

Abstract
Purpose:
To characterize changes in the soft tissue sarcoma tumor immune microenvironment induced by standard neoadjuvant therapy with the goal of informing neoadjuvant immunotherapy trial design.
Experimental Design:
Paired pre- and post-neoadjuvant therapy specimens were retrospectively identified for 32 patients with soft tissue sarcomas and analyzed by three modalities: multiplexed immunohistochemistry, NanoString, and RNA sequencing with ImmunoPrism analysis.
Results:
All 32 patients, representing a variety of soft tissue sarcoma histologic subtypes, received neoadjuvant radiotherapy and 21 (66%) received chemotherapy prior to radiation therapy (RT). The most prevalent immune cells in the tumor before neoadjuvant therapy were myeloid cells (45% of all immune cells) and B cells (37%), with T (13%) and NK (natural killer) cells (5%) also present. Neoadjuvant therapy significantly increased the total immune cells infiltrating the tumors across all histologic subtypes for patients receiving neoadjuvant RT with or without chemotherapy. An increase in the percentage of monocytes and macrophages, particularly M2 macrophages, B cells, and CD4+ T cells was observed post-neoadjuvant therapy. Upregulation of genes and cytokines associated with antigen presentation was also observed, and a favorable pathological response (≥90% necrosis post-neoadjuvant therapy) was associated with an increase in monocytic infiltrate. Upregulation of the T cell checkpoint TIM3 and downregulation of OX40 were observed post-treatment.
Conclusions:
Standard neoadjuvant therapy induces both immunostimulatory and immunosuppressive effects within a complex sarcoma microenvironment dominated by myeloid and B cells. This work informs ongoing efforts to incorporate immune checkpoint inhibitors and novel immunotherapies into the neoadjuvant setting for soft tissue sarcomas.
Keywords: Soft tissue sarcomas, radiotherapy, neoadjuvant therapy, tumor immune microenvironment
INTRODUCTION
Following curative-intent resection, up to half of patients with large, high-grade soft tissue sarcomas (STS) will recur, often with metastatic disease, after which survival is severely limited [1–4]. Radiotherapy (RT) decreases the risk of local recurrence after surgical resection for localized STS of the extremity and trunk [5, 6]. However, neither peri-operative RT nor chemotherapy have been convincingly demonstrated to improve overall survival [6, 7]. Therefore, development of more effective therapies is needed. Immune checkpoint inhibitors (ICI) have transformed the treatment of selected inflamed solid tumors. For example, targeting the programmed cell death protein 1 (PD-1) pathway has markedly improved long-term survival for a subset of patients with advanced melanoma and non-small cell lung cancer [8, 9]. However, most metastatic cancer patients, including those with STS, do not significantly benefit [10–14]. Response rate varies among STS subtypes, with UPS, liposarcoma and synovial sarcoma representing the highest response to immunotherapy [10].
The impact of neoadjuvant therapy on antitumor immunity is under active investigation for multiple cancer types including STS [15–21], colorectal [22, 23], esophageal [17, 18] and non-small cell lung [24] cancers. Defining the immune landscape of STS and the immunologic effects of standard cytotoxic radio- and chemotherapies is of critical importance for integrating immunotherapies into the STS treatment paradigm. In vitro and in vivo experiments in animal models have established the mechanisms by which radiation induces activation of the innate and adaptive immune system [25]. Ionizing radiation may activate the cGAS/STING pathway to promote type-I interferon-dependent antigen uptake and cross-presentation of antigens by dendritic cells to drive adaptive antitumor T cell immunity [26]. Neoadjuvant therapy combinations that optimally induce an inflamed tumor microenvironment (TME) with type-I immunity features and predominance of CD8+, CD4+ Th1 T cells and M1-type macrophages may improve response rates to immune checkpoint inhibition and favorably impact survival. Developing a granular understanding of which specific immune cell populations in the TME are modified by RT or chemotherapies is needed to rationally select combinations of cytotoxic- and immune-therapies that are likely to synergize in the neoadjuvant setting on future clinical trials [27–29].
Sarcomas present a unique opportunity to assess changes to the TME induced by chemo- and radiotherapy using paired patient tissue samples obtained at the time of diagnostic biopsy (pre-neoadjuvant therapy) and surgical resection (post-neoadjuvant therapy). In this project, we sought to better define the immune response to STS and determine how it is modified by neoadjuvant therapy. In a cohort of 32 STS patients spanning multiple histologic subtypes, with paired pre- and post-treatment tissue, we used multiple orthogonal methods to assay immune markers at both the transcriptional and protein expression level.
MATERIALS AND METHODS
Case Selection
Retrospective review of an institutional database was performed to identify patients who underwent curative intent resection of histologically confirmed STS following neoadjuvant RT, with or without neoadjuvant chemotherapy, at the University of Washington (UW) / Seattle Cancer Care Alliance (SCCA) from 2007–2014. All patients (n=32), both female and male, received conventionally fractionated radiotherapy (median dose: 50 Gray; range: 48 – 50.4 Gy). For those receiving chemotherapy (n=21), 16 (76%) received AIM (anthracycline (doxorubicin), ifosfamide and mesna), 3 (14%) received VDC/IE (vincristine, doxorubicin and cyclophosphamide/ ifosfamide and etoposide), 1 (5%) received ifosfamide alone and 1 (5%) received gemcitabine and docetaxel. Information regarding chemotherapy administration by histological subtype can be found in Supplemental Table S1.
Patients included in the study had accessible pre- and post-treatment tumor tissue (i.e., pre-neoadjuvant treatment core needle biopsy and surgical resection specimen, respectively) obtained at our institution. Patients with non-metastasizing histologies (e.g., desmoid fibromatosis, chordoma), Kaposi sarcomas, and primary bone sarcomas were excluded. The Ewing sarcoma included were soft tissue tumors and all analyses were completed on soft tissue samples. Eligible patients with sufficient tissue archived for analysis were then identified for further histologic and transcriptomic analyses as detailed below. Clinical and pathologic information was collected from patient charts. Tumor size was defined as the largest single dimension of the primary tumor and was extracted from radiology reports. Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) tumor grade and microscopic margin status were extracted from pathology reports. Histologic diagnoses were categorized as undifferentiated pleomorphic sarcoma (UPS), synovial sarcoma (SS), Ewing or Ewing-like sarcoma (ES), leiomyosarcoma (LMS), well/dedifferentiated liposarcoma (LPS), myxoid round cell Liposarcoma (MRCL), or other. Pathological response was assessed by board-certified anatomic pathologists with subspecialty training in bone and soft tissue Pathology. We defined a favorable or major pathologic response as ≥ 90% tumor necrosis or hyalinization on surgical pathology following neoadjuvant therapy. All analyses were performed on de-identified data. The study was approved as a minimal-risk protocol by the Cancer Consortium Institutional Review Board at Fred Hutchinson Cancer Research Center. All investigations were performed according to the principles expressed in the Declaration of Helsinki.
Multiplex Immunohistochemistry (mIHC) and Image Cytometry
Hematoxylin and eosin-stained (H&E) tissue sections and formalin-fixed paraffin-embedded (FFPE) blocks were retrieved from the Pathology archives at UW. H&E slides from the entire resection specimen of each eligible case were reviewed by a board-certified bone and soft tissue pathologist. Cases deemed to have adequate viable tissue before and after neoadjuvant therapy were then selected for further analysis. Necrotic areas in the sections were excluded from the mIHC analysis.
Multiplex immunohistochemistry (mIHC) was performed as described previously [30]. Briefly, FPPE tissues were stained on a Leica BOND Rx autostainer (Leica, Buffalo Grove, IL) using the Akoya Opal Multiplex IHC assay (Akoya Biosciences, Menlo Park, CA) with high stringency washes after the secondary antibody and Opal fluor applications using TBST (0.05M Tris, 0.3M NaCl, and 0.1% Tween-20, pH 7.2–7.6). Primary antibodies were incubated at room temperature for 1 hour followed by the OPAL Polymer HRP Mouse plus Rabbit (PerkinElmer, Hopkington, MA). Two different panels were used for mIHC, each of them including 6 different primary antibodies. Panel 1 includes: CD3, FOXP3, CD68/CD163, CD206, PD1 and VISTA. Panel 2 includes: CD4, CD8, CD33, CD66b, HLA-DR and Ki67 (Supplemental Table S2). Slides were mounted with ProLong Gold and images were acquired with the Akoya Vectra 3.0 Automated or Polaris Automated Imaging System. Images were spectrally unmixed using Akoya Phenoptics inForm software and explored as multi-image TIFFs for analysis in HALO software (Indica Labs, Corrales, NM). Nuclear staining in HALO was used to detect individual cells for the multi-spectral images analysis using automated cell counting. Review of tissue staining, selection of regions of interest, segmentation for cell definition and training algorithm for analysis was supervised by a clinically trained pathologist. Microsoft Excel (RRID:SCR_016137) was used to convert the exported object data csv files into a format compatible with FlowJo 10 (RRID:SCR_008520, Becton Dickinson), by multiplying the fluorescent intensity of each marker on each object by a factor of 106 to convert decimals to integer form. The processed csv files were transformed into fcs files and analyzed in FlowJo according to standard methods, hence allowing for quantitative multiplex analysis.
