 In contrast with metal-based SERS, the CT contribution has been demonstrated to be dominant in a semiconductor SERS spectrum. In this section, we introduce the enhancement mechanism of semiconductor SERS with some appropriate examples.
5.4.1. CT Resonance
Similar to that with metal-based SERS, the CT between a semiconductor and an adsorbate can produce an enhanced Raman scattering signal.382 The CT models for semiconductor-enhanced Raman scattering can also be divided into three categories, namely, ground-state CT, electronic resonant transition, and excited-state CT. The first two types correspond to the CT models proposed for metal-based SERS, while the excited-state CT for semiconductor SERS is more complex. Therefore, we will further discuss this last type by introducing excited-state CT contributions to semiconductor SERS.
The excited-state CT model for metal-based SERS is understood to be based on the vibronic coupling theory, which involves the vibronic coupling of the molecular states to the Fermi level of the metal states. In semiconductors, the Fermi level does not correspond to an actual energy level and cannot participate in CT. Instead, semiconductors have a partially filled VB and an almost empty CB separated by a band gap, the energy levels of which may function similarly to the Fermi level of the metals involved in the CT processes. In addition, the defect states in semiconductors play a crucial role in CT because they either provide free electrons or bind electrons to produce a new CT pathway. The CT contributions to semiconductor-enhanced Raman scattering are derived from the vibronic coupling of the CBs and/or VBs with the molecular excited state and/or ground state.
Understanding the pathways of the CT is crucial to explain the contributions of the CT to semiconductor-enhanced Raman scattering. The CT pathways between the semiconductor and adsorbed molecule are highly dependent on their energy band structures, and can be approximately divided into the following six categories376,377 (Figure 40):
-
(1)
Resonant excitation of the CT complex. In some cases, the strong electronic coupling between the semiconductor and adsorbed molecule results in the formation of a new CT complex (Figure 40A). When the excitation energy of the laser matches the transition energy of the CT complex, the resonant excitation of the CT complex leads to a resonance Raman scattering-like enhancement for semiconductor SERS. Notably, this resonant excitation is different from the molecular resonance.
-
(2)
Molecular HOMO to CB. Electrons are transferred from the HOMO of the molecule to the CB of the semiconductor (Figure 40B). The electron occupying the ground state of the molecule is directly excited, by the incident light, from the HOMO to an energy level in the semiconductor that corresponds to the CB. Therefore, the excited electron immediately transitions back to a ground vibrational energy level of the molecule, and a Raman photon is subsequently released. The SERS intensity is borrowed from an allowed molecular transition, μmol, by the molecule-to-semiconductor transition, μCT, through the HT coupling term hcl, as explained in eqs 10a and 10b.
-
(3)
CT complex to CB. Electrons are transferred from the CT complex to the CB of the semiconductor (Figure 40C). The CT complex is formed by the strong chemical bond between the molecule and semiconductor, thus providing a new electron transfer pathway for the CT process. The molecular polarizability is enhanced by such a CT transition, leading to an increase in the SERS signals. The SERS intensity is borrowed from the allowed molecular transition, μmol, by the CT complex-to-semiconductor transition, μCT, through the HT coupling term, hcl.
-
(4)
VB to molecular LUMO. Electrons are transferred from the VB of the semiconductor to the LUMO of the molecule (Figure 40D). The electrons in the VB are first excited from the semiconductor to the high-energy level in the molecule that corresponds to the HOMO, and thereafter quickly transition back to the VB of the semiconductor, releasing a Raman phonon with the molecule at a vibrational ground state. The SERS intensity is borrowed from an allowed molecular transition, μmol, by the semiconductor-to-molecule transition, μCT, through the HT coupling term, hvh.
-
(5)
Surface state to molecular LUMO. Electrons are transferred from the surface state of the semiconductor to the LUMO of the molecule (Figure 40E). The electron is first excited from the VB to the surface defect, resulting in the formation of a surface state at the semiconductor surface. Subsequently, the electron is further excited from the surface state to the HOMO of the molecule. The SERS intensity is borrowed from the allowed molecular transition, μmol, from the surface-state-to-semiconductor transition, μCT, through the HT coupling term, hsh.
-
(6)
Molecular excited state to CB. Electrons are transferred from the excited state of the molecule, and electron injection occurs from the photoexcited molecule to the semiconductor (Figure 40F). This type of CT model usually occurs in the dye-molecule-adsorbed-semiconductor systems. Generally, the dye molecules can be easily excited using visible light and have a more negative potential than that of the CB of the semiconductor. Thus, the electrons are initially excited from the HOMO to a higher energy level in the dye by the incident light energy, which corresponds to the LUMO. Subsequently, the electron is injected, through resonant tunneling, into a matching energy level in the semiconductor that corresponds to the CB. Thereafter, the electron transitions back to a ground vibrational energy level of the molecule and releases a Raman photon with the molecule at a vibrational ground state. This CT process gives rise to Raman enhancement, which is similar to the resonance Raman scattering mechanism.
Figure 40.

Mechanism of a photoinduce CT process (blue line) and coupling scheme (purple line) in semiconductor–molecule systems. (A) Resonant excitation of the CT complex, (B) molecular HOMO-to-CB, (C) CT complex-to-CB, (D) VB-to-molecular LUMO, (E) surface state-to-molecular LUMO, and (F) molecular excited state-to-CB transitions. Adapted with permission from ref (377). Copyright 2018 World Scientific Publishing Europe Ltd.
According to the six CT models proposed above, several strategies for designing semiconductor substrates have been proposed as follows:
-
(1)
Introducing surface defect states by doping or nonstoichiometric synthesis to promote CT interactions at the substrate–molecule interface.370,383−385 The doping of transition metals into semiconductors introduces a large quantity of the doped element into the bandgap of semiconductors and is an effective way to adjust surface defect states. A series of metal-doped TiO2 materials (M–TiO2, M = Fe, Co, Ni, Cu, Mn, and Zn) was synthesized by Zhao et al. First, the SERS activity of small (≤10 nm) anatase TiO2 NPs was investigated using three common molecular probes, namely, 4-MBA, 4-MPy, and 4-ATP. The three molecules have similar Raman scattering cross-sections, whereas the enhanced abilities of their TiO2 NPs are different. The observed enhanced sequence was 4-MBA > 4-MPy > 4-ATP, which is in accordance with the attractive electron ability of the functional groups in the three molecules. Considering the energy levels of TiO2 and the molecules, the enhancement was attributed to the TiO2-to-molecule CT contribution, or specifically, the surface-defect-state-to-molecular LUMO CT transition. After doping with different transition metals, the authors discovered that the SERS performance of M-doped TiO2 NPs largely depended on the doping quantity and intrinsic nature of the doping ions. The SERS enhancement capacity of the 3% Ni-doped TiO2 NPs was four or more times higher than that of the undoped TiO2 NPs. This increased enhancement capacity occurs because a suitable dopant prohibits the recombination of photoinduced carriers and promotes the TiO2-to-molecule CT transition. However, the excessive dopant becomes the recombination center of the photoinduced carriers, which prohibit the TiO2-to-molecule CT transition. In addition, the SERS performance of TiO2 NPs comprising different doping metals was found to be associated with the ionic radius of the transition metals, the mechanism of which needs further investigation. A similar effect was observed for a sequence of transition-metal-doped ZnO NPs. These studies clearly indicate that the appropriate introduction of surface defects by transition-metal doping can improve the SERS performance of semiconductors.
Nonstoichiometry is common among transition metal oxides, such as TiOx (1.998 ≤ x ≤ 2.000), VOx (0.79 ≤ x ≤ 1.29), NixO (0.999 ≤ x ≤ 1.000), and WOx (2.50 ≤ x ≤ 3.00). The stoichiometric composition of these semiconductors leads to the formation of intrinsic point defects (vacancies, ant-sites, and interstitials). The correlation between the nonstoichiometry of the transition metal and SERS performance is vital for the rational design of SERS-active semiconductors. For example, the SERS behavior of R6G on nonstoichiometric W18O49 NWs was revealed by Cong et al.386 The synthesized W18O49 sample contains a large number of oxygen vacancies, which can be modulated by simple annealing of the W18O49 sample in an Ar/H2 atmosphere. They observed that the oxygen vacancies in W18O49 strengthened the vibronic coupling interaction of the substrate–R6G and promoted the CT transition between the W18O49 NWs and adsorbed R6G molecules. Thus, SERS sensitivity can be observed in nonstoichiometric W18O49 NWs. Furthermore, the downsizing of WO3-x to ultrathin two-dimensional nanosheets could further improve the vibronic coupling effect, because the abundant surface defect states on the nanosheet surface promote CT activity at the substrate–molecular interface.383
-
(2)
Increasing the electronic density of states by utilizing amorphous semiconductor materials to promote the CT efficiency.379,387 Amorphous semiconductor materials exhibit two characteristic features: structural disorder and quasi-equilibrium. Thus, the electron energy band structure of amorphous semiconductors is essentially identical to that of crystalline semiconductors. However, the disordered structure of the amorphous semiconductors results in localized electronic states within the energy gap. The electronic density of states in amorphous semiconductor materials can improve vibronic coupling in substrate–molecule systems. This was demonstrated by Guo et al. based on their study of the SERS activity of amorphous ZnO nanocages.388 The first principle DFT simulation revealed that the CT process in the amorphous ZnO nanocages was stronger than that in their crystalline structures. This was attributed to the weaker electron constraint of the metastable electronic states on the surface electrons in the amorphous structures. Such a weak constraint on the surface electrons strengthens the coupling interaction of ZnO with surface adsorbates, even with sulfhydryl adsorbates. An unusual π-interaction between Zn and S in amorphous ZnO nanocages was verified by X-ray absorption near-edge structure (XANES) characterization. The strong coupling effect efficiently improved the contribution of the CT to SERS enhancement. Based on this finding, two-dimensional amorphous TiO2 nanosheets containing abundant oxygen defects were further designed by the same group.387 Compared to their crystalline counterparts, the higher electronic density of states of the amorphous TiO2 nanosheets strongly affect the electron cloud distribution of the adsorbed molecules and endow a larger SERS contribution from the interfacial CT transitions.
5.4.2. Mie Resonance
High EM fields are also sustained by high-refractive-index dielectric nanostructures, referred to as Mie resonance modes or whispering gallery modes (WGMs). Specifically, the Mie resonance, is a special type of morphology-dependent optical resonance localized close to the external boundary of a dielectric spherical particle, which is expected to be based on the Mie theory. However, in modern optics and photonics, Mie resonance is a generic term that refers to morphology-dependent optical resonances. The Mie resonance modes can be engineered by adjusting the particle size, wavelength, and refractive index. This type of resonance can confine free-space light into nanoscale volumes, exhibiting intriguing optical magnetic and electric resonance modes in the visible or near-IR spectral regions. If the Mie resonance modes match the incident laser, the enlarged local EM field significantly improves the SERS signal.
The EM field intensity at a spherical particle surface can be described by the near-field scattering efficiency (QNF) proposed by Messinger et al.389 The QNF is qualitatively similar to the scattering efficiency (Qscat), and can be used to evaluate the ability of a spherical particle to convert an incident electric-field intensity into an electric near-field intensity. In particular, for near dielectric sphere surfaces, the amplitude of the near-field scattering intensity can be much larger than that of the far-field scattering, leading to a local field enhancement. For a spherical particle, QNF may be determined from the equation
| 30 |
where hn(2) is the Hankel function of the second type, x = πd/λ is the size parameter, d is the diameter of the spherical particle, and λ is the wavelength of the incident light. In addition, an and bn are the Mie coefficients, which can be deduced using the complex Riccati–Bessel functions ψ and ζ:
| 31a |
| 31b |
where m is the complex refractive index of the spherical particle. The complex refractive index is a fundamental physical parameter of a material that changes with the wavelength of incident light. For a specific semiconductor material, the near-field scattering intensity depends mainly on the size of the spherical particles and the incident light wavelength. According to the classical EM field theory, the EF of SERS is approximately proportional to the second power of the amplitude enhancement of the near-field scattering intensity, QNF2. Thus, to produce a high electric-field contribution for SERS enhancement, the design of a SERS-active semiconductor substrate can proceed from the near-field scattering efficiency.382
First, semiconductor materials with high near-field scattering efficiency typically must have a high refractive index and small imaginary parts of their dielectric functions across a broad spectral range. The imaginary part is associated with dielectric losses, which represent the loss factor or energy-absorption capacity. For instance, Figure 41A shows the near-field scattering efficiency of CdSe calculated using different imaginary parts of the refractive index (0.356 and 0.010). The large imaginary part broadens the spectral lines and greatly weakens the scattering efficiency.382 This is because a semiconductor with a large imaginary part experiences loss or absorption of the incident photon, and subsequently weakens the near-field Mie scattering of the incident photon. For a selected semiconductor material, optimal dimensions are essential for highly efficient Mie resonances.390 In contrast to plasmonic metal materials, a strong Mie resonance is usually observed in semiconductors having diameters in the submicron range. For example, the map of the near-field scattering efficiency for spherical ZnO particles as a function of the wavelength and particle diameter was calculated based on the Mie theory (Figure 41B).380 The extrema shown in this map are related to the various transverse electric (TE) and transverse magnetic (TM) resonant modes.390 This occurs because when the incident light resonates with the TEn (or TMn) mode, the Mie coefficient an (or bn) appearing in the QNF expression increases substantially, leading to a resonant peak in the QNF. This allows us to estimate the required particle size for the most effective exploitation of near-field scattering with a specific incident photon wavelength. Clearly, the TE and TM resonant modes become more pronounced for larger spherical ZnO particles with short-wavelength excitations, and particles with a diameter of >100 nm appear to be suitable for producing a high QNF. Accordingly, to achieve semiconductor-enhanced Raman scattering dominated by a high electric-field contribution, the diameter of the ZnO particles should be larger than 100 nm.
Figure 41.

(A) Near-field scattering efficiency as a function of x = 2πa/λ for CdSe, calculated using different imaginary parts of the refractive index (0.356 and 0.010). Adapted with permission from ref (382). Copyright 2014 American Chemical Society. (B) Dependence on the incident wavelength and ZnO microsphere diameter for the near-field scattering efficiency. Adapted with permission from ref (380). Copyright 2019 Wiley-VCH Verlag.
The near-field Mie scattering theory was first applied to explain the enhanced Raman spectra of copper Pc adsorbed on GaP NPs.353 The results revealed that the EF increased with increasing average particle size. The obtained dependence of the EFs on the particle diameter was in accordance with the calculated dependence of QNF2. This size range of GaP NPs corresponded to the tail of the TE1 resonance, in which QNF monotonically increased along with the particle diameter increase. Another related work was reported by Rodriguez et al., who demonstrated that Si NPs exhibit a huge SERS effect owing to their Mie resonances.381 The simulation results demonstrated that the electric field induced by the Mie resonance for spherical Si exhibits the same or a larger intensity than that for Au plasmons at particular particle sizes. A high SERS of p-aminobenzoic acid on Si NPs was observed by the excitation of the different Mie resonance modes. Different methods, including mechanical milling, pulsed laser ablation in liquid, and gas phase synthesis, have been developed for the fabrication of Si NPs. However, a simple and suitable method for the large-scale preparation of high-quality Si NPs for practical application is yet to be developed. Other semiconductors, such as GaP, TiO2, ZnO, and ZnS, also meet the requirements for producing strong Mie resonances in the visible region, and more importantly, these semiconductors can be easily synthesized in the laboratory.
The multiple scattering effect can be interpreted as a succession of independent, single Mie-like scattering (morphology-dependent optical resonances) events that enhance material–light interactions.391 Alessandri et al. proposed the use of a modulated multiple scattering effect for the construction of plasmon-free semiconductor SERS substrates.392 Core/shell resonators based on SiO2/TiO2 beads (T-rex) were fabricated by the deposition of an ultrathin (≤5 nm) anatase-TiO2 layer on a SiO2 NP surface. The refractive index differences between the SiO2 core and TiO2 shell layer resulted in the total internal reflection of light within the TiO2 shell layer. Meanwhile, the electric field originating from the multiple light scattering effect was demonstrated to enhance Raman scattering for the analytes adsorbed at the T-rex surface. Similarly, a TiO2 inverse opal photonic microarray was reported by Zhang et al. for improved SERS sensitivity.393 The highest SERS sensitivity could be achieved by appropriately utilizing the slow light effect of a photonic microarray. TiO2 inverse opals with a cavity diameter of 225 nm exhibited the highest sensitivity. This is because the multiple laser scattering in the cavity improved the light–matter interaction, which provides significantly more opportunities for the stimulation of Raman scattering. Furthermore, they observed enhanced Raman scattering of methylene blue over chiral carbonaceous nanotubes modified with titania nanocrystals.394 The enhanced Raman scattering was ascribed to the multiple light scattering effect induced by the birefringence in the unique helical structure.
5.4.3. Plasmon Resonance
The plasmon resonance collective excitation of free electrons under illumination produces highly localized electric fields on the surface of the particle as explained in Section 1.2.1. The plasmon resonance frequency is typically calculated from the classical Drude mode, which can be expressed as
| 32 |
where N is the electron density (or P for holes), e is the elementary charge, m* is the carrier effective mass, and ε∞ is the high-frequency dielectric constant. According to this equation, plasmon resonances at visible frequencies occur only on electron-dense (1022–1023 cm–3) metal surfaces. The very low carrier density in the CB results in a plasmon resonance frequency of the semiconductors in the infrared region. The electron density (n) of the valence electrons in solids is typically in the range of 1022–1024 cm–3, and thus, the VB plasmon is expected to lie within the ultraviolet region, instead of the visible range.395 The plasmon resonance frequency of semiconductors does not match the excitation energy used for Raman measurements and cannot account for the SERS enhancement.
Carrier-doped semiconductors provide a new class of nanomaterials with surface plasmons in the NIR region, and their resonance frequencies can be tuned by controlling the free carrier density via chemical doping. Nonstoichiometric synthesis strategies,396,397 for example, indium tin oxide (ITO), Al-doped zinc oxide (AZO), Cu2–xS, Cu2–xSe, WO3–x, and MoO3–x, have been demonstrated to exhibit NIR surface plasmon modes.396,398 A few proof-of-concept works reported that the plasmon resonance frequencies can be further tuned into the visible region, and observed the enhanced Raman signal of these plasmonic semiconductors.399−401 However, the enhancement mechanism in these systems cannot be fully ascribed to plasmon resonance enhancement.402 In most cases, it is still difficult to observe the enhanced Raman signal on these plasmonic semiconductors with visible responses, without the assistance of the CT contribution.
5.4.4. Exciton Resonance
An electron is capable of being excited from the VB into the CB, resulting in a strongly correlated electron–hole state, namely an exciton. The exciton Bohr radius is regarded as the distance between an electron and hole pair, and the quantum effect of semiconductors is related to this radius. When the particle size of a semiconductor is less than the intrinsic exciton Bohr radius, the quantum confinement effect becomes apparent and gives rise to unique optical properties. For an exciton resonance process, a vibrational quantum of energy is transferred to the vibrational level of the molecule, and a Raman photon is radiated from the molecule at some vibrational state. Numerous studies have demonstrated that the Raman EF depends on the size of the semiconductor NPs. For example, 5 nm CdSe quantum dots exhibit an EF as high as 105 under 488 nm excitation, which is significantly greater than that of the original contribution from traditional CT resonance.403 This additional enhancement is considered to be derived from the contribution of the exciton resonance, because the excitation energy is similar to the exciton resonance. The relationship between the exciton resonance and SERS needs to be further studied.
5.4.5. Synergistic Effect
As mentioned above, various mechanisms, including the CT, Mie, exciton, and plasmon resonance mechanisms, have been discovered to exhibit enhanced contribution to semiconductor-enhanced Raman scattering. However, most current semiconductor substrates were designed based on a single enhancement mechanism. The coupling of multiple resonances to create a synergistic effect is a new direction to further increase the semiconductor SERS performance. Such a synergistic effect can be integrated and optimized from two perspectives: The first is more focused on the improvement of the Mie resonance by engineering the morphology of the semiconductor substrate, while the second focuses on improving the CT resonance by adjusting the energy band of the semiconductor substrate.380,404−407
For the first case, Ji et al. demonstrated that a synergistic effect between the Mie and CT resonances can be gained by the ingenious design of spherical ZnO superstructures (Figure 42A).380 In this work, a ZnO superstructure with a uniform diameter in the range of 200–500 nm, formed by aggregation of ∼13 nm primary single crystallites, was designed for exploring the Mie resonance scattered near-field enhancement effect for SERS enhancement and its synergistic effect with CT. The existence of the Mie scattering effect in these ZnO superstructures was confirmed by the extinction spectra, which were identical to those observed from Mie theory simulations. By engineering the superstructure size and excitation wavelength to induce Mie resonances, the Mie resonance contribution to the SERS enhancement was well studied. The results of the SERS spectra and theoretical simulations, including near-field scattering efficiency and finite element method simulations, indicated that the Mie resonance induced by the secondary superstructure could enhance the electric near-field, leading to an EM contribution for SERS enhancement. Meanwhile, the primary nanocrystallites in these ZnO superstructures yielded a highly effective CT contribution for SERS enhancement, which was verified by ultraviolet photoemission spectroscopy (UPS) and the CT degree. The synergistic effect of the CT and Mie resonances increased the EF by up to 105. This SERS performance is one of the highest reported sensitivities among SERS substrates used for the enhancement of nonresonant molecules. Notably, even without the photoinduced CT contribution, the EF can reach up to 103, owing to the synergistic effect of Mie resonance and static chemical enhancement. This implies that semiconductor superstructures may be used as a universal SERS-active substrate for different analytes.
Figure 42.

(A) Diagram illustrating the microstructure of a ZnO superstructure comprising closely packed nanocrystallites. Comparison of the EF and near-field scattering efficiency observed on ZnO superstructures with different diameters under 532 nm excitation. Inset shows the corresponding SEM and electric-field distribution images of the ZnO superstructures. Adapted with permission from ref (380). Copyright 2019 Wiley-VCH Verlag. (B) Mechanism of the synergistic resonance strategy on hollow multishelled V2O5 microstructures. Adapted with permission from ref (404). Copyright 2021 Wiley-VCH Verlag. (C) Mechanism of the synergistic resonance strategy on the Mo-doped Ta2O5 nanosheets. Adapted with permission from ref (405). Copyright 2019 Wiley-VCH Verlag.
Based on this finding, Ji et al. further demonstrated a new type of multiple synergistic resonant strategy by utilizing a unique double-shelled hollow spherical V2O5 microstructure as a model (Figure 42B).404 Three types of V2O5 microstructures, namely double-shelled hollow spherical, single-shelled hollow spherical, and lamellar microstructures, were fabricated and compared. The multiple CT contributions of these V2O5 microstructures were verified by structure and defect characterization. Furthermore, symmetry analysis of the vibrational mode observed in the SERS spectrum well explains the origin of the CT coupling of the HT selection rules. More importantly, the significance of the double-shelled hollow spherical microstructure was further investigated by finite elemental analysis. The results revealed that in contrast to the lamellar microstructure, light is confined and guided inside the shell instead of passing directly through the shell, indicating that reflections by multiple convex shells can provide an efficient resonance absorption for CT and exciton enhancements. Meanwhile, the coupling of Mie resonances from outer and inner shells strongly reinforces the magnitude of theEM near-field, leading to a high EM contribution for SERS enhancement. The synergy of all these factors contributes to a significant increase in the semiconductor SERS sensitivity.
For the second case, Mo-doped Ta2O5 nanosheets were synthesized by Yang et al. to optimize the semiconductor SERS performance (Figure 42C).405 Notably, Ta2O5 is a typical dielectric material with high refractive index and low losses in the visible region, which also satisfy the requirements of Mie resonance. The Mie resonance-induced electric field distribution of the Ta2O5 nanosheets was simulated by the FDTD method. The maximum electric field EF was only nine at the angular positions of the two Ta2O5 nanosheets under irradiation with a 532 nm laser. This relative week electric field contribution is not surprising, because the sheet structure cannot support a strong Mie resonance. However, the CT contribution was significantly boosted by the energy band adjustment through Mo-doping. The DFT results indicated that doping of the Mo atom could substantially lower the band gap of Ta2O5. The experimental results demonstrated that the Ta2O5 nanosheets with 15% Mo doping concentration showed the most suitable energy band structural match between methyl violet and the doped Ta2O5, leading to the best CT contribution for SERS. The lowest detection limit of methyl violet on the Mo-doped Ta2O5 substrate was as low as 9 × 10–9 M under resonant excitation of the methyl violet with a 532 nm laser. The high enhancement performance can be attributed to the synergistic effect of the Mie, CT, and molecular resonances. Similarly, this group also reported another two types of two-dimensional materials, namely a nanocanyon-like SnS2 structure and an MXene, to couple the Mie and CT resonances for SERS detection of coronavirus (see the detailed discussion in Section 5.5.1).406,407
5.5. Applications of Semiconductor-Enhanced Raman Scattering
Along with the rapid development of nanoscience and the ever-deepening understanding of the enhancement mechanism, the SERS performance on semiconducting materials has been greatly improved, and the EFs for some specific semiconductors were reported to be comparable to those of noble metals without HSs.386,405 These advances pave the way for the direct study of molecular information on semiconductor surfaces by SERS technology, which offers a new possibility to extend SERS applications in several semiconductor-based fields. These applications can be roughly divided into four categories and are discussed as following.
5.5.1. Biochemistry
Semiconducting materials are biocompatible and have frequently been proposed as ideal surfaces for the immobilization of biomolecules. Combined with semiconductor-enhanced Raman scattering, detailed molecular-level information can be obtained from a semiconductor surface to investigate the structure and function of biomolecules. For instance, Rajh et al. performed SERS measurements to study the adsorption of biologically active enediol molecules, such as dopamine (DA), salicylate, acetylacetone, and sodium benzoate, on a colloidal TiO2 NP surface.408−411 The colloidal TiO2 NPs comprised undercoordinated defect sites, which exhibit high reactivity toward the binding of monocyclic aromatic enediol ligands to the NP surface.408 Compared with the unmodified TiO2 NPs, the strong vibronic coupling between the TiO2 NPs and enediol molecules shifted the onset of absorption to the visible region, leading to the formation of hybrid CT-NP complexes. According to the wavelength-dependent Raman intensities of these hybrid CT-NP complexes, Rajh et al. observed that a molecules-to-TiO2 CT contributed to the SERS enhancement. DA was selected as the model molecule for the intensive study of the SERS spectrum of enediol molecules on TiO2 NPs. The results revealed that all the enhanced vibrations were associated with nontotally symmetric ring vibrations of the DA molecules, further confirming that the enhancement is due to the CT mechanism via HT vibronic coupling. The SERS intensity exhibited an asymptotic relationship with DA, indicating that there was a specific saturation effect for electron injection from DA to the CB of the TiO2 NPs. Following this work, the researchers further theoretically studied the effect of size, solvent, and pH on the observed Raman activities by performing computational investigations for TiO2 and its complexes with both catechol and DA.410 Furthermore, Lee et al. studied the pH-dependent configuration of l-dihydroxyphenylalanine (L-DOPA) at the rutile TiO2–water interface.411 They reported that as the pH and surface coverage increased, the intensity of the C=O stretching vibrational mode decreased, while that of the C=C stretching mode increased. These results indicate that the adsorption of L-DOPA on TiO2 NPs from a nearly perpendicular conformation translated into a parallel conformation as the pH and surface coverage increased. The orientation of L-DOPA on the TiO2 NPs may be critical for the condensation reactions of amino acids on mineral surfaces.
EC SERS is a powerful method for studying the structure and reaction dynamics of redox proteins immobilized on electrodes.415 The metal electrode surface must be modified with an amphiphilic monolayer to improve biocompatibility.416 However, monolayers may weaken the electronic interactions between the redox-active biomolecule and metal material. Semiconducting materials are good candidates for studying and simulating the redox reactions of proteins using SERS. For example, Hildebrandt and Weidinger’s group applied a nanostructured TiO2 electrode to probe the electron-transfer process of Cyt b5 (Figure 43A).412 The electrode surface comprised half-ellipsoidal TiO2, which was fabricated by the simple anodization of Ti foils. The aspect ratio of the half-ellipsoidal TiO2 can be modulated by the anode voltage, and a strong electric field enhancement for SERS was observed in the gap between the two adjacent half ellipsoids when the TiO2 electrodes were anodized at 20 V. TiO2 electrodes can be used directly for the biocompatible immobilization of proteins without further modifications, compared with metal electrodes. The electron transfer dynamics of Cyt b5 was investigated by monitoring the characteristic oxidation marker ν4 mode. Based on this finding, this group extended their studies to a new type of TiO2 electrode with nanotubular geometry (Figure 43B).413 For this nanostructure, the high electric field contribution to SERS was attributed to the multiple scattering and interference effects within the nanotubes. The SERS signal of Cyt b5 was approximately 10 times higher than that observed on the TiO2 electrodes with half-ellipsoidal geometries. Furthermore, both studies found that a lower resistivity benefits the probing of the electron transfer dynamics of redox proteins on semiconductor electrodes.
Figure 43.

(A) SEM images and electric field distribution of the synthesized half ellipsoidal TiO2 nanostructures and the molar ratios of oxidized (squares) and reduced (circles) Cyt b5 on the TiO2 electrodes. Adapted with permission from ref (412). Copyright 2013 Wiley-VCH Verlag. (B) SEM images and calculated electric field for the TiO2 nanotube electrodes and SERS spectra of Cyt b5 adsorbed on the fabricated TiO2 nanotube electrodes with different annealing temperatures. Adapted with permission from ref (413). Copyright 2018 Wiley-VCH Verlag. (C) SEM and HRTEM images of the K2Ti6O13 NWs; structure of the K2Ti6O13 NWs; schematic diagram of the photoinduced CT mechanism; Raman and SERS spectra of cytochrome c (Cyt c), the spectra from bottom to top are for the oxidized form of Cyt c (532 nm laser), reduced form of Cyt c (532 nm laser), Fe3+ Cyt c on K2Ti6O13 (532 nm laser), Fe3+ Cyt c on K2Ti6O13 (633 nm laser), and reduced form of Cyt c (633 nm laser). Adapted with permission from ref (414). Copyright 2021 Elsevier.
A related study based on CT SERS was performed on semiconducting K2Ti6O13 NWs by Han et al. (Figure 43C).414 The SERS of 4-MBA on K2Ti6O13 NWs was used as a model system to study the enhancement mechanism. The DFT calculations revealed that the CT from K2Ti6O13 to 4-MBA is as high as 2.28 e, which is ten times larger than that observed in the 4-MBA-amorphous ZnO system.388 These results indicated that SERS on K2Ti6O13 NWs is dominated by the highly efficient CT mechanism. The synthesized K2Ti6O13 NWs were ideal biocompatible substrates for protein immobilization. The SERS signal corresponding to the reduced form of Cyt c was clearly observed when Cyt c was attached to the surface of the K2Ti6O13 NWs under 532 nm laser excitation. However, Cyt c remained in its oxidized form at a lower excitation energy (633 nm). Molecular dynamics simulations further revealed that the CT from K2Ti6O13 to the Fe center of heme was realized because of the lysine residues of Cyt c. This work further demonstrates that semiconductor-enhanced Raman scattering has immense potential for studying the electron transfer process at protein–semiconductor interfaces.
In addition to small biological molecules and proteins, semiconductor-enhanced Raman scattering has been used to detect coronaviruses.406,407 Very recently, Yang et al. reported two types of MXene nanosheets (Nb2C and Ta2C MXenes) that could be used to detect severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) S protein.406 Nb2C and Ta2C MXenes exhibited high SERS activity for methylene blue and methyl violet, respectively. The SERS spectra of the SARS-CoV-2 S protein were dominated by the Raman vibrational modes of three amino acids, namely tyrosine (Tyr), tryptophan (Trp), and phenylalanine (Phe). Interestingly, the Raman signals of the SARS-CoV-2 S protein obtained on Ma2C MXenes appear to be different from those obtained on Au NPs. The authors attributed this difference to the difference in the SERS mechanism and extent of CT between the two SERS substrates. Furthermore, another example of SARS-CoV-2 detection system was proposed by the same group.407 In the latter study, SnS2 microspheres with a unique nanocanyon morphology were designed for the identification of various physical forms of SARS-CoV-2 through the principal component analysis of the SERS data.
5.5.2. Photoelectrochemistry
Recently, DSSCs fabricated from low-cost semiconducting materials have displayed commercial potential as alternatives to traditional Si photovoltaics. The anchoring mechanism between the semiconductor and dye directly influences the electron transfer and performance of the dye-sensitized photoanode. SERS studies of N719 dye-adsorbed TiO2 photoanodes have been performed by Lee et al., to reveal the bonding mechanism between the N719 anchoring groups (COO– and COOH) and the TiO2 surface groups (Ti–O, Ti–OH, Ti–OH2).417 Qiu et al. prepared Ag@TiO2 NPs to study the potential-dependent SERS spectra at TiO2-N719 dye interfaces.418 Both groups observed that the N719 dye chemically binds to TiO2 through a bidentate carboxylate (COO–) linkage; however, ester-like linkage cannot be excluded. Notably, these studies were only performed on separate electrodes or independent interfaces, rather than on a complete solar cell.
Very recently, Mao et al. first applied SERS to dynamically monitor the interface evolution of a complete DSSC under operando conditions (Figure 44).419 In this work, the DSSC system comprised N719-modified TiO2 nanotube arrays as the photoanode, Pt-coated F-doped Sn-oxide (FTO) glass as the counter electrode, and four electrolytes (KI/I2, LiI/I2, Et36, and Et23). The interface evolution of TiO2/N719/liquid-electrolyte DSSCs could be comprehensively assessed by the characteristic Raman bands of TiO2, dye N719, and the KI/I2 electrolyte in the DSSC under open-circuit, short-circuit, and load conditions. In the Raman spectra, the C=C stretching modes of N719 associated with 2,2′-bipyridyl (bpy) rings were observed on the TiO2 nanotube electrode surface. Notably, the researchers did not clarify the enhanced Raman mechanism of N719 on TiO2 nanotube arrays. Considering the specific structure of the TiO2 electrode and the excitation energy, the enhancement may be ascribed to the synergistic effect of the Mie, CT, and molecular resonances.
Figure 44.

(A) Schematic representation of the DSSC analysis by operandor resonance Raman spectroscopy combined with I–V testing. (B) SERS spectra of the DSSC under open circuit (OC) and short circuit (SC) conditions. Adapted with permission from ref (419). Copyright 2020 Wiley-VCH Verlag.
