

Abstract
The endocannabinoid system (ECS) plays a very important role in numerous physiological and pharmacological processes, such as those related to the central nervous system (CNS), including learning, memory, emotional processing, as well pain control, inflammatory and immune response, and as a biomarker in certain psychiatric disorders. Unfortunately, the half-life of the natural ligands responsible for these effects is very short. This perspective describes the potential role of the inhibitors of the enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MGL), which are mainly responsible for the degradation of endogenous ligands in psychic disorders and related pathologies. The examination was carried out considering both the impact that the classical exogenous ligands such as Δ9-tetrahydrocannabinol (THC) and (−)-trans-cannabidiol (CBD) have on the ECS and through an analysis focused on the possibility of predicting the potential toxicity of the inhibitors before they are subjected to clinical studies. In particular, cardiotoxicity (hERG liability), probably the worst early adverse reaction studied during clinical studies focused on acute toxicity, was predicted, and some of the most used and robust metrics available were considered to select which of the analyzed compounds could be repositioned as possible oral antipsychotics.
Keywords: endocannabinoid system, FAAH inhibitors, MGL inhibitors, repositioning, drug-likeness, hERG, ligand efficiency metrics
1. Introduction
Clinical and social psychologists seldom use an ancient Hindu parable to illustrate the seductive potential of our perspective on a complex problem—the well-known story of the blind men and the elephant [Briefly, six wise men were blind and wanted to learn about an animal that they had never met before—an elephant. They went through an organoleptic analysis. Depending on the part of the animal body they touched, each of them produced a different definition (e.g., ‘huge, wrinkled fan,’ said the experimenter who had touched the elephant ears; ‘a big rope,’ said the wise man who had touched the elephant swinging tail; and so on) and a vehement disputation originated since each wise man suspected the others as dishonest persons. All six were telling the truth, but each of them had touched only one part of the animal and therefore knew only that part of the truth] [1]. When a complex problem is concerned, we generally know only a part of the truth while the whole story remains mostly unknown. Mental illness is a complex problem since the human brain is a holistic apparatus: our attempt to explain mental disorders at the synaptic level makes us resemble the wise men who wanted to infer the elephant by analyzing only a part of its body. Anyway, when looking for new drugs to treat mental illness, the best rational way still starts from an alleged biochemical defect occurring at one or several types of synaptic gaps.
The dopamine hypothesis has been the leading theory of most psychotic disorders, and the currently used typical and atypical antipsychotics share a common mechanism of action in antagonism of the dopamine D2 receptor [2]. The remarkable case of clozapine, the only atypical antipsychotic effective in psychoses that are otherwise treatment-resistant [3], indicates that multipotent antipsychotics acting at many different receptor sites in the brain may represent a more efficacious treatment than existing drugs [4]. Numerous lines of evidence have highlighted the possible involvement of diverse neurotransmitter pathways, including glutamate, serotonin, γ-aminobutyric acid, adenosine, and acetylcholine [5]. Frequently polypharmacy is prescribed to face the most severe psychotic syndromes despite controversial results about the efficacy of the approach [6,7] and related safety concern [8,9], obviously higher in older patients [10].
In 1997, a “cannabinoid hypothesis of schizophrenia” was also suggested [11], and the increasing body of evidence supporting this new paradigm has been brilliantly reviewed [12]. The endocannabinoid system (ECS) is one of the most relevant neurotransmitter systems in the brain and plays a pivotal role in the regulation of cognitive abilities, mood, stress, and sleep [13]. Relatively fewer explored targets for antipsychotic treatment could be found in ECS. Thinking about cannabinoids, our minds run to the well-known pro-psychotic properties of ∆9-tetrahydrocannabinol (THC, Figure 1), the main psychoactive ingredient of cannabis, which acts as an agonist on cannabinoid (CB) receptors (CBR). In addition to its pro-psychotic potential, THC causes an undesirable behavioral tetrad, that is, analgesia, catalepsy, hypothermia, and hypolocomotion. THC synthetic analogs, both agonists and antagonists [14,15], or recreational drugs—the so-called NPS (new psychoactive substances) [16,17]—are generally tainted with severe side effects. The worst is that the activation of CBR of type 1 (CB1R) in the central nervous system (CNS) by xenobiotics can lead to irreversible effects [18]. On the other hand, (−)-trans-cannabidiol (CBD, Figure 1), one of cannabis’ main secondary metabolites, seems to be endowed with antipsychotic properties useful to protect against the pro-psychotic effects of THC: depending on its composition, cannabis would act either as Mister Hyde (i.e., a risk factor for psychosis) or as Doctor Jekyll (i.e., an antipsychotic). The hypothesis has been formulated that CBD could be an antipsychotic, with benefits in preventing psychotic disorders, whatever the cause (endogenous or THC-induced) [19].
Figure 1.
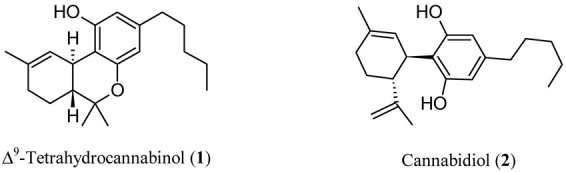
Chemical structures of Δ9-tetrahydrocannabinol (THC, 1) and cannabidiol (CBD, 2).
In a randomized, double-blind controlled clinical trial, CBD exerted antipsychotic properties comparable to the reference drug amisulpride [20,21]. Interestingly, the reduction of psychotic symptoms was significantly associated with an increase in the serum concentrations of N-arachidonoylethanolamine (anandamide, AEA, Figure 2), which is the most important endogenous ligand of CBR, and this outcome was found only in patients treated with CBD. The results indicated that, at least in part, the antipsychotic activity of CBD was due to the inhibition of the enzymes physiologically devoted to the degradation of AEA [22], thus acting as an indirect agonist. This finding agrees with the observation that both increased availability of CB1R and upregulation of AEA seem beneficial, although the underlying mechanisms are mostly elusive. The evidence supporting the possible protective role of AEA in schizophrenia has been reviewed [23].
Figure 2.
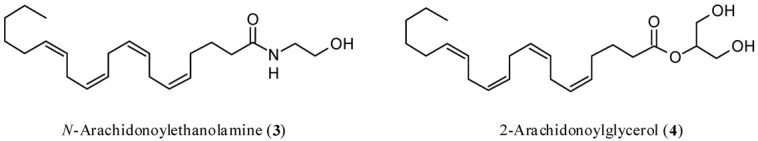
Chemical structures of N-arachidonoylethanolamine (anandamide, AEA, 3) and 2-arachidonoylglycerol (2-AG, 4).
The inhibition of enzymes responsible for the degradation of the ECS endogenous ligands might overcome the above obstacles to the systemic use of exogenous substances acting on ECS. EC degradation inhibitors would provide focused action, where and when necessary, by acting as endogenous CBRs ligand modulators. Since the available clinical data on EC potentiators as antipsychotics are relatively scarce and essentially limited to CBD, we conceived this perspective as an attempt to envision which of the EC degradation most considered inhibitors so far might be repositioned as possible oral antipsychotics based on their predicted drug-likeness and safety. After a dutiful short survey on the ECS architecture and functions, we shall review the most studied, clinically relevant EC metabolizing enzyme inhibitors reporting their corresponding main pharmacological activities. Then, we shall predict ADMET properties and calculate structure descriptors generally related to drug-likeness and probability of success as CNS acting agents for each of them using some of the most robust medicinal chemistry predicting tools today available.
The purpose of this narrative review is to select some of the analyzed compounds as either candidates for repositioning as orally active antipsychotics or starting compounds for structural simplification/optimization studies to reduce toxicity and improve selectivity.
The information presented in this perspective was acquired through consultation of the PubMed®, Reaxys®, and SciFinder® Scholar databases.
2. Endocannabinoid System
The exhaustive description of ECS architecture and functioning is beyond the scope of this work; more details can be found elsewhere [24,25]. The right functioning of the ECS is related to the natural balance established between its main components, which are CBRs, the endogenous ligands binding them, and the enzymes involved in the synthesis, transport, and degradation of ECs. A disruption of the physiological activity of this system (i.e., modifications in the expression of receptors or the functions of enzymes) is associated with various pathologies. This situation, therefore, is the basis for therapeutic pharmacological opportunities founded on drugs able to interact naturally with ECS [26,27,28,29,30].
The discovery of CBRs and the main endogenous ligands is relatively recent, as the first one, CB1R, was identified in the second half of the 1980s [31], while the second receptor, namely CB2R, was discovered a few years later [32]. The two targets differ in their corresponding main functions, signaling processes, and structural aspects [33,34]. Their signal neurobiology and tissue distribution are also different, being the CB1R mainly expressed in the CNS (mostly in the basal ganglia, cerebellum, cortex, and hippocampus), whereas CB2R is particularly present in the immune system (mostly in B-cells and natural killers) [35]. Overall, it is demonstrated that CBR, through their activation, performs a key role in inducing activation or depression of neurotransmission by the inhibition of adenylate cyclase, which determines a decrease in cyclic adenosine monophosphate levels, or, only in the case of CB1R, by the coupling with ion channels [35,36]. A careful analysis of the above characteristics, in particular those related to the different tissue distribution, is important when envisioning a pharmacological therapy aiming at a selectivity of action and the consequent reduction of undesired effects.