NanoString Transcriptomic Analysis
Bulk tumor RNA was extracted from FFPE tissue as described previously [31]. Briefly, 5 µm thick FFPE sections were deparaffinized in xylene, rehydrated in ethanol, and then lysed on the slide by adding 10–50 mL PKD buffer (Qiagen Inc., Gaithersburg, MD). Tissue was then transferred to a 1.5 mL Eppendorf tube and incubated with Proteinase K (Roche Molecular Systems, Inc., Branchburg, NJ) for 15 minutes at 55 °C followed by incubation at 80 °C for 15 minutes. Final RNA was quantified within the tissue lysate using a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA) and then stored at −80 °C until gene expression profiling was performed using the NanoString nCounter. A custom 800-gene NanoString codeset Panel, including 789 test genes and 11 housekeeping genes, was used for the analysis. NanoString nCounter (NanoString Technologies, Seattle, WA) analysis was then performed as previously described [31]. Briefly, 50 ng RNA per sample was mixed with a 3’ biotinylated capture probe and 5’ reporter probe tagged with a fluorescent barcode from the desired gene expression code set following the manufacturer’s instructions. Hybridized samples were run using the high-sensitivity protocol for the NanoString nCounter, and samples were scanned at maximum scan resolution using the NanoString nCounter Digital Analyzer. Data was quantile normalized and nSolver software (NanoString Technologies) was used to calculate raw counts prior to quantile normalization. Exploratory pathway analysis was conducted with KEGG Mapper Search tool (RRID:SCR_012773, https://www.genome.jp/kegg/tool/map_pathway1.html) in May 2021. Significant genes by Bonferroni (p <0.05/778) up- or down-regulated by at least 1.5-fold change when comparing post- to pre- neoadjuvant therapy were used in the analysis.
Cofactor Transcriptomic Analysis
Unstained, unmounted FFPE sections cut sequentially from the same specimen block were processed for RNA extraction using the Prism Extraction Kit (Cofactor Genomics, San Francisco, CA), following the manufacturer’s suggested protocol. Bioanalyzer or TapeStation assay (Agilent, Santa Clara, CA) and Qubit RNA HS or BR Assay (ThermoFisher, Waltham, MA) were used to evaluate total RNA quality and quantity. RNA concentration and quantity (in ng/ μL and total ng) and quality (DV200, % of fragments above 200 bp) were evaluated. Total RNA was processed by Cofactor Genomics’ laboratory (St. Louis, MO) using the TruSeq RNA Exome Library Prep Kit (Illumina, San Diego, CA) following the standard protocol for FFPE material to generate whole exome libraries; 40 – 100 ng of RNA was used as input depending on sample quality. Libraries were sequenced as single-end 75 base pair reads on a NextSeq500 (Illumina, San Diego, CA) following the manufacturer’s protocols. Samples were analyzed using the ImmunoPrism analysis pipeline (Version 1.0) which delivers a standardized report including expression characterization and immune cell quantification of 18 analytes [30, 32]. Using the ImmunoPrism algorithm, RNA sequencing data is compared to a database of gene expression models of specific immune cells. These models were built using purified immune cell populations, isolated based on canonical cell-surface markers, followed by machine-learning methods to identify multigenic expression patterns from whole-transcriptome data associated with specific immune cells [32, 33].
Statistical Analyses
Comparison of matched pre- and post-treatment samples was performed using two-tailed paired Student’s t-test. Data were analyzed using GraphPad Prism 9 for Windows 64-bit, Version 9.0.0 (RRID:SCR_002798, GraphPad Software, Inc., La Jolla, CA). A p-value <0.05 was considered significant. Quantile normalized NanoString data were log2 transformed using R version 4.0.2. 778 genes were obtained for analysis after filtering out genes with small variation by Standard Deviation (SD) <0.5. Fold change of mean value of post-treatment over mean value of pre-treatment, and paired t test p-value were output. To adjust for multiple comparisons, we used the Benjamini-Hochberg or Bonferroni correction as indicated, defining significance as either a Benjamini-Hochberg q-value <0.05, or a p-value <6.43e-5, i.e., the Bonferroni cut-off of 0.05 divided by 778 test genes.
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Raw data for this study were generated at Fred Hutchinson Cancer Research Center (mIHC), Merck & Co., Inc., Kenilworth, NJ, USA (NanoString), and Cofactor Genomics (RNA sequencing with ImmunoPrism analysis). Derived data supporting the findings of this study are available from the corresponding author upon request.
RESULTS
Patient clinicopathologic characteristics and experimental design
Patients with localized STS treated with neoadjuvant radiation (with or without preceding chemotherapy) and curative intent surgical resection were retrospectively identified in an institutional database. Of these, 32 patients had adequate matched pre- and post-treatment tissue for analysis with mIHC (n=19), NanoString (n=32) and/or RNA sequencing with Cofactor Genomics’ ImmunoPrism analysis (n=26). The patients’ clinical and pathological characteristics are presented in Table 1 and organized by the assays that were performed. Median age was 49 years (range 24–77), and 59% of the cohort was male. Most tumors were in the extremity (81%), >5 cm (81%) in size and intermediate-to-high grade. UPS was the most common histology (44%). Most patients had a negative microscopic surgical margin, i.e., R0 resection (n=27, 84%). After obtaining a diagnostic biopsy (pre-treatment specimen), 66% of patients received (n=21) neoadjuvant chemotherapy followed by RT, while 34% (n=11) received neoadjuvant RT alone before proceeding to definitive surgical resection (post-treatment specimen). Cases with sufficient matched pre- and post-treatment tissue samples were selected for analysis with mIHC, NanoString and RNA sequencing followed by ImmunoPrism analysis as schematized in Figure 1.
Table 1.
Clinicopathologic characteristics of patients undergoing neoadjuvant therapy followed by curative intent resection of localized soft tissue sarcoma.
| Variable | Multiplex IHC (N = 19) |
NanoString (N = 32) |
ImmunoPrism (N = 26) |
|---|---|---|---|
| No. (%) | No. (%) | No. (%) | |
| Age* | 52.8 | 48.5 | 48.5 |
| (25.3–76.7) | (23.7–76.7) | (23.7–76.7) | |
| Sex | |||
| Male | 12 (63) | 19 (59) | 17 (65) |
| Female | 7 (37) | 13 (41) | 9 (35) |
| Location | |||
| Extremity | 17 (90) | 26 (81) | 21 (81) |
| Central | 1 (5) | 3 (9) | 3 (12) |
| Retroperitoneal | 1 (5) | 3 (9) | 2 (8) |
| Tumor Size | |||
| ≤5 cm | 2 (11) | 6 (19) | 4 (15) |
| >5 cm | 17 (89) | 26 (81) | 22 (85) |
| Grade (FNCLCC System) | |||
| 1 | 1 (5) | 5 (16) | 4 (15) |
| 2 | 8 (42) | 13 (41) | 10 (39) |
| 3 | 9 (47) | 13 (41) | 11 (42) |
| Unknown | 1 (5) | 1 (3) | 1 (4) |
| Histologic Type | |||
| Undifferentiated pleomorphic sarcoma | 12 (63) | 14 (44) | 12 (46) |
| Synovial Sarcoma | 2 (11) | 3 (9) | 2 (8) |
| Ewing Sarcoma | - | 3 (9) | 2 (8) |
| Leiomyosarcoma | 2 (11) | 2 (6) | 2 (8) |
| Myxoid/Round Cell Liposarcoma | 3 (16) | 3 (9) | 3 (12) |
| Other† | - | 7 (22) | 5 (19) |
| Preoperative Chemotherapy | |||
| Yes | 15 (79) | 21 (66) | 18 (69) |
| No | 4 (21) | 11 (34) | 8 (31) |
FNCLCC: French Federation of Comprehensive Cancer Centers.
Age at diagnosis is displayed as median (min-max) in years.
Other histologies: 2 myxofibrosarcomas; 1 each of fibroblastic-myofibroblastic sarcoma, sclerosing epithelioid fibrosarcoma, solitary fibrous tumor, sarcoma with epithelioid features suggestive of MPNST, and low grade fibromyxoid sarcoma
Figure 1.
