

Abstract
Nitrogenase catalyzes the multi-electron reduction of dinitrogen to ammonia. Electron transfer in the catalytic protein (MoFeP) proceeds through a unique [8Fe-7S] cluster (P-cluster) to the active site (FeMoco). In the reduced, all-ferrous (PN) state, the P-cluster is coordinated by six cysteine residues. Upon two-electron oxidation to the P2+ state, the P-cluster undergoes conformational changes in which a highly conserved oxygen-based residue (a Ser or a Tyr) and a backbone amide additionally ligate the cluster. Previous studies of Azotobacter vinelandii (Av) MoFeP revealed that when the oxygen-based residue, βSer188, was mutated to a non-coordinating residue, Ala, the P-cluster became redox-labile and reversibly lost two of its eight Fe centers. Surprisingly, the Av strain with a MoFeP variant that lacked the serine ligand (Av βSer188Ala MoFeP) displayed the same diazotrophic growth and in vitro enzyme turnover rates as wild-type Av MoFeP, calling into question the necessity of this conserved ligand for nitrogenase function. Based on these observations, we hypothesized that βSer188 plays a role in protecting the P-cluster under non-ideal conditions. Here, we investigated the protective role of βSer188 both in vivo and in vitro by characterizing the ability of Av βSer188Ala cells to grow under suboptimal conditions (high oxidative stress or Fe limitation) and by characterizing the ability of Av βSer188Ala MoFeP to be mismetallated in vitro. Our results demonstrate that βSer188 (1) increases Av cell survival upon exposure to oxidative stress in the form of hydrogen peroxide, (2) is necessary for efficient Av diazotrophic growth under Fe-limiting conditions, and (3) may protect the P-cluster from metal exchange in vitro. Taken together, our findings suggest a structural adaptation of nitrogenase to protect the P-cluster via Ser ligation, which is a previously unidentified functional role of the Ser residue in redox proteins and adds to the expanding functional roles of non-Cys ligands to FeS clusters.
Graphical Abstract
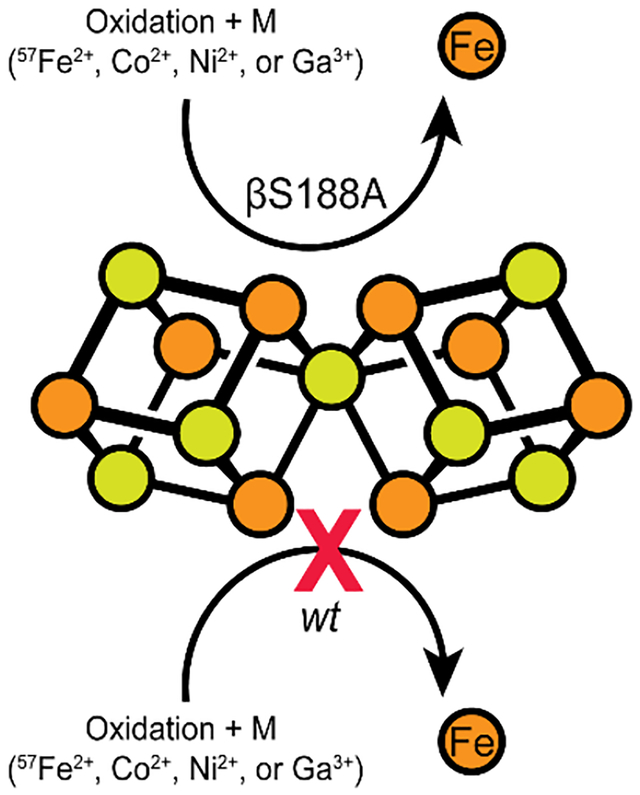
Introduction
Nitrogenase is the only known enzyme capable of catalyzing the reduction of atmospheric dinitrogen (N2) into ammonia (NH3) (Equation 1).1–4
| (1) |
The most widely studied version of nitrogenase is the Mo-nitrogenase which comprises two component proteins: the iron-protein (FeP), which serves as the reductase/ATPase, and the molybdenum-iron protein (MoFeP) which performs N2 reduction.5 The multi-electron reduction of N2 to NH3 is both thermodynamically and kinetically challenging,6 requiring precise coordination of electron and proton transfer events within the nitrogenase complex to preferentially reduce N2 over protons (H+).2 Electron transfer (ET) to the active site begins at the [4Fe-4S] cluster in FeP, proceeds through the intermediary [8Fe-7S] P-cluster in MoFeP, and ends at the site of catalysis, the iron-molybdenum-cofactor (FeMoco).1 Many successive cycles of one- (or possibly two-)7–9 ET steps are required for the reduction of a single N2 molecule, with each turnover cycle requiring ATP-gated association and dissociation of the FeP-MoFeP complex.
The distinct structure and composition of FeMoco are ultimately responsible for this cofactor’s ability to efficiently reduce N2. Yet, nitrogenase is also unique in its use of an expanded (i.e., the P-cluster)10–11 rather than a canonical Fe-S cluster (i.e., [2Fe-2S], [3Fe-4S], [4Fe-4S])12–14 as an electron relay center, implying that the P-cluster may not merely act as a passive one-electron transfer conduit. Aside from its composition, the P-cluster is also distinct in terms of its coordinating ligands (Figure 1a).15 In canonical Fe-S clusters, every Fe atom is coordinated to a terminal side-chain ligand from the protein.14 In contrast, the P-cluster is coordinated by both bridging and terminal cysteine (Cys) residues, which enables the cluster to undergo substantial structural changes in an oxidation-state-dependent manner.15 In the dithionite (DT)-reduced, all-ferrous state (PN), the P-cluster is coordinated by six Cys (four terminal and two bridging) residues (Figure 1a, 1c top).15 Upon oxidation by one electron to reach the P1+ state, a serine (Ser) residue (βS188 in Azotobacter vinelandii (Av) MoFeP) ligates an Fe center (Fe6), resulting in the opening of one cubane half of the cluster proximal to the β-subunit of MoFeP.16–17 Oxidation by one more electron to reach the P2+ state (also termed POX) is accompanied by the coordination of another Fe center (Fe5) by the backbone amidate N of a bridging Cys (αC88) (Figure 1c).15–16 Interestingly, βS188 is not strictly conserved: many nitrogenases contain an alanine (Ala) in position β188 (Av numbering).18 Yet, all but one of such nitrogenases contain a tyrosine (Tyr) in an alternative position (β99, Av numbering), which acts similarly to βS188 as a redox-switchable ligand, as recently demonstrated in the case of MoFeP from Gluconacetobacter diazotrophicus (Gd).18 The strict conservation of a covariant Ser or Tyr residue as a P-cluster ligand across all known nitrogenase sequences (with only one exception; see Conclusions) strongly suggests that presence of a hard, O-based ligand is important for nitrogenase function.18 It has been proposed that the binding of FeP to MoFeP during catalysis might trigger the coordination of the Ser or Tyr ligand to the P-cluster, thereby decreasing the reduction potential of the cluster and triggering ET from the P-cluster to FeMoco.1, 18–20 Alternatively, FeP-MoFeP complexation may induce conformational changes near FeMoco to increase its reduction potential and drive gated ET from the P-cluster. In this scenario, which is supported by recent cryoEM structures of the FeP-MoFeP complex isolated during catalytic turnover,21 Ser- or Tyr-ligation would serve to stabilize the resulting oxidized P-cluster, thus serving as a redox ratchet (rather than a trigger).
Figure 1. Examples of coordinatively unsaturated Fe-S clusters including the nitrogenase P-cluster and the [4Fe-4S] active site of aconitase.
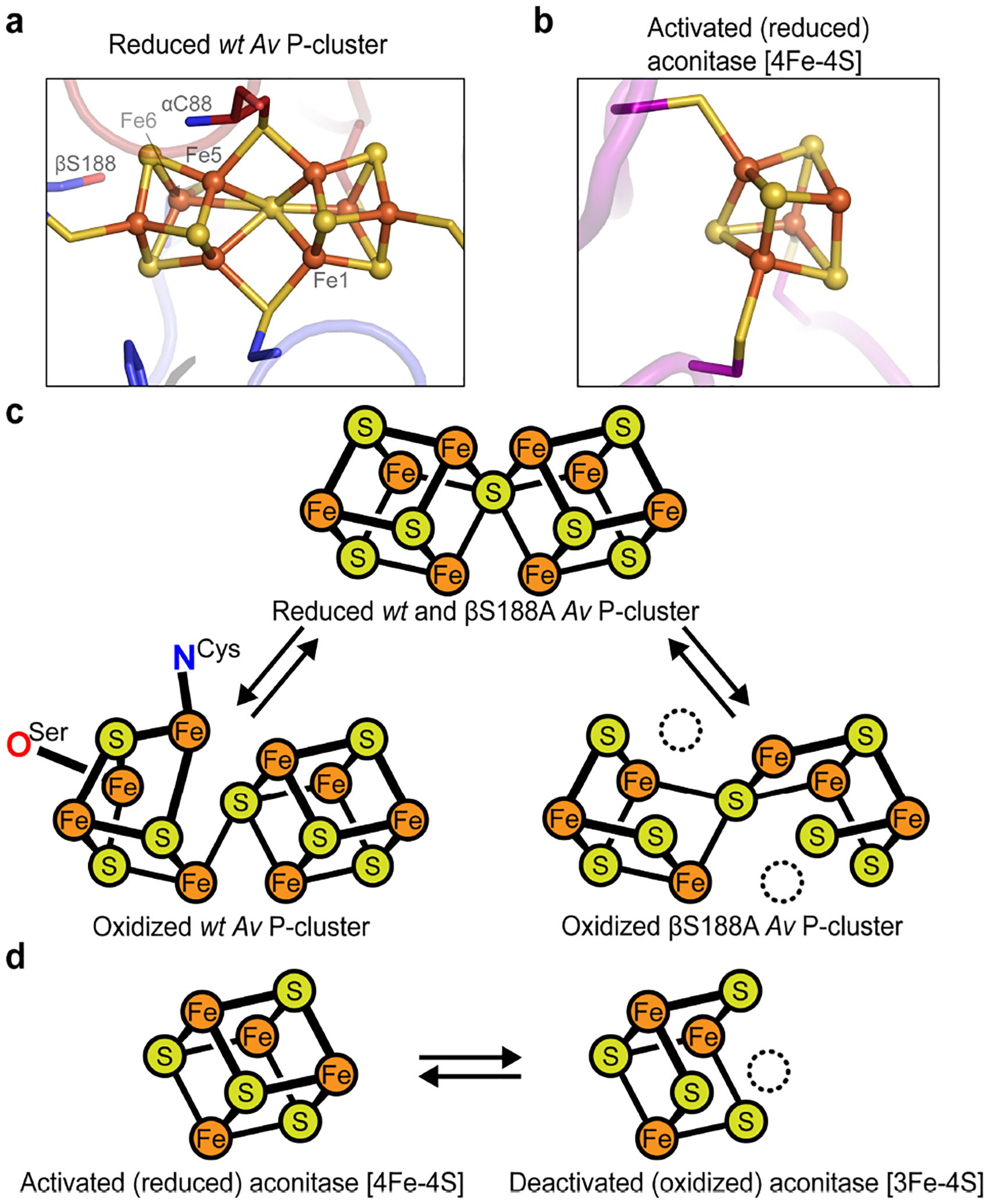
(a) The fully-reduced Av nitrogenase P-cluster (PN) is coordinated by four terminal and two bridging Cys residues. (PDB ID: 3MIN) (b) Activated aconitase contains a coordinatively unsaturated [4Fe-4S] cluster. The non-Cys-ligated Fe is coordinated to a water molecule (not shown). (PDB ID: 6ACN) (c) Two-electron oxidation of the Av MoFeP P-cluster results in coordination by βS188 and the backbone amide of αC88. The βS188A Av MoFeP mutant contains two redox-labile Fe centers (dashed circles, Fe1 and Fe5). (d) The [4Fe-4S] cluster in aconitase contains one redox-labile Fe center (dashed circle).