Raman spectroscopy was employed to simultaneously obtain the vibrational information on the TiO2 and KI/I2 electrolytes in the DSSC system. They found that the phonon vibrational modes of TiO2 and the C=C stretching modes of N719 moved in the opposite direction when the potential was higher than the open-circuit voltage (VOC = 0.5 V) of the cell. In the Raman spectra, the shift of the peaks toward the lower or higher wavenumbers is related to the chemical bond length (migration of electron clouds). Thus, a shorter bond length (high electron cloud density) leads to a shift to a higher wavenumber and vice versa. The electron injection from TiO2 to N719 can be directly reflected in the Raman spectra by the analysis of the corresponding Raman peaks. Mao et al. reported that the Raman position and intensity of the C=C stretching mode at 1546 cm–1 exhibited a similar shape to the photocurrent–voltage (I–V) curves of the DSSC, when the applied potential was smaller than the VOC of the cell. This can be used as a measurable indicator to predict the power conversion efficiency. Thus, this work demonstrated a new possibility for the semiquantitative analysis of the DSSC performance using an in situ SERS technique under operando conditions.
5.5.3. Catalytic Chemistry
SERS has become a useful and practical method for in situ surface studies of catalytic reactions. Semiconducting materials are widely used in the catalysis of photochemical reactions. To study the photochemical reaction at a semiconductor surface, shell-isolated metal NPs must be placed at the semiconductor surface to produce a detectable SERS signal. Thus, shell-isolated NP-enhanced Raman spectroscopy has proven to be a powerful technique that can be used to reveal the catalytic mechanism and dynamics of the reaction. However, this method does not always work, because the shell-isolated metal NPs cannot be placed onto the catalytically active site. Semiconductor-enhanced Raman scattering provides a new possibility for the in situ and direct study of the reaction mechanism and kinetics at the solution–semiconductor interface.
Recently, Song et al. reported an enzyme-catalyzed kinetic model on reduced MnCo2O4 (R-MnCo2O4) nanotubes using the semiconductor SERS method.420 Reduced MnCo2O4 (R-MnCo2O4) nanotubes were synthesized via an electrospinning process followed by calcination and reduction using NaBH4. X-ray photoelectron spectroscopy (XPS) revealed that abundant oxygen vacancies were introduced into the lattice of the R-MnCo2O4 nanotubes during the reduction process. Additionally, the electric field contribution from the hollow tubular structure was confirmed by the numerical simulation of the near-field distribution of the Mie scattering via the FDTD method. Compared with the MnCo2O4 nanotubes, the R-MnCo2O4 nanotubes exhibit significantly higher SERS activity, attributed to the synergistic effect of the high-efficiency CT caused by the oxygen vacancies and the electric field induced by the Mie resonances. In addition, R-MnCo2O4 nanotubes display favorable oxidase-like catalytic activity and can thus be used as a platform for in situ SERS monitoring of enzyme-catalyzed reactions. The Michaelis–Menten parameter Km (affinity between the enzyme and substrate) and Vm (maximum reaction rate) were estimated from the characteristic SERS peaks of oxidized TMB. The Vm values obtained from the SERS-kinetics and UV–vis-kinetics models were similar; however, the Km values measured by the SERS-kinetics model were much lower than those measured by the UV–vis-kinetics model. These results further demonstrated that SERS is highly sensitive to the reaction occurring at the solution–catalyst interface, and the kinetic parameters obtained from the SERS-kinetics model were more accurate than those obtained from the UV–vis-kinetics model.
Aside from the study of enzyme-catalysis reactions, semiconductor SERS can also be used for photocatalytic systems. Alessandri et al. monitored the ultraviolet light-induced degradation process of methylene on core/shell SiO2/TiO2 microspheres.392 Further, Song et al. investigated the degradation kinetics of methylene blue on reduced graphene oxide/AgCo composite nanosheets surfaces.421 Inspired by the natural photocatalytic property of the semiconductors, Yang et al. fabricated a self-cleaning mesoporous TiO2 substrate via the sol-hydrothermal method.422 The superior SERS performance was mainly attributed to the surface oxygen vacancies, which provided many active sites for molecular adsorption and broadened the donor energy level distribution for the CT. The adsorbed analytes on mesoporous TiO2 can be degraded into small inorganic molecules using ultraviolet light irradiation. Thus, mesoporous TiO2 can be repeatedly used in SERS detection without losing its activity. Similarly, Sampath et al. reported carbon-doped TiO2 substrates that can be used as visible-light-assisted recyclable SERS substrates owing to carbon doping.423
Mujica et al. investigated the preactivation mechanism of CO2 on a 3-aminosalicylic acid (3-ASA) functional anatase TiO2 NP surface.424 The as-produced 3-ASA was adsorbed onto the TiO2 surface by forming complexes with the Ti atoms through the adjacent OH groups, and the amino group was close to the TiO2 surface. The ligand-to-metal CT (LMCT) from 3-ASA to TiO2 lowers the effective band gap of the TiO2-3-ASA conjugates and results in a shift of the excitation threshold to the visible region. In the presence of CO2, the excitation threshold of the TiO2-3-ASA conjugate undergoes a further bathochromic shift. The nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) results indicated that electrons were photoexcited from 3-ASA into the CB of TiO2, and thereafter transferred to the neighboring CO2 site along with the deprotonation of the amine. These preactivation processes are directly or indirectly reflected in the SERS spectra. For instance, no significant changes were observed in the vibrational modes associated with the N–H bonds in the presence or absence of CO2, confirming that CO2 does not directly bond with the amino group. Furthermore, some vibrational modes displayed significant changes, which may be associated with the CT. Combined with the theoretical calculation for the assignment of the vibrations, the preactivation mechanism of CO2 on semiconductors can be further revealed using SERS.
5.5.4. Chemical Sensing
Recently, the performance of SERS-active semiconductor substrates has improved. However, the universality and sensitivity are the two main factors that still limit the practical application of semiconductor-enhancement Raman scattering for quantitative analysis.425 Ji et al. proposed a turn-off SERS strategy for the quantitative detection of inorganic ions, including CrO42– and PO42–.426,427 First, a sensing system based on a cocatalysis mechanism was developed using alizarin red S (ARS) dye-sensitized colloidal TiO2 NPs as the SERS reporter.427 The strong electronic coupling between ARS and the TiO2 NPs leads to the formation of a CT complex, providing a highly efficient CT pathway from the ARS-TiO2 complex to the CB of the TiO2 NPs. A high SERS enhancement of ARS was observed on the TiO2 NP surface, which borrows intensity from the allowed transitions within the ARS-TiO2 complex through HT vibronic coupling. In the presence of CrO42–, the ARS-sensitized TiO2 NPs can function as an active catalyst to induce the self-degradation of the adsorbed Raman probes. This is because the excited electrons are captured by CrO42– ions due to the suitable redox potential of the Cr(VI)/Cr(V) couple. The SERS intensity of ARS was inversely proportional to that of the CrO42– ions, which can be used to quantitatively determine the concentration of CrO42– ions in aqueous environments.
In the example with PO42– ions, the turn-off SERS method was proposed based on the first-layer effect of the CT mechanism.426 Under low pH conditions, the binding constants for pyrocatechol and PO42– ions on colloidal TiO2 NPs were similar. The preabsorbed PO42– ions on the TiO2 NPs surface inhibited the chelation interaction between pyrocatechol and the TiO2 NPs. According to the first-layer effect, in which only the molecule adsorbed directly on the substrate surface can be enhanced by the CT mechanism, the SERS signal of pyrocatechol decreases with increasing PO42– ion concentration. The sensitivities of these two turn-off SERS methods were sufficiently high to determine the level of the drinking water standard. More importantly, the stability and reproducibility can be markedly improved by employing semiconductor NPs as the SERS-active substrate.
SiO2@TiO2 (defined as T-red) NPs have been demonstrated by Alessandri et al. to have a strong Mie scattering effect because of the total internal reflection and multiple scattering of light at the core/shell and shell/environment interfaces.392 The authors observed that the T-red NPs with a high SERS effect allowed the detection of CO2 adsorbed on the TiO2 surface.428 Typical Raman peaks of CO2 were observed at 1388 and 1286 cm–1. In addition, the Raman intensity of the CO2 peaks was inversely related to the temperature, which is in accordance with previous findings on the adsorption and thermal desorption of CO2 bound to the TiO2 surfaces. The high sensitivity indicates that these T-red beads have immense potential for the development of SERS-based sensing systems for monitoring environmental CO2 over a wide temperature range. Furthermore, the researchers proposed a multimodal analytical method that employs T-red NPs to combine SERS and surface-assisted laser desorption/ionization mass spectrometry (SALDI-MS).429 As mentioned in Section 5.4.2, the unique structure of T-red NPs provides a plasmon-free SERS enhancement for the adsorbed molecule due to the Mie-resonance-induced electric field contribution. The T-red NPs serve as a background-free matrix for mass analysis. The proposed multimodal SERS/SALDI-MS platform combines the advantages of mass spectrometry and vibrational spectroscopy with signal-enhancing abilities, which can obtain the molecular fingerprint and mass of the same substrate. For example, structurally related compounds, such as amphetamine and ephedrine can be identified using SALDI-MS, while regioisomers, such as caffeine and cocaine can be distinguished using SERS.
6. Making a Strong Bridge between the Mechanism Studies and Applications: Current Status and Future Prospects
Although SERS and TERS applications have significantly advanced in the past few years, it is still rather difficult to find such applications in the real world. It is important to consider why SERS and TERS have not been used for practical applications. Indeed, numerous applications have been proposed, yet none of them are subjected to real practical uses. In this section, we will try to bridge the mechanism studies and applications at the nanometer scale. In particular, we will describe how the mechanism studies can support the real applications of SERS and TERS. The success of advanced applications is closely related to the theoretical aspects, more specifically, to the better prediction and optimization of the EF, by considering both the EM and chemical enhancement mechanisms (see Section 1.2), relevant secondary processes, and quantum effects (see Section 3). Interpretation of SERS and TERS data must take into consideration temporal and spatial variations as a consequence of the inherent localization to surface plasmon resonances. To realize the practical applications of SERS and TERS, we therefore have to consider both high sensitivity and high reproducibility. Finally, in this section we highlight recent breakthroughs in biological and biomedical applications.
6.1. Relation between the SERS/TERS Mechanism and Applications
Plasmon-enhanced Raman spectroscopy is governed by the optical response of a molecule interacting with an optical resonator, which represents a plasmonic nanostructure, in analogy to cavity quantum optics. The keys to understanding the EF of plasmon-enhanced Raman spectroscopy, which value is expressed as the square of a radiative portion of the Purcell factor as described in eq 5, are the quality factor and mode volume of plasmonic resonators.25
In both SERS and TERS, the optical response is nascent from molecules in the vicinity of plasmonic nanostructures. In addition to the spectral response by SERS/TERS, TERS also offers the advantage of imaging the optical response. The electric field and LSP resonance (LSPR) wavelength are highly sensitive to the sizes, shapes, and coupling modes of nanostructures, which may lead to poor reproducibility as the structure of plasmonic NPs varies by nanometer-scale distance. Indeed, it is still quite challenging to acquire high sensitivity, uniformity, and reproducibility simultaneously.12
In the case of TERS, strong experimental and theoretical support are required to achieve an extraordinarily high spatial resolution. This is partly because of the relatively long measurement time required for TERS imaging. The thermal drift and thermal diffusion as well as the poor yield, instability and the short lifetime of TERS-active tips present challenges for TERS sensitivity and reproducibility,430 particularly for experiments at ambient. New tip designs and new experimental strategies can improve the measurement throughput by increasing the signal-to-noise ratio. The concept of using isolated molecules as nanoscopic probes of their immediate local environments is one of the major motivations that has led to the rise of SM-microscopy and -spectroscopy.17 As discussed in Section 1.2.1, HSs exhibit extremely high electric field enhancement (108–1011).431 The measurement of target molecules localized in HSs has enabled SM sensitivity for a variety of resonant and nonresonant molecules.44,45 In fact, only a small portion of the SERS surface can be considered as HS sites.432 It was reported that 80% of the total SERS signals originate from 35% of the Ag sphere surface and 0.6% of the Ag dimer surface containing the majority of the HS sites.47 The theoretical calculations showed that if the distance (gap) between two NPs increases from ∼ 2 to 10 nm, the maximum EF in the HS drops rapidly. Furthermore, the EF decreases by orders of magnitude over a few nanometers away from the HS, as shown in Figure 45A for Ag NPs, 90 nm in diameter, under 514.5 nm excitation.50 Moreover, SM-EF exhibits long-tail behavior, wherein the slope in the high enhancement region defines a long-tail distribution (with a cutoff of the HS) that completely dominates the statistics (Figure 45B).431
Figure 45.

(A) EF calculated for the dimer of two spheres and the corresponding isolated single Ag NP (90 nm diameter). The color scale is logarithmic, and EFs outside the range 10°–108 are shown in dark blue and dark red, respectively. Adapted with permission from ref (50). Copyright 2000 The American Physical Society. (B) Long-tail probability distribution of EFs in SERS. Reproduced with permission from ref (431). Copyright 2009 American Chemical Society.
To achieve successful SM-SERS detection, there is no longer a big challenge to further increase the EF, since a plasmon coupled system, such as a NP dimer or NP over film, can provide SM sensitivity.47 On the other hand, it is necessary to bring the molecule of interest to the HS and minimize the interference of other species in the sample from blocking the site. Therefore, it is highly important to rationally design the surface structure of the HS in such a way that only the molecules of interest will be trapped. Under ambient conditions, the molecules can easily diffuse into and out of the HS, particularly in the case of ultralow concentration or weak adsorption of the target molecule. If a single target molecule is replaced by another species in the HS, the SERS spectrum will be completely altered. Moreover, changes in the orientation of the adsorbed molecules can also lead to spectral changes.434 The analyte photodecomposition and presence of carbonous bands from impurities and air contamination can also interfere with the signal of the target molecule.435 All these effects lead to significant variation and intensity fluctuations in the SERS signals acquired under SM conditions.
To overcome these problems, the acquisition time can be shortened to the time scale of the stay-time of the target molecule on the surface. The desired SERS spectra can be acquired and those containing only noise background or irrelevant signals from impurities can be excluded. Such an approach has enhanced the signal–noise ratio 10-fold by using a short acquisition time in the millisecond scale.436 Another possibility is to use an internal standard (another molecule or isotope of the target molecule) to correct the intensity fluctuations.437,438 A common way is to adsorb the internal standard molecules with a comparable Raman cross-section as the target molecule on the surface. However, this strategy does not work well in the case of highly competitive adsorption of the target molecules with the internal standard molecules. A more reliable approach was developed by embedding the internal standard molecules between the core and shell of the NP, while the target molecules were adsorbed on the bare shell surface.439 Therefore, the competitive adsorption between the internal standard and target could be avoided. This label-free approach does not require the comparable Raman cross-section of the internal standard and target molecules. Brolo et al. proposed a relatively simple concept called “digital SERS” to correct spectral fluctuations under SM conditions.440 In this approach, each event in a signal intensity fluctuation trajectory was digitized based on an intensity threshold. A calibration curve was then constructed, which considered the total digital counts for each concentration. This strategy was demonstrated on antibiotic ciprofloxacin adsorbed onto a Au surface (estimated 10 molecules/μm2).441 A 50 × 50 μm area of the surface was scanned with 5 μm steps, leading to 121 SERS spectra. The signal intensity fluctuation trajectory (spatially dependent SERS spectra) was analyzed using chemometrics.442 The events with a measurable SERS response were attributed a value of “1,” while the white areas of the regions were labeled as having “0” SERS intensity. Figure 46 shows the results for 14 pM ciprofloxacin. In the digital maps (Figure 46a, b), blue represents the detection of the normal molecule, while red represents its isotope. The two ciprofloxacin species were detected simultaneously in a single event (purple). The SERS digital counts, obtained by counting the total number of SM events for a given concentration, were then used to generate the analytical calibration curve (Figure 46c). The counting fractional error in basic digital statistics decreases with increasing number of counts. Therefore, the digital procedure can provide quantification with a reasonable level of precision, as long as a large area is mapped. This digital method relies on the assumption that the SERS response originates from a SM.
Figure 46.
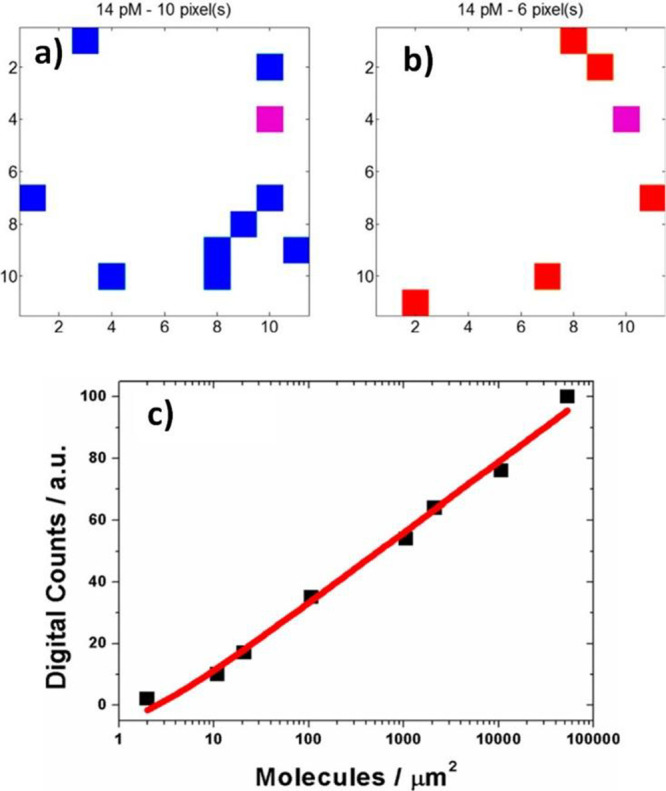
(a, b) Digital SERS maps for an isotopologue mixture of ciprofloxacin. The colors are related to the detection of normal (blue) and isotopically substituted (red) ciprofloxacin. The two ciprofloxacin species were detected simultaneously in a single event (purple). (c) Analytical calibration curve from the digital SERS procedure. Adapted with permission from ref (440). Copyright 2018 American Chemical Society.
In the case of surface-enhanced resonance Raman scattering (SERRS), HSs are attractive sites for directly investigating the strong coupling between the plasmon and molecular exciton resonances, as explained in Section 3.1. It was shown that the small mode volume of the HSs is important for both the large EM EF and the coupling energy.17 Theoretical evidence for the strong coupling at SERRS HSs of the dimers can be obtained by classical electromagnetism and the FDTD method. Both methods showed anticrossing of the coupled resonance by experimentally obtained parameters under near-SM conditions. Unique SERRS HSs, such as one-dimensional HSs, that are 104 times longer than conventional HSs, are potentially useful sites for examining the intermolecular photochemistry of multiple molecules strongly coupled with plasmons. SM strong coupling may be difficult to realize in vibrational strong coupling using plasmons, owing to the small oscillator strength of the vibrational transition compared to that of the electronic transition.
In practice, the gap between the NPs is difficult to control. It is ideal to produce a “hot surface” rather than HSs. The “hot surface” ensures that the molecules can always be maximally enhanced while they diffuse over the SERS substrate. It requires a SERS substrate with a high density of HSs with uniform SERS enhancement, which is key to the reproducibility of the SERS technique.12 It can also be fulfilled in the TERS technique when HSs are formed between the tip and metal surface. In the case of the TERS experiment, when the tip is in contact with the substrate (discussed further below), the optical signal originates from a 2D area with a diameter of one to a few nanometers. In the monolayer–submonolayer coverage regimes, it is reasonable to assume that the signal arises from one (or very few) molecule(s). Van Duyne’s group proved the SERS sensitivity of TERS by using isotopologues of rhodamine 6G (R6G).443 They showed that the vibrational signature of only one R6G isotopologue could be observed at a given time in multiple TERS spectra.
It is necessary to emphasize that the sensitivity and spatial resolution of the TERS technique are linked: The smaller the area that is probed by the TERS tip, the fewer molecules are under the tip; thus, at the extreme (resolution of 1 nm) there is only a SM. Therefore, there are parallels between ultrasensitive and SM-SERS on the one hand and high spatial resolution TERS on the other. The hybrid FDTD-DFT simulations can be used to forward simulate both ultrasensitive and ensemble-averaged TERS spectral images.444 Increasing the number of molecules and/or the effective TERS probing volume reduces the dimensionality and complexity of the problem. Recently, since TERS can function in an aqueous solution,445 it was proposed that this technique shows great potential in complex biological systems. However, the use of TERS to study living cells or the intracellular environment is yet to be reported. The main drawback is the slow imaging speed and weak enhancement by solely using the tip, without the strong LSPR coupling between the tip and sample.
Complementary to TERS, there is a major development of a combination of Raman with super-resolution techniques. Fujita et al. were able to improve the spatial resolution 1.4-fold using a structured line illumination (SLI) technique for Raman imaging while retaining the fingerprint information.446 To improve the time resolution of this technique, wide-field structured illumination microscopy was developed by using a spatial light modulator (SLM) to provide a faster image rate operated at a specific Raman peak.447 Willets et al. successfully mapped the SERS HSs of Ag-NP aggregates with a 5 nm resolution.448 They used the SM-SERS signal and point spread function to locate the HS with high precision. Lindquist et al. used the blinking behavior of the SERS signal, in analogy to the fluorescence signal in stochastic optical reconstruction microscopy, to reconstruct super-resolution images of the fibril on the SERS-active nanohole chip.449 By altering the phase profile of the laser using a rotating optical diffuser, most of the HSs spread across the plasmonic surface were excited, rendering the SERS image of a collagen fiber with a spatial resolution of 10 nm. These two methods rely on the diffusion of the molecules in and out of the HSs.
6.2. Innovative Applications and Future Directions of SERS and TERS at the Nanometer Scale
SERS/TERS techniques combined with significant progress in the development of enhancing nanomaterials and nanosubstrates, in conjunction with the related instrumentation, have resulted in an explosion of their applications. Practical and sensitive diagnostics and cost-effective techniques that are capable of simultaneously detecting the presence of several biomarkers (i.e., multiplex detection) associated with specific diseases at the point-of-care (POC) are highly desirable. From SM detection through to in vivo studies, the variety of examples that profit from SERS detection is immense.
In this section, we summarize the recent bioapplications that employ noble plasmonic metal nanostructures. We also discuss ultralow concentration molecular sensing by SM-SERS and its possible quantification. Here we concentrate on the study of EC and electrocatalytic processes at the nanometer scale. We also point to a new area that combines the power of SERS fingerprinting at the SM level with modern concepts of catalysis, such as hot-electron-driven chemistry. TERS applications profit from high spatial resolution, therefore we show how TERS can serve to explore the structure of biological systems and image 2D and even 3D materials. Finally, we overview breakthrough biomedical applications of SERS/TERS including (i) surgery guidance for the detection of tumor margins, (ii) optical fiber-guided visualization of superficial diseased tissues within the interior of the body, and (iii) detection of circulating tumor cells (CTCs).
6.2.1. Ultralow Concentration Molecular Sensing with High Spatial Resolution
SERS and TERS biomolecular studies also include the detection of the most important biomolecules such as nucleic acids and proteins.450 The importance of DNA sequencing is steadily increasing in biomedical research. However, to enable DNA sequencing, one should also address SM sensitivity and spatial resolution.
Treffer et al. proposed the use of TERS to detect individual nucleobases on calf thymus ss-DNA with an arbitrary sequence.451 They demonstrated that the four nucleobases exhibited distinctly different vibrational signatures that could in principle be used for DNA and RNA sequencing.286 Later, Najjar et al. were able to read the DNA of an l-phage virus with TERS.452 Although the authors revealed vibrational modes originating from DNA nucleobases and the DNA backbone, the reported 9 nm spatial resolution significantly limited the base-by-base readout necessary for DNA sequencing. Nanopore sensing can realize label-free detection of DNA by resolving the intimate interactions between the ionic fluids and molecules mechanically confined inside the pore.453 Chen et al. combined SERS with nanopore fluidics in an aqueous environment for SM identification and sequencing of ss-DNA.454Figure 47a depicts the concept of such detection. Instead of the conventional round plasmonic nanopores, they used elongated plasmonic nanoslits where the ss-DNA was temporarily adsorbed. The color background represents the localization of the optical field inside the nanoslit. The black curve represents the longitudinal profile of the SERS amplification near the Au surface, which provides local sensing at the subnanometer scale. The few DNA bases present inside this black curve contribute to the SERS signals, while the others (semitransparent) outside this region do not. Figure 47b shows the temporal color map of the nanoslit SERS of a 50-mers ss-DNA oligonucleotide 5′-poly(dA)48dCdG-3′ (1 × 10–8 M in a 10 mM KNO3 solution). A voltage of +0.3 V was applied between 50 and 550 s. Each spectrum was acquired for 0.5 s. The different colored dash-line frames indicate the characteristic Raman bands of G (green), A (blue), and C (orange), respectively. Figure 47c shows the temporal traces of the band intensity of the nucleobases marked in Figure 47b. The persistent signal in the trace of A indicates the voltage-driven adsorption of the ss-DNA inside the nanoslit. On the other hand, the spikes in the traces of G and C indicate asynchronous molecular identifications of distinct nucleobases during the stochastic fluctuations of the DNA strand. The insets show six representative spectra. The asynchronism of the spikes from adjacent nucleobases indicates sensing at subnanometer resolution.
Figure 47.

(a) Schema of temporarily adsorbed ss-DNA inside the nanoslit. Objects are not at the same scale. (b) Temporal contour map of the nanoslit SERS of the 50-mers ss-DNA oligonucleotide 5′-poly(dA) 48 dCdG-3. The differently colored dash-line frames indicate the characteristic Raman bands of G (green), A (blue), and C (orange), respectively. (c) Temporal traces of the band intensity of the nucleobases marked in panel b. The insets show six representative spectra. Reproduced with permission from ref (454). Copyright 2018 The Authors.
Pashaee et al. used TERS to distinguish between plasmid-free and plasmid-embedded DNA molecules,455 owing to the stronger TERS signals from plasmid DNA which contribute additional nucleic acids from the plasmid. Deckert’s group attempted to reach ∼0.5 nm spatial resolution using TERS for sequencing specifically designed ss-DNA.271 However, such high-resolution TERS proved extremely difficult to reproduce.
TERS was used to probe the amino acid composition and protein secondary structure on the surface of insulin fibrils with ∼1.5 nm spatial resolution.456 The propensity of various amino acids to be on the fibril surface and specific surface secondary structure elements were evaluated. The β-sheet areas were rich in cysteine and aromatic amino acids, such as Phe and Tyr, whereas proline was only found in α-helical and unordered protein clusters. It was also shown that carboxyl, amino, and imino groups are nearly equally distributed over the β-sheet and α-helix/unordered regions. Later, TERS served to image hydrophobic and hydrophilic domains on the surface of insulin fibrils, and to reveal clusters of specific amino acids, such as cysteine and proline.457 It was observed that the surface of insulin fibrils is composed of a mixture of α-helix and disordered protein secondary structures, while the core of the fibrils exclusively consists of β-sheets. The structural organization of two synthetic mutants of Aβ1–42 fibrils was studied using TERS.458 TERS spectra were measured from the surface of normal Aβ1–42 fibrils and fibrils formed from a peptide with leucine-to-threonine mutation at the 34th residue (L34T) fibrils. The comparison of these spectra with those collected from the surfaces of highly toxic oligomers (oG37C) determined that the secondary structures of the Aβ1–42 and L34T fibrils consisted of parallel β-sheets, while that of oG37C comprised antiparallel β-sheets. The structural organization of the Aβ1–42 fibrils was further elucidated by TERS to reveal the spatial distribution of their secondary structure (β-sheet and turn/random coil) in fibrillar species at different stages of maturation.459 It was possible to image amyloid-β plaques on the surface of neuronal spines using TERS.460 TERS was also used to probe the surface organization of insulin fibril polymorphs with different topologies, i.e., tape-like and twisted fibrils.461
Viruses can be identified and classified by SERS. In the case of label-free detection, obtained SERS spectra contain mainly spectral information about capsid (protein shell) of the virus due to Raman bands of amino acids.462 Higher sensitivity and selectivity can be achieved by TERS which was demonstrated in the case of detection of a single tobacco mosaic virus at a molecular level.463
6.2.2. Monitoring of Electrochemical (EC) and Electrocatalytic Processes at the Nanometer Scale
The SERS technique can be used to investigate heterogeneous chemical reactions with high sensitivity. SERS is suitable to explore EC reactions, which are the basic processes in electrocatalysis, solar energy conversion, energy storage in batteries, and photosynthesis. EC SERS is advantageous over EC detection techniques because SM detection (EC-SM-SERS) is achievable, and the signal does not need more amplification. Moreover, EC-SERS has shown great potential for studying electrochemistry at the nanometer scale if it is combined with super-resolution or scanning probe microscopy techniques. From a practical point of view, SERS is ideally suited to study nanoscale electrochemistry because the Raman-enhancing metallic nanosubstrate serves as the working electrode material.
“Hot electrons” generated through plasmonic excitations in metal nanostructures show great promise for efficiently driving EC reactions by thermal heating, such as in solar energy utilization.464 Plasmonic metal nanostructures that act as light absorbers can generate “hot electrons” under specific light conditions. Moreover, HSs with local high EM fields are efficient in generating “hot carriers.” The narrower and more intensive plasmonic resonances exhibit significantly larger rates of “hot electron” generation. The interactions between the plasmonic metal and surrounding environment determine the lifetime and injection mechanism of the “hot electrons” together with the extraction efficiency. The plasmon-induced “hot carriers” may transfer from the plasmonic core to the metallic catalysts to influence the EC reaction. To avoid the potential interference of the plasmonic core with the catalysts and related electrocatalytic reactions, an inert shell is utilized to isolate the plasmonic core in shell-isolated NPs (SHINERS).465 The thickness of the shell must be carefully controlled to be less than 3 nm, to provide sufficient plasmonic enhancement for the species adsorbed on the catalysts.466 Such isolation is particularly important for photocatalytic reactions where plasmon-mediated “hot carriers” have a significant impact.
One typical EC reaction is the conversion of 4-ATP to p,p′-DMAB, which has been unambiguously demonstrated to be triggered by “hot carriers.” The conversion and selectivity of this reaction can be greatly influenced by the properties of the plasmonic nanomaterials and reaction conditions.467,468 It has also been reported that “hot electrons” can promote the reduction of 4-NTP to 4-ATP in the absence of conventional chemical reducing agents.469,470 Another example is the plasmon catalyzed dimerization of 4-NBT to DMAB.471 Ultrafast SERS measurements were collected at various pump/probe time delays and processed by subtracting SERS spectra recorded with the pump pulse blocked from identical spectra recorded with the pump pulse on. Through analysis of the change in the 4-NBT/DMAB amplitude ratio between the ground-state SER spectrum and transient Fano line shapes, the authors concluded that the transient responses interrogate a specific subset of Ag NP HSs. The increased presence of DMAB in the HSs that produced the transient response is evidence that the plasmon-driven photochemical dimerization of 4-NBT to DMAB is the result of plasmon-generated “hot electrons.” These probe reactions allow researchers to use SERS to investigate the fundamental role of “hot electrons” in situ in plasmon-induced photocatalysis.
EC-SERS showed excellent potential for label-free detection of biologically relevant analytes, such as nucleic acids and their components and aptamers, in aqueous media. Karaballi et al. reported the first EC-SERS DNA aptasensor capable of the direct detection of small DNA strands with biomarker value for Mycobacterium tuberculosis detection in urine samples.472 The characteristic SERS fingerprint of adenine was considered for direct detection since adenine was only present in the target DNA strand. At low cathodic potentials (−1 V vs Ag/AgCl), the Raman bands could be attributed solely to the probe, reaching a limit of detection (LOD) of 280 mg target strand/mL. The EC-SERS was successfully explored for miRNA-155 quantification.473 Ss-RNA was labeled with methylene blue (redox and Raman reporter) and immobilized on a AuNPs@ITO substrate. Based on the target strand displacement, an extremely low LOD (60 × 10–18 M) could be achieved for the selected label molecule (methylene blue). Using a different approach, sensitive detection of miRNA-21 was reported on a highly reusable, electrochemically fabricated substrate.474 The scheme of the SERS substrate and fabrication of the biosensor are shown in Figure 48A,B. The 2′-hydroxymethyl-3,4-thylenedioxythiophene (EDOT-OH) was electropolymerized to form PEDOT-OH, which simultaneously acted as part of the SERS substrate and as a Raman probe. PEDOT–OH undergoes the catalytic reaction described in Figure 48C. The Raman intensities of PEDOT-OH in the reduction (Red) and oxidation (Ox) states were different (Figure 48D). When oxidized, the signal of the PEDOT-OH (Ox) on the electrode surface could be restored by applying a reduction voltage. A Raman enhanced substrate displaying signal changes was constructed in this way and could be recycled quickly and efficiently (at least 15 times). Combined with a double-amplification strategy, the SERS platform could detect miRNA-21 in the range of 100 fM–1 μM.
Figure 48.

(A) Construction of a SERS substrate, (B) fabrication process of the biosensor, (C) catalytic reaction, and (D) comparison of the Raman signal between PEDOT-OH (Red) and PEDOT-OH (Ox). See text for more details. Reproduced with permission from ref (474). Copyright 2021 Elsevier.
EC-SERS was successfully employed to detect biomarkers in diagnostic assays. Uric acid is an important diagnostic biomarker for many health issues. An EC-SERS sensor for uric acid detection was described for the first time by Goodall et al.475 They reported that under negative potentials, uric acid forms dianions at the metal–solution interface. This adsorption mode provided the strongest EC-assisted SERS signal, and the observed minimal interference was attributed to urine simulant constituents (including urea). Hernandez et al. recorded the SERS spectra of uric acid during the oxidation step of an Ag electrode and used chemometric data processing for quantification.476 Huang and Hsiao developed an EC-SERS chip integrated with a microfluidic reservoir.477 A nanocone array polycarbonate substrate decorated with gold nanospheres was used, wherein the substrate showed a strong EF and excellent EC-SERS uniformity with an relative standard deviation (RSD) of only 1.41%.
The first EC-SERS study for bacterial screening was reported by Lynk et al.478 The recorded signal for two different strains of bacteria (Gram-negative Escherichia coli and Gram-positive Bacillus megaterium) was stronger at the anodic potentials (−1 V vs Ag/AgCl); however, their spectra remained quite similar, and characteristic Raman bands could not be allocated for direct detection. This trend is also an impediment to the attempts of regular SERS discrimination of various bacteria. Although from a qualitative viewpoint the recorded Raman spectra were highly reproducible, significant variability in the band intensities was observed. Both the substrate assembling process (drop-casting of Ag NPs on SPE) and the natural dynamics in the number of colonies forming units (CFU) present in the bacterial culture during the incubation time (>16 h) were responsible for these band intensity fluctuations. Even if EC-SERS provided improved signal intensities and spectral richness compared to sole SERS, the technique still needs further optimization to become a reliable tool in routine bacterial screening. Later, a dual functional sequential EC-SERS sensor was developed for the diagnosis of the pathogenic bacteria Pseudomonas aeruginosa, responsible for severe nosocomial infections.479 Considering pyocyanin as a biomarker (a virulence factor that is both redox- and Raman-active), the diagnosis and monitoring of the infection progress were demonstrated. EC-SERS sensors were also used for the rapid detection of protein biomarkers in body fluids. Hassanain et al. reported the first dual sequential EC-SERS sensor for the direct detection of cystatin C, involved in the clinical diagnosis and monitoring of Alzheimer’s disease and renal failure.480 Cystatin C was isolated from blood plasma samples using a thiol-ended antibody-based extractor chip. The disulfide bond of the captured protein was then chemically reduced and assembled on a gold-coated silicon nanopillar substrate, which was further used for sensitive SERS quantification down to 1 pM.