The main and most studied CBR endogenous ligands are AEA [37] and 2-arachidonoylglycerol (2-AG) [38,39] (Figure 2).
Both ligands are produced on demand from membrane phospholipids to satisfy contingent physiological needs due to intense neuronal activation [40,41]. AEA and 2-AG act through a retrograde or non-retrograde signaling pathway. Their half-life is short (a few minutes) as a rapid carrier-mediated diffusion occurs in the cells where they are metabolized [40,41]. It is very interesting to consider that ECs-mediated retrograde signaling is involved in the excitatory or inhibitory processes related to the modulation of neurotransmitters, such as glutamate or γ-aminobutyric acid [41,42,43,44], through short-term and long-term neuroplasticity (taking some seconds and some minutes, respectively) physiological processes [45,46]. The first is involved in processes such as depolarization-induced suppression of inhibition and depolarization-induced suppression of excitation through the inhibition of voltage-gated Ca2+ channels, whereas the second one leads to the long-term depression phenomenon through a CB1R repeated stimulation of these brain circuits. Consequently, CBR has to be considered a potential drug target for the prevention and treatment of neurologic pathologies, in particular, in the case of CNS involvement [47].
AEA is biosynthesized by the N-acyl phosphatidylethanol-selective phospholipase D [48]. It acts as a total or partial agonist of the CB1R and has low activity toward CB2R [49]. AEA comes up against rapid degradation due to its capture by a transporter [50,51], as occurs in the extracellular space of brain neurons and astrocytes [29], which is followed by the degradative action mainly carried out by fatty acid amide hydrolase (FAAH) [52,53,54,55], a homodimer integral membrane protein. The functional component of the enzyme consists of a catalytic triad formed by the amino acids Lys142-Ser217-Ser241, with the latter determining the nucleophilic attack on the electrophilic carbonyl group of AEA through the hydroxy group [56].
The biosynthesis of 2-AG begins with diacylglycerols and hydrolysis operated by the diacylglycerol lipase isoform α or β [57,58]. It acts as a full agonist of both CB1R and CB2R [59]. In addition, in the case of 2-AG, therefore, the molecule is captured by a transporter with characteristics identical or similar to those shown by AEA [60], which causes internalization and subsequent hydrolysis mainly by monoacylglycerol lipase (MGL) [61,62,63], an enzyme belonging to the α/β hydrolase superfamily. The mechanism involves the participation of various amino acids, which contribute to the initial preparatory phase aimed at catalytic activity by the Ser122-Asp239-His269 triad, where the serine residue is responsible for the nucleophilic action towards the carbonyl group of the substrate [64,65].
Taken together, the ECS certainly constitutes a reference model for drug discovery endeavors aimed at finding ideal molecules without the undesirable effects caused by the direct activation of CBRs [66]. With this goal, a fundamental role is played by compounds able to inhibit the enzymes that degrade natural ligands.
3. FAAH and MGL Inhibitors
Of particular interest is the discovery of the first FAAH enzyme inhibitors dates back just over twenty years ago, while those of MGL appeared a few years later. Since then, there have been numerous studies that have made it possible to expand the panorama of molecules available to the scientific community. The compounds that have emerged over time have the common need to be excellent substrates for enzymes. Therefore, all contain a reactive group able to favor the nucleophilic reaction. However, they differ from each other not only for the type of inhibition expressed (reversible vs. irreversible) but, above all, for the structural characteristics, which allow the opportunity to have various classes, and for potential therapeutic applications. A distinct description of the classes of FAAH and MGL inhibitors will be made here, in particular on those owing a therapeutic potential related to pathologies that may have a direct or indirect influence on psyche disorders. For anything not covered here, please refer to some excellent previously reported reviews [29,66,67,68,69,70].
The first study on the topic was published about twenty years ago, when covalent and irreversible carbamate FAAH inhibitors, such as the pharmacological tool URB597 (Figure 3), showed relevant anxiolytic-like and antidepressant-like properties related to the indirect activation of CB1R [71]. This discovery then opened the way to numerous further related opportunities, such as the possibility of using molecules of the URB series for new experimental models for depression [72,73], but also to test compounds belonging to different classes. The covalent and slow reversible piperazine-urea-based inhibitor JNJ-42165279 (Figure 3) [74] has been characterized and tested in clinical trials for the treatment of social anxiety disorders [75,76] and major depression. On the other hand, the covalent and irreversible piperidine-urea-based inhibitor PF-04457845 (Figure 3) [77] was able to attenuate the anxiety-inducing effect and entered clinical trials [78]. Finally, the reversible carbamate SSR411298 (Figure 3) exerts anxiolytic-like and antidepressant-like effects [79], but it fails clinical studies on major depression disorders [65].
Figure 3.

The 2D structures of selected FAAH inhibitors.
FAAH inhibitors could be considered to face nicotine or cannabis withdrawal syndromes. With regard to tobacco and cannabis disorders, URB597 has proved useful in reducing nicotine reward, preventing the reinstatement of nicotine use [80], and regulating acute and protracted nicotine withdrawal [81]. PF-04457845 was tested in the treatment of cannabis withdrawal and dependence in men [82].
Potential applications for neuropathic and inflammatory pain have been demonstrated with URB597 [83], which had also passed Phase I clinical trials several years ago (data not available), and URB937 (Figure 3) [84,85,86], which showed peripheral FAAH inhibition without the involvement of CNS. On the other hand, PF-04457845 produced potent CBR-dependent antinociceptive effects in both inflammatory and non-inflammatory arthritic pain models [87], but in related phase II clinical trials, it was not effective [88]. Moreover, JNJ-42165279 showed analgesic properties in a neuropathic pain model [74]. The piperidine-carbamate ASP8477 [89] and the azetidine-urea V158866 derivatives (Figure 3) [90] demonstrated significant analgesic effects in animal models of pain, but even in this case, the phase II clinical trials did not give the expected results [65]. For an overview of the influence of ECS in pain regulation, see [91], while for a FAAH inhibitor clinical perspective, see [92].
The study of MGL inhibitors has also led to the discovery of molecules with interesting profiles. Firstly, concerning anxiety and related disorders, it was observed that the covalent and irreversible piperidine-carbamate JZL184 (Figure 4) [93] attenuates anxiety-like behavior [94,95,96] and exerts an antidepressant-like effect [97].
Figure 4.

The 2D structures of selected FAAH inhibitors.
The first results on the potential of 2-AG in the field of pain were obtained in CB1R-dependent stress-induced analgesia employing the covalent and partial reversible carbamate inhibitor URB602 (Figure 4) [42], a molecule with the functional portion reversed in comparison to that present in FAAH inhibitors of the URB series. In the following years, other pharmacological tools have contributed to confirming the importance of inhibition of the MGL enzyme as a therapeutically relevant strategy. It was demonstrated that JZL184 induces a CB1R-mediated antinociceptive effect [93,98], whereas the covalent and irreversible piperazine-carbamate MJN110 (Figure 4) [99] alleviates mechanical allodynia [100] and exerts an antihyperalgesic effect through the increase in 2-AG levels [101], as well as the covalent and irreversible piperidine-carbamate KML29 (Figure 4) [102], which produces an antinociceptive activity in pain models [103]. Lastly, experiments carried out with covalent and irreversible pyridazine-carbamate SAR127303 (Figure 4) confirmed not only the ability to produce an analgesic effect in inflammation and pain models but also attenuate the symptoms of epilepsy [104]. The covalent and irreversible piperazine-carbamate ABX-1431 (Figure 4) [105] is effective in pain and neurological diseases and other disorders, such as Tourette syndrome [106], for which phase I clinical studies have been efficacious [65].
Other important results obtained by MGL inhibitors are related to the role of ECS in neuroprotection, which was demonstrated by using URB602 [107] and confirmed some years later through other drugs (for example, see [108]). A similar action is exerted by the covalent and irreversible inhibitor pyrrolidine-carbamate PF-06795071 (Figure 4) in reducing neuroinflammation markers in animal models [109].
Finally, it is interesting to consider some exemplary compounds reported as inhibitors of both enzymes. Indeed, covalent piperazine-carbamate-based inhibitors JZL195 [110] and SA-57 [111] (Figure 5) reduce inflammation-induced allodynia at a dose that does not cause side effects [112,113].
Figure 5.

The 2D structures of the selected FAAH/MGL inhibitors.
4. Primum Non Nocere
Some of the compounds reviewed in the previous sections entered clinical trials and were generally well tolerated being; the only adverse events the lack of efficacy or the appearance of fewer adverse reactions [66,67]. Thus, they might be expected to be relatively safe as antipsychotics. However, FAAH and MGL inhibitors should be carefully designed and go through accurate biochemical profiling and preclinical investigation before entering further clinical trials. The tragic outcome in the phase I clinical trial on the alleged FAAH inhibitor BIA10-2474 (Figure 6) (one dead volunteer and four others showing severe adverse reactions) does reinforce the caveat [68], but the relative negative outcomes may be puzzling [114]; however, in this specific scenario the misconduct of the trial must certainly be considered [115]. In any case, even where the inhibitors are still in the pipeline, we cannot let our guard down if those inhibitors are candidates for repositioning towards new clinical aims. Some of the FAAH and MGL inhibitors are under clinical evaluation for peripheral clinical activities targeting the peripheral tissues and the spinal cord; higher active doses might be expected for purported inhibitors acting on the brain. In view of the above, human toxicity may be unpredictable from a non-clinical toxicological perspective [116].