Ser- or Tyr-coordinated Fe-S clusters are very rare, and the few established cases are not involved in electron relay.22–24 To elucidate the functional role of these oxygenic ligands to the P-cluster, we previously characterized a Av MoFeP mutant that lacked βS188 (βS188A MoFeP) and found that this residue was required to maintain the redox-dependent structural integrity of the P-cluster.19 Notably, the oxidation of βS188A MoFeP led to the labilization and loss of two of the Fe centers (Fe1 and Fe5) of the P-cluster, yielding a cluster composed of two [3Fe-4S] units that shared a bridging sulfide (Figure 1c).19 Surprisingly, we also observed that the redox-instability of the P-cluster did not affect the diazotrophic growth rate of the βS188A Av strain, and the βS188A MoFeP mutant was as catalytically active as wild-type (wt) MoFeP for N2 reduction in vitro.19 These results called into question whether the Ser (or Tyr) ligand to the P-cluster was actually required for nitrogenase catalysis in Av.
The redox-labile βS188A P-cluster is reminiscent of some biological [4Fe-4S] clusters that can lose an Fe to form [3Fe-4S] clusters (Figure 1b,d). Such labile Fe-S clusters are found in some ferredoxins (including Pyrococcus furiosus Fd, Desulfovibrio vulgaris Fd I, and Desulfovibrio africanus Fd III)25 and in a family of enzymes that use the cluster as a catalytic Lewis acid site (e.g., dihydroxy-acid dehydratase,26 fumarases A and B,27 and aconitases28–29). In all of these clusters, only three of the Fe centers are Cys-ligated. In the case of the ferredoxins, the fourth Fe is either coordinated by a non-Cys residue (e.g., Asp),25 or, in the case of Lewis acidic active sites, by an aqua ligand.30 Analogous to the βS188A P-cluster, these dynamic clusters are prone to oxidative damage: upon oxidation to [4Fe-4S]3+, the non-Cys-ligated Fe dissociates from the cluster, leading to its inactivation (Figure 1d).25, 27, 31–35 In addition to their sensitivity to oxidation, some of these clusters also suffer from demetallation when the cellular labile Fe pool (LIP) is low in concentration36 and from mismetallation when they are reconstituted in the presence of other metal ions.25, 37–42
Based on the similarities between the βS188A P-cluster and the redox-labile [4Fe-4S] clusters in other redox proteins, we hypothesized that (1) the Av βS188 P-cluster ligand may serve to protect the P-cluster from oxidative damage and mismetallation, and (2) this role may not have been evident under the ideal, Fe-replete conditions previously used to measure βS188A Av’s diazotrophic growth rate and in vitro catalytic activity.19 To investigate this hypothesis, we probed in this study whether βS188A Av cells were more prone to oxidative stress than wt cells and measured the growth rate and in vivo activity of Av βS188A under non-ideal, Fe-limited conditions. Additionally, we examined the capacity of the βS188A P-cluster to be heterometallated in vitro by crystallographic, spectroscopic, and analytical methods. Our results provide strong evidence for the previously unconsidered role of the Ser ligand in protecting the structural and functional integrity of the P-cluster under environmental duress. They also show that the P-cluster is inherently more dynamic than originally envisioned, capable of undergoing metal exchange reactions upon oxidation.
Results and Discussion
In vivo effects of the βS188A mutation
We initially investigated what, if any, protective roles the Ser ligand to the P-cluster may provide in vivo. To this end, we carried out an oxidative stress test of Av βS188A cells in which we measured cell survival after 30 minutes of exposure to 5 mM hydrogen peroxide (H2O2) using previously reported procedures, which had used 15 mM H2O2 (Figure 2a).43–44 Increasing catalase activity, which catalyzes the decomposition of H2O2 into H2O and O2, has been shown to augment the cellular efficiency of nitrogenase in the diazotroph Rhizobium leguminosarum, indicating that oxidative stress and nitrogenase activity are intertwined.45 If the Ser ligand is protective of the wt P-cluster during in vivo catalysis, we hypothesized that βS188A MoFeP would be more prone to oxidative stress and release Fe from the P-cluster when grown diazotrophically, thereby resulting in lower survival rates than wt MoFeP. Indeed, we found that the survival frequency of the H2O2-treated βS188A Av cells was only 7% of that of wt Av cells, indicating that the mutant cells were significantly more prone to oxidative stress when grown diazotrophically.
Figure 2. In vivo experiments to examine the physiological role of βS188 ligand to the P-cluster in Av MoFeP.
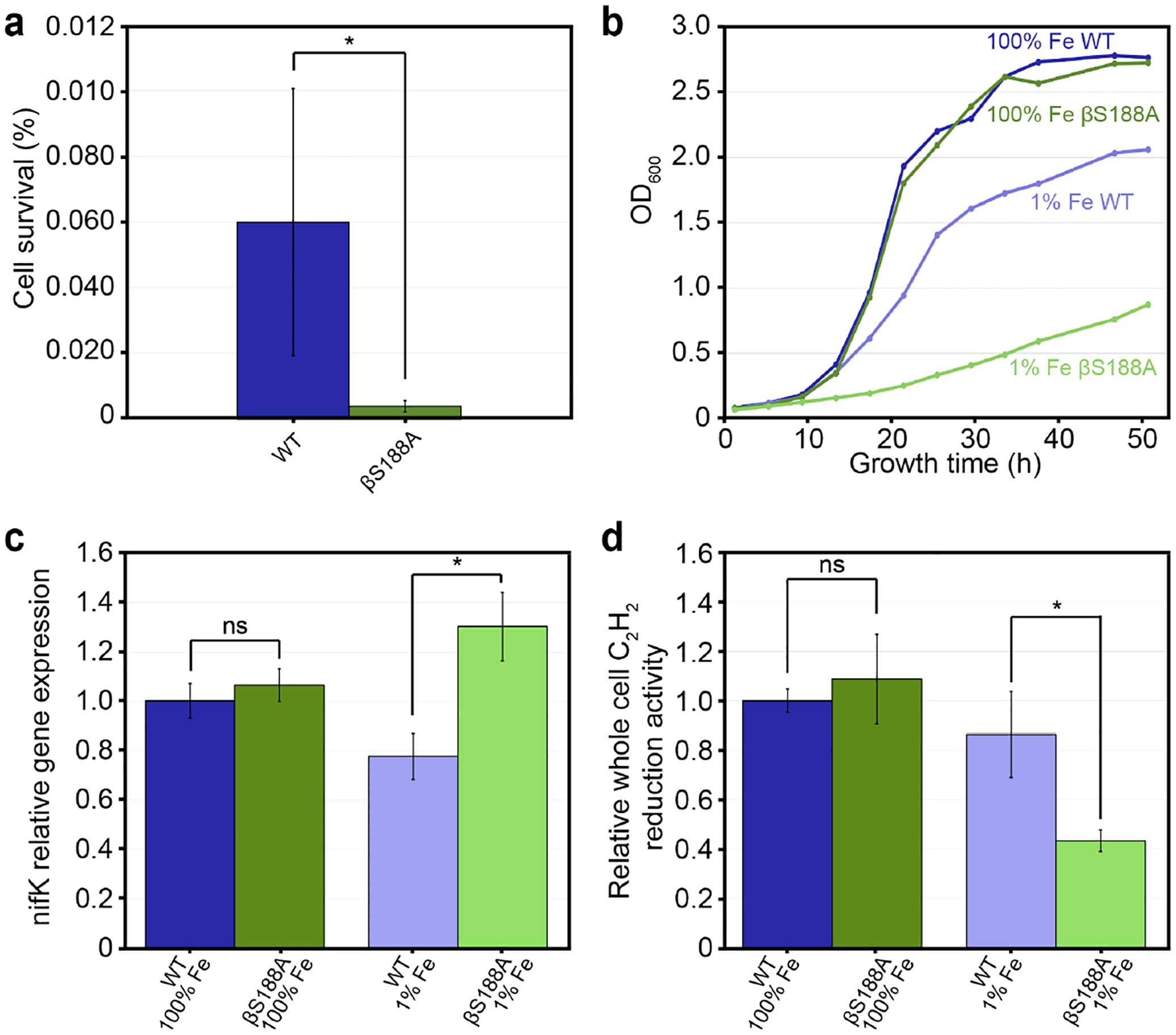
The color scheme for the variants is as follows: wt (dark and light green) and βS188A MoFeP (dark and light blue). Error bars represent one standard deviation of experiments performed at least in triplicate. Statistical significance is represented by “ns” (not significant) or * (p-value < 0.05). (a) Oxidative stress test of Av cells represented as percent cell survival determined via colony forming units (CFUs) before and after 30 min exposure to 5 mM H2O2. Cells were grown diazotrophically with 35 μM Fe (100% Fe). The experiment was performed in biological triplicate. (b) Growth curves of Av wt and Av βS188A cells in cultures with 100% and 1% Fe (0.4 μM). (c) RT-qPCR of nifK gene expression of wt and βS188A Av grown diazotrophically with varying amounts of Fe. Expression levels were normalized to the expression level of reference gene rho, and the data is presented relative to wt Av cells grown diazotrophically with 100% Fe. The experiment was carried out in technical triplicate of biological triplicates. (d) Av cell cultures used for RT-qPCR in (c) were also used for whole cell C2H2 reduction assays (biological triplicate). C2H4 produced was measured and normalized to both the relative nifK transcript levels and the OD600 of the cells, then presented relative to wt Av grown with 100% Fe.
When culturing Av cells, the medium is typically supplemented with Fe to allow for rapid cell growth.46 The Burke’s medium used in our protocols contains 35 μM Fe, hereafter referred to as 100% Fe. If the Fe centers of the βS188A MoFeP P-cluster are labilized during catalysis under physiological conditions, the high Fe concentration in the 100% Fe medium may be beneficial either by maintaining a large cellular LIP (which in turn could enable fast reconstitution of the labile P-cluster during in vivo catalysis) or by providing excess Fe to enable higher levels of MoFeP expression. To probe these possibilities, we investigated the diazotrophic growth rates of wt and βS188A Av cells in media with varying concentrations of Fe (Figures 2b and S1). Under 100% Fe growth conditions, both wt and βS188A Av cells displayed identical doubling times (3.9 h). Reducing the Fe concentration in the growth medium to 0.4 μM (hereafter referred to as 1% Fe) significantly slowed down the growth rates of both Av strains. However, βS188A cells experienced a more precipitous reduction in growth rate than wt cells with doubling times of 12.7 h and 6.3 h, respectively. The slower growth rate of Av βS188A cells relative to wt cells under Fe-limited growth conditions could potentially arise from differences in the expression levels of MoFeP and/or differences in the in vivo activity of βS188A and wt MoFeP.