EC-SERS provides valuable insights into potential distributions of SM electrochemistry but is typically performed on NP aggregates with poorly defined or heterogeneous geometries. Correlating EC behavior with specific sites on an electrode surface requires both a controllable, well-defined HS and nanoscale spatial resolution/characterization. EC tip-enhanced Raman spectroscopy (EC-TERS) can provide molecular fingerprint information with nanometer-scale spatial resolution (sub-5 nm).430,481 The pioneering work of EC-TERS was reported by Kurouski et al.482 For this study, Nile blue (NB) was absorbed into an ITO film that was used as a working electrode, while Pt and Ag/AgCl were used as counter and reference electrodes, respectively. Next, a Au-coated AFM tip was positioned on the working electrode, and changes in the TERS spectra were then monitored as a function of the applied potential. The overall intensities of the spectra decreased with decreasing potential. When the potential was swept from negative back to zero, 75% of the initial spectral intensity was recovered, demonstrating the reversibility of the redox reaction of NB under the AFM tip, as monitored by TERS. The observed step-like behavior of TERS suggested that redox reactions of only a few NB molecules were probed at those surface sites—six, based on the estimate of the average number of molecules under the tip. This work demonstrated the possibility of EC-TERS in monitoring few-molecule electrochemistry and probing variations in local coverage, otherwise inaccessible by SERS. Later, the same group showed that EC-TERS could be used to measure nanoscale variations in the formal potential (E00) of a surface-bound redox couple.483 Independently, Ren’s group leveraged EC-TERS to monitor the arrangements and elucidate the protonation states of 4-(pyridin-4-yl)biphenyl-4-yl)methanethiol (4-PBT) molecules on a Au(111) surface.484 They developed a new EC-STM-TERS setup with a tilted sample plate relative to the incident laser light. The group used a water immersion objective with a short working distance and high numerical aperture in their setup to significantly improve the signal-to-noise ratio of the spectra.485 This novel imaging approach was used to investigate the redox properties of hydroquinone. The results revealed that more than half of the hydroquinone molecules could not be reversibly oxidized. It appeared that it was the synergistic effect of the negative potential and laser illumination, rather than the tip LSPR, that led to such an irreversible reaction.
Using EC-TERS, the Domke’s group examined the adsorption geometry and chemical reactivity of adenine on Au(111) as a function of the applied potential.486 They demonstrated that protonated physisorbed adenine adopted a tilted orientation at low potentials, while it was vertically adsorbed near the point of zero charge. A further increase in the potential induces adenine deprotonation and the reorientation to a planar configuration. Touzalin et al. acquired EC-TERS spectra using an objective immersed into a very thin layer of liquid and a slightly bent tip to illuminate the tip with an objective located in air above the sample.487 With this experimental setup, they could track the progressive conversion of the nitro group of 4-NTP to its amino counterpart (4-ATP). The same group reported EC-TERS imaging of a Au surface functionalized with sodium sulfate with 8 nm spatial resolution.488 Kumar et al. revealed the possibility of EC-TERS in situ imaging of catalysis at liquid–solid interfaces with nanoscale spatial resolution.489 They monitored the plasmon-assisted oxidation of 4-ATP using the highly robust, durable, and chemically inert zirconia-protected TERS probe. The improved lifetime and stability of the probes in a liquid environment extended the potential scope of TERS as a nanoanalytical tool. These studies demonstrated the strength of EC-TERS in the nanoscale characterization of potential-driven reactions that are taking place in battery materials and at electrocatalytic sites.
Later, Wang and Kurouski reported the first 3D-TERS, which could be achieved by active plasmon zones located on a shaft of the scanning probe.490 They utilized 3D-TERS to monitor the reduction of 4-NTP molecules catalyzed by Au microplates. This approach is described in Figure 49. The HSs were located at both the shaft and apex (a) and only at the apex (b) of the TERS probe (Figure 49a,b). The excitation wavelength was 671 nm. The 4-NTP molecules underwent reduction on the Au plateau regions corresponding to Au(111) with high catalytic activity but not on the Au(110)/(100) regions with low catalytic activity. The reduction is illustrated in the Raman spectra (Figure 49g,l) by the formation of a N=N band between two 4-NTP molecules (Figure 49a,b). The results showed that 3D-TERS could distinguish the location of both species with high resolution (Figure 49c,d,h,i).
Figure 49.

(a, b) Proposed schemes showing the HSs located at both the shaft and apex (a) and only at the apex (b) of the TERS probe. (c, d), (h, i) High-resolution TERS images of the distribution of the −NO2 (c, h) and N=N groups (d, i) obtained with TERS active (4) and inactive (5) probes, respectively. (e, j) Corresponding overlap images. (f, k) Zoomed-in TERS images of the white rectangle position in panels e and j, respectively. (g, l) Typical TERS spectra extracted from the marked position in panels f and k. Reproduced with permission from ref (490). Copyright 2018 American Chemical Society.
6.2.3. Biomedical Analysis at the Nanometer Scale
Over the past decade, the number of medical SERS applications has increased rapidly.7,450 Three perspective clinical implementations of SERS have been recently developed: (i) Surgery guidance for the detection of tumor margins, (ii) optical fiber-guided visualization of superficial diseased tissues within the body, and (iii) detection of CTCs.
Delineation of tumor margins is very important in cancer treatment because incomplete tumor resections lead to its recurrence. The breakthrough demonstration of intraoperative detection of tumor margins by SERS was reported by Kircher et al.491 They combined static SERS imaging and real-time SERS NP detection using a hand-held Raman scanner during surgery. This was accomplished in a genetic mouse model that closely mimics the pathology of human glioblastoma multiforme. The SERS nanotags, each comprising a Au NP, Raman reporter molecule (RRM) 4,4′-dipyridyl, and silica shell, were allowed to circulate for 24 h in the mouse to ensure that they accumulated in the tumors. Tumor tissue was detected in situ with the hand-held Raman scanner using a 785 nm laser line as excitation. A SERS signal of the RRM was only observed when a tumor was present, while no SERS signal appeared in normal tissue. Importantly, in 10 of the mice, the hand-held scanner could identify small tumor spots that were not detected on the Raman images. This is attributable to the flexibility of angulating the hand-held scanner to probe any area in the operating bed, even those located at the lateral margins underneath overlying normal brain tissue. One of the limitations of this approach is that it was performed on brain tissue samples that had been fixed in paraformaldehyde, which are not fully representative of the actual surgical environment. Another potential limitation is that depth penetration with conventional Raman spectroscopy imaging techniques is in the order of a few mm.
Duan et al. introduced a ratiometrically pH-responsive SERS probe delineating acidic margins by determining the pH values of tumor cutting (Figure 50).492−494 The SERS sensors consisted of Au nanostars and a heptamethine cyanine derivative (IR7) as the RRM, affording pH-dependent Raman spectra. Rat brain bearing C6 orthotopic glioblastoma allografts (the most aggressive type of brain tumor) was imaged by magnetic resonance imaging (MRI) before surgery (Figure 50A,B). Figure 50D shows sequential white light images (left panel), intraoperatively collected Raman spectra (middle panel), and a pH topographic map (right panel) of the resection bed. Yellow dashed boxes mark the area with detectable SERRS signals, while star symbols represent the points with the highest acidity. The authors reported good correlation between tumor acidity and malignancy and that the surgery remarkably extended the survival of the animal models and minimized their postsurgical complications. Recently, the same group improved their previous approach and developed a SERS navigation system for intraoperatively delineating the acidic margin of glioma without the administration of an exogenous probe.493 The system was composed of a SERS chip that senses the sample acidity via pH-responsive Raman signals, a hand-held Raman scanner that collects the Raman signal of the sample placed on the SERS chips, and a homemade deep learning model that determines the sample pH by automatically processing the Raman spectra. A trace amount of pure water (≈0.4 μL) in the pipet tip was in contact with suspicious tissue at the tumor cutting edge for 2–4 s. A water droplet was aspirated and dripped onto a homemade pH-sensitive SERS chip. The Raman spectra of the aqueous sample on the SERS chip were acquired by a hand-held Raman scanner equipped with a 785 nm laser. The pH map of the tumor cutting edges was intraoperatively delineated with the assistance of a deep learning model by automatically analyzing the Raman spectra. With the guidance of the pH map, acidic tissues with pH values less than 7.0 were excised. Rat models bearing orthotopic C6 glioblastoma allografts were used and the therapeutic efficacies were evaluated by contrast-enhanced MRI. The results showed that the acidic regions were distributed heterogeneously and predominately located in the tumor margins. The overall survival of the animal models post-SERS system-guided surgery significantly increased in comparison to the conventional strategy used in clinical practice. Therefore, this SERS system holds the promise of accelerating the clinical transition of acidic margin-guided surgery for solid tumors with an infiltrative nature.
Figure 50.

Acidic margin-guided brain tumor surgery in live rat models. (A) Preoperative MRI image of rat brain bearing C6 orthotopic allografts. The tumor is indicated by a white arrow. (B) In vivo chemical exchange saturation transfer (CEST)-MRI pH map of a rat brain model. (C) Illustration indicating the surgical location before the craniotomy. (D) Sequential white light images (left panel), intraoperatively collected Raman spectra (middle panel), and a pH topographic map (right panel) of the resection bed. Reproduced with permission from ref (494). Copyright 2020 Royal Society of Chemistry.
Endoscopic imaging is another diagnostic tool for minimally invasive access to tissues located deep within the body. In this case, the limitation of most optical techniques is associated with the depth of penetration. Zavaleta et al. first reported SERS-based multiplexing using commercial SERS tags consisting of 10 different RRMs adsorbed onto a Au core (60 nm diameter) and then coated with silica.495,496 The Raman endoscopic device was equipped with a flexible fiber bundle with a centrally located single-mode fiber for illumination at 785 nm and an additional 36 surrounding multimode fibers for light collection. The technique was tested on excised human tissue samples and showed excellent sensitivity and multiplexing capabilities, detecting a 326 fM concentration of NPs and unmixing 10 variations of colocalized SERS tags. Importantly, the Raman endoscope was designed for efficient use over working distances ranging from 1 to 10 mm. Later, the same group reported the in vivo application of a miniature and noncontact Raman endoscopic device that can be inserted through the accessory channel of a clinical endoscope.497 The endoscope was retracted from the gastrointestinal tract, and the SERS signal from the SERS tags was acquired from the lumen. This device enabled the rapid scanning of topologically complex luminal surfaces of hollow organs (e.g., colon and esophagus). Multiplex detection was possible by quantitative analysis of the concentrations of a panel of present SERS tags. One human study demonstrated the speed and simplicity of this technique. With a similar approach, Liu et al. topically administered a multiplexed cocktail of SERS tags to a rat esophageal tumor model.498 The orthotopic esophageal tumor model was constructed by surgically attaching a tumor xenograft that was previously implanted subcutaneously in the flank of a nude mouse. Antibody-conjugated SERS NPs were topically applied on the lumenal surface of the rat esophagus to target the epidermal growth factor receptors EGFR and HER2, and a miniature spectral endoscope featuring rotational scanning and axial pull-back was employed to comprehensively image the NPs bound on the lumen of the esophagus. The results are shown in Figure 51. Figure 51a is a zoomed-in view of the surgically exposed cervical esophagus with three tumor implants. Figure 51b shows the reference spectrum of the SERS NPs, while Figure 51c shows the background spectrum of esophagus tissues and raw (mixed) spectra acquired from NP-stained normal esophagus tissue and A431 tumor implants. Spectral demultiplexing reveals that the respective concentrations of the EGFR-NPs, HER2-NP epidermal growth factor receptor, and isotype-NPs are 21.4, 21.5, and 20.6 pM on the normal esophagus (blue spectrum) and 33.1, 24.7, and 18.2 pM on the A431 tumor (red spectrum). Ratiometric images show the concentration ratios of EGFR-NPs vs isotype-NPs (Figure 51d) and HER2-NPs vs isotype-NPs (Figure 51e). These results demonstrate that multiplexed ratiometric imaging of targeted vs untargeted SERS NPs not only reveals the location of the receptor-positive tumors but also quantifies the EGFR and HER2 expression levels, in agreement with the flow cytometry results (right-side plots in Figure 51d,e).
Figure 51.

In vivo endoscopic molecular imaging performed with multiplexed SERS NPs delivered via oral gavage. (a) Photograph of a surgically exposed rat esophagus implanted with three tumor xenografts. (b) Reference spectrum of the SERS NPs that were mixed together and topically applied into the rat esophagus in this study. (c) Background spectrum of esophagus tissue and raw (mixed) spectra acquired from NP-stained normal esophagus tissue and A431 tumor implants. (d, e) Images showing the concentration ratio of (d) EGFR-NPs vs isotype-NPs and (e) HER2-NPs vs isotype-NPs. Right-side plots show the correlation between the image-derived intensities from various tissue types (normal esophagus and three tumors) and the corresponding fluorescence ratio (targeted-NP vs isotype-NP) from flow-cytometry experiments with the cell lines used to generate the various tumor xenografts. All values in the figures are presented as mean ± standard deviation; R > 0.95. Reproduced for permission from ref (498). Copyright 2015 The Optical Society of America.
Other implementations of endoscopy include the use of plasmonic NW waveguides for remote excitation499 and the assessment of gastric and colon disease through individual and multiplex imaging.497,500 Currently, the main limitation to this approach is the minimum optical fiber diameter that can be employed, owing to constraints in the design and implementation of the excitation and detection optics.
Recently developed liquid biopsy technologies have enabled the discovery of many liquid biopsy-based cancer biomarkers including CTCs. When targeting CTCs, it is necessary to design simpler enrichment methods based on complex microfluidic devices and to ensure the ability to capture cells by recognizing cell surface biomarkers. A variety of newly developed SERS platforms and tags can be used to solve this issue.501,502
SERS was successfully applied for the detection of CTCs in human blood. Sha et al. employed commercial SERS tags comprising one or more Au NPs with a submonolayer of RRMs and protective silica coating.503 Magnetic NPs (MNPs) functionalized with antiepithelial cell adhesion molecule (EpCAM) antibodies immobilized on the surface served to capture breast cancer SK-BR-3 cells spiked in human blood. Subsequently, the captured EpCAM-overexpessing cancer cells were detected with SERS tags using near-IR excitation. SK-BR-3 cells were spiked directly into whole blood prior to a short incubation of 30 min with magnetic bead-EpCAM and SERS tag-her2 conjugates. A magnet served to concentrate the magnetic beads on the side of the tube where the SERS signal was collected. A strong SERS signal was detected when SK-BR-3 cells were spiked into whole blood, indicating that a signal can be directly acquired in whole blood without washing or additional handling steps. The detection limit was less than 10 cells per mL with 99.7% confidence. The LOD in blood approximated 50 cells per mL. Wang et al. successfully identified CTCs in the peripheral blood of patients with squamous cell carcinoma of the head and neck.504 They employed Au NPs with RRM functionalized with a thiolated PEG layer to stabilize the NPs and minimize nonspecific interaction with blood cells. Approximately 15% of the PEG molecules comprised carboxyl functional groups for conjugation with the N-terminus of EGF peptide, and the detection limit was in the range of 5–50 tumor cells per mL of blood. Shi et al. proposed a combination of magnetic enrichment and multiplex detection of CTCs using targeted MNPs and Au NP tags.505 Both Au NPs and MNPs were targeted to folate receptor, which is overexpressed on many cancer cells. The results showed that only cells targeted with both Au NPs and MNPs exhibited an increasing SERS signal due to the magnetic accumulation of CTCs. The authors demonstrated a SERS imaging system with 1 pM sensitivity and 2.5 mm penetration depth. A magnetic trapping system could effectively trap cells at flow velocities ranging from 0.2 to 12 cm·s–1. In addition, discrete SERS signals were detected in the magnetic trapping zone from flowing dual-NP labeled HeLa cells mixed with rat blood, indicative of single-cell detection. Nima et al. presented a scheme for the multiplex detection of CTCs in whole human blood using tunable, silver-decorated, Au NRs as SERS multispectral contrast agents.506 The Au NRs were functionalized with four RRMs and four antibody breast cancer markers–anti-EpCAM, anti-IGF-1, receptor β, anti-CD44, anti-Keratin18, together with the leukocyte-specific marker anti-CD45. More than 2 orders of magnitude of SERS signal enhancement was observed from these hybrid nanosystems compared to the signal of conventional Au NRs. Using SERS, Bhamidipati et al. were able to quantify the expression of prostate-specific membrane antigen (PSMA) in tissue microarrays obtained from 34 patients with varying stages of prostate cancer.507
Reza et al. developed a SERS immunoassay on a simple microfluidic channel integrated with a SERS detection system for simultaneous in situ identification of single CTCs, to unravel intercellular heterogeneity in their protein expression levels during the progression of disease and response to the treatment.508 The schema of such an approach is described in Figure 52. First, CTCs were isolated from the patient’s blood (Figure 52a) and then passed through the microfluidic channel for single-cell SERS analysis (Figure 52b). The SERS nanotags were prepared from Au NPs conjugated with target antibodies and RRMs. The SK-MEL-28 melanoma cell line was used for monitoring the expression of a panel of cancer-specific surface protein biomarkers. The dot plot in Figure 52c shows the heterogeneity of single cells in expressing three melanoma surface proteins: MCSP (black), MCAM (red), and LNGFR (blue). Integrating a label-free EC system with the device, they also monitored the expression of an intracellular protein (BRAFV600E) under drug treatment. The average SERS intensities proved the treatment response over the tested period (Figure 52d). Figure 52e,f respectively illustrates EC lysis of CTCs and intracellular BRAF protein detection using disabling positional vertigo (DPV). Figure 52g shows treatment monitoring with a single CTC analysis identifying resistant CTCs, indicating disease progression. They performed a longitudinal study with 15 samples from five different melanoma patients who underwent therapy. The main finding was that the average expression of receptor proteins in a patient fails to determine the therapy response, particularly when the disease progresses. However, a single CTC analysis with this device demonstrated a high level of intercellular heterogeneity in the receptor expression profiles of patient-derived CTCs and identified heterogeneity within the CTCs. More importantly, a fraction of the CTCs still showed a high expression of these receptor proteins during and after therapy, indicating the presence of resistant CTCs, which may evolve after a certain time to progress the disease.
Figure 52.

Graphical representation of the platform combining microfluidics, SERS, and electrochemistry for the analysis of CTC heterogeneity during treatment. See text for more details. Adapted with permission from ref (508). Copyright 2021 American Chemical Society.
Cho et al. reported a SERS sensor combined with a microfluidic chip for simultaneous multiplex detection, isolation, and analysis of circulating cancer stem cells and various subtypes of CTCs in breast cancer cells.509 The simultaneous detection of targets (CD133, EpCAM, EGFR, HER2, and MUC1) was achieved by five different combinations of SERS nanotags, prepared by conjugating AuNPs with different RRMs and different detection antibodies along with biotinylated dsDNA. The isolation was performed using a microfluidic chip system functionalized with avidin–biotin and the restriction enzyme digestion of the dsDNA linker from the SERS nanotag. Wilson Jr. et al. demonstrated a combined microfluidic platform-based immunomagnetic enrichment with a SERS detection system using magnetic multicolor SERS nanotags for the simultaneous and quantitative detection of CTCs.510 They monitored four surface protein biomarkers (i.e., EpCAM, HER2, CD44, and IGF1R) on a single breast cancer cell for analyzing a CTC subpopulation in a clinical sample and monitoring cancer metastasis. The on-chip multiplexing assay was performed by synthesizing IO/Au core/shell NP SERS nanotags with four organic dyes, namely QSY21, BHQ3, QXL680, and IR740, conjugated with different RRMs and a target antibody for targeting four different biomarkers in each individual cell. Signal deconvolution was performed to analyze the levels of each target surface protein biomarker.
Imaging the spatial organization of cell surfaces is possible by TERS. The Deckert group used TERS to detect hemozoin crystals (produced by the parasite responsible for malaria, Plasmodium falciparum) inside red blood cells and determine the oxidation state of iron ions in hemozoin.511 Xiao et al. showed that TERS can be employed to provide chemical information on specific ligand–receptor binding sites of the integrin αvβ3 molecule in a cancer cell membrane.512 They obtained distinct Raman signals using gold NPs (located on the substrate) functionalized with three different peptide ligands. The structural organization of the cell membrane was probed by TERS revealing individual protein and lipid domains on the surface of the eukaryotic cell and oligosaccharides on the cell surface.513
7. Perspectives
This review has outlined recent advances in SERS and TERS at the nanometer scale, from fundamentals to innovative applications. SERS and TERS have opened a variety of exciting research fields and enormous number of applications have been proposed. However, there are many new directions that we have to explore. In this last section, we discuss future development of SERS and TERS. First, general perspectives of SERS and TERS studies from the viewpoint of SERS and TERS mechanisms are described. Then, we present future outlooks on two more specific topics, Ångström-resolved TERS and semiconductor-enhanced Raman scattering. The section is concluded with a perspective on SERS and TERS applications.
7.1. General Perspectives of SERS and TERS Studies from the Viewpoint of SERS and TERS Mechanisms
The frontiers of SERS and TERS mechanisms will continue to advance by exploring various phenomena related to light–molecule interaction inside HSs with improving technologies of spectroscopies. The keys of EM and the chemical mechanism are plasmon resonance confining EM field inside HS and CT resonance forming between a metal surface and a molecule, respectively. Both theoretical and experimental evidence for these mechanisms has been provided by many researchers. However, such experimental data also include the breakdowns of assumptions in the mechanisms. For the EM mechanism, the breakdown of weak coupling in EM enhancement inside a HS will be important to quantitatively understand EM EFs. For the chemical mechanism, the assumption of a weak electronic interaction between molecules and metal surface, indicating that the electronic structure of molecule does not change by forming CT complexes, should be modified to correctly understand SERS spectral properties. By the way, one can expect that both EM and chemical mechanisms will be merged by controlling coupling energy between a plasmon and a molecular excitation. In other words, the resonance energy of the coupled state will be controllable, resulting in the reversible switching from nonresonant to resonant SERS or vice versa. TERS, which can be applicable for tuning a mode volume of a HS by changing the tip position, will be a powerful tool to realize such reversible switching.
The integrated SERS HSs with various functions will appear in near future. For the tightly confining EM field, the HSs utilize coupled plasmon resonance, i.e., the dipole–dipole coupled plasmon mode for a NP dimer gap, the coupled plasmon mode between local and surface plasmon for a gap between NP and a substrate surface, the Fano plasmon mode between the dipoles and higher order plasmons for gaps inside a NP cluster. There are advantage and disadvantage in each HS. For example, Fano plasmon exhibits the considerable large EM enhancement due to the small radiation loss, but the SERS signal needs to be extracted through radiative plasmon modes. Thus, the combination among these plasmon modes is attractive to optimize EM enhancement of HSs. We consider that the combination between propagating plasmons, localized plasmons, and their coupled plasmon is useful to construct large-scale integration of plasmonic circuits for detection, identification, and propagation of SERS signals.
Using various HSs, analysis of SERS spectral fluctuations has revealed both physical and chemical properties on the SM level. Such analysis of SERS fluctuations will be useful for evaluating subnanometer mechanical and chemical changes inside SMs, e.g., the dynamics of tentative defects inside a SM. One can expect that such an analysis will reveal and create new pass ways of chemical reactions for various applications, e.g., EC and photochemical reactions.
Unique phenomena beyond the assumption of classical spectroscopies, e.g., strong coupling, ultrafast SEF, vibrational pumping, field gradient effect and so on, have been discovered in SERS and TERS HSs. The origin of these phenomena is the tightly confined EM field in HSs, resulting in ultrafast interaction between a plasmon and a molecular exciton, ultrafast de-excitation, ultrafast excitation, ultrainhomogeneous structure of EM field. The unified theory, which can comprehensively treat the above phenomena, will be developed with the assistance of numerical calculations such as TDDFT. The application of these phenomena will contribute to polariton chemistry by manipulating the chemical properties of hybridized state between molecular excitons and plasmons inside HSs. SERS and TERS HSs will continue to create new optical, physical, and chemical technologies.
7.2. Future Directions of Ångström-Resolved TERS
As we have discussed in Section 4, the Ångström-resolved TERS has been developed as a powerful tool to unravel chemical structures in various systems at the single-bond level. However, in addition to the development of scanning Raman picoscopy itself for structural reconstruction described at the end of Section 4, there are still many challenges remaining, and we would like to suggest several potential directions in the future development of Ångström-resolved TERS.
-
(1)
First comes the tip quality issue. As described in Section 4, the tip quality is absolutely crucial for performing TERS experiments, which is viewed as the bottleneck limiting the development of TERS. Such a requirement is even more important for Ångström-resolved TERS studies. Although the TERS community has now reached a consensus that an atomic-scale protrusion is required at the tip apex for Ångström-resolved TERS experiments, very few are known about the exact configuration of the atomistic structure. Further experimental and theoretical studies are in high demands in order to figure out the exact apex configuration for TERS-active tips. Scientists also need to find ways on how to fabricate it through chemical or physical approaches in a controlled and reliable manner, hopefully also cost-effective. In addition, for Ångström-resolved TERS experiments to be carried out in ambient conditions, one also has to find a way on how to keep the atomistic structure at the tip apex stable, probably protecting it by coating a special thin layer of oxides or nitrides.
-
(2)
Molecular complexity issue. What we have demonstrated above for Ångström-resolved TERS is mainly to identify single chemical groups or bonds from simple planar molecular systems. One of the directions to go for the application of this technique is to extend from the simple planar molecules to more complex molecules such as three-dimensional structures and even twisted biomolecules including DNA/RNA chains and proteins. The detection of these complex molecular systems may need further efforts in improving the vertical resolving ability of TERS as well as data processing methods. The former may be achieved by allowing an apex-protected tip inserting into the soft molecule under study, while the latter may be improved again with the aid of artificial intelligence (AI) techniques. The combination of both may offer a tomographic TERS technique for the structure reconstruction of complex molecules.
-
(3)
Subsurface sensitivity issue. 2D materials have been considered as the ideal sample systems for TERS, and the advances described in Section 4 already showcase some of its applications in this field. Many more studies are expected to occur in this direction, with focus more on surface defects, surface strains associated with subsurface structures, and local reconstructions associated with Moiré lattice. Through Ångström-resolved TERS, the last may provide deep insights into the van der Waals interactions between 2D layers such as in graphene, transition-metal dichalcogenides and their twisted or nontwisted heterostructures. It is worth noting that, to make TERS as a practical and widely used tool for investigating 2D materials and subsurface structures, the fabrication of highly active TERS tips is again crucial. It is highly desirable to have a TERS tip that the tip plasmonic enhancement alone is strong enough so that we can get away from the requirement of using plasmonic metal substrates or even gap-mode plasmons.
-
(4)
Time-resolved studies. So far, the TERS technique is limited in temporal resolution because of the slow collection of Raman spectra. Therefore, one clear trend in the field is to move from static to dynamic studies. Further combination with ultrafast spectroscopic techniques including fs-lasers may enable the TERS technique to have simultaneous ultrahigh spatial-temporal resolution. Such a powerful capability will help to reveal the dynamic mechanism of physical processes such as surface reactions, photocatalysis, and carrier transport as well as exciton and phonon decay.
-
(5)
Nonlinear studies and others. Most of the TERS studies to date are generally based on the linear response of SMs to the local electric field, in which magnetic components and the field gradients as well as the chirality of the local field are generally ignored. Although these effects may already reflect themselves in the previous experimental results and have been accounted for to some extent, it certainly deserves further experimental and theoretical studies, so that one can get deep understanding about the nonlinear responses of molecules and other low-dimensional materials within an atomically confined local EM fields, such as the contribution of higher-order terms from quadruples and magnetic dipoles to the Raman scattering from the SM. Another development for the TERS technique in recent years is EC TERS, which not only offers an important parameter via the potential to tune the energy level of the system under study, but also extend the application of TERS from air and UHV to liquid environments. Pushing the spatial resolution of EC TERS down to the Ångström level might be challenging, but could provide more microscopic insights into the reaction mechanisms.
7.3. New Directions of Semiconductor-Enhanced Raman Scattering
As described in Section 5, semiconductor-enhanced Raman scattering has not only made remarkable progress in mechanism studies, but also demonstrated notable achievements in practical applications. The EF of semiconductor SERS was reported to be comparable to metal-based SERS in some works. Indeed, the EF just represents the enhancement ability of a semiconductor substrate for a specific molecule, especially when the resonant excitation of a dye molecule is involved in the SERS measurement. It is strongly recommended to test the semiconductor SERS performance under a nonresonant excitation condition. Instead of the EF, the lowest detectable concentration is more reliable to determine the enhancement performance.
At present, the sensitivity of semiconductor SERS is no longer a major constraint for semiconductor SERS. One of the most urgent problems is how to improve the enhancement universality of semiconductor substrates. Most existing investigations have focused on vacancy defect-mediated methods based on the CT mechanism; the universality of such semiconductor substrates is difficult to achieve due to the requirement of energy level matching between substrate and analytes. Similar with metal SERS, the construction of the electric field contribution for semiconductor SERS could expand the universality of semiconductor SERS. Recent works have demonstrated that Mie resonance could enhance the electric near-field for semiconductor SERS, which offers a new theoretical framework for designing SERS-active semiconductor substrates with extensive adaptability. According to this theoretical framework, a semiconductor material with a high reflective index will be the ideal material for generating high electric field contributions to semiconductor SERS. Furthermore, the coupling of a multiresonance effect, such as CT, Mie resonance, exciton resonance, and plasmon resonance, is a promising direction for the design of SERS-active semiconductor substrates with synergistic contributions.
Plasmon resonance of semiconductor nanocrystals provides a new probability to construct SERS-active semiconductor substrates. However, the so-called plasmon resonance contribution for semiconductor SERS still needs to be further investigated, and the following point should be carefully considered: (1) visible spectral bands shown in the electronic spectrum may be attributed to the plasmon resonances or the intraband excitations of carriers. Carrier density test is useful to distinguish the two physical phenomenon; (2) the highly doped and nonstoichiometric semiconductors are not always stable during the specimen preparation and measurement processes; (3) the enhancement universality of the proposed semiconductor substrates should be verified with multiple molecules under a nonresonant excitation.
The introduction of the electric field contribution into semiconductor SERS creates a better vision for us to comprehensively study the surface/interface problem of semiconductor materials. Coupling the newly developed electric field contribution with the CT mechanism would open new opportunities in in situ characterization of semiconductor-based catalysis and energy conversion systems. Besides, the biocompatibility of semiconductor materials makes semiconductor SERS uniquely qualified to be used for biological systems. We may have the chance to obtain the native structure and function of a protein and a nucleic acid in cell and/or in vivo by an in situ semiconductor SERS technique. Meanwhile, the chemical stability of semiconductor materials is another noticeable advantage. This property is quite suitable for the SERS analysis of the environmental mediums, such as corrosive, oxidized, and hyperthermia mediums. Notable, in some cases, semiconductor SERS based on the CT mechanism only produces an enhancement for a specific molecule, which may become an advantage for the analysis of a target analyte in a complex system. With its rapid progress, we believe that semiconductor SERS will certainly play a bigger role in further development of the SERS technique.
7.4. Perspective in SERS and TERS Applications
SERS and TERS spectroscopies are powerful techniques to probe simple as well as complex molecules, in contact or close to a plasmonic substrate, usually based on the noble metals but also on semiconductor or hybrid materials. SERS/TERS combined with significant progress in the development of enhancing nanomaterials and nanosubstrates, as well as related instrumentation, has resulted in an explosion of their applications.
Concerning photochemical and photocatalytic studies, SERS possesses unique advantages because both SERS and “hot electrons” are excited by light, which avoids interference from other excitation sources and improves the signal intensity. On the other hand, basic knowledge about the lifetime, yield and how “hot electrons” initiate and accelerate chemical reactions is still limited. Moreover, the spatial and temporal resolution of SERS could still not completely satisfy the needs of the in situ study of plasmon-induced photochemical processes. A closer integration of experimental results with theoretical calculations is needed to better understand the complexity of the EC-SERS technique that could become a standard analytical method. In comparison to SERS, an additional benefit of TERS is its extraordinary spatial resolution, around a few nanometers in the ambient and down to the subnanometer scale under low-temperature and UHV conditions.
The sensitivity and spatial resolution of TERS are linked: the smaller the area that is probed by the TERS tip, the fewer molecules are under the tip; at the extreme (resolution of 0.2–1 nm) there is only a SM. The hybrid FDTD-density functional theory simulations can be used to forward simulate both ultrasensitive and ensemble-averaged TERS spectral images.
Applications of SERS in biosensing (both direct and using molecular labels and nanotags) have been rapidly growing during the past decades. The first main requirement for a SERS/TERS biosensor is that it should possess high sensitivity to detect trace-level concentrations of analytes even at low sample volumes and in complex matrixes. Second, it should be capable of providing quantitative or semiquantitative analytical information on a target analyte with high specificity and spectral reproducibility. Last but not least is that such a biosensor platform should be cheap to produce, have a fast readout, and be portable. The development of advanced techniques has enabled fast, low-cost, reproducible, and large-scale production of nanosubstrates and nanotags with rationally designed and tailored morphologies with high SERS response, for general as well as target-oriented sensing applications. The advanced medical applications include surgery guidance for the detection of tumor margins, optical fiber-guided visualization of superficial diseased tissues and detection of CTCs.
The drawback of SERS/TERS in becoming a widespread clinical diagnostics technology is the use of highly diluted samples in complex matrixes and the need for sophisticated instrumentation. Therefore, development of better SERS probes, such as SERS tags combined with microfluidic systems is highly demanding. Moreover, AI can be implemented to analyze the Raman spectra generated from complex samples for disease diagnosis. In the case of in vivo applications, limited tissue penetration depth has hindered implementation in full-body or deep tissues. Therefore, it is better to know if the laser beam can access the diseased area. The POC fiber sensors for body fluid analysis provide a superior sensitivity in comparison with planar/NPs platforms, owing to the increased interaction length between the guided light and analyte. On the other hand, the continuous improvement and miniaturization of Raman spectrometers and optical fibers could be incorporated for a faster and cost-effective signal readout. The transfer of SERS from laboratories to clinical routines is not a trivial task, but we can expect that SERS/TERS will play an important role in medical diagnosis and treatment in the coming years.