Figure 6.
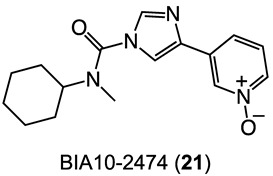
The 2D structure of BIA10-2474 (21).
Despite what was reported above, medicinal chemists must, to the best of their ability, predict possible toxic liabilities. Several chemoinformatic tools are available to run a preliminary analysis for drug toxicity prediction and usually refer to numerous safety-related properties [117]. To have an overview of the safety of the clinically relevant compounds chosen as candidates for repositioning, we started our analysis with cardiotoxicity (hERG liability), probably the worst early adverse reaction studied during clinical trials focused on acute toxicity [118,119]. To this aim, we employed a recently published ligand-based classifier based on the application of different machine learning algorithms and proved to outperform many predictors commonly used in the literature [120]. More specifically, the model has been developed by using an IC50 value equal to 10 μM as the threshold for discerning blockers from non-blockers and applies a consensus strategy based on two different algorithms, namely SVM (Support Vector Machine) and BRF (Balanced Random Forest), to provide the requested predictions. Importantly, all the compounds under investigation fall within the applicability domain of the model [121], hence supporting the reliability of the performed calculations. Table 1 shows the obtained results. Interestingly, none of the considered FAAH inhibitors was predicted as hERG blockers. Noteworthy, these data are in full agreement with the experimental literature available for URB597 [122], JNJ42165279 [74], PF-04457845 [109], and V158866 [90]. As far as the MGL and FAAH/MGL inhibitors are concerned, only three of them were predicted as able to induce hERG-related cardiotoxicity, namely JZL184, ABX-1431, and JZL195. Importantly, these data are again in agreement with the available literature as Grice et al. recently showed that ABX1431 is responsible for a significant hERG activity (IC50 = 7 μM) in vitro [105] while McAllister et al. provided experimental evidence that PF-06795071 is not a hERG blocker [109]. To the best of our knowledge, no data are instead available for JZL184 and JZL195. In summary, this preliminary investigation focused on the prediction of hERG-mediated cardiotoxicity indicates that it would be wise not to consider it an ideal candidate for repurposing JZL184, ABX-1431, and JZL195.
Table 1.
Results returned by the ligand-based classifier recently published by Delre et al. [120]. Notice that an IC50 = 10 μM was used as the threshold.
| Target Enzyme | Inhibitor | Predicted hERG Blocking Ability |
|---|---|---|
| FAAH | URB597 (5) | Non-blocker |
| URB937 (6) | Non-blocker | |
| JNJ-42165279 (7) | Non-blocker | |
| PF-04457845 (8) | Non-blocker | |
| SSR411298 (9) | Non-blocker | |
| ASP8477 (10) | Non-blocker | |
| V158866 (11) | Non-blocker | |
| MGL | JZL184 (12) | Blocker |
| KML29 (13) | Non-blocker | |
| URB602 (14) | Non-blocker | |
| MJN110 (15) | Non-blocker | |
| SAR127303 (16) | Non-blocker | |
| ABX-1431 (17) | Blocker | |
| PF-06795071(18) | Non-blocker | |
| FAAH/MGL | JZL195 (19) | Blocker |
| SA-57 (20) | Non-blocker |
5. Molecular ‘Beauty’ and ‘Ugly’ Compounds
Medicinal chemists look at their designed and synthesized compounds just like loving moms staring at their kids and generally do not resist the temptation to anthropomorphize their products. When speaking about the drug structural features that may be related to a higher probability of success, they refer to chemical ‘beauty’ (generally linked to a relatively high number of sp3 hybridized carbon atoms, Csp3), while ‘ugly’ molecules are tainted with some structural defect mostly related to ‘obesity’ (that is, high lipophilicity) that generally reduces that chance. Anthropomorphized jargon in drug design and discovery, while probably oversimplifying concepts, may be useful to make them easily appreciated, reinforce issues, and surrogate long periphrases. At worst, anthropomorphizing drug design is only a bit cryptic for non-aficionados. This is why the above esthetic evaluations have been somewhat codified in dozens of alternative or complementary metrics [123] that, through back-of-the-envelope-calculations allow both naïf and expert scholars to have a rapid outlook of favorable predicted pharmacokinetic (i.e., oral bioavailability) and pharmacodynamic (i.e., efficiency) properties of the designed compound [124,125].
As a first step, we computed the Quantitative Estimate of Drug-likeness (QED), which is an integrative score widely used to estimate the drug-likeness of a given small molecule [126]. In particular, good drug candidates are expected to return a QED > 0.6. The obtained data are reported in Table 2. As expected, most of the known inhibitors exceed this threshold. Few exceptions are represented by one FAAH inhibitor (PF-04457845), three MGL inhibitors (JZL184, KML29, and ABX-1431), and one dual FAAH/MGL inhibitor (JZL195). Noteworthy, all the compounds already predicted as hERG blockers returned low (< 0.6) QED values, thus supporting the idea whereby these inhibitors are not the best candidates as antipsychotics.
Table 2.
Drug-likeness and ADME properties were computed for the compounds under investigation. Notice that the QED values were obtained by using an in-house script based on the paper by Bickerton et al. [126], while the software program QikProp, available from the Schrodinger suite 2022-4 [127], was employed to compute all the other relevant ADME properties. QED values > 0.6 are highlighted in bold.
| Compound | QED | CNS | QPP Caco * |
QPlogBB ** | Human Oral Absorption |
Human Percent Oral Absorption |
Rule of Three |
|
|---|---|---|---|---|---|---|---|---|
| FAAH Inhibitors | URB597 (5) | 0.89 | −2 | 142.196 | −1.755 | 3 | 79.101 | 0 |
| URB937 (6) | 0.78 | −2 | 331.772 | −1.286 | 3 | 89.452 | 0 | |
| JNJ-42165279 (7) | 0.84 | 2 | 373.210 | 0.496 | 3 | 87.382 | 0 | |
| PF-04457845 (8) | 0.57 | 0 | 509.513 | −0.661 | 1 | 100.000 | 1 | |
| SSR-411298 (9) | 0.66 | −2 | 65.327 | −1.995 | 3 | 68.495 | 0 | |
| ASP8477 (10) | 0.94 | 0 | 1139.010 | −0.644 | 3 | 100.000 | 0 | |
| V158866 (11) | 0.78 | −1 | 964.142 | −0.878 | 3 | 100.000 | 0 | |
| MGL Inhibitors |
JZL184 (12) | 0.39 | −2 | 374.020 | −1.335 | 2 | 72.038 | 1 |
| KML29 (13) | 0.55 | 1 | 3100.818 | 0.221 | 1 | 100.000 | 1 | |
| URB602 (14) | 0.84 | 0 | 3551.379 | −0.089 | 3 | 100.000 | 1 | |
| MJN110 (15) | 0.65 | 1 | 130.559 | −0.400 | 3 | 85.779 | 0 | |
| SAR127303 (16) | 0.64 | 0 | 549.445 | −0.372 | 3 | 100.000 | 0 | |
| ABX-1431 (17) | 0.53 | 2 | 1125.247 | 1.180 | 1 | 89.621 | 1 | |
| PF-06795071 (18) | 0.80 | 0 | 1152.477 | −0.297 | 1 | 100.000 | 1 | |
| FAAH/ MGL Inhibitors |
JZL195 (19) | 0.41 | 0 | 131.320 | −0.842 | 3 | 88.922 | 0 |
| SA-57 (20) | 0.90 | −1 | 538.896 | −0.741 | 3 | 96.006 | 0 | |
| BIA10-2474 (21) | 0.63 | 0 | 854.649 | −0.429 | 3 | 94.821 | 0 | |
| CBD (2) | 0.51 | 0 | 2405.636 | −0.496 | 3 | 100.000 | 2 |
* QPPCaco: Predicted apparent Caco-2 cell permeability in nm/sec. ** QPlogBB: Logarithm of BBB predicted partition coefficient.
Obviously, to have an antipsychotic activity, a given drug must be able to act in the CNS. Furthermore, oral administration is highly desirable. Keeping this in mind, we also computed several descriptors related to the CNS activity as well as oral adsorption in humans using QikProp [125] as a software program, namely: (i) CNS, able to provide an estimation of the CNS activity on a −2 (inactive) to +2 (active) scale; (ii) QPPCaco, providing an estimation of the Caco-2 cell permeability in nm/sec (i.e., ability to cross the gut-blood barrier; <25 poor, >500 great); (iii) QPlogBB, providing an estimation of the brain/blood partition coefficient; (iv) HumanOralAbsorption, providing a score indicating a low (1), medium (2) and high (3) qualitative human oral absorption; (v) PercentHumanOralAbsorption, providing a score based on multiple linear regression on 0 to 100% scale (>80% is high; <25% is poor) and (vi) RuleOfThree; which is the number of violations of the so-called Jorgensen’s rule of three [128]. In particular, compounds responsible for no violations are expected to be orally available. The picture that emerged from data reported in Table 2 puts forward JNJ-42165279 (7) as the best candidate for oral antipsychotics. Remarkably, this FAAH inhibitor is predicted to have: (i) a high CNS activity (CNS = 2); (ii) good Caco-2 cell (QPPCaco = 373.210) and blood-brain barrier (QPlogBB = 0.496) permeabilities and (iii) very high oral adsorption in humans (HumanOralAdsorption = 3, PercentHumanOralAbsorption = 87.382 and RuleOfThree = 0). Noteworthy, JNJ42165279 is also responsible for a QED > 0.6 (Table 2) and has been predicted as not able to induce cardiotoxicity (Table 1). Although proven to block hERG [128], worth mentioning is herein also ABX1431 (17), which is the only compound [together with JNJ-42165279 (7)] predicted as responsible for a high CNS activity (CNS = 2). Finally, interesting data were also provided by MJN110 (15), predicted to be responsible for significant CNS activity (CNS = 1) and high oral availability (Table 2).