To determine relative βS188A and wt MoFeP expression levels, we quantified MoFeP mRNA transcripts via quantitative reverse transcription PCR (RT-qPCR) of the nifK gene, which codes for the β-subunit of MoFeP. The values obtained were then normalized relative to the reference gene rho, a transcription termination factor that is constitutively expressed and has been used previously for Av RT-qPCR normalization (Figure 2c).47 As a negative control, nifK transcript levels were measured under non-diazotrophic growth conditions, and not surprisingly, MoFeP expression was found to be downregulated for both wt and βS188A Av strains (Figure S2). We found that when grown diazotrophically under 100% Fe, βS188A and wt Av cells expressed MoFeP at the same level, suggesting that their identical growth rates resulted from both wt and βS188A Av MoFeP functioning with the same catalytic efficiency (Figure 2c). Thus, even if the βS188A P-cluster loses Fe during in vivo catalysis, reconstitution of the P-cluster is not rate-limiting under these conditions, which can be ascribed to the large LIP.
In contrast, wt and βS188A Av cells grown diazotrophically in the presence of 1% Fe exhibited nifK transcription levels that were 78% and 130% that of wt cells grown in 100% Fe media, respectively. The increase in βS188A’s nifK expression taken together with the strain’s slower growth rate under Fe-limiting conditions suggests that βS188A MoFeP does not function as efficiently as wt MoFeP under Fe-limited growth conditions. Thus, the cells are likely compensating via upregulation of MoFeP gene expression. This change in expression levels can be attributed to βS188A having slower P-cluster reconstitution at 1% Fe than at 100% Fe, and therefore, P-cluster reconstitution may become the rate-limiting step for N2 fixation.
In vivo catalytic efficiencies of wt and βS188A MoFeP were probed by measuring whole cell acetylene (C2H2) reduction activity. The quantity of ethylene (C2H4) produced was normalized to the amount of MoFeP transcripts and the OD600 of the cells (Figure 2d). βS188A MoFeP from the 100% Fe culture and wt MoFeP from both 1% and 100% Fe cultures all had nearly identical cellular C2H2 reduction activities. In contrast, βS188A MoFeP in cells grown under 1% Fe displayed substantially reduced activity (<50% of wt), suggesting that βS188A MoFeP is not as catalytically efficient as the wt protein under limiting Fe conditions. Based on these results, we conclude that (1) βS188A MoFeP requires a larger LIP than wt MoFeP to operate efficiently under physiological conditions, and thus, (2) the P-cluster of βS188A MoFeP must be compositionally labile during catalysis under physiological conditions.
Dynamic, redox-dependent metal exchange by the βS188A P-cluster
Having determined the role of βS188 in maintaining the structural and functional integrity of the P-cluster under in vivo conditions, we investigated if the absence of this Ser ligand could also lead to dynamic metal exchange with the environment. We were interested in this possibility not only because it could furnish site-specifically metal-substituted variants of the P-cluster with unusual/useful electronic properties (as shown in the case of FeMoco),48–49 but also because it could help demonstrate that a possible physiological role of βS188 may be to reduce the vulnerability of the P-cluster to mismetallation. We previously determined via X-ray crystallography that the two missing Fe centers in the oxidized βS188A P-cluster could be replenished upon reduction with DT to obtain the fully reconstituted the P-cluster.19 This reconstitution did not require the addition of excess Fe, leading us to propose that some of the MoFeP in solution could be “cannibalized” for its Fe content. This behavior is similar to the redox-labile [4Fe-4S] cluster of aconitase that can scavenge Fe from other protein-bound clusters upon re-reduction.28, 50 Aconitase can also be reconstituted via the addition of excess reduced Fe to solution, minimizing the amount of protein clusters that are scavenged for Fe.50
We first investigated the ability of purified βS188A MoFeP to uptake Fe from solution (rather than from other MoFeP molecules) by reconstituting the oxidized P-cluster with 57Fe2+, followed by the characterization of the isotopically labeled P-cluster by Mössbauer spectroscopy (Figure 3, green spectrum). The resulting spectrum is best fit by three quadrupole doublets (Figure S3, Table 1). Recording the spectrum at 54 mT clearly established that no FeMoco signals were present in the spectrum based on comparison to 57Fe isotopically labeled FeMoco in wt Av MoFeP,51 providing evidence that the incorporation of 57Fe was specific to the P-clusters. McLean et al. termed the spectral components D (composed of D1 and D2), Fe2+, and S, and determined that they accounted for 10, 4 and 2 Fe sites (4.2 K), respectively (Figure 3, black spectrum).52 The D components in the spectrum are in close agreement with those observed for the 57Fe-labeled P-clusters in MoFeP from Klebsiella pneumoniae (Kp),52 indicating that 57Fe was indeed incorporated into the P-clusters. Minor differences in isomer shifts and quadrupole splittings could arise from slight differences in the P-cluster environment in Kp and Av. Other Kp P-cluster doublets (S and Fe2+ sites) were not observed in the βS188A spectrum, however, as the parameters for D and S are quite similar, some contribution from the S component cannot be fully discounted. Regardless, our findings indicate that 57Fe may not incorporate uniformly into every site in the P-cluster, or that the differences between the Kp and βS188A Av P-cluster environments may significantly shift the signals of some of the metal sites. We also observed an additional feature (Table 1, Figure 3, green spectrum, labeled as “adventitious 57Fe2+”) which closely matches the reported spectrum of aquated 57FeSO4 and is consistent with adventitious 57Fe2+ not associated with MoFeP.53 To see if complete loss of the same 57Fe-labeled Fe centers (presumably Fe1 and Fe5) occurred during each redox cycle, we performed a subsequent round of oxidation and reconstitution with the original Mössbauer sample, this time with natural-abundance Fe2+ (Figure 3, blue spectrum, Table S1). The peak intensities decreased significantly, but the features were not completely lost, demonstrating substantial but incomplete dissociation of 57Fe from the βS188A P-clusters upon oxidation. The ratio of the peak areas in both spectra were nearly identical (Table S1). This observation may have two possible origins: incomplete loss of Fe from positions Fe1 and Fe5 during oxidation (Figure 1a) or shuffling of metals within the P-cluster during the reconstitution process.
Figure 3. Mössbauer spectra of Av βS188A MoFeP reconstituted with 57Fe-enriched or natural-abundance Fe in comparison to Kp MoFeP (previously reported).
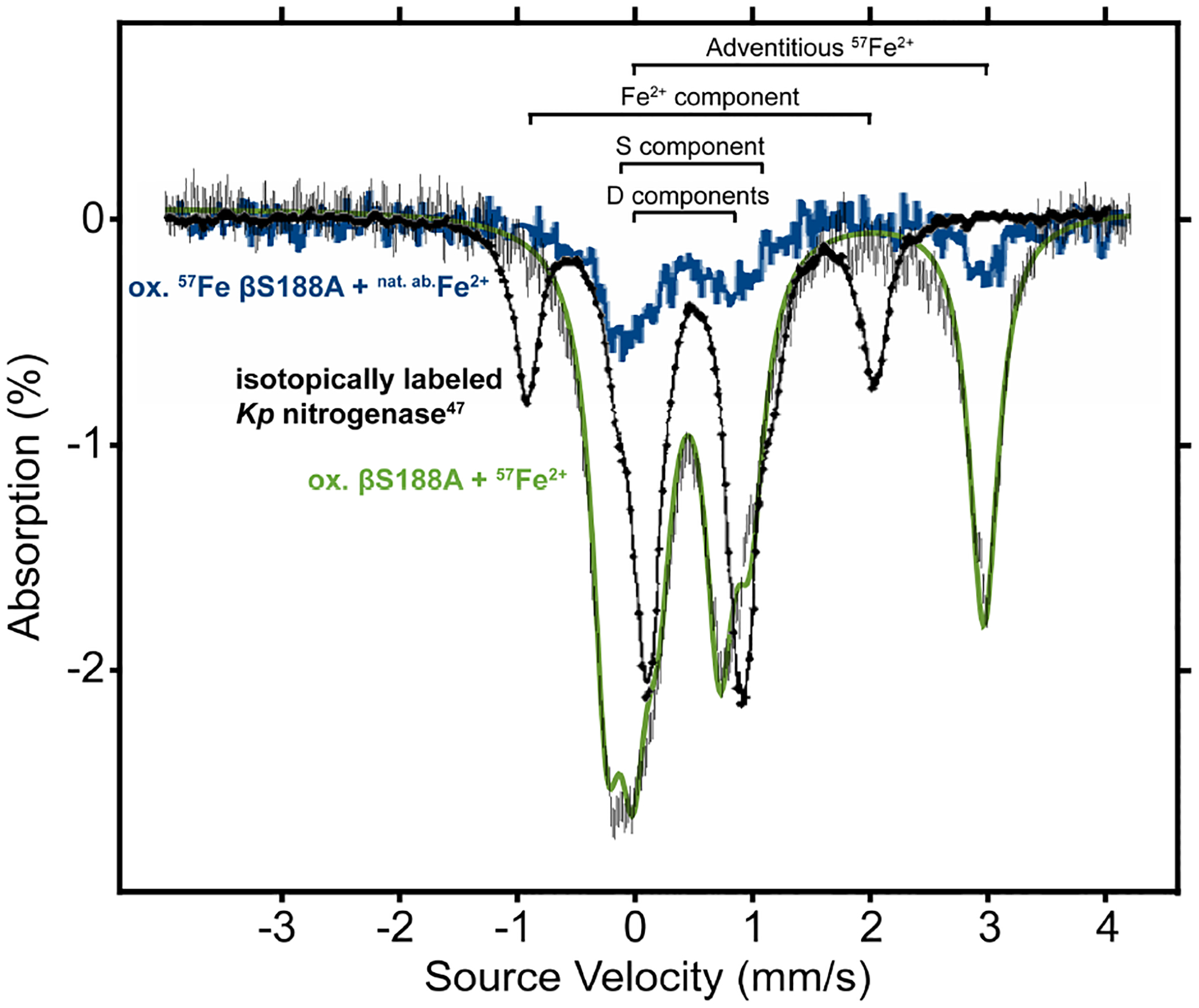
. Spectra were recorded at 80 K and 54 mT. Oxidized βS188A was first reconstituted with 57Fe2+ (green trace), followed by a second oxidation and reconstitution with natural abundance Fe2+ (blue trace). The spectrum of Kp MoFeP with 57Fe-labelled P-clusters (black trace, 4.2 K, concentration unknown) is included as a comparison and adapted with permission under a Creative Commons (CC-BY 4.0) License from reference 52. Copyright 1987, Else-vier.
Table 1.