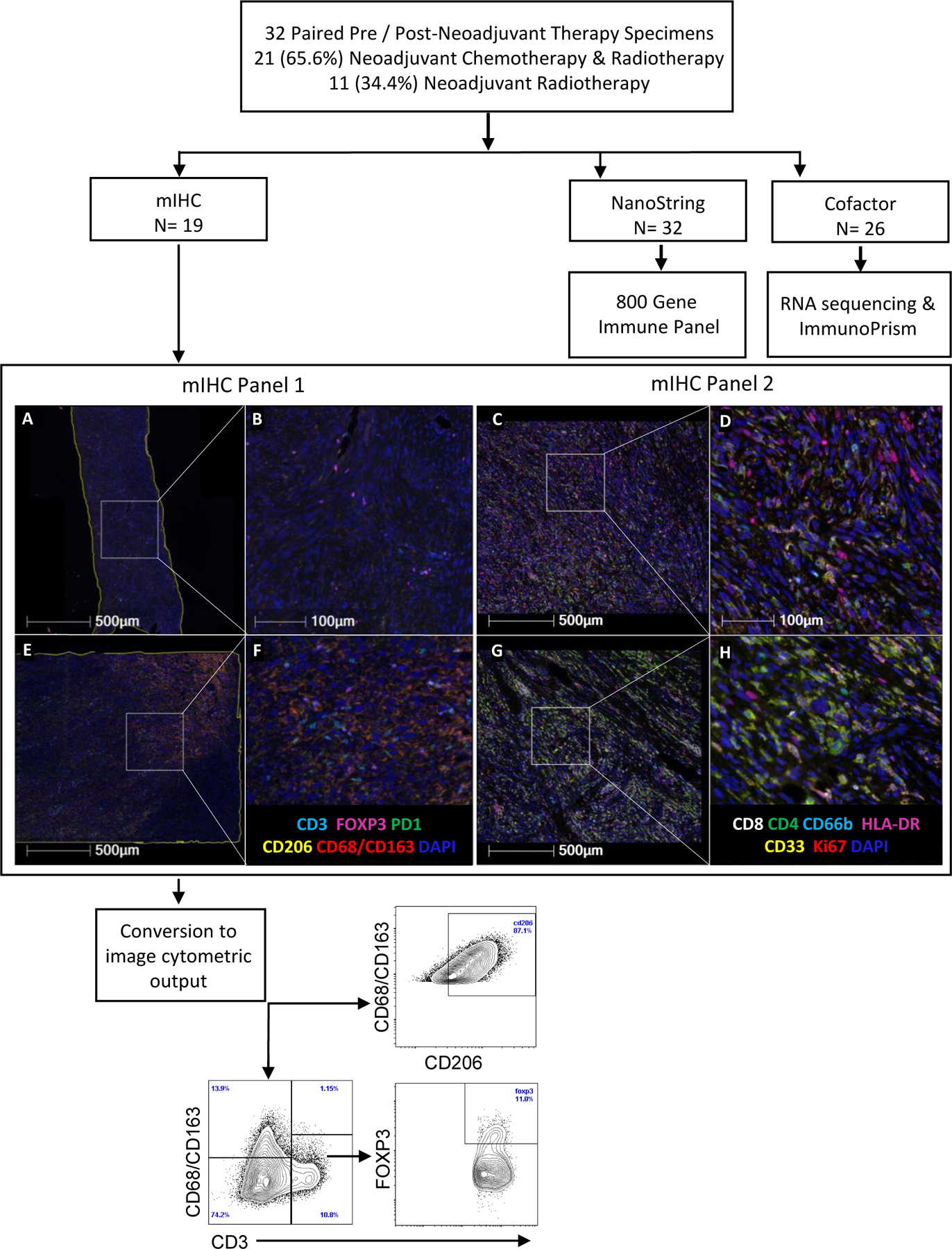
Experimental design. Multiplexed immunohistochemistry (mIHC), NanoString (800 Gene Immune Panel) and RNA sequencing with analysis by ImmunoPrism platform were performed on paired soft tissue sarcoma tissue samples before and after neoadjuvant therapy. Schema indicates the number of paired samples analyzed by each technology. Two mIHC panels were performed with the indicated markers above (in addition to VISTA which is not shown in Panel 1). Example mIHC of undifferentiated pleomorphic sarcoma tumors paired pre- (A-D) and post-neoadjuvant chemoradiation (E-H) at low (A, C, E, G) and high (B, D, F, H) magnification, with an example of the flow cytometric analysis shown below.
Neoadjuvant therapy increases immune cell infiltration in sarcomas
Prior to treatment, myeloid cells (monocytes and macrophages) and B cells were the most common immune cells in the TME, constituting respectively 45.3% and 36.5% of the total immune cells; T cells constituted 13.4% of immune cells, while NK cells represented 4.9% of total immune cells by Cofactor’s ImmunoPrism analysis. The percentage of all ImmunoPrism-defined cell populations are presented as a percentage of total cells in Supp. Table S3.
RNA sequencing followed by ImmunoPrism analysis demonstrated a statistically significant (p=0.0002) 1.7-fold increase in the percentage of total immune cell infiltrate pre- (mean % of total cells ± standard deviation (SD): 24.04 ± 12.37) versus post- (mean % of total cells ± SD: 40.19 ± 17.92) neoadjuvant treatment (Fig. 2A). This statistically significant difference in the percentage of total immune infiltrate pre- versus post- neoadjuvant treatment was observed for both patients receiving RT alone (n=11; mean % of total cells ± SD: pre: 29.50 ± 9.94 versus post: 37.25 ± 10.90, p=0.029) and chemotherapy and RT (n=21; mean % of total cells ± SD: pre: 21.61 ± 12.81 versus post: 41.50 ± 20.43, p=0.0009), and therefore data from patients receiving RT and chemotherapy and RT has been combined in subsequent analysis. No statistically significant difference in the total immune infiltrates post-neoadjuvant therapy (p=0.587) was observed between those patients receiving RT alone and those receiving chemotherapy and RT.
Figure 2.
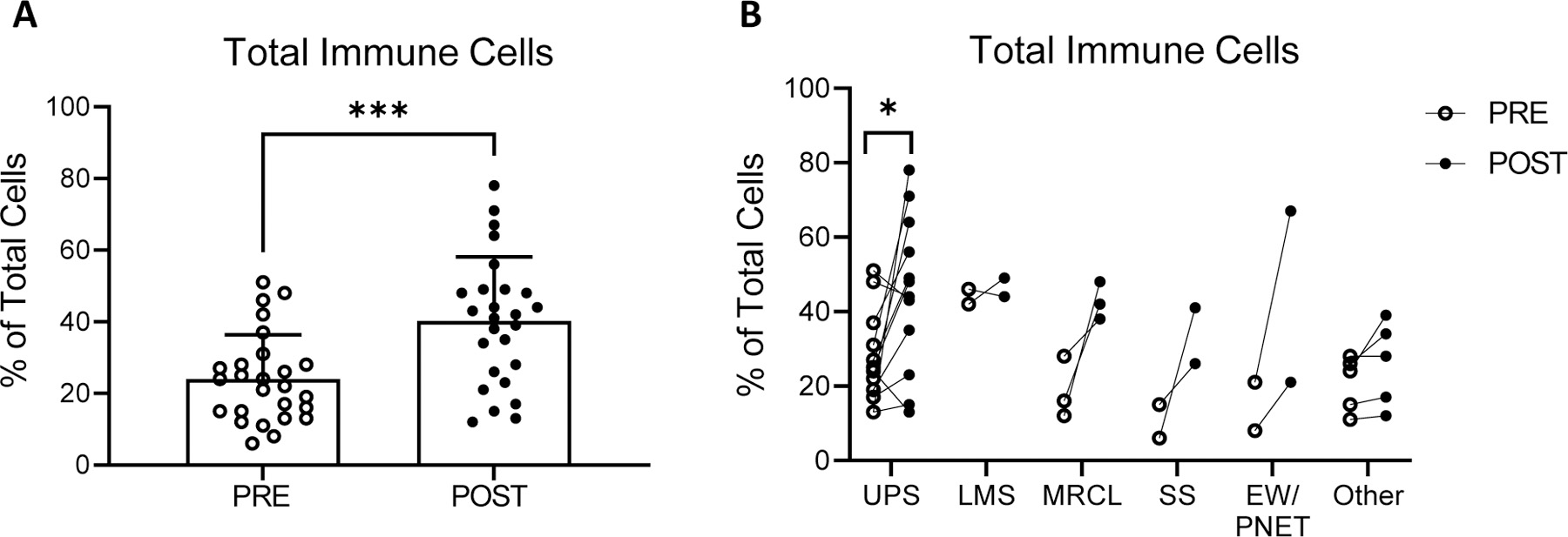
Tumor infiltrating immune cells pre- and post- neoadjuvant therapy identified by RNA sequencing with analysis by Cofactor ImmunoPrism. (A) Percentage of immune infiltrating cells for all pooled sarcoma subtypes. Individual values, mean and standard deviations are shown. (B) Percentage of immune infiltrating cells by sarcoma subtype. UPS: Undifferentiated pleomorphic sarcoma (n=12); LMS: Leiomyosarcoma (n=2); MRCL: Myxoid/round cell liposarcoma (n=3); SS: Synovial Sarcoma (n=2); EW: Ewing sarcoma (n=2) and other (n=5). Comparisons for statistical differences in means were tested using two-tailed paired t-tests: * p-value < 0.05, *** p-value <0.001.