Finally, we predicted whether the compounds object of the present investigation reacts with one (or more) cytochrome P450 isoforms (among the most important ones, namely CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4). The prediction was performed using as software program CypReact, recently published by Tian et al. [129] and available in the recently published web platform for de novo design DeLA-Drug [130]. The obtained results are shown in Table 3.
Table 3.
Metabolic liability is predicted for all the investigated compounds. The prediction is performed using as software program CypReact.
| Compound | P450 Isoforms Predicted to React with the Ligand | |
|---|---|---|
| FAAH Inhibitors | URB597 (5) | 1A2 2C8 2C9 2C19 2D6 3A4 |
| URB937 (6) | 1A2 2B6 2C8 2C9 2C19 2D6 3A4 | |
| JNJ-42165279 (7) | 01A2 2C8 2C9 2C19 2D6 3A4 | |
| PF-04457845 (8) | 1A2 2D6 3A4 | |
| SSR-411298 (9) | 1A2 2B6 2C8 2C9 2C19 2D6 3A4 | |
| ASP8477 (10) | 1A2 2B6 2C8 2C9 2C19 2D6 3A4 | |
| V158866 (11) | 2C8 2D6 3A4 | |
| MGL Inhibitors |
JZL184 (12) | 1A2 2C8 2C19 2D6 3A4 |
| KML29 (13) | 1A2 2B6 2C9 2C19 2D6 3A4 | |
| URB602 (14) | 1A2 2B6 2C8 2C9 2C19 2D6 3A4 | |
| MJN110 (15) | 1A2 2B6 2C8 2C9 2C19 2D6 3A4 | |
| SAR127303 (16) | 1A2 2C8 2C9 2C19 2D6 3A4 | |
| ABX-1431 (17) | 1A2 2C8 2C9 2C19 2D6 3A4 | |
| PF-06795071 (18) | 1A2 2B6 2C8 2C9 2C19 2D6 2|E1 3A4 | |
| FAAH/ MGL Inhibitors |
JZL195 (19) | None |
| SA-57 (20) | None | |
| BIA10-2474 (21) | 1A2 2C8 2C9 2C19 2D6 3A4 | |
| CBD (2) | 1A2 2B6 2C9 2C19 2D6 3A4 |
All FAAH and MGL selective inhibitors should be substrates of several cytochrome P450 isoforms and thus possibly act as metabolic auto-/heteroinducers. Possible adverse events due to drug-drug metabolic interactions should be considered. Interestingly, both FAAH/MGL inhibitors 19 and 20 were predicted as poor substrates of all cytochrome P450 isoforms, thus performing as the best candidate for repositioning when metabolic liability is concerned.
A complementary analysis may be obtained using three of the most used and robust metrics to classify the selected inhibitors.
(1) As an indicator of promiscuity liability (i.e., the propensity to target not only enzymes but also receptors), the property forecast index has been proposed [131]. This index may be easily obtained as the sum of clogP and number of aromatic rings for each stated compound; as a rule of thumb, property forecast index should be kept below five to reduce possible promiscuity.
(2) CNS penetration may be forecasted from the CNS multiparameter optimization descriptor obtained by a linear combination of six parameters, each weighted one if in the desirable range, 0 when in the undesirable range, and scaled when falling between the targeted values (CNS multiparameter optimization desirability ≥ score, using a scale of 0–6) [132].
(3) As a ligand efficiency evaluator, the lipophilic efficient index is generally considered the most robust descriptor and represents a measure that correlates potency and lipophilicity. It is obtained from the difference between a measure of the potency of the ligand and its clogP and, therefore, can be considered as a measure of the gain in potency net of the (non-specific) entropic contribution due to the mere increase in lipophilicity [133]. An efficient inhibitor should locate its lipophilic efficient index between 5 and 7.
In Figure 7, the result of the above analysis is graphically summarized. The most efficient inhibitors endowed with the highest CNS tropism are represented by points in the colored parallelepiped. The highest-ranking compound was BIA10-2474 (21). Thus, compounds ASP8477 (10), V158866 (11), and SA-57 (20), while displaying high efficiency and good CNS penetration, should be considered carefully for possible toxicity issues stemming from their profiles that seem close to the one of BIA10-2474 (21). Interestingly, compounds JNJ-42165279 (7) and MJN110 (15) are located near the high efficiency/selectivity/CNS permeation volume and may be considered a good compromise between efficiency and safety requirements. It is worth noting that CBD (2) is the less efficient inhibitor, highly promiscuous, and presents acceptable CNS permeation properties. This outcome agrees with what has been reported so far on CBD pharmacology.
Figure 7.

Ligand efficiency metric analysis on the selected inhibitors 2, 5–21. Compounds falling within the colored volume are expected to display the highest efficiency, selectivity, and brain permeation. See text and Table S1 for details.
6. Discussion
A relatively lower incidence of psychosis is found in Spain and Italy than in Northern Europe [134]. This evidence parallels the higher risk of developing schizophrenia among those born and brought up in cities than in rural settings [135]. The question is open if the green color itself may condition mental health [136]. It is perceived to be associated with life itself, and green color perception may boost the placebo effect when administering drugs acting on the CNS, such as tranquilizing medicines. It has been suggested that patients with schizophrenia should have more contact with green [137]. Ironically, it has been insinuated that a certain kind of ‘contact’ with ‘green’ would be mostly deleterious to our mind, with a clear winking to cannabis abuse in western cities as a triggering agent of psychosis [138]. Cannabis seems to have also deleterious long-term effects [139], thus dooming both adolescents and adults to the use of antipsychotics with related cardiometabolic liabilities due to adverse drug effects [140]. However, the ambiguity of cannabis makes it potentially both harmful to the heart [141] and beneficial in cardiovascular diseases [142]. The drug may have clinical applications for the treatment of autism spectrum disorder [143], and CBD showed antipsychotic potential. Since the intervention on ECS degrading enzymes is generally considered more suitable for ECS physiological balance restoration than the use of agents directly acting on CBR, we have tried to infer the possibility of repositioning some known endocannabinoid degradation enzyme inhibitors as potential antipsychotics, as considered by Navarrete et al. [144].
Based on the performed predictions focused on CNS activity, oral adsorption, and ligand efficiency, the FAAH inhibitor JNJ-42165279 (7, Figure 3) seems to be the best candidate for this purpose, followed by the MGL inhibitor MJN110 (15, Figure 4). Other interesting inhibitors [e.g., ASP8477 (10), V158866 (11), ABX-1431 (17), and SA-57 (20)] may be considered as starting points for structural simplification/optimization endeavors to reduce possible hERG liability (17) and selectively favoring FAAH/MGL over other targets. None of the stated compounds is chiral, while chirality has been related to clinical success [145] and toxicity reduction [146]. Chiral analogs of the stated compounds could be easily designed and prepared to take advantage of chirality in the quest for safe and efficient inhibitors of FAAH and/or MGL as orally effective antipsychotics. Given the predicted favorable metabolic profile of SA-57 (20), lactamide (R = Me, Figure 8) and mandelamide (R = Ph, Figure 8) pyridyl carbamates could be considered as suitable starting points.
Figure 8.
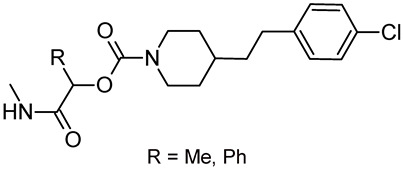
Possible chiral analogs of SA-57 (20).
Acknowledgments
Thanks are due to CNRS for financial support (J.P.M.). One of the authors [G.L. (Giovanni Lentini)] is indebted to Alberto Lentini for helpful hints about Excel use and data visualization in R (Plotly package).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines11020469/s1, Table S1: SMILES, Inhibiting Potency Values, Literature Sources of Data, and Ligand Efficiency Metrics for Compounds 2–21.
