

Abstract
Cellular metabolism generates molecular damage affecting all levels of biological organization. Accumulation of this damage over time is thought to play a central role in the aging process, but damage manifests in diverse molecular forms complicating its assessment. Insufficient attention has been paid to date to the role of molecular damage in aging-related phenotypes, particularly in humans, in part because of the difficulty in measuring its various forms. Recently, omics approaches have been developed that begin to address this challenge, because they are able to assess a sizeable proportion of age-related damage at the level of small molecules, proteins, RNA, DNA, organelles and cells. This review describes the concept of molecular damage in aging and discusses its diverse aspects from theoretical models to experimental approaches. Measurement of multiple types of damage enables studies of the role of damage in human aging outcomes and lays a foundation for testing interventions to reduce the burden of molecular damage, opening new approaches to slowing aging and reducing its consequences.
Introduction
Molecular damage has long been thought to be a part of the aging process 1,2. As organisms grow older, more and more endogenous and exogenous damage accumulates, as only some of this damage can be cleared or repaired 3. The role of cumulative damage in aging, particularly human aging, has not been thoroughly assessed, in part, because of limitations in and unfamiliarity with methods for measuring it. Understanding the role that molecular damage plays in aging and aging-related outcomes requires the ability to simultaneously quantify many diverse forms of damage that accumulate throughout life in cells and tissues and assess their associations with age, aging-related diseases, healthspan, and longevity.
Intimate relationship between damage and aging
Place of damage in theoretical and mechanistic understanding of aging
Aging has many faces 4. It is characterized by increased mortality, functional decline, and exponential increases in the incidence of degenerative diseases. At the cellular level, aging may be represented by the” Hallmarks of Aging”2. However, what ultimately drives all these processes is damage that changes functionally relevant biomolecules in ways that deleteriously affect cellular and organismal processes. The age-related damage comes in myriad forms such as by-products of metabolism, mutations, epimutations, errors in transcription and translation, organelle damage, and other known and undiscovered forms of damage. Damage can be reasonably viewed as a cornerstone of aging -- its essence and its cause. Given its centrality, one would expect age-related molecular damage to be a major focus of aging research, but it has not been the case due, in part, to the complexity of the aging process and the lack of methods to tackle damage accumulation in toto as opposed to its individual forms. Damage begins to accumulate in organisms early in life, even before birth, and in mammals, it occurs transgenerationally. This is clear from the analyses of somatic and germline mutations, patterns of telomere shortening, DNA methylation changes, and other factors 5. As damaging mutations are inherited from parents, some damage is already present at the onset of life, and the burden of these mutations is associated with reduced lifespan and healthspan 6. Molecular damage also manifests as cellular aging. For example, forms of intracellular molecular damage can induce cell senescence.7 Cell senescence itself can accelerate the accumulation of intracellular damage. Thus, senescent cells both carry and induce molecular damage that contributes to aging of the whole organism.
Interestingly, some organisms such as the hydra fully control damage accumulation by diluting it by constant cell division and cell renewal. In contrast, organisms with essential non-renewable cells and structures (e.g. neurons, skeleton), such as all mammals, inevitably age. Some of the age-related damage in these organisms is random, but some is not. Specific forms of damage are generated by specific metabolic processes and functions. As these processes are programmed through their respective genes, these same genes superficially appear to be programmed to generate damage. While these programmatic features are evident, they have not evolved with the purpose of aging.
The dominant theories of aging (Fig. 1, Box 1) all recognize either the primacy of damage or its critical importance in the aging process, albeit in different contexts.
Figure 1. Molecular damage is a central element of aging theories.
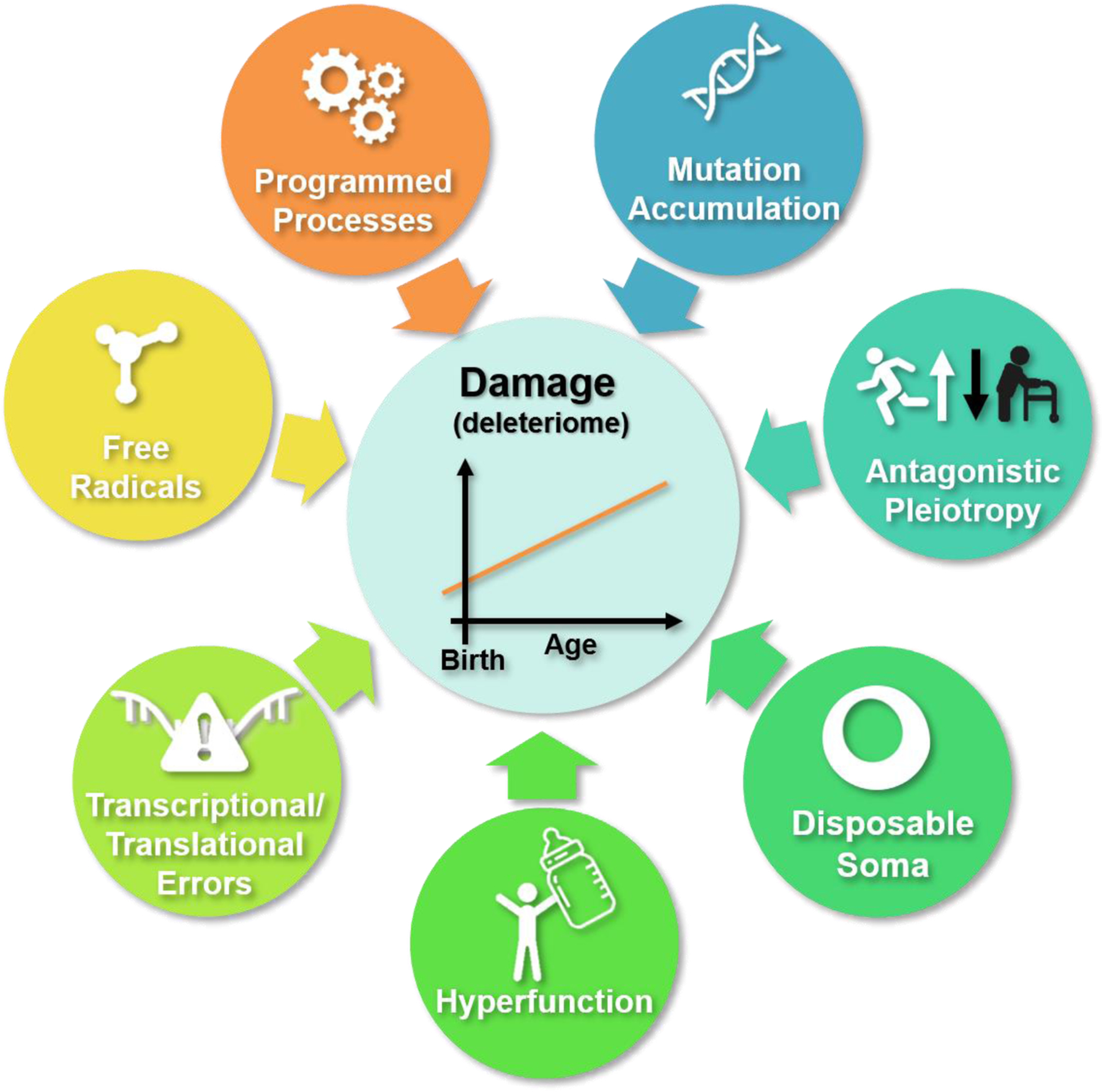
While the existing theories do not agree on the nature of aging, they all consider cumulative damage as a central factor. Molecular damage accumulates throughout lifespan beginning early in life, in mammals before birth, and never reaches zero.
BOX 1. How damage is viewed by different aging concepts.
The programmed theory of aging envisions a genetically defined, evolutionarily conserved program whose purpose is to force organisms towards the aging path over time and ultimately eliminate older organisms from the population to preserve resources for the younger individuals 108. Damage is viewed as a mediator of such aging program.
Antagonistic pleiotropy and mutation accumulation theories 109–112 consider aging as non-programmed and posit that it occurs due to the accumulation of germline mutations that are either neutral (in mutation accumulation) or beneficial (in antagonistic pleiotropy) in the pre- and early reproductive life, but are deleterious in the late life. As these mutations have no deleterious effects on fitness in reproductively-active organisms, they cannot be eliminated by natural selection from the population and are passed to the next generation. These evolutionary theories typically consider damage accumulation as a key factor in progressing through age, although the molecular basis of these theories is not well defined113.
The disposal soma theory of aging 114 offers an evolutionary basis for the buildup of global age-related damage, suggesting that, as resources are limited organisms have to invest them in both reproduction and maintenance, the maintenance part is imperfect, and therefore damage accumulates.
The hyperfunction theory of aging 115–117 suggests that aging is caused by the persistence of the overactivity of genes generating excessive functions that lead to damage, rather than the other way around. While this model gives the damage a secondary role, the overactivity of genes and damage may be said to work hand-in-hand to bring about aging phenotypes.