Next, we examined changes induced by neoadjuvant therapy in total immune cells infiltrate by histologic subtype. A significant increase in the percentage of total immune cell infiltrate was observed in the UPS cohort (mean % of total cells ± SD, pre: 27.25 ± 12.53; post: 44.92 ± 20.86; p=0.021; Fig 2B). Most other sarcoma subtypes, which represented small sample sizes, showed an increased total immune cell infiltration post-treatment though small samples sizes limit the power to detect statistically significant differences. Confirming previous reports [31, 34, 35], translocation-associated tumors demonstrated a significantly lower percentage of total immune cell infiltrate prior to neoadjuvant therapy (mean % of total cells ± SD, translocation-driven sarcomas: 16.75 ± 8.36; not translocation-driven: 27.28 ± 12.65; p=0.043). Following neoadjuvant therapy, total immune cell infiltrate increased in both non-translocation and translocation-associated histologies, and no significant difference in the percentage of total immune cell infiltrate was observed relative to translocation status (mean % of total cells ± SD, translocation-driven sarcomas: 38.88 ± 14.60; not translocation-driven: 40.78 ± 19.57; p=0.808). To increase the power of subsequent analyses, all sarcoma histologic subtypes were pooled to further characterize the changes in the TME induced by neoadjuvant therapy.
Neoadjuvant therapy induces an increase in monocyte-lineage cells including CD206+ macrophages in the sarcoma TME
The myeloid compartment of the TME is described in Figure 3. Macrophages, defined as CD68+/CD163+ cells in mIHC, showed a 1.8-fold increase from pre- to post-treatment (mean ± SD % of total cells: pre: 7.05 ± 11.56; post: 12.87 ± 13.92; p=0.055; Fig. 3A). A significant increase in the percentage of CD206+ M2 macrophages was observed after neoadjuvant therapy (mean % of total cells ± SD, pre: 4.85 ± 10.46; post: 9.84 ± 13.51; p=0.031; Fig. 3B). CD33 and CD66b markers were also assayed for mMDSC, (CD33+/CD66−) and polymorphonuclear MDSC (pmnMDSC) / neutrophils (CD33+/CD66b+), and no statistically significant changes were observed before and after neoadjuvant therapy (Supp. Table S4). Comprehensive mIHC data are presented by sarcoma subtype in Supp. Fig. S1 and in Supp. Tables S4 & S5.
Figure 3.
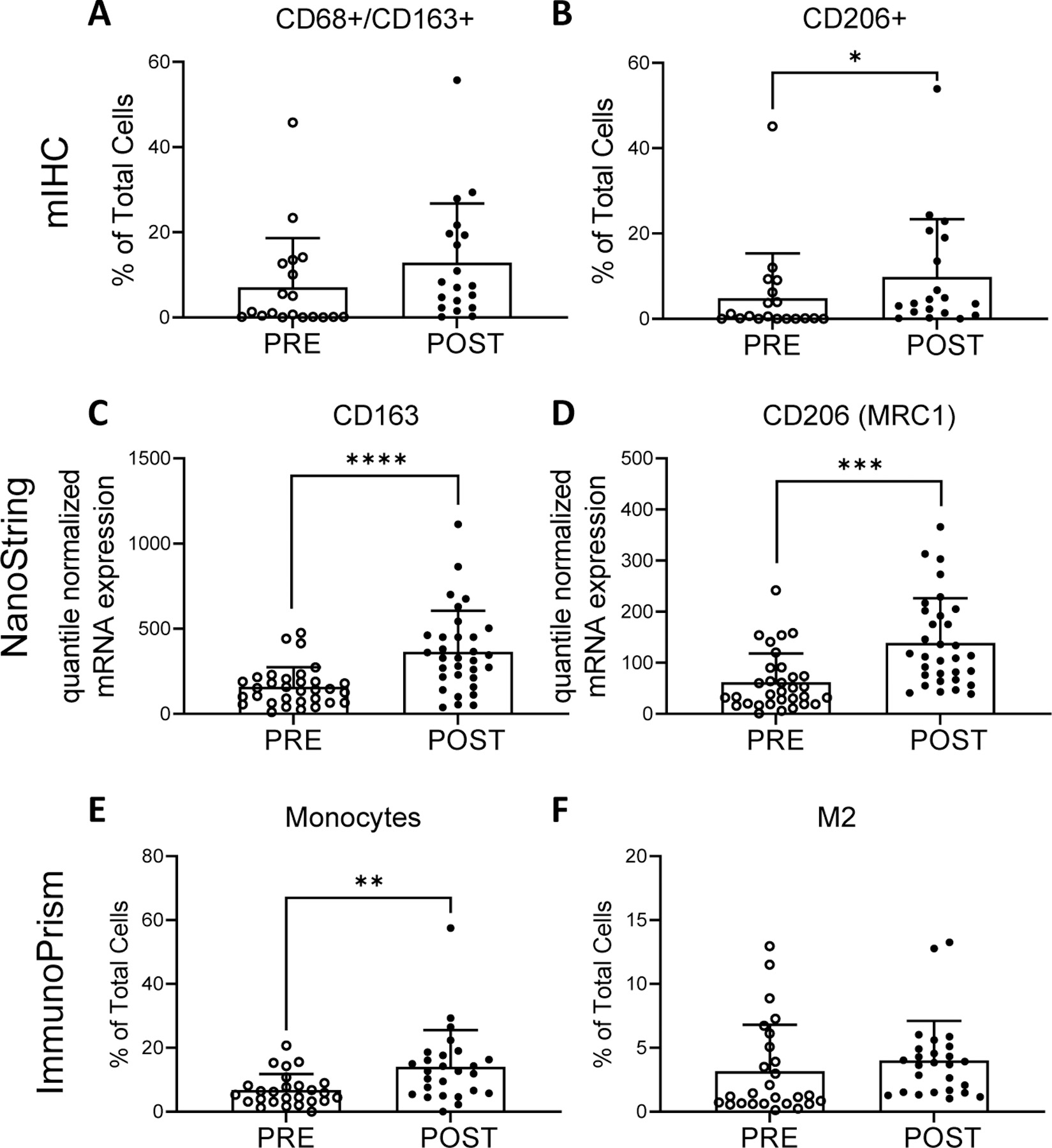
Myeloid cells pre- and post- neoadjuvant therapy identified by multiplex immunohistochemistry (mIHC) (top row), NanoString quantile normalized mRNA expression (middle row), and RNA sequencing with analysis by ImmunoPrism (bottom row). mIHC: percentage of CD68+/CD163+ cells (A) and CD206+ as percentage of total cells (B). NanoString: quantile normalized expression values for CD163 (C) and CD206 (MRC1) (D) genes. ImmunoPrism: percentage of monocytes (E) and M2 macrophages (F). Individual values, mean and standard deviations are shown. mIHC and ImmunoPrism statistical differences were tested using two-tailed paired t-tests: * p-value <0.05, ** p-value <0.01. NanoString: differences were tested with paired t-tests with Benjamini-Hochberg adjusted p-values (q-values). *** q-value < 0.001, **** q-value <0.0001.
In agreement with mIHC findings, macrophage marker gene expression was significantly increased in NanoString analysis for M2 gene markers CD163 (mean quantile normalized values ± SD, pre: 159.16 ± 114.89; post: 364.38 ± 241.23; q<0.0001; Fig. 3C), and CD206 (MRC1) (pre: 62.00 ± 56.12; post: 139.00 ± 87.37; q=0.0002; Fig. 3D). Analysis with ImmunoPrism showed a significant (p=0.002) increase in the monocyte population from pre- to post-neoadjuvant therapy (mean % of total cells ± SD, pre: 6.74 ± 5.01; post: 14.06 ± 11.48; Fig. 3E) with no significant changes in the populations defined as M1 (p=0.504; not shown) or M2 (p=0.426; Fig. 3F) macrophages. M2 macrophages were prevalent in the TME and constituted on average 3.18% (± 3.6) of total cell population, while M1 macrophages constituted 0.65% (± 0.87) of total cells (Supp. Table S3). Comprehensive ImmunoPrism data are presented by sarcoma subtype in Supp. Fig. S2 and Supp. Table S3. Additional transcriptional changes are discussed below.
Neoadjuvant therapy increases CD4+ T cells while decreasing CD3+FOXP3+ regulatory-T cells in the sarcoma TME
Tumor infiltrating lymphocytes (TILs) in the sarcoma TME are characterized in Figure 4. A non-statistically significant increase in the percentage of CD3+ cells was observed pre- to post-treatment (mean % of total cells ± SD, pre: 3.33 ± 3.90; post: 5.63 ± 4.49; p=0.069; Fig. 4A) by mIHC. Further characterization demonstrated a significant 2.9-fold increase in CD4+ cells (mean % of total cells ± SD, pre: 1.68 ± 1.92; post: 4.80 ± 4.97; p=0.015; Fig. 4B). We observed a significant 2.3-fold decrease in CD3+FOXP3+ T-regulatory cells (Treg) (mean % of CD3+ cells ± SD, pre: 9.63 ± 12.46; post: 4.17 ± 5.12; p=0.023; Fig. 4C). No significant change was observed in the percentage of CD8+ cells before and after treatment (p=0.175). Comprehensive mIHC data are presented by sarcoma subtype in Supp. Fig. S1 and in Supp. Tables S4 & S5.
Figure 4.
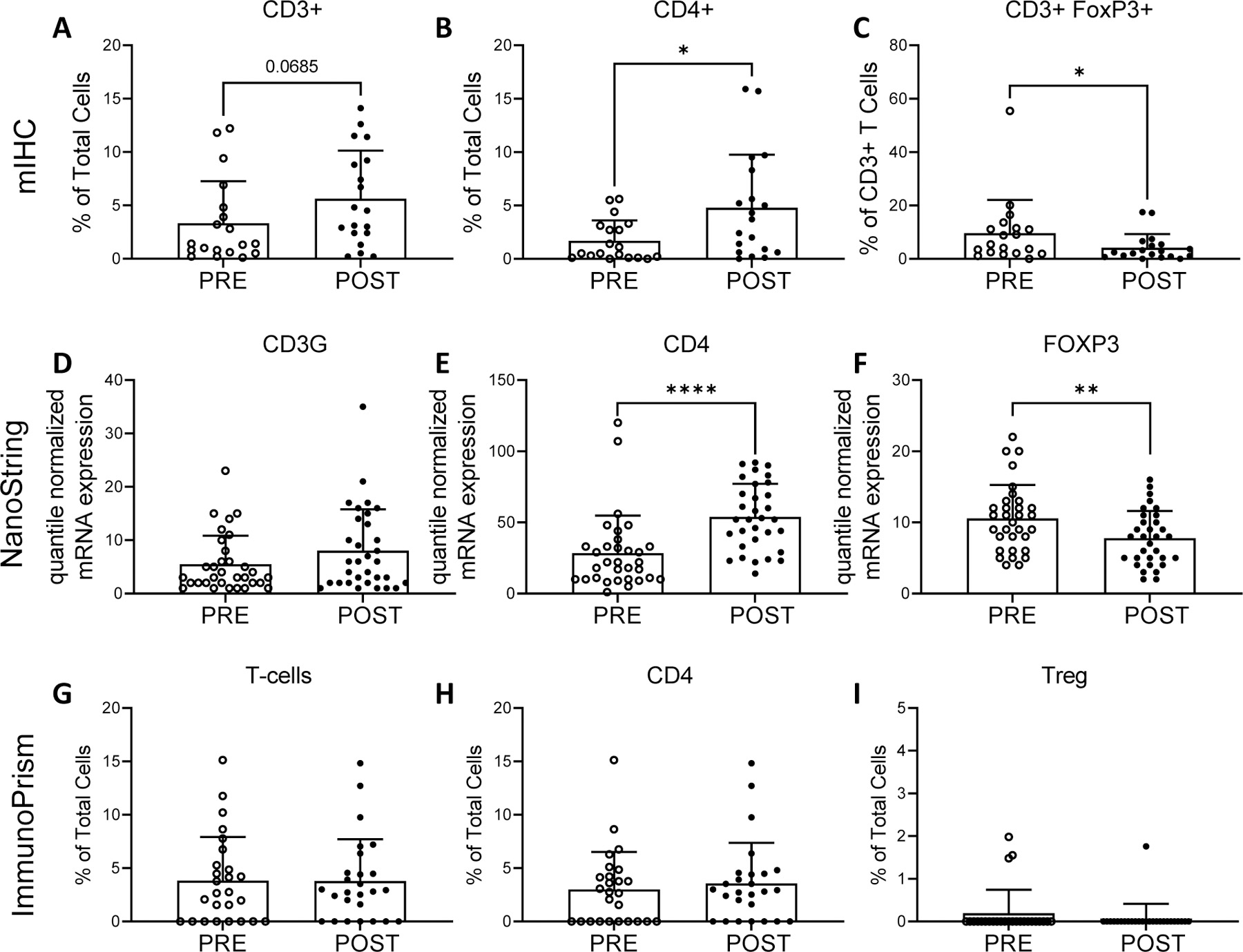
Tumor infiltrating T cells pre- and post- neoadjuvant therapy identified by mIHC (top row), NanoString quantile normalized mRNA expression (middle row), and RNA sequencing with analysis by ImmunoPrism (bottom row). mIHC: percentage of CD3+ cells (A), CD4+ cells (B) and FOXP3+ T cells (C). NanoString: quantile normalized expression values for CD3G (D), CD4 (E) and FOXP3 (F) genes. ImmunoPrism: percentage of T cells (CD3) (G), CD4 cells (H) and T regulatory cells (Treg) (I). Individual values, mean and standard deviations are shown. mIHC and ImmunoPrism statistical differences were tested using two-tailed paired t-tests: * p-value <0.05. Nanostring: differences were tested with paired t-tests with Benjamini-Hochberg adjusted p-values (q-values). ** q-value < 0.01, **** q-value <0.0001.
Gene expression analyses were performed with the NanoString and ImmunoPrism platforms. Supporting the mIHC data, NanoString showed a significant increase in CD4 expression (mean quantile normalized values ± SD, pre: 28.44 ± 26.36; post: 53.88 ± 23.30; q=0.0001; Fig. 4E), accompanied by significantly decreased FOXP3 expression (pre: 10.56 ± 4.71; post: 7.78 ± 3.84; q=0.009; Fig. 4F). No statistically significant changes were observed in CD3G (q=0.262; Fig. 4D) or CD8A (q=0.982) expression. ImmunoPrism did not show a significant change in the percentage of T cells (p=0.979; Fig. 4G) or CD4+ T cells (p=0.605; Fig. 4H). Limited detection of Treg (Fig. 4I) and CD8+ T cells was consistent with the low percentage of CD3+FOXP3+ and CD8+ cells observed in most mIHC samples. Comprehensive ImmunoPrism data are presented by sarcoma subtype in Supp. Fig. S2 and Supp. Table S3.
Neoadjuvant therapy induces changes in immune activation status and checkpoint molecule expression
Changes in CD4+ and CD8+ T cell activation pre- versus post-treatment were evaluated by mIHC quantification of activation and immune checkpoint markers, including HLA-DR, Ki67, VISTA and PD-1. No significant changes in the expression of any of these proteins was observed in either CD4+ or CD8+ T cells (Supp. Table S5). The percentage of PD-1+CD3+ cells was not significantly changed from pre- to post- neoadjuvant therapy by mIHC (Supp. Fig. S3A).
The following activation and/or checkpoint control genes were included in the ImmunoPrism analysis: PD1, PD-L1, CTLA4, OX40, TIM3, BTLA, ICOS, CD47, IDO and ARG1. A significant increase pre- to post-treatment was observed in the expression of the inhibitory T cell co-receptor TIM3 (p= 0.0002; mean ± SD: pre: 15.92 ± 13.62 transcripts per million (TPM); post: 38.15 ± 27.68 TPM), together with a significant decrease in the activating T cell co-receptor OX40 (p=0.032; pre: 4.62 ± 4.85 TPM; post: 2.15 ± 2.13 TPM) (Supp. Fig. S3B). No statistically significant changes were observed in other genes. NanoString analysis supported the ImmunoPrism expression data with a significant increase in TIM3 (HAVCR2) expression (fold change (FC)=1.34, q=0.005; Supp. Fig. S3C) and decrease in OX40 (TNFRSF4) (FC=0.45, q=0.003; Supp. Fig. S3D). NanoString analysis also demonstrated mixed changes in other immune-stimulating and inhibitory markers, with significant decreases in the coinhibitory ligand CD276 (B7-H3) and inhibitory receptor ADORA2A (adenosine A2A receptor), but increased expression of the coinhibitory ligand PDCD1LG2 (PD-L2) (Supp. Fig. S3C).
Neoadjuvant therapy increases expression of antigen presentation-related pathways in the sarcoma TME
Next, we investigated how changes observed in the TME associated with outcome utilizing pathologic response to neoadjuvant treatment as a surrogate outcome. Patients with favorable pathologic response (n=9, 35%) demonstrated a significantly greater increase in tumor-infiltrating monocytes (p=0.034), measured by ImmunoPrism, than their counterparts without such a pathologic response (n=17, 65%; Fig. 5A). No significant difference in the ImmunoPrism-defined monocyte population was observed pre-treatment between patients with favorable and unfavorable pathological responses to neoadjuvant treatment (p=0.93). Favorable pathological response rates were not significantly different (p=0.19; Fisher’s exact test), for patients receiving chemotherapy and RT compared to RT alone. An association between pathologic response and pre-treatment percentages or post-treatment changes was not observed in any other immune cell types, including T cells, B cells, M1 macrophages, M2 macrophages and NK-cells. In concordance with the cytotoxic effects of neoadjuvant treatment, genes associated with cell cycle progression were downregulated (Fig. 5B). A significant decrease in Ki67+ non-immune cells (CD4− CD8− HLA-DR− CD33− CD66b− % of total cells, pre: 10.08 ± 12.19; post: 2.30 ± 2.50; p=0.010; Supp. Table S5) was observed in mIHC, likely indicating that neoadjuvant therapy killed proliferating tumor cells as intended.
Figure 5.
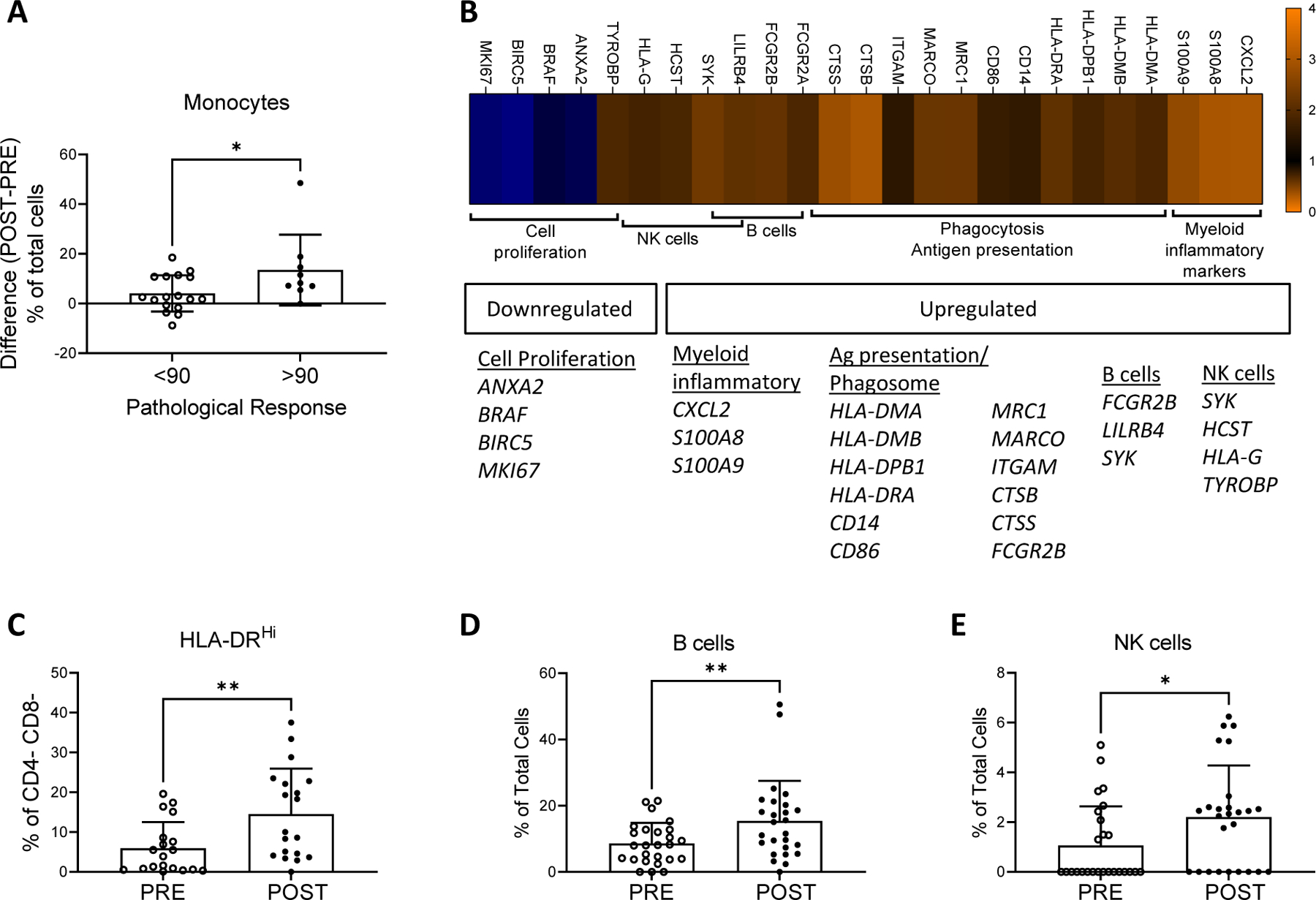
Changes in gene expression pathways after neoadjuvant therapy. (A) Difference in the percentage of monocytes (post – pre neoadjuvant therapy) identified by ImmunoPrism in tumors with a favorable pathological response (≥90% necrosis) vs. <90% necrosis. (B) Heatmap with differentially regulated genes pre- vs. post- neoadjuvant therapy analyzed by NanoString followed by clustering with KEGG pathway analysis. Genes significantly up- or down-regulated (fold change >1.5 for upregulation, <0.67 for downregulation with a Bonferroni corrected p-value <6.43e-5) pre- vs. post- neoadjuvant therapy were analyzed in KEGG to identify gene pathways. Immune-oncology relevant pathways with >3 genes identified are shown. (C) Percentage of HLA-DRHi + non-T cells (CD4- CD8- cells) pre- and post- neoadjuvant therapy identified by mIHC. (D) Percentage of B cells pre- and post- neoadjuvant therapy identified by ImmunoPrism. (E) Percentage of NK cells pre- and post- neoadjuvant therapy identified by ImmunoPrism. Testing for differences in mean levels was performed using two-tailed paired t-tests: * p-value <0.05, ** p-value <0.01.
NanoString data were used to investigate immunological pathways altered by therapy. Sixty-five genes from the custom, immune-oncology 800-gene NanoString panel had significantly altered expression pre- versus post-neoadjuvant therapy, using Bonferroni correction for multiple comparisons. Forty-three genes were upregulated (fold change >1.5) and 22 genes were down-regulated (fold change <0.67) (Supp. Fig. S4). These 65 genes were evaluated in KEGG Gene Ontology enrichment analysis to identify pathways of interest (Fig. 5B). Several genes and cytokines related to antigen presentation and the phagosome were enriched and upregulated post-treatment, including HLA-II genes, the costimulatory ligand CD86 and cathepsin proteases (CTSB, CTSS). Increased expression of monocyte (CD14) and macrophage (MRC1, MARCO) related genes was observed. Similarly, a significant increase in the frequency of HLA-DRhi CD4−/CD8− cells (% of total cells, pre: 5.95 ± 6.56; post: 14.54 ± 11.39; p=0.003, Fig. 5C) by mIHC is presumed to be from antigen-presenting cells (APCs) such as myeloid or B cells.
Genes associated with B and NK cells were also enriched and upregulated per KEGG Gene Ontology enrichment analysis of the NanoString expression data (Fig. 5B). B cells were significantly upregulated by neoadjuvant therapy (Fig. 5D; mean % of total cells ± SD, pre: 8.60 ± 6.23; post: 15.41 ± 12.13; p=0.006) and were the second most common tumor-infiltrating immune cell type (39.8% of all immune cells) post- neoadjuvant therapy, as determined by ImmunoPrism assays. An increase in NK cells from pre- to post-neoadjuvant therapy was also observed (Fig. 5E; % of total cells, pre: 1.06 ± 1.57; post: 2.21 ± 2.07; p=0.010) by ImmunoPrism analysis. B and NK cell markers were not assessed by mIHC, and putative B (CD19, CD20) and NK cell (CD56) genes were not included in the NanoString panel.
DISCUSSION
Standard neoadjuvant therapy induced heterogeneous inflammatory effects in the sarcoma TME, including increased myeloid cells, B cells, and CD4+ cells, with corresponding transcriptional changes reflecting enhanced phagocytosis, immune trafficking, and antigen presentation. These findings were broadly applicable to patients receiving either neoadjuvant RT alone or chemotherapy preceding RT for diverse sarcoma subtypes. As previously reported, genomically complex sarcoma subtypes, e.g., LMS and UPS, had greater immune cell infiltration than those driven by oncogenic chromosome translocations, e.g., SS and MRCL [31, 34–36], and neoadjuvant therapy increased immune infiltration significantly in both sarcoma groups. Data were consistent across three different platforms incorporating transcription and protein level expression data. These data contributed to the design of a neoadjuvant study of pembrolizumab and radiation in locally advanced STS patients (NCT03338959), and are particularly relevant to the ongoing national, randomized SARC032 trial of neoadjuvant radiation with or without neoadjuvant and adjuvant pembrolizumab [20]. In establishing real-world changes in the human sarcoma TME following radiation with or without chemotherapy, we have defined baseline TME changes against which the effects of novel combinations of therapies can be compared. Such results also deepen our understanding of how cytotoxic therapies may synergize with immunotherapies and how best to integrate immunotherapy with RT and chemotherapy in future clinical trials. In our cohort of STS patients, both antecedent and post-neoadjuvant treatment immune infiltrates were dominated by two cell lineages involved in professional antigen presentation: monocytes/macrophages and B cells.
Our findings show that neoadjuvant therapy significantly increases monocyte-lineage cells and CD206+ (M2-type) macrophages in the sarcoma TME, and our baseline findings prior to neoadjuvant therapy are consistent with published reports on myeloid cells in STS [19, 30, 34]. A significant increase in the pre- to post-neoadjuvant intratumoral monocyte-lineage cell frequency was observed in tumors with a favorable pathological response (>90% tumor necrosis or hyalinization) compared to those without such a response. Pathological complete response has been associated with improved survival in sarcoma patients following neoadjuvant therapy and resection [37, 38]. Our findings suggest that a subset of intratumoral myeloid cells could be stimulating anti-sarcoma immune responses and contributing to neoadjuvant therapy effects. Tumor-associated macrophages (TAMs) and related tumor-infiltrating myeloid cells are functionally diverse and can elicit acute inflammation, stimulating T cells in an MHC class II/antigen-specific manner, or, conversely, terminate immune responses and mediate tissue remodeling and repair [39–41]. Multiple ongoing studies are attempting to target macrophages in metastatic STS using either standard treatments such as doxorubicin and trabectedin (NCT03074318) or novel treatments such as anti-CD47 (NCT04886271) [14]. Our findings support further pre-clinical investigation and clinical study of TAM-targeted therapies for STS. Establishing type-I antitumor immunity early in the disease course may lead to improved survival outcomes.
B cells were the second most abundant immune cell within the STS TME at baseline and were significantly enriched by neoadjuvant therapy. Although the role of B cells in antitumor immunity remains controversial [42], recent studies have found that B cells, especially when organized in tertiary lymphoid structures (TLS), played a critical role in the response to ICI [43, 44]. In STS, an increased density of B cells and TLS was associated with improved survival and higher response rates to ICI [45]. Neoadjuvant therapy also increased the percentage of CD4+ T cells and decreased CD3+FOXP3+ cells, suggesting enrichment of helper T cells (Th) capable of stimulating antigen-specific CD8+ cytotoxic T cells. Increased CD4+ T cell infiltration has been previously reported following neoadjuvant treatment for UPS [46]. Our study expands these findings to a larger and more diverse cohort of STS patients.
More broadly surveying immune costimulatory and coinhibitory pathways, we found that neoadjuvant therapy had a net inhibitory effect on T cell activation pathways, including a transcriptional level increase in the inhibitory receptor TIM3, an increase in PD-L2 expression, and a decrease in the stimulatory receptor OX40. TIM3 blockade is being increasingly studied in anti-PD-1-refractory solid tumor patients [47], but has not been studied extensively in STS. Our results support further investigation of the role of TIM3 in STS, including verification of TIM3 upregulation by neoadjuvant therapy in larger cohorts, confirmation of TIM3 protein expression in STS, and histology-specific studies. We observed no difference in PD-1 or PD-L1 expression following neoadjuvant therapy. This contrasts with a study by Patel et al., in which they reported an increase in PD-L1 expression (p=0.056) [48]. Taken together, these data emphasize the need for correlative analyses from ongoing trials of PD-1 inhibition and neoadjuvant radiation in STS (NCT03092323, NCT03338959, NCT03307616, NCT03463408, NCT03116529) [21, 49]. Higher dimensional profiling (e.g., single-cell RNA sequencing analyses, surface proteomics or spatial transcriptomics) and functional analyses of the myeloid, B cell and T cell components of the STS TME may provide more insight into the phenotype, clonality, and functional state of these populations and improve our understanding of how antigen presentation in sarcomas direct the functionality of CD4 helper and CD8 effector T cells.
In conclusion, valuable matched pre- and post-neoadjuvant treatment STS samples were assayed with multiple, complementary genomic and immunohistological techniques to characterize immunologic effects of neoadjuvant therapy across a broad spectrum of histologic and molecular STS subtypes. Our study is limited by its retrospective nature, sample size, heterogeneity in the STS subtypes enrolled, and heterogeneity in treatment received (RT alone or chemotherapy preceding RT), recognizing a selection bias for patients receiving chemotherapy based on STS histology. Nevertheless, these data strongly support further histology-specific clinical and laboratory research to explore future trials of novel myeloid- and B cell-targeted therapies in combination with cytotoxic neoadjuvant therapy in STS. Importantly, our analyses highlight emerging inhibitory immune checkpoints, such as TIM3, for which there are antibody therapies being tested in clinical trials [47], and blockade of which may bolster anti-tumor immunity in conjunction with neoadjuvant therapy in STS patients. Our findings also support the study of novel immunotherapy agents stimulating type-I immunity, e.g., oncolytic viruses (NCT02923778, NCT03069378), vaccines (NCT01803152) or toll-like receptors (TLR) (NCT02180698), in the neoadjuvant setting for STS and may enhance the efficacy of established or novel ICIs. This work serves to focus future clinical trial designs on the most relevant immune checkpoints, cell types, and pathways to target in combination with neoadjuvant cytotoxic therapy in STS. More broadly, our work may also help to inform the investigation of other rare solid tumor types for which preoperative cytotoxic therapy is standard, but ICI is not efficacious.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Defining the immunologic effects of standard cytotoxic radio- and chemotherapies is of critical importance for integrating immunotherapies into current cytotoxic treatments. We have used multiple, complementary methodologies to characterize the changes in the tumor immune microenvironment pre- versus post- neoadjuvant therapy, in a cohort of 32 soft tissue sarcoma (STS) patients spanning multiple histologic subtypes. The total immune cell infiltration of tumors increased after neoadjuvant therapy and was dominated by professional antigen presenting cells, namely myeloid and B cells. Neoadjuvant therapy upregulated genes and cytokines associated with antigen presentation, increased the prevalence of CD4+ T cells, and upregulated the T cell inhibitory checkpoint receptor TIM3. Our findings are relevant to interpret ongoing trials that combine immunotherapies with neoadjuvant cytotoxic therapies and serves to focus future clinical trial designs on the most relevant immune checkpoints, cell types, and pathways to target in combination with neoadjuvant cytotoxic therapy.
ACKNOWLEDGMENTS
We thank members of the Fred Hutchinson Cancer Research Center Experimental Histopathology Core Facility for assistance with multiplex immunohistochemistry and image analysis. We thank Dr. Venu Pillarisetty for helpful discussions, and Ms. Silvia Christian for administrative support. This work was supported by CA180380 and the Sarcoma Foundation of America (S. Pollack). NanoString assays were performed by Merck & Co., Inc., Kenilworth, NJ, USA. RNA sequencing with ImmunoPrism was performed by Cofactor Genomics. S. Pollack was also supported by R01CA244872, CA180380, a grant from the V Foundation, and the Seattle Translational Tumor Research (STTR). The Experimental Histopathology Shared Resource at the Fred Hutchinson Cancer Research Center was supported by the NIH P30 CA015704 Cancer Center Support Grant.
Footnotes
Conflicts of Interest: The authors have disclosed they have significant relationships with, or financial interest in, the following commercial companies pertaining to this article: PHG received research funding from Gilead Sciences, Inc. outside the submitted work. BL is a paid consultant from Cofactor Genomics, Inc., the company that developed and produces the ImmunoPrism® reagent kit and informatics tools used in this article. JE, KF, and NL are employed by Cofactor Genomics, Inc., which funded and performed the RNA sequencing and ImmunoPrism analysis herein but did not otherwise play a role in the conceptualization, analysis, or presentation of the work herein. LDC received research funding paid to the institution from Eli Lilly, AADi, BluePrint Medicine, Iterion, Gradalis, Philogen, Advenchen Laboratories, and CBA Pharma; LDC has received honoraria or has served on advisory boards for Daichi Sankyo, BluePrint Medicines and Regeneron. JC and RHP are employed by Sensei Biotherapeutics, Inc., which did not play any role in the conceptualization, analysis, or presentation of the work herein. EM, WMB, and TM are employed by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA which funded and performed NanoString nCounter gene expression analysis herein but did not otherwise play a role in the conceptualization, analysis, or presentation of the work herein. SMP reported research funding from Merck during the conduct of the study; research funding from EMD Serono, Incyte, Presage, Janssen, OncoSec, and Juno and consulting, honoraria, and advisory activity with Deciphera, Aadi, Epizyme, Springworks, GlaxoSmithKline, Obsidian, T-knife, Daiichi Sankyo, and Blueprint Medicine, outside the submitted work. MJW reports consulting, honoraria, and advisory activity from Epizyme, Adaptimmune, and Deciphera. The remaining authors declare no potential conflicts of interest.
REFERENCES
- 1.Beane JD, et al. , Efficacy of adjuvant radiation therapy in the treatment of soft tissue sarcoma of the extremity: 20-year follow-up of a randomized prospective trial. Ann Surg Oncol, 2014. 21(8): p. 2484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daigeler A, et al. , Long-term outcome after local recurrence of soft tissue sarcoma: a retrospective analysis of factors predictive of survival in 135 patients with locally recurrent soft tissue sarcoma. Br J Cancer, 2014. 110(6): p. 1456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothermundt C, et al. , What is the role of routine follow-up for localised limb soft tissue sarcomas? A retrospective analysis of 174 patients. Br J Cancer, 2014. 110(10): p. 2420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savina M, et al. , Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med, 2017. 15(1): p. 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkert MR, et al. , Comparison of local recurrence with conventional and intensity-modulated radiation therapy for primary soft-tissue sarcomas of the extremity. J Clin Oncol, 2014. 32(29): p. 3236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nussbaum DP, et al. , Preoperative or postoperative radiotherapy versus surgery alone for retroperitoneal sarcoma: a case-control, propensity score-matched analysis of a nationwide clinical oncology database. Lancet Oncol, 2016. 17(7): p. 966–975. [DOI] [PubMed] [Google Scholar]
- 7.Bajpai J and Susan D, Adjuvant chemotherapy in soft tissue sarcomas...Conflicts, consensus, and controversies. South Asian J Cancer, 2016. 5(1): p. 15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlino MS, Larkin J, and Long GV, Immune checkpoint inhibitors in melanoma. Lancet, 2021. 398(10304): p. 1002–1014. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi L and Garassino MC, Pembrolizumab plus Chemotherapy in Lung Cancer. N Engl J Med, 2018. 379(11): p. e18. [DOI] [PubMed] [Google Scholar]
- 10.Tawbi HA, et al. , Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol, 2017. 18(11): p. 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boye K, et al. , Pembrolizumab in advanced osteosarcoma: results of a single-arm, open-label, phase 2 trial. Cancer Immunol Immunother, 2021. 70(9): p. 2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saerens M, et al. , Immune checkpoint inhibitors in treatment of soft-tissue sarcoma: A systematic review and meta-analysis. Eur J Cancer, 2021. 152: p. 165–182. [DOI] [PubMed] [Google Scholar]
- 13.D’Angelo SP, et al. , Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol, 2018. 19(3): p. 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollack SM, et al. , Assessment of Doxorubicin and Pembrolizumab in Patients With Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol, 2020. 6(11): p. 1778–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jagodinsky JC, Harari PM, and Morris ZS, The Promise of Combining Radiation Therapy With Immunotherapy. Int J Radiat Oncol Biol Phys, 2020. 108(1): p. 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weichselbaum RR, et al. , Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol, 2017. 14(6): p. 365–379. [DOI] [PubMed] [Google Scholar]
- 17.Noma T, et al. , Immunoscore Signatures in Surgical Specimens and Tumor-Infiltrating Lymphocytes in Pretreatment Biopsy Predict Treatment Efficacy and Survival in Esophageal Cancer. Ann Surg, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christina Svensson M, et al. , T cells, B cells, and PD-L1 expression in esophageal and gastric adenocarcinoma before and after neoadjuvant chemotherapy: relationship with histopathological response and survival. Oncoimmunology, 2021. 10(1): p. 1921443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dufresne A, et al. , Specific immune landscapes and immune checkpoint expressions in histotypes and molecular subtypes of sarcoma. Oncoimmunology, 2020. 9(1): p. 1792036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.A Randomized Trial of Pembrolizumab & Radiotherapy Versus Radiotherapy in High-Risk Soft Tissue Sarcoma of the Extremity. https://clinicaltrials.gov/ct2/show/NCT03092323. [DOI] [PMC free article] [PubMed]
- 21.Roland CL, et al. , Preliminary results of a phase II study of neoadjuvant checkpoint blockade for surgically resectable undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated liposarcoma (DDLPS). Journal of Clinical Oncology, 2020. 38(15_suppl): p. 11505–11505. [Google Scholar]
- 22.Yasui K, et al. , Effect of preoperative chemoradiotherapy on the immunological status of rectal cancer patients. J Radiat Res, 2020. 61(5): p. 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CC, et al. , The Effects of Neoadjuvant Treatment on the Tumor Microenvironment in Rectal Cancer: Implications for Immune Activation and Therapy Response. Clin Colorectal Cancer, 2020. 19(4): p. e164–e180. [DOI] [PubMed] [Google Scholar]
- 24.Goff PH, et al. , Radiation and Modulation of the Tumor Immune Microenvironment in Non-Small Cell Lung Cancer. Semin Radiat Oncol, 2021. 31(2): p. 133–139. [DOI] [PubMed] [Google Scholar]
- 25.Golden EB, Marciscano AE, and Formenti SC, Radiation Therapy and the In Situ Vaccination Approach. Int J Radiat Oncol Biol Phys, 2020. 108(4): p. 891–898. [DOI] [PubMed] [Google Scholar]
- 26.Deng L, et al. , STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity, 2014. 41(5): p. 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pages F, et al. , International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet, 2018. 391(10135): p. 2128–2139. [DOI] [PubMed] [Google Scholar]
- 28.Pages F, et al. , Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene, 2010. 29(8): p. 1093–102. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Zhao B, and Wang X, Tumor infiltrating immune cells (TIICs) as a biomarker for prognosis benefits in patients with osteosarcoma. BMC Cancer, 2020. 20(1): p. 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder BA, et al. , CD4+ T cell and M2 macrophage infiltration predict dedifferentiated liposarcoma patient outcomes. J Immunother Cancer, 2021. 9(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollack SM, et al. , T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer, 2017. 123(17): p. 3291–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaFranzo NA, Flanagan KC, and Quintanilha D, Predictive Immune Modeling of Solid Tumors. J Vis Exp, 2020(156). [DOI] [PubMed] [Google Scholar]
- 33.James NE, et al. , Immune Modeling Analysis Reveals Immunologic Signatures Associated With Improved Outcomes in High Grade Serous Ovarian Cancer. Front Oncol, 2021. 11: p. 622182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dancsok AR, et al. , Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology, 2020. 9(1): p. 1747340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dancsok AR, et al. , Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod Pathol, 2019. 32(12): p. 1772–1785. [DOI] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research Network. Electronic address, e.d.s.c. and N. Cancer Genome Atlas Research, Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell, 2017. 171(4): p. 950–965 e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eilber FC, et al. , Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol, 2001. 19(13): p. 3203–9. [DOI] [PubMed] [Google Scholar]
- 38.MacDermed DM, et al. , Primary tumor necrosis predicts distant control in locally advanced soft-tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys, 2010. 76(4): p. 1147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biswas SK and Mantovani A, Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol, 2010. 11(10): p. 889–96. [DOI] [PubMed] [Google Scholar]
- 40.Cheng S, et al. , A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell, 2021. 184(3): p. 792–809 e23. [DOI] [PubMed] [Google Scholar]
- 41.Mulder K, et al. , Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity, 2021. 54(8): p. 1883–1900 e5. [DOI] [PubMed] [Google Scholar]
- 42.Silina K, et al. , Manipulation of tumour-infiltrating B cells and tertiary lymphoid structures: a novel anti-cancer treatment avenue? Cancer Immunol Immunother, 2014. 63(7): p. 643–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helmink BA, et al. , B cells and tertiary lymphoid structures promote immunotherapy response. Nature, 2020. 577(7791): p. 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrita R, et al. , Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature, 2020. 577(7791): p. 561–565. [DOI] [PubMed] [Google Scholar]
- 45.Petitprez F, et al. , B cells are associated with survival and immunotherapy response in sarcoma. Nature, 2020. 577(7791): p. 556–560. [DOI] [PubMed] [Google Scholar]
- 46.Keung EZ, et al. , Analysis of the immune infiltrate in undifferentiated pleomorphic sarcoma of the extremity and trunk in response to radiotherapy: Rationale for combination neoadjuvant immune checkpoint inhibition and radiotherapy. Oncoimmunology, 2018. 7(2): p. e1385689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolf Y, Anderson AC, and Kuchroo VK, TIM3 comes of age as an inhibitory receptor. Nature Reviews Immunology, 2020. 20(3): p. 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel KR, et al. , Increase in PD-L1 expression after pre-operative radiotherapy for soft tissue sarcoma. Oncoimmunology, 2018. 7(7): p. e1442168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mowery YM, et al. , SU2C-SARC032: A phase II randomized controlled trial of neoadjuvant pembrolizumab with radiotherapy and adjuvant pembrolizumab for high-risk soft tissue sarcoma. Journal of Clinical Oncology, 2018. 36(15_suppl): p. TPS11588–TPS11588. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Raw data for this study were generated at Fred Hutchinson Cancer Research Center (mIHC), Merck & Co., Inc., Kenilworth, NJ, USA (NanoString), and Cofactor Genomics (RNA sequencing with ImmunoPrism analysis). Derived data supporting the findings of this study are available from the corresponding author upon request.