Author Contributions
Conceptualization, A.D. and G.L. (Giovanni Lentini); software, G.F.M., P.D., G.L. (Giuseppe Lamanna), M.C.L., M.S. and G.L. (Giovanni Lentini); validation, G.F.M., P.D., G.L. (Giuseppe Lamanna), M.C.L. and M.S.; formal analysis, G.F.M., M.M.C., P.D., G.L. (Giuseppe Lamanna), M.C.L., M.S. and G.L. (Giovanni Lentini); writing—original draft preparation, G.F.M., M.M.C., A.D. and G.L. (Giovanni Lentini); writing—review and editing, G.F.M., M.M.C., J.-P.M., S.M., A.D. and G.L. (Giovanni Lentini); supervision, G.L. (Giovanni Lentini); funding acquisition, G.F.M., S.M., A.D. and G.L. (Giovanni Lentini) All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research was supported by the National Research Council of Italy (G.F.M., P.D., G.L. (Giuseppe Lamanna), M.C.L., M.S.), University of Bari Aldo Moro (M.M.C., G.La, G.L. (Giovanni Lentini)), FCT-Fundação para a Ciência e a Tecnologia (Base Fund UIDB/00674/2020 and Programmatic Fund UIDP/00674/2020, Portuguese Government Funds) and ARDITI-Agência Regional para o Desenvolvimento da Investigação Tecnologia e Inovação through the project M1420-01-0145-FEDER-000005-CQM + (Madeira 14–20 Program) (S.M.), and the University of Urbino Carlo Bo (A.D.). The Ph.D. fellowship of Dr. Giuseppe Lamanna was co-funded by Chiesi Farmaceutici S.p.A. under the program “Dottorato Industriale CNR XXXVI ciclo.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Snyder C.R., Ford C.E., Harris R.N. The Effects of theoretical perspective on the analysis of coping with negative life events. In: Snyder C.R., Ford C.E., editors. Coping with Negative Life Events: Clinical and Social Psychological Perspectives. Plenum Press; New York, NY, USA: 1987. pp. 3–13. [DOI] [Google Scholar]
- 2.Spark D.L., Fornito A., Langmead C.J., Stewart G.D. Beyond antipsychotics: A twenty-first century update for preclinical development of schizophrenia therapeutics. Transl. Psychiatry. 2022;12:147. doi: 10.1038/s41398-022-01904-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González-Rodríguez A., Monreal J.A., Natividad M., Seeman M.V. Seventy years of treating delusional disorder with antipsychotics: A historical perspective. Biomedicines. 2022;10:3281. doi: 10.3390/biomedicines10123281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meltzer H.Y. An overview of the mechanism of action of clozapine. J. Clin. Psychiatry. 1994;55((Suppl. SB)):47–52. [PubMed] [Google Scholar]
- 5.Sotiropoulos M.G., Poulogiannopoulou E., Delis F., Dalla C., Antoniou K., Kokras N. Innovative screening models for the discovery of new schizophrenia drug therapies: An integrated approach. Expert Opin. Drug Discov. 2021;16:791–806. doi: 10.1080/17460441.2021.1877657. [DOI] [PubMed] [Google Scholar]
- 6.Azorin J.-M., Simon N. Antipsychotic polypharmacy in schizophrenia: Evolving evidence and rationale. Expert Opin. Drug Metab. Toxicol. 2020;16:1175–1186. doi: 10.1080/17425255.2020.1821646. [DOI] [PubMed] [Google Scholar]
- 7.Faden J., Kiryankova-Dalseth N., Barghini R., Citrome L. Does antipsychotic combination therapy reduce the risk of hospitalization in schizophrenia? Exp. Opin. Pharmacother. 2021;22:635–646. doi: 10.1080/14656566.2020.1847274. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J., Liu S., Wolf C.A., Wolber G., Parr M.K., Bureik M. Changes in alprazolam metabolism by CYP3A43 mutants. Biomedicines. 2022;10:3022. doi: 10.3390/biomedicines10123022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bulatova N., Altaher N., BaniMustafa R., Al-Saleh A., Yasin H., Zawiah M., Khalefah H., Ghilan M., Al-Lahham A., Hudaib M., et al. The effect of antipsychotics and their combinations with other psychotropic drugs on electrocardiogram intervals other than QTc among Jordanian adult outpatients. Biomedicines. 2023;11:13. doi: 10.3390/biomedicines11010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gebauer E.-M., Lukas A. Prescriptions of antipsychotics in younger and older geriatric patients with polypharmacy, their safety, and the impact of a pharmaceutical-medical dialogue on antipsychotic use. Biomedicines. 2022;10:3127. doi: 10.3390/biomedicines10123127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emrich H.M., Leweke F.M., Schneider U. Towards a cannabinoid hypothesis of schizophrenia: Cognitive impairments due to dysregulation of the endogenous cannabinoid system. Pharmacol. Biochem. Behav. 1997;56:803–807. doi: 10.1016/S0091-3057(96)00426-1. [DOI] [PubMed] [Google Scholar]
- 12.Leweke F.M., Mueller J.K., Lange B., Rohleder C. Therapeutic potential of cannabinoids in psychosis. Biol. Psychiatry. 2016;79:604–612. doi: 10.1016/j.biopsych.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 13.Graczyk M., Łukowicz M., Dzierzanowski T. Prospects for the use of cannabinoids in psychiatric disorders. Front. Psychiatry. 2021;12:620073. doi: 10.3389/fpsyt.2021.620073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An D., Peigneur S., Hendrickx L.A., Tytgat J. Targeting cannabinoid receptors: Current status and prospects of natural products. Int. J. Mol. Sci. 2020;21:5064. doi: 10.3390/ijms21145064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deventer M.H., Van Uytfanghe K., Vinckier I.M.J., Reniero F., Guillou C., Stove C.P. Cannabinoid receptor activation potential of the next generation, generic ban evading OXIZID synthetic cannabinoid receptor agonists. Drug Test. Anal. 2022;14:1565–1575. doi: 10.1002/dta.3283. [DOI] [PubMed] [Google Scholar]
- 16.Shafi A., Berry A.J., Sumnall H., Wood D.M., Tracy D.K. New psychoactive substances: A review and updates. Ther. Adv. Psychopharmacol. 2020;10:2045125320967197. doi: 10.1177/2045125320967197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markham J., Sparkes E., Boyd R., Chen S., Manning J.J., Finlay D., Lai F., McGregor E., Maloney C.J., Gerona R.R., et al. Defining steric requirements at CB1 and CB2 cannabinoid receptors using synthetic cannabinoid receptor agonists 5F-AB-PINACA, 5F-ADB-PINACA, PX-1, PX-2, NNL-1, and their analogues. ACS Chem. Neurosci. 2022;13:1281–1295. doi: 10.1021/acschemneuro.2c00034. [DOI] [PubMed] [Google Scholar]
- 18.Chang H.-A., Dai W., Hu S.S.-J. Sex differences in cocaine-associated memory: The interplay between CB1, mGluR5, and estradiol. Psychoneuroendocrinology. 2021;133:105366. doi: 10.1016/j.psyneuen.2021.105366. [DOI] [PubMed] [Google Scholar]
- 19.Morrison P.D., Murray R.M. The antipsychotic landscape: Dopamine and beyond. Ther. Adv. Psychopharmacol. 2018;8:127–135. doi: 10.1177/2045125317752915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leweke F.M. Anandamide dysfunction in prodromal and established psychosis. Curr. Pharm. Des. 2012;18:5188–5193. doi: 10.2174/138161212802884843. [DOI] [PubMed] [Google Scholar]
- 21.Rohleder C., Müller J.K., Lange B., Leweke F.M. Cannabidiol as a potential new type of an antipsychotic. A critical review of the evidence. Front. Pharmacol. 2016;7:422. doi: 10.3389/fphar.2016.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Criscuolo E., De Sciscio M.L., Fezza F., Maccarrone M. In silico and in vitro analysis of major cannabis-derived compounds as fatty acid amide hydrolase inhibitors. Molecules. 2021;26:48. doi: 10.3390/molecules26010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leweke F.M., Piomelli D., Pahlisch F., Muhl D., Gerth C.W., Hoyer C., Klosterkötter J., Hellmich M., Koethe D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maccarrone M. Missing pieces to the endocannabinoid puzzle. Trends Mol. Med. 2020;26:263–272. doi: 10.1016/j.molmed.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Duranti A., Beldarrain G., Álvarez A., Sbriscia M., Carloni S., Balduini W., Alonso-Alconada D. The endocannabinoid system as a target for neuroprotection/neuroregeneration in perinatal hypoxic–ischemic brain injury. Biomedicines. 2023;11:28. doi: 10.3390/biomedicines11010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mangiatordi G.F., Intranuovo F., Delre P., Abatematteo F.S., Abate C., Niso M., Creanza T.M., Ancona N., Stefanachi A., Contino M. Cannabinoid receptor subtype 2 (CB2R) in a multitarget approach: Perspective of an innovative strategy in cancer and neurodegeneration. J. Med. Chem. 2020;63:14448–14469. doi: 10.1021/acs.jmedchem.0c01357. [DOI] [PubMed] [Google Scholar]
- 27.Ren S., Wang Z., Zhang Y., Chen N. Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—Focusing on FAAH/MAGL inhibitors. Acta Pharmacol. Sin. 2020;41:1263–1271. doi: 10.1038/s41401-020-0385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe H., Toyang N., Steele B., Bryant J., Ngwa W. The endocannabinoid system: A potential target for the treatment of various diseases. Int. J. Mol. Sci. 2021;22:9472. doi: 10.3390/ijms22179472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piomelli D., Mabou Tagne A. Endocannabinoid-based therapies. Annu. Rev. Pharmacol. Toxicol. 2022;62:483–507. doi: 10.1146/annurev-pharmtox-052220-021800. [DOI] [PubMed] [Google Scholar]
- 30.Intranuovo F., Brunetti L., DelRe P., Mangiatordi G.F., Stefanachi A., Laghezza A., Niso M., Leonetti F., Loiodice F., Ligresti A., et al. Development of N-(1-adamantyl)benzamides as novel anti-inflammatory multitarget agents acting as dual modulators of the cannabinoid CB2 receptor and fatty acid amide hydrolase. J. Med. Chem. 2022;66:235–250. doi: 10.1021/acs.jmedchem.2c01084. [DOI] [PubMed] [Google Scholar]