Acknowledgments
This work was supported by grant 22-16667S from the Czech Science Foundation (M.P.), a JSPS KAKENHI Grant-in-Aid for Scientific Research (C), number 21K04935, the National Natural Science Foundation of China (Grant No. 21790352), Chinese Academy of Sciences (Grant No. XDB36000000), and Anhui Initiative in Quantum Information Technologies (Grant No. AHY090000).
Glossary
List of Abbreviations
- ABT
4-aminobenzenethiol
- AFM
atomic force microscopy
- AI
artificial intelligence
- ARS
alizarin red S
- 3-ASA
3-aminosalicylic acid
- 4-ATP
4-aminothiophenol
- AuNC
Au nanocube
- AZO
Al-doped zinc oxide
- BPE
1,2-di(4-pyridyl) ethylene
- bpy
2,2′-bipyridyl
- CB
conduction band
- CB[n]
Cucurbit[n]urils
- CEM
classical electromagnetic model
- CEST
chemical exchange saturation transfer
- CFU
colonies forming units
- CoTPP
Co(II)-tetraphenyl porphyrin
- CT
charge-transfer
- CT-SERS
CT-mechanism-based SERS
- CTC
circulating tumor cell
- DA
dopamine
- DBP
tetraphenyldibenzoperiflanthene
- DD
coupled plasmon, dipole–dipole coupled plasmon
- DMAB
4,4′-dimercaptoazobenzene
- DNP
5,15-(dinaphthalene)-10,20-(dibromo)-porphyrin
- DPP
5,15-(diphenyl)-10,20-(dibromo)-porphyrin
- DQ
coupled plasmon, dipole–quadrupole coupled plasmon
- DSSC
dye-sensitized solar cell
- ERS
electronic Raman scattering
- EC
electrochemical
- EDOT–OH
2′-hydroxymethyl-3,4-thylenedioxythiophene
- EF
enhancement factor
- EM
electromagnetic
- EpCAM
antiepithelial cell adhesion molecule
- EPR
electron paramagnetic resonance
- FC
Franck–Condon
- FePc
4-MPy-iron phthalocyanine
- FDTD
finite-difference time-domain
- FTO
F-doped Sn-oxide
- GFR
gradient-field Raman
- HABT
hydroxylaminobenzenethiol
- Hb
hemoglobin
- HNP
hybrid nanoparticle
- HOMO
highest occupied molecular orbital
- HS
hotspot
- HT
Herzberg–Teller
- IR
infrared
- ITO
indium tin oxide
- J-agg
J-aggregate
- L-DOPA
l-dihydroxyphenylalanine
- LMCT
ligand-to-metal CT
- LSP
localized surface plasmon
- LSPR
localized surface plasmon resonance
- LUMO
lowest unoccupied molecular orbital
- MBA
4-mercaptobenzoic acid
- MgP
Mg-porphine
- μPEC-SERS
microphotoelectrochemical-SERS
- metal-4-MBA
metal-4-mercaptobenzoic acid
- MNP
Magnetic nanoparticle
- MPBA
p-mercaptophenylboronic acid
- 4-MPH
4-mercaptophenol
- 4-MPy
p-mercaptopyridine
- MM
multiple molecule
- MPP
5-(phenyl)-10,20-(dibromo)-porphyrin
- MRI
magnetic resonance imaging
- NB
Nile blue
- NBT
4-nitrobenzenethiol
- NCP
nanocavity plasmon
- NIR
near-infrared
- NMR
nuclear magnetic resonance
- NP
nanoparticle
- 4-NTP
4-nitrothiophenol
- NW
nanowire
- 1D HS
one-dimensional hotspot
- 4-PBT
4-(pyridin-4-yl)biphenyl-4-yl)methanethiol
- Pc
phthalocyanine
- p-MA
para-mercaptoaniline
- PIC
phenyl isocyanide
- Phe
phenylalanine
- PMCR
plasmon-mediated chemical reaction
- POC
point-of-care
- PSMA
prostate-specific membrane antigen
- PTCA
3,4,9,10-perylene tetracarboxylic acid
- QCM
quantum-corrected model
- QD
quantum dot
- QED
quantum electrodynamics
- RBM
radial-breathing mode
- R6G
Rhodamine 6G
- RRM
Raman reporter molecule
- SALDI-MS
surface-assisted laser desorption/ionization mass spectrometry
- SEA
surface-enhanced absorption
- SECARS
surface-enhanced coherent anti-Stokes Raman scattering
- SEF
surface-enhanced fluorescence
- SEHRS
surface-enhanced hyper Raman scattering
- SERS
surface-enhanced Raman scattering
- SERRS
surface-enhanced resonance Raman scattering
- SEM
scanning electron microscopy
- SFM
scanning force microscopy
- SHINERS
plasmonic core in shell-isolated NPs
- SLI
structured line illumination
- SLM
spatial light modulator
- SM
single-molecule
- SPM
scanning probe microscope
- SPP
surface plasmon polariton
- SPR
surface plasmon resonance
- SRP
scanning Raman picoscopy
- STM
scanning tunneling microscopy
- SWNT
single-walled carbon nanotube
- TDDFT
time-dependent density-functional theory
- TE
transverse electric
- TEPL
tip-enhanced photoluminescence
- TERS
tip-enhanced Raman scattering
- TFIP
1,1,1-trifluoro-2-propanol
- TM
transverse magnetic
- TPP
tetraphenyl porphyrin
- TPY
4′-(N-piperazinyl)terpyridine
- TPY-DTC
4′-(N-piperazinyl)terpyridine dithiocarbamate
- Trp
tryptophan
- Tyr
tyrosine
- UHV
ultrahigh-vacuum
- UME
ultramicroelectrode
- UPS
ultraviolet photoemission spectroscopy
- VB
valence band
- VRS
vacuum Rabi splitting
- WGMs
whispering gallery modes
- XANES
X-ray absorption near-edge structure
- XPS
X-ray photoelectron spectroscopy
Biographies
Dr. Tamitake Itoh is currently a chief senior researcher of National Institute of Advanced Industrial Science and Technology (AIST) in Japan. He completed his Ph.D. (2002) degree at Osaka University under the supervision of Profs. Hiroshi Masuhara and Tsuyoshi Asahi. As a postdoctoral research fellow, he worked at Kwansei Gakuin University from 2002 to 2005. In April 2005, he became a researcher in AIST. In April 2017, he was promoted as a senior researcher and in 2020 as a chief senior researcher. In 2017 for an additional post he was appointed as a visiting professor at Kwansei Gakuin University. His research theme is surface enhanced Raman spectroscopy at ensemble- and single-plasmonic nanoparticle levels.
Marek Procházka received his MSc. degree in Biophysics from Charles University, Faculty of Mathematics and Physics (1994) and Ph.D. degree in Biophysics from Charles University, Faculty of Mathematics and Physics, Prague, Czech Republic and University Pierre et Marie Curie, Paris VI, France (1997). He became an assistant professor (1997), associate professor (2006), and a full professor (2019) at Institute of Physics, Charles University, Faculty of Mathematics and Physics, Prague, Czech Republic. He spent his Marie Curie Individual Fellowship in Laboratoire de Physicochimie Biomoléculaire et Cellulaire, Université Paris VI (2011–2012). His current interests are focused on Raman/SERS spectroscopy of biologically important molecules, designing/testing new metallic nanoparticles/nanostructures for SERS, plasmon-enhanced spectroscopies, and biosensing.
Zhen-Chao Dong received his B.S. from Sichuan University in 1983, M.S. from Xiamen University in 1987, and Ph.D. from Fujian Institute on the Structure of Matter, Chinese Academy of Sciences in 1990. He then went to Iowa State University as a postdoctoral research associate in 1992. From 1996 to 2004, he worked at National Institute for Materials Science (NIMS) in Japan as a senior researcher and sub-theme leader. Since 2004, he is a full professor in Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China. His recent research interests are in the field of single-molecule optoelectronics and nanoplasmonics, particularly on STM based single-molecule Raman scattering, single-molecule electroluminescence and photoluminescence as well as energy transfer at the single-molecule level, all achieving spectro-microscopic imaging down to the subnanometer scale.
Wei Ji received his Bachelor’s (2008) and Doctoral (2013) degrees from Jilin University. From October 2013, he worked in the chemistry department of Kwansei Gakuin University as a postdoctoral fellow of the Japan Society for the Promotion of Science (JSPS). In November 2015, he, as an associate professor, started to work at Dalian University of Technology. Since July 2021, he has been a professor at Northeast Forestry University. His current research is concerned with SERS mechanism with specific interest in the CT mechanism of semiconductor and metal/semiconductor systems, and the development of SERS-based sensing methods for biological and environmental applications.
Dr. Yuko S. Yamamoto is currently an associate professor in Japan Advanced Institute of Science and Technology (2017−). She received her M.S. in Chemistry (2003), worked in a company, and restarted her research activities in the laboratory of Yukihiro Ozaki, focusing on Raman spectroscopy (2007–2011). She then obtained her Ph.D. in Chemistry (2011) and worked on SERS under the supervision of Prof. Tamitake Itoh (2011–2016). She is currently mentoring students in the field of molecular spectroscopy and is mastering management science under the guidance of Mr. James Skinner (2021−). Her current interests are SERS, molecular spectroscopy, sustainable materials, and management science.
Yao Zhang received his B.S. from Shanxi University in 2008, M.S. from Institute of Solid Physics, Chinese Academy of Sciences in 2011, and Ph.D. from the University of Science and Technology of China in 2014. From 2014 to 2017, he joined Prof. Javier Aizpurua’s group as the postdoctoral research associate in the Center for Material Physics (CSIC – UPV/EHU and DIPC) in Donostia-San Sebastián in Spain. Since 2018, he is a professor in Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China. His research interests are in the field of nano-optics and molecular spectroscopies, focusing on the plasmonic properties of metallic nanostructures, the interactions between single molecules and plasmons and plasmon-enhanced spectroscopies.
Yukihiro Ozaki received his Ph.D. in 1978 from Osaka University, Japan. After he had spent two and a half years at National Research Council, Canada as a research associate, he joined the Jikei University School of Medicine. In 1989 he moved to Kwansei Gakuin University, Japan as an associate professor in the Department of Chemistry. Since 1993, he was a professor in the Department of Chemistry until 2018. Currently, he is a professor emeritus of Kwansei Gakuin University, a guest research fellow of Toyota Physical and Chemical Research Institute, a guest professor of Kobe University, and a guest principal researcher of RIKEN. Dr. Ozaki is involved in studies of a wide range of molecular spectroscopy, ranging from far-ultraviolet to far-infrared/Terahertz spectroscopy and also Raman spectroscopy. He is a rather rare scientist who has been concerned with both electronic spectroscopy and vibrational spectroscopy. He has received several awards including Chemical Society of Japan Award (2017), The Medal with Purple Ribbon (2018), Pittsburgh Spectroscopy Award (2019), Charles Mann Award (2020), Karl Norris Award (2021), and Ioannes Marcus Marci Medal (2022).
Author Contributions
CRediT: Tamitake Itoh conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing-original draft, writing-review & editing; Marek Prochazka conceptualization, funding acquisition, investigation, supervision, validation, writing-original draft, writing-review & editing; Zhen-Chao Dong conceptualization, funding acquisition, investigation, methodology, resources, supervision, validation, visualization, writing-original draft, writing-review & editing; Wei Ji investigation, validation, writing-original draft, writing-review & editing; Yuko S. Yamamoto investigation, validation, writing-original draft; Yao Zhang investigation, validation, writing-original draft; Yukihiro Ozaki conceptualization, investigation, supervision, validation, writing-review & editing.
The authors declare no competing financial interest.
References
- Fleischmann M.; Hendra P. J.; Mcquillan A. J. Raman-Spectra of Pyridine Adsorbed at a Silver Electrode. Chem. Phys. Lett. 1974, 26, 163–166. 10.1016/0009-2614(74)85388-1. [DOI] [Google Scholar]
- Albrecht M. G.; Creighton J. A. Anomalously Intense Raman-Spectra of Pyridine at a Silver Electrode. J. Am. Chem. Soc. 1977, 99, 5215–5217. 10.1021/ja00457a071. [DOI] [Google Scholar]
- Jeanmaire D. L.; Van Duyne R. P. Surface Raman Spectroelectrochemistry: Part I. Heterocyclic, Aromatic, and Aliphatic Amines Adsorbed on the Anodized Silver Electrode. J. Electroanal. Chem. 1977, 84, 1–20. 10.1016/S0022-0728(77)80224-6. [DOI] [Google Scholar]
- Stöckle R. M.; Suh Y. D.; Deckert V.; Zenobi R. Nanoscale Chemical Analysis by Tip-enhanced Raman Spectroscopy. Chem. Phys. Lett. 2000, 318, 131–136. 10.1016/S0009-2614(99)01451-7. [DOI] [Google Scholar]
- Anderson M. S. Locally Enhanced Raman Spectroscopy with an Atomic Force Microscope. Appl. Phys. Lett. 2000, 76, 3130–3132. 10.1063/1.126546. [DOI] [Google Scholar]
- Hayazawa N.; Inouye Y.; Sekkat Z.; Kawata S. Metallized Tip Amplification of Near-field Raman Scattering. Opt. Commun. 2000, 183, 333–336. 10.1016/S0030-4018(00)00894-4. [DOI] [Google Scholar]
- Langer J.; Jimenez de Aberasturi D.; Aizpurua J.; Alvarez-Puebla R. A.; Auguié B.; Baumberg J. J.; Bazan G. C.; Bell S. E.; Boisen A.; Brolo A. G.; et al. Present and Future of Surface-Enhanced Raman Scattering. ACS Nano 2020, 14, 28–117. 10.1021/acsnano.9b04224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C.; Xu M.; Xu L. J.; Wei T.; Ma X.; Zheng X. S.; Hu R.; Ren B. Surface-Enhanced Raman Spectroscopy for Bioanalysis: Reliability and Challenges. Chem. Rev. 2018, 118, 4946–4980. 10.1021/acs.chemrev.7b00668. [DOI] [PubMed] [Google Scholar]
- Xu K.; Zhou R.; Takei K.; Hong M. Toward Flexible Surface-Enhanced Raman Scattering (SERS) Sensors for Point-of-Care Diagnostics. Adv. Sci. 2019, 6, 1900925. 10.1002/advs.201900925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. F.; Sun D. W.; Pu H. B.; Wei Q. Y. Surface Enhanced Raman Spectroscopy (SERS): A Novel Reliable Technique for Rapid Detection of Common Harmful Chemical Residues. Trends Food Sci. Technol. 2018, 75, 10–22. 10.1016/j.tifs.2018.02.020. [DOI] [Google Scholar]
- Fan M.; Andrade G. F. S.; Brolo A. G. A Review on Recent Advances in the Applications of Surface-Enhanced Raman Scattering in Analytical Chemistry. Anal. Chim. Acta 2020, 1097, 1–29. 10.1016/j.aca.2019.11.049. [DOI] [PubMed] [Google Scholar]
- Bell S. E. J.; Charron G.; Cortes E.; Kneipp J.; de la Chapelle M. L.; Langer J.; Prochazka M.; Tran V.; Schlucker S. Towards Reliable and Quantitative Surface-Enhanced Raman Scattering (SERS): From Key Parameters to Good Analytical Practice. Angew. Chem., Int. Ed. Engl. 2020, 59, 5454–5462. 10.1002/anie.201908154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rico E.; Alvarez-Puebla R. A.; Guerrini L. Direct Surface-Enhanced Raman Scattering (SERS) Spectroscopy of Nucleic Acids: From Fundamental Studies to Real-Life Applications. Chem. Soc. Rev. 2018, 47, 4909–4923. 10.1039/C7CS00809K. [DOI] [PubMed] [Google Scholar]
- Tang H. B.; Zhu C. H.; Meng G. W.; Wu N. Q. Review-Surface-Enhanced Raman Scattering Sensors for Food Safety and Environmental Monitoring. J. Electrochem. Soc. 2018, 165, B3098–B3118. 10.1149/2.0161808jes. [DOI] [Google Scholar]
- Baumberg J. J.; Aizpurua J.; Mikkelsen M. H.; Smith D. R. Extreme Nanophotonics from Ultrathin Metallic Gaps. Nat. Mater. 2019, 18, 668–678. 10.1038/s41563-019-0290-y. [DOI] [PubMed] [Google Scholar]
- Haran G.; Chuntonov L. Artificial Plasmonic Molecules and Their Interaction with Real Molecules. Chem. Rev. 2018, 118, 5539–5580. 10.1021/acs.chemrev.7b00647. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S. Between Plasmonics and Surface-enhanced Resonant Raman Spectroscopy: Toward Single-Molecule Strong Coupling at a Hotspot. Nanoscale 2021, 13, 1566–1580. 10.1039/D0NR07344J. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Duan S.; Radjenovic P. M.; Tian Z.-Q.; Li J.-F. Core-Shell Nanostructure-Enhanced Raman Spectroscopy for Surface Catalysis. Acc. Chem. Res. 2020, 53, 729–739. 10.1021/acs.accounts.9b00545. [DOI] [PubMed] [Google Scholar]
- Hess C. New Advances in Using Raman Spectroscopy for the Characterization of Catalysts and Catalytic Reactions. Chem. Soc. Rev. 2021, 50, 3519–3564. 10.1039/D0CS01059F. [DOI] [PubMed] [Google Scholar]
- Li Z. D.; Kurouski D. Nanoscale Structural Characterization of Plasmon-driven Reactions. Nanophotonics 2021, 10, 1657–1673. 10.1515/nanoph-2020-0647. [DOI] [Google Scholar]
- Su H. S.; Feng H. S.; Wu X.; Sun J. J.; Ren B. Recent Advances in Plasmon-Enhanced Raman Spectroscopy for Catalytic Reactions on Bifunctional Metallic Nanostructures. Nanoscale 2021, 13, 13962–13975. 10.1039/D1NR04009J. [DOI] [PubMed] [Google Scholar]
- Chen H.; Das A.; Bi L.; Choi N.; Moon J. I.; Wu Y.; Park S.; Choo J. Recent Advances in Surface-enhanced Raman Scattering-Based Microdevices for Point-of-Care Diagnosis of Viruses and Bacteria. Nanoscale 2020, 12, 21560–21570. 10.1039/D0NR06340A. [DOI] [PubMed] [Google Scholar]
- Tahir M. A.; Dina N. E.; Cheng H.; Valev V. K.; Zhang L. Surface-Enhanced Raman Spectroscopy for Bioanalysis and Diagnosis. Nanoscale 2021, 13, 11593–11634. 10.1039/D1NR00708D. [DOI] [PubMed] [Google Scholar]
- Ding S. Y.; You E. M.; Tian Z. Q.; Moskovits M. Electromagnetic Theories of Surface-Enhanced Raman Spectroscopy. Chem. Soc. Rev. 2017, 46, 4042–4076. 10.1039/C7CS00238F. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Ozaki Y. Plasmon-Enhanced Spectroscopy of Absorption and Spontaneous Emissions Explained Using Cavity Quantum Optics. Chem. Soc. Rev. 2017, 46, 3904–3921. 10.1039/C7CS00155J. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y. S.; Itoh T. Why and How Do the Shapes of Surface-Enhanced Raman Scattering Spectra Change? Recent Progress from Mechanistic Studies. J. Raman Spectrosc. 2016, 47, 78–88. 10.1002/jrs.4874. [DOI] [Google Scholar]
- Yamamoto Y. S.; Ozaki Y.; Itoh T. Recent Progress and Frontiers in the Electromagnetic Mechanism of Surface-Enhanced Raman Scattering. J. Photochem. Photobiol. C: Photochem. Rev. 2014, 21, 81–104. 10.1016/j.jphotochemrev.2014.10.001. [DOI] [Google Scholar]
- Dovzhenko D. S.; Ryabchuk S. V.; Rakovich Y. P.; Nabiev I. R. Light-Matter Interaction in the Strong Coupling Regime: Configurations, Conditions, and Applications. Nanoscale 2018, 10, 3589–3605. 10.1039/C7NR06917K. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Esteban R.; Borisov A. G.; Baumberg J. J.; Nordlander P.; Lezec H. J.; Aizpurua J.; Crozier K. B. Quantum Mechanical Effects in Plasmonic Structures with Subnanometre Gaps. Nat. Commun. 2016, 7, 11495. 10.1038/ncomms11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. F.; Wu D. Y.; Zhu H. P.; Zhao L. B.; Liu G. K.; Ren B.; Tian Z. Q. Surface-Enhanced Raman Spectroscopic Study of p-Aminothiophenol. Phys. Chem. Chem. Phys. 2012, 14, 8485–8497. 10.1039/c2cp40558j. [DOI] [PubMed] [Google Scholar]
- Payton J. L.; Morton S. M.; Moore J. E.; Jensen L. A Hybrid Atomistic Electrodynamics-quantum Mechanical Approach for Simulating Surface-Enhanced Raman Scattering. Acc. Chem. Res. 2014, 47, 88–99. 10.1021/ar400075r. [DOI] [PubMed] [Google Scholar]
- Demirel G.; Usta H.; Yilmaz M.; Celik M.; Alidagi H. A.; Buyukserin F. Surface-Enhanced Raman Spectroscopy (SERS): An Adventure from Plasmonic Metals to Organic Semiconductors as SERS Platforms. J. Mater. Chem. C 2018, 6, 5314–5335. 10.1039/C8TC01168K. [DOI] [Google Scholar]
- Ji W.; Zhao B.; Ozaki Y. Semiconductor Materials in Analytical Applications of Surface-Enhanced Raman Scattering. J. Raman Spectrosc. 2016, 47, 51–58. 10.1002/jrs.4854. [DOI] [Google Scholar]
- Wang H.; Liu Y.; Rao G.; Wang Y.; Du X.; Hu A.; Hu Y.; Gong C.; Wang X.; Xiong J. Coupling Enhancement Mechanisms, Materials, and Strategies for Surface-Enhanced Raman Scattering Devices. Analyst 2021, 146, 5008–5032. 10.1039/D1AN00624J. [DOI] [PubMed] [Google Scholar]
- Shvalya V.; Filipic G.; Zavasnik J.; Abdulhalim I.; Cvelbar U. Surface-enhanced Raman Spectroscopy for Chemical and Biological Sensing Using Nanoplasmonics: The Relevance of Interparticle Spacing and Surface Morphology. Appl. Phys. Rev. 2020, 7, 031307. 10.1063/5.0015246. [DOI] [Google Scholar]
- Hang Y.; Boryczka J.; Wu N. Visible-light and Near-infrared Fluorescence and Surface-enhanced Raman Scattering Point-of-Care Sensing and Bio-imaging: A Review. Chem. Soc. Rev. 2022, 51, 329–375. 10.1039/C9CS00621D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S.; Zhang Y.; Ahmed S.; Abdulraheem M. I.; Hu J.; Tahir M. N.; Raghavan V. Applied Surface Enhanced Raman Spectroscopy in Plant Hormones Detection, Annexation of Advanced Technologies: A review. Talanta 2022, 236, 122823. 10.1016/j.talanta.2021.122823. [DOI] [PubMed] [Google Scholar]
- Ding S.-Y.; Yi J.; Li J.-F.; Ren B.; Wu D.-Y.; Panneerselvam R.; Tian Z.-Q. Nanostructure-based Plasmon-Enhanced Raman Spectroscopy for Surface Analysis of Materials. Nat. Rev. Mater. 2016, 1, 16021. 10.1038/natrevmats.2016.21. [DOI] [Google Scholar]
- Li J.-F.; Zhang Y.-J.; Ding S.-Y.; Panneerselvam R.; Tian Z.-Q. Core-Shell Nanoparticle-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117, 5002–5069. 10.1021/acs.chemrev.6b00596. [DOI] [PubMed] [Google Scholar]
- Zhang Z. L.; Xu P.; Yang X. Z.; Liang W. J.; Sun M. T. Surface Plasmon-Driven Photocatalysis in Ambient, Aqueous and High-Vacuum Monitored by SERS and TERS. J. Photochem. Photobiol. C-Photochem. Rev. 2016, 27, 100–112. 10.1016/j.jphotochemrev.2016.04.001. [DOI] [Google Scholar]
- Moskovits M. Surface-Enhanced Spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. 10.1103/RevModPhys.57.783. [DOI] [Google Scholar]
- Campion A.; Kambhampati P. Surface-Enhanced Raman Scattering. Chem. Soc. Rev. 1998, 27, 241–250. 10.1039/a827241z. [DOI] [Google Scholar]
- Zrimsek A. B.; Chiang N.; Mattei M.; Zaleski S.; McAnally M. O.; Chapman C. T.; Henry A. I.; Schatz G. C.; Van Duyne R. P. Single-Molecule Chemistry with Surface- and Tip-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117, 7583–7613. 10.1021/acs.chemrev.6b00552. [DOI] [PubMed] [Google Scholar]
- Nie S.; Emory S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 275, 1102–1106. 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Perelman L. T.; Itzkan I.; Dasari R.; Feld M. S. Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. 10.1103/PhysRevLett.78.1667. [DOI] [Google Scholar]
- Gruenke N. L.; Cardinal M. F.; McAnally M. O.; Frontiera R. R.; Schatz G. C.; Van Duyne R. P. Ultrafast and Nonlinear Surface-Enhanced Raman Spectroscopy. Chem. Soc. Rev. 2016, 45, 2263–2290. 10.1039/C5CS00763A. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Etchegoin P. G.. Principles of Surface-Enhanced Raman Spectroscopy and Related Plasmonic Effects. Elsevier, 2009; pp 1–663. [Google Scholar]
- Moskovits M. Surface-Roughness and Enhanced Intensity of Raman-scattering by Molecules Adsorbed on Metals. J. Chem. Phys. 1978, 69, 4159–4161. 10.1063/1.437095. [DOI] [Google Scholar]
- Xu H. X.; Bjerneld E. J.; Kall M.; Borjesson L. Spectroscopy of Single Hemoglobin Molecules by Surface Enhanced Raman Scattering. Phys. Rev. Lett. 1999, 83, 4357–4360. 10.1103/PhysRevLett.83.4357. [DOI] [Google Scholar]
- Xu H.; Aizpurua J.; Kall M.; Apell P. Electromagnetic Contributions to Single-Molecule Sensitivity in Surface-Enhanced Raman Scattering. Phys. Rev. E 2000, 62, 4318–4324. 10.1103/PhysRevE.62.4318. [DOI] [PubMed] [Google Scholar]
- Yoshida K.; Itoh T.; Tamaru H.; Biju V.; Ishikawa M.; Ozaki Y. Quantitative Evaluation of Electromagnetic Enhancement in Surface-Enhanced Resonance Raman Scattering from Plasmonic Properties and Morphologies of Individual Ag Nanostructures. Phys. Rev. B 2010, 81, 115406. 10.1103/PhysRevB.81.115406. [DOI] [Google Scholar]
- Dubey A.; Mishra R.; Cheng C. W.; Kuang Y. P.; Gwo S.; Yen T. J. Demonstration of a Superior Deep-UV Surface-Enhanced Resonance Raman Scattering (SERRS) Substrate and Single-Base Mutation Detection in Oligonucleotides. J. Am. Chem. Soc. 2021, 143, 19282–19286. 10.1021/jacs.1c09762. [DOI] [PubMed] [Google Scholar]
- Lombardi J. R.; Birke R. L.; Lu T. H.; Xu J. Charge-Transfer Theory of Surface Enhanced Raman Spectroscopy: Herzberg-Teller Contributions. J. Chem. Phys. 1986, 84, 4174–4180. 10.1063/1.450037. [DOI] [Google Scholar]
- Otto A.; Mrozek I.; Grabhorn H.; Akemann W. Surface-Enhanced Raman Scattering. J. Phys.-Condens. Matter 1992, 4, 1143–1212. 10.1088/0953-8984/4/5/001. [DOI] [Google Scholar]
- Arenas J. F.; Soto J.; Tocon I. L.; Fernandez D. J.; Otero J. C.; Marcos J. I. The Role of Charge-Transfer States of the Metal-adsorbate Complex in Surface-Enhanced Raman scattering. J. Chem. Phys. 2002, 116, 7207–7216. 10.1063/1.1450542. [DOI] [Google Scholar]
- Benz F.; Schmidt M. K.; Dreismann A.; Chikkaraddy R.; Zhang Y.; Demetriadou A.; Carnegie C.; Ohadi H.; De Nijs B.; Esteban R.; et al. Single-Molecule Optomechanics in “Picocavities. Science 2016, 354, 726–729. 10.1126/science.aah5243. [DOI] [PubMed] [Google Scholar]
- Wessel J. Surface-Enhanced Optical Microscopy. J. Opt. Soc. Am. B 1985, 2, 1538–1541. 10.1364/JOSAB.2.001538. [DOI] [Google Scholar]
- Pettinger B.; Picardi G.; Schuster R.; Ertl G. Surface Enhanced Raman Spectroscopy: Towards Single Molecule Spectroscopy. Electrochem. 2000, 68, 942–949. 10.5796/electrochemistry.68.942. [DOI] [Google Scholar]
- Verma P. Tip-Enhanced Raman Spectroscopy: Technique and Recent Advances. Chem. Rev. 2017, 117, 6447–6466. 10.1021/acs.chemrev.6b00821. [DOI] [PubMed] [Google Scholar]
- Wang X.; Huang S. C.; Huang T. X.; Su H. S.; Zhong J. H.; Zeng Z. C.; Li M. H.; Ren B. Tip-Enhanced Raman spectroscopy for Surfaces and Interfaces. Chem. Soc. Rev. 2017, 46, 4020–4041. 10.1039/C7CS00206H. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Zhang Y.; Dong Z. C.; Jiang S.; Zhang C.; Chen L. G.; Zhang L.; Liao Y.; Aizpurua J.; Luo Y.; et al. Chemical Mapping of a Single Molecule by Plasmon-Enhanced Raman Scattering. Nature 2013, 498, 82–86. 10.1038/nature12151. [DOI] [PubMed] [Google Scholar]
- Wang D. S.; Kerker M. Enhanced Raman Scattering by Molecules Adsorbed at the Surface of Colloidal Spheroids. Phys. Rev. B 1981, 24, 1777–1790. 10.1103/PhysRevB.24.1777. [DOI] [Google Scholar]
- Aravind P. K.; Nitzan A.; Metiu H. The Interaction between Electromagnetic Resonances and its Role in Spectroscopic Studies of Molecules Adsorbed on Colloidal Particles or Metal Spheres. Surf. Sci. 1981, 110, 189–204. 10.1016/0039-6028(81)90595-1. [DOI] [Google Scholar]
- Inoue M.; Ohtaka K. Surface Enhanced Raman-Scattering by Metal Spheres. 1. Cluster Effect. J. Phys. Soc. Jpn. 1983, 52, 3853–3864. 10.1143/JPSJ.52.3853. [DOI] [Google Scholar]
- Bohren C. F.; Huffman D. R.. Absorption and Scattering of Light by Small Particles; Wiley, 1983; pp 1–530. [Google Scholar]
- Fox M.Quantum Optics: An Introduction; Oxford University Press, 2006; pp 1–378. [Google Scholar]
- Andreani L. C.; Panzarini G.; Gerard J. M. Strong-Coupling Regime for Quantum Boxes in Pillar Microcavities: Theory. Phys. Rev. B 1999, 60, 13276–13279. 10.1103/PhysRevB.60.13276. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S. Reproduction of Surface-Enhanced Resonant Raman Scattering and Fluorescence Spectra of a Strong Coupling System Composed of a Single Silver Nanoparticle Dimer and a Few Dye Molecules. J. Chem. Phys. 2018, 149, 244701. 10.1063/1.5061816. [DOI] [PubMed] [Google Scholar]
- Savage K. J.; Hawkeye M. M.; Esteban R.; Borisov A. G.; Aizpurua J.; Baumberg J. J. Revealing the Quantum Regime in Tunnelling Plasmonics. Nature 2012, 491, 574–577. 10.1038/nature11653. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Crozier K. B. Quantum Mechanical Limit to Plasmonic Enhancement as Observed by Surface-Enhanced Raman Scattering. Nat. Commun. 2014, 5, 5228. 10.1038/ncomms6228. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Iga M.; Tamaru H.; Yoshida K.; Biju V.; Ishikawa M. Quantitative Evaluation of Blinking in Surface Enhanced Resonance Raman Scattering and Fluorescence by Electromagnetic Mechanism. J. Chem. Phys. 2012, 136, 024703. 10.1063/1.3675567. [DOI] [PubMed] [Google Scholar]
- Jensen L.; Aikens C. M.; Schatz G. C. Electronic Structure Methods for Studying Surface-Enhanced Raman Scattering. Chem. Soc. Rev. 2008, 37, 1061–1073. 10.1039/b706023h. [DOI] [PubMed] [Google Scholar]
- Dirac P. A. M. The Quantum Theory of the Emission and Absorption of Radiation. Proc. R. Soc. London A: Math. Phys. Eng. Sci. 1927, 114, 243–265. 10.1098/rspa.1927.0039. [DOI] [Google Scholar]
- Born M.; Oppenheimer J. R. Zur Quantentheorie der Molekeln. Ann. Phys. 1927, 389, 457–484. 10.1002/andp.19273892002. [DOI] [Google Scholar]
- Allemand C. D. Depolarization Ratio Measurements in Raman Spectrometry. Appl. Spectrosc. 1970, 24, 348–353. 10.1366/000370270774371552. [DOI] [Google Scholar]
- Albrecht A. C. Theory of Raman Intensities. J. Chem. Phys. 1961, 34, 1476–1484. 10.1063/1.1701032. [DOI] [Google Scholar]
- Watanabe H.; Hayazawa N.; Inouye Y.; Kawata S. DFT Vibrational Calculations of Rhodamine 6G Adsorbed on Silver: Analysis of Tip-Enhanced Raman Spectroscopy. J. Phys. Chem. B 2005, 109, 5012–5020. 10.1021/jp045771u. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Li X.; Fan Q.; Zhang X.; Zheng J. Charge Transfer between Metal Nanoparticles Interconnected with a Functionalized Molecule Probed by Surface-Enhanced Raman Spectroscopy. Angew. Chem., Int. Ed. Engl. 2006, 45, 3970–3973. 10.1002/anie.200504419. [DOI] [PubMed] [Google Scholar]
- Osawa M.; Matsuda N.; Yoshii K.; Uchida I. Charge-Transfer Resonance Raman Process in Surface-Enhanced Raman-Scattering from p-Aminothiophenol Adsorbed on Silver- Herzberg-Teller contribution. J. Phys. Chem. 1994, 98, 12702–12707. 10.1021/j100099a038. [DOI] [Google Scholar]
- Brolo A. G.; Irish D. E.; Lipkowski J. Surface-Enhanced Raman Spectra of Pyridine and Pyrazine Adsorbed on a Au(210) Single-Crystal Electrode. J. Phys. Chem. B 1997, 101, 3906–3909. 10.1021/jp970340f. [DOI] [Google Scholar]
- Centeno S. P.; Lopez-Tocon I.; Arenas J. F.; Soto J.; Otero J. C. Selection Rules of the Charge Transfer Mechanism of Surface-Enhanced Raman Scattering: The Effect of the Adsorption on the Relative Intensities of Pyrimidine Bonded to Silver Nanoclusters. J. Phys. Chem. B 2006, 110, 14916–14922. 10.1021/jp0621373. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Jensen L.; Schatz G. C. Pyridine-Ag20 cluster: A Model System for Studying Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 2006, 128, 2911–2919. 10.1021/ja0556326. [DOI] [PubMed] [Google Scholar]
- Lombardi J. R.; Birke R. L. A Unified Approach to Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2008, 112, 5605–5617. 10.1021/jp800167v. [DOI] [Google Scholar]
- Halas N. J.; Lal S.; Chang W. S.; Link S.; Nordlander P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem. Rev. 2011, 111, 3913–3961. 10.1021/cr200061k. [DOI] [PubMed] [Google Scholar]
- Nam J. M.; Oh J. W.; Lee H.; Suh Y. D. Plasmonic Nanogap-Enhanced Raman Scattering with Nanoparticles. Acc. Chem. Res. 2016, 49, 2746–2755. 10.1021/acs.accounts.6b00409. [DOI] [PubMed] [Google Scholar]
- Li G.-C.; Zhang Q.; Maier S. A.; Lei D. Plasmonic Particle-on-Film Nanocavities: A Versatile Platform for Plasmon-Enhanced Spectroscopy and Photochemistry. Nanophotonics 2018, 7, 1865–1889. 10.1515/nanoph-2018-0162. [DOI] [Google Scholar]
- Simoncelli S.; Roller E. M.; Urban P.; Schreiber R.; Turberfield A. J.; Liedl T.; Lohmuller T. Quantitative Single-Molecule Surface-Enhanced Raman Scattering by Optothermal Tuning of DNA Origami-Assembled Plasmonic Nanoantennas. ACS Nano 2016, 10, 9809–9815. 10.1021/acsnano.6b05276. [DOI] [PubMed] [Google Scholar]
- Zhan P.; Wen T.; Wang Z. G.; He Y.; Shi J.; Wang T.; Liu X.; Lu G.; Ding B. DNA Origami Directed Assembly of Gold Bowtie Nanoantennas for Single-Molecule Surface-Enhanced Raman Scattering. Angew. Chem., Int. Ed. Engl. 2018, 57, 2846–2850. 10.1002/anie.201712749. [DOI] [PubMed] [Google Scholar]
- Garai M.; Gao N.; Xu Q.-H. Single-Particle Spectroscopic Studies on Two-Photon Photoluminescence of Coupled Au Nanorod Dimers. J. Phys. Chem. C 2018, 122, 23102–23110. 10.1021/acs.jpcc.8b07094. [DOI] [Google Scholar]
- Xiang Q.; Li Z.; Zheng M.; Liu Q.; Chen Y.; Yang L.; Jiang T.; Duan H. Sensitive SERS Detection at the Single-Particle Level Based on Nanometer-Separated Mushroom-Shaped Plasmonic Dimers. Nanotechnology 2018, 29, 105301. 10.1088/1361-6528/aaa691. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Shuai Z.; Shen J.; Zhang L.; Chen S.; Song C.; Zhao B.; Fan Q.; Wang L. Plasmonic Heterodimers with Binding Site-Dependent Hot Spot for Surface-Enhanced Raman Scattering. Small 2018, 14, e1800669 10.1002/smll.201800669. [DOI] [PubMed] [Google Scholar]
- Qiu J.; Xie M.; Lyu Z.; Gilroy K. D.; Liu H.; Xia Y. General Approach to the Synthesis of Heterodimers of Metal Nanoparticles through Site-Selected Protection and Growth. Nano Lett. 2019, 19, 6703–6708. 10.1021/acs.nanolett.9b03167. [DOI] [PubMed] [Google Scholar]
- Tiwari S.; Khandelwal U.; Sharma V.; Kumar G. V. P. Single Molecule Surface Enhanced Raman Scattering in a Single Gold Nanoparticle-Driven Thermoplasmonic Tweezer. J. Phys. Chem. Lett. 2021, 12, 11910–11918. 10.1021/acs.jpclett.1c03450. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Maruoka K.; Ikegami K.; Uesugi A.; Isono Y. Dynamic Surface-Enhanced Raman Spectroscopy of DNA Oligomer with a Single Hotspot from a Gold Nanoparticle Dimer. Opt. Lett. 2022, 47, 373–376. 10.1364/OL.441580. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yoshikawa H.; Yoshida K.; Biju V.; Ishikawa M. Evaluation of Electromagnetic Enhancement of Surface Enhanced Hyper Raman Scattering Using Plasmonic Properties of Binary Active Sites in Single Ag Nanoaggregates. J. Chem. Phys. 2009, 130, 214706. 10.1063/1.3146788. [DOI] [PubMed] [Google Scholar]
- Brown L. V.; Sobhani H.; Lassiter J. B.; Nordlander P.; Halas N. J. Heterodimers: Plasmonic Properties of Mismatched Nanoparticle Pairs. ACS Nano 2010, 4, 819–832. 10.1021/nn9017312. [DOI] [PubMed] [Google Scholar]
- Ye J.; Wen F.; Sobhani H.; Lassiter J. B.; Van Dorpe P.; Nordlander P.; Halas N. J. Plasmonic Nanoclusters: Near Field Properties of the Fano Resonance Interrogated with SERS. Nano Lett. 2012, 12, 1660–1667. 10.1021/nl3000453. [DOI] [PubMed] [Google Scholar]
- Yoshida K.-I.; Itoh T.; Biju V.; Ishikawa M.; Ozaki Y. Experimental Evaluation of the Twofold Electromagnetic Enhancement Theory of Surface-Enhanced Resonance Raman Scattering. Phys. Rev. B 2009, 79, 085419. 10.1103/PhysRevB.79.085419. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Kitahama Y.; Balachandran J. One-Dimensional Plasmonic Hotspots Located Between Silver Nanowire Dimers Evaluated by Surface-Enhanced Resonance Raman scattering. Phys. Rev. B 2017, 95, 115441. 10.1103/PhysRevB.95.115441. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Balachandran J. Propagation Mechanism of Surface Plasmons Coupled with Surface-Enhanced Resonant Raman Scattering Light through a One-Dimensional Hotspot along a Silver Nanowire Dimer Junction. Phys. Rev. B 2021, 103, 245425. 10.1103/PhysRevB.103.245425. [DOI] [Google Scholar]
- Fang Y. R.; Wei H.; Hao F.; Nordlander P.; Xu H. X. Remote-Excitation Surface-Enhanced Raman Scattering Using Propagating Ag Nanowire Plasmons. Nano Lett. 2009, 9, 2049–2053. 10.1021/nl900321e. [DOI] [PubMed] [Google Scholar]
- Hutchison J. A.; Centeno S. P.; Odaka H.; Fukumura H.; Hofkens J.; Uji I. H. Subdiffraction Limited, Remote Excitation of Surface Enhanced Raman Scattering. Nano Lett. 2009, 9, 995–1001. 10.1021/nl8030696. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S. Recent Topics on Single-Molecule Fluctuation Analysis Using Blinking in Surface-Enhanced Resonance Raman Scattering: Clarification by the Electromagnetic Mechanism. Analyst 2016, 141, 5000–5009. 10.1039/C6AN00936K. [DOI] [PubMed] [Google Scholar]
- Li Y.; Hu H.; Jiang W.; Shi J.; Halas N. J.; Nordlander P.; Zhang S.; Xu H. Duplicating Plasmonic Hotspots by Matched Nanoantenna Pairs for Remote Nanogap Enhanced Spectroscopy. Nano Lett. 2020, 20, 3499–3505. 10.1021/acs.nanolett.0c00434. [DOI] [PubMed] [Google Scholar]
- Aravind P. K.; Metiu H. The Effects of the Interaction between Resonances in the Electromagnetic Response of a Sphere-Plane Structure - Applications to Surface Enhanced Spectroscopy. Surf. Sci. 1983, 124, 506–528. 10.1016/0039-6028(83)90806-3. [DOI] [Google Scholar]
- Nordlander P.; Prodan E. Plasmon Hybridization in Nanoparticles Near Metallic Surfaces. Nano Lett. 2004, 4, 2209–2213. 10.1021/nl0486160. [DOI] [Google Scholar]
- Le F.; Lwin N. Z.; Steele J. M.; Kall M.; Halas N. J.; Nordlander P. Plasmons in the Metallic Nanoparticle - Film System as a Tunable Impurity Problem. Nano Lett. 2005, 5, 2009–2013. 10.1021/nl0515100. [DOI] [PubMed] [Google Scholar]
- Ciraci C.; Hill R. T.; Mock J. J.; Urzhumov Y.; Fernandez-Dominguez A. I.; Maier S. A.; Pendry J. B.; Chilkoti A.; Smith D. R. Probing the Ultimate Limits of Plasmonic Enhancement. Science 2012, 337, 1072–1074. 10.1126/science.1224823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz F.; Chikkaraddy R.; Salmon A.; Ohadi H.; de Nijs B.; Mertens J.; Carnegie C.; Bowman R. W.; Baumberg J. J. SERS of Individual Nanoparticles on a Mirror: Size Does Matter, but so Does Shape. J. Phys. Chem. Lett. 2016, 7, 2264–2269. 10.1021/acs.jpclett.6b00986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie C.; Griffiths J.; de Nijs B.; Readman C.; Chikkaraddy R.; Deacon W. M.; Zhang Y.; Szabo I.; Rosta E.; Aizpurua J.; et al. Room-Temperature Optical Picocavities below 1 nm(3) Accessing Single-Atom Geometries. J. Phys. Chem. Lett. 2018, 9, 7146–7151. 10.1021/acs.jpclett.8b03466. [DOI] [PubMed] [Google Scholar]
- Shin H. H.; Yeon G. J.; Choi H. K.; Park S. M.; Lee K. S.; Kim Z. H. Frequency-Domain Proof of the Existence of Atomic-Scale SERS Hot-Spots. Nano Lett. 2018, 18, 262–271. 10.1021/acs.nanolett.7b04052. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Xiao T.-H.; Wu Y.; Li W.; Zeng Q.-G.; Long L.; Li Z.-Y. Roadmap for Single-Molecule Surface-Enhanced Raman Spectroscopy. Adv. Photonics 2020, 2, 014002. 10.1117/1.AP.2.1.014002. [DOI] [Google Scholar]
- Huang J. A.; Mousavi M. Z.; Giovannini G.; Zhao Y.; Hubarevich A.; Soler M. A.; Rocchia W.; Garoli D.; De Angelis F. Multiplexed Discrimination of Single Amino Acid Residues in Polypeptides in a Single SERS Hot Spot. Angew. Chem., Int. Ed. Engl. 2020, 59, 11423–11431. 10.1002/anie.202000489. [DOI] [PubMed] [Google Scholar]
- Fan J. A.; Wu C.; Bao K.; Bao J.; Bardhan R.; Halas N. J.; Manoharan V. N.; Nordlander P.; Shvets G.; Capasso F. Self-Assembled Plasmonic Nanoparticle Clusters. Science 2010, 328, 1135–1138. 10.1126/science.1187949. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhen Y. R.; Neumann O.; Day J. K.; Nordlander P.; Halas N. J. Coherent Anti-Stokes Raman Scattering with Single-Molecule Sensitivity Using a Plasmonic Fano Resonance. Nat. Commun. 2014, 5, 4424. 10.1038/ncomms5424. [DOI] [PubMed] [Google Scholar]
- Kleinman S. L.; Sharma B.; Blaber M. G.; Henry A. I.; Valley N.; Freeman R. G.; Natan M. J.; Schatz G. C.; Van Duyne R. P. Structure Enhancement Factor Relationships in Single Gold Nanoantennas by Surface-Enhanced Raman Excitation Spectroscopy. J. Am. Chem. Soc. 2013, 135, 301–308. 10.1021/ja309300d. [DOI] [PubMed] [Google Scholar]
- Chen J.; Gan F.; Wang Y.; Li G. Plasmonic Sensing and Modulation Based on Fano Resonances. Adv. Opt. Mater. 2018, 6, 1701152. 10.1002/adom.201701152. [DOI] [Google Scholar]
- Rahmani M.; Luk’yanchuk B.; Hong M. Fano Resonance in Novel Plasmonic Nanostructures. Laser Photon. Rev. 2013, 7, 329–349. 10.1002/lpor.201200021. [DOI] [Google Scholar]
- Luk’yanchuk B.; Zheludev N. I.; Maier S. A.; Halas N. J.; Nordlander P.; Giessen H.; Chong C. T. The Fano Resonance in Plasmonic Nanostructures and Metamaterials. Nat. Mater. 2010, 9, 707–715. 10.1038/nmat2810. [DOI] [PubMed] [Google Scholar]
- Michaels A. M.; Nirmal M.; Brus L. E. Surface Enhanced Raman Spectroscopy of Individual Rhodamine 6G Molecules on Large Ag Nanocrystals. J. Am. Chem. Soc. 1999, 121, 9932–9939. 10.1021/ja992128q. [DOI] [Google Scholar]
- Constantino C. J. L.; Lemma T.; Antunes P. A.; Aroca R. Single-Molecule Detection Using Surface-Enhanced Resonance Raman Scattering and Langmuir-Blodgett Monolayers. Anal. Chem. 2001, 73, 3674–3678. 10.1021/ac0101961. [DOI] [PubMed] [Google Scholar]
- Weiss A.; Haran G. Time-Dependent Single-Molecule Raman Scattering as a Probe of Surface Dynamics. J. Phys. Chem. B 2001, 105, 12348–12354. 10.1021/jp0126863. [DOI] [Google Scholar]
- Habuchi S.; Cotlet M.; Gronheid R.; Dirix G.; Michiels J.; Vanderleyden J.; De Schryver F. C.; Hofkens J. Single-Molecule Surface Enhanced Resonance Raman Spectroscopy of the Enhanced Green Fluorescent Protein. J. Am. Chem. Soc. 2003, 125, 8446–8447. 10.1021/ja0353311. [DOI] [PubMed] [Google Scholar]
- Ward D. R.; Grady N. K.; Levin C. S.; Halas N. J.; Wu Y.; Nordlander P.; Natelson D. Electromigrated Nanoscale Gaps for Surface-Enhanced Raman Spectroscopy. Nano Lett. 2007, 7, 1396–1400. 10.1021/nl070625w. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Meyer M.; Etchegoin P. G. Proof of Single-Molecule Sensitivity in Surface Enhanced Raman Scattering (SERS) by Means of a Two-Analyte Technique. J. Phys. Chem. B 2006, 110, 1944–1948. 10.1021/jp054732v. [DOI] [PubMed] [Google Scholar]
- Dieringer J. A.; Lettan R. B. 2nd; Scheidt K. A.; Van Duyne R. P. A Frequency Domain Existence Proof of Single-Molecule Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2007, 129, 16249–16256. 10.1021/ja077243c. [DOI] [PubMed] [Google Scholar]
- Stranahan S. M.; Willets K. A. Super-Resolution Optical Imaging of Single-Molecule SERS Hot Spots. Nano Lett. 2010, 10, 3777–3784. 10.1021/nl102559d. [DOI] [PubMed] [Google Scholar]
- Kitahama Y.; Tanaka Y.; Itoh T.; Ozaki Y. Power-Law Statistics in Blinking SERS of Thiacyanine Adsorbed on a Single Silver Nanoaggregate. Phys. Chem. Chem. Phys. 2010, 12, 7457–7460. 10.1039/c000824a. [DOI] [PubMed] [Google Scholar]
- Etchegoin P. G.; Le Ru E. C. Resolving Single Molecules in Surface-Enhanced Raman Scattering within the Inhomogeneous Broadening of Raman Peaks. Anal. Chem. 2010, 82, 2888–2892. 10.1021/ac9028888. [DOI] [PubMed] [Google Scholar]
- Kim N. H.; Hwang W.; Baek K.; Rohman M. R.; Kim J.; Kim H. W.; Mun J.; Lee S. Y.; Yun G.; Murray J.; et al. Smart SERS Hot Spots: Single Molecules Can Be Positioned in a Plasmonic Nanojunction Using Host-Guest Chemistry. J. Am. Chem. Soc. 2018, 140, 4705–4711. 10.1021/jacs.8b01501. [DOI] [PubMed] [Google Scholar]
- Willets K. A. Super-Resolution Imaging of SERS Hot Spots. Chem. Soc. Rev. 2014, 43, 3854–3864. 10.1039/C3CS60334B. [DOI] [PubMed] [Google Scholar]
- Titus E. J.; Weber M. L.; Stranahan S. M.; Willets K. A. Super-Resolution SERS Imaging Beyond the Single-Molecule Limit: An Isotope-Edited Approach. Nano Lett. 2012, 12, 5103–5110. 10.1021/nl3017779. [DOI] [PubMed] [Google Scholar]
- Olson A. P.; Spies K. B.; Browning A. C.; Soneral P. A. G.; Lindquist N. C. Chemically Imaging Bacteria with Super-Resolution SERS on Ultra-Thin Silver Substrates. Sci. Rep. 2017, 7, 9135. 10.1038/s41598-017-08915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Chen M.; Zhanghao K.; Zhang X.; Jing Z.; Gao J.; Zhang M. Q.; Jin D.; Dai Z.; Xi P.; et al. Polarization-Based Super-Resolution Imaging of Surface-Enhanced Raman Scattering Nanoparticles with Orientational Information. Nanoscale 2018, 10, 19757–19765. 10.1039/C8NR04808H. [DOI] [PubMed] [Google Scholar]
- de Albuquerque C. D. L.; Schultz Z. D. Super-resolution Surface-Enhanced Raman Scattering Imaging of Single Particles in Cells. Anal. Chem. 2020, 92, 9389–9398. 10.1021/acs.analchem.0c01864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway C. M.; Etchegoin P. G.; Le Ru E. C. Ultrafast Nonradiative Decay Rates on Metallic Surfaces by Comparing Surface-Enhanced Raman and Fluorescence Signals of Single Molecules. Phys. Rev. Lett. 2009, 103, 063003. 10.1103/PhysRevLett.103.063003. [DOI] [PubMed] [Google Scholar]
- Xu H.; Wang X. H.; Persson M. P.; Xu H. Q.; Kall M.; Johansson P. Unified Treatment of Fluorescence and Raman Scattering Processes Near Metal Surfaces. Phys. Rev. Lett. 2004, 93, 243002. 10.1103/PhysRevLett.93.243002. [DOI] [PubMed] [Google Scholar]
- Johansson P.; Xu H.; Käll M. Surface-Enhanced Raman Scattering and Fluorescence Near Metal Nanoparticles. Phys. Rev. B 2005, 72, 035427. 10.1103/PhysRevB.72.035427. [DOI] [Google Scholar]
- Xu H.; Kall M. Surface-Plasmon-Enhanced Optical Forces in Silver Nanoaggregates. Phys. Rev. Lett. 2002, 89, 246802. 10.1103/PhysRevLett.89.246802. [DOI] [PubMed] [Google Scholar]
- Shimizu K. T.; Neuhauser R. G.; Leatherdale C. A.; Empedocles S. A.; Woo W. K.; Bawendi M. G. Blinking Statistics in Single Semiconductor Nanocrystal Quantum Dots. Phys. Rev. B 2001, 63, 205316. 10.1103/PhysRevB.63.205316. [DOI] [Google Scholar]
- Kitahama Y.; Nishiyama Y.; Ozaki Y. Blinking Surface-Enhanced Raman Scattering and Fluorescence From a Single Silver Nanoaggregate Simultaneously Analyzed by Bi-Color Intensity Ratios and a Truncated Power Law. J. Phys. Chem. C 2018, 122, 22106–22113. 10.1021/acs.jpcc.8b06920. [DOI] [Google Scholar]
- Lindquist N. C.; Brolo A. G. Ultra-High-Speed Dynamics in Surface-Enhanced Raman Scattering. J. Phys. Chem. C 2021, 125, 7523–7532. 10.1021/acs.jpcc.0c11150. [DOI] [Google Scholar]
- Zong C.; Chen C. J.; Wang X.; Hu P.; Liu G. K.; Ren B. Single-Molecule Level Rare Events Revealed by Dynamic Surface-Enhanced Raman Spectroscopy. Anal. Chem. 2020, 92, 15806–15810. 10.1021/acs.analchem.0c02936. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Kasera S.; Rowland M. J.; del Barrio J.; Scherman O. A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- Ai Q.; Zhou J.; Guo J.; Pandey P.; Liu S.; Fu Q.; Liu Y.; Deng C.; Chang S.; Liang F.; et al. Observing Dynamic Molecular Changes at Single-Molecule Level in a Cucurbituril Based Plasmonic Molecular Junction. Nanoscale 2020, 12, 17103–17112. 10.1039/D0NR03360J. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Lee T. C.; Scherman O. A.; Esteban R.; Aizpurua J.; Huang F. M.; Baumberg J. J.; Mahajan S. Precise Subnanometer Plasmonic Junctions for SERS within Gold Nanoparticle Assemblies Using Cucurbit[n]uril ″Glue″. ACS Nano 2011, 5, 3878–3887. 10.1021/nn200250v. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Coulston R. J.; Biedermann F.; Mahajan S.; Baumberg J. J.; Scherman O. A. In situ SERS Monitoring of Photochemistry within a Nanojunction Reactor. Nano Lett. 2013, 13, 5985–5990. 10.1021/nl403164c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigle D. O.; Kasera S.; Herrmann L. O.; Palma A.; de Nijs B.; Benz F.; Mahajan S.; Baumberg J. J.; Scherman O. A. Observing Single Molecules Complexing with Cucurbit[7]uril through Nanogap Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 704–710. 10.1021/acs.jpclett.5b02535. [DOI] [PubMed] [Google Scholar]
- Lee H. K.; Lee Y. H.; Koh C. S. L.; Phan-Quang G. C.; Han X.; Lay C. L.; Sim H. Y. F.; Kao Y. C.; An Q.; Ling X. Y. Designing Surface-Enhanced Raman Scattering (SERS) Platforms beyond Hotspot Engineering: Emerging Opportunities in Analyte Manipulations and Hybrid Materials. Chem. Soc. Rev. 2019, 48, 731–756. 10.1039/C7CS00786H. [DOI] [PubMed] [Google Scholar]
- Brus L. Noble Metal Nanocrystals: Plasmon Electron Transfer Photochemistry and Single-Molecule Raman Spectroscopy. Acc. Chem. Res. 2008, 41, 1742–1749. 10.1021/ar800121r. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Lee K. S.; Shin H. H.; Koo J. J.; Yeon G. J.; Kim Z. H. Single-Molecule Surface-Enhanced Raman Scattering as a Probe of Single-Molecule Surface Reactions: Promises and Current Challenges. Acc. Chem. Res. 2019, 52, 3008–3017. 10.1021/acs.accounts.9b00358. [DOI] [PubMed] [Google Scholar]
- Xie W.; Schlucker S. Surface-Enhanced Raman Spectroscopic Detection of Molecular Chemo- and Plasmo-Catalysis on Noble Metal Nanoparticles. Chem. Commun. 2018, 54, 2326–2336. 10.1039/C7CC07951F. [DOI] [PubMed] [Google Scholar]
- Lombardi J. R.; Birke R. L.; Haran G. Single Molecule SERS Spectral Blinking and Vibronic Coupling. J. Phys. Chem. C 2011, 115, 4540–4545. 10.1021/jp111345u. [DOI] [Google Scholar]
- Itoh T.; Hashimoto K.; Biju V.; Ishikawa M.; Wood B. R.; Ozaki Y. Elucidation of Interaction between Metal-Fee Tetraphenylporphine and Surface Ag Atoms through Temporal Fluctuation of Surface-Enhanced Resonance Raman Scattering and Background-Light Emission. J. Phys. Chem. B 2006, 110, 9579–9585. 10.1021/jp0609939. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Libisch F.; Large N.; Neumann O.; Brown L. V.; Cheng J.; Lassiter J. B.; Carter E. A.; Nordlander P.; Halas N. J. Hot Electrons Do the Impossible: Plasmon-Induced Dissociation of H2 on Au. Nano Lett. 2013, 13, 240–247. 10.1021/nl303940z. [DOI] [PubMed] [Google Scholar]
- Yu S.; Wilson A. J.; Kumari G.; Zhang X.; Jain P. K. Opportunities and Challenges of Solar-Energy-Driven Carbon Dioxide to Fuel Conversion with Plasmonic Catalysts. ACS Energy Lett. 2017, 2, 2058–2070. 10.1021/acsenergylett.7b00640. [DOI] [Google Scholar]
- Ingram D. B.; Linic S. Water Splitting on Composite Plasmonic-Metal/Semiconductor Photoelectrodes: Evidence for Selective Plasmon-Induced Formation of Charge Carriers Near the Semiconductor Surface. J. Am. Chem. Soc. 2011, 133, 5202–5205. 10.1021/ja200086g. [DOI] [PubMed] [Google Scholar]
- Oshikiri T.; Ueno K.; Misawa H. Plasmon-Induced Ammonia Synthesis through Nitrogen Photofixation with Visible Light Irradiation. Angew. Chem., Int. Ed. Engl. 2014, 53, 9802–9805. 10.1002/anie.201404748. [DOI] [PubMed] [Google Scholar]
- Christopher P.; Xin H.; Linic S. Visible-Light-Enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures. Nat. Chem. 2011, 3, 467–472. 10.1038/nchem.1032. [DOI] [PubMed] [Google Scholar]
- Zhan C.; Chen X. J.; Huang Y. F.; Wu D. Y.; Tian Z. Q. Plasmon-Mediated Chemical Reactions on Nanostructures Unveiled by Surface-Enhanced Raman Spectroscopy. Acc. Chem. Res. 2019, 52, 2784–2792. 10.1021/acs.accounts.9b00280. [DOI] [PubMed] [Google Scholar]
- Li S.; Miao P.; Zhang Y.; Wu J.; Zhang B.; Du Y.; Han X.; Sun J.; Xu P. Recent Advances in Plasmonic Nanostructures for Enhanced Photocatalysis and Electrocatalysis. Adv. Mater. 2021, 33, e2000086 10.1002/adma.202000086. [DOI] [PubMed] [Google Scholar]
- Cai Z. F.; Merino J. P.; Fang W.; Kumar N.; Richardson J. O.; De Feyter S.; Zenobi R. Molecular-Level Insights on Reactive Arrangement in On-Surface Photocatalytic Coupling Reactions Using Tip-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2022, 144, 538–546. 10.1021/jacs.1c11263. [DOI] [PubMed] [Google Scholar]
- Huang Y. F.; Wang W.; Guo H. Y.; Zhan C.; Duan S.; Zhan D.; Wu D. Y.; Ren B.; Tian Z. Q. Microphotoelectrochemical Surface-Enhanced Raman Spectroscopy: Toward Bridging Hot-Electron Transfer with a Molecular Reaction. J. Am. Chem. Soc. 2020, 142, 8483–8489. 10.1021/jacs.0c02523. [DOI] [PubMed] [Google Scholar]
- de Nijs B.; Benz F.; Barrow S. J.; Sigle D. O.; Chikkaraddy R.; Palma A.; Carnegie C.; Kamp M.; Sundararaman R.; Narang P.; et al. Plasmonic Tunnel Junctions for Single-Molecule Redox Chemistry. Nat. Commun. 2017, 8, 994. 10.1038/s41467-017-00819-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczerbinski J.; Gyr L.; Kaeslin J.; Zenobi R. Plasmon-Driven Photocatalysis Leads to Products Known from E-beam and X-ray-Induced Surface Chemistry. Nano Lett. 2018, 18, 6740–6749. 10.1021/acs.nanolett.8b02426. [DOI] [PubMed] [Google Scholar]
- Ferrari A. C.; Basko D. M. Raman Spectroscopy as a Versatile Tool for Studying the Properties of Graphene. Nat. Nanotechnol. 2013, 8, 235–246. 10.1038/nnano.2013.46. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Biju V.; Tamaru H.; Wakida S.-I. Fluctuating Single sp2 Carbon Clusters at Single Hotspots of Silver Nanoparticle Dimers Investigated by Surface-Enhanced Resonance Raman Scattering. AIP Advances 2015, 5, 127113. 10.1063/1.4937936. [DOI] [Google Scholar]
- Hertzog M.; Wang M.; Mony J.; Borjesson K. Strong Light-Matter Interactions: A New Direction within Chemistry. Chem. Soc. Rev. 2019, 48, 937–961. 10.1039/C8CS00193F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro R. F.; Martinez-Martinez L. A.; Du M.; Campos-Gonzalez-Angulo J.; Yuen-Zhou J. Polariton Chemistry: Controlling Molecular Dynamics with Optical Cavities. Chem. Sci. 2018, 9, 6325–6339. 10.1039/C8SC01043A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbesen T. W. Hybrid Light-Matter States in a Molecular and Material Science Perspective. Acc. Chem. Res. 2016, 49, 2403–2412. 10.1021/acs.accounts.6b00295. [DOI] [PubMed] [Google Scholar]
- Galego J.; Garcia-Vidal F. J.; Feist J. Suppressing Photochemical Reactions with Quantized Light Fields. Nat. Commun. 2016, 7, 13841. 10.1038/ncomms13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Ueno K.; Oshikiri T.; Sun Q.; Sasaki K.; Misawa H. Enhanced Water Splitting under Modal Strong Coupling Conditions. Nat. Nanotechnol. 2018, 13, 953–958. 10.1038/s41565-018-0208-x. [DOI] [PubMed] [Google Scholar]
- Torma P.; Barnes W. L. Strong Coupling between Surface Plasmon Polaritons and Emitters: A Review. Rep. Prog. Phys. 2015, 78, 013901. 10.1088/0034-4885/78/1/013901. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Etchegoin P. G.; Grand J.; Felidj N.; Aubard J.; Levi G. Mechanisms of Spectral Profile Modification in Surface-Enhanced Fluorescence. J. Phys. Chem. C 2007, 111, 16076–16079. 10.1021/jp076003g. [DOI] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Itzkan I. I.; Dasari R. R.; Feld M. S. Population Pumping of Excited Vibrational States by Spontaneous Surface-Enhanced Raman Scattering. Phys. Rev. Lett. 1996, 76, 2444–2447. 10.1103/PhysRevLett.76.2444. [DOI] [PubMed] [Google Scholar]
- Maher R. C.; Galloway C. M.; Le Ru E. C.; Cohen L. F.; Etchegoin P. G. Vibrational Pumping in Surface Enhanced Raman Scattering (SERS). Chem. Soc. Rev. 2008, 37, 965–79. 10.1039/b707870f. [DOI] [PubMed] [Google Scholar]
- Wei H.; Yan X.; Niu Y.; Li Q.; Jia Z.; Xu H. Plasmon-Exciton Interactions: Spontaneous Emission and Strong Coupling. Adv. Funct. Mater. 2021, 31, 2100889. 10.1002/adfm.202100889. [DOI] [Google Scholar]
- Wu X.; Gray S. K.; Pelton M. Quantum-Dot-Induced Transparency in a Nanoscale Plasmonic Resonator. Opt. Express 2010, 18, 23633–23645. 10.1364/OE.18.023633. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Tamaru H.; Biju V.; Wakida S.-i.; Ozaki Y. Single-Molecular Surface-Enhanced Resonance Raman Scattering as a Quantitative Probe of Local Electromagnetic Field: The Case of Strong Coupling between Plasmonic and Excitonic Resonance. Phys. Rev. B 2014, 89, 195436. 10.1103/PhysRevB.89.195436. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Okamoto T. Anti-crossing Property of Strong Coupling System of Silver Nanoparticle Dimers Coated with Thin Dye Molecular Films Analyzed by Electromagnetism. J. Chem. Phys. 2020, 152, 054710. 10.1063/1.5133875. [DOI] [PubMed] [Google Scholar]
- Wersall M.; Cuadra J.; Antosiewicz T. J.; Balci S.; Shegai T. Observation of Mode Splitting in Photoluminescence of Individual Plasmonic Nanoparticles Strongly Coupled to Molecular Excitons. Nano Lett. 2017, 17, 551–558. 10.1021/acs.nanolett.6b04659. [DOI] [PubMed] [Google Scholar]
- Neuman T.; Aizpurua J.; Esteban R. Quantum Theory of Surface-Enhanced Resonant Raman Scattering (SERRS) of Molecules in Strongly Coupled Plasmon-Exciton Systems. Nanophotonics 2020, 9, 295–308. 10.1515/nanoph-2019-0336. [DOI] [Google Scholar]
- Rossi T. P.; Shegai T.; Erhart P.; Antosiewicz T. J. Strong Plasmon-Molecule Coupling at the Nanoscale Revealed by First-Principles Modeling. Nat. Commun. 2019, 10, 3336. 10.1038/s41467-019-11315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppin R. Optical-Absorption of a Coated Sphere above a Substrate. Physica A 1991, 178, 195–205. 10.1016/0378-4371(91)90080-V. [DOI] [Google Scholar]
- Murata N.; Hata R.; Ishihara H. Crossover between Energy Transparency Resonance and Rabi Splitting in Antenna-Molecule Coupled Systems. J. Phys. Chem. C 2015, 119, 25493–25498. 10.1021/acs.jpcc.5b08590. [DOI] [Google Scholar]
- Zengin G.; Gschneidtner T.; Verre R.; Shao L.; Antosiewicz T. J.; Moth-Poulsen K.; Käll M.; Shegai T. Evaluating Conditions for Strong Coupling between Nanoparticle Plasmons and Organic Dyes Using Scattering and Absorption Spectroscopy. J. Phys. Chem. C 2016, 120, 20588–20596. 10.1021/acs.jpcc.6b00219. [DOI] [Google Scholar]
- Chikkaraddy R.; de Nijs B.; Benz F.; Barrow S. J.; Scherman O. A.; Rosta E.; Demetriadou A.; Fox P.; Hess O.; Baumberg J. J. Single-Molecule Strong Coupling at Room Temperature in Plasmonic Nanocavities. Nature 2016, 535, 127–130. 10.1038/nature17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton M.; Storm S. D.; Leng H. Strong Coupling of Emitters to Single Plasmonic Nanoparticles: Exciton-Induced Transparency and Rabi Splitting. Nanoscale 2019, 11, 14540–14552. 10.1039/C9NR05044B. [DOI] [PubMed] [Google Scholar]
- Zengin G.; Johansson G.; Johansson P.; Antosiewicz T. J.; Kall M.; Shegai T. Approaching the Strong Coupling Limit in Single Plasmonic Nanorods Interacting with J-aggregates. Sci. Rep. 2013, 3, 3074. 10.1038/srep03074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengin G.; Wersall M.; Nilsson S.; Antosiewicz T. J.; Kall M.; Shegai T. Realizing Strong Light-Matter Interactions between Single-Nanoparticle Plasmons and Molecular Excitons at Ambient Conditions. Phys. Rev. Lett. 2015, 114, 157401. 10.1103/PhysRevLett.114.157401. [DOI] [PubMed] [Google Scholar]
- Munkhbat B.; Wersall M.; Baranov D. G.; Antosiewicz T. J.; Shegai T. Suppression of Photo-Oxidation of Organic Chromophores by Strong Coupling to Plasmonic Nanoantennas. Sci. Adv. 2018, 4, eaas9552 10.1126/sciadv.aas9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Han Z.; Huang Y.; Liang K.; Wang X.; Wu F.; Qi X.; Shang Y.; Yu L.; Ding B. Strong Plasmon-Exciton Coupling in Bimetallic Nanorings and Nanocuboids. J. Mater. Chem. C 2020, 8, 7672–7678. 10.1039/D0TC01837F. [DOI] [Google Scholar]
- Zhang W.; You J. B.; Liu J.; Xiong X.; Li Z.; Png C. E.; Wu L.; Qiu C. W.; Zhou Z. K. Steering Room-Temperature Plexcitonic Strong Coupling: A Diexcitonic Perspective. Nano Lett. 2021, 21, 8979–8986. 10.1021/acs.nanolett.1c02248. [DOI] [PubMed] [Google Scholar]
- Liu R.; Zhou Z. K.; Yu Y. C.; Zhang T.; Wang H.; Liu G.; Wei Y.; Chen H.; Wang X. H. Strong Light-Matter Interactions in Single Open Plasmonic Nanocavities at the Quantum Optics Limit. Phys. Rev. Lett. 2017, 118, 237401. 10.1103/PhysRevLett.118.237401. [DOI] [PubMed] [Google Scholar]
- Nagasawa F.; Takase M.; Murakoshi K. Raman Enhancement via Polariton States Produced by Strong Coupling between a Localized Surface Plasmon and Dye Excitons at Metal Nanogaps. J. Phys. Chem. Lett. 2014, 5, 14–19. 10.1021/jz402243a. [DOI] [PubMed] [Google Scholar]
- Huang J.; Traverso A. J.; Yang G.; Mikkelsen M. H. Real-Time Tunable Strong Coupling: From Individual Nanocavities to Metasurfaces. ACS Photonics 2019, 6, 838–843. 10.1021/acsphotonics.8b01743. [DOI] [Google Scholar]
- Schlather A. E.; Large N.; Urban A. S.; Nordlander P.; Halas N. J. Near-Field Mediated Plexcitonic Coupling and Giant Rabi Splitting in Individual Metallic Dimers. Nano Lett. 2013, 13, 3281–3286. 10.1021/nl4014887. [DOI] [PubMed] [Google Scholar]
- Santhosh K.; Bitton O.; Chuntonov L.; Haran G. Vacuum Rabi splitting in a plasmonic cavity at the single quantum emitter limit. Nat. Commun. 2016, 7, 11823. 10.1038/ncomms11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller E. M.; Argyropoulos C.; Hogele A.; Liedl T.; Pilo-Pais M. Plasmon-Exciton Coupling Using DNA Templates. Nano Lett. 2016, 16, 5962–5966. 10.1021/acs.nanolett.6b03015. [DOI] [PubMed] [Google Scholar]
- Gross H.; Hamm J. M.; Tufarelli T.; Hess O.; Hecht B. Near-Field Strong Coupling of Single Quantum Dots. Sci. Adv. 2018, 4, eaar4906 10.1126/sciadv.aar4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Wang Y.; Liu M.; Zhu H.; Chen O.; Zou S.; Zhao J. Colloidal Assembly of Au-Quantum Dot-Au Sandwiched Nanostructures with Strong Plasmon-Exciton Coupling. J. Phys. Chem. Lett. 2020, 11, 2449–2456. 10.1021/acs.jpclett.0c00110. [DOI] [PubMed] [Google Scholar]
- Bitton O.; Gupta S. N.; Houben L.; Kvapil M.; Krapek V.; Sikola T.; Haran G. Vacuum Rabi Splitting of a Dark Plasmonic Cavity Mode Revealed by Fast Electrons. Nat. Commun. 2020, 11, 487. 10.1038/s41467-020-14364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng H.; Szychowski B.; Daniel M. C.; Pelton M. Strong Coupling and Induced Transparency at Room Temperature with Single Quantum Dots and Gap Plasmons. Nat. Commun. 2018, 9, 4012. 10.1038/s41467-018-06450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Chen Y. H.; Qin J.; Zhao D.; Ding B.; Blaikie R. J.; Qiu M. Mode Modification of Plasmonic Gap Resonances Induced by Strong Coupling with Molecular Excitons. Nano Lett. 2017, 17, 3246–3251. 10.1021/acs.nanolett.7b00858. [DOI] [PubMed] [Google Scholar]
- Ojambati O. S.; Chikkaraddy R.; Deacon W. D.; Horton M.; Kos D.; Turek V. A.; Keyser U. F.; Baumberg J. J. Quantum Electrodynamics at Room Temperature Coupling a Single Vibrating Molecule with a Plasmonic Nanocavity. Nat. Commun. 2019, 10, 1049. 10.1038/s41467-019-08611-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen J. M.; Tserkezis C.; Cai Q.; Li L. H.; Kim J. M.; Lee G.; Yi G. R.; Hendren W. R.; Santos E. J. G.; Bowman R. M.; et al. Strong Coupling of Carbon Quantum Dots in Plasmonic Nanocavities. ACS Appl. Mater. Interfaces 2020, 12, 19866–19873. 10.1021/acsami.0c03312. [DOI] [PubMed] [Google Scholar]
- Li L.; Wang L.; Du C.; Guan Z.; Xiang Y.; Wu W.; Ren M.; Zhang X.; Tang A.; Cai W.; Xu J. Ultrastrong Coupling of CdZnS/ZnS Quantum Dots to Bonding Breathing Plasmons of Aluminum Metal-Insulator-Metal Nanocavities in Near-Ultraviolet Spectrum. Nanoscale 2020, 12, 3112–3120. 10.1039/C9NR08048A. [DOI] [PubMed] [Google Scholar]
- Cade N. I.; Ritman-Meer T.; Richards D. Strong Coupling of Localized Plasmons and Molecular Excitons in Nanostructured Silver Films. Phys. Rev. B 2009, 79, 241404(R) 10.1103/PhysRevB.79.241404. [DOI] [Google Scholar]
- Kato F.; Minamimoto H.; Nagasawa F.; Yamamoto Y. S.; Itoh T.; Murakoshi K. Active Tuning of Strong Coupling States between Dye Excitons and Localized Surface Plasmons via Electrochemical Potential Control. ACS Photonics 2018, 5, 788–796. 10.1021/acsphotonics.7b00841. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Okamoto T. Absorption Cross-Section Spectroscopy of a Single Strong-Coupling System between Plasmon and Molecular Exciton Resonance Using a Single Silver Nanoparticle Dimer Generating Surface-Enhanced Resonant Raman Scattering. Phys. Rev. B 2019, 99, 235409. 10.1103/PhysRevB.99.235409. [DOI] [Google Scholar]
- Itoh T.; Yamamoto Y. S.; Tamaru H.; Biju V.; Murase N.; Ozaki Y. Excitation Laser Energy Dependence of Surface-Enhanced Fluorescence Showing Plasmon-Induced Ultrafast Electronic Dynamics in Dye Molecules. Phys. Rev. B 2013, 87, 235408. 10.1103/PhysRevB.87.235408. [DOI] [Google Scholar]
- Bergfield J. P.; Hendrickson J. R. Signatures of Plexcitonic States in Molecular Electroluminescence. Sci. Rep. 2018, 8, 2314. 10.1038/s41598-018-19382-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G.; Luo Y. Electroluminescence of Molecules in a Scanning Tunneling Microscope: Role of Tunneling Electrons and Surface Plasmons. Phys. Rev. B 2011, 84, 205419. 10.1103/PhysRevB.84.205419. [DOI] [PubMed] [Google Scholar]
- Cho C. H.; Aspetti C. O.; Park J.; Agarwal R. Silicon Coupled with Plasmon Nanocavity Generates Bright Visible Hot-Luminescence. Nat. Photonics 2013, 7, 285–289. 10.1038/nphoton.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C. H.; Aspetti C. O.; Turk M. E.; Kikkawa J. M.; Nam S. W.; Agarwal R. Tailoring Hot-Exciton Emission and Lifetimes in Semiconducting Nanowires via Whispering-Gallery Nanocavity Plasmons. Nat. Mater. 2011, 10, 669–675. 10.1038/nmat3067. [DOI] [PubMed] [Google Scholar]
- Bingi J.; S V.; Warrier A. R.; Vijayan C. Plasmonically Tunable Blue-Shifted Emission from Coumarin 153 in Ag Nanostructure Random Media: A Demonstration of Fast Dynamic Surface-Enhanced Fluorescence. Plasmonics 2014, 9, 349–355. 10.1007/s11468-013-9631-x. [DOI] [Google Scholar]
- Aspetti C. O.; Cho C. H.; Agarwal R.; Agarwal R. Studies of Hot Photoluminescence in Plasmonically Coupled Silicon via Variable Energy Excitation and Temperature-Dependent Spectroscopy. Nano Lett. 2014, 14, 5413–5422. 10.1021/nl502606q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gökbulut B.; Topcu G.; Demir M. M.; Inci M. N. Plasmon-Induced Spectral Tunability of Perovskite Nanowires. Opt. Mater. 2021, 122, 111702. 10.1016/j.optmat.2021.111702. [DOI] [Google Scholar]
- Dong Z. C.; Zhang X. L.; Gao H. Y.; Luo Y.; Zhang C.; Chen L. G.; Zhang R.; Tao X.; Zhang Y.; Yang J. L.; et al. Generation of Molecular Hot Electroluminescence by Resonant Nanocavity Plasmons. Nat. Photonics 2010, 4, 50–54. 10.1038/nphoton.2009.257. [DOI] [Google Scholar]
- Chen G.; Li X. G.; Zhang Z. Y.; Dong Z. C. Molecular Hot Electroluminescence Due to Strongly Enhanced Spontaneous Emission Rates in a Plasmonic Nanocavity. Nanoscale 2015, 7, 2442–2449. 10.1039/C4NR06519K. [DOI] [PubMed] [Google Scholar]
- Chong M. C.; Sosa-Vargas L.; Bulou H.; Boeglin A.; Scheurer F.; Mathevet F.; Schull G. Ordinary and Hot Electroluminescence from Single-Molecule Devices: Controlling the Emission Color by Chemical Engineering. Nano Lett. 2016, 16, 6480–6484. 10.1021/acs.nanolett.6b02997. [DOI] [PubMed] [Google Scholar]
- Tian X.-J.; Kong F.-F.; Yu Y.-J.; Jing S.-H.; Zhang X.-B.; Liao Y.; Zhang Y.; Zhang Y.; Dong Z.-C. Plasmon-Enhanced S-2 Electroluminescence from the High-Lying Excited State of a Single Porphyrin Molecule. Appl. Phys. Lett. 2020, 117, 243301. 10.1063/5.0027291. [DOI] [Google Scholar]
- Crim F. F. Bond-Selected Chemistry: Vibrational State Control of Photodissociation and Bimolecular Reaction. J. Phys. Chem. 1996, 100, 12725–12734. 10.1021/jp9604812. [DOI] [Google Scholar]
- Laubereau A.; Kaiser W. Vibrational Dynamics of Liquids and Solids Investigated by Picosecond Light Pulses. Rev. Mod. Phys. 1978, 50, 607–665. 10.1103/RevModPhys.50.607. [DOI] [Google Scholar]
- Haslett T. L.; Tay L.; Moskovits M. Can Surface-Enhanced Raman Scattering Serve as a Channel for Strong Optical Pumping?. J. Chem. Phys. 2000, 113, 1641–1646. 10.1063/1.481952. [DOI] [Google Scholar]
- Brolo A. G.; Sanderson A. C.; Smith A. P. Ratio of the Surface-Enhanced Anti-Stokes Scattering to the Surface-Enhanced Stokes-Raman Scattering for Molecules Adsorbed on a Silver Electrode. Phys. Rev. B 2004, 69, 045424. 10.1103/PhysRevB.69.045424. [DOI] [Google Scholar]
- Maher R. C.; Etchegoin P. G.; Le Ru E. C.; Cohen L. F. A Conclusive Demonstration of Vibrational Pumping under Surface Enhanced Raman Scattering Conditions. J. Phys. Chem. B 2006, 110, 11757–11760. 10.1021/jp060306d. [DOI] [PubMed] [Google Scholar]
- Maher R. C.; Cohen L. F.; Le Ru E. C.; Etchegoin P. G. On the Experimental Estimation of Surface Enhanced Raman Scattering (SERS) Cross Sections by Vibrational Pumping. J. Phys. Chem. B 2006, 110, 19469–19478. 10.1021/jp0626521. [DOI] [PubMed] [Google Scholar]
- Etchegoin P. G.; Le Ru E. C.; Maher R. C.; Cohen L. F. Enhancement Factor Averaging and the Photostability of Probes in SERS Vibrational Pumping. Phys. Chem. Chem. Phys. 2007, 9, 4923–4929. 10.1039/b706395d. [DOI] [PubMed] [Google Scholar]
- Galloway C. M.; Le Ru E. C.; Etchegoin P. G. Single-Molecule Vibrational Pumping in SERS. Phys. Chem. Chem. Phys. 2009, 11, 7372–7380. 10.1039/b904638k. [DOI] [PubMed] [Google Scholar]
- Kozich V.; Werncke W. The Vibrational Pumping Mechanism in Surface-Enhanced Raman Scattering: A Subpicosecond Time-Resolved Study. J. Phys. Chem. C 2010, 114, 10484–10488. 10.1021/jp101219e. [DOI] [Google Scholar]
- Roelli P.; Galland C.; Piro N.; Kippenberg T. J. Molecular Cavity Optomechanics as a Theory of Plasmon-Enhanced Raman Scattering. Nat. Nanotechnol. 2016, 11, 164–169. 10.1038/nnano.2015.264. [DOI] [PubMed] [Google Scholar]
- Pozzi E. A.; Zrimsek A. B.; Lethiec C. M.; Schatz G. C.; Hersam M. C.; Van Duyne R. P. Evaluating Single-Molecule Stokes and Anti-Stokes SERS for Nanoscale Thermometry. J. Phys. Chem. C 2015, 119, 21116–21124. 10.1021/acs.jpcc.5b08054. [DOI] [Google Scholar]
- Schmidt M. K.; Esteban R.; Benz F.; Baumberg J. J.; Aizpurua J. Linking Classical and Molecular Optomechanics Descriptions of SERS. Faraday Discuss. 2017, 205, 31–65. 10.1039/C7FD00145B. [DOI] [PubMed] [Google Scholar]
- Schmidt M. K.; Esteban R.; Gonzalez-Tudela A.; Giedke G.; Aizpurua J. Quantum Mechanical Description of Raman Scattering from Molecules in Plasmonic Cavities. ACS Nano 2016, 10, 6291–6298. 10.1021/acsnano.6b02484. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Esteban R.; Boto R. A.; Urbieta M.; Arrieta X.; Shan C.; Li S.; Baumberg J. J.; Aizpurua J. Addressing Molecular Optomechanical Effects in Nanocavity-Enhanced Raman Scattering Beyond the Single Plasmonic Mode. Nanoscale 2021, 13, 1938–1954. 10.1039/D0NR06649D. [DOI] [PubMed] [Google Scholar]
- Crampton K. T.; Fast A.; Potma E. O.; Apkarian V. A. Junction Plasmon Driven Population Inversion of Molecular Vibrations: A Picosecond Surface-Enhanced Raman Spectroscopy Study. Nano Lett. 2018, 18, 5791–5796. 10.1021/acs.nanolett.8b02438. [DOI] [PubMed] [Google Scholar]
- Ward D. R.; Corley D. A.; Tour J. M.; Natelson D. Vibrational and Electronic Heating in Nanoscale Junctions. Nat. Nanotechnol. 2011, 6, 33–38. 10.1038/nnano.2010.240. [DOI] [PubMed] [Google Scholar]
- Ayars E. J.; Hallen H. D.; Jahncke C. L. Electric Field Gradient Effects in Raman Spectroscopy. Phys. Rev. Lett. 2000, 85, 4180–4183. 10.1103/PhysRevLett.85.4180. [DOI] [PubMed] [Google Scholar]
- Duan S.; Tian G.; Ji Y.; Shao J.; Dong Z.; Luo Y. Theoretical Modeling of Plasmon-Enhanced Raman Images of a Single Molecule with Subnanometer Resolution. J. Am. Chem. Soc. 2015, 137, 9515–9518. 10.1021/jacs.5b03741. [DOI] [PubMed] [Google Scholar]
- Sass J. K.; Neff H.; Moskovits M.; Holloway S. Electric-Field Gradient Effects on the Spectroscopy of Adsorbed Molecules. J. Phys. Chem. 1981, 85, 621–623. 10.1021/j150606a002. [DOI] [Google Scholar]
- Moskovits M.; DiLella D. P. Surface Enhanced Raman Spectroscopy of Benzene and Benzene-d6 Adsorbed on Silver. J. Chem. Phys. 1980, 73, 6068–6075. 10.1063/1.440142. [DOI] [Google Scholar]
- Moskovits M.; DiLella D. P. Intense Quadrupole Transitions in the Spectra of Molecules Near Metal Surfaces. J. Chem. Phys. 1982, 77, 1655–1660. 10.1063/1.444008. [DOI] [Google Scholar]
- Takase M.; Ajiki H.; Mizumoto Y.; Komeda K.; Nara M.; Nabika H.; Yasuda S.; Ishihara H.; Murakoshi K. Selection-Rule Breakdown in Plasmon-Induced Electronic Excitation of an Isolated Single-Walled Carbon Nanotube. Nat. Photonics 2013, 7, 550–554. 10.1038/nphoton.2013.129. [DOI] [Google Scholar]
- Aikens C. M.; Madison L. R.; Schatz G. C. The Effect of Field Gradient on SERS. Nat. Photonics 2013, 7, 508–510. 10.1038/nphoton.2013.153. [DOI] [Google Scholar]
- Jorio A.; Mueller N. S.; Reich S. Symmetry-Derived Selection Rules for Plasmon-Enhanced Raman Scattering. Phys. Rev. B 2017, 95, 155409. 10.1103/PhysRevB.95.155409. [DOI] [Google Scholar]
- Kim H. Y.; Kim D. S. Selection Rule Engineering of Forbidden Transitions of a Hydrogen Atom Near a Nanogap. Nanophotonics 2018, 7, 229–236. 10.1515/nanoph-2017-0037. [DOI] [Google Scholar]
- Neuman T.; Esteban R.; Casanova D.; Garcia-Vidal F. J.; Aizpurua J. Coupling of Molecular Emitters and Plasmonic Cavities beyond the Point-Dipole Approximation. Nano Lett. 2018, 18, 2358–2364. 10.1021/acs.nanolett.7b05297. [DOI] [PubMed] [Google Scholar]
- Liu P.; Chulhai D. V.; Jensen L. Single-Molecule Imaging Using Atomistic Near-Field Tip-Enhanced Raman Spectroscopy. ACS Nano 2017, 11, 5094–5102. 10.1021/acsnano.7b02058. [DOI] [PubMed] [Google Scholar]
- Chen X.; Liu P.; Hu Z.; Jensen L. High-Resolution Tip-Enhanced Raman Scattering Probes Sub-Molecular Density Changes. Nat. Commun. 2019, 10, 2567. 10.1038/s41467-019-10618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera N.; Kaminer I.; Zhen B.; Joannopoulos J. D.; Soljacic M. Shrinking Light to Allow Forbidden Transitions on the Atomic Scale. Science 2016, 353, 263–269. 10.1126/science.aaf6308. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Dong Z.-C.; Aizpurua J. Theoretical Treatment of Single-Molecule Scanning Raman Picoscopy in Strongly Inhomogeneous Near Fields. J. Raman Spectrosc. 2021, 52, 296–309. 10.1002/jrs.5991. [DOI] [Google Scholar]
- Wang X.; Huang S.; Hu S.; Yan S.; Ren B. Fundamental Understanding and Applications of Plasmon-Enhanced Raman Spectroscopy. Nat. Rev. Phys. 2020, 2, 253–271. 10.1038/s42254-020-0171-y. [DOI] [Google Scholar]
- Gersten J. I. The Effect of Surface Roughness on Surface Enhanced Raman Scattering. J. Chem. Phys. 1980, 72, 5779–5780. 10.1063/1.439002. [DOI] [Google Scholar]
- Kolhatkar G.; Plathier J.; Ruediger A. Nanoscale Investigation of Materials, Chemical Reactions, and Biological Systems by Tip Enhanced Raman Spectroscopy-A Review. J. Mater. Chem. C 2018, 6, 1307–1319. 10.1039/C7TC05688E. [DOI] [Google Scholar]
- Zhang Y.; Yang B.; Ghafoor A.; Zhang Y.; Zhang Y. F.; Wang R. P.; Yang J. L.; Luo Y.; Dong Z. C.; Hou J. G. Visually Constructing the Chemical Structure of a Single Molecule by Scanning Raman Picoscopy. Nat. Sci. Rev. 2019, 6, 1169–1175. 10.1093/nsr/nwz180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Crampton K. T.; Tallarida N.; Apkarian V. A. Visualizing Vibrational Normal Modes of a Single Molecule with Atomically Confined Light. Nature 2019, 568, 78–82. 10.1038/s41586-019-1059-9. [DOI] [PubMed] [Google Scholar]
- Jaculbia R. B.; Imada H.; Miwa K.; Iwasa T.; Takenaka M.; Yang B.; Kazuma E.; Hayazawa N.; Taketsugu T.; Kim Y. Single-Molecule Resonance Raman Effect in a Plasmonic Nanocavity. Nat. Nanotechnol. 2020, 15, 105–110. 10.1038/s41565-019-0614-8. [DOI] [PubMed] [Google Scholar]
- Aizpurua J.; Apell S. P.; Berndt R. Role of Tip Shape in Light Emission from the Scanning Tunneling Microscope. Phys. Rev. B 2000, 62, 2065–2073. 10.1103/PhysRevB.62.2065. [DOI] [Google Scholar]
- Wang Y. H.; Zheng S.; Yang W. M.; Zhou R. Y.; He Q. F.; Radjenovic P.; Dong J. C.; Li S.; Zheng J.; Yang Z. L.; et al. In situ Raman Spectroscopy Reveals the Structure and Dissociation of Interfacial Water. Nature 2021, 600, 81–85. 10.1038/s41586-021-04068-z. [DOI] [PubMed] [Google Scholar]
- Wang R. P.; Yang B.; Fu Q.; Zhang Y.; Zhu R.; Dong X. R.; Zhang Y.; Wang B.; Yang J. L.; Luo Y.; et al. Raman Detection of Bond Breaking and Making of a Chemisorbed Up-Standing Single Molecule at Single-Bond Level. J. Phys. Chem. Lett. 2021, 12, 1961–1968. 10.1021/acs.jpclett.1c00074. [DOI] [PubMed] [Google Scholar]
- Xu J. Y.; Zhu X.; Tan S. J.; Zhang Y.; Li B.; Tian Y. Z.; Shan H.; Cui X. F.; Zhao A. D.; Dong Z. C.; et al. Determining Structural and Chemical Heterogeneities of Surface Species at the Single-Bond Limit. Science 2021, 371, 818–822. 10.1126/science.abd1827. [DOI] [PubMed] [Google Scholar]
- Hell S. W.; Wichmann J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780–782. 10.1364/OL.19.000780. [DOI] [PubMed] [Google Scholar]
- Moerner W. E.; Kador L. Optical Detection and Spectroscopy of Single Molecules in a Solid. Phys. Rev. Lett. 1989, 62, 2535. 10.1103/PhysRevLett.62.2535. [DOI] [PubMed] [Google Scholar]
- Betzig E. Proposed Method for Molecular Optical Imaging. Opt. Lett. 1995, 20, 237–239. 10.1364/OL.20.000237. [DOI] [PubMed] [Google Scholar]
- Kazuma E.; Jung J.; Ueba H.; Trenary M.; Kim Y. Real-Space and Real-Time Observation of a Plasmon-Induced Chemical Reaction of a Single Molecule. Science 2018, 360, 521–526. 10.1126/science.aao0872. [DOI] [PubMed] [Google Scholar]
- Zhong J.; Jin X.; Meng L.; Wang X.; Su H.-S.; Yang Z.; Williams C. T.; Ren B. Probing the Electronic and Catalytic Properties of a Bimetallic Surface with 3 nm Resolution. Nat. Nanotechnol. 2017, 12, 132. 10.1038/nnano.2016.241. [DOI] [PubMed] [Google Scholar]
- Su H. S.; Feng H. S.; Zhao Q. Q.; Zhang X. G.; Sun J. J.; He Y.; Huang S. C.; Huang T. X.; Zhong J. H.; Wu D. Y.; et al. Probing the Local Generation and Diffusion of Active Oxygen Species on a Pd/Au Bimetallic Surface by Tip-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2020, 142, 1341–1347. 10.1021/jacs.9b10512. [DOI] [PubMed] [Google Scholar]
- Gadelha A. C.; Ohlberg D. A.; Rabelo C.; Neto E. G.; Vasconcelos T. L.; Campos J. L.; Lemos J. S.; Ornelas V.; Miranda D.; Nadas R.; et al. Localization of Lattice Dynamics in Low-Angle Twisted Bilayer Graphene. Nature 2021, 590, 405–409. 10.1038/s41586-021-03252-5. [DOI] [PubMed] [Google Scholar]
- Sheng S. X.; Wu J. B.; Cong X.; Li W. B.; Gou J.; Zhong Q.; Cheng P.; Tan P. H.; Chen L.; Wu K. H. Vibrational Properties of a Monolayer Silicene Sheet Studied by Tip-Enhanced Raman Spectroscopy. Phys. Rev. Lett. 2017, 119, 196803. 10.1103/PhysRevLett.119.196803. [DOI] [PubMed] [Google Scholar]
- He Z.; Han Z. H.; Kizer M.; Linhardt R. J.; Wang X.; Sinyukov A. M.; Wang J. Z.; Deckert V.; Sokolov A. V.; Hu J.; et al. Tip-Enhanced Raman Imaging of Single-Stranded DNA with Single Base Resolution. J. Am. Chem. Soc. 2019, 141, 753–757. 10.1021/jacs.8b11506. [DOI] [PubMed] [Google Scholar]
- Deckert Gaudig T.; Kämmer E.; Deckert V. Tracking of Nanoscale Structural Variations on a Single Amyloid Fibril with Tip-Enhanced Raman Scattering. J. Biophotonics 2012, 5, 215–219. 10.1002/jbio.201100142. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Zhang X. B.; Wang H. F.; Zhang Y.; Jiang S.; Hu C. R.; Zhang Y.; Luo Y.; Dong Z. C. Distinguishing Individual DNA Bases In a Network by Non Resonant Tip Enhanced Raman Scattering. Angew. Chem., Int. Ed. Engl. 2017, 56, 5561–5564. 10.1002/anie.201702263. [DOI] [PubMed] [Google Scholar]
- Abbe E. Beiträge zur Theorie des Mikroskops und der Mikroskopischen Wahrnehmung. Arch. Mikroskop. Anatom. 1873, 9, 413–468. 10.1007/BF02956173. [DOI] [Google Scholar]
- Liu Y.; Zhang X. Metamaterials: A New Frontier of Science and Technology. Chem. Soc. Rev. 2011, 40, 2494–2507. 10.1039/c0cs00184h. [DOI] [PubMed] [Google Scholar]
- Barnes W. L.; Dereux A.; Ebbesen T. W. Surface Plasmon Subwavelength Optics. Nature 2003, 424, 824–830. 10.1038/nature01937. [DOI] [PubMed] [Google Scholar]
- Pohl D. W.; Denk W.; Lanz M. Optical Stethoscopy: Image Recording with Resolution λ/20. Appl. Phys. Lett. 1984, 44, 651–653. 10.1063/1.94865. [DOI] [Google Scholar]
- Lewis A.; Isaacson M.; Harootunian A.; Muray A. Development of a 500 Å Spatial Resolution Light Microscope: I. Light is Efficiently Transmitted Through λ/16 Diameter Apertures. Ultramicroscopy 1984, 13, 227–231. 10.1016/0304-3991(84)90201-8. [DOI] [Google Scholar]
- Zhang W. H.; Fang Z. Y.; Zhu X. Near-Field Raman Spectroscopy with Aperture Tips. Chem. Rev. 2017, 117, 5095–5109. 10.1021/acs.chemrev.6b00337. [DOI] [PubMed] [Google Scholar]
- Hermann R. J.; Gordon M. J. Nanoscale Optical Microscopy and Spectroscopy Using Near-Field Probes. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 365–387. 10.1146/annurev-chembioeng-060817-084150. [DOI] [PubMed] [Google Scholar]
- Anderson N.; Hartschuh A.; Cronin S.; Novotny L. Nanoscale Vibrational Analysis of Single-Walled Carbon Nanotubes. J. Am. Chem. Soc. 2005, 127, 2533–2537. 10.1021/ja045190i. [DOI] [PubMed] [Google Scholar]
- Ichimura T.; Fujii S.; Verma P.; Yano T.; Inouye Y.; Kawata S. Subnanometric Near-Field Raman Investigation in the Vicinity of a Metallic Nanostructure. Phys. Rev. Lett. 2009, 102, 186101. 10.1103/PhysRevLett.102.186101. [DOI] [PubMed] [Google Scholar]
- Steidtner J.; Pettinger B. Tip-Enhanced Raman Spectroscopy and Microscopy on Single Dye Molecules with 15 nm Resolution. Phys. Rev. Lett. 2008, 100, 236101. 10.1103/PhysRevLett.100.236101. [DOI] [PubMed] [Google Scholar]
- Stadler J.; Schmid T.; Zenobi R. Nanoscale Chemical Imaging Using Top-Illumination Tip-Enhanced Raman Spectroscopy. Nano Lett. 2010, 10, 4514–4520. 10.1021/nl102423m. [DOI] [PubMed] [Google Scholar]
- Yano T.-A.; Verma P.; Saito Y.; Ichimura T.; Kawata S. Pressure-Assisted Tip-Enhanced Raman Imaging at a Resolution of a Few Nanometres. Nat. Photonics 2009, 3, 473–477. 10.1038/nphoton.2009.74. [DOI] [Google Scholar]
- Treffer R.; Lin X.; Bailo E.; Deckert-Gaudig T.; Deckert V. Distinction of Nucleobases-a Tip-Enhanced Raman Approach. Beilstein. J. Nanotechnol. 2011, 2, 628–637. 10.3762/bjnano.2.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Chen G.; Ghafoor A.; Zhang Y. F.; Zhang Y.; Zhang Y.; Luo Y.; Yang J. L.; Sandoghdar V.; Aizpurua J.; et al. Sub-Nanometre Resolution in Single-Molecule Photoluminescence Imaging. Nat. Photonics 2020, 14, 693–699. 10.1038/s41566-020-0677-y. [DOI] [Google Scholar]
- Zhu J. Z.; Chen G.; Ijaz T.; Li X. G.; Dong Z. C. Influence of an Atomistic Protrusion at the Tip Apex on Enhancing Molecular Emission in Tunnel Junctions: A Theoretical Study. J. Chem. Phys. 2021, 154, 214706. 10.1063/5.0048440. [DOI] [PubMed] [Google Scholar]
- Urbieta M.; Barbry M.; Zhang Y.; Koval P.; Sánchez-Portal D.; Zabala N.; Aizpurua J. Atomic-Scale Lightning Rod Effect in Plasmonic Picocavities: A Classical View to a Quantum Effect. ACS Nano 2018, 12, 585–595. 10.1021/acsnano.7b07401. [DOI] [PubMed] [Google Scholar]
- Rendell R.; Scalapino D. Surface Plasmons Confined by Microstructures on Tunnel Junctions. Phys. Rev. B 1981, 24, 3276. 10.1103/PhysRevB.24.3276. [DOI] [Google Scholar]
- Becker S. F.; Esmann M.; Yoo K.; Gross P.; Vogelgesang R.; Park N.; Lienau C. Gap-Plasmon-Enhanced Nanofocusing Near-Field Microscopy. ACS Photonics 2016, 3, 223–232. 10.1021/acsphotonics.5b00438. [DOI] [Google Scholar]
- Marchesin F.; Koval P.; Barbry M.; Aizpurua J.; Sanchez-Portal D. Plasmonic Response of Metallic Nanojunctions Driven by Single Atom Motion: Quantum Transport Revealed in Optics. ACS Photonics 2016, 3, 269–277. 10.1021/acsphotonics.5b00609. [DOI] [Google Scholar]
- Barbry M.; Koval P.; Marchesin F.; Esteban R.; Borisov A. G.; Aizpurua J.; Sánchez-Portal D. Atomistic Near-Field Nanoplasmonics: Reaching Atomic-Scale Resolution in Nanooptics. Nano Lett. 2015, 15, 3410–3419. 10.1021/acs.nanolett.5b00759. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhou Q.; Zhang P.; Chen X. W. Bright optical eigenmode of 1 nm 3 mode volume. Phys. Rev. Lett. 2021, 126, 257401. 10.1103/PhysRevLett.126.257401. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Fernandez-Dominguez A.; Wiener A.; Maier S. A.; Pendry J. Surface Plasmons and Nonlocality: A Simple Model. Phys. Rev. Lett. 2013, 111, 093901. 10.1103/PhysRevLett.111.093901. [DOI] [PubMed] [Google Scholar]