Features observed in the Mössbauer spectrum of 57Fe-reconstituted βS188A MoFeP.
| Feature | ΔEq (mm/s) | δ (mm/s) | Relative area (%) |
|---|---|---|---|
| Di | 0.779 | 0.589 | 23.8 |
| D2 | 0.735 | 0.352 | 39.6 |
| Adventitious 57Fe2+ | 3.203 | 1.363 | 36.6 |
X-ray crystallographic analysis of heterometallated βS188A P-clusters
After establishing via Mӧssbauer spectroscopy that the βS188A P-cluster can be reconstituted with 57Fe2+ from solution, we investigated if the cluster could be replaced with non-Fe metal ions (i.e., heterometals) upon oxidation in vitro. To this end, we determined the crystal structures of βS188A MoFeP reconstituted with Ga3+, Ni2+, and Co2+ (Figures 4 and S4, Table S2). To prepare these crystals, βS188A MoFeP was oxidized, reconstituted with the heterometal, and then crystallized in the presence of DT after removal of excess heterometal ions from the protein solution. We previously confirmed through X-ray crystallography19 that the oxidized βS188A P-cluster could be fully reconstituted (i.e., 8.0 Fe atoms per P-cluster) upon crystallization in the presence of DT without addition of metal ions (Figure 4a). In contrast, the three heterometal-reconstituted βS188A MoFeP crystal structures crystallized in the presence of DT revealed P-clusters with a total metal occupancy of 5.7 to 7.4 metal atoms (integrated over the entire P-cluster) (Figure 4, Table 2). The occupied metal sites included positions that are normally vacant in the oxidized βS188A P-cluster (i.e., M1 and M5, where M refers to any metal ion), although the occupancy of these positions was not unity.
Figure 4.
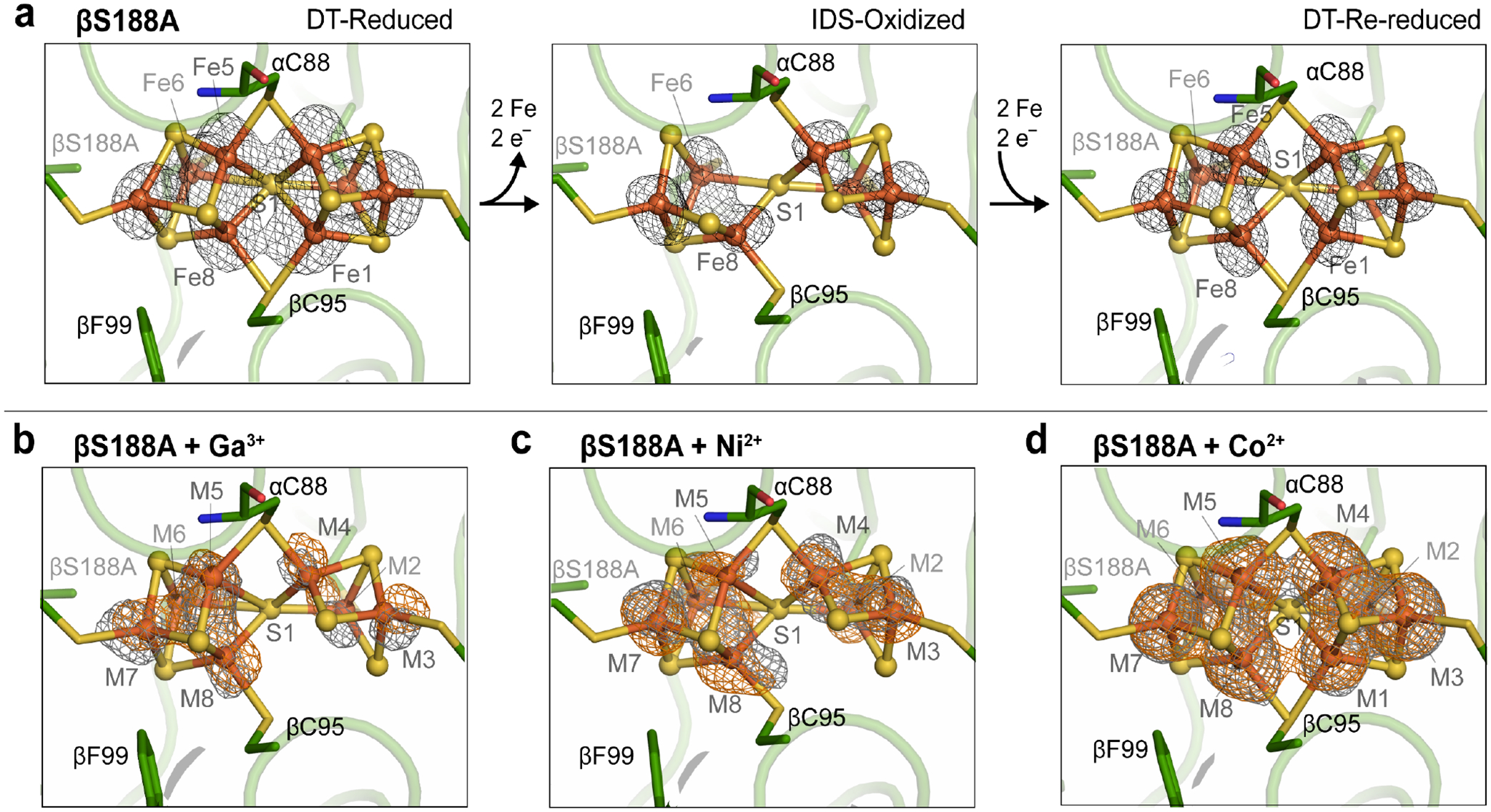
Crystal structures of Av βS188A MoFeP P-clusters. (a) Redox states of the βS188A P-clusters without heterometal: DT-reduced (left, PDB ID: 6O7L), IDS-oxidized (middle, PDB ID: 6O7S), and DT-re-reduced (right, PDB ID:6O7Q). Anomalous electron density difference maps depicting the location of the Fe-centers are shown in black mesh.19 (b,c,d) Heterometallated βS188A P-clusters. Anomalous electron density difference maps (contoured at 4.5 σ) determined using X-ray diffraction data collected above and below the heterometal K-edge are shown in orange and gray mesh, respectively. Inorganic sulfides are depicted as yellow spheres and metals as orange spheres (modelled as Fe). (b) 2.2-Å resolution structure of Av βS188A P-cluster reconstituted with Ga3+ contoured at 4.0 σ. (Above Ga K-edge: 10379 eV, Fe f” = 2.1, Ga f” = 3.9; below Ga K-edge: 10357 eV, Fe f” = 2.1, Ga f” = 0.5) (c) 2.0-Å resolution structure of Av βS188A P-cluster reconstituted with Ni2+ contoured at 4.5 σ. (Above Ni K-edge: 8350 eV, Fe f” = 3.0, Ni f” = 3.9; below Ni K-edge: 8228 eV, Fe f” = 3.1, Ni f” = 0.5) (d) 2.0-Å resolution structure of Av βS188A P-cluster reconstituted with Co2+ contoured at 4.5 σ. (Above Co K-edge: 7730 eV, Fe f” = 3.4, Co f” = 3.9; below Co K-edge: 7690 eV, Fe f” = 3.4, Co f” = 0.5).
Table 2.
Crystallographic occupancies of each metal site in the re-reduced, heterometallated βS188A P-clusters
| Metal site | DT-re-reduced βS188A | βS188A + Ga3+ | βS188A + Ni2+ | βS188A + Co2+ |
|---|---|---|---|---|
| M1 | 1.00 | 0.00 | 0.00 | 0.68 |
| M2 | 1.00 | 0.87 | 0.74 | 0.92 |
| M3 | 1.00 | 0.97 | 0.80 | 0.97 |
| M4 | 1.00 | 0.89 | 0.74 | 1.00 |
| M5 | 1.00 | 0.58 | 0.61 | 0.90 |
| M6 | 1.00 | 0.98 | 0.99 | 0.94 |
| M7 | 1.00 | 1.00 | 1.00 | 0.99 |
| M8 | 1.00 | 0.95 | 0.83 | 0.96 |
| Total | 8.00 | 6.24 | 5.71 | 7.36 |
The incomplete occupancy of the metal sites in the presence of DT (unlike DT-re-reduced βS188A MoFeP without added metals) strongly suggests that the P-clusters are indeed heterometallated. Interestingly, the analysis of the metal-specific anomalous electron density maps determined both above and below the K-edge of the heterometal indicated that each metal-bound site displayed at least some fractional occupancy of Fe, and thus the heterometal contributed only partially to the metal sites (Figure 4). Such partial occupancies suggest a shuffling of the heterometal and Fe within the cluster. Due to confounding factors such as the partial occupancy of heterometals and <1.0 total metal occupancy at each site, it was not possible to determine the absolute or relative occupancies of Fe or the heterometal at these positions. Furthermore, crystal structures inherently represent an average of the molecules in the crystals, indicating that the model may represent a multitude of P-cluster variations as well as clusters possibly damaged by the metal-substitution protocol. Therefore, in the structural models deposited into the Protein Data Bank, we treated all occupied metal sites in the P-cluster as Fe-only. Total occupancy at each site was determined by refinement of metal at each site after refining B-factors, whereby B-factors were held constant at values relative to the inorganic sulfides.
We next investigated if mismetallation affects ET during catalysis by measuring the specific activity of the mismetallated MoFeP for the reduction of acetylene (C2H2) to ethylene (C2H4) (Table 3). Previously, we determined that the specific C2H2 reduction activities of wt and βS188A MoFeP were 1560 and 1440 nmol of C2H4 per min per mg MoFeP, respectively. After the heterometallation treatment, wt MoFeP largely retained its maximal specific activity (81 – 92%). We attribute the small loss in wt MoFeP activity to the deactivation/degradation of a small fraction of the protein during the oxidation and metal-soaking procedure. In contrast, βS188A MoFeP lost most of its activity upon the same treatment; oxidized βS188A MoFeP retained only 29% of its maximal activity compared to the as-isolated, DT-reduced βS188A MoFeP. Reconstitution with Fe2+ led to a slight recovery of catalytic activity (38% of maximal activity), indicating that the reconstitution of the P-cluster with the remetallation protocol is not a high-yielding process and that the oxidized βS188A P-cluster is more prone to degradation than wt oxidized P-cluster. Reconstitution with Co2+ showed a similar improvement in activity of βS188A MoFeP (implying that Co2+ may be a functional substitute for Fe2+ in the P-cluster), while both Ga3+ and Ni2+ displayed low activities comparable to that of the untreated, oxidized βS188A MoFeP. Taken together, these activity assays further emphasize the protective role of βS188 in preventing mismetallation of the Av P-cluster and thus maintaining catalytic activity even after redox cycling in the presence of heterometals.
Table 3.
Percent specific activities of MoFeP variants for C2H2 reduction relative to DT-reduced, as-isolated wt MoFeP.
| MoFeP variant | Metal used for soaking after oxidation of MoFeP | ||||
|---|---|---|---|---|---|
| No metal | Fe2+ | Ga3+ | Ni2+ | Co2+ | |
| wt MoFeP | 81 ± 4% | 83 ± 4% | 92 ± 5% | 90 ± 4% | 89 ± 8% |
| βS188A MoFeP | 29 ± 2% | 38 ± 2% | 24 ± 2% | 25 ± 4% | 38 ± 2% |
We also examined the incorporation of heterometals into the P-cluster using inductively coupled plasma mass spectrometry (ICP-MS). The results, shown in Table S3, confirm that 57Fe and heterometals can be incorporated into MoFeP only in the case of the βS188A variant but not wt MoFeP. We must also note that there were very large sample-dependent variations in the amounts of heterometals associated with MoFeP, which we interpret as further evidence for the highly dynamic nature of the oxidized βS188A P-cluster. We observed that the composition of the reconstituted βS188A P-cluster was dependent on the identity of the added heterometal. When the cluster was reconstituted with Ga3+ or Ni2+, only seven of the eight P-cluster metal sites were populated by a metal ion (Figure 4b and 4c). On the other hand, incubation with Co2+ led to the occupation of all eight sites (Figure 4d). The higher efficiency of Co2+ to metallate the P-cluster may be attributed to its similarity to Fe2+ in terms of ionic radius and propensity to form tetrathiolate complexes with tetrahedral symmetry.54
EPR analysis of heterometallated βS188A P-clusters
To further characterize the heterometallated P-clusters of βS188A, we collected electron paramagnetic resonance (EPR) spectra after reconstitution of the cluster with 57Fe2+ and heterometals (Ga3+, Ni2+, and Co2+) (Figure 5). βS188A and wt MoFeP were oxidized with indigo disulfonate (IDS) followed by the addition of 57Fe2+ or the heterometals. Previously established S = 3/2 features (g ≈ 4.3, 3.7, and 2.0) arising from FeMoco were present in all samples as expected.55–57 Reconstitution of βS188A MoFeP with 57Fe2+, Co2+, and Ni2+ resulted in near identical features in the g ≈ 5 region (Figure 5b) which were not detected in the wt samples (Figure 5a). Similar features have been observed in synthetic NiFe3S4 cubane clusters58–59 and in Pyrococcus furiosus Fd that has been mismetallated with Cd39 or Ni.40 These observations are again consistent with a heterometal-associated perturbation of the βS188A P cluster that does not occur in the wt MoFeP, in agreement with the crystallographic results (Figure 4). The possibility that heterometals may associate with FeMoco rather than the P-cluster was eliminated by the fact that the heterometal-treated wt MoFeP does not feature a g ≈ 5 EPR signal (Figure 5a).
Figure 5. X-band EPR spectra of wt and βS188A MoFeP after oxidation and soaking with heterometals collected at 4 K.
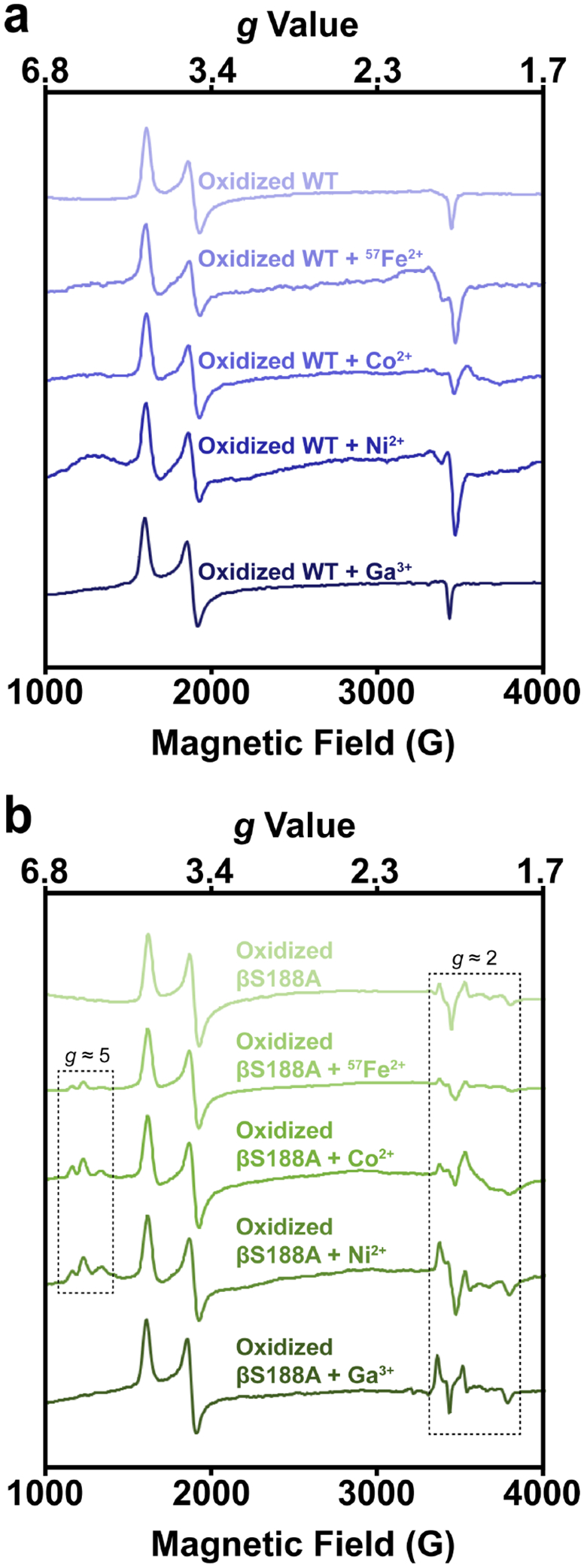
(a) IDS-oxidized wt Av MoFeP (top) soaked with 57Fe2+, Co2+, Ni2+, and Ga3+ (top to bottom). (b) IDS-oxidized βS188A Av MoFeP (top) soaked with 57Fe2+, Co2+, Ni2+, and Ga3+ (top to bottom). Reconstitution with 57Fe2+, Co2+, and Ni2+ resulted in new features in the g ≈ 5 region. All metal-treated βS188A samples had features in the g ≈ 2 region that closely resembled those for the oxidized βS188A species, but the features varied in intensity. Oxidized wt and βS188A spectra without added metals (top) are reproduced from reference 19 for comparison.
The finding that the new EPR features were similar in all cases of heterometallated βS188A MoFeP species‒regardless of the identity of the heterometal‒was unexpected. We tentatively ascribe this observation to a perturbation of the many close, low-energy electronic states of the P-cluster60 by heterometal substitution or changes in the population of a previously established S = 5/2 excited state of the cluster.61 It is also possible that the metal substitution protocols result in damaged forms of the βS188A P-cluster with similar EPR signatures. Further in-depth spectroscopic studies will be required to identify the origin of these signals and understand their similarities among the various heterometals. In contrast to other heterometals, reconstitution of βS188A with Ga3+ did not produce the g ≈ 5 signals, which may be rationalized by its frontier orbitals having much more p and less d character than the other heterometals used, resulting in substantially different electronic properties from the other heterometallic P-clusters. EPR spectra of all the reconstituted βS188A MoFeP species had additional features in the g ≈ 2 region that closely resemble oxidized, metal-deficient βS188A MoFeP (Figure 5b),19 and unusual P-cluster features seen in FeMoco-deficient MoFeP.62 Of note, these features were weakest in the samples reconstituted with 57Fe2+ and Co2+, possibly due to the ability of these ions to occupy all eight P-cluster metal sites, creating a structurally more symmetrical and electronically more isotropic spin system.
As a control, EPR spectra were collected on both wt and βS188A MoFeP samples in which the IDS oxidation step was omitted (Figure S5). All wt spectra displayed only the expected FeMoco signals (Figure S5a). Ni2+, Co2+, and 57Fe2+ were unable to incorporate into the reduced P-cluster as evidenced by the lack of features in the g ≈ 2 and g ≈ 5 regions (Figure S5b), confirming the full occupancy of all eight metal sites by iron in the PN state. Unexpectedly, the Ga3+ treatment of the reduced βS188A MoFeP resulted in g ≈ 2 features (Figure S5b), suggesting that Ga3+ may be capable of perturbing the electronic state of the βS188A P-cluster through an unidentified mechanism.
Conclusions
In this work, we have examined the potential role of a Ser ligand in maintaining the structural and functional integrity of the nitrogenase P-cluster under environmental duress. Our studies have demonstrated that (1) the growth of βS188A Av cultures is adversely affected by suboptimal growth conditions (i.e., Fe-limited growth media and oxidative stress) to a much greater extent than wt Av cells, (2) βS188A MoFeP has significantly lower in vivo catalytic activity than wt MoFeP, and (3) the oxidized [6Fe-7S] P-cluster of βS188A can be reconstituted with various exogenous metals (57Fe2+, Co2+, Ni2+, Ga3+) in vitro. Importantly, these heterometals are found to be distributed throughout the P-cluster rather than being localized to the positions of the displaced Fe centers. These findings highlight the role of the Ser ligand in protecting the P-cluster under stress conditions in vivo and demonstrate that this cluster is compositionally dynamic and readily capable of exchanging its atomic constituents in vitro. This attribute is shared by FeMoco, Fe-Vco, and the [4Fe-4S] cluster of FeP which have also been shown to undergo substitution of their sulfur components under catalytically relevant conditions.63–66
Why has evolution selected a nearly invariant O-based ligand (Ser or Tyr) to the P-cluster when a similarly negatively charged and more common Cys could have potentially served the same role? We previously suggested that the hard, O-based Ser or Tyr side chains may be preferred over the soft, Cys thiolate due their strong preferences for binding Fe3+ over Fe2+, allowing them to serve as redox-dependent switches that may be an integral component of the ET mechanism to the active site.1, 18–20 This proposal is supported by the fact that the cysteine-substituted Av βS188C MoFeP mutant is only ~60% as catalytically active as the wt protein for H+ reduction,11 and potentially even less active for the reduction of N2. Our current results with βS188A MoFeP indicate that the role of the Ser ligand to the P-cluster may extend beyond acting merely as a redox switch and expand upon the known mechanisms that diazotrophic organisms have evolved to protect nitrogenase from oxidative stress.67–70 It is reasonable to assume that a Tyr may fulfill an analogous functional role in nitrogenases lacking a Ser ligand to the P-cluster such as the previously characterized MoFeP from Gd.18 Interestingly, there is only one known diazotrophic microbe that contains neither a Ser nor a Tyr P-cluster ligand, Methanococcus aeolicus Nankai-3.18 This archaeon is a strict anaerobe that was isolated from marine sediments near the Nankai Trough,71 an environment with unusually high Fe availability,72 which may explain the ability of this organism to thrive without the protection of Ser or Tyr ligand to the P-cluster.
Materials and Methods
Reagents.
All reagents were obtained from Fischer Scientific, Sigma-Aldrich, VWR International, Praxair, Roche Diagnostics, or New England Biolabs unless otherwise noted.
Growth media.
All Av cultures were grown in Burke’s medium (BM+) containing 2.0% sucrose, 0.9 mM CaCl2, 1.67 mM MgSO4, 35 μM FeSO4, 2 μM Na2Mo2O4, 10 mM K3PO4 pH 7.5, and 10 mM NH4Cl. Diazotrophic media (BM‒) lacked NH4Cl. Media with varying concentrations of iron are referred to as X% Fe, relative to 100% Fe (35 μM). Solid Burke’s media contained 23 g/L agar. All growths were completed at 30°C and shaken at 200 rpm, unless otherwise noted. All OD600 values were measured using a cuvette with a 1 cm path length, unless otherwise noted.
Mutagenesis of A. vinelandii genome.