- 31.Devane W.A., Dysarz F.A., Johnson M.R., Melvin L.S., Howlett A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 32.Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 33.Shahbazi F., Grandi V., Banerjee A., Trant J.F. Cannabinoids and cannabinoid receptors: The story so far. IScience. 2020;23:101301. doi: 10.1016/j.isci.2020.101301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang X., Liu Z., Li X., Wang J., Li L. Cannabinoid receptors in myocardial injury: A brother born to rival. Int. J. Mol. Sci. 2021;22:6886. doi: 10.3390/ijms22136886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz B. Neurobiology of cannabinoid receptor signaling. Dialogues Clin. Neurosci. 2020;22:207–222. doi: 10.31887/DCNS.2020.22.3/blutz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felder C.C., Joyce K.E., Briley E.M., Mansouri J., Mackie K., Blond O., Lai Y., Ma A.L., Mitchell R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- 37.Devane W.A., Hanuš L., Breuer A., Pertwee R.G., Stevenson L.A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 38.Mechoulam R., Ben-Shabat S., Hanus L., Ligumsky M., Kaminski N.E., Schatz A.R., Gopher A., Almog S., Martin B.R., Compton D.R., et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-D. [DOI] [PubMed] [Google Scholar]
- 39.Sugiura T., Kondo S., Sukagawa A., Nakane S., Shinoda A., Itoh K., Yamashita A., Waku K. 2-Arachidonoylgylcerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 40.Piomelli D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 41.Augustin S.M., Lovinger D.M. Functional Relevance of endocannabinoid-dependent synaptic plasticity in the central nervous system. ACS Chem. Neurosci. 2018;9:2146–2161. doi: 10.1021/acschemneuro.7b00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diana M.A., Marty A. Endocannabinoid-mediated short-term synaptic plasticity: Depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br. J. Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hohmann A.G., Suplita R.L., Bolton N.M., Neely M.H., Fegley D., Mangieri R., Krey J.F., Walker J.M., Holmes P.V., Crystal J.D., et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 44.Castillo P.E., Younts T.J., Chávez A.E., Hashimotodani Y. Endocannabinoid Signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alger B.E. Retrograde Signaling in the regulation of synaptic transmission: Focus on endocannabinoids. Prog. Neurobiol. 2002;68:247–286. doi: 10.1016/S0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- 46.Makara J.K., Mor M., Fegley D., Szabó S.I., Kathuria S., Astarita G., Duranti A., Tontini A., Tarzia G., Rivara S., et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- 47.Estrada J.A., Contreras I. Endocannabinoid receptors in the CNS: Potential drug targets for the prevention and treatment of neurologic and psychiatric disorders. Curr. Neuropharmacol. 2020;18:769–787. doi: 10.2174/1570159X18666200217140255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Marzo V., Fontana A., Cadas H., Schinelli S., Cimino G., Schwartz J.C., Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 49.Hillard C.J., Campbell W.B. Biochemistry and pharmacology of arachidonylethanolamide, a putative endogenous cannabinoid. J. Lipid Res. 1997;38:2383–2398. doi: 10.1016/S0022-2275(20)30024-9. [DOI] [PubMed] [Google Scholar]
- 50.Hillard C.J., Edgemond W.S., Jarrahian A., Campbell W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- 51.Beltramo M., Stella N., Calignano A., Lin S.Y., Makriyannis A., Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 52.Desarnaud F., Cadas H., Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J. Biol. Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 53.Hillard C.J., Wilkison D.M., Edgemond W.S., Campbell W.B. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim. Biophys. Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-S. [DOI] [PubMed] [Google Scholar]
- 54.Ueda N., Kurahashi Y., Yamamoto S., Tokunaga T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- 55.Cravatt B.F., Giang D.K., Mayfield S.P., Boger D.L., Lerner R.A., Gilula N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 56.Patricelli M.P., Cravatt B.F. Clarifying the catalytic roles of conserved residues in the amidase signature family. J. Biol. Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]
- 57.Farooqui A.A., Rammohan K.W., Horrocks L.A. Isolation, characterization, and regulation of diacylglycerol lipases from the bovine brain. Ann. N. Y. Acad. Sci. 1989;559:25–36. doi: 10.1111/j.1749-6632.1989.tb22596.x. [DOI] [PubMed] [Google Scholar]
- 58.Bisogno T., Howell F., Williams G., Minassi A., Cascio M.G., Ligresti A., Matias I., Schiano-Moriello A., Paul P., Williams E.-J., et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugiura T., Waku K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem. Phys. Lipids. 2000;108:89–106. doi: 10.1016/S0009-3084(00)00189-4. [DOI] [PubMed] [Google Scholar]
- 60.Beltramo M., Piomelli D. Carrier-mediated transport and enzymatic hydrolysis of the endogenous cannabinoid 2-arachidonylglycerol. NeuroReport. 2000;11:1231–1235. doi: 10.1097/00001756-200004270-00018. [DOI] [PubMed] [Google Scholar]
- 61.Tornqvist H., Belfrage P. Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J. Biol. Chem. 1976;251:813–819. doi: 10.1016/S0021-9258(17)33857-7. [DOI] [PubMed] [Google Scholar]
- 62.Prescott S.M., Majerus P.W. Characterization of 1,2-diacylglycerol hydrolysis in human platelets. Demonstration of an arachidonoyl-monoacylglycerol intermediate. J. Biol. Chem. 1983;258:764–769. doi: 10.1016/S0021-9258(18)33114-4. [DOI] [PubMed] [Google Scholar]
- 63.Farooqui A.A., Taylor W.A., Horrocks L.A. Separation of bovine brain mono- and diacylglycerol lipases by heparin sepharose affinity chromatography. Biochem. Biophys. Res. Commun. 1984;122:1241–1246. doi: 10.1016/0006-291X(84)91225-7. [DOI] [PubMed] [Google Scholar]
- 64.Karlsson M., Contreras J.A., Hellman U., Tornqvist H., Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 65.Labar G., Bauvois C., Borel F., Ferrer J.-L., Wouters J., Lambert D.M. Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. ChemBioChem. 2010;11:218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 66.van Egmond N., Straub V.M., van der Stelt M. Targeting endocannabinoid signaling: FAAH and MAG lipase inhibitors. Annu. Rev. Pharmacol. Toxicol. 2021;61:441–463. doi: 10.1146/annurev-pharmtox-030220-112741. [DOI] [PubMed] [Google Scholar]
- 67.Tuo W., Leleu-Chavain N., Spencer J., Sansook S., Millet R., Chavatte P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem. 2017;60:4–46. doi: 10.1021/acs.jmedchem.6b00538. [DOI] [PubMed] [Google Scholar]
- 68.Tripathi R.K.P. A perspective review on fatty acid amide hydrolase (FAAH) inhibitors as potential therapeutic agents. Eur. J. Med. Chem. 2020;188:111953. doi: 10.1016/j.ejmech.2019.111953. [DOI] [PubMed] [Google Scholar]
- 69.Fazio D., Criscuolo E., Piccoli A., Barboni B., Fezza F., Maccarrone M. Advances in the discovery of fatty acid amide hydrolase inhibitors: What does the future hold? Exp. Opin. Drug Discov. 2020;15:765–778. doi: 10.1080/17460441.2020.1751118. [DOI] [PubMed] [Google Scholar]
- 70.Kashyap A., Kumar S., Dutt R. A Review on Structurally Diversified Synthesized Molecules as Monoacylglycerol Lipase Inhibitors and their Therapeutic uses. Curr. Drug Res. Rev. 2022;14:96–115. doi: 10.2174/2589977514666220301111457. [DOI] [PubMed] [Google Scholar]
- 71.Kathuria S., Gaetani S., Fegley D., Valiño F., Duranti A., Tontini A., Mor M., Tarzia G., Rana G.L., Calignano A., et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 72.Gobbi G., Bambico F.R., Mangieri R., Bortolato M., Campolongo P., Solinas M., Cassano T., Morgese M.G., Debonnel G., Duranti A., et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Nat. Acad. Sci. USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bortolato M., Mangieri R.A., Fu J., Kim J.H., Arguello O., Duranti A., Tontini A., Mor M., Tarzia G., Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 74.Keith J.M., Jones W.M., Tichenor M., Liu J., Seierstad M., Palmer J.A., Webb M., Karbarz M., Scott B.P., Wilson S.J., et al. Preclinical Characterization of the FAAH Inhibitor JNJ-42165279. ACS Med. Chem. Lett. 2015;6:1204–1208. doi: 10.1021/acsmedchemlett.5b00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paulus M.P., Stein M.B., Simmons A.N., Risbrough V.B., Halter R., Chaplan S.R. The effects of FAAH inhibition on the neural basis of anxiety-related processing in healthy male subjects: A randomized clinical trial. Neuropsychopharmacology. 2021;46:1011–1019. doi: 10.1038/s41386-020-00936-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmidt M.E., Liebowitz M.R., Stein M.B., Grunfeld J., Van Hove I., Simmons W.K., Van Der Ark P., Palmer J.A., Saad Z.S., Pemberton D.J., et al. The effects of inhibition of fatty acid amide hydrolase (FAAH) by JNJ-42165279 in social anxiety disorder: A double-blind, randomized, placebo-controlled proof-of-concept study. Neuropsychopharmacology. 2021;46:1004–1010. doi: 10.1038/s41386-020-00888-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson D.S., Stiff C., Lazerwith S.E., Kesten S.R., Fay L.K., Morris M., Beidler D., Liimatta M.B., Smith S.E., Dudley D.T., et al. Discovery of PF-04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med. Chem. Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mayo L.M., Asratian A., Lindé J., Morena M., Haataja R., Hammar V., Augier G., Hill M.N., Heilig M. Elevated anandamide, enhanced recall of fear extinction, and attenuated stress responses following inhibition of fatty acid amide hydrolase: A randomized, controlled experimental medicine trial. Biol. Psychiatry. 2020;87:538–547. doi: 10.1016/j.biopsych.2019.07.034. [DOI] [PubMed] [Google Scholar]