The free radical theory of aging 14,118,119 suggests that molecular damage results from the reactivity of partially reduced molecular oxygen produced by metabolic processes. However, free radicals are only one potential damage source, so this theory has somewhat grown out of favor.
A more recent deleteriome 47 concept suggests that endogenous damage is a by-product of imperfect metabolic processes and results from most if not all biological functions. In this model, damage is the consequence of life and exists in the myriad of forms 15,120. As the number of these damage forms is greater than the means of protection against it, damage accumulates. The deleteriome encompasses molecular damage as well as other age-related deleterious changes at all levels of biological organization.
Measurements of damage and inferences about causes of aging
Considering the diverse forms of age-related damage, how can global damage be measured? Individual methods are currently available that detect only a subset of molecular damage. Omics approaches, described in this review, may cover more damage than methods that measure individual forms of damage and, therefore, better describe the aging process in the whole organism. The use of omics methods has led to important insights into the behavior of cumulative damage in model organisms. For example, metabolite profiling that quantified changes in non-targeted metabolites was used to follow age-associated trajectories of more than ten thousand metabolites in fruit flies subjected to control and dietary restriction diets 8. These analyses revealed that aging is associated with increased metabolite diversity and the rise in low-abundance molecules. Further improvements in sensitivity of mass-spectrometry (MS) may even better represent the aging metabolome 8.
Increased chemical diversity with age, as measured by various omics methods, may be a marker of cumulative damage. The reactivity of cellular metabolites far exceeds their metabolic functions 9–11 and is very common in biology, with some unwanted reactions having proper names (e.g. Michael addition, Amadori rearrangement, and Pictet-Spengler reaction). A more complete description of aging would include numerous types of endogenous damage.
Cumulative molecular damage may even influence the aging process through diet 12, as some of this damage may be ingested by organisms together with nutrients. In one study, species-specific culture media and diets were employed that incorporated molecular extracts of young and old organisms 12. In each model organism tested (budding yeast, fruit flies, mice), the “old” diet or medium shortened the lifespan of one or both sexes compared to the control that used the “young” diet or medium. This finding suggests that age-associated cumulative damage is deleterious, is causally linked with aging, and may affect lifespan through diet. This also suggests that age-accelerating environmental exposures might be identified through their effects on damage accumulation.
The exact contributions of particular damage forms to aging are often challenging to determine; however, one study attempted to address this issue by subjecting budding yeast to multiple rounds of replicative aging and assessing de novo mutations in daughters of mothers of different ages 13. As expected, mutations increased with age, but their low numbers, < 1 per lifespan, excluded their causal role in aging. Conceptually, these findings should also apply to other damage types, suggesting a causal role of cumulative damage, as opposed to individual damage types, in organismal aging.
This view on the importance of cumulative damage in aging is also illustrated by the free radical theory of aging 14. This concept has been useful in defining the contribution of oxidative damage to aging, but an increasing number of studies contradicted both its centrality and sufficiency to aging mechanisms 15. Although reactive oxygen species (ROS) are prototypical damaging by-products, their contribution to aging is governed by the metabolic organization of the cell, its protective systems, and genotype. These factors are controlled by natural selection, and like dietary and genetic interventions that extend lifespan, they influence aging outcomes through changes related to the composition of cumulative damage and the rates of accumulation in its various forms. Therefore, oxidative damage, like other specific damage types viewed in isolation or even in combination, does not represent a major cause of aging. Rather, inevitable accumulation of damage in the form of numerous molecular species helps define the true root of aging through their combinatorial effect.
Omics methods can quantify and integrate the impact of age-related damage. But since life is sustained by complex and dynamic systems, making it difficult to interpret age-related changes. Such associations, as revealed by omics studies, may include responses to damage and neutral changes, in addition to the damage itself. Moreover, this balance of damaging, neutral and adaptive changes unequally applies to particular damage forms, e.g. mutations and epigenetic changes, and RNA-seq, proteomic, and metabolomic patterns. Thus, caution should be taken when inferring causal relationships between molecular profiles or other measurements of biological damage and aging.
DNA Damage
Somatic mutations
With age, DNA accumulates somatic mutations driven by DNA replication and transcription 16,17. With recent advances of single-cell sequencing technologies, it is now possible to better distinguish mutations from sequencing errors and other noise. An analysis of single neurons from people aged 4 months to 82 years revealed that mutation abundance is increased in older individuals in all brain regions18. Importantly, the same types of mutations (point mutations) are increased in the brains of patients with xeroderma pigmentosum and Cockayne syndrome, diseases caused by a defect in nucleotide excision repair and characterized by premature neurodegeneration19. This provides a strong correlation between mutation load and age-related disease. Mutations accumulate most rapidly during early and late life 20. Most mutations in the nuclear and mitochondrial genomes are either neutral or, less frequently, detrimental to cell function.
Mutation accumulation varies across cell types. A single-cell multiple displacement amplification analysis of liver cells from humans aged 5 months and 77 years revealed that mutations are significantly increased with age in hepatocytes, while in adult liver stem cells, the mutation rate is much lower 21. Age-related mutations accumulate in specific patterns across the genome, as revealed by cancer genomics 22–24. In addition to driving mutagenesis, DNA damage that cannot be replicated drives other outcomes, such as cell death or senescence 25. DNA strand breaks and stalled replication forks are potent drivers of these cell fates if not rapidly repaired.
Somatic DNA damage
Evidence that supports the notion that DNA damage plays a causal role in aging includes: i) all organisms have multiple highly conserved DNA repair mechanisms that have evolved to protect the nuclear genome, illustrating the importance of maintaining genome integrity; ii) genetic defects that interfere with the ability to repair DNA damage cause accelerated aging; iii) the vast majority of diseases of accelerated aging have a component of genome instability or intolerance of genotoxic stress; iv) skin in areas of the body exposed to the sun and UV radiation, without exception, ages more rapidly than non-exposed skin; v) cancer therapy with genotoxic agents dramatically accelerates aging and the onset of age-related diseases in cancer survivors 26, and vi) DNA damage is a potent driver of cellular senescence, which has been established to play a causal role in aging and many age-related diseases27. Despite this compelling case for endogenous and exogenous DNA damage driving aging, formal proof of how, when, and where somatic DNA damage does so is lacking. The reason for these gaps in knowledge is that it is challenging to measure endogenous DNA lesions, metabolites that damage nucleic acids, and DNA repair capacity in tissues.
Cross-sectional studies show increased DNA damage in old organisms compared to young. This includes increased abundance of oxidative DNA lesions (e.g., 8-oxodG 28 or cyclopurine adducts 29) and perhaps single-strand breaks 30. There is also increased expression of surrogate markers of genotoxic stress in older organisms relative to young, such as modified histones (γH2AX) and DNA damage response (DDR) proteins (e.g., p53, p21 or GADD45), as well as evidence for activation of the DDR (e.g., increased phospho-p53, phospho-ATM, and 53BP1 nuclear foci). It is also well established that in aged mammals, expression and activity of antioxidants are reduced compared to young organisms 31, with a concomitant increase in ROS 32, providing increased opportunity for DNA damage to occur.
DNA repair is so efficient that a cross-sectional measure of DNA damage is thought to reflect only the preceding few weeks of exposure to genotoxins, whether endogenous or exogenous 33. Thus, it remains unclear if increased DNA damage in older organism is due to a wane in DNA repair capacity with aging 26.
Perhaps the biggest challenge to directly quantifying somatic DNA damage is that cells often respond robustly to genotoxic stress. They activate the DDR, which initially halts cell cycle progression to allow time for DNA repair. This prevents replication of DNA adducts, somatic mutations, and the risk of transformation and carcinogenesis. Acute activation of the DDR is advantageous and a potent tumor suppressor mechanism. However, chronic activation of the DDR, either due to repeated episodes of genotoxic stress or complex DNA lesions that are irreparable, leads to activation of cell fate decisions 34. While there are a variety of cell fates, which are currently difficult to distinguish based on specific markers, genotoxic stress clearly drives apoptosis 35 and cellular senescence 36,37. This exacerbates the challenges of measuring DNA damage because if DNA damage triggers apoptosis, the cells with the damage are dead and eliminated. In contrast, if DNA damage drives senescence, the cell will persist, DNA damage is measurable, and surrogate markers of genotoxic stress become amplified (γH2AX, 53BP1 foci, p53 and p21 levels) along with cellular senescence biomarkers (p16, p21, β-galactosidase and the senescence-associated secretory phenotype factors). In addition, senescence rewires cellular metabolism, which can cause amplification of sources of genotoxic stress 31. Thus, the absence of detectable DNA damage in a tissue does not exclude it having been there, while the presence of surrogate marks of DNA damage in the absence of detecting adducts may point to other causes of senescence rather than genotoxic stress.