- Atkin J. M.; Raschke M. B. Optical Spectroscopy Goes Intramolecular. Nature 2013, 498, 44–45. 10.1038/498044a. [DOI] [PubMed] [Google Scholar]
- Li X.; Xiao D.; Zhang Z. Landau Damping of Quantum Plasmons in Metal Nanostructures. New J. Phys. 2013, 15, 023011. 10.1088/1367-2630/15/2/023011. [DOI] [Google Scholar]
- Kale M. J.; Christopher P. Plasmons at the Interface. Science 2015, 349, 587–588. 10.1126/science.aac8522. [DOI] [PubMed] [Google Scholar]
- Khurgin J.; Tsai W. Y.; Tsai D. P.; Sun G. Landau Damping and Limit to Field Confinement and Enhancement in Plasmonic Dimers. ACS Photonics 2017, 4, 2871–2880. 10.1021/acsphotonics.7b00860. [DOI] [Google Scholar]
- Gonçalves P.; Christensen T.; Rivera N.; Jauho A.-P.; Mortensen N. A.; Soljačić M. Plasmon-Emitter Interactions at the Nanoscale. Nat. Commun. 2020, 11, 1–13. 10.1038/s41467-019-13820-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tserkezis C.; Mortensen N. A.; Wubs M. How Nonlocal Damping Reduces Plasmon-Enhanced Fluorescence in Ultranarrow Gaps. Phys. Rev. B 2017, 96, 085413. 10.1103/PhysRevB.96.085413. [DOI] [Google Scholar]
- Alcaraz Iranzo D.; Nanot S.; Dias E. J.; Epstein I.; Peng C.; Efetov D. K.; Lundeberg M. B.; Parret R.; Osmond J.; Hong J. Y.; et al. Probing the Ultimate Plasmon Confinement Limits with a van der Waals Heterostructure. Science 2018, 360, 291–295. 10.1126/science.aar8438. [DOI] [PubMed] [Google Scholar]
- Duan S.; Tian G.; Luo Y. Theory for Modeling of High Resolution Resonant and Nonresonant Raman Images. Chem. Theory Comput. 2016, 12, 4986–4995. 10.1021/acs.jctc.6b00592. [DOI] [PubMed] [Google Scholar]
- Kawata S.; Verma P. Optical Nano-Imaging of Materials: Peeping Through Tip-Enhanced Raman Scattering. CHIMIA Inter J. Chem. 2006, 60, 770–776. 10.2533/chimia.2006.770. [DOI] [Google Scholar]
- Meng L.; Yang Z.; Chen J.; Sun M. Effect of Electric Field Gradient on Sub-Nanometer Spatial Resolution of Tip-Enhanced Raman Spectroscopy. Sci. Rep. 2015, 5, 9240. 10.1038/srep09240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Chen X.; Ye H.; Jensen L. Resolving Molecular Structures with High-Resolution Tip-Enhanced Raman Scattering Images. ACS Nano 2019, 13, 9342–9351. 10.1021/acsnano.9b03980. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Chen B. Q.; Li Z. Y. Optical Origin of Subnanometer Resolution in Tip-Enhanced Raman Mapping. J. Phys. Chem. C 2015, 119, 11858–11871. 10.1021/acs.jpcc.5b02653. [DOI] [Google Scholar]
- Latorre F.; Kupfer S.; Bocklitz T.; Kinzel D.; Trautmann S.; Gräfe S.; Deckert V. Spatial Resolution of Tip-Enhanced Raman Spectroscopy-DFT Assessment of the Chemical Effect. Nanoscale 2016, 8, 10229–10239. 10.1039/C6NR00093B. [DOI] [PubMed] [Google Scholar]
- Long D. A.; The Raman effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; Wiley Chichester, 2002; pp 1–584. [Google Scholar]
- McHale J. L.Molecular spectroscopy; CRC Press, 2017; pp 1–475. [Google Scholar]
- Jiang S.; Zhang Y.; Zhang R.; Hu C. R.; Liao M. H.; Luo Y.; Yang J. L.; Dong Z. C.; Hou J. G. Distinguishing Adjacent Molecules on a Surface Using Plasmon-Enhanced Raman Scattering. Nat. Nanotechnol. 2015, 10, 865–869. 10.1038/nnano.2015.170. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Zhang X. B.; Zhang Y.; Hu C. R.; Zhang R.; Zhang Y.; Liao Y.; Smith Z. J.; Dong Z. C.; Hou J. G. Subnanometer-Resolved Chemical Imaging via Multivariate Analysis of Tip-Enhanced Raman Maps. Light. Sci. Appl. 2017, 6, e17098 10.1038/lsa.2017.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N.; Chen X.; Goubert G.; Chulhai D. V.; Chen X.; Pozzi E. A.; Jiang N.; Hersam M. C.; Seideman T.; Jensen L.; et al. Conformational Contrast of Surface-Mediated Molecular Switches Yields Ångstrom-Scale Spatial Resolution in Ultrahigh Vacuum Tip-Enhanced Raman Spectroscopy. Nano Lett. 2016, 16, 7774–7778. 10.1021/acs.nanolett.6b03958. [DOI] [PubMed] [Google Scholar]
- Li L.; Schultz J. F.; Mahapatra S.; Liu X.; Shaw C.; Zhang X.; Hersam M. C.; Jiang N. Angstrom-Scale Spectroscopic Visualization of Interfacial Interactions in an Organic/Borophene Vertical Heterostructure. J. Am. Chem. Soc. 2021, 143, 15624–15634. 10.1021/jacs.1c04380. [DOI] [PubMed] [Google Scholar]
- Huang T.; Cong X.; Wu S.; Lin K.; Yao X.; He Y. H.; Wu J. B.; Bao Y. F.; Huang S. C.; Wang X.; et al. Probing the Edge-Related Properties of Atomically Thin MoS2 at Nanoscale. Nat. Commun. 2019, 10, 5544. 10.1038/s41467-019-13486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Hammud A.; Wolf M.; Kumagai T. Atomic Point Contact Raman Spectroscopy of a Si(111)-7 × 7 Surface. Nano Lett. 2021, 21, 4057–4061. 10.1021/acs.nanolett.1c00998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Cirera B.; Sun Y.; Hamada I.; Müller M.; Hammud A.; Wolf M.; Kumagai T. Dramatic Enhancement of Tip-Enhanced Raman Scattering Mediated by Atomic Point Contact Formation. Nano Lett. 2020, 20, 5879–5884. 10.1021/acs.nanolett.0c01791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhang R.; Jiang S.; Zhang Y.; Dong Z. C. Probing Adsorption Configurations of Small Molecules on Surfaces by Single Molecule Tip Enhanced Raman Spectroscopy. ChemPhysChem 2019, 20, 37–41. 10.1002/cphc.201800861. [DOI] [PubMed] [Google Scholar]
- Lee J.; Tallarida N.; Chen X.; Jensen L.; Apkarian V. A. Microscopy with a Single-Molecule Scanning Electrometer. Sci. Adv. 2018, 4, eaat5472 10.1126/sciadv.aat5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Duan S.; Wen B.; Li S.; Kathiresan M.; Xie L.; Chen S.; Anema J. R.; Mao B.; Luo Y.; et al. Observation of Inhomogeneous Plasmonic Field Distribution in a Nanocavity. Nat. Nanotechnol. 2020, 15, 922–926. 10.1038/s41565-020-0753-y. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang Y.; Dong Z. C. Scanning Raman picoscopy: Ångström-Resolved Tip-Enhanced Raman Spectromicroscopy. Chin. J. Chem. Phys. 2021, 34, 1–14. 10.1063/1674-0068/cjcp2102027. [DOI] [Google Scholar]
- Zhang C.; Gao B.; Chen L. G.; Meng Q. S.; Yang H.; Zhang R.; Tao X.; Gao H. Y.; Liao Y.; Dong Z. C. Fabrication of Silver Tips for Scanning Tunneling Microscope Induced Luminescence. Rev. Sci. Instrum. 2011, 82, 083101. 10.1063/1.3617456. [DOI] [PubMed] [Google Scholar]
- Ghafoor A.; Yang B.; Yu Y. J.; Zhang Y. F.; Zhang X. B.; Chen G.; Zhang Y.; Zhang Y.; Dong Z. C. Site-Dependent TERS Study of a Porphyrin Molecule on Ag (100) at 7 K. Chin. J. Chem. Phys. 2019, 32, 287–291. 10.1063/1674-0068/cjcp1812280. [DOI] [Google Scholar]
- Li H.; Zhang Y. F.; Zhang X. B.; Farrukh A.; Zhang Y.; Zhang Y.; Dong Z. C. Probing the Deformation of [12] Cycloparaphenylene Molecular Nanohoops Adsorbed on Metal Surfaces by Tip-Enhanced Raman Spectroscopy. J. Chem. Phys. 2020, 153, 244201. 10.1063/5.0033383. [DOI] [PubMed] [Google Scholar]
- Kneipp K. Surface-Enhanced Raman Scattering. Phys. Today 2007, 60, 40–46. 10.1063/1.2812122. [DOI] [Google Scholar]
- Otto A. The ’Chemical’ (Electronic) Contribution to Surface-Enhanced Raman Scattering. J. Raman Spectrosc. 2005, 36, 497–509. 10.1002/jrs.1355. [DOI] [Google Scholar]
- Valley N.; Greeneltch N.; Van Duyne R. P.; Schatz G. C. A Look at the Origin and Magnitude of the Chemical Contribution to the Enhancement Mechanism of Surface-Enhanced Raman Spectroscopy (SERS): Theory and Experiment. J. Phys. Chem. Lett. 2013, 4, 2599–2604. 10.1021/jz4012383. [DOI] [Google Scholar]
- Morton S. M.; Jensen L. Understanding the Molecule-Surface Chemical Coupling in SERS. J. Am. Chem. Soc. 2009, 131, 4090–4098. 10.1021/ja809143c. [DOI] [PubMed] [Google Scholar]
- Jensen L.; Zhao L. L.; Schatz G. C. Size-Dependence of the Enhanced Raman Scattering of Pyridine Adsorbed on Agn (n = 2–8, 20) Clusters. J. Phys. Chem. C 2007, 111, 4756–4764. 10.1021/jp067634y. [DOI] [Google Scholar]
- Lombardi J. R.; Birke R. L. A Unified View of Surface-Enhanced Raman Scattering. Acc. Chem. Res. 2009, 42, 734–742. 10.1021/ar800249y. [DOI] [PubMed] [Google Scholar]
- Ma H.; Chen Y.; Wang H.; Wang X.; Zhang X.; Han X.; He C.; Zhao B. Charge-Transfer Effect on Surface-Enhanced Raman Spectroscopy in Ag/PTCA: Herzberg-Teller Selection Rules. J. Phys. Chem. C 2017, 121, 25788–25794. 10.1021/acs.jpcc.7b07281. [DOI] [Google Scholar]
- Chenal C.; Birke R. L.; Lombardi J. R. Determination of the Degree of Charge-Transfer Contributions to Surface-Enhanced Raman Spectroscopy. ChemPhysChem 2008, 9, 1617–1623. 10.1002/cphc.200800221. [DOI] [PubMed] [Google Scholar]
- Ji W.; Xue X. X.; Ruan W. D.; Wang C. X.; Ji N.; Chen L.; Li Z. S.; Song W.; Zhao B.; Lombardi J. R. Scanned Chemical Enhancement of Surface-enhanced Raman Scattering Using a Charge-Transfer Complex. Chem. Commun. 2011, 47, 2426–2428. 10.1039/C0CC03697H. [DOI] [PubMed] [Google Scholar]
- Ji W.; Kitahama Y.; Xue X. X.; Zhao B.; Ozaki Y. Generation of Pronounced Resonance Profile of Charge-Transfer Contributions to Surface-Enhanced Raman Scattering. J. Phys. Chem. C 2012, 116, 2515–2520. 10.1021/jp209947p. [DOI] [Google Scholar]
- Ji W.; Kitahama Y.; Han X. X.; Xue X. X.; Ozaki Y.; Zhao B. pH-Dependent SERS by Semiconductor-Controlled Charge-Transfer Contribution. J. Phys. Chem. C 2012, 116, 24829–24836. 10.1021/jp308805n. [DOI] [Google Scholar]
- Sun L.; Bai F. Q.; Zhang H. X. Theoretical Investigation of Chemically Enhanced Mechanism of SERS Spectroscopy for Ag/MPH/TiO2 System. Acta Phys.-Chim. Sin. 2011, 27, 1335–1340. 10.3866/PKU.WHXB20110602. [DOI] [Google Scholar]
- Rajh T.; Chen L. X.; Lukas K.; Liu T.; Thurnauer M. C.; Tiede D. M. Surface Restructuring of Nanoparticles: An Efficient Route for Ligand-Metal Oxide Crosstalk. J. Phys. Chem. B 2002, 106, 10543–10552. 10.1021/jp021235v. [DOI] [Google Scholar]
- Alvarez-Puebla R. A.; Liz-Marzán L. M. SERS Detection of Small Inorganic Molecules and Ions. Angew. Chem., Int. Ed. Engl. 2012, 51, 11214–11223. 10.1002/anie.201204438. [DOI] [PubMed] [Google Scholar]
- Ji W.; Li L.; Zhang Y.; Wang X.; Ozaki Y. Recent Advances in Surface-Enhanced Raman Scattering-Based Sensors for the Detection of Inorganic Ions: Sensing Mechanism and Beyond. J. Raman Spectrosc. 2021, 52, 468–481. 10.1002/jrs.5975. [DOI] [Google Scholar]
- Guerrini L.; Alvarez-Puebla R. A. Surface-Enhanced Raman Scattering Sensing of Transition Metal Ions in Waters. ACS Omega 2021, 6, 1054–1063. 10.1021/acsomega.0c05261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoutsi D.; Guerrini L.; Hermida-Ramon J. M.; Giannini V.; Liz-Marzán L. M.; Wei A.; Alvarez-Puebla R. A. Simultaneous SERS Detection of Copper and Cobalt at Ultratrace Levels. Nanoscale 2013, 5, 5841–5846. 10.1039/c3nr01518a. [DOI] [PubMed] [Google Scholar]
- Ling X.; Zhang J. First-Layer Effect in Graphene-Enhanced Raman Scattering. Small 2010, 6, 2020–2025. 10.1002/smll.201000918. [DOI] [PubMed] [Google Scholar]
- Ling X.; Zhang J. Interference Phenomenon in Graphene-Enhanced Raman Scattering. J. Phys. Chem. C 2011, 115, 2835–2840. 10.1021/jp111502n. [DOI] [Google Scholar]
- Lee Y. K.; Jung C. H.; Park J.; Seo H.; Somorjai G. A.; Park J. Y. Surface Plasmon-Driven Hot Electron Flow Probed with Metal-Semiconductor Nanodiodes. Nano Lett. 2011, 11, 4251–4255. 10.1021/nl2022459. [DOI] [PubMed] [Google Scholar]
- Lee S. J.; Moskovits M. Visualizing Chromatographic Separation of Metal Ions on a Surface-Enhanced Raman Active Medium. Nano Lett. 2011, 11, 145–150. 10.1021/nl1031309. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Ai J.; Kumar P.; You E.; Zhou X.; Liu X.; Tian Z.; Bouř P.; Duan Y.; Han L.; et al. Enantiomeric Discrimination by Surface-Enhanced Raman Scattering-Chiral Anisotropy of Chiral Nanostructured Gold Films. Angew. Chem., Int. Ed. Engl. 2020, 59, 15226–15231. 10.1002/anie.202006486. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yu Z.; Ji W.; Tanaka Y.; Sui H.; Zhao B.; Ozaki Y. Enantioselective Discrimination of Alcohols by Hydrogen Bonding: A SERS Study. Angew. Chem., Int. Ed. Engl. 2014, 53, 13866–13870. 10.1002/anie.201407642. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhao X.; Yu Z.; Xu Z.; Zhao B.; Ozaki Y. A Chiral-Label-Free SERS Strategy for the Synchronous Chiral Discrimination and Identification of Small Aromatic Molecules. Angew. Chem., Int. Ed. Engl. 2020, 59, 19079–19086. 10.1002/anie.202007771. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Liu J.; Zhao X.; Yang C.; Ozaki Y.; Xu Z.; Zhao B.; Yu Z. A Chiral Signal-Amplified Sensor for Enantioselective Discrimination of Amino Acids Based on Charge Transfer-Induced SERS. Chem. Commun. 2019, 55, 9697–9700. 10.1039/C9CC04665H. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Liu J.; Ozaki Y.; Xu Z.; Zhao B. Effect of TiO2 on Altering Direction of Interfacial Charge Transfer in a TiO2-Ag-MPY-FePc System by SERS. Angew. Chem., Int. Ed. Engl. 2019, 58, 8172–8176. 10.1002/anie.201900589. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhao B.; Li P.; Han X. X.; Ozaki Y. Charge Transfer at the TiO2/N3/Ag Interface Monitored by Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2017, 121, 5145–5153. 10.1021/acs.jpcc.7b00153. [DOI] [Google Scholar]
- Wang X.; Li P.; Han X. X.; Kitahama Y.; Zhao B.; Ozaki Y. An Enhanced Degree of Charge Transfer in Dye-Sensitized Solar Cells with a ZnO-TiO2/N3/Ag Structure as Revealed by Surface-Enhanced Raman Scattering. Nanoscale 2017, 9, 15303–15313. 10.1039/C7NR03839A. [DOI] [PubMed] [Google Scholar]
- Hayashi S.; Koh R.; Ichiyama Y.; Yamamoto K. Evidence for Surface-Enhanced Raman Scattering on Nonmetallic Surfaces: Copper Phthalocyanine Molecules on GaP Small Particles. Phys. Rev. Lett. 1988, 60, 1085–1088. 10.1103/PhysRevLett.60.1085. [DOI] [PubMed] [Google Scholar]
- Loo B. H. Observation of the Surface Enhanced Raman Scattering Effect from the Semiconductor—Electrolyte Interface. J. Electroanal. Chem. Interfacial Electrochem. 1982, 136, 209–213. 10.1016/0022-0728(82)87037-X. [DOI] [Google Scholar]
- Yamada H.; Yamamoto Y. Surface Enhanced Raman Scattering (SERS) of Chemisorbed Species on Various Kinds of Metals and Semiconductors. Surf. Sci. 1983, 134, 71–90. 10.1016/0039-6028(83)90312-6. [DOI] [Google Scholar]
- Zhang P.; Wang Y.; He T.; Zhang B.; Wang X.; Xin H.; Liu F.-c. SERS of Pyridine, 1,4-Dioxane and 1-Ethyl-3-methyl-2-thiacyanine Iodide Adsorbed on Alfa-Fe2O3 Colloids. Chem. Phys. Lett. 1988, 153, 215–222. 10.1016/0009-2614(88)85215-1. [DOI] [Google Scholar]
- Alessandri I.; Lombardi J. R. Enhanced Raman Scattering with Dielectrics. Chem. Rev. 2016, 116, 14921–14981. 10.1021/acs.chemrev.6b00365. [DOI] [PubMed] [Google Scholar]
- Wang Y.-X.; Wang Y.-F.; Gao Y.; Sun Z.-H.; Zhao C.; Hu H.-L.; Xu W.-Q.; Wang Z.-C.; Zhao B. Surface Enhanced Raman Spectroscopy of 4-Mercaptopyridine Molecules on Pb3O4 Nanoparticles. Chem. Res. Chin. Univ. 2006, 22, 388–389. 10.1016/S1005-9040(06)60123-2. [DOI] [Google Scholar]
- Wang Y.; Hu H.; Jing S.; Wang Y.; Sun Z.; Zhao B.; Zhao C.; Lombardi J. R. Enhanced Raman Scattering as a Probe for 4-Mercaptopyridine Surface-Modified Copper Oxide Nanocrystals. Anal. Sci. 2007, 23, 787–791. 10.2116/analsci.23.787. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Sun Z.; Wang Y.; Hu H.; Zhao B.; Xu W.; Lombardi J. R. Surface-enhanced Raman Scattering on Mercaptopyridine-Capped US Microclusters. Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2007, 66, 1199–1203. 10.1016/j.saa.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang J.; Jia H.; Li M.; Zeng J.; Yang B.; Zhao B.; Xu W.; Lombardi J. R. Mercaptopyridine Surface-Functionalized CdTe Quantum Dots with Enhanced Raman Scattering Properties. J. Phys. Chem. C 2008, 112, 996–1000. 10.1021/jp077467h. [DOI] [Google Scholar]
- Wang Y.; Ruan W.; Zhang J.; Yang B.; Xu W.; Zhao B.; Lombardi J. R. Direct Observation of Surface-Enhanced Raman Scattering in ZnO Nanocrystals. J. Raman Spectrosc. 2009, 40, 1072–1077. 10.1002/jrs.2241. [DOI] [Google Scholar]
- Wang Y. F.; Sun Z. H.; Hu H. L.; Jing S. Y.; Zhao B.; Xu W. Q.; Zhao C.; Lombardi J. R. Raman Scattering Study of Molecules Adsorbed on ZnS Nanocrystals. J. Raman Spectrosc. 2007, 38, 34–38. 10.1002/jrs.1570. [DOI] [Google Scholar]
- Ling X.; Xie L. M.; Fang Y.; Xu H.; Zhang H. L.; Kong J.; Dresselhaus M. S.; Zhang J.; Liu Z. F. Can Graphene be Used as a Substrate for Raman Enhancement?. Nano Lett. 2010, 10, 553–561. 10.1021/nl903414x. [DOI] [PubMed] [Google Scholar]
- Xue X.; Ji W.; Mao Z.; Li Z.; Ruan W.; Zhao B.; Lombardi J. R. Effects of Mn Doping on Surface Enhanced Raman Scattering Properties of TiO2 Nanoparticles. Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2012, 95, 213–217. 10.1016/j.saa.2012.04.101. [DOI] [PubMed] [Google Scholar]
- Xue X.; Ji W.; Mao Z.; Ruan W.; Zhao B.; Ma S.; Lombardi J. R. Study of Enhanced Raman Scattering for Molecules Adsorbed on Cu-doped TiO2 Nanoparticles. Sci. Sin. Chim. 2011, 41, 398–402. 10.1360/032010-679. [DOI] [Google Scholar]
- Xue X.; Ruan W.; Yang L.; Ji W.; Xie Y.; Chen L.; Song W.; Zhao B.; Lombardi J. R. Surface-Enhanced Raman Scattering of Molecules Adsorbed on Co-doped ZnO Nanoparticles. J. Raman Spectrosc. 2012, 43, 61–64. 10.1002/jrs.2988. [DOI] [Google Scholar]
- Xue X. X.; Ji W.; Mao Z.; Li Z. S.; Guo Z. N.; Zhao B.; Zhao C. SERS Study of Co-doped TiO2 Nanoparticles. Chem. Res. Chin. Univ. 2013, 29, 751–754. 10.1007/s40242-013-3051-5. [DOI] [Google Scholar]
- Xue X. X.; Ji W.; Mao Z.; Zhao C.; Zhao B.; Lombardi J. R. Simultaneous Enhancement of Phonons Modes with Molecular Vibrations Due to Mg Doping of a TiO2 Substrate. RSC Adv. 2013, 3, 20891–20895. 10.1039/c3ra43780a. [DOI] [Google Scholar]
- Yang L.; Jiang X.; Yang M. Improvement of Surface-Enhanced Raman Scattering Performance for Broad Band Gap Semiconductor Nanomaterial (TiO2): Strategy of Metal doping. Appl. Phys. Lett. 2011, 99, 111114. 10.1063/1.3638467. [DOI] [Google Scholar]
- Ling X.; Moura L. G.; Pimenta M. A.; Zhang J. Charge-Transfer Mechanism in Graphene-Enhanced Raman Scattering. J. Phys. Chem. C 2012, 116, 25112–25118. 10.1021/jp3088447. [DOI] [Google Scholar]
- Ling X.; Wu J.; Xu W.; Zhang J. Probing the Effect of Molecular Orientation on the Intensity of Chemical Enhancement Using Graphene-Enhanced Raman Spectroscopy. Small 2012, 8, 1365–1372. 10.1002/smll.201102223. [DOI] [PubMed] [Google Scholar]
- Ling X.; Zhang J. Investigation of the Adsorption Behavior of PbPc on Graphene by Raman Spectroscopy. Acta Phys.-Chim. Sin. 2012, 28, 2355–2362. 10.3866/PKU.WHXB201208242. [DOI] [Google Scholar]
- Ling X.; Wu J.; Xie L.; Zhang J. Graphene-Thickness-Dependent Graphene-Enhanced Raman Scattering. J. Phys. Chem. C 2013, 117, 2369–2376. 10.1021/jp310564d. [DOI] [Google Scholar]
- Huang S.; Ling X.; Liang L.; Song Y.; Fang W.; Zhang J.; Kong J.; Meunier V.; Dresselhaus M. S. Molecular Selectivity of Graphene-Enhanced Raman Scattering. Nano Lett. 2015, 15, 2892–2901. 10.1021/nl5045988. [DOI] [PubMed] [Google Scholar]
- Han X. X.; Ji W.; Zhao B.; Ozaki Y. Semiconductor-Enhanced Raman Scattering: Active Nanomaterials and Applications. Nanoscale 2017, 9, 4847–4861. 10.1039/C6NR08693D. [DOI] [PubMed] [Google Scholar]
- Ji W.; Han X. X.; Zhao B.. Charge-Transfer-Induced Enhancement of Raman Scattering Based on Semiconductors. In Recent Developments in Plasmon-Supported Raman Spectroscopy: 45 Years of Enhanced Raman Signals; World Scientific Publishing Europe Ltd., 2018; pp 451–482. [Google Scholar]
- Yang B.; Jin S.; Guo S.; Park Y.; Chen L.; Zhao B.; Jung Y. M. Recent Development of SERS Technology: Semiconductor-Based Study. ACS Omega 2019, 4, 20101–20108. 10.1021/acsomega.9b03154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Guo L. SERS Activity of Semiconductors: Crystalline and Amorphous Nanomaterials. Angew. Chem., Int. Ed. Engl. 2020, 59, 4231–4239. 10.1002/anie.201913375. [DOI] [PubMed] [Google Scholar]
- Ji W.; Li L.; Song W.; Wang X.; Zhao B.; Ozaki Y. Enhanced Raman Scattering by ZnO Superstructures: Synergistic Effect of Charge Transfer and Mie Resonances. Angew. Chem., Int. Ed. Engl. 2019, 58, 14452–14456. 10.1002/anie.201907283. [DOI] [PubMed] [Google Scholar]
- Rodriguez I.; Shi L.; Lu X.; Korgel B. A.; Alvarez-Puebla R. A.; Meseguer F. Silicon Nanoparticles as Raman Scattering Enhancers. Nanoscale 2014, 6, 5666–5670. 10.1039/C4NR00593G. [DOI] [PubMed] [Google Scholar]
- Lombardi J. R.; Birke R. L. Theory of Surface-Enhanced Raman Scattering in Semiconductors. J. Phys. Chem. C 2014, 118, 11120–11130. 10.1021/jp5020675. [DOI] [Google Scholar]
- Song G.; Gong W.; Cong S.; Zhao Z. Ultrathin Two-Dimensional Nanostructures: Surface Defects for Morphology-Driven Enhanced Semiconductor SERS. Angew. Chem., Int. Ed. Engl. 2021, 60, 5505–5511. 10.1002/anie.202015306. [DOI] [PubMed] [Google Scholar]
- Zheng Z.; Cong S.; Gong W.; Xuan J.; Li G.; Lu W.; Geng F.; Zhao Z. Semiconductor SERS Enhancement Enabled by Oxygen Incorporation. Nat. Commun. 2017, 8, 1993. 10.1038/s41467-017-02166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G.; Cong S.; Zhao Z. Defect Engineering in Semiconductor-Based SERS. Chem. Sci. 2022, 13, 1210–1224. 10.1039/D1SC05940H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong S.; Yuan Y.; Chen Z.; Hou J.; Yang M.; Su Y.; Zhang Y.; Li L.; Li Q.; Geng F.; et al. Noble Metal-Comparable SERS Enhancement from Semiconducting Metal Oxides by Making Oxygen Vacancies. Nat. Commun. 2015, 6, 7800. 10.1038/ncomms8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Shi W.; Wang S.; Zhao H.; Lin J.; Yang Z.; Chen M.; Guo L. Two-dimensional Amorphous TiO2 Nanosheets Enabling High-efficiency Photo-induced Charge Transfer for Excellent SERS Activity. J. Am. Chem. Soc. 2019, 141, 5856–5862. 10.1021/jacs.9b00029. [DOI] [PubMed] [Google Scholar]
- Wang X.; Shi W.; Jin Z.; Huang W.; Lin J.; Ma G.; Li S.; Guo L. Remarkable SERS Activity Observed from Amorphous ZnO Nanocages. Angew. Chem., Int. Ed. Engl. 2017, 56, 9851–9855. 10.1002/anie.201705187. [DOI] [PubMed] [Google Scholar]
- Messinger B. J.; von Raben K. U.; Chang R. K.; Barber P. W. Local Fields at the Surface of Noble-Metal Microspheres. Phys. Rev. B 1981, 24, 649–657. 10.1103/PhysRevB.24.649. [DOI] [Google Scholar]
- Bohren C. F.; Huffman D. R.. Absorption and Scattering of Light by Small Partices.; Wiley-Verlag, 1998; pp 1–530. [Google Scholar]
- Swanson N. L.; Billard D. B. Multiple Scattering Efficiency and Optical Extinction. Phys. Rev. E 2000, 61, 4518–4522. 10.1103/PhysRevE.61.4518. [DOI] [PubMed] [Google Scholar]
- Alessandri I. Enhancing Raman Scattering without Plasmons: Unprecedented Sensitivity Achieved by TiO2 Shell-Based Resonators. J. Am. Chem. Soc. 2013, 135, 5541–5544. 10.1021/ja401666p. [DOI] [PubMed] [Google Scholar]
- Qi D.; Lu L.; Wang L.; Zhang J. Improved SERS Sensitivity on Plasmon-Free TiO2 Photonic Microarray by Enhancing Light-Matter Coupling. J. Am. Chem. Soc. 2014, 136, 9886–9889. 10.1021/ja5052632. [DOI] [PubMed] [Google Scholar]
- Qiu B.; Xing M.; Yi Q.; Zhang J. Chiral Carbonaceous Nanotubes Modified with Titania Nanocrystals: Plasmon-Free and Recyclable SERS Sensitivity. Angew. Chem., Int. Ed. Engl. 2015, 54, 10643–10647. 10.1002/anie.201505319. [DOI] [PubMed] [Google Scholar]
- Jain P. K. Plasmon-in-a-Box: On the Physical Nature of Few-Carrier Plasmon Resonances. J. Phys. Chem. Lett. 2014, 5, 3112–3119. 10.1021/jz501456t. [DOI] [PubMed] [Google Scholar]
- Faucheaux J. A.; Stanton A. L. D.; Jain P. K. Plasmon Resonances of Semiconductor Nanocrystals: Physical Principles and New Opportunities. J. Phys. Chem. Lett. 2014, 5, 976–985. 10.1021/jz500037k. [DOI] [PubMed] [Google Scholar]
- Lounis S. D.; Runnerstrom E. L.; Llordés A.; Milliron D. J. Defect Chemistry and Plasmon Physics of Colloidal Metal Oxide Nanocrystals. J. Phys. Chem. Lett. 2014, 5, 1564–1574. 10.1021/jz500440e. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Jain P. K.; Ewers T.; Alivisatos A. P. Localized Surface Plasmon Resonances Arising from Free Carriers in Doped Quantum Dots. Nat. Mater. 2011, 10, 361–366. 10.1038/nmat3004. [DOI] [PubMed] [Google Scholar]
- Li W.; Zamani R.; Rivera Gil P.; Pelaz B.; Ibáñez M.; Cadavid D.; Shavel A.; Alvarez-Puebla R. A.; Parak W. J.; Arbiol J.; et al. CuTe Nanocrystals: Shape and Size Control, Plasmonic Properties, and Use as SERS Probes and Photothermal Agents. J. Am. Chem. Soc. 2013, 135, 7098–7101. 10.1021/ja401428e. [DOI] [PubMed] [Google Scholar]
- Li Y.; Bai H.; Zhai J.; Yi W.; Li J.; Yang H.; Xi G. Alternative to Noble Metal Substrates: Metallic and Plasmonic Ti3O5 Hierarchical Microspheres for Surface Enhanced Raman Spectroscopy. Anal. Chem. 2019, 91, 4496–4503. 10.1021/acs.analchem.8b05282. [DOI] [PubMed] [Google Scholar]
- Liu W.; Bai H.; Li X.; Li W.; Zhai J.; Li J.; Xi G. Improved Surface-Enhanced Raman Spectroscopy Sensitivity on Metallic Tungsten Oxide by the Synergistic Effect of Surface Plasmon Resonance Coupling and Charge Transfer. J. Phys. Chem. Lett. 2018, 9, 4096–4100. 10.1021/acs.jpclett.8b01624. [DOI] [PubMed] [Google Scholar]
- Hou X.; Luo X.; Fan X.; Peng Z.; Qiu T. Plasmon-Coupled Charge Transfer in WO3-x Semiconductor Nanoarrays: Toward Highly Uniform Silver-Comparable SERS Platforms. Phys. Chem. Chem. Phys. 2019, 21, 2611–2618. 10.1039/C8CP07305H. [DOI] [PubMed] [Google Scholar]
- Livingstone R.; Zhou X. C.; Tamargo M. C.; Lombardi J. R.; Quagliano L. C.; Jean-Mary F. Surface Enhanced Raman Spectroscopy of Pyridine on CdSe/ZnBeSe Quantum Dots Crown by Molecular Beam Epitaxy. J. Phys. Chem. C 2010, 114, 17460–17464. 10.1021/jp105619m. [DOI] [Google Scholar]
- Ji W.; Li L.; Guan J.; Mu M.; Song W.; Sun L.; Zhao B.; Ozaki Y. Hollow Multi-Shelled V2O5 Microstructures Integrating Multiple Synergistic Resonances for Enhanced Semiconductor SERS. Adv. Opt. Mater. 2021, 9, 2101866. 10.1002/adom.202101866. [DOI] [Google Scholar]
- Yang L.; Peng Y.; Yang Y.; Liu J.; Huang H.; Yu B.; Zhao J.; Lu Y.; Huang Z.; Li Z.; et al. A Novel Ultra-Sensitive Semiconductor SERS Substrate Boosted by the Coupled Resonance Effect. Adv. Sci. 2019, 6, 1900310. 10.1002/advs.201900310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Lin C.; Long L.; Masaki T.; Tang M.; Yang L.; Liu J.; Huang Z.; Li Z.; Luo X.; et al. Charge-Transfer Resonance and Electromagnetic Enhancement Synergistically Enabling MXenes with Excellent SERS Sensitivity for SARS-CoV-2 S Protein Detection. Nano-Micro Lett. 2021, 13, 52. 10.1007/s40820-020-00565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Lin C.; Li Y.; Gao Y.; Wang J.; He J.; Huang Z.; Liu J.; Luo X.; Yang Y. Identifying Infectiousness of SARS-CoV-2 by Ultra-Sensitive SnS2 SERS Biosensors with Capillary Effect. Matter 2022, 5, 694–709. 10.1016/j.matt.2021.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci A.; Gosztola D.; Schiller T.; Dimitrijevic N. M.; Mujica V.; Martin D.; Rajh T. SERS of Semiconducting Nanoparticies (TiO2 Hybrid Composites). J. Am. Chem. Soc. 2009, 131, 6040–6041. 10.1021/ja808277u. [DOI] [PubMed] [Google Scholar]
- Hurst S. J.; Fry H. C.; Gosztola D. J.; Rajh T. Utilizing Chemical Raman Enhancement: A Route for Metal Oxide Support-Based Biodetection. J. Phys. Chem. C 2011, 115, 620–630. 10.1021/jp1096162. [DOI] [Google Scholar]
- Tarakeshwar P.; Finkelstein-Shapiro D.; Hurst S. J.; Rajh T.; Mujica V. Surface-Enhanced Raman Scattering on Semiconducting Oxide Nanoparticles: Oxide Nature, Size, Solvent, and pH Effects. J. Phys. Chem. C 2011, 115, 8994–9004. 10.1021/jp202590e. [DOI] [Google Scholar]
- Lee N.; Hummer D. R.; Sverjensky D. A.; Rajh T.; Hazen R. M.; Steele A.; Cody G. D. Speciation of l-DOPA on Nanorutile as a Function of pH and Surface Coverage Using Surface-Enhanced Raman Spectroscopy (SERS). Langmuir 2012, 28, 17322–17330. 10.1021/la303607a. [DOI] [PubMed] [Google Scholar]
- Han X. X.; Köhler C.; Kozuch J.; Kuhlmann U.; Paasche L.; Sivanesan A.; Weidinger I. M.; Hildebrandt P. Potential-Dependent Surface-Enhanced Resonance Raman Spectroscopy at Nanostructured TiO2: A Case Study on Cytochrome b5. Small 2013, 9, 4175–4181. 10.1002/smll.201301070. [DOI] [PubMed] [Google Scholar]
- Öner I. H.; Querebillo C. J.; David C.; Gernert U.; Walter C.; Driess M.; Leimkühler S.; Ly K. H.; Weidinger I. M. High Electromagnetic Field Enhancement of TiO2 Nanotube Electrodes. Angew. Chem., Int. Ed. Engl. 2018, 57, 7225–7229. 10.1002/anie.201802597. [DOI] [PubMed] [Google Scholar]
- Chen L.; Tang J.; Ma H.; Jin S.; Xue X.; Han X. X.; Jung Y. M. High-Efficiency Charge Transfer on SERS-Active Semiconducting K2Ti6O13 Nanowires Enables Direct Transition of Photoinduced Electrons to Protein Redox Centers. Biosens. Bioelectron. 2021, 191, 113452. 10.1016/j.bios.2021.113452. [DOI] [PubMed] [Google Scholar]
- Murgida D. H.; Hildebrandt P. Disentangling Interfacial Redox Processes of Proteins by SERR Spectroscopy. Chem. Soc. Rev. 2008, 37, 937–945. 10.1039/b705976k. [DOI] [PubMed] [Google Scholar]
- Murgida D. H.; Hildebrandt P. Electron-Transfer Processes of Cytochrome c at Interfaces. New Insights by Surface-Enhanced Resonance Raman Spectroscopy. Acc. Chem. Res. 2004, 37, 854–861. 10.1021/ar0400443. [DOI] [PubMed] [Google Scholar]
- Lee K. E.; Gomez M. A.; Elouatik S.; Demopoulos G. P. Further Understanding of the Adsorption Mechanism of N719 Sensitizer on Anatase TiO2 Films for DSSC Applications Using Vibrational Spectroscopy and Confocal Raman Imaging. Langmuir 2010, 26, 9575–9583. 10.1021/la100137u. [DOI] [PubMed] [Google Scholar]
- Qiu Z.; Zhang M.; Wu D.-Y.; Ding S.-Y.; Zuo Q.-Q.; Huang Y.-F.; Shen W.; Lin X.-D.; Tian Z.-Q.; Mao B.-W. Raman Spectroscopic Investigation on TiO2-N719 Dye Interfaces Using Ag@TiO2 Nanoparticles and Potential Correlation Strategies. ChemPhysChem 2013, 14, 2217–2224. 10.1002/cphc.201300381. [DOI] [PubMed] [Google Scholar]
- Mao Z.; Ye Y.; Lv H.; Han X. X.; Park Y.; Zang L.; Zhao B.; Jung Y. M. Direct Dynamic Evidence of Charge Separation in a Dye-Sensitized Solar Cell Obtained under Operando Conditions by Raman Spectroscopy. Angew. Chem., Int. Ed. Engl. 2020, 59, 10780–10784. 10.1002/anie.201915824. [DOI] [PubMed] [Google Scholar]
- Wen S.; Ma X.; Liu H.; Chen G.; Wang H.; Deng G.; Zhang Y.; Song W.; Zhao B.; Ozaki Y. Accurate Monitoring Platform for the Surface Catalysis of Nanozyme Validated by Surface-Enhanced Raman-Kinetics Model. Anal. Chem. 2020, 92, 11763–11770. 10.1021/acs.analchem.0c01886. [DOI] [PubMed] [Google Scholar]
- Ma X.; Guo Y.; Jin J.; Zhao B.; Song W. Bi-functional Reduced Graphene Oxide/AgCo Composite Nanosheets: An Efficient Catalyst and SERS Substrate for Monitoring the Catalytic Reactions. RSC Adv. 2017, 7, 41962–41969. 10.1039/C7RA07216C. [DOI] [Google Scholar]
- Yang L.; Yin D.; Shen Y.; Yang M.; Li X.; Han X.; Jiang X.; Zhao B. Mesoporous Semiconducting TiO2 with Rich Active Sites as a Remarkable Substrate for Surface-Enhanced Raman Scattering. Phys. Chem. Chem. Phys. 2017, 19, 18731–18738. 10.1039/C7CP03399K. [DOI] [PubMed] [Google Scholar]
- Kiran V.; Sampath S. Enhanced Raman Spectroscopy of Molecules Adsorbed on Carbon-Doped TiO2 Obtained from Titanium Carbide: A Visible-Light-Assisted Renewable Substrate. ACS Appl. Mater. Interfaces 2012, 4, 3818–3828. 10.1021/am300349k. [DOI] [PubMed] [Google Scholar]
- Finkelstein-Shapiro D.; Petrosko S. H.; Dimitrijevic N. M.; Gosztola D.; Gray K. A.; Rajh T.; Tarakeshwar P.; Mujica V. CO2 Preactivation in Photoinduced Reduction via Surface Functionalization of TiO2 Nanoparticles. J. Phys. Chem. Lett. 2013, 4, 475–479. 10.1021/jz3020327. [DOI] [PubMed] [Google Scholar]
- Lombardi J. R. The Theory of Surface-Enhanced Raman Scattering on Semiconductor Nanoparticles: Toward the Optimization of SERS Sensors. Faraday Discuss. 2017, 205, 105–120. 10.1039/C7FD00138J. [DOI] [PubMed] [Google Scholar]
- Ji W.; Song W.; Tanabe I.; Wang Y.; Zhao B.; Ozaki Y. Semiconductor-Enhanced Raman Scattering For Highly Robust SERS Sensing: The Case of Phosphate Analysis. Chem. Commun. 2015, 51, 7641–7644. 10.1039/C5CC02395E. [DOI] [PubMed] [Google Scholar]
- Ji W.; Wang Y.; Tanabe I.; Han X.; Zhao B.; Ozaki Y. Semiconductor-Driven ″turn-off″ Surface-Enhanced Raman Scattering Spectroscopy: Application in Selective Determination of Chromium(vi) in Water. Chem. Sci. 2015, 6, 342–348. 10.1039/C4SC02618G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontempi N.; Carletti L.; De Angelis C.; Alessandri I. Plasmon-Free SERS Detection of Environmental CO2 on TiO2 Surfaces. Nanoscale 2016, 8, 3226–3231. 10.1039/C5NR08380J. [DOI] [PubMed] [Google Scholar]
- Alessandri I.; Vassalini I.; Bertuzzi M.; Bontempi N.; Memo M.; Gianoncelli A. RaMassays”: Synergistic Enhancement of Plasmon-Free Raman Scattering and Mass Spectrometry for Multimodal Analysis of Small Molecules. Sci. Rep. 2016, 6, 34521. 10.1038/srep34521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurouski D.; Dazzi A.; Zenobi R.; Centrone A. Infrared and Raman chemical imaging and spectroscopy at the nanoscale. Chem. Soc. Rev. 2020, 49, 3315–3347. 10.1039/C8CS00916C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackie E. J.; Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy of Nonresonant Molecules. J. Am. Chem. Soc. 2009, 131, 14466–14472. 10.1021/ja905319w. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Seong N.-H.; Dlott D. D. Measurement of the Distribution of Site Enhancements in Surface-Enhanced Raman Scattering. Science 2008, 321, 388–392. 10.1126/science.1159499. [DOI] [PubMed] [Google Scholar]
- Wu D. Y.; Li J. F.; Ren B.; Tian Z. Q. Electrochemical Surface-Enhanced Raman Spectroscopy of Nanostructures. Chem. Soc. Rev. 2008, 37, 1025–1041. 10.1039/b707872m. [DOI] [PubMed] [Google Scholar]
- Yeo B. S.; Schmid T.; Zhang W. H.; Zenobi R. A Strategy to Prevent Signal Losses, Analyte Decomposition, and Fluctuating Carbon Contamination Bands in Surface-Enhanced Raman Spectroscopy. Appl. Spectrosc. 2008, 62, 708–713. 10.1366/000370208784658165. [DOI] [PubMed] [Google Scholar]
- Asiala S. M.; Schultz Z. D. Label-Free in situ Detection of Individual Macromolecular Assemblies by Surface Enhanced Raman Scattering. Chem. Commun. 2013, 49, 4340–4342. 10.1039/C2CC37268A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S. E. J.; Sirimuthu N. M. S. Quantitative Surface-Enhanced Raman Spectroscopy. Chem. Soc. Rev. 2008, 37, 1012–1024. 10.1039/b705965p. [DOI] [PubMed] [Google Scholar]
- Zhang D. M.; Xie Y.; Deb S. K.; Davison V. J.; Ben-Amotz D. Isotope Edited Internal Standard Method for Quantitative Surface-Enhanced Raman Spectroscopy. Anal. Chem. 2005, 77, 3563–3569. 10.1021/ac050338h. [DOI] [PubMed] [Google Scholar]
- Shen W.; Lin X.; Jiang C. Y.; Li C. Y.; Lin H. X.; Huang J. T.; Wang S.; Liu G. K.; Yan X. M.; Zhong Q. L.; et al. Reliable Quantitative SERS Analysis Facilitated by Core-Shell Nanoparticles with Embedded Internal Standards. Angew. Chem., Int. Ed. Engl. 2015, 54, 7308–7312. 10.1002/anie.201502171. [DOI] [PubMed] [Google Scholar]
- de Albuquerque C. D. L.; Sobral R. G.; Poppi R. J.; Brolo A. G. Digital Protocol for Chemical Analysis at Ultralow Concentrations by Surface-Enhanced Raman Scattering. Anal. Chem. 2018, 90, 1248–1254. 10.1021/acs.analchem.7b03968. [DOI] [PubMed] [Google Scholar]
- Hong K. Y.; de Albuquerque C. D. L.; Poppi R. J.; Brolo A. G. Determination of Aqueous Antibiotic Solutions Using SERS Nanogratings. Anal. Chim. Acta 2017, 982, 148–155. 10.1016/j.aca.2017.05.025. [DOI] [PubMed] [Google Scholar]
- Bohn J. E.; Le Ru E. C.; Etchegoin P. G. A Statistical Criterion for Evaluating the Single-Molecule Character of SERS Signals. J. Phys. Chem. C 2010, 114, 7330–7335. 10.1021/jp908990v. [DOI] [Google Scholar]
- Sonntag M. D.; Klingsporn J. M.; Garibay L. K.; Roberts J. M.; Dieringer J. A.; Seideman T.; Scheidt K. A.; Jensen L.; Schatz G. C.; Van Duyne R. P. Single-Molecule Tip-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2012, 116, 478–483. 10.1021/jp209982h. [DOI] [Google Scholar]
- Bhattarai A.; Joly A. G.; Hess W. P.; El-Khoury P. Z. Visualizing Electric Fields at Au(111) Step Edges via Tip-Enhanced Raman Scattering. Nano Lett. 2017, 17, 7131–7137. 10.1021/acs.nanolett.7b04027. [DOI] [PubMed] [Google Scholar]
- Schmid T.; Yeo B. S.; Leong G.; Stadler J.; Zenobi R. Performing Tip-Enhanced Raman Spectroscopy in Liquids. J. Raman Spectrosc. 2009, 40, 1392–1399. 10.1002/jrs.2387. [DOI] [Google Scholar]
- Watanabe K.; Palonpon A. F.; Smith N. I.; Chiu L. D.; Kasai A.; Hashimoto H.; Kawata S.; Fujita K. Structured Line Illumination Raman Microscopy. Nat. Commun. 2015, 6, 10095. 10.1038/ncomms10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. K.; Wang S. Q.; Zhang Y. Q.; Yang Y.; Fang H.; Zhu S. W.; Yuan X. C. Structured Illumination for Wide-Field Raman Imaging of Cell Membranes. Opt. Commun. 2017, 402, 221–225. 10.1016/j.optcom.2017.06.021. [DOI] [Google Scholar]
- Weber M. L.; Willets K. A. Correlated Super-Resolution Optical and Structural Studies of Surface-Enhanced Raman Scattering Hot Spots in Silver Colloid Aggregates. J. Phys. Chem. Lett. 2011, 2, 1766–1770. 10.1021/jz200784e. [DOI] [Google Scholar]
- Olson A. P.; Ertsgaard C. T.; Elliott S. N.; Lindquist N. C. Super-Resolution Chemical Imaging with Plasmonic Substrates. ACS Photonics 2016, 3, 329–336. 10.1021/acsphotonics.5b00647. [DOI] [Google Scholar]
- Procházka M.Surface-Enhanced Raman Spectroscopy - Bioanalytical, Biomolecular and Medical Applications; Springer International Publishing, 2016; pp 1–221. [Google Scholar]
- Treffer R.; Deckert V. Recent Advances in Single-Molecule Sequencing. Curr. Opin. Biotechnol. 2010, 21, 4–11. 10.1016/j.copbio.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Najjar S.; Talaga D.; Schue L.; Coffinier Y.; Szunerits S.; Boukherroub R.; Servant L.; Rodriguez V.; Bonhommeau S. Tip-Enhanced Raman Spectroscopy of Combed Double-Stranded DNA Bundles. J. Phys. Chem. C 2014, 118, 1174–1181. 10.1021/jp410963z. [DOI] [Google Scholar]
- Venkatesan B. M.; Bashir R. Nanopore Sensors for Nucleic Acid Analysis. Nat. Nanotechnol. 2011, 6, 615–624. 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- Chen C.; Li Y.; Kerman S.; Neutens P.; Willems K.; Cornelissen S.; Lagae L.; Stakenborg T.; Van Dorpe P. High Spatial Resolution Nanoslit SERS for Single-Molecule Nucleobase Sensing. Nat. Commun. 2018, 9, 1733. 10.1038/s41467-018-04118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashaee F.; Tabatabaei M.; Caetano F. A.; Ferguson S. S. G.; Lagugne-Labarthet F. Tip-Enhanced Raman Spectroscopy: Plasmid-Free vs. Plasmid-Embedded DNA. Analyst 2016, 141, 3251–3258. 10.1039/C6AN00350H. [DOI] [PubMed] [Google Scholar]
- Kurouski D.; Deckert-Gaudig T.; Deckert V.; Lednev I. K. Structure and Composition of Insulin Fibril Surfaces Probed by TERS. J. Am. Chem. Soc. 2012, 134, 13323–13329. 10.1021/ja303263y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert-Gaudig T.; Kurouski D.; Hedegaard M. A. B.; Singh P.; Lednev I. K.; Deckert V. Spatially Resolved Spectroscopic Differentiation of Hydrophilic and Hydrophobic Domains on Individual Insulin Amyloid Fibrils. Sci. Rep. 2016, 6, 33575. 10.1038/srep33575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhommeau S.; Talaga D.; Hunel J.; Cullin C.; Lecomte S. Tip-Enhanced Raman Spectroscopy to Distinguish Toxic Oligomers from Aβ1-42 Fibrils at the Nanometer Scale. Angew. Chem., Int. Ed. Engl. 2017, 56, 1771–1774. 10.1002/anie.201610399. [DOI] [PubMed] [Google Scholar]
- Lipiec E.; Perez-Guaita D.; Kaderli J.; Wood B. R.; Zenobi R. Direct Nanospectroscopic Verification of the Amyloid Aggregation Pathway. Angew. Chem., Int. Ed. Engl. 2018, 57, 8519–8524. 10.1002/anie.201803234. [DOI] [PubMed] [Google Scholar]
- Tabatabaei M.; Caetano F. A.; Pashee F.; Ferguson S. S. G.; Lagugne-Labarthet F. Tip-Enhanced Raman Spectroscopy of Amyloid Beta at Neuronal spines. Analyst 2017, 142, 4415–4421. 10.1039/C7AN00744B. [DOI] [PubMed] [Google Scholar]
- Kurouski D.; Deckert-Gaudig T.; Deckert V.; Lednev I. K. Surface Characterization of Insulin Protofilaments and Fibril Polymorphs Using Tip-Enhanced Raman Spectroscopy (TERS). Biophys. J. 2014, 106, 263–271. 10.1016/j.bpj.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao P. D.; Huang T. Q.; Liu X. M.; Wu T. Q. Surface-Enhanced Raman Spectroscopy of Insect Nuclear Polyhedrosis Virus. J. Raman Spectrosc. 2001, 32, 227–230. 10.1002/jrs.665. [DOI] [Google Scholar]
- Cialla D.; Deckert-Gaudig T.; Budich C.; Laue M.; Moller R.; Naumann D.; Deckert V.; Popp J. Raman to the Limit: Tip-Enhanced Raman Spectroscopic Investigations of a Single Tobacco Mosaic Virus. J. Raman Spectrosc. 2009, 40, 240–243. 10.1002/jrs.2123. [DOI] [Google Scholar]
- Yang J. L.; Wang H. J.; Zhang H.; Tian Z. Q.; Li J. F. Probing Hot Electron Behaviors by Surface-Enhanced Raman Spectroscopy. Cell. Rep. Phys. Sci. 2020, 1, 100184. 10.1016/j.xcrp.2020.100184. [DOI] [Google Scholar]
- Li J. F.; Huang Y. F.; Ding Y.; Yang Z. L.; Li S. B.; Zhou X. S.; Fan F. R.; Zhang W.; Zhou Z. Y.; Wu D. Y.; et al. Shell-Isolated Nanoparticle-Enhanced Raman Spectroscopy. Nature 2010, 464, 392–395. 10.1038/nature08907. [DOI] [PubMed] [Google Scholar]
- Wang Y. H.; Wei J.; Radjenovic P.; Tian Z. Q.; Li J. F. In Situ Analysis of Surface Catalytic Reactions Using Shell-Isolated Nanoparticle-Enhanced Raman Spectroscopy. Anal. Chem. 2019, 91, 1675–1685. 10.1021/acs.analchem.8b05499. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Merk V.; Hermanns A.; Unger W. E. S.; Kneipp J. Role of Metal Cations in Plasmon-Catalyzed Oxidation: A Case Study of p-Aminothiophenol Dimerization. ACS Catal. 2017, 7, 7803–7809. 10.1021/acscatal.7b02700. [DOI] [Google Scholar]
- Wang J. L.; Ando R. A.; Camargo P. H. C. Controlling the Selectivity of the Surface Plasmon Resonance Mediated Oxidation of p-Aminothiophenol on Au Nanoparticles by Charge Transfer from UV-excited TiO2. Angew. Chem., Int. Ed. Engl. 2015, 54, 6909–6912. 10.1002/anie.201502077. [DOI] [PubMed] [Google Scholar]
- Xie W.; Schlucker S. Hot Electron-Induced Reduction of Small Molecules on Photorecycling Metal Surfaces. Nat. Commun. 2015, 6, 7570. 10.1038/ncomms8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes E.; Xie W.; Cambiasso J.; Jermyn A. S.; Sundararaman R.; Narang P.; Schlucker S.; Maier S. A. Plasmonic Hot Electron Transport Drives Nano-Localized Chemistry. Nat. Commun. 2017, 8, 14880. 10.1038/ncomms14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt N. C.; Keller E. L.; Frontiera R. R. Ultrafast Surface-Enhanced Raman Probing of the Role of Hot Electrons in Plasmon-Driven Chemistry. J. Phys. Chem. Lett. 2016, 7, 3179–3185. 10.1021/acs.jpclett.6b01453. [DOI] [PubMed] [Google Scholar]
- Karaballi R. A.; Nel A.; Krishnan S.; Blackburn J.; Brosseau C. L. Development of an Electrochemical Surface-Enhanced Raman Spectroscopy (EC-SERS) Aptasensor for Direct Detection of DNA Hybridization. Phys. Chem. Chem. Phys. 2015, 17, 21356–21363. 10.1039/C4CP05077K. [DOI] [PubMed] [Google Scholar]
- Lee T.; Mohammadniaei M.; Zhang H.; Yoon J.; Choi H. K.; Guo S. J.; Guo P. X.; Choi J. W. Single Functionalized pRNA/Gold Nanoparticle for Ultrasensitive MicroRNA Detection Using Electrochemical Surface-Enhanced Raman Spectroscopy. Adv. Sci. 2020, 7, 1902477. 10.1002/advs.201902477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. L.; Wu C. J.; Huang S. Q.; Luo X. L.; Yuan R.; Yang X. A Novel SERS Substrate with High Reusability for Sensitive Detection of miRNA 21. Talanta 2021, 228, 122240. 10.1016/j.talanta.2021.122240. [DOI] [PubMed] [Google Scholar]
- Goodall B. L.; Robinson A. M.; Brosseau C. L. Electrochemical-Surface Enhanced Raman Spectroscopy (E-SERS) of Uric Acid: A Potential Rapid Diagnostic Method for Early Preeclampsia Detection. Phys. Chem. Chem. Phys. 2013, 15, 1382–1388. 10.1039/C2CP42596C. [DOI] [PubMed] [Google Scholar]
- Hernandez S.; Perales-Rondon J. V.; Heras A.; Colina A. Determination of Uric Acid in Synthetic Urine by Using Electrochemical Surface Oxidation Enhanced Raman Scattering. Anal. Chim. Acta 2019, 1085, 61–67. 10.1016/j.aca.2019.07.057. [DOI] [PubMed] [Google Scholar]
- Huang C. Y.; Hsiao H. C. Integrated EC-SERS Chip with Uniform Nanostructured EC-SERS Active Working Electrode for Rapid Detection of Uric Acid. Sensors 2020, 20, 7066. 10.3390/s20247066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynk T. P.; Sit C. S.; Brosseau C. L. Electrochemical Surface-Enhanced Raman Spectroscopy as a Platform for Bacterial Detection and Identification. Anal. Chem. 2018, 90, 12639–12646. 10.1021/acs.analchem.8b02806. [DOI] [PubMed] [Google Scholar]
- Do H.; Kwon S. R.; Fu K. Y.; Morales-Soto N.; Shrout J. D.; Bohn P. W. Electrochemical Surface-Enhanced Raman Spectroscopy of Pyocyanin Secreted by Pseudomonas aeruginosa Communities. Langmuir 2019, 35, 7043–7049. 10.1021/acs.langmuir.9b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanain W. A.; Izake E. L.; Ayoko G. A. Spectroelectrochemical Nanosensor for the Determination of Cystatin C in Human Blood. Anal. Chem. 2018, 90, 10843–10850. 10.1021/acs.analchem.8b02121. [DOI] [PubMed] [Google Scholar]
- Huang S. C.; Bao Y. F.; Wu S. S.; Huang T. X.; Sartin M. M.; Wang X.; Ren B. Electrochemical Tip-Enhanced Raman Spectroscopy: An in Situ Nanospectroscopy for Electrochemistry. Annu. Rev. Phys. Chem. 2021, 72, 213–234. 10.1146/annurev-physchem-061020-053442. [DOI] [PubMed] [Google Scholar]
- Kurouski D.; Mattei M.; Van Duyne R. P. Probing Redox Reactions at the Nanoscale with Electrochemical Tip-Enhanced Raman Spectroscopy. Nano Lett. 2015, 15, 7956–7962. 10.1021/acs.nanolett.5b04177. [DOI] [PubMed] [Google Scholar]
- Mattei M.; Kang G.; Goubert G.; Chulhai D. V.; Schatz G. C.; Jensen L.; Van Duyne R. P. Tip-Enhanced Raman Voltammetry: Coverage Dependence and Quantitative Modeling. Nano Lett. 2017, 17, 590–596. 10.1021/acs.nanolett.6b04868. [DOI] [PubMed] [Google Scholar]
- Zeng Z. C.; Huang S. C.; Wu D. Y.; Meng L. Y.; Li M. H.; Huang T. X.; Zhong J. H.; Wang X.; Yang Z. L.; Ren B. Electrochemical Tip-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2015, 137, 11928–11931. 10.1021/jacs.5b08143. [DOI] [PubMed] [Google Scholar]
- Huang S. C.; Ye J. Z.; Shen X. R.; Zhao Q. Q.; Zeng Z. C.; Li M. H.; Wu D. Y.; Wang X.; Ren B. Electrochemical Tip-Enhanced Raman Spectroscopy with Improved Sensitivity Enabled by a Water Immersion Objective. Anal. Chem. 2019, 91, 11092–11097. 10.1021/acs.analchem.9b01701. [DOI] [PubMed] [Google Scholar]
- Sabanes N. M.; Ohto T.; Andrienko D.; Nagata Y.; Domke K. F. Electrochemical TERS Elucidates Potential-Induced Molecular Reorientation of Adenine/Au(111). Angew. Chem., Int. Ed. Engl. 2017, 56, 9796–9801. 10.1002/anie.201704460. [DOI] [PubMed] [Google Scholar]
- Touzalin T.; Joiret S.; Maisonhaute E.; Lucas I. T. Complex Electron Transfer Pathway at a Microelectrode Captured by in Situ Nanospectroscopy. Anal. Chem. 2017, 89, 8974–8980. 10.1021/acs.analchem.7b01542. [DOI] [PubMed] [Google Scholar]
- Touzalin T.; Joiret S.; Lucas I. T.; Maisonhaute E. Electrochemical Tip-Enhanced Raman Spectroscopy Imaging with 8 nm Lateral Resolution. Electrochem. Commun. 2019, 108, 106557. 10.1016/j.elecom.2019.106557. [DOI] [Google Scholar]
- Kumar N.; Wondergem C. S.; Wain A. J.; Weckhuysen B. M. In Situ Nanoscale Investigation of Catalytic Reactions in the Liquid Phase Using Zirconia-Protected Tip-Enhanced Raman Spectroscopy Probes. J. Phys. Chem. Lett. 2019, 10, 1669–1675. 10.1021/acs.jpclett.8b02496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Kurouski D. Elucidation of Tip-Broadening Effect in Tip-Enhanced Raman Spectroscopy (TERS): A Cause of Artifacts or Potential for 3D TERS. J. Phys. Chem. C 2018, 122, 24334–24340. 10.1021/acs.jpcc.8b09455. [DOI] [Google Scholar]
- Karabeber H.; Huang R. M.; Iacono P.; Samii J. M.; Pitter K.; Holland E. C.; Kircher M. F. Guiding Brain Tumor Resection Using Surface-Enhanced Raman Scattering Nanoparticles and a Hand-Held Raman Scanner. ACS Nano 2014, 8, 9755–9766. 10.1021/nn503948b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L. M.; Duan W. J.; Li X. W.; Wang C.; Jin Z. Y.; Zhai Y. T.; Cao C.; Chen L.; Xu W. J.; Liu Y.; et al. Surface-Enhanced Resonace Raman Scattering - Guided Brain Tumor Surgery Showing Prognostic Benefit in Rat Models. ACS Appl. Mater. Interfaces 2019, 11, 15241–15250. 10.1021/acsami.9b00227. [DOI] [PubMed] [Google Scholar]
- Jin Z. Y.; Yue Q.; Duan W. J.; Sui A.; Zhao B. T.; Deng Y. H.; Zhai Y. T.; Zhang Y. W.; Sun T.; Zhang G. P.; et al. Intelligent SERS Navigation System Guiding Brain Tumor Surgery by Intraoperatively Delineating the Metabolic Acidosis. Adv. Sci. 2022, 9, e2104935 10.1002/advs.202270043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W. J.; Yue Q.; Liu Y.; Zhang Y. F.; Guo Q. H.; Wang C.; Yin S. J.; Fan D. D.; Xu W. J.; Zhuang J. X.; et al. A pH Ratiometrically Responsive Surface Enhanced Resonance Raman Scattering Probe for Tumor Acidic Margin Delineation and Image-Guided Surgery. Chem. Sci. 2020, 11, 4397–4402. 10.1039/D0SC00844C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavaleta C. L.; Garai E.; Liu J. T. C.; Sensarn S.; Mandella M. J.; Van de Sompel D.; Friedland S.; Van Dam J.; Contag C. H.; Gambhir S. S. A Raman-Based Endoscopic Strategy for Multiplexed Molecular Imaging. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10062–10063. 10.1073/pnas.1211309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai E.; Sensarn S.; Zavaleta C. L.; Van de Sompel D.; Loewke N. O.; Mandella M. J.; Gambhir S. S.; Contag C. H. High-Sensitivity, Real-Time, Ratiometric Imaging of Surface-Enhanced Raman Scattering Nanoparticles with a Clinically Translatable Raman Endoscope Device. J. Biomed. Opt. 2013, 18, 096008. 10.1117/1.JBO.18.9.096008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai E.; Sensarn S.; Zavaleta C. L.; Loewke N. O.; Rogalla S.; Mandella M. J.; Felt S. A.; Friedland S.; Liu J. T. C.; Gambhir S. S.; et al. A Real-Time Clinical Endoscopic System for Intraluminal, Multiplexed Imaging of Surface-Enhanced Raman Scattering Nanoparticles. PLoS One 2015, 10, e0123185 10.1371/journal.pone.0123185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. W.; Kang S.; Khan A.; Bao P. Q.; Liu J. T. C. In vivo Multiplexed Molecular Imaging of Esophageal Cancer via Spectral Endoscopy of Topically Applied SERS Nanoparticles. Biomed. Opt. Express 2015, 6, 3714–3723. 10.1364/BOE.6.003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; De Keersmaecker H.; Su L.; Kenens B.; Rocha S.; Fron E.; Chen C.; Van Dorpe P.; Mizuno H.; Hofkens J.; et al. Live-Cell SERS Endoscopy Using Plasmonic Nanowire Waveguides. Adv. Mater. 2014, 26, 5124–5128. 10.1002/adma.201401237. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Takeshita N.; Ho K. Y. Raman Spectroscopy for the Endoscopic Diagnosis of Esophageal, Gastric, and Colonic Diseases. Clin. Endosc. 2016, 49, 404–407. 10.5946/ce.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Y.; Mi X.; Tan X. Y.; Xiang R. Recent Progress on Liquid Biopsy Analysis using Surface-Enhanced Raman Spectroscopy. Theranostics 2019, 9, 491–525. 10.7150/thno.29875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugasundaram K. B.; Li J. R.; Sina A. I.; Wuethrich A.; Trau M. Toward Precision Oncology: SERS Microfluidic Systems for Multiplex Biomarker Analysis in Liquid Biopsy. Mater. Adv. 2022, 3, 1459–1471. 10.1039/D1MA00848J. [DOI] [Google Scholar]
- Sha M. Y.; Xu H. X.; Natan M. J.; Cromer R. Surface-Enhanced Raman Scattering Tags for Rapid and Homogeneous Detection of Circulating Tumor Cells in the Presence of Human Whole Blood. J. Am. Chem. Soc. 2008, 130, 17214–17215. 10.1021/ja804494m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Qian X. M.; Beitler J. J.; Chen Z. G.; Khuri F. R.; Lewis M. M.; Shin H. J. C.; Nie S. M.; Shin D. M. Detection of Circulating Tumor Cells in Human Peripheral Blood Using Surface-Enhanced Raman Scattering Nanoparticles. Cancer Res. 2011, 71, 1526–1532. 10.1158/0008-5472.CAN-10-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W.; Paproski R. J.; Moore R.; Zemp R. Detection of Circulating Tumor Cells Using Targeted Surface-Enhanced Raman Scattering Nanoparticles and Magnetic Enrichment. J. Biomed. Opt. 2014, 19, 056014. 10.1117/1.JBO.19.5.056014. [DOI] [PubMed] [Google Scholar]
- Nima Z. A.; Mahmood M.; Xu Y.; Mustafa T.; Watanabe F.; Nedosekin D. A.; Juratli M. A.; Fahmi T.; Galanzha E. I.; Nolan J. P.; et al. Circulating Tumor Cell Identification by Functionalized Silver-Gold Nanorods with Multicolor, Super-Enhanced SERS and Photothermal Resonances. Sci. Rep. 2015, 4, 4752. 10.1038/srep04752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamidipati M.; Cho H. Y.; Lee K. B.; Fabris L. SERS-Based Quantification of Biomarker Expression at the Single Cell Level Enabled by Gold Nanostars and Truncated Aptamers. Bioconjugate Chem. 2018, 29, 2970–2981. 10.1021/acs.bioconjchem.8b00397. [DOI] [PubMed] [Google Scholar]
- Reza K. K.; Dey S.; Wuethrich A.; Wang J.; Behren A.; Antaw F.; Wang Y. L.; Ibn Sina A.; Trau M. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano 2021, 15, 11231–11243. 10.1021/acsnano.0c10008. [DOI] [PubMed] [Google Scholar]
- Cho H. Y.; Hossain M. K.; Lee J. H.; Han J.; Lee H. J.; Kim K. J.; Kim J. H.; Lee K. B.; Choi J. W. Selective Isolation and Noninvasive Analysis of Circulating Cancer Stem Cells Through Raman Imaging. Biosens. Bioelectron. 2018, 102, 372–382. 10.1016/j.bios.2017.11.049. [DOI] [PubMed] [Google Scholar]
- Wilson R. E.; O’Connor R.; Gallops C. E.; Kwizera E. A.; Noroozi B.; Morshed B. I.; Wang Y. M.; Huang X. H. Immunomagnetic Capture and Multiplexed Surface Marker Detection of Circulating Tumor Cells with Magnetic Multicolor Surface-Enhanced Raman Scattering Nanotags. ACS Appl. Mater. Interfaces 2020, 12, 47220–47232. 10.1021/acsami.0c12395. [DOI] [PubMed] [Google Scholar]
- Wood B. R.; Bailo E.; Khiavi M. A.; Tilley L.; Deed S.; Deckert-Gaudig T.; McNaughton D.; Deckert V. Tip-Enhanced Raman Scattering (TERS) from Hemozoin Crystals within a Sectioned Erythrocyte. Nano Lett. 2011, 11, 1868–1873. 10.1021/nl103004n. [DOI] [PubMed] [Google Scholar]
- Xiao L. F.; Bailey K. A.; Wang H.; Schultz Z. D. Probing Membrane Receptor-Ligand Specificity with Surface- and Tip-Enhanced Raman Scattering. Anal. Chem. 2017, 89, 9091–9099. 10.1021/acs.analchem.7b01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treffer R.; Bohme R.; Deckert-Gaudig T.; Lau K.; Tiede S.; Lin X. M.; Deckert V. Advances in TERS (Tip-Enhanced Raman scattering) for Biochemical Applications. Biochem. Soc. Trans. 2012, 40, 609–614. 10.1042/BST20120033. [DOI] [PubMed] [Google Scholar]