- 79.Griebel G., Stemmelin J., Lopez-Grancha M., Fauchey V., Slowinski F., Pichat P., Dargazanli G., Abouabdellah A., Cohen C., Bergis O.E. The selective reversible FAAH inhibitor, SSR411298, restores the development of maladaptive behaviors to acute and chronic stress in rodents. Sci. Rep. 2018;8:2416. doi: 10.1038/s41598-018-20895-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Justinova Z., Panlilio L.V., Moreno-Sanz G., Redhi G.H., Auber A., Secci M.E., Mascia P., Bandiera T., Armirotti A., Bertorelli R., et al. Effects of fatty acid amide hydrolase (FAAH) inhibitors in non-human primate models of nicotine reward and relapse. Neuropsychopharmacology. 2015;40:2185–2197. doi: 10.1038/npp.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cippitelli A., Astarita G., Duranti A., Caprioli G., Ubaldi M., Stopponi S., Kallupi M., Sagratini G., Rodrìguez de Fonseca F., Piomelli D., et al. Endocannabinoid regulation of acute and protracted nicotine withdrawal: Effect of FAAH inhibition. PLoS ONE. 2011;6:e28142. doi: 10.1371/journal.pone.0028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D’Souza D.C., Cortes-Briones J., Creatura G., Bluez G., Thurnauer H., Deaso E., Bielen K., Surti T., Radhakrishnan R., Gupta A., et al. Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: A double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry. 2019;6:35–45. doi: 10.1016/S2215-0366(18)30427-9. [DOI] [PubMed] [Google Scholar]
- 83.Russo R., LoVerme J., Rana G.L., Compton T.R., Parrott J., Duranti A., Tontini A., Mor M., Tarzia G., Calignano A., et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J. Pharmacol. Exp. Ther. 2007;322:236–342. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 84.Clapper J.R., Moreno-Sanz G., Russo R., Guijarro A., Vacondio F., Duranti A., Tontini A., Sanchini S., Sciolino N.R., Spradley J.M., et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moreno-Sanz G., Duranti A., Melzig L., Fiorelli C., Ruda G.F., Colombano G., Mestichelli P., Sanchini S., Tontini A., Mor M., et al. Synthesis and structure–activity relationship studies of O-biphenyl-3-yl carbamates as peripherally restricted fatty acid amide hydrolase inhibitors. J. Med. Chem. 2013;56:5917–5930. doi: 10.1021/jm4007017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vozella V., Ahmed F., Choobchian P., Merrill C.B., Zibardi C., Tarzia G., Mor M., Duranti A., Tontini A., Rivara S., et al. Pharmacokinetics, pharmacodynamics and safety studies on URB937, a peripherally restricted fatty acid amide hydrolase inhibitor, in rats. J. Pharm. Pharmacol. 2019;71:1762–1773. doi: 10.1111/jphp.13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahn K., Smith S.E., Liimatta M.B., Beidler D., Sadagopan N., Dudley D.T., Young T., Wren P., Zhang Y., Swaney S., et al. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huggins J.P., Smart T.S., Langman S., Taylor L., Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 89.Watabiki T., Tsuji N., Kiso T., Ozawa T., Narazaki F., Kakimoto S. In vitro and in vivo pharmacological characterization of ASP8477: A novel highly selective fatty acid amide hydrolase inhibitor. Eur. J. Pharmacol. 2017;815:42–48. doi: 10.1016/j.ejphar.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 90.Pawsey S., Wood M., Browne H., Donaldson K., Christie M., Warrington S. Safety, tolerability and pharmacokinetics of FAAH inhibitor V158866: A double-blind, randomised, placebo-controlled phase I study in healthy volunteers. Drugs RD. 2016;16:181–191. doi: 10.1007/s40268-016-0127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Finn D.P., Haroutounian S., Hohmann A.G., Krane E., Soliman N., Rice A.S.C. Cannabinoids, the endocannabinoid system, and pain: A review of preclinical studies. Pain. 2021;162((Suppl. S1)):S5–S25. doi: 10.1097/j.pain.0000000000002268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santoso A.D., De Ridder D. Fatty acid amide hydrolase: An integrative clinical perspective. Cannabis Cannabinoid Res. 2022 doi: 10.1089/can.2021.0237. [DOI] [PubMed] [Google Scholar]
- 93.Long J.Z., Li W., Booker L., Burston J.J., Kinsey S.G., Schlosburg J.E., Pavón F.J., Serrano A.M., Selley D.E., Parsons L.H., et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sciolino N.R., Zhou W., Hohmann A.G. Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol. Res. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bluett R.J., Báldi R., Haymer A., Gaulden A.D., Hartley N.D., Parrish W.P., Baechle J., Marcus D.J., Mardam-Bey R., Shonesy B.C., et al. Endocannabinoid signalling modulates susceptibility to traumatic stress exposure. Nat. Commun. 2017;8:14782. doi: 10.1038/ncomms14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ivy D., Palese F., Vozella V., Fotio Y., Yalcin A., Ramirez G., Mears D., Wynn G., Piomelli D. Cannabinoid CB2 receptors mediate the anxiolytic-like effects of monoacylglycerol lipase inhibition in a rat model of predator-induced fear. Neuropsychopharmacology. 2020;45:1330–1338. doi: 10.1038/s41386-020-0696-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Z., Wang W., Zhong P., Liu S.J., Long J.Z., Zhao L., Gao H.-Q., Cravatt B.F., Liu Q.-S. Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus. 2015;25:16–26. doi: 10.1002/hipo.22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kinsey S.G., Wise L.E., Ramesh D., Abdullah R., Selley D.E., Cravatt B.F., Lichtman A.H. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1–mediated antinociceptive and gastroprotective effects. J. Pharmacol. Exp. Ther. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang J.W., Cognetta A.B.I., Niphakis M.J., Cravatt B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013;8:1590–1599. doi: 10.1021/cb400261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Niphakis M.J., Cognetta A.B.I., Chang J.W., Buczynski M.W., Parsons L.H., Byrne F., Burston J.J., Chapman V., Cravatt B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013;4:1322–1332. doi: 10.1021/cn400116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wilkerson J.L., Niphakis M.J., Grim T.W., Mustafa M.A., Abdullah R.A., Poklis J.L., Dewey W.L., Akbarali H., Banks M.L., Wise L.E., et al. The selective monoacylglycerol lipase inhibitor MJN110 produces opioid-sparing effects in a mouse neuropathic pain model. J. Pharmacol. Exp. Ther. 2016;357:145–156. doi: 10.1124/jpet.115.229971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chang J.W., Niphakis M.J., Lum K.M., Cognetta A.B., Wang C., Matthews M.L., Niessen S., Buczynski M.W., Parsons L.H., Cravatt B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012;19:579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ignatowska-Jankowska B.M., Ghosh S., Crowe M.S., Kinsey S.G., Niphakis M.J., Abdullah R.A., Tao Q., O’Neal S.T., Walentiny D.M., Wiley J.L., et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2014;171:1392–1407. doi: 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Griebel G., Pichat P., Beeské S., Leroy T., Redon N., Jacquet A., Françon D., Bert L., Even L., Lopez-Grancha M., et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015;5:7642. doi: 10.1038/srep07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cisar J.S., Weber O.D., Clapper J.R., Blankman J.L., Henry C.L., Simon G.M., Alexander J.P., Jones T.K., Ezekowitz R.A.B., O’neill G.P., et al. Identification of ABX-1431, a Selective inhibitor of monoacylglycerol lipase and clinical candidate for treatment of neurological disorders. J. Med. Chem. 2018;61:9062–9084. doi: 10.1021/acs.jmedchem.8b00951. [DOI] [PubMed] [Google Scholar]
- 106.Jiang M., van der Stelt M. Activity-based protein profiling delivers selective drug candidate ABX-1431, a monoacylglycerol lipase inhibitor, to control lipid metabolism in neurological disorders. J. Med. Chem. 2018;61:9059–9061. doi: 10.1021/acs.jmedchem.8b01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carloni S., Alonso-Alconada D., Girelli S., Duranti A., Tontini A., Piomelli D., Hilario E., Alvarez A., Balduini W. Pretreatment with the monoacylglycerol lipase inhibitor URB602 protects from the long-term consequences of neonatal hypoxic–ischemic brain injury in rats. Pediatr. Res. 2012;72:400–406. doi: 10.1038/pr.2012.91. [DOI] [PubMed] [Google Scholar]