The nitrogenase βS188A MoFeP mutant from a previous study was used in these experiments.19. βS188A ΔnifB Av was generated by inserting a kanamycin resistance cassette into nifB using a plasmid (PDB218) kindly provided by V. Cash and D. Dean. This plasmid contained the cassette flanked by the beginning and end of the nifB gene. βS188A Av cells were made competent for transformation using an established procedure73–74 by growing to OD600 = 0.5 in 50 mL 0% Fe BM+ media, shaking at 150 rpm. As a result of Fe-starvation, cell cultures were fluorescent green due to excreted siderophores. 50 μL suspensions of competent cells were transformed in 50 μL transformation buffer solutions (20 mM MOPS, pH 7.5, 20 mM MgSO4) containing 5 μg of PDB218. Transformation of Av cells using this protocol results in genomic modifications via double homologous recombination of the plasmid and the genome. Transformants were screened for kanamycin resistance on solid BM+ media containing 5 μg/mL kanamycin (BM+ Kan). Transformants were also streaked on solid BM‒ to ensure that the N2 fixation ability was lost. Multiple passes of the transformants on solid BM+ Kan media were required for complete loss of N2 fixation ability. The final transformant was verified with sequencing of the nifB region of the genome.
A. vinelandii culture growth.
Av cells were grown beginning with a 10- mL BM‒ starter culture in a 500 mL Erlenmeyer flask until the cells reached a density of OD600 1.5‒2.0. 15 mL of the starter culture was used to inoculate 1.0 L of BM‒ media in a 2.8-L flask (ΔnifB strains were grown in BM+ Kan for all growth steps). Once the cells were densely grown (OD600 > 1.5), 400 mL of the culture was used to inoculate 50 L of BM+ media with only 3 mM NH4Cl in a 60-L fermenter (New Brunswick Scientific). Cells were monitored for derepression of nitrogenase (indicated by a spike in dissolved oxygen) and harvested 4 h later. Cells were concentrated to <6 L using a Pellicon 2 Tangential flow membrane (Eppendorf), then pelleted by centrifugation at 5000 rpm and 4°C. Wet cell pellets of ΔnifB strains were ~50 g, and cell pellets of wt and βS188A weighed >100 g. Cell pellets were stored at −80°C until cell lysis and protein purification.
Protein purification.
All cell lysis and purification steps were performed anaerobically on a Schlenk line under ultra-high-purity Ar or in a Coy anaerobic chamber under 95% Ar/5% H2 (<2 ppm O2). All buffers and columns were prepared under anaerobic conditions. Cell pellets were thawed overnight at 4°C, then resuspended in a ~ 150 mL buffer solution, termed Eq (50 mM TRIS, pH 7.75, 200 mM NaCl, 5 mM DT, 0.1 mg/mL DNase I). The suspension was lysed at 16,000 psi N2 using a microfluidizer (Microfluidics), and the lysate (dark brown) was centrifuged at 12,000 rpm for 75 min under anaerobic conditions. The resulting dark brown supernatant was loaded onto a DEAE Sepharose column (equilibrated with Eq) and washed overnight with 1.5 L of Eq. Nitrogenase component proteins were eluted using a linear NaCl gradient (200 to 500 mM NaCl at 2.5 mL/min for 1.0 L, 50 mM TRIS, pH 7.75, 5 mM DT). MoFeP and FeP eluted at conductivities of ~25 (~23 for the ΔnifB strains) and ~30 mS/cm, respectively. Fractions containing tge protein of interest were identified via denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Fractions were then pooled and diluted 1.8-fold with a salt-free buffer solution (50 mM TRIS, pH 7.75, 0 mM NaCl, 5 mM DT). Proteins were independently concentrated on a DEAE column that was equilibrated with a salt-free buffer solution and rapidly eluted with a high-salt buffer solution (50 mM TRIS, pH 7.75, 500 mM NaCl, 5 mM DT). Proteins were further purified by gel filtration on a Sepharose 200 column (GE Healthcare), using a high-salt buffer solution (50 mM TRIS, pH 8.0, 500 mM NaCl, 5 mM DT). Pure protein containing fractions were identified via SDSPAGE. The fractions were combined and concentrated to ~10 mg/mL with an Amicon concentrator (EMD/Millipore, ~20 psi 95% Ar/ 5% H2, 30 kDa membrane for FeP, 100 kDa membrane for MoFeP). Concentrated proteins were stored in ~500 μL aliquots in cryovials in liquid N2. Protein concentrations were determined using both Bradford assays and Fe-chelation assays (6.2 M guanidine-HCl, 2 mM 2,2-bipyridine, 10% glacial acetic acid) at 522 nm with an extinction coefficient of 8650 M−1 cm−1.
Preparation of heterometallic- and 57Fe-incorporated-P-cluster MoFeP.
MoFeP was desalted over a 10-DG gravity flow column (Bio-Rad) that had been equilibrated with a buffer solution (50 mM TRIS, pH 8.0, 500 mM NaCl) to remove DT. The protein was concentrated to ~50 μM in 30 kDa Microcon centrifugal filters (EMD/Millipore) and then oxidized with 2.5 mM IDS for 1 h. The protein solution was run over another 10-DG gravity flow column that had been equilibrated with a buffer solution (50 mM TRIS, pH 8.0, 500 mM NaCl) to remove IDS. The protein was concentrated to ~50 μM in 30 kDa Microcon centrifugal filters (EMD/Millipore), followed by addition of 1 mM metal (M = NiSO4, CoCl2, GaCl3, or 57FeSO4). After a 1-h incubation, excess metal was removed by exchanging the protein into a fresh buffer solution (50 mM TRIS, pH 8.0, 500 mM NaCl) with 30 kDa Microcon centrifugal filters (EMD/Millipore) and concentrated to ~50 μM. The first desalting and the IDS-oxidation steps were omitted during the preparation of the control EPR samples with reduced protein. All wt and βS188A MoFeP samples were prepared in parallel.
Nitrogenase MoFeP C2H4 activity assays.
Protein stocks were prepared as described in the previous section. All assays were carried out in triplicate under ultra-high-purity Ar on a Schlenk line, and all samples were prepared in a Coy anaerobic chamber under 95% Ar / 5% H2 (O2 < 2ppm). Reactions contained 1.00 mL degassed ATP regeneration solution (50 mM TRIS, pH 8.0, 10 mM MgCl2, 0.125 mg/mL creatine kinase, 5 mM Na2ATP, 300 mM phosphocreatine, and 13 mM DT) in a 10 mL vial. 1.0 mL of C2H2 (room temperature, atmospheric pressure) was added to the headspace of each reaction vial. Vials were shaken in a water bath at 30 °C for 10 min before addition of protein. Proteins were added using gas-tight Hamilton syringes to a final concentration of 0.2 μM MoFeP and 40 μM FeP. Reduction of C2H2 proceeded for 15 min at 30°C and was quenched with 0.4 mL of 4 M NaCl. The product, C2H4, was quantified via gas chromatography on an SRI 8610C instrument with 0.2 mL injections of the headspace into a HayeSep N packed column (SRI Instruments) and detection with a FID detector. A standard curve of C2H4 was determined daily prior to each assay.
Mössbauer spectroscopy of MoFeP.
The first Mössbauer sample was prepared as described above, followed by the concentration of the sample to 1 mM (400 μL final volume). After initial data collection, the sample was diluted to 125 μM. The sample was then re-metalated with natural abundance Fe2+ as described above, followed by the concentration of the sample back down to 1 mM. Mössbauer samples were prepared and flash frozen in liquid N2 in a Coy anaerobic chamber under 95% Ar/5% H2 (<2ppm O2). Mössbauer spectra were recorded on a SEE Co. spectrometer in constant acceleration mode and transmission geometry at 80 K. A Janis SVT-300T dewar was used with a 54 mT magnetic field applied parallel to the propagation of the γ-beam. Isomer shifts were determined relative to the centroid of a metallic foil of a α-Fe at room temperature.
Crystallographic data collection and refinement.
Crystals of heterometallated βS188A MoFeP were prepared using the sitting drop vapor diffusion method at room temperature under 95% Ar/5% H2 (O2 <2 ppm) in a Coy anaerobic chamber. All crystals were dark brown in color. Crystallization reservoirs each contained 500 μL of precipitation solution and crystallization wells contained 2 μL of the precipitation solution and 2 μL of the protein stock solution (125 μM). Crystals appeared within a few days and took up to two weeks to mature. Ni- and Co-substituted βS188A MoFeP crystals were grown from the same samples that were used in the EPR experiments. The crystal of the Ga-substituted variant was obtained using a sample prepared separately from the EPR samples. All proteins were crystallized in the presence of 10 mM DT. Crystals were cryoprotected with perfluoropolyether and flash frozen in liquid N2 in a Coy anaerobic chamber. All data were collected using multi-wavelength synchrotron radiation at either SSRL beamline 9–2 (Ga- and Co-substituted structures) or ALS beamline 5.0.2 (Ni-substituted structure).
Data sets were integrated and scaled using iMosflm and Aimless, followed by molecular replacement (search model PDB 2MIN) with Phaser-MR of the PHENIX suite. Structures were refined with phenix.refine and Coot, using riding hydrogens added with phenix.reduce. Data collection and refinement parameters and statistics can be found in Table S2. All figures of crystal structures were produced with PyMOL. Integration with MAPMAN (Uppsala Software Factory) to assign partial occupancy of each metal (Fe and heterometal) at each metal site in the P-cluster was attempted.
Electron paramagnetic resonance spectroscopy of MoFeP.
Samples were prepared as described in the “Preparation of heterometallic- and 57Fe-incorporated-P-cluster MoFeP” sections above. Proteins were concentrated to 50 μM using 30 kDa Microcon centrifugal filters (EMD/Millipore). All data were collected on a Bruker EMX spectrometer. A helium cryostat was used to maintain temperatures in the range of 5−10 K. The modulation frequency was 100.0 kHz and the modulation amplitude was 9.8 G during the recording of the spectra. The microwave frequency was ~9.62 GHz. Microwave power for each spectrum is indicated in figure captions. All spectra in figures were background subtracted.
H2O2 oxidative stress survival test of A. vinelandii.
Av cell cultures (wt and βS188A MoFeP) were grown in both BM+ and BM− media (200 rpm, 30°C) in biological triplicate until OD600 ~ 0.8. Dilutions of cells were plated on solid BM+ or BM− media (same as liquid growth conditions) to determine colony forming units (CFUs) present in the cultures prior to H2O2 exposure. Cultures were centrifuged (5,000 rpm, 4°C) to sediment the cells. The cell pellets were resuspended in fresh media (BM+ and BM− media corresponding to initial growth conditions) containing 5 mM Suprapur H2O2 (EMD Millipore) and shaken at 200 rpm and 30°C for 30 min. Cells were pelleted again via centrifugation (5,000 rpm, 4°C), then resuspended in fresh media (BM+ and BM− media corresponding to initial growth conditions) lacking H2O2. A dilution series of cells were plated to determine CFUs present after oxidative stress by H2O2. Colonies were manually counted 3–5 days after plating. The plates used for colony counting contained between 30–500 colonies.
Growth curves of A. vinelandii.
All cultures used for growth curves were prepared in 125-mL flasks (pre-washed with nitric acid) containing 25 mL BM− media (varying Fe %) and were shaken at 200 rpm, 30°C. wt and βS188A Av cells were initially Fe-starved by conducting four passes of the cells on solid 0% Fe BM+ media, followed by one 25-mL liquid growth in 0% Fe BM+ in a 125 mL flask until cells reached OD600 >1.0. Cells were fluorescent green due to secreted siderophores due to Fe starvation. Each variant (wt and βS188A MoFeP) of the Fe-starved cultures was used to inoculate five flasks of BM− media with varying Fe concentrations (1%, 10%, 25%, 50%, and 100% Fe) to an initial OD600 of ~0.1. All media and culture growths were prepared in parallel. 200-μL aliquots were sterilely removed from each flask approximately every 4 h over the course of ~50 h to measure OD600.
Extraction of total RNA from Av cells and RT-qPCR of MoFeP (nifK) transcripts.
Av cells (wt and βS188A) were grown in biological triplicate to an OD600 of ~1.0 in 25 mL 100% and 1% Fe BM− media with shaking at 200 rpm and 30 °C. 3 mL of cell suspensions were removed and added to 6 mL of RNAprotect Bacteria Reagent (Qiagen), immediately vortexed for 5 s and then incubated at room temperature for 5 min. Cells were centrifuged (5,000x g) for 10 min at room temperature, yielding a small, white pellet. The supernatant was removed, and pellets were frozen at −80 °C until RNA purification (total time frozen <1 week).
Total RNA purification was carried out using the RNeasy Mini Kit (Qiagen) using the prescribed protocols for enzymatic lysis of bacteria and purification of total RNA from bacterial lysate. Successful extraction of RNA was confirmed by visualizing rRNA bands with a 1% RNA agarose gel and quantified by UV-vis spectroscopy. Reverse transcription (RT) was carried out with Superscript III Reverse Transcriptase, random 9-mer primers, and 1 μg total RNA. cDNA products were confirmed via a smear on a 1% DNA agarose gel.
qPCR of the cDNA products was carried out using 10 ng of cDNA, Sybr dye, and Phusion HF polymerase in 20 μL total reaction volume in a Stratagene Mx3000 qPCR thermalcycler. qPCR reactions were carried out in technical triplicate of the biological replicates. Two housekeeping genes, gyrA and rho, were amplified as internal controls. However, gyrA provided inconsistent results. Data was thus normalized the rho reference gene. The following primers were used with an annealing temperature of 54 °C, resulting in an amplicons with a lengths of ~100 base pairs:
Forward nifK primer: CGAGACCTACCTGGGCAAC
Reverse nifK primer: CACTTCTTCCGGATCGGAGA
Forward rho primer: GGAAATGGCCGAACAGATGG
Reverse rho primer: GATTTCCTCGCCGCTTTTCG
Whole cell C2H4 activity assays.
Whole cell activity assays were performed with the same Av cell cultures that were used for total RNA extraction and RT-qPCR analysis. Immediately after removing a portion of cells for RNA extraction, 1.0 mL of whole cell suspensions was removed and placed in a 10-mL vial under air and sealed with a septum. 1.0 mL of C2H2 (room temperature, atmospheric pressure) was transferred into the headspace of the vial via a gastight Hamilton syringe. Vials were shaken in a 30 °C water bath for 95 min. The product, C2H4, was quantified by gas chromatography (SRI 8610C) with an FID detector, using 0.2-mL injections of the headspace into a HayeSep N packed column (SRI Instruments). A standard curve of C2H4 was prepared daily prior to each experiment. In vivo relative specific activity of MoFeP was calculated using the relative quantities of nifK mRNA transcripts determined from RT-qPCR.
Supplementary Material
ACKNOWLEDGMENT
We thank C. Owens, J. Zhu, C.J. Yu, N. Avakyan, A. Kakkis, and R. Yu for critical discussions, V. Cash and D. Dean (Va. Tech) for generously providing the Av nifK knockout strain DJ200 and the plasmid for creating the ΔnifB strain, and E. Lavorando for assistance with RT-qPCR experiments. This work was supported by the National Institutes of Health (Grants R01-GM099813 to F.A.T. and R01-GM101390 to M.T.G.) and by NASA (80NSSC18M0093; ENIGMA: Evolution of Nanomachines in Geospheres and Microbial Ancestors, NASA Astrobiology Institute Cycle 8 to F.A.T.). Additional support was provided by an NIH Molecular Biophysics Training Grant to H.L.R. (T32-GM008326) and an NIH Ruth Kirschstein Postdoctoral Fellowship to J.R. (F32-GM120981).
Footnotes
Supporting Information. The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Diazotrophic growth curves (Figure S1); RT-qPCR analyses of nifK gene expression (Figure S2); Mössbauer spectrum of Av βS188A MoFeP reconstituted with 57Fe (Figure S3); 2Fo–Fc omit maps of heterometal-reconstituted βS188A MoFeP (Figure S4); X-band EPR spectra of heterometal-treated βS188A MoFeP (Figure S5); Mössbauer fitting parameters for βS188A MoFeP (Table S1); X-ray data collection and refinement statistics (Table S2); ICP-MS analysis of heterometal-treated MoFeP variants (Table S3).
The authors declare no competing financial interests. Coordinate and structure factor files have been deposited into the RCSB databank under the following PDB IDs: 8E3T, 8E3U, 8E3V.
REFERENCES
- 1.Rutledge HL; Tezcan FA, Electron Transfer in Nitrogenase. Chem. Rev 2020, 120, 5158–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seefeldt LC; Yang ZY; Lukoyanov DA; Harris DF; Dean DR; Raugei S; Hoffman BM, Reduction of Substrates by Nitrogenases. Chem. Rev 2020, 120, 5082–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman BM; Lukoyanov D; Yang ZY; Dean DR; Seefeldt LC, Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson FB; Burris RH, A nitrogen pressure of 50 atmospheres does not prevent evolution of hydrogen by nitrogenase. Science 1984, 224, 1095–7. [DOI] [PubMed] [Google Scholar]
- 5.Einsle O; Rees DC, Structural Enzymology of Nitrogenase Enzymes. Chem. Rev 2020, 120, 4969–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howard JB; Rees DC, Structural basis of biological nitrogen fixation. Chem. Rev 1996, 96, 2965–2982. [DOI] [PubMed] [Google Scholar]
- 7.Erickson JA; Nyborg AC; Johnson JL; Truscott SM; Gunn A; Nordmeyer FR; Watt GD, Enhanced efficiency of ATP hydrolysis during nitrogenase catalysis utilizing reductants that form the all-ferrous redox state of the Fe protein. Biochemistry 1999, 38, 14279–85. [DOI] [PubMed] [Google Scholar]
- 8.Lowery TJ; Wilson PE; Zhang B; Bunker J; Harrison RG; Nyborg AC; Thiriot D; Watt GD, Flavodoxin hydroquinone reduces Azotobacter vinelandii Fe protein to the all-ferrous redox state with a S = 0 spin state. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy RWF; Knight E, Reductant-Dependent Adenosine Triphosphatase of Nitrogen-Fixing Extracts of Azotobacter vinelandii. Biochim. Biophys. Acta 1966, 122, 520–&. [Google Scholar]
- 10.Chan JM; Christiansen J; Dean DR; Seefeldt LC, Spectroscopic evidence for changes in the redox state of the nitrogenase P-cluster during turnover. Biochemistry 1999, 38, 5779–5785. [DOI] [PubMed] [Google Scholar]
- 11.Danyal K; Dean DR; Hoffman BM; Seefeldt LC, Electron Transfer within Nitrogenase: Evidence for a Deficit-Spending Mechanism. Biochemistry 2011, 50, 9255–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson DC; Dean DR; Smith AD; Johnson MK, Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem 2005, 74, 247–81. [DOI] [PubMed] [Google Scholar]
- 13.Cammack R, Iron-Sulfur Clusters in Enzymes - Themes and Variations. Adv. Inorg. Chem 1992, 38, 281–322. [Google Scholar]
- 14.Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y, Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem. Rev 2014, 114, 4366–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters JW; Stowell MH; Soltis SM; Finnegan MG; Johnson MK; Rees DC, Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36, 1181–7. [DOI] [PubMed] [Google Scholar]
- 16.Cao L; Borner MC; Bergmann J; Caldararu O; Ryde U, Geometry and Electronic Structure of the P-Cluster in Nitrogenase Studied by Combined Quantum Mechanical and Molecular Mechanical Calculations and Quantum Refinement. Inorg. Chem 2019. [DOI] [PubMed] [Google Scholar]
- 17.Keable SM; Zadvornyy OA; Johnson LE; Ginovska B; Rasmussen AJ; Danyal K; Eilers BJ; Prussia GA; LeVan AX; Raugei S; Seefeldt LC; Peters JW, Structural characterization of the P1+ intermediate state of the P-cluster of nitrogenase. J. Biol. Chem 2018, 293, 9629–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Owens CP; Katz FE; Carter CH; Oswald VF; Tezcan FA, Tyrosine-Coordinated P-Cluster in G diazotrophicus Nitrogenase: Evidence for the Importance of O-Based Ligands in Conformationally Gated Electron Transfer. J. Am. Chem. Soc 2016, 138, 10124–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutledge HL; Rittle J; Williamson LM; Xu WA; Gagnon DM; Tezcan FA, Redox-Dependent Metastability of the Nitrogenase P-Cluster. J. Am. Chem. Soc 2019, 141, 10091–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seefeldt LC; Hoffman BM; Peters JW; Raugei S; Beratan DN; Antony E; Dean DR, Energy Transduction in Nitrogenase. Acc. Chem. Res 2018, 51, 2179–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rutledge HL; Cook BD; Nguyen HPM; Herzik MA Jr.; Tezcan FA, Structures of the nitrogenase complex prepared under catalytic turnover conditions. Science 2022, 377, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berkovitch F; Nicolet Y; Wan JT; Jarrett JT; Drennan CL, Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 2004, 303, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaughlin MI; Lanz ND; Goldman PJ; Lee KH; Booker SJ; Drennan CL, Crystallographic snapshots of sulfur insertion by lipoyl synthase. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 9446–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolet Y; Rohac R; Martin L; Fontecilla-Camps JC, X-ray snapshots of possible intermediates in the time course of synthesis and degradation of protein-bound Fe4S4 clusters. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 7188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holm RH, Trinuclear Cuboidal and Heterometallic Cubane-Type Iron-Sulfur Clusters - New Structural and Reactivity Themes in Chemistry and Biology. Adv. Inorg. Chem 1992, 38, 1–71. [Google Scholar]
- 26.Brown OR; Smyk-Randall E; Draczynska-Lusiak B; Fee JA, Dihydroxy-acid dehydratase, a [4Fe-4S] cluster-containing enzyme in Escherichia coli: effects of intracellular superoxide dismutase on its inactivation by oxidant stress. Arch. Biochem. Biophys 1995, 319, 10–22. [DOI] [PubMed] [Google Scholar]
- 27.Flint DH; Emptage MH; Guest JR, Fumarase-a from Escherichia-Coli - Purification and Characterization as an Iron Sulfur Cluster Containing Enzyme. Biochemistry 1992, 31, 10331–10337. [DOI] [PubMed] [Google Scholar]
- 28.Beinert H; Kennedy MC; Stout CD, Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev 1996, 96, 2335–2373. [DOI] [PubMed] [Google Scholar]
- 29.Robbins AH; Stout CD, Structure of Activated Aconitase - Formation of the [4Fe-4S] Cluster in the Crystal. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 3639–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauble H; Kennedy MC; Beinert H; Stout CD, Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry 1992, 31, 2735–48. [DOI] [PubMed] [Google Scholar]
- 31.Flint DH; Tuminello JF; Emptage MH, The Inactivation of Fe-S Cluster Containing Hydro-Lyases by Superoxide. J. Biol. Chem 1993, 268, 22369–22376. [PubMed] [Google Scholar]
- 32.Imlay JA, Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol 2006, 59, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 33.Imlay JA, Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem 2008, 77, 755–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imlay JA, The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol 2013, 11, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo CF; Mashino T; Fridovich I, Alpha,Beta-Dihydroxyisovalerate Dehydratase - a Superoxide-Sensitive Enzyme. J. Biol. Chem 1987, 262, 4724–4727. [PubMed] [Google Scholar]
- 36.Varghese S; Tang Y; Imlay JA, Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J. Bacteriol 2003, 185, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martic M; Jakab-Simon IN; Haahr LT; Hagen WR; Christensen HEM, Heterometallic [AgFe3S4] ferredoxin variants: synthesis, characterization, and the first crystal structure of an engineered heterometallic iron-sulfur protein. J. Biol. Inorg. Chem 2013, 18, 261–276. [DOI] [PubMed] [Google Scholar]
- 38.Finnegan MG; Conover RC; Park JB; Zhou ZH; Adams MWW; Johnson MK, Electronic, Magnetic, Redox, and Ligand-Binding Properties of (MFe3S4) Clusters (M=Zn, CO, Mn) in Pyrococcus-Furiosus Ferredoxin. Inorg. Chem 1995, 34, 5358–5369. [Google Scholar]
- 39.Staples CR; Dhawan IK; Finnegan MG; Dwinell DA; Zhou ZH; Huang HS; Verhagen MFJM; Adams MWW; Johnson MK, Electronic, magnetic, and redox properties of [MFe3S4] clusters (M = Cd, Cu, Cr) in Pyrococcus furiosus ferredoxin. Inorg. Chem 1997, 36, 5740–5749. [DOI] [PubMed] [Google Scholar]
- 40.Conover RC; Park JB; Adams MWW; Johnson MK, Formation and Properties of a NiFe3S4 Cluster in Pyrococcus-Furiosus Ferredoxin. J. Am. Chem. Soc 1990, 112, 4562–4564. [Google Scholar]
- 41.Fu WG; Telser J; Hoffman BM; Smith ET; Adams MWW; Finnegan MG; Conover RC; Johnson MK, Interaction of Tl+ and Cs+ with the [Fe3s4] Cluster of Pyrococcus-Furiosus Ferredoxin - Investigation by Resonance Raman, MCD, EPR, and Endor Spectroscopy. J. Am. Chem. Soc 1994, 116, 5722–5729. [Google Scholar]
- 42.Faridoon KY; Zhuang HY; Sykes AG, Kinetic Studies on the Reaction of M2+ Ions with Aconitase Fe3S40 To Give Fe3MS42+ Clusters (M = Fe, Mn, Co). Inorg. Chem 1994, 33, 2209–2212. [Google Scholar]
- 43.Sandercock JR; Page WJ, RpoS expression and the general stress response in Azotobacter vinelandii during carbon and nitrogen diauxic shifts. J. Bacteriol 2008, 190, 946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Rojas A; Kim JJ; Johnston PR; Makarova O; Eravci M; Weise C; Hengge R; Rolff J, Non-lethal exposure to H2O2 boosts bacterial survival and evolvability against oxidative stress. PLoS Genet. 2020, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orikasa Y; Nodasaka Y; Ohyama T; Okuyama H; Ichise N; Yumoto I; Morita N; Wei M; Ohwada T, Enhancement of the nitrogen fixation efficiency of genetically-engineered Rhizobium with high catalase activity. J. Biosci. Bioeng 2010, 110, 397–402. [DOI] [PubMed] [Google Scholar]
- 46.Page WJ; Huyer M, Derepression of the Azotobacter-Vinelandii Siderophore System, Using Iron-Containing Minerals to Limit Iron Repletion. J. Bacteriol 1984, 158, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noguez R; Segura D; Moreno S; Hernandez A; Juarez K; Espin G, Enzyme I NPr, NPr and IIA Ntr are involved in regulation of the poly-beta-hydroxybutyrate biosynthetic genes in Azotobacter vinelandii. J. Mol. Microbiol. Biotechnol 2008, 15, 244–54. [DOI] [PubMed] [Google Scholar]
- 48.Srisantitham S; Badding ED; Suess DLM, Postbiosynthetic modification of a precursor to the nitrogenase iron-molybdenum cofactor. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2015361118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Badding E; Srisantitham S; Lukoyanov D; Hoffman BM; Suess D, Connecting the Geometric and Electronic Structures of the Nitrogenase Iron–Molybdenum Cofactor through Site-Selective Labeling. 2022, ChemRxiv; Cambridge Open Engage: Cambridge; https://chemrxiv.org/engage/chemrxiv/articledetails/61e1a5d46afbef3cfb64777d (accessed on Aug. 15, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy MC; Emptage MH; Dreyer JL; Beinert H, The role of iron in the activation-inactivation of aconitase. J. Biol. Chem 1983, 258, 11098–105. [PubMed] [Google Scholar]
- 51.Yoo SJ; Angove HC; Papaefthymiou V; Burgess BK; Münck E, Mössbauer Study of the MoFe Protein of Nitrogenase from Azotobacter vinelandii Using Selective 57Fe Enrichment of the M-Centers. J. Am. Chem. Soc 2000, 122, 4926–4936. [Google Scholar]
- 52.McLean PA; Papaefthymiou V; Orme-Johnson WH; Münck E, Isotopic hybrids of nitrogenase. Mossbauer study of MoFe protein with selective 57Fe enrichment of the P-cluster. J. Biol. Chem 1987, 262, 12900–3. [PubMed] [Google Scholar]
- 53.Rittle J; Field MJ; Green MT; Tezcan FA, An efficient, step-economical strategy for the design of functional metalloproteins. Nat. Chem 2019, 11, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasak M; Kagi JH, Metal thiolate clusters in cobalt(II)-metallothionein. Proc. Natl. Acad. Sci. U. S. A 1981, 78, 6709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Stappen C; Decamps L; Cutsail GE; Bjornsson R; Henthorn JT; Birrell JA; DeBeer S, The Spectroscopy of Nitrogenases. Chem. Rev 2020, 120, 5005–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munck E; Rhodes H; Orme-Johnson WH; Davis LC; Brill WJ; Shah VK, Nitrogenase. VIII. Mossbauer and EPR spectroscopy. The MoFe protein component from Azotobacter vinelandii OP. Biochim. Biophys. Acta 1975, 400, 32–53. [DOI] [PubMed] [Google Scholar]
- 57.Rawlings J; Shah VK; Chisnell JR; Brill WJ; Zimmermann R; Münck E; Orme-Johnson WH, Novel metal cluster in the iron-molybdenum cofactor of nitrogenase. Spectroscopic evidence. J. Biol. Chem 1978, 253, 1001–4. [PubMed] [Google Scholar]
- 58.Ciurli S; Ross PK; Scott MJ; Yu SB; Holm RH, Synthetic Nickel-Containing Heterometal Cubane-Type Clusters with Nife3q4 Cores (Q = S, Se). J. Am. Chem. Soc 1992, 114, 5415–5423. [Google Scholar]
- 59.Zhou J; Scott MJ; Hu ZG; Peng G; Munck E; Holm RH, Synthesis and Comparative Reactivity and Electronic Structural Features of [MFe3S4]Z+ Cubane-Type Clusters (M = Fe, CO, Ni). J. Am. Chem. Soc 1992, 114, 10843–10854. [Google Scholar]
- 60.Li Z; Guo S; Sun Q; Chan GK, Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations. Nat. Chem 2019. [DOI] [PubMed] [Google Scholar]
- 61.Tittsworth RC; Hales BJ, Detection of EPR Signals Assigned to the 1-Equiv-Oxidized P-Clusters of the Nitrogenase MoFe-Protein from Azotobacter vinelandii. J. Am. Chem. Soc 1993, 115, 9763–9767. [Google Scholar]
- 62.Ribbe MW; Hu Y; Guo M; Schmid B; Burgess BK, The FeMoco-deficient MoFe protein produced by a nifH deletion strain of Azotobacter vinelandii shows unusual P-cluster features. J. Biol. Chem 2002, 277, 23469–76. [DOI] [PubMed] [Google Scholar]
- 63.Spatzal T; Perez KA; Howard JB; Rees DC, Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. Elife 2015, 4, e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang W; Lee CC; Jasniewski AJ; Ribbe MW; Hu Y, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase. Science 2020, 368, 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O, A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–+. [DOI] [PubMed] [Google Scholar]
- 66.Buscagan TM; Kaiser JT; Rees DC, Selenocyanate derived Se-incorporation into the Nitrogenase Fe protein cluster. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schlesier J; Rohde M; Gerhardt S; Einsle O, A Conformational Switch Triggers Nitrogenase Protection from Oxygen Damage by Shethna Protein II (FeSII). J. Am. Chem. Soc 2016, 138, 239–47. [DOI] [PubMed] [Google Scholar]
- 68.Medina MS; Bretzing KO; Aviles RA; Chong K; Espinoza A; Garcia CNG; Katz BB; Kharwa RN; Hernandez A; Lee JL; Lee TM; Strul MW; Lo Verde C; Wong EY; Owens CP, CowN sustains nitrogenase turnover in the presence of the inhibitor carbon monoxide. J. Biol. Chem 2021, 100501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mus F; Colman DR; Peters JW; Boyd ES, Geobiological feedbacks, oxygen, and the evolution of nitrogenase. Free Radical Biol. Med 2019, 140, 250–259. [DOI] [PubMed] [Google Scholar]
- 70.Varghese F; Kabasakal BV; Cotton CAR; Schumacher J; Rutherford AW; Fantuzzi A; Murray JW, A low-potential terminal oxidase associated with the iron-only nitrogenase from the nitrogen-fixing bacterium Azotobacter vinelandii. J. Biol. Chem 2019, 294, 9367–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kendall MM; Liu Y; Sieprawska-Lupa M; Stetter KO; Whitman WB; Boone DR, Methanococcus aeolicus sp. nov., a mesophilic, methanogenic archaeon from shallow and deep marine sediments. Int. J. Syst. Evol. Microbiol 2006, 56, 1525–1529. [DOI] [PubMed] [Google Scholar]
- 72.Torres ME; Cox T; Hong W-L; McManus J; Sample JC; Destrigneville C; Gan HM; Gan HY; Moreau JW, Crustal fluid and ash alteration impacts on the biosphere of Shikoku Basin sediments, Nankai Trough, Japan. Geobiology 2015, 13, 562–580. [DOI] [PubMed] [Google Scholar]
- 73.Glick BR; Brooks HE; Pasternak JJ, Transformation of Azotobacter vinelandii with plasmid DNA. J. Bacteriol 1985, 162, 276–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Page WJ; Von Tigerstrom M, Optimal Conditions for Transformation of Azotobacter vinelandii. J. Bacteriol 1979, 139, 1058–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.