LC-MS/MS/MS is the most sensitive and specific method to measure endogenous DNA damage though it is laborious and expensive 38. It also requires prior knowledge about what to measure (what is/are the DNA adduct(s) of interest?, where to look [in what tissue?] and when [at what age?]). Thus, a tiered approach to assessing genotoxic stress is recommended, starting with (1) evidence for genotoxic stress based on the cellular response to DNA damage, then (2) measures of endpoints that inform about whether cells are in a state of increased oxidative stress implying increased DNA damage. With these criteria met, DNA lesion quantification can proceed to create a correlation between DNA damage levels and aging or age-related diseases (Supplemental Table 1).
Routine measurement of DNA damage levels will require the identification of stable DNA lesions that can be quantitated by LC-MS/MS/MS or with antibodies. Currently, the only antibodies to detect endogenous DNA adducts commercially available are against 8-oxodG 39 and M1G (pyrimido[1,2-a]purin-10(3H)-one). Measuring the adductome is a promising emerging technology. 40 This allows the quantitation of the total burden of DNA damage in tissues or cells without identifying the adducts themselves. Ultimately, longitudinal studies are needed to determine if various DNA adducts accumulate over time in vivo or increase towards the end of life in association with disease. It will also be important to determine if the DNA damage measured in cells or bodily fluids that are readily collected from humans and model organisms reflect the DNA damage burden of the entire soma.
Mitochondrial DNA damage
Mitochondrial oxidative phosphorylation (OXPHOS) supplies 90% of molecular energy, and is dependent upon the coordinated expression and interaction of genes encoded in the nuclear and mitochondrial genomes. Human mitochondrial DNA (mtDNA), a maternally inherited 16,569 bp genome containing genes critical to OXPHOS, commonly exhibits a mixture of standard and mutated states known as heteroplasmy 41. Unlike somatic DNA, there are many copies of mitochondrial DNA in a cell. Heteroplasmic mutations and rearrangements of mtDNA are not repaired and, therefore, accumulate with aging in various tissues42. Consequently, elderly adults exhibit reduced activity of OXPHOS enzymes in postmitotic tissues. In general, organs with the highest ATP requirements and the lowest regenerative capacities, such as the brain, heart, retina, auditory neuroepithelia, and skeletal muscle, are the most sensitive to the effects of mtDNA mutations.
Because mitochondria perform diverse functions in different tissues, specific mutations in mtDNA lead to mitochondrial diseases resulting in a broad spectrum of abnormalities. Many of these diseases result from high heteroplasmy loads (>80% burden of pathologic mtDNA mutation) and cause neurologic, sensory, movement, metabolic, and cardiopulmonary impairments. For example, one study found that mtDNA heteroplasmy sequenced at 20 loci known to be linked to inherited mitochondrial diseases was associated with reduced cognition, vision, hearing, and mobility 43. Another study found that increasing heteroplasmy in a single locus, m.3243A > G. was associated with multiple features of aging including poorer cognitive performance decreased grip strength and increased risk of mortality 44.
Thus, the accumulation of mutations in mDNA with age at specific sites in the mitochondrial genome, where mutations can be quantified, provides a measurement of the proportion of mitochondria having a mutation at that locus.
Epigenetic damage
The epigenome is another target for omics approaches 45. Many chemical changes in the epigenome with aging, similar to changes in the genome, can be broadly conceived as “damage” 46,47. Epimutations also resemble chemical modifications of other molecules, e.g. metabolites and proteins. Some studies assessed cumulative epigenetic damage in the form of Shannon entropy where the degree of predictability and levels of randomness are assessed. An increase in entropy at a CpG site with advancing age would mean its methylation status becomes harder to predict. Given that aging is accompanied by an accelerated increase in Shannon entropy, this is consistent with damage accumulation 48. Interestingly, these entropy effects differed for the CpG sites that increased, decreased, or remain unchanged in methylation status with age. Many sites trailed behind, whereas some followed or even exceeded the entropy trajectory and altered the developmental DNA methylation pattern. Thus, DNA methylome remodeling may play a role in aging and provides a framework to better understand the molecular changes in aging.
An important advance in the field associated with epigenetic changes has been the analysis of DNA methylation at hundreds of sites, which may serve as a ‘clock’ of organismal aging 49,50,51. The best-known human clocks include the Horvath pan-tissue clock trained based on chronological age 49, the Hannum blood clock 52, the PhenoAge clock trained based on blood biochemistry parameters associated with disease 53, and the GrimAge clock trained on future mortality 54,55. Numerous human clocks have also been developed for various tissues and developmental stages. The epigenetic clocks have been widely used in recent years to address various questions in the aging field, such as the difference in aging rates among tissues and species, association with factors that affect the aging process, and association with various age-related diseases 51,56. The difference between chronological and biological (epigenetic) age assessed by these clocks, is termed ‘age acceleration’, and it has been consistently associated with mortality in human cohort studies 53,54.
The epigenetic clocks studies have also been extended to other organisms, most notably mice, where both single tissue and multi-tissue clocks are available that are trained based on chronological age 57–61. In addition, universal epigenetic clocks have been developed to assess the biological age across species 62. The mouse clocks were applied to test the effect of longevity interventions (e.g. calorie restriction, rapamycin, growth hormone receptor deficiency) on aging, as well as the conversion of somatic cells to induced pluripotent stem cells, which showed an age close to zero 49,58,63,64. Some interventions showed the potential of rewinding the epigenetic age 65,66.
Thus, far, age-related changes in the epigenome have been quantified primarily using human Illumina DNA methylation chips and by subjecting mouse samples to reduced representation bisulfite sequencing. Like other omics studies, DNA methylation may be used to assess age-related damage, but the contribution of damage to the observed epigenetic changes is currently unknown. Indeed, even though it is expected that many methylation changes are damaging, some of them may be adaptive.
RNA damage
Biomolecules are not stable over time; their inherent instability leads to degradation and loss of function. Like other biomolecules in the cell, various RNA species, e.g. mRNA, tRNA, rRNA and various small RNAs, get damaged with age, which includes chemical modifications, transcriptional errors, cross-links, unwanted RNA breaks and other forms of damage. Recent work described changes in splicing patterns as a function of age across model organisms 67. By analyzing more than thousand human samples, another study found ~50,000 splicing events that correlated with age in a tissue-specific manner 68. There is evidence that age-related changes in splicing are functionally related to processes that have been implicated in lifespan control. In nematodes, splicing factor Sfa-1 mediates the lifespan-extending effect of caloric restriction, and its overexpression was shown to extend lifespan69. Other forms of damage to RNA include variance in gene expression, expression of repetitive elements, and erroneously expressed genes in tissues.
In contrast to DNA damage, there have been less focus on damage to RNA. However, it may be assessed by RNA-seq, which is among the most common omics methods, and in the future this approach RNA damage may extend even to single cells. For example, one study identified differentially expressed “dark matter” transcripts that do not map to exons 70. RNA-seq, as well as microarray profiling, are commonly used to assess changes in the expression of genes with age, and some of these changes also reflect damage. Several comparative studies have searched for conserved molecular signatures of aging as defined by sets of genes whose expression changes with age across tissues and organisms. For example, a study comparing mice, rats and humans found common genes that were differentially expressed with age in certain tissues 71,72. However, such studies do not distinguish between changes that are damaging and those that representing compensatory, adaptive responses to damage. With the release of large-scale RNA-seq datasets, such as human GTEx73 and mouse Tabula Muris74, it may now be possible to examine patterns of RNA damage across cell types and tissues.
Protein damage
The chemistry of protein damage
Like other biomolecules, the proteome is under constant attack by reactive molecules such as oxygen, glucose and water 75. Spontaneous oxidation of methionine, cysteine, histidine, and tyrosine by non-enzymatic pathways leads to dysfunctional protein products. There are also pathways where aspartic acid and asparagine residues racemize, isomerize, and deamidate into dysfunctional products (Fig. 2). These serve as prototypes for how spontaneous, non-enzymatic damage to biomolecules contributes to aging 11. Many forms of protein damage can be recognized by enzymes that repair that damage, such as reductase-mediated repair of oxidation damage of methionine sulfoxide residues 76–79. But there are also many damage forms, for which there is no protection.
Figure 2. Asparagine and aspartate residues in proteins may be damaged (red) and repaired (green).
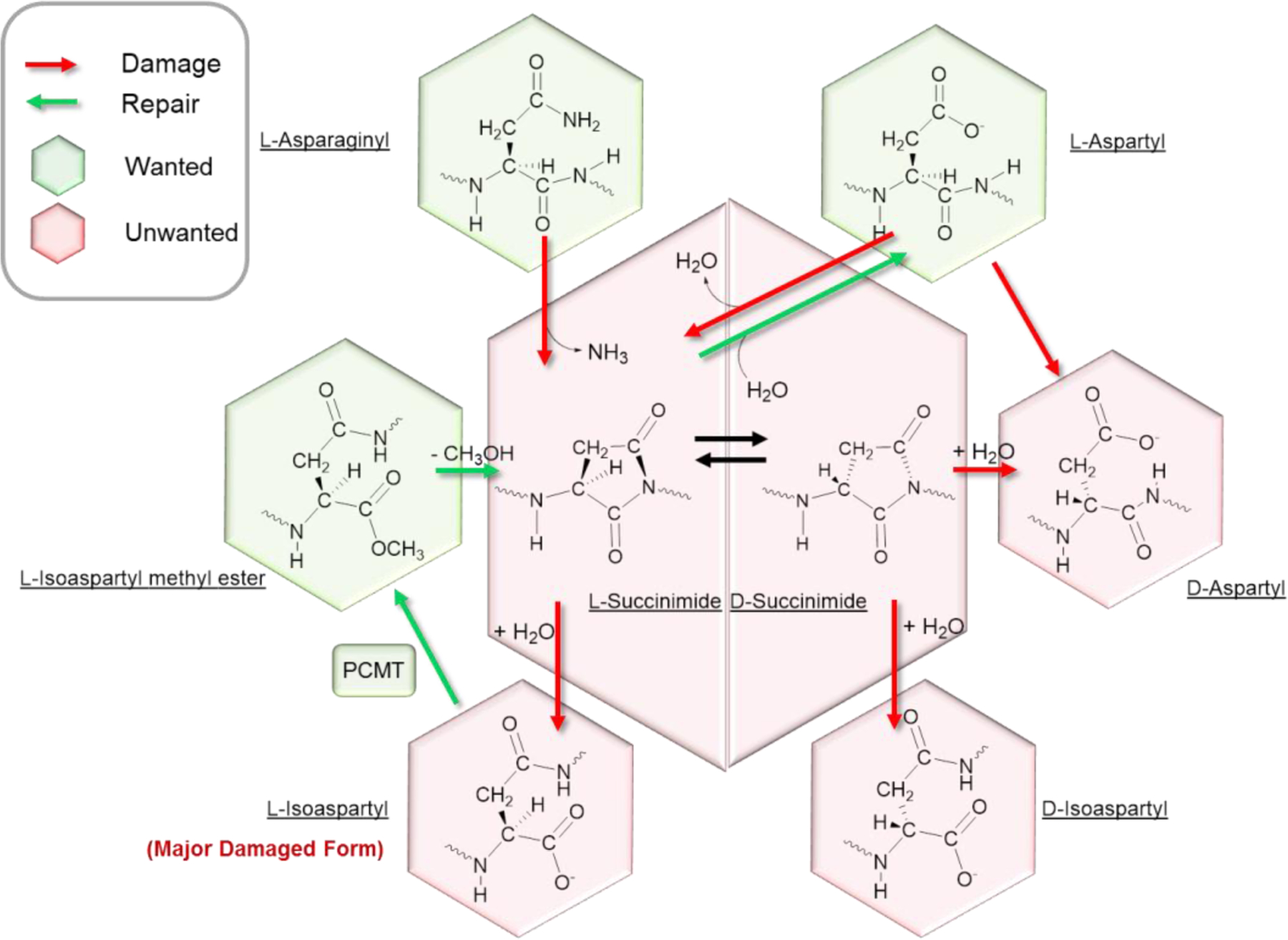
Some forms of damage are generated spontaneously, and some may be repaired by designated enzymes, such as PCMT.
Posttranslational modifications (PTM) of proteins
PTMs are an understudied area of protein damage with aging. They include, but are not limited to, oxidation, glycation, formylation, methylation, phosphorylation, SUMOylation, ubiquitination, and crosslinking. Many PTMs have been observed and quantified but many more, ‘putative’ PTMs have been predicted to exist or are yet to be discovered entirely 80. Some well-known PTMs have important roles in normal biological functions, but these same modifications, however, may also represent damage when occurring at incorrect positions in proteins where instead of supporting function, they disrupt it. For example, oxidation of a particular methionine residue in actin supports depolymerization of the protein81, whereas methionine sulfoxidation at other residues would represent damage82. An even more common situation is represented by non-regulatory PTMs, which are modifications that have no biological function. For example, formation of methionine sulfone residues has no function but is damaging as this modification cannot be repaired. Like other forms of damage, post-translational age-related damage to proteins contributes to cellular and tissue dysfunction that characterize the aging process.
Measuring specific post translational modifications (PTM) of proteins
Mass spectrometry (MS) is the method of choice to study and quantify PTMs and the most commonly measured modifications are phosphorylation, oxidation and methylation. The identification of protein oxidation requires only a peptide database search to include oxidation in the variable modification list to identify these PTMs. Although computationally expensive, a vast landscape of protein oxidation can be defined, albeit at the lack of specificity, because oxidation may occur during sample handling. Like phosphorylation, carboxymethyllysine is measured using - specific enrichment based MS methods detection83. A large cohort analysis of young and old male subjects (n = 1000), found that carboxymethyllysine increased with age84. Protein methylation can also be assessed by simple HPLC analysis-driven approaches, but the identified site of methylation must be carefully confirmed to prevent false aging related identifications from sample handling and processing. Although methylation has been defined in blood proteins, this is not performed routinely and reliable identification remains difficult.
For most other low abundance PTMs, enrichment steps to affinity purify peptides with specific PTMs preserved is standard. For example, phosphorylation, measured primarily on serine threonine, and tyrosine amino acids, are affinity purified by using metal catalyzed or antibody-bound supports to bind phosphorylated peptides specifically. Other antibody-based approaches are useful for identification of ubiquitinated peptides85. Finally, as proteins age, their propensity to develop cross-links increases and a number of proteins can be measured by MS and quantified computationally86.
Repair of protein damage
The potential importance of repair mechanisms in aging can be illustrated by the case of asparagine and aspartic acid residues in proteins. L-Asparaginyl and L-aspartyl residues are crucial to the structures and functions of many proteins, but these desirable residues can nonenzymatically transform to forms that represent the molecular damage. The prevalent L-isoaspartyl form may be particularly toxic to cells and therefore requires a strategy for its removal. Indeed, the PCMT protein repair methyltransferase converts L-isoaspartyl residues back to L-aspartyl residues via the L-isoaspartyl methyl ester (Fig. 2). In Caenorhabditis elegans, overexpression of PCMT extends lifespan, whereas in mouse models, PCMT knockout caused seizures and dramatically shortened lifespan 87. In this model, damaged aspartyl/asparaginyl residues accumulated in brain to a greater amount than in other tissues. But the response was also unexpected: blocking repair increased brain size and improved performance in spatial learning (hippocampus) and rotarod (cerebellar) tests. This is an example of how blocking a repair pathway leads to toxic effects, but also induces compensatory changes that may be beneficial. It also illustrates the complexity and surprising results of translating a single chemical change to phenotypic changes that tends to occur, particularly in mammals.
Treatments that enhance repair or proteolysis may be novel approaches to slowing the development of Alzheimer’s disease 88. It should be noted that generation of spontaneous and inflicted molecular damage is the inherent property of biological systems. The most dangerous molecules produced by these reactions may be repaired by designated enzymatic and other cellular systems, but during aging, these and other forms still accumulate. Moreover, the pathways leading to the synthesis of these enzymes are themselves susceptible to damage with time resulting in an exponential rate of accumulation of damage which may contribute to explaining the exponential rise of phenotypic damage with age.
Proteostasis and autophagy
Proteins that cannot be repaired may be cleared by the “Proteostasis Network.” Decline in cellular proteostasis is one of the hallmarks of aging2 and the consequent failure to clear damaged proteins may contribute to the accumulation of intracellular damage with aging.
This Proteostasis Network includes molecular chaperones and two intracellular proteolytic systems, the ubiquitin-proteasome (UPS) and the autophagic/lysosomal system 89. Chaperones identify misfolded proteins and promote their refolding or triage them for degradation through the UPS or lysosomes. Chaperones also prevent protein intermediates or partially folded states from undergoing hydrophobic collapse into globular conformations in the crowded cell environment. When protein intermediates aggregate, they can become stuck in a state of large configurational entropy 90,91. Some aggregates contain exposed, reactive side chains, which leads to nucleation of further aggregation or development into amyloid fibrils.
The degradation of intracellular components (proteins and organelles) by lysosomes is termed autophagy. There are several types of autophagy that coexist in all mammalian cells: macroautophagy, chaperone-mediated autophagy (CMA) and endosomal microautophagy (eMI) 89. Macroautophagy includes cellular sequestration and delivery to lysosomes inside double membrane vesicles (autophagosomes) of proteins and organelles including mitochondria (mitophagy), lipids (lipophagy), aggregates of proteins (aggregophagy), and pathogens (xenophagy), among others. In CMA and eMI, selective proteins are targeted one by one to lysosomes or late endosomes, respectively, for their degradation once internalized in their lumen. The lysosome contains enzymes that digest these biologic products into constituents, including amino acids and fatty acids, that can be recycled back to the cytosol and reused in synthesis of other proteins or used as energy substrates. The consequences of decline in autophagy with aging, in the face of continued generation of damage, include loss of cellular quality control and accumulation of damaged biological structures, as well as an energetic compromise and deregulation of important cellular functions such, as adaptation to stress or defense against pathogens.
Lysosomes are the organelles where all autophagy degradation takes place. Therefore, data on changes in lysosomal number and properties also inform on autophagic function. A combination of pH probes (e.g. Lysotracker or Lysosensor) that can quantify the degree of acidification of lysosomes or probes that quantify enzymatic activity (e.g. Magic-red®), with structural lysosomal components such as LAMPs, can be used to detect abnormalities on the lysosomal system that result in lower autophagic flux. An indirect assessment of the robustness/activity of the lysosomal system is the detection of accumulation of lipofuscin, which reflects a general failure of lysosomal digestion of damaged material 92. These auto-fluorescent yellow-brown pigment lipid-containing granules are composed of undigestible residues from the enzymatic digestion of macromolecules and organelles that accumulate in lysosomes (Fig. 3). Cross-linking of the undegraded protein products with lipids results in autofluorescence allowing for number and area of these granules to be quantified using direct fluorescence microscopy without need for additional staining. The quantity of lipofuscin increases with aging and, although an indirect measurement of autophagic function, quantification of this pigment has proven of value in fixed tissues where functional analysis is not possible.
Figure 3. Autofluorescent lipofuscin granules (green) in the liver of an old mouse.
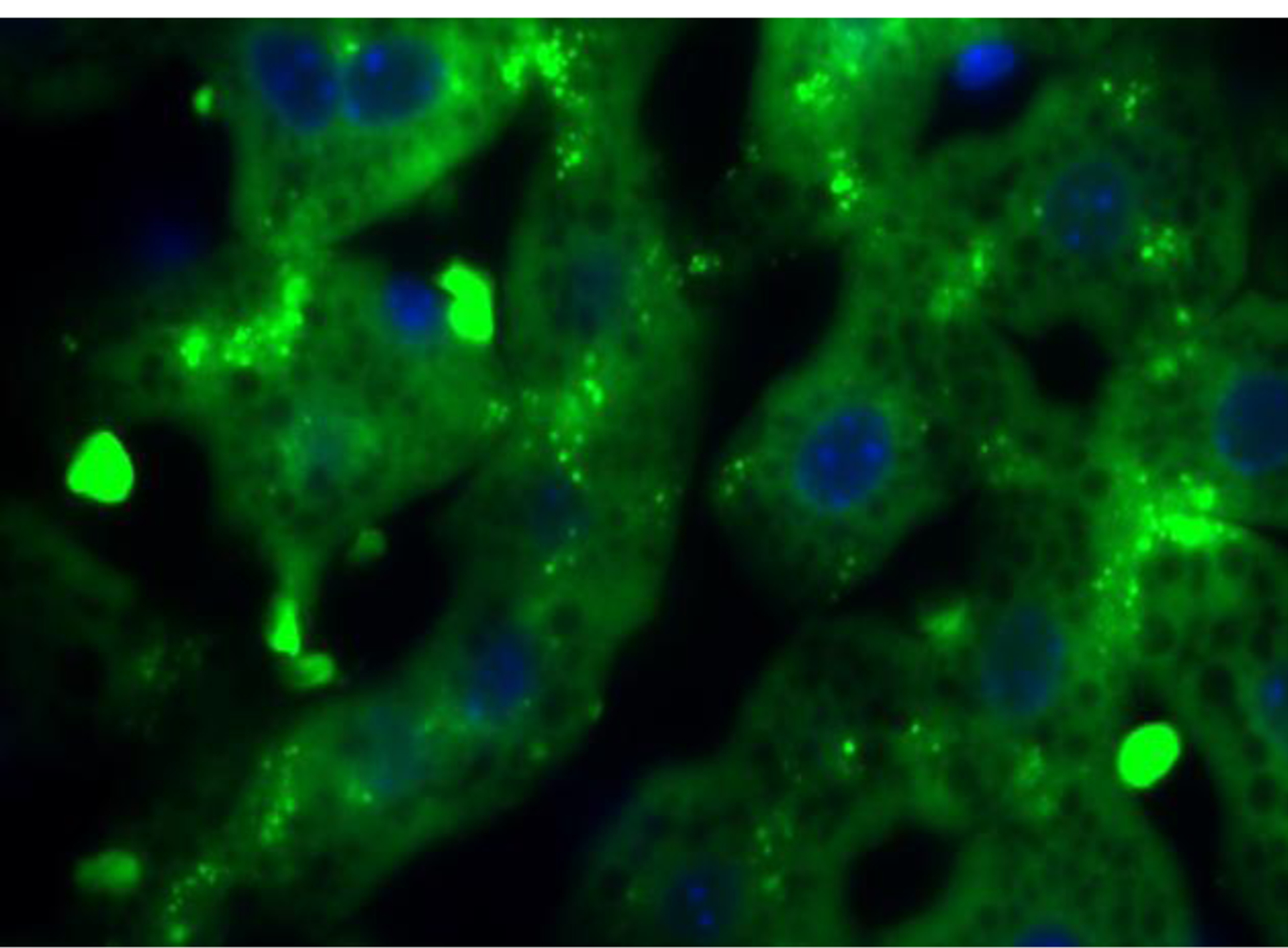
Information on the steady-state of autophagy can be determined in frozen or fixed samples by measurement of changes in both genes and proteins involved in the autophagic process and image-based procedures, such as immunohistochemistry and electron microscopy. Gene transcription is used to quantify changes in both macroautophagy and CMA as well as in the lysosomal system 93. Importantly, analysis of proteins, lysosomal cargos and receptors that deliver cargo to autophagosomes are required to fully characterize the status of a cell’s autophagy 94. The appropriate degree of activity of the autophagy system depends on the state of the cell: high levels of autophagy may be an appropriate response to increased rate of damage, or an inappropriate activation that may itself damage components of the cell.
Measuring the accumulation of damaged proteins could also provide an indirect measurement of the activity of the proteostasis network status. The accumulation of modified proteins (damaged or bearing unwanted PTMs) and the metabolic disbalance and subsequent alterations in metabolite levels can be assayed by proteomics and metabolomics. The failure of macroautophagy leads to morphological and functional changes in organelles such as swollen and deformed mitochondria, that can be recognized by electron microscopy. The best measurements of failing or insufficient autophagy depend on the type of autophagy, but in general, due to the multi-step nature of the process and the complexity of its regulation, experts in this area agree on the value of combining information coming from different quantitative approaches 95.
There have been few applications of measurements of autophagic activity to human longevity and aging. It was found that offspring of centenarians have better proteostasis and preserved macroautophagy in peripheral T cells than their non-centenarian spouses, and that this correlates directly with their immune function 96. Appreciation of the value of using multiple approaches to quantifying autophagy and gain information on the dynamics of this process, even in already collected samples, provides the opportunity to directly investigate the role of its decline or failure in aging and aging related conditions. To provide meaningful results, however, they must be interpreted in relationship to each other to characterize the dynamic process of autophagy.
Metabolite damage
Forms of metabolite damage
Metabolites are chemicals. Anything that can happen to metabolites in the laboratory can also happen to them in cells 97: e.g. the various types of oxidation, but also hydration, hydrolysis, nitrosylation, isomerization, and other chemical reactions. In a test tube, important metabolic intermediates such as THF-polyglutamate 98 or NADPH 99 are chemically unstable. Indeed, many essential metabolites, from bis- and diphosphates to aldehydes, gain their critical functions in metabolism from their inherent chemical reactivity. In addition, classic metabolic enzymes are not perfect e.g., glyceraldehyde-3-phosphate dehydrogenase can inadvertently hydrate NADH into its useless form NADHX 100. Other enzymes show reaction promiscuity in addition to substrate ambiguity 101, building up unwanted metabolite pools over time. To keep metabolites in their native chemical states serving their metabolic functions in cells, a plethora of helper enzymes, called damage control enzymes, are required. During aging, the defense system exhibited by such damage repair enzymes may become less effective. Hence, the extent of dysfunctional and damaged metabolites will change over time and become a readout as well as contributor to the accelerated effects of aging.
Even the best studied genomes, i.e. those of humans, mice, nematodes, fruit flies and budding yeast, have numerous genes of unknown function. Interestingly, many of more recently functionally characterized genes code for repair proteins 102. Virtually all enzymes exhibit side activities that often represent less than 0.1 % of the classical activity, and even sometimes < 10−6 for the most specific enzymes. The products generated by side-activities have been neglected until recently when it was realized that a new category of enzymes, metabolite repair and clearance enzymes, serve to destroy the most important side products and avoid their accumulation, which might otherwise be toxic, causing disease.
An example of a side activity is the production of L-2-hydroxyglutarate by L-malate dehydrogenase and lactate dehydrogenase, two abundant enzymes. Their apparently tiny side activity (<10−6 compared to the regular activity) leads to the daily production of grams of L-2-hydroxyglutarate in humans. An FAD-linked mitochondrial enzyme reconverts L-2-hydroxyglutarate to alpha-ketoglutarate, avoiding its accumulation, which is toxic particularly to the brain. The metabolic disease L-2-hydroxyglutaric aciduria, which is due to inactivating mutations in the repair enzyme, leads to progressive neurodegeneration and increased incidence of brain tumors.
In glycolysis, there appear to be at least as many distinct repair reactions as the 11 classical reactions of glycolysis 103. This huge diversity of side products related to glycolysis suggests that hundreds, and probably thousands of different side-products may be formed in cells. It is likely that only some of them are eliminated by repair and clearance enzymes. But at least for those that are eliminated, it is possible to evaluate their potential toxicity in cell-based experiments. Such experiments have shown that some of the side-products are indeed extremely toxic.
Clearly, many more defects of metabolite repair exist that contribute to aging. Some recognized metabolite repair diseases manifest early in life, as the consequences of protein-truncating variants in these genes are usually quite severe at that time. Other metabolite repair diseases, such as L-2-hydroxyglutaric aciduria, may be diagnosed later in life (probably due to mutations that do not completely inactivate L-2-hydroxyglutarate dehydrogenase). It is clear that many types of metabolite damage are not fully dealt with by enzymes of metabolite repair. This is particularly true for the damage that occurs at an extremely low rate and gives rise to products that only very slowly accumulate in cells and organs.
Measuring metabolic damage and repair
Non-targeted metabolomics is the best tool to study the extent of buildup of damaged metabolites over time. Here, a combination of assays using liquid chromatography with high resolution mass spectrometry (LC-HRMS) has been shown to identify and quantify over 1,000 metabolites in large cohort studies. In addition, non-targeted metabolomics utilizes large databases and novel software to annotate unidentified metabolites by their most likely chemical structures, vastly expanding the possibility to find and identify novel damaged metabolites. For example, he most classic version of metabolite damage is lipid oxidation, often induced by ROS that lead to hydroxylated, epoxidated, or highly reactive short chain lipids 104, including 4-hydroxynonenal (HNE) 105. Lipid peroxidation generates electrophilic compounds, typically containing aldehydes, which form ethenoadducts with DNA, a type of DNA damage. These DNA adducts formed with lipid peroxidation products result in exocyclic additions to DNA bases 106, disrupting transcription at its own site and to adjacent pyrimidine bases. When DNA repair mechanisms fail to detect or resolve these adducts, an incorrect base may be substituted, conceiving a mutagenic event. The accumulation of lipid peroxidation products can therefore reflect both a subset of damaged metabolites and a pool of adducts with the capacity to damage other macromolecules, namely DNA and proteins.
Future directions
Damage accumulates with aging at every level, from small molecules to cells. There have been many studies on specific damage types in aging, but a remarkably few studies on the role of global damage in aging. This is due, in large part, to the lack of methods to accurately assess many types of damage. However, this has been changing with the development of new omics methods, increased sensitivity of existing methods, and the development of computational tools for big data analyses. Many of these methods are discussed in this review and have already found applications in assessing age-related molecular damage.
The time is ripe for systematic studies of the role of damage in human aging, which could proceed with omics assays and methods that are already feasible in assessing a more sizeable fraction of damage (Table 1), especially in blood and biopsy tissues from existing human cohort studies. The association of candidate measurements of damage with chronological and/or biological age is a first step that is missing for almost all assays of damage included in this review. Progress can be accelerated by developing methods to measure damage burden in ways that encompass several kinds of damage, e.g. applying multi-omics approaches. Because cross-sectional associations with age often underestimate associations with aging, it would be ideal to describe the change in these markers of damage in serial specimens from longitudinal studies. Those measurements and types of damage that change with age would be candidates to test for associations with phenotypes of aging, such as weakness, slowness, and declining cognitive performance, and, using biobanks from cohort studies. Frailty, as assessed by accumulated health deficits, is conceptually well aligned with premise that molecular damage accumulation drives aging107. In turn, this would allow the testing of hypotheses that the markers of types of damage predict the incidence of degenerative diseases, such as cardiac, vascular, mobility disability, frailty, dementia, and non-cancer mortality.
Table 1.
Types of age-related molecular damage and approaches to its assessment.
| Class of molecule or process | Type of damage | Measurement |
|---|---|---|
| RNA | Transcriptional errors and RNA editing errors | RNAseq and whole genome or whole exome sequencing |
| Splicing errors | RNAseq | |
| Metabolites | By-products of metabolism | Targeted and untargeted metabolite profiling |
| Exogenous molecular species | Targeted and untargeted metabolite profiling | |
| Proteins | Post-translational modifications | Mass-spectrometry |
| Genome | Structural damage | Genome sequencing |
| Somatic mutations | Single cell genome sequencing | |
| Clonal expansions | Clonality assays | |
| mtDNA heteroplasmy | Mitochondrial genome sequencing | |
| Epigenome | Hypomethylation, hypermethylation of DNA | Whole genome bisulfite sequencing, reduced representation bisulfite sequencing, DNA methylation arrays |
| Autophagy failure | Decreased clearance of damaged molecules and organelles | Quantification of lipofuscin granules |
| Proteomics |
It may be possible to reduce the accumulation of damage with aging by slowing its generation or increasing systems of repair, clearance or replacement of damage. This approach would reduce the risk of many aging-related degenerative diseases. Reduction in the burden of damage – the deleteriome – might also mediate the benefits of treatments that reduce the risk of aging-related conditions. Testing these possibilities will require the development and validation of an array of measurements of types of molecular damage that play an important role in aging and its consequences.
Supplementary Material
Acknowledgments
The review is based in part on a workshop, Biological Damage and Human Aging held in December 2019, sponsored by the Longevity Consortium with support from the National Institute of Aging. Supported by NIH AG021332 (SK), NSF MCB-1714569, Life Extension Foundation, Inc., and the Elizabeth and Thomas Plott Chair in Gerontology (SGC), NIH AG064223, AG067782, AG065403 and AG047200 (VNG), NIH HL121023 and AG032498 (GT), and NIA U19AG023122 (the Longevity Consortium).
References
- 1.Kirkwood TB & Austad SN Why do we age? Nature 408, 233–238, doi: 10.1038/35041682 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217, doi: 10.1016/j.cell.2013.05.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stadtman ER Protein oxidation and aging. Science 257, 1220–1224, doi: 10.1126/science.1355616 (1992). [DOI] [PubMed] [Google Scholar]
- 4.Rando TA & Wyss-Coray T Asynchronous, contagious and digital aging. Nature Aging 1, 29–35, doi: 10.1038/s43587-020-00015-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinzina ED, Podolskiy DI, Dmitriev SE & Gladyshev VN Patterns of Aging Biomarkers, Mortality, and Damaging Mutations Illuminate the Beginning of Aging and Causes of Early-Life Mortality. Cell reports 29, 4276–4284 e4273, doi: 10.1016/j.celrep.2019.11.091 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Shindyapina AV et al. Germline burden of rare damaging variants negatively affects human healthspan and lifespan. eLife 9, doi: 10.7554/eLife.53449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogrodnik M, Salmonowicz H & Gladyshev VN Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 18, e12841, doi: 10.1111/acel.12841 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avanesov AS et al. Age- and diet-associated metabolome remodeling characterizes the aging process driven by damage accumulation. eLife 3, e02077, doi: 10.7554/eLife.02077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golubev AG [The other side of metabolism]. Biokhimiia 61, 2018–2039 (1996). [PubMed] [Google Scholar]
- 10.Golubev A, Hanson AD & Gladyshev VN A Tale of Two Concepts: Harmonizing the Free Radical and Antagonistic Pleiotropy Theories of Aging. Antioxidants & Redox Signaling 29, 1003–1017, doi: 10.1089/ars.2017.7105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golubev A, Hanson AD & Gladyshev VN Non-enzymatic molecular damage as a prototypic driver of aging. The Journal of biological chemistry 292, 6029–6038, doi: 10.1074/jbc.R116.751164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SG et al. Age-associated molecular changes are deleterious and may modulate life span through diet. Science Advances 3, e1601833, doi: 10.1126/sciadv.1601833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaya A, Lobanov AV & Gladyshev VN Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell 14, 366–371, doi: 10.1111/acel.12290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harman D Aging: a theory based on free radical and radiation chemistry. J Gerontol 11, 298–300, doi: 10.1093/geronj/11.3.298 (1956). [DOI] [PubMed] [Google Scholar]
- 15.Gladyshev VN The free radical theory of aging is dead. Long live the damage theory! Antioxidants & Redox Signaling 20, 727–731, doi: 10.1089/ars.2013.5228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamatoyannopoulos JA et al. Human mutation rate associated with DNA replication timing. Nat Genet 41, 393–395, doi: 10.1038/ng.363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lodato MA et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98, doi: 10.1126/science.aab1785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559, doi: 10.1126/science.aao4426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lodato MA & Walsh CA Genome aging: somatic mutation in the brain links age-related decline with disease and nominates pathogenic mechanisms. Human Molecular Genetics 28, R197–R206, doi: 10.1093/hmg/ddz191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podolskiy DI, Lobanov AV, Kryukov GV & Gladyshev VN Analysis of cancer genomes reveals basic features of human aging and its role in cancer development. Nature communications 7, 12157, doi: 10.1038/ncomms12157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brazhnik K et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Science Advances 6, eaax2659, doi: 10.1126/sciadv.aax2659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollstein M, Alexandrov LB, Wild CP, Ardin M & Zavadil J Base changes in tumour DNA have the power to reveal the causes and evolution of cancer. Oncogene 36, 158–167, doi: 10.1038/onc.2016.192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB & Stratton MR Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Current opinion in genetics & development 24, 52–60, doi: 10.1016/j.gde.2013.11.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101, doi: 10.1038/s41586-020-1943-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freitas AA & de Magalhaes JP A review and appraisal of the DNA damage theory of ageing. Mutat Res 728, 12–22, doi: 10.1016/j.mrrev.2011.05.001 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Niedernhofer LJ et al. Nuclear Genomic Instability and Aging. Annu Rev Biochem 87, 295–322, doi: 10.1146/annurev-biochem-062917-012239 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Baker DJ et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189, doi: 10.1038/nature16932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacob KD, Noren Hooten N, Trzeciak AR & Evans MK Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech Ageing Dev 134, 139–157, doi: 10.1016/j.mad.2013.02.008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Clauson CL, Robbins PD, Niedernhofer LJ & Wang Y The oxidative DNA lesions 8,5’-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 11, 714–716, doi: 10.1111/j.1474-9726.2012.00828.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beerman I Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin Hematol 54, 12–18, doi: 10.1053/j.seminhematol.2016.11.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson AR et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox biology 17, 259–273, doi: 10.1016/j.redox.2018.04.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui H, Kong Y & Zhang H Oxidative stress, mitochondrial dysfunction, and aging. Journal of signal transduction 2012, 646354, doi: 10.1155/2012/646354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfohl-Leszkowicz A in Advances in Molecular Toxicology Vol. 2 Advances in Molecular Toxicology (ed Fishbein James C.) 183–239 (Elsevier, 2008). [Google Scholar]
- 34.Ioannidou A, Goulielmaki E & Garinis GA DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front Genet 7, 187, doi: 10.3389/fgene.2016.00187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roos WP & Kaina B DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332, 237–248, doi: 10.1016/j.canlet.2012.01.007 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Kang C et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612, doi: 10.1126/science.aaa5612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F & Jackson SP Human cell senescence as a DNA damage response. Mech Ageing Dev 126, 111–117, doi: 10.1016/j.mad.2004.09.034 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Farmer PB et al. DNA adducts: mass spectrometry methods and future prospects. Toxicol Appl Pharmacol 207, 293–301, doi: 10.1016/j.taap.2004.12.020 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Guthrie OW Localization and distribution of neurons that co-express xeroderma pigmentosum-A and epidermal growth factor receptor within Rosenthal’s canal. Acta Histochem 117, 688–695, doi: 10.1016/j.acthis.2015.10.001 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Carra A et al. Targeted High Resolution LC/MS(3) Adductomics Method for the Characterization of Endogenous DNA Damage. Frontiers in chemistry 7, 658, doi: 10.3389/fchem.2019.00658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinto M & Moraes CT Mechanisms linking mtDNA damage and aging. Free radical biology & medicine 85, 250–258, doi: 10.1016/j.freeradbiomed.2015.05.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carelli V & Chan DC Mitochondrial DNA: impacting central and peripheral nervous systems. Neuron 84, 1126–1142, doi: 10.1016/j.neuron.2014.11.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tranah GJ et al. Mitochondrial DNA Heteroplasmy Associations With Neurosensory and Mobility Function in Elderly Adults. The journals of gerontology. Series A, Biological sciences and medical sciences 70, 1418–1424, doi: 10.1093/gerona/glv097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tranah GJ et al. Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality. Scientific reports 8, 11887, doi: 10.1038/s41598-018-30255-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Booth LN & Brunet A The Aging Epigenome. Molecular cell 62, 728–744, doi: 10.1016/j.molcel.2016.05.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinclair DA & Oberdoerffer P The ageing epigenome: damaged beyond repair? Ageing Res Rev 8, 189–198, doi: 10.1016/j.arr.2009.04.004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gladyshev VN Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15, 594–602, doi: 10.1111/acel.12480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sziraki A, Tyshkovskiy A & Gladyshev VN Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell 17, e12738, doi: 10.1111/acel.12738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horvath S DNA methylation age of human tissues and cell types. Genome biology 14, R115, doi: 10.1186/gb-2013-14-10-r115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson AA et al. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res 15, 483–494, doi: 10.1089/rej.2012.1324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nature reviews. Genetics 19, 371–384, doi: 10.1038/s41576-018-0004-3 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Hannum G et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell 49, 359–367, doi: 10.1016/j.molcel.2012.10.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levine ME et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591, doi: 10.18632/aging.101414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu AT et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327, doi: 10.18632/aging.101684 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belsky DW et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife 9, doi: 10.7554/eLife.54870 (2020). [DOI] [PMC free article] [PubMed]
- 56.Bell CG et al. DNA methylation aging clocks: challenges and recommendations. Genome biology 20, 249, doi: 10.1186/s13059-019-1824-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petkovich DA et al. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell metabolism 25, 954–960 e956, doi: 10.1016/j.cmet.2017.03.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meer MV, Podolskiy DI, Tyshkovskiy A & Gladyshev VN A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675, doi: 10.7554/eLife.40675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stubbs TM et al. Multi-tissue DNA methylation age predictor in mouse. Genome biology 18, 68, doi: 10.1186/s13059-017-1203-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson MJ et al. A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854, doi: 10.18632/aging.101590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang T et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome biology 18, 57, doi: 10.1186/s13059-017-1186-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu AT et al. Universal DNA methylation age across mammalian tissues. bioRxiv, 2021.2001.2018.426733, doi: 10.1101/2021.01.18.426733 (2021). [DOI]
- 63.Lu Y et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129, doi: 10.1038/s41586-020-2975-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olova N, Simpson DJ, Marioni RE & Chandra T Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 18, e12877, doi: 10.1111/acel.12877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fahy GM et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 18, e13028, doi: 10.1111/acel.13028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horvath S et al. Reversing age: dual species measurement of epigenetic age with a single clock. bioRxiv, 2020.2005.2007.082917, doi: 10.1101/2020.05.07.082917 (2020). [DOI]
- 67.Bhadra M, Howell P, Dutta S, Heintz C & Mair WB Alternative splicing in aging and longevity. Hum Genet 139, 357–369, doi: 10.1007/s00439-019-02094-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang K et al. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Scientific reports 8, 10929, doi: 10.1038/s41598-018-29086-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heintz C et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 541, 102–106, doi: 10.1038/nature20789 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wood SH, Craig T, Li Y, Merry B & de Magalhaes JP Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age (Dordrecht, Netherlands) 35, 763–776, doi: 10.1007/s11357-012-9410-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Magalhaes JP, Curado J & Church GM Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25, 875–881, doi: 10.1093/bioinformatics/btp073 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palmer D, Fabris F, Doherty A, Freitas AA & de Magalhaes JP Ageing transcriptome meta-analysis reveals similarities and differences between key mammalian tissues. Aging 13, 3313–3341, doi: 10.18632/aging.202648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Consortium GT The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585, doi: 10.1038/ng.2653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tabula Muris C et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372, doi: 10.1038/s41586-018-0590-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grassi L & Cabrele C Susceptibility of protein therapeutics to spontaneous chemical modifications by oxidation, cyclization, and elimination reactions. Amino Acids 51, 1409–1431, doi: 10.1007/s00726-019-02787-2 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Lourenco Dos Santos S, Petropoulos I & Friguet B The Oxidized Protein Repair Enzymes Methionine Sulfoxide Reductases and Their Roles in Protecting against Oxidative Stress, in Ageing and in Regulating Protein Function. Antioxidants (Basel) 7, 191, doi: 10.3390/antiox7120191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mishra PKK & Mahawar M PIMT-Mediated Protein Repair: Mechanism and Implications. Biochemistry (Mosc) 84, 453–463, doi: 10.1134/S0006297919050018 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Weinert BT, Moustafa T, Iesmantavicius V, Zechner R & Choudhary C Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J 34, 2620–2632, doi: 10.15252/embj.201591271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van Schaftingen E, Collard F, Wiame E & Veiga-da-Cunha M Enzymatic repair of Amadori products. Amino Acids 42, 1143–1150, doi: 10.1007/s00726-010-0780-3 (2012). [DOI] [PubMed] [Google Scholar]
- 80.Khoury GA, Baliban RC & Floudas CA Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Scientific reports 1, doi: 10.1038/srep00090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee BC et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Molecular cell 51, 397–404, doi: 10.1016/j.molcel.2013.06.019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fedorova M, Kuleva N & Hoffmann R Identification of cysteine, methionine and tryptophan residues of actin oxidized in vivo during oxidative stress. Journal of proteome research 9, 1598–1609, doi: 10.1021/pr901099e (2010). [DOI] [PubMed] [Google Scholar]
- 83.Wang R et al. Affinity Purification of Methyllysine Proteome by Site-Specific Covalent Conjugation. Analytical chemistry 90, 13876–13881, doi: 10.1021/acs.analchem.8b02796 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Rankin NJ et al. High-throughput quantification of carboxymethyl lysine in serum and plasma using high-resolution accurate mass Orbitrap mass spectrometry. Annals of clinical biochemistry 56, 397–407, doi: 10.1177/0004563219830432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Akimov V et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nature structural & molecular biology 25, 631–640, doi: 10.1038/s41594-018-0084-y (2018). [DOI] [PubMed] [Google Scholar]
- 86.Hoopmann MR et al. Kojak: efficient analysis of chemically cross-linked protein complexes. Journal of proteome research 14, 2190–2198, doi: 10.1021/pr501321h (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim E, Lowenson JD, MacLaren DC, Clarke S & Young SG Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proceedings of the National Academy of Sciences of the United States of America 94, 6132–6137, doi: 10.1073/pnas.94.12.6132 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Warmack RA et al. Structure of amyloid-beta (20–34) with Alzheimer’s-associated isomerization at Asp23 reveals a distinct protofilament interface. Nature communications 10, 3357, doi: 10.1038/s41467-019-11183-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaushik S & Cuervo AM Proteostasis and aging. Nat Med 21, 1406–1415, doi: 10.1038/nm.4001 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Bartlett AI & Radford SE An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nature structural & molecular biology 16, 582–588, doi: 10.1038/nsmb.1592 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M & Hartl FU Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82, 323–355, doi: 10.1146/annurev-biochem-060208-092442 (2013). [DOI] [PubMed] [Google Scholar]
- 92.Hohn A & Grune T Lipofuscin: formation, effects and role of macroautophagy. Redox biology 1, 140–144, doi: 10.1016/j.redox.2013.01.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sardiello M et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477, doi: 10.1126/science.1174447 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Juste YR & Cuervo AM Analysis of Chaperone-Mediated Autophagy. Methods Mol Biol 1880, 703–727, doi: 10.1007/978-1-4939-8873-0_47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klionsky DJ et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222, doi: 10.1080/15548627.2015.1100356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raz Y et al. Activation-Induced Autophagy Is Preserved in CD4+ T-Cells in Familial Longevity. The journals of gerontology. Series A, Biological sciences and medical sciences 72, 1201–1206, doi: 10.1093/gerona/glx020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lerma-Ortiz C et al. ‘Nothing of chemistry disappears in biology’: the Top 30 damage-prone endogenous metabolites. Biochem Soc Trans 44, 961–971, doi: 10.1042/BST20160073 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Chen L, Ducker GS, Lu W, Teng X & Rabinowitz JD An LC-MS chemical derivatization method for the measurement of five different one-carbon states of cellular tetrahydrofolate. Anal Bioanal Chem 409, 5955–5964, doi: 10.1007/s00216-017-0514-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang J et al. Determination of the Cytosolic NADPH/NADP Ratio in Saccharomyces cerevisiae using Shikimate Dehydrogenase as Sensor Reaction. Scientific reports 5, 12846, doi: 10.1038/srep12846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Niehaus TD et al. Plants utilize a highly conserved system for repair of NADH and NADPH hydrates. Plant Physiol 165, 52–61, doi: 10.1104/pp.114.236539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tawfik DS Enzyme promiscuity and evolution in light of cellular metabolism. FEBS J 287, 1260–1261, doi: 10.1111/febs.15296 (2020). [DOI] [PubMed] [Google Scholar]
- 102.Linster CL, Van Schaftingen E & Hanson AD Metabolite damage and its repair or pre-emption. Nature chemical biology 9, 72–80, doi: 10.1038/nchembio.1141 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Bommer GT, Van Schaftingen E & Veiga-da-Cunha M Metabolite Repair Enzymes Control Metabolic Damage in Glycolysis. Trends in biochemical sciences 45, 228–243, doi: 10.1016/j.tibs.2019.07.004 (2020). [DOI] [PubMed] [Google Scholar]
- 104.Piedrafita G, Keller MA & Ralser M The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions. Biomolecules 5, 2101–2122, doi: 10.3390/biom5032101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuiper HC, Miranda CL, Sowell JD & Stevens JF Mercapturic acid conjugates of 4-hydroxy-2-nonenal and 4-oxo-2-nonenal metabolites are in vivo markers of oxidative stress. The Journal of biological chemistry 283, 17131–17138, doi: 10.1074/jbc.M802797200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marnett LJ & Plastaras JP Endogenous DNA damage and mutation. Trends in genetics : TIG 17, 214–221, doi: 10.1016/s0168-9525(01)02239-9 (2001). [DOI] [PubMed] [Google Scholar]
- 107.Rockwood K & Mitnitski A Frailty in relation to the accumulation of deficits. The journals of gerontology. Series A, Biological sciences and medical sciences 62, 722–727, doi: 10.1093/gerona/62.7.722 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Longo VD, Mitteldorf J & Skulachev VP Programmed and altruistic ageing. Nature reviews. Genetics 6, 866–872, doi: 10.1038/nrg1706 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Williams GC Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 11, 398–411, doi: 10.1111/j.1558-5646.1957.tb02911.x (1957). [DOI] [Google Scholar]
- 110.Medawar PB An unsolved problem of biology (College, 1952). [Google Scholar]
- 111.Hamilton WD The moulding of senescence by natural selection. J Theor Biol 12, 12–45, doi: 10.1016/0022-5193(66)90184-6 (1966). [DOI] [PubMed] [Google Scholar]
- 112.Ronce O & Promislow D Kin competition, natal dispersal and the moulding of senescence by natural selection. Proceedings. Biological sciences 277, 3659–3667, doi: 10.1098/rspb.2010.1095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de Magalhaes JP Programmatic features of aging originating in development: aging mechanisms beyond molecular damage? FASEB journal : official publication of the Federation of American Societies for Experimental Biology 26, 4821–4826, doi: 10.1096/fj.12-210872 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kirkwood TB Evolution of ageing. Nature 270, 301–304, doi: 10.1038/270301a0 (1977). [DOI] [PubMed] [Google Scholar]
- 115.Blagosklonny MV Aging: ROS or TOR. Cell cycle (Georgetown, Tex.) 7, 3344–3354, doi: 10.4161/cc.7.21.6965 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Blagosklonny MV Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell cycle (Georgetown, Tex.) 5, 2087–2102, doi: 10.4161/cc.5.18.3288 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Blagosklonny MV Paradoxes of aging. Cell cycle (Georgetown, Tex.) 6, 2997–3003, doi: 10.4161/cc.6.24.5124 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Vina J, Borras C, Abdelaziz KM, Garcia-Valles R & Gomez-Cabrera MC The free radical theory of aging revisited: the cell signaling disruption theory of aging. Antioxidants & Redox Signaling 19, 779–787, doi: 10.1089/ars.2012.5111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miquel J, Economos AC, Fleming J & Johnson JE Jr. Mitochondrial role in cell aging. Exp Gerontol 15, 575–591, doi: 10.1016/0531-5565(80)90010-8 (1980). [DOI] [PubMed] [Google Scholar]
- 120.Gladyshev VN The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends in genetics : TIG 29, 506–512, doi: 10.1016/j.tig.2013.05.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Watson CJ et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449–1454, doi: 10.1126/science.aay9333 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Holstege H et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Research 24, 733–742, doi: 10.1101/gr.162131.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498, doi: 10.1056/NEJMoa1408617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zink F et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752, doi: 10.1182/blood-2017-02-769869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Evans MA, Sano S & Walsh K Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu Rev Pathol 15, 419–438, doi: 10.1146/annurev-pathmechdis-012419-032544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jaiswal S et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121, doi: 10.1056/NEJMoa1701719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847, doi: 10.1126/science.aag1381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sano S et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res 123, 335–341, doi: 10.1161/CIRCRESAHA.118.313225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sano S et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol 71, 875–886, doi: 10.1016/j.jacc.2017.12.037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sano S et al. JAK2 (V617F) -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci 4, 684–697, doi: 10.1016/j.jacbts.2019.05.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, doi: 10.1172/jci.insight.135204 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cai Z et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell stem cell 23, 833–849 e835, doi: 10.1016/j.stem.2018.10.013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bick AG et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 141, 124–131, doi: 10.1161/CIRCULATIONAHA.119.044362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131, doi: 10.1056/NEJMoa1707914 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Sano S, Wang Y & Walsh K Somatic mosaicism: implications for the cardiovascular system. European heart journal 41, 2904–2907, doi: 10.1093/eurheartj/ehz907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.