- 108.Carloni S., Crinelli R., Palma L., Álvarez F.J., Piomelli D., Duranti A., Balduini W., Alonso-Alconada D. The synthetic cannabinoid URB447 reduces brain injury and the associated white matter demyelination after hypoxia-ischemia in neonatal rats. ACS Chem. Neurosci. 2020;11:1291–1299. doi: 10.1021/acschemneuro.0c00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McAllister L.A., Butler C.R., Mente S., O’Neil S.V., Fonseca K.R., Piro J.R., Cianfrogna J.A., Foley T.L., Gilbert A.M., Harris A.R., et al. Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation. J. Med. Chem. 2018;61:3008–3026. doi: 10.1021/acs.jmedchem.8b00070. [DOI] [PubMed] [Google Scholar]
- 110.Long J.Z., Nomura D.K., Vann R.E., Walentiny D.M., Booker L., Jin X., Burston J.J., Sim-Selley L.J., Lichtman A.H., Wiley J.L., et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Nat. Acad. Sci. USA. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niphakis M.J., Johnson D.S., Ballard T.E., Stiff C., Cravatt B.F. O-Hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2012;3:418–426. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anderson W.B., Gould M.J., Torres R.D., Mitchell V.A., Vaughan C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacol. 2014;81:224–230. doi: 10.1016/j.neuropharm.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 113.Wilkerson J.L., Ghosh S., Mustafa M., Abdullah R.A., Niphakis M.J., Cabrera R., Maldonado R.L., Cravatt B.F., Lichtman A.H. The endocannabinoid hydrolysis inhibitor SA-57: Intrinsic antinociceptive effects, augmented morphine-induced antinociception, and attenuated heroin seeking behavior in mice. Neuropharmacol. 2017;114:156–167. doi: 10.1016/j.neuropharm.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rocha J.-F., Santos A., Gama H., Moser P., Falcão A., Pressman P. Safety, Tolerability, and Pharmacokinetics of FAAH Inhibitor BIA 10-2474: A double-blind, randomized, placebo-controlled study in healthy volunteers. Clin. Pharmacol. Ther. 2022;111:391–403. doi: 10.1002/cpt.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaur R., Sidhu P., Singh S. What failed BIA 10–2474 Phase I clinical trial? Global speculations and recommendations for future Phase I trials. J. Pharmacol. Pharmacother. 2016;7:120–126. doi: 10.4103/0976-500X.189661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hayes A.W., Weber K., Moser P., Soares-da-Silva P. Non-clinical toxicology evaluation of BIA 10-2474. Crit. Rev. Toxicol. 2021;51:65–75. doi: 10.1080/10408444.2020.1867821. [DOI] [PubMed] [Google Scholar]
- 117.Ferreira L.L.G., Andricopulo A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today. 2019;24:1157–1165. doi: 10.1016/j.drudis.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 118.Cavalluzzi M.M., Imbrici P., Gualdani R., Stefanachi A., Mangiatordi G.F., Lentini G., Nicolotti O. Human ether-à-go-go-related potassium channel: Exploring SAR to improve drug design. Drug Discov. Today. 2020;25:344–366. doi: 10.1016/j.drudis.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 119.Creanza T.M., Delre P., Ancona N., Lentini G., Saviano M., Mangiatordi G.F. Structure-Based Prediction of hERG-Related Cardiotoxicity: A Benchmark Study. J. Chem. Inf. Model. 2021;61:4758–4770. doi: 10.1021/acs.jcim.1c00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Delre P., Lavado G.J., Lamanna G., Saviano M., Roncaglioni A., Benfenati E., Mangiatordi G.F., Gadaleta D. Ligand-based prediction of hERG-mediated cardiotoxicity based on the integration of different machine learning techniques. Front. Pharmacol. 2022;13:951083. doi: 10.3389/fphar.2022.951083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gadaleta D., Mangiatordi G.F., Catto M., Carotti A., Nicolotti O. Applicability domain for QSAR models: Where theory meets reality. Int. J. Quant. Struct. Prop. Relatsh. 2016;1:45–63. doi: 10.4018/IJQSPR.2016010102. [DOI] [Google Scholar]
- 122.Grillo A., Chemi G., Brogi S., Brindisi M., Relitti N., Fezza F., Fazio D., Castelletti L., Perdona E., Wong A., et al. Development of novel multipotent compounds modulating endocannabinoid and dopaminergic systems. Eur. J. Med. Chem. 2019;183:111674. doi: 10.1016/j.ejmech.2019.111674. [DOI] [PubMed] [Google Scholar]
- 123.Cavalluzzi M.M., Mangiatordi G.F., Nicolotti O., Lentini G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017;12:1087–1104. doi: 10.1080/17460441.2017.1365056. [DOI] [PubMed] [Google Scholar]
- 124.Mignani S., Rodrigues J., Tomas H., Jalal R., Singh P.P., Majoral J.-P., Vishwakarma R.A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: How far can they be simplified? Drug Discov. Today. 2018;23:605–615. doi: 10.1016/j.drudis.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 125.Scott J.S., Waring M.J. Practical application of ligand efficiency metrics in lead optimisation. Bioorg. Med. Chem. 2018;26:3006–3015. doi: 10.1016/j.bmc.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 126.Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schrödinger Release 2022-4: QikProp. Schrödinger, LLC; New York, NY, USA: 2021. [Google Scholar]
- 128.Jorgensen W.L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tian S., Djoumbou-Feunang Y., Greiner R., Wishart D.S. CypReact: A software tool for in silico reactant prediction for human cytochrome P450 enzymes. J. Chem. Inf. Model. 2018;58:1282–1291. doi: 10.1021/acs.jcim.8b00035. [DOI] [PubMed] [Google Scholar]
- 130.Creanza T.M., Lamanna G., Delre P., Contino M., Corriero N., Saviano M., Mangiatordi G.F., Ancona N. DeLA-Drug: A deep learning algorithm for automed design of druglike analogues. J. Chem. Inf. Model. 2022;62:1411–1424. doi: 10.1021/acs.jcim.2c00205. [DOI] [PubMed] [Google Scholar]
- 131.Young R.J., Green D.V.S., Luscombe C.N., Hill A.P. Getting physical in drug discovery II: The impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discov. Today. 2011;16:822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 132.Wager T.T., Hou X., Verhoest P.R., Villalobos A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Leeson P.D., Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 134.Jongsma H.E., Gayer-Andreson C., Lasalvia A., Quattrone D., Mulè A., Szoke A., Selten J.-P., Turner C., Arango C., Tarriicone I., et al. Treated incidence of psychotic disorders in the multinational EU–GEI study. JAMA Psychiatry. 2018;75:36–46. doi: 10.1001/jamapsychiatry.2017.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McGrath J., Saha S., Welham J., El Saadi O., MacCauley C., Chant D. A systematic review of the incidence of schizophrenia: The distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Medicine. 2004;2:13. doi: 10.1186/1741-7015-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lentini G., Milani G., Habtemariam S. The therapeutic power of green. Schizophr. Res. 2019;210:310. doi: 10.1016/j.schres.2019.06.024. [DOI] [PubMed] [Google Scholar]
- 137.Tao B., Xu S., Pan X., Gao Q., Wang W. Personality trait correlates of color preference in schizophrenia. Transl. Neurosci. 2015;6:174–178. doi: 10.1515/tnsci-2015-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Murray R.M. Is the grass greener for those at risk of psychosis? Schizophr. Res. 2018;201:35–36. doi: 10.1016/j.schres.2018.04.041. [DOI] [PubMed] [Google Scholar]
- 139.Portugalov A., Akirav I. Do adolescent exposure to cannabinoids and early adverse experience interact to increase the risk of psychiatric disorders: Evidence from rodent models. Int. J. Mol. Sci. 2021;22:730. doi: 10.3390/ijms22020730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Matera E., Cristofano G., Furente F., Marzulli L., Tarantini M., Margari L., Piarulli F.M., De Giacomo A., Petruzzelli M.G. Glucose and lipid profiles predict anthropometric changes in drug-naïve adolescents starting treatment with risperidone or sertraline: A pilot study. Biomedicines. 2023;11:48. doi: 10.3390/biomedicines11010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weresa J., Pędzińska-Betiuk A., Mińczuk K., Malinowska B., Schlicker E. Why do marijuana and synthetic cannabimimetics induce acute myocardial infarction in healthy young people? Cells. 2022;11:1142. doi: 10.3390/cells11071142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Navarrete C., Garcia-Martin A., DeMesa J., Muñoz E. Cannabinoids in metabolic syndrome and cardiac fibrosis. Curr. Hypertens. Rep. 2020;22:98. doi: 10.1007/s11906-020-01112-7. [DOI] [PubMed] [Google Scholar]
- 143.da Silva E.A., Medeiros W.M.B., Torro N., de Sousa J.M.M., de Almeida I.B.C.M., da Costa F.B., Pontes K.M., Nunes E.L.G., da Rosa M.D., de Albuquerque K.L.G.D. Cannabis and cannabinoid use in autism spectrum disorder: A systematic review. Trends Psychiatry Psychother. 2022;44:e20200149. doi: 10.47626/2237-6089-2020-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Navarrete F., García-Gutiérrez M.S., Jurado-Barba R., Rubio G., Gasparyan A., Austrich-Olivares A., Manzaranes J. Endocannabinoid system components as potential biomarkers in psychiatry. Mol. Psychiatry. 2020;11:315. doi: 10.3389/fpsyt.2020.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lovering F., Bikker J., Humblet C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 146.Lentini G., Cavalluzzi M.M., Habtemariam S. COVID-19, chloroquine repurposing, and cardiac safety concern: Chirality might help. Molecules. 2020;25:1834. doi: 10.3390/molecules25081834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable.