

Abstract
Loss of Krev interaction-trapped-1 (KRIT1) expression leads to the development of cerebral cavernous malformations (CCM), a disease in which abnormal blood vessel formation compromises the structure and function of the blood–brain barrier. The role of KRIT1 in regulating endothelial function is well-established. However, several studies have suggested that KRIT1 could also play a role in regulating nonendothelial cell types and, in particular, immune cells. In this study, we generated a mouse model with neutrophil-specific deletion of KRIT1 in order to investigate the effect of KRIT1 deficiency on neutrophil function. Neutrophils isolated from adult Ly6Gtm2621(cre)Arte Krit1flox/flox mice had a reduced ability to attach and spread on the extracellular matrix protein fibronectin and exhibited a subsequent increase in migration. However, adhesion to and migration on ICAM-1 was unchanged. In addition, we used a monomeric, fluorescently-labelled fragment of fibronectin to show that integrin activation is reduced in the absence of KRIT1 expression, though β1 integrin expression appears unchanged. Finally, neutrophil migration in response to lipopolysaccharide-induced inflammation in the lung was decreased, as shown by reduced cell number and myeloperoxidase activity in lavage samples from Krit1PMNKO mice. Altogether, we show that KRIT1 regulates neutrophil adhesion and migration, likely through regulation of integrin activation, which can lead to altered inflammatory responses in vivo.
Keywords: cell migration, cerebral cavernous malformations, inflammation, integrins
Graphical Abstract

Loss of KRIT1 expression in neutrophils has no effect on adhesion to and migration on ICAM-1, but causes a loss of the neutrophil attachment and spreading on fibronectin and a reduction in fibronectin-binding integrin activity. This may explain why, though KRIT1 deficient neutrophils migrate faster ex vivo, migration of KRIT1 ko neutrophils into the lung in response to lipopolysaccharide was decreased. Thus, we conclude that KRIT1 regulates neutrophil behavior.
Introduction
Cerebral cavernous malformation (CCM) is a vascular disease characterized by the formation of abnormal blood vessels, which exhibit a loss of barrier function [1–3]. CCM is thought to occur due to loss of function mutations in one of three genes: Krit1 (CCM1), CCM2 and CCM3 (PDCD10) [4–7], which form a heterotrimeric complex [8]. Loss of CCM protein expression in endothelial cells promotes proliferation [9] and resistance to apoptosis [10], and leads to higher levels of oxidative stress [11,12]. However, it is still unclear how the loss of these proteins leads to the development of CCM. Currently, defects in endothelial cell–cell junction integrity are thought to be the primary drivers of CCM formation and clinical presentation due to leakage of blood from the capillaries into the brain tissue.
On the other hand, we and others have shown that KRIT1 is widely expressed and that the expression level of KRIT1 in nonendothelial cells is often higher than in endothelial cells [13]. Furthermore, KRIT1 and the other CCM proteins have been shown to play important roles in nonendothelial cell types and tissues, which may or may not be related to their role as pathogenic originators of CCM. For example, we previously demonstrated that heterozygous loss of KRIT1 increases the oncogene-driven development of intestinal adenocarcinoma, likely due to decreased epithelial cell–cell contact [14]. This suggests that KRIT1 acts as an epithelial tumour suppressor, and that Krit1 haploinsufficiency could have an important clinical impact despite not leading to the formation of CCM lesions. Related studies have shown that loss of KRIT1 in intestinal epithelial cells decreases epithelial barrier function and increases cation selectivity [15] and that loss of CCM3 in mouse gut epithelium results in disruption of the colonic mucus barrier [16]. Furthermore, studies in zebrafish have pointed to a role for CCM proteins in cardiac development [17–19]. In addition, loss of KRIT1 in fibroblasts was shown to alter NADPH oxidase signalling and promote increased production of reactive oxygen species [20], which parallels a similar pathway we described in endothelial cells [21]. Another cell type in which CCM proteins have been shown to function is in astrocytes/neuroglia. Mice with CCM3-deficient astrocytes/neuroglia have been shown to develop CCM-like lesions [22]. Moreover, feedback mechanisms between endothelial cells and astrocytes or endothelial cells and pericytes in the brain milieu have been suggested to enhance angiogenic signalling in CCM lesions [23,24]. While the number of studies is limited, the data strongly suggest that KRIT1 and the other CCM proteins are highly likely to have important signalling functions in nonendothelial cell types.
In our prior studies, we sought to determine what effect KRIT1-depletion would have on the response to inflammation. To accomplish this, we used a global heterozygous KRIT1 knockout mouse (Krit1+/−) to assess changes in vascular physiology and inflammation due to Krit1 haploinsufficiency, and discovered that heterozygous mice were more sensitive to inflammatory stimuli and exhibited an enhanced response, including increased edema formation and cell infiltration [25]. Given that these animals were KRIT1-deficient in all cell types, we were unable to definitively point to endothelial KRIT1 expression as being responsible for these phenotypes, leaving open the possibility of a role for other cell types, particularly leukocytes (including neutrophils). Leukocyte adhesion and migration are regulated by the GTPase Rap1 [26,27], which promotes integrin activation and adhesion to various substrates, including ICAM-1 [28,29] and fibronectin [29,30]. KRIT1 is an effector of Rap1 and has been shown to mediate specific Rap1-dependent functions in endothelial cells [31]. KRIT1 also binds to the β1 integrin regulatory protein ICAP1α, which is expressed in most cell types [32,33]. KRIT1 has been reported to regulate ICAP1α expression and compete with β1 integrin for binding to ICAP1α [34–37]. These studies support the hypothesis that KRIT1 could regulate leukocyte adhesion in general, yet a more recent study reported that CCM3 regulates the release of the ‘reserved’ pool of granules from neutrophils due to its interaction with the serine/threonine kinase-24 (STK24) [38]. Though the impact of the interaction of CCM3 with STK24 vs. with KRIT1/CCM2 is still in debate [39–41], these findings demonstrate that CCM proteins can regulate neutrophil-specific functions. Together, the role of KRIT1 in limiting the response to inflammatory stimuli [25] and the high expression of KRIT1 in neutrophils and other myeloid cells (data not shown), combined with the potential for KRIT1 to regulate neutrophil adhesion via binding to Rap1 [31] or ICAP1α [32–37], led us to explore a potential role for KRIT1 in regulating neutrophil function.
We therefore generated neutrophil-specific KRIT1 knockout mice by breeding Krit1flox/flox mice with Ly6Gtm2621(cre)Arte animals, which express Cre recombinase downstream of the Ly6G promoter. By isolating neutrophils from these mice, we show that loss of KRIT1 in neutrophils reduces neutrophil adhesion and spreading on a fibronectin matrix, but not when plated on ICAM-1. This reduced adhesion likely contributes to increased migration of KRIT1-deficient neutrophils on fibronectin and corresponds with a significant attenuation of integrin activity. Finally, we observe that in vivo, loss of KRIT1 results in decreased neutrophil recruitment to the inflamed lung tissue of mature Ly6G mice. Thus, we conclude that KRIT1 is a regulator of integrin activity, cell adhesion and cell migration in neutrophils.
Results
Generation of neutrophil-specific KRIT1 knockout mice
Neutrophils mature in the bone marrow before circulating in the blood, where they await activation by inflammatory stimuli [42]. With a half-life of approximately 1 day, primary neutrophils are short-lived, terminally differentiated cells not amenable to cell culture [43–45]. Neutrophil-like cell culture models are derived from neoplastic tissues, require lengthy culture times to induce a neutrophil-like phenotype and fail to replicate true neutrophil phenotypes [46,47]. Therefore, in order to probe the role of KRIT1 in neutrophils, we generated a novel mouse model that lacks expression of KRIT1 in mature neutrophils by crossing the existing Krit1flox/flox transgenic line [48] with Ly6Gtm2621(cre)Arte mice [49] in which Cre recombinase is inserted into the neutrophil-specific Ly6G locus. The Ly6G-Cre transgene also contains the red fluorescent reporter tdTomato [49], which allowed us to quantify recombination in mature neutrophils via flow cytometry. Blood and whole bone marrow samples from adult Ly6Gtm2621(cre)Arte Krit1flox/flox mice (Krit1PMNKO) vs. Krit1flox/flox mice (Krit1PMNWT) clearly showed that only Krit1PMNKO blood and bone marrow–expressed tdTomato (Fig. 1A). As seen in Fig. 1B, we were also able to detect the Ly6G-Cre transgene (Ly6G Cre) in both ear tissue and purified bone marrow–derived neutrophils (PMN) by PCR; however, recombination of Krit1, which generates the Krit1D allele, was only detected in the purified PMN indicating specific deletion of KRIT1 in mature neutrophils. Semi-quantitative RT-PCR showed that Krit1 mRNA was reduced in Krit1PMNKO neutrophils by ~ 50% (Fig. 1C); however, protein expression, as shown by western blot, was almost completely lost (> 85% reduction, Fig. 1D). The overall health of the Krit1PMNKO animals was indistinguishable from control animals. Krit1PMNKO animals demonstrated normal haematopoiesis as assessed using complete blood count (CBC) analysis; numbers of white blood cells (WBC 7.3 × 109), lymphocytes (LYM 6.4 × 109), monocytes (MON 0.25 × 109), neutrophils (NEU 0.69 × 109), platelets (PLT 4.93 × 1011) and red blood cells (RBC 8.87 × 1012) observed in Krit1PMNKO were comparable to their Krit1PMNWT littermates (Fig. 1E). In addition, we reliably recovered similar total numbers of neutrophils from each genotype using the Easy-Sep Mouse Neutrophil Enrichment kit from STEMCELL Technologies (WT 4.47 ± 0.32 × 106 vs. KO 4.35 ± 0.52 × 106, data not shown) Thus, we used mature neutrophils isolated from these animals for further analysis.
Fig. 1.

Ly6Gtm2621(cre)Arte Krit1flox/flox mice (Krit1PMNKO) display normal haematopoiesis despite neutrophil-specific reduction of KRIT1. (A) Representative FACS analysis of Krit1PMNWT and Krit1PMNKO mouse blood and bone marrow for tdTomato and Ly6G expression. (B) Representative genotyping gel of ear tissue and isolated neutrophils from Krit1PMNWT and Krit1PMNKO mice. Ly6G Cre-Cre transgene; Ly6G WT wildtype Ly6g allele; KRIT1 D-recombined Krit1 allele (exons 4–8 deleted). (C) Krit1 mRNA levels were measured by qPCR in isolated Krit1PMNWT and Krit1PMNKO mouse neutrophils. Data shown are mean Krit1 mRNA levels ± SEM, relative to Gapdh and normalized to Krit1PMNWT, from n = 8 independent experiments. ****P < 0.0001 by the unpaired t-test. (D) Representative western blot of neutrophil lysate from Krit1PMNWT and Krit1PMNKO mice. Marker lane was adjusted to allow marker bands to be easily visible, and black line denotes boundary of adjustment. (E) Blood collected from Krit1PMNWT and Krit1PMNKO mice was analysed using an Abaxis VetScan HM5 for white blood cell (WBC), lymphocyte (LYM), monocyte (MON), neutrophil (NEU), platelet (PLT) and red blood cell (RBC) counts. n = 3 mice/genotype, all between-genotype comparisons were not significant as determined by one-way ANOVA.
Loss of KRIT1 reduces neutrophil adhesion to fibronectin
While the loss of KRIT1 did not appear to affect neutrophil viability or the recoverable population, based on its role as a Rap1 effector [31] and a regulator of ICAP1α [32–37], we hypothesized that loss of KRIT1 could affect neutrophil behaviour and function. As mentioned earlier, neutrophils will circulate in the bloodstream until activation triggers their adherence to and migration through the endothelium to the source of inflammation [42,50]. As our previous study indicated a potential role for KRIT1 in leukocyte extravasation [25], we first investigated whether loss of KRIT1 affected neutrophil adhesion to the endothelially expressed surface protein and β2 integrin ligand ICAM-1. Krit1PMNKO or Krit1PMNWT neutrophils we loaded with calcein-AM to facilitate cell counting, then were treated with ±10 nm phorbol myristate acetate (PMA) and allowed to adhere to plates coated with 5 μg·mL−1 immobilized recombinant mouse ICAM-1 for 20 min. While treatment with PMA increased adhesion to ICAM-1, we detected no difference between Krit1PMNKO and Krit1PMNWT neutrophils in their ability to adhere to recombinant mouse ICAM-1 (Fig. 2A).
Fig. 2.
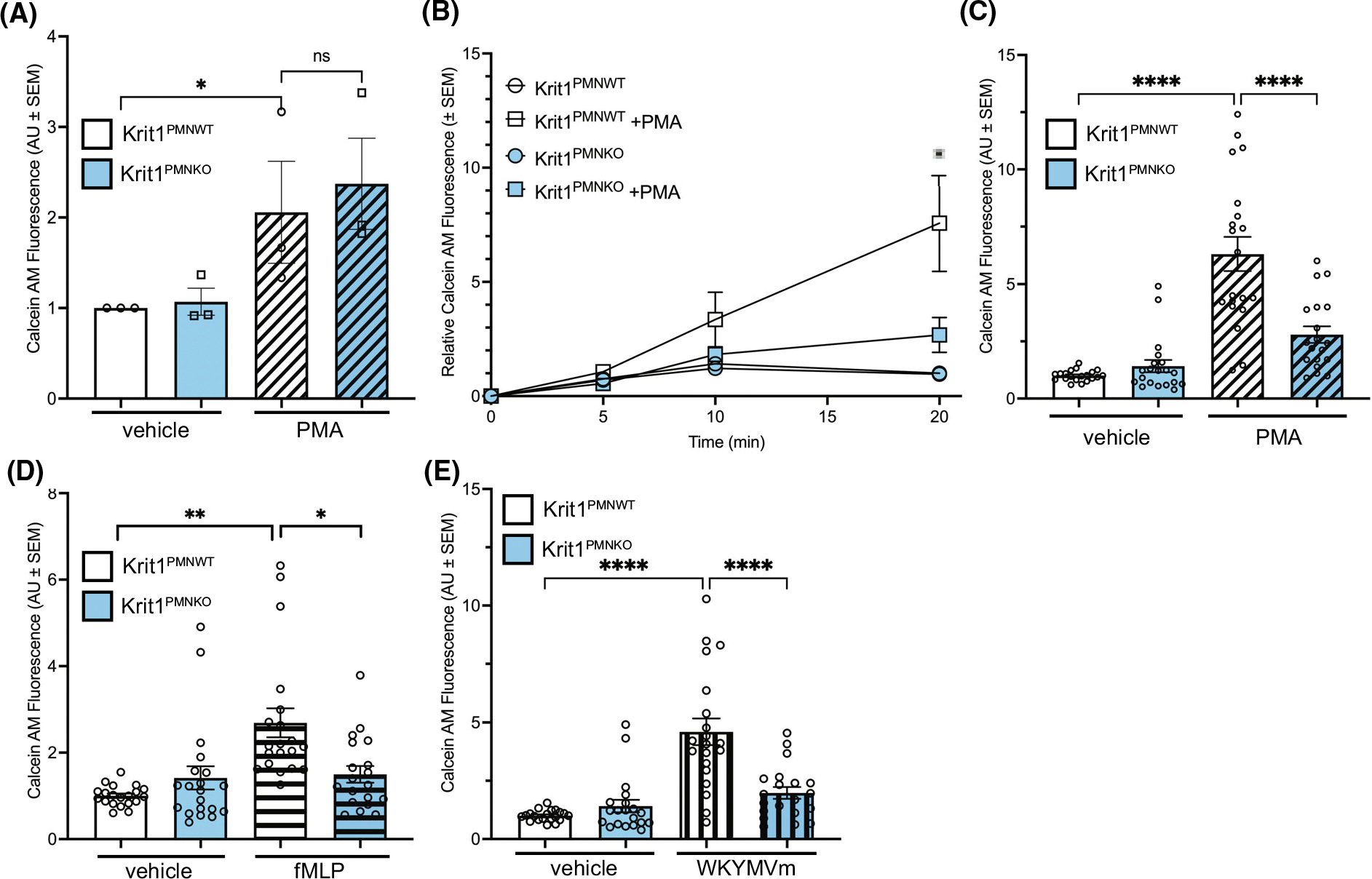
Krit1PMNKO neutrophils have reduced adhesion to fibronectin. (A) Adhesion of vehicle and 10 nm PMA-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils plated on 5 μg·mL−1 ICAM-1 after 20 min. Data shown are mean calcein-AM fluorescence, ±SEM, n = 3. (B) Time course of adhesion of 10 nm PMA-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils to 10 μg·mL−1 fibronectin. Data shown are mean calcein-AM fluorescence normalized to Krit1PMNWT vehicle: 20 min, ±SEM, n = 4. *P < 0.001 by the two-way ANOVA with the Tukey’s post hoc testing. (C) Adhesion of vehicle or 10 nm PMA-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils to fibronectin (10 μg·mL−1) after 20 min. Data shown are mean calcein-AM fluorescence, ±SEM, n = 20. ****P < 0.001 by the one-way ANOVA and Tukey’s post hoc testing. Alternative stimuli were also used: (D) Adhesion of vehicle or 250 nm fMLP-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils to fibronectin after 20 min. Data shown are mean calcein-AM fluorescence, ±SEM, n = 20. **P < 0.01 and *P < 0.05 by the one-way ANOVA and Tukey’s post hoc testing. (E) Adhesion of vehicle or 1 μm WKYMVm-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils to fibronectin after 20 min. Data shown are mean calcein-AM fluorescence, ±SEM, n = 20. ****P < 0.0001 by the one-way ANOVA and Tukey’s post hoc testing.
Following extravasation through the endothelial layer, neutrophils adhere to extracellular matrix (ECM) proteins in the endothelial basement membrane and interstitial tissue. While these matrices are comprised of a complex mix of matrix proteins, the glycoprotein fibronectin is an important component of the extravascular ECM. Therefore, we plated vehicle and PMA-treated KRIT1-deficient neutrophils on plates coated with 10 μg·mL−1 fibronectin and allowed them to adhere for 5, 10 and 20 min prior to washing off unattached cells. After 10 min of adhesion, we could detect a difference between PMA-treated and vehicle-treated cells, and between PMA-treated Krit1PMNWT and Krit1PMNKO neutrophils (Fig. 2B). After 20 min, KRIT1-deficient neutrophils demonstrated a 56% reduction in PMA-induced adhesion (Fig. 2B,C). To determine whether this effect was specific to PMA treatment, we also treated Krit1PMNWT and Krit1PMNKO neutrophils with 250 nm N-formylmethionyl-leucyl-phenylalanine (fMLP) or 1 μM of the chemoattractant peptide Trp-Lys-Tyr-Met-Val-Met (WKYMVm) and measured the number of cells adhering after 20 min. Krit1PMNKO cells treated with fMLP showed a 44% reduction in adhesion vs. Krit1PMNWT (Fig. 2D), and Krit1PMNKO cells treated with WKYMVm exhibited a 57% decrease in adhesion compared with Krit1PMNWT (Fig. 2E), indicating that the loss of KRIT1 expression reduces neutrophil adhesion to fibronectin independent of the type of stimulus.
Loss of KRIT1 reduces neutrophil spreading on fibronectin
We then wanted to characterize the adhesion deficiency in Krit1PMNKO neutrophils, therefore we examined the ability of Krit1PMNKO or Krit1PMNWT cells to adhere and spread on fibronectin-coated glass coverslips. Neutrophils were allowed to adhere for an hour in the presence of 10 nm PMA, 1 μg·mL−1 lipopolysaccharide (LPS), 250 nm fMLP, 1 μm WKYMVm or vehicle, then were fixed and stained with fluorescein isothiocyanate (FITC)-phalloidin to visualize the actin cytoskeleton. Cell area was then quantified using metamorph. Minimal differences in the number of cells attached after 1 h were seen between groups, suggesting that the Krit1PMNKO cells can overcome their adhesion deficiency given sufficient time (data not shown). No significant differences in spreading were seen in fMLP or WKYMVm treated cells (data not shown). However, when we examined the distribution of cell areas of adhered Krit1PMNWT and Krit1PMNKO neutrophils using frequency analysis, vehicle-treated Krit1PMNKO neutrophils exhibited a reduction in cell area vs. vehicle-treated Krit1PMNWT neutrophils (P = 0.0156, Fig. 3A), though cells from both genotypes remained rounded (Fig. 3B,C). This trend also held true with PMA- or LPS-treated Krit1PMNKO neutrophils, whose distribution trended toward smaller cell areas (PMA P = 0.0102, Fig. 3D; LPS P = 1.28 × 10−6, Fig. 3G) and were significantly less spread (Fig. 3E,F,H,I) when compared to Krit1PMNWT neutrophils with the same treatment.
Fig. 3.

Krit1PMNKO neutrophils show reduced cell spreading on fibronectin. (A) Frequency distribution of average cell area of unstimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils plated on 10 μg·mL−1 fibronectin after 1 h. P = 0.0156 by the Kolmogorov–Smirnov test. Representative images of FITC-phalloidin-stained Krit1PMNWT (B) and Krit1PMNKO (C) neutrophils. n = 3 mice /genotype, 10 images per mouse. Scale bar = 50 μm. (D) Frequency distribution of average cell area of 10 nm PMA-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils plated on 10 μg·mL−1 fibronectin after 1 h. P = 0.0102 by the Kolmogorov–Smirnov test. Representative images of FITC-phalloidin-stained Krit1PMNWT (E) and Krit1PMNKO (F) neutrophils. n = 3 mice/genotype, 10 images per mouse. Scale bar = 50 μm. (G) Frequency distribution of average cell area of 1 μg·mL−1 LPS-stimulated Krit1PMNWT and Krit1PMNKO mouse neutrophils plated on 10 μg·mL−1 fibronectin after 1 h. P = 1.28 × 10−6 by the Kolmogorov–Smirnov test. Representative images of FITC-phalloidin-stained Krit1PMNWT (H) and Krit1PMNKO (I) neutrophils. n = 3 mice/genotype, 10 images per mouse. Scale bar = 50 μm. (J) Krit1PMNKO neutrophils show increased phosphorylation of myosin light chain. Krit1PMNWT vs. Krit1PMNKO neutrophil phospho-MLC staining intensity per cell, ±SEM, n = 4 mice/genotype, n = 5 (WT + vehicle), 17 (WT + PMA), 14 (WT + LPS), 14 (KO + vehicle), 19 (KO + PMA), 11 (KO + LPS) fields/condition. *P < 0.05, ****P < 0.0001 by the Brown–Forsythe ANOVA and Games–Howell post hoc testing.
Studies in endothelial cells have shown that KRIT1 is a negative regulator of RhoA and its effector Rho kinase (ROCK); loss of KRIT1 expression leads to increased RhoA/ROCK activity, phosphorylation of myosin light chain (MLC) and increased actin-myosin contractility [51,52]. In neutrophils, RhoA/ROCK has been shown to regulate adhesion and migration on multiple substrates [53–57]. Therefore, we examined the effect of KRIT1 depletion on RhoA/ROCK activity in neutrophils by measuring phospo-MLC levels using immunofluorescent microscopy. Similar to the spreading assays above, neutrophils were allowed to adhere to fibronectin-coated coverslips for an hour in the presence of 10 nm PMA, 1 μg·mL−1 LPS or vehicle then were fixed and stained with anti-pMLC antibody followed by Alexa 568 labelled goat anti-rabbit secondary antibody. Images were captured using a Leica Stellaris 5 laser scanning confocal microscope at 63× magnification, and individual cell fluorescence intensity was quantified using imaris analysis software. Vehicle-treated Krit1PMNKO neutrophils displayed significantly increased pMLC fluorescence compared with vehicle-treated Krit1PMNWT neutrophils, though this difference was reduced by PMA treatment (Fig. 3J). This suggests that KRIT1 deficient neutrophils exhibit increases in RhoA/ROCK activity similar to those observed in endothelial cells; however, this difference may not explain the reduced spreading and adhesion observed in these cells in response to PMA.
Loss of KRIT1 enables increased neutrophil migration on fibronectin
After transmigrating through the endothelium, neutrophils will migrate through the basement membrane and interstitial connective tissue toward sources of inflammation [50]. Both of these migration events require reversible adhesion to the ECM proteins via integrin cell adhesion receptors [58–60]. Reduced adhesion to ECM components has been shown to both reduce and amplify neutrophil migration based on the cellular context. Therefore, we investigated the ability of Krit1PMNKO and Krit1PMNWT neutrophils to migrate on cell culture dishes coated with either 5 μg·mL−1 ICAM-1 or 10 μg·mL−1 fibronectin. Migration was stimulated by the addition of 250 nM fMLP to the tissue culture media prior to imaging. Migratory behaviour was captured over 100 min using live-cell video microscopy. volocity software was used to quantify several migration-related parameters from individual cells isolated from n = 6/group Krit1PMNWT or Krit1PMNKO mice.
As expected based on our adhesion assay, the ability of Krit1PMNKO cells to migrate on ICAM-1 was similar to that of Krit1PMNWT neutrophils (Fig. 4A–E). However, loss of KRIT1 increased neutrophil migration on fibronectin. The distribution of total track length (distance migrated) of each cell is shown as a violin plot of cells from each animal (Figs 4A and 5A). While Krit1PMNKO neutrophils on fibronectin showed a trend toward longer track length, the overall average track length was not significantly different (Fig. 5B). However, the KRIT1 deficient neutrophils had significantly higher cell velocity (distance/time, Fig. 5C). Careful examination of these data suggest that the explanation for this discrepancy is that the Krit1PMNKO neutrophils exhibited a higher likelihood of leaving the frame of imaging (15.31% vs. 7.06%) during the time-course of migration, which lowered the average track length. In addition, Krit1PMNKO neutrophils also exhibited an increase in persistence/directionality as calculated by the meandering index (Fig. 5D) and shown by an increase in cell displacement (Fig. 5E). In sum, these data indicate that the loss of KRIT1 can increase fMLP-stimulated cell migration velocity and improves cellular persistence, likely related to the defect in neutrophil adhesion.
Fig. 4.

Krit1PMNKO neutrophils display normal migration on ICAM-1. Krit1PMNWT and Krit1PMNKO mouse neutrophils were plated in chamber wells coated with 10 μg·mL−1 ICAM-1 then stimulated with 250 nm fMLP. The migration of individual neutrophils was tracked using time-lapse video microscopy and then analysed using volocity software. (A) Violin plots of total track length per cell for each animal, representing data point frequency distribution between minimum and maximum values, with quartiles indicated by dotted lines and median indicated by a dashed line. n = 41–72 cells per mouse. (B) Average cell track length. Data shown are mean of total track length for each genotype, ±SEM, n = 6. (C) Average cell velocity (total track length/time) ± SEM, n = 6. (D) Average meandering index ± SEM, n = 6. (E) Average cell displacement (distance between origin and endpoint) ± SEM, n = 6 mice. Data were analysed by the one-way ANOVA (A) or unpaired t-test (B–E); no significant differences were detected.
Fig. 5.

Krit1PMNKO neutrophils display increased motility on fibronectin. Krit1PMNWT and Krit1PMNKO mouse neutrophils were plated in chamber wells coated with 10 μg·mL−1 fibronectin then stimulated with 250 nm fMLP. The migration of individual neutrophils was tracked using time-lapse video microscopy and then analysed using volocity software. (A) Violin plots of total track length per cell for each animal, representing data point frequency distribution between minimum and maximum values, with quartiles indicated by dotted lines, and median indicated by a dashed line. n = 24–91 cells per mouse. (B) Average cell track length. Data shown are mean of total track length for each genotype, ±SEM, n = 6. P = 0.0526 by the unpaired t-test. (C) Average cell velocity (total track length/time) ± SEM, n = 6. *P < 0.05 by the unpaired t-test. (D) Average meandering index ± SEM, n = 6. *P < 0.05 by the unpaired t-test. (E) Average cell displacement (distance between origin and endpoint) ± SEM, n = 6 mice. *P < 0.05 by the unpaired t-test.
Loss of KRIT1 reduces integrin activation
Altered neutrophil adhesion and migration on fibronectin could be driven by any of a number of changes to adhesion/migration signalling. In order to determine the level at which loss of KRIT1 leads to defects in neutrophil adhesion and migration, we first tested whether loss of KRIT1 expression led to differences in integrin activity or expression. Using an Alexa488-labelled monomeric fragment of fibronectin containing the integrin-binding RGD sequence, fibronectin 9–11 (Fn9–11) [61] we measured integrin activation in neutrophils from Krit1PMNKO and Krit1PMNWT mice using flow cytometry. Activation was calculated as an index of maximum activation (+Mn) and minimum activation (+EGTA) controls. Though neutrophils express both fibronectin receptors α5β1 and αvβ3, fibronectin-dependent migration has been shown to depend heavily on β1 integrin [62,63]. Therefore, we also measured total β1 integrin expression by FACS. KRIT1-deficient neutrophils exhibited a 53% decrease in integrin activation compared with Krit1PMNWT (Fig. 6A). There was not a significant difference in β1 integrin expression (Fig. 6B), which supports the hypothesis that KRIT1 contributes to integrin activation in neutrophils. Furthermore, we infer that the reduction in integrin activation underlies the reduction in adhesion to fibronectin and enhanced migration of KRIT1-depleted neutrophils.
Fig. 6.

Krit1PMNKO neutrophils have decreased fibronectin-binding integrin activation. (A) PMA-treated Krit1PMNWT and Krit1PMNKO neutrophils were incubated with fluorescently-labelled monomeric FN-9–11 to assess integrin activation. Data shown are (columns) average activation index ± SEM; individual data points are matched by dashed lines, n = 4. *P = 0.003 by the unpaired t-test. (B) β1 integrin expression in Krit1PMNWT and Krit1PMNKO neutrophils was analysed by flow cytometry. Data shown are average integrin expression, normalized to wildtype neutrophils, ±SEM, n = 8.
Loss of KRIT1 decreases neutrophil infiltration to lung tissue
Finally, changes in integrin activity and subsequent adhesion in our in vitro studies suggest that the loss of KRIT1 expression could affect the neutrophil response to inflammation in vivo. Therefore, we wanted to assess whether loss of KRIT1 expression altered neutrophil recruitment in vivo. While there are many models of in vivo leukocyte recruitment, we elected to use LPS-induced neutrophil recruitment into the lung as our in vivo model as it is a predominantly neutrophil-driven response. Intratracheal administration of 5 mg·kg−1 LPS in saline has been shown to stimulate neutrophil migration into the lung [64], which can be quantified following recovery of the cell-rich lung exudate by bronchoalveolar lavage (BAL). LPS or vehicle (saline) was administered by intratracheal aspiration/instillation 24 h prior to lavage. Infiltrates were collected by flushing the lungs with saline and analysed for total cell concentration (cells·mL−1). The specific presence of activated neutrophils was quantified using a myeloperoxidase activity assay. LPS treatment triggered a large increase in the presence of cells in the lavage fluid of 8-week-old mice, indicating a robust inflammatory response (Fig. 7A). The myeloperoxidase activity in the lavage fluid (a measure of neutrophil activation) was also increased by LPS treatment but was unchanged in Krit1PMNKO mice compared with Krit1PMNWT mice (Fig. 7B). Interestingly, the serendipitous assessment of a slightly older animal cohort (12-weeks) exposed a deficit in the neutrophil response in these animals. Twelve-week-old Krit1PMNKO animals had significantly fewer cells in the lavage fluid (Fig. 7C) and a significant reduction in myeloperoxidase activity compared to Krit1PMNWT mice (Fig. 7D). This result is surprising, as the difference in age between these animal cohorts is minor. Nevertheless, the neutrophil function is known to decline with age, stress levels, inflammation and frailty in humans, and our data suggest that the expression of KRIT1 could be a modifier of these processes.
Fig. 7.

Krit1PMNKO mice have reduced cell infiltration to inflamed lung tissue. Krit1PMNWT and Krit1PMNKO mice were given saline or 5 mg·kg−1 LPS by inhalation 24 h prior bronchoalveolar lavage with 2 × 1 mL lavage buffer. (A) Raw cell counts per mL of lavage fluid recovered from 8-week-old animals; ±SEM, n = 3–6. (B) Myeloperoxidase activity (U·L−1) in lavage fluid from 8-week-old animals; ±SEM, n = 3–6. (C) Raw cell counts per mL of lavage fluid recovered from 12-week-old animals; ±SEM, n = 3–6. P = 0.004 by the Brown–Forsythe ANOVA, **P < 0.01 and *P < 0.05 by the unpaired t-test with Welch’s correction. (B) Myeloperoxidase activity (U·L−1) in lavage fluid from 12-week-old animals; ±SEM, n = 3–6. P < 0.0001 by the Brown–Forsythe ANOVA, *P < 0.05 by the unpaired t-test with Welch’s correction.
Discussion
The goal of this study was to characterize the phenotype of neutrophil-specific KRIT1 knockout animals and KRIT1-deficient neutrophils. We found that loss of KRIT1 in mature neutrophils did not affect viability, fertility or normal haematopoiesis (Fig. 1), neither did the loss of KRIT1 in neutrophils affect adhesion to ICAM-1, putatively through β2 mediated integrins. However, KRIT1-deficient neutrophils did exhibit reduced adhesion to fibronectin, an important neutrophil ligand in the vascular basement membrane and interstitial space (Fig. 2). The reduction in adhesion corresponded to a reduction in cell spreading on fibronectin, in both the absence and presence of inflammatory stimuli but did not directly correlate with increased pMLC staining (indicative of increased RhoA/ROCK signalling and cellular contractility) as only unstimulated Krit1PMNKO neutrophils exhibited an increase pMLC staining vs. Krit1PMNWT cells (Fig. 3). Furthermore, KRIT1-deficient neutrophils induced to migrate on fibronectin migrated faster, and in a more directional manner, than wildtype cells (Fig. 5). This may be due to changes in integrin activation, as we observed reduced integrin activation in Krit1PMNKO as measured by flow cytometry (Fig. 6). Finally, we observed that these defects manifested as decreased neutrophil infiltration into the inflamed lung tissue in vivo, albeit only in older animals (Fig. 7).
Previous studies have largely focused on KRIT1 as an endothelial protein due to the primary role of endothelial KRIT1 (and the other CCM proteins) in the vascular disorder Cerebral Cavernous Malformations. However, several studies have already pointed to roles for nonendothelial cells in CCM formation [14–16,20,22]. Notably, we previously found that bone marrow–derived cells regulate increased endothelial permeability in Krit1 heterozygous mice, as engraftment of wildtype bone marrow restored the increased microvascular permeability in KRIT1-deficient mice to control levels [25]. As increased permeability is tightly linked to CCM development, severity and progression, this suggests that loss of KRIT1 expression in neutrophils or other bone marrow–derived cells may play a role in endothelial dysfunction and CCM disease pathogenesis. Indeed, CCM lesions from human patients regularly contain leukocytes, including B-cells/plasma cells, T-cells and neutrophils [65,66]. Recently, it was shown that neutrophils make up the most pronounced increase in immune cells in an endothelial-specific CCM3 knockout model, which was accompanied by the deposition of neutrophil extracellular traps (NETs) [67]. However, it remains unclear whether the recruitment of leukocytes is involved in CCM formation or is a general response to vascular injury. More studies will be needed to fill in such gaps in knowledge and clarify the role haematopoietic cells in CCM. This study, which suggests that KRIT1 functions in postextravasation neutrophil adhesion, which can result in improper trafficking to sites of inflammation, is the first step in that direction.
Our data also suggests that KRIT1 is a key modifier of neutrophil behaviour, a finding that could impact our understanding of the inflammatory response in general. Specifically, we show that loss of KRIT1 reduces neutrophil adhesion to fibronectin due to reduced integrin activity. Neutrophils are thought to bind to fibronectin through activation of α5β1 integrin, and to some extent αvβ3 integrin, though some have reported a role for α4β1 and even β2 integrins in neutrophil binding to fibronectin [62,63,68–71]. Future studies will closely examine the role of KRIT1 in regulating the activation of these integrin isoforms. Several potential regulatory mechanisms are supported by the literature. For example, KRIT1 binds to the small GTPase Rap1, an integrin regulator involved in the recruitment of talin-1 to the β integrin cytoplasmic tail [26,31,72]. This function has been ascribed to the interaction of Rap1 with its effector RIAM [73–75] or to direct interaction with talin [76]. Other Rap1 effectors reported to modify integrin signalling include RAPL [77,78], Mst1 [79], PKD [80], Rasip1 [81,82] and Radil [83,84], though their involvement varies depending on cell and tissue context. Though we recently reported that Rap1 binding is not required for endothelial KRIT1 to maintain barrier function [85], we speculate that KRIT1 could modify Rap1-mediated integrin activation by competing with other Rap1 effectors for binding to Rap1. On the other hand, based on data from endothelial cells, it has been proposed that Rap1 binding to KRIT1 facilitates the inactivation of RhoA/ROCK [31,51,52]. As RhoA/ROCK activity is also important for neutrophil function [53–57], that KRIT1 could also regulate RhoA in neutrophils is an attractive hypothesis. Our data suggest that KRIT1 deficient neutrophils exhibit increased baseline RhoA/ROCK activity, as shown by increased pMLC staining in vehicle-treated cells. However, PMA strongly increases pMLC levels in both Krit1PMNWT and Krit1PMNKO neutrophils and, though PMA-treated Krit1PMNKO neutrophils exhibit a trend toward higher pMLC levels, the difference between genotypes is not significant. These data suggest that KRIT1 depletion-dependent increased RhoA/ROCK activity and subsequent cell contractility could contribute to the decrease in cell spreading in vehicle-treated Krit1PMNKO cells (Fig. 3A–C) but is unlikely to underlie the decreased spreading or adhesion of PMA-treated Krit1PMNKO cells (Figs 3D–F and 2B,C). Alternatively, KRIT1 also binds to the β1 integrin inhibitory protein ICAP-1α. ICAP1α binds to KRIT1 and also was reported to limit KRIT1 interaction with microtubules [32,86]. However, ICAP1α can also bind to the cytoplasmic tail of β1 integrins [36,37], where it acts as a competitive inhibitor of talin binding [87]. In endothelial cells, studies support conflicting roles for KRIT1 in regulating ICAP1α, as KRIT1 expression has been reported to promote β1 integrin activation by sequestering ICAP1α away from the integrin [35] and to inhibit β1 integrin activation by increasing the stability of ICAP1α protein [34]. As ICAP1α is an important regulator of integrin activity and turnover [87], it is feasible that KRIT1 expression could modify this critical pathway in neutrophils.
The reduction in integrin activity and adhesion to fibronectin we observed in KRIT1-deficient neutrophils would be expected to drive changes in neutrophil migration. In fact, we did observe increased neutrophil migration speed and directionality on a fibronectin matrix in vitro. It is well established that changes in the cell migration machinery, including integrin activity, have biphasic effects on cell migratory behaviour, thus this disparity is not unexpected. However, in vivo, neutrophil recruitment into the lung was unaffected or reduced. The discrepancy between the in vitro and in vivo models is likely due to differences in the complex tissue microenvironment of the lung, which requires cells to migrate through two cellular barriers (endothelial and epithelial) and interstitial tissue in order to be collected in the lavage fluid. Additionally, while administration of LPS via aspiration is designed to induce local lung inflammation, it is possible that, given the 24 h treatment time frame, it could also stimulate some level of systemic inflammation, which could alter neutrophil phenotype in unexpected ways. More intriguing is the serendipitous finding that neutrophil infiltration is reduced in 12-week-old but not 8-week-old Krit1PMNKO mice. Aging is known to reduce neutrophil infiltration, but most studies of neutrophil aging in animal models have been performed by comparing ‘elderly’ (18–24-month-old) animals to 3-month-old ‘young adult’ controls [88,89]. While we were unable to find precedent for differences in the neutrophil function between juvenile and adult mice, the development of the immune system is still ongoing at 8 weeks of age [90]. Indeed, in models of juvenile asthma, differences in immune cell infiltration and other inflammatory responses have been observed between 100-, 40- and 18-day-old mice [91]. Thus, our data, while demonstrating that loss of KRIT1 can affect neutrophil function in vivo, could also point to interesting developmental changes in the mouse lung or in the neutrophil function that could be explored in future work.
In conclusion, we have developed a new mouse model, which demonstrates that KRIT1 regulates neutrophil adhesion and migration through integrin activation and that this affects immune responses in the host organism. While much remains to be done in order to understand the underlying mechanism by which KRIT1 regulates neutrophil adhesion, this is an important step toward understanding how KRIT1 expression in immune cells could impact the response to inflammation. Further study of KRIT1’s role in neutrophils will enhance our understanding of its impact on CCM disease and more.
Materials and methods
Mouse models
Mice with a conditional (floxed) allele for Krit1, a germline deleted Krit1 allele, and the constitutively active neutrophil Cre recombinase (Ly6Gtm2621(cre)Arte) have been described [48,49]. The presence of the Ly6G-Cre transgene, the conditional Krit1 allele and recombination at the conditional Krit1 allele were validated by PCR (Ly6G-WT: F-GGTTTTATCTGTGCAGCCC, R-GAGGTCCAAGAGACTTTCTGG; Ly6G-Cre: F-ACGTCCAGACACAGCATAGG, R-GAGGTCCAAGAGACTTTCTGG; Krit1 D: F-TACACCAGGCTACTTGGCTTCAC, R-AAACCAGCAGTCTCAACTAATCGG). As the Ly6G-Cre transgene only weakly drives Cre-expression, heterozygous Ly6G-Cre animals did not exhibit significant loss of KRIT1 expression (data not shown). Therefore, it was necessary to breed mice homozygous for both the floxed Krit1 allele and the Ly6G-Cre recombinase (Krit1PMNKO). Cre-negative mice containing homozygous floxed Krit1 gene alleles (Krit1PMNWT) were used as controls and were either littermates derived from a Ly6G+/−/Krit1fl/fl × Ly6G+/−/Krit1fl/fl cross or from Ly6G−/−/Krit1fl/fl breeding pairs. No differences in phenotype were observed between littermate and nonlittermate controls, therefore these data are combined in our analyses. Mice were bred and maintained under standard conditions in the University of Rochester animal facilities, which are accredited by the American Association for Accreditation of Laboratory Animal Care. All protocols were approved by the institutional review board.
Cell isolation
Blood was collected retro-orbitally for complete blood count (CBC) analysis using an Abaxis (Union City, CA, USA) VetScan HM5. In brief, mice between 8 and 10 weeks of age were anaesthetised, and a micro-capillary tube was inserted into the venous retro-orbital sinus behind the eye. Blood was collected by applying light but firm pressure to penetrate the vessels. Collected blood was heparinized and transferred into a cuvette for CBC analysis.
Neutrophils were isolated from the bone marrow of 8- to 10-week-old animals using a Mouse Neutrophil Enrichment Kit (StemCell Technologies, Vancouver, BC, Canada). In brief, bone marrow from the femurs and tibia was flushed out with saline and then resuspended in EasySep buffer (Hanks’ Balanced Salt Solution (HBSS) w/o Ca2+ or Mg2+, 2% fetal bovine serum (FBS), 1 mm ethylenediaminete-traacetic acid (EDTA)). The cells were then incubated with an antibody cocktail for negative selection, followed by a cocktail of biotinylated secondary antibodies, and then incubated with streptavidin-coated magnetic beads. This cell suspension was exposed to a magnet, which removed all non-neutrophil cells. The purity of the remaining cell population was measured by FACS analysis of Ly6G expression (wildtype cells) or tdTomato expression (knockout cells, Fig. 1A) and consistently exceeded 80%. Following isolation, neutrophils were resuspended in L-15 media (Gibco Leibovitz L-15 phenol-free medium, 2 mg·mL−1 glucose) and used for experiments within 1 h.
Antibodies
Polyclonal rabbit anti-KRIT1 (Abcam, Cambridge, UK) was used at a dilution of 1 : 1000 for western blotting. Horseradish peroxidase (HRP)-linked donkey anti-rabbit IgG secondary antibody (Cell Signalling Technology, Danvers, MA, USA) was used at a dilution of 1 : 1000. Monoclonal rabbit anti-phospho-myosin light chain 2 (Cell Signalling Technology) and goat anti-rabbit IgG, Alexa Fluor 568 (Invitrogen, Waltham, MA, USA) were used at a dilution of 1 : 500 for immunocytochemistry. BV786 Rat anti-Mouse Ly6G and Ly6C (BD Biosciences, Franklin Lakes, NJ, USA), Allophycocyanin (APC) Rat anti-Mouse Ly6G (BD Biosciences) and/or APC Rat anti-Mouse Ly6C (BD Biosciences) antibodies were used at a concentration of 200 μg·mL−1, and/or FITC Rat anti-Mouse CD4 (BD Biosciences) antibody was used at a concentration of 500 μg·mL−1 to stain mouse neutrophils prior to FACS analysis. Armenian hamster anti-mouse CD29 (BD Biosciences) and FITC-goat anti-hamster IgG (ThermoFisher Scientific, Waltham, MA, USA) were used at 500 μg·mL−1 to stain mouse neutrophils before FACS analysis.
RNA isolation and semi-quantitative RT-PCR
RNA was isolated using Trizol (Life Technologies, Waltham, MA, USA) extraction according to the manufacturer’s instructions. Complementary DNA was obtained using Lunascript RT Supermix (New England BioLabs, Ipswich, MA, USA) and oligo dT primers. Amplifications were run on a 7000 real-time PCR system (Applied Biosystems/ThermoFisher) using Taqman gene expression assays (Krit1, Mm00459502; Gapdh, Mm99999915). Each value was calculated using the comparative Ct method and normalized to Gapdh internal control. All samples were run in at least triplicate.
Western blotting
Neutrophil lysates were prepared by lysing cells in lysis buffer (50 mmol·L−1 Tris pH 7.4, 150 mmol·L−1 NaCl, 0.5% NP-40, 5 mmol·L−1 MgCl2) containing complete mini EdTA-free protease inhibitor tablet (Roche, Basel, Switzerland) and phosphatase inhibitor cocktail (final concentration 100 μm Na4P2O7, 1mm NaF, 1 mm Na3VO4, 300 μm β-glycerophosphate, 2 mm imidazole, 4 mm sodium tartrate). Protein quantification was performed using a bicinchoninic acid assay kit (Thermo Fisher), and equivalent amounts of total cell protein were separated by SDS/PAGE on precast 7.5% Tris–HCl gels (BioRad, Hercules, CA, USA). Following transfer to 0.45 μm nitrocellulose, blots were blocked in 5% nonfat dry milk (NFDM) in 1× TBS plus 0.1% Tween 20 (TBST). Primary rabbit anti-KRIT1 antibody (Abcam) was diluted in TBST plus 5% NFDM and the membrane was incubated in primary antibody overnight at 4 °C. The membrane was then incubated with anti-rabbit-HRP secondary antibody for 1 h at room temperature before detection using SuperSignal West Femto enhanced chemiluminescent substrate (ThermoFisher Scientific). Densitometry was performed using a BioRad ChemiDoc Imaging System to quantify band intensity.
Neutrophil adhesion, spreading and migration assays
To assess the ability of neutrophils to adhere to different substrates, glass-bottom 96-well plates were coated with 20 μg·mL−1 recombinant Protein A (ThermoFisher Scientific) overnight at 4 °C then at room temperature for 2 h followed by 5 μg·mL−1 recombinant ICAM-1 (R&D Systems, Minneapolis, MN, USA) for 2 h at room temperature [92] or coated with 10 μg·mL−1 human fibronectin (isolated from human plasma) for 2 h at 37 °C [93]. Prior to the adhesion assay, neutrophils were loaded with calcein-AM (1 μg·mL−1, Invitrogen) at 37 °C for 15 min prior to treatment with 10 nm PMA [94,95], 250 nm fMLP [96], 1 μm WKYMVm [97] or vehicle (phosphate-buffered saline, PBS). The cells were then plated onto the precoated plate and allowed to adhere for the times indicated. At the end of the adhesion period, the plates were rinsed twice with 1× PBS to wash off any nonadherent neutrophils and then covered with 100 μL 1× PBS. Blank wells were prepared by adding 100 μL 1× PBS to wells that had been coated but received no cells; positive control wells were prepared by adding 3 × 104 calcein-AM-treated neutrophils (suspended in 100 μL 1× PBS) to control wells immediately prior to measurement of calcein-AM fluorescence. The fluorescence of the labelled cells was read at excitation/emission wavelengths of 485/528 nm using a BioTek (Winooski, VT, USA) Synergy H4 plate reader.
To quantify cell spreading and phosphorylation of MLC, glass coverslips were coated with 10 μg·mL−1 fibronectin for 2 h at 37 °C. Neutrophils were then plated on the coated coverslips and immediately treated with 10 nm PMA, 250 nm fMLP, 1 μm WKYMVm, 1 μg·mL−1 LPS [98] or vehicle (PBS) and allowed to adhere and spread for 1 h. Cells were then fixed with 10% formalin (Fisher) and permeabilized with 0.2% Triton-X-100 (Research Products International, Mount Prospect, IL, USA) before staining with FITC-phalloidin (Enzo, New York, NY, USA) or primary rabbit anti-pMLC antibody followed by Alexa Fluor 568 goat anti-rabbit IgG. Cells were then counterstained with Hoechst 33258 before the coverslips were mounted on slides using Prolong Gold antifade reagent (Invitrogen). Phalloidin-stained neutrophil images were captured using an Olympus IX81 microscope using a 40×/0.60 Ph2 objective and a Hamamatsu (Hamamatsu City, Japan) ORCA-ER camera. Cell area was quantified by dividing the FITC-phalloidin fluorescent area by the total number of cells to get the average cell area per field using metamorph software (Metamorph Inc, Nashville, TN, USA). pMLC antibody-stained neutrophil images were captured using a Stellaris 5 laser scanning confocal microscope (Leica, Wetzlar, Germany) using a 63×/1.4 n.a. oil immersion objective. Individual cell pMLC staining intensity was obtained using imaris image visualization & analysis Software (Oxford Instruments, Abingdon, UK).
Neutrophil migration was quantified in cells plated on 20 μg·mL−1 recombinant Protein A plus 5 μg·mL−1 ICAM-1, or 10 μg·mL−1 fibronectin-coated chamber wells (Nunc, Rochester, NY, USA). Isolated neutrophils were added and allowed to adhere for 20 min in the presence of 250 nm fMLP before the media was changed to remove nonadherent cells (refreshed media still contained 250 nm fMLP). Cells were then imaged on an IX70 inverted microscope (Olympus, Tokyo, Japan) with a 10×/0.40 Ph1 objective. Digital images were acquired every 30 s for up to 100 min using a digital camera (MicroPublisher 3.3, QImaging, Sarasota, FL, USA). volocity software (Quorum Technologies, Lewes, UK) was used to track individual cells across the image sets and analyse the data.
Bronchoalveolar lavage
Bronchoalveolar lavage was performed as previously described [64,99]. In brief, 24 h prior to lavage, 5 mg·kg−1 LPS (Sigma, Burlington, MA, USA) was intratracheally administered to Krit1PMNWT and Krit1PMNKO mice. Prior to lavage, mice were euthanized with 100 mg sodium pentobarbital (Sleepaway: Euthasol Euthanasia Solution, Virbac, Carros, France). The trachea was exposed, and a catheter was introduced into the trachea to allow for the introduction and removal of lavage fluid. The lungs were rinsed twice with 1 mL 1× PBS containing 0.1 mm EDTA and the lavage fluid combined. The concentration of cells in the lavage fluid was measured using an Invitrogen Countess auto cell counter. Analysis of myeloperoxidase activity in 20 μL of lavage fluid was done using an EnzyFluo assay kit (BioAssay Systems, Hayward, CA, USA) and quantified on a BioTek Synergy H4 plate reader.
Flow cytometry
Blood, collected retro-orbitally, was resuspended in 1× PBS with heparin and centrifuged at 300 × g for 10 min to collect the buffy coat. Buffy coat and whole bone marrow, collected by flushing the femur and tibia, were resuspended in HBSS without calcium and magnesium plus 20 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 5% FBS and 1 mm EDTA. Approximately 5 × 105 buffy coat or bone marrow cells were incubated with BV786 Rat anti-Mouse Ly6G and Ly6C (BD Biosciences), APC Rat anti-Mouse Ly6G (BD Biosciences), APC Rat anti-Mouse Ly6C (BD Biosciences) and/or FITC Rat anti-Mouse CD4 (BD Biosciences) antibodies. FACS was performed on an LSRII flow cytometer, and data were analysed using flojo software (FloJo, Ashland, OR, USA).
To measure integrin activity, neutrophils isolated from Krit1PMNWT and Krit1PMNKO mice were resuspended in Tyrode buffer (100 mm HEPES with 170 mm NaCl, 12 mm NaHCO3, 2.6 mm KCl, 1.5 mL MgCl, 0.5 mm CaCl2, 1 mg·mL−1 dextrose and 1 mg·mL−1 bovine serum albumin) and 5 × 106 cells per condition were aliquoted into polystyrene tubes. Individual aliquots were treated with 10 mm EDTA, 1 mm MnCl2, 10 nm PMA or vehicle for 10 min prior to the addition of Alexa488-labelled Fn9-11, generated as previously described [61]. Integrin expression was measured by staining Krit1PMNWT and Krit1PMNKO neutrophils with Armenian hamster anti-mouse CD29 (BD Biosciences) followed by FITC-goat anti-hamster IgG (ThermoFisher Scientific). Flow cytometry was performed on an LSRII flow cytometer, and data were analysed using flojo. Activation index was calculated as described in Konstandin et al. [100].
Statistics
Statistical analysis (i.e. one-way ANOVA with appropriate post hoc testing) was performed using prism software (version 9.0, GraphPad Software Inc, San Diego, CA, USA). Statistical significance was assessed assuming a 0.05 significance level and a two-sided alternative hypothesis.
Acknowledgements
Ly6Gtm2621(cre)Arte was a generous gift from Matthias Gunzer, Institute of Experimental Immunology & Imaging, Center of Medical Biotechnology (ZMB), University of Duisburg-Essen. The authors would like to acknowledge the technical assistance of the URMC Flow Cytometry Shared Resource, the URMC Center for Advanced Light Microscopy and Nanoscopy, and Dr. Denise Hocking and Dr. David Dean (University of Rochester Medical Center) for the use of their equipment. This work was supported by grants from the National Institutes of Health (HL141131 and HD098576 to AJG).
Abbreviations
- ICAM-1
intercellular adhesion molecule-1
- KRIT1
Krev interaction-trapped-1
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16627.
Data availability statement
All data and materials from this work are available upon reasonable request.
References
- 1.Chan AC, Li DY, Berg MJ, Whitehead KJ. Recent insights into cerebral cavernous malformations: animal models of CCM and the human phenotype. FEBS J. 2010;277:1076–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. 2001;71:188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Revencu N, Vikkula M. Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet. 2006;43:716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15–13 and 3q25.2–27. Hum Mol Genet. 1998;7:1851–8. [DOI] [PubMed] [Google Scholar]
- 5.Dubovsky J, Zabramski JM, Kurth J, Spetzler RF, Rich SS, Orr HT, et al. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet. 1995;4:453–8. [DOI] [PubMed] [Google Scholar]
- 6.Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. Genetics of cavernous angiomas. Lancet Neurol. 2007;6:237–44. [DOI] [PubMed] [Google Scholar]
- 7.Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, et al. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet. 2007;80:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343–55. [DOI] [PubMed] [Google Scholar]
- 9.Shenkar R, Sarin H, Awadallah NA, Gault J, Kleinschmidt-DeMasters BK, Awad IA. Variations in structural protein expression and endothelial cell proliferation in relation to clinical manifestations of cerebral cavernous malformations. Neurosurgery. 2005;56:343–54. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Wu Q, Fass M, Xu JF, You C, Muller O, et al. In vitro characterization of the angiogenic phenotype and genotype of the endothelia derived from sporadic cerebral cavernous malformations. Neurosurgery. 2011;69:722–31. discussion 731–2. [DOI] [PubMed] [Google Scholar]
- 11.Antognelli C, Trapani E, Delle Monache S, Perrelli A, Daga M, Pizzimenti S, et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic Biol Med. 2018;115:202–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goitre L, Balzac F, Degani S, Degan P, Marchi S, Pinton P, et al. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE. 2010;5:e11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kehrer-Sawatzki H, Wilda M, Braun VM, Richter HP, Hameister H. Mutation and expression analysis of the KRIT1 gene associated with cerebral cavernous malformations (CCM1). Acta Neuropathol. 2002;104:231–40. [DOI] [PubMed] [Google Scholar]
- 14.Glading AJ, Ginsberg MH. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis Model Mech. 2010;3:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Li Y, Zou J, Polster SP, Lightle R, Moore T, et al. The cerebral cavernous malformation disease causing gene KRIT1 participates in intestinal epithelial barrier maintenance and regulation. FASEB J. 2019;33:2132–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang AT, Sullivan KR, Hong CC, Goddard LM, Mahadevan A, Ren A, et al. Distinct cellular roles for PDCD10 define a gut-brain axis in cerebral cavernous malformation. Sci Transl Med. 2019;11:eaaw3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donat S, Lourenco M, Paolini A, Otten C, Renz M, Abdelilah-Seyfried S. Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. Elife. 2018;7:e28939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogan BM, Bussmann J, Wolburg H, Schulte-Merker S. ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet. 2008;17:2424–32. [DOI] [PubMed] [Google Scholar]
- 19.Rosen JN, Sogah VM, Ye LY, Mably JD. ccm2-like is required for cardiovascular development as a novel component of the Heg-CCM pathway. Dev Biol. 2013;376:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finetti F, Schiavo I, Ercoli J, Zotta A, Boda E, Retta SF, et al. KRIT1 loss-mediated upregulation of NOX1 in stromal cells promotes paracrine pro-angiogenic responses. Cell Signal. 2020;68:109527. [DOI] [PubMed] [Google Scholar]
- 21.Goitre L, DiStefano PV, Moglia A, Nobiletti N, Baldini E, Trabalzini L, et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci Rep. 2017;7:8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louvi A, Chen L, Two AM, Zhang H, Min W, Gunel M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci USA. 2011;108:3737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Ramirez MA, Lai CC, Soliman SI, Hale P, Pham A, Estrada EJ, et al. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J Clin Invest. 2021;131:e139570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulz GB, Wieland E, Wustehube-Lausch J, Boulday G, Moll I, Tournier-Lasserve E, et al. Cerebral cavernous malformation-1 protein controls DLL4-Notch3 signaling between the endothelium and pericytes. Stroke. 2015;46:1337–43. [DOI] [PubMed] [Google Scholar]
- 25.Corr M, Lerman I, Keubel JM, Ronacher L, Misra R, Lund F, et al. Decreased Krev interaction-trapped 1 expression leads to increased vascular permeability and modifies inflammatory responses in vivo. Arterioscler Thromb Vasc Biol. 2012;32:2702–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bromberger T, Klapproth S, Rohwedder I, Zhu L, Mittmann L, Reichel CA, et al. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood. 2018;132:2754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latasiewicz J, Artz A, Jing D, Blanco MP, Currie SM, Avila MV, et al. HS1 deficiency impairs neutrophil recruitment in vivo and activation of the small GTPases Rac1 and Rap1. J Leukoc Biol. 2017;101:1133–42. [DOI] [PubMed] [Google Scholar]
- 28.Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung KJ, Mitroulis I, Wiessner JR, Zheng YY, Siegert G, Sperandio M, et al. A novel pathway of rapid TLR-triggered activation of integrin-dependent leukocyte adhesion that requires Rap1 GTPase. Mol Biol Cell. 2014;25:2948–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Schwartz BR, Tupper J, Lin N, Winn RK, Harlan JM. The GTPase Rap1 regulates phorbol 12-myristate 13-acetate-stimulated but not ligand-induced beta 1 integrin-dependent leukocyte adhesion. J Biol Chem. 2002;277:40893–900. [DOI] [PubMed] [Google Scholar]
- 31.Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beraud-Dufour S, Gautier R, Albiges-Rizo C, Chardin P, Faurobert E. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 2007;274:5518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521–31. [DOI] [PubMed] [Google Scholar]
- 34.Faurobert E, Rome C, Lisowska J, Manet-Dupe S, Boulday G, Malbouyres M, et al. CCM1-ICAP-1 complex controls beta1 integrin-dependent endothelial contractility and fibronectin remodeling. J Cell Biol. 2013;202:545–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Draheim KM, Zhang R, Calderwood DA, Boggon TJ. Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol Cell. 2013;49:719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001;10:2953–60. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Basu S, Rigamonti D, Dietz HC, Clatterbuck RE. Krit1 modulates beta 1-integrin-mediated endothelial cell proliferation. Neurosurgery. 2008;63:571–8. discussion 578. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Tang W, Zhang H, Niu X, Xu Y, Zhang J, et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev Cell. 2013;27:215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Padarti A, Qu Y, Sheng S, Abou-Fadel J, Badr A, et al. Alternatively spliced isoforms reveal a novel type of PTB domain in CCM2 protein. Sci Rep. 2019;9:15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Zhang R, Zhang H, He Y, Ji W, Min W, et al. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J Biol Chem. 2010;285:24099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Padarti A, Jiang X, Abou-Fadel J. Redefining PTB domain into independently functional dual cores. Biochem Biophys Res Commun. 2020;524:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Day RB, Link DC. Regulation of neutrophil trafficking from the bone marrow. Cell Mol Life Sci. 2012;69:1415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eash KJ, Means JM, White DW, Link DC. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood. 2009;113:4711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–93. [DOI] [PubMed] [Google Scholar]
- 45.Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004;104:565–71. [DOI] [PubMed] [Google Scholar]
- 46.Klinker JF, Wenzel-Seifert K, Seifert R. G-protein-coupled receptors in HL-60 human leukemia cells. Gen Pharmacol. 1996;27:33–54. [DOI] [PubMed] [Google Scholar]
- 47.Saunders CA, Majumdar R, Molina Y, Subramanian BC, Parent CA. Genetic manipulation of PLB-985 cells and quantification of chemotaxis using the underagarose assay. Methods Cell Biol. 2019;149:31–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mleynek TM, Chan AC, Redd M, Gibson CC, Davis CT, Shi DS, et al. Lack of CCM1 induces hypersprouting and impairs response to flow. Hum Mol Genet. 2014;23:6223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. 2015;12:445–52. [DOI] [PubMed] [Google Scholar]
- 50.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17:413–23. [DOI] [PubMed] [Google Scholar]
- 51.Liu JJ, Stockton RA, Gingras AR, Ablooglu AJ, Han J, Bobkov AA, et al. A mechanism of Rap1-induced stabilization of endothelial cell–cell junctions. Mol Biol Cell. 2011;22:2509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207:881–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francis SA, Shen X, Young JB, Kaul P, Lerner DJ. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood. 2006;107:1627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galvao I, Athayde RM, Perez DA, Reis AC, Rezende L, de Oliveira VLS, et al. ROCK inhibition drives resolution of acute inflammation by enhancing neutrophil apoptosis. Cells. 2019;8:964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Itakura A, Aslan JE, Kusanto BT, Phillips KG, Porter JE, Newton PK, et al. p21-Activated kinase (PAK) regulates cytoskeletal reorganization and directional migration in human neutrophils. PLoS ONE. 2013;8:e73063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li M, Lyu X, Liao J, Werth VP, Liu ML. Rho Kinase regulates neutrophil NET formation that is involved in UVB-induced skin inflammation. Theranostics. 2022;12:2133–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yagi Y, Otani H, Ando S, Oshiro A, Kawai K, Nishikawa H, et al. Involvement of Rho signaling in PAR2-mediated regulation of neutrophil adhesion to lung epithelial cells. Eur J Pharmacol. 2006;536:19–27. [DOI] [PubMed] [Google Scholar]
- 58.Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17:638–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lerman YV, Kim M. Neutrophil migration under normal and sepsis conditions. Cardiovasc Hematol Disord Drug Targets. 2015;15:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDowall A, Inwald D, Leitinger B, Jones A, Liesner R, Klein N, et al. A novel form of integrin dysfunction involving beta1, beta2, and beta3 integrins. J Clin Invest. 2003;111:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramos JW, DeSimone DW. Xenopus embryonic cell adhesion to fibronectin: position-specific activation of RGD/synergy site-dependent migratory behavior at gastrulation. J Cell Biol. 1996;134:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blystone SD, Graham IL, Lindberg FP, Brown EJ. Integrin alpha v beta 3 differentially regulates adhesive and phagocytic functions of the fibronectin receptor alpha 5 beta 1. J Cell Biol. 1994;127:1129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindbom L, Werr J. Integrin-dependent neutrophil migration in extravascular tissue. Semin Immunol. 2002;14:115–21. [DOI] [PubMed] [Google Scholar]
- 64.Mutlu GM, Machado-Aranda D, Norton JE, Bellmeyer A, Urich D, Zhou R, et al. Electroporation-mediated Gene Transfer of the Na+,K+-ATPase Rescues Endotoxin-induced Lung Injury. Am J Respir Crit Care Med. 2007;176:582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagiwara K, Khaskhely NM, Uezato H, Nonaka S. Mast cell “densities” in vascular proliferations: a preliminary study of pyogenic granuloma, portwine stain, cavernous hemangioma, cherry angioma, Kaposi’s sarcoma, and malignant hemangioendothelioma. J Dermatol. 1999;26:577–86. [DOI] [PubMed] [Google Scholar]
- 66.Shi C, Shenkar R, Du H, Duckworth E, Raja H, Batjer HH, et al. Immune response in human cerebral cavernous malformations. Stroke. 2009;40:1659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yau ACY, Globisch MA, Onyeogaziri FC, Conze LL, Smith R, Jauhiainen S, et al. Inflammation and neutrophil extracellular traps in cerebral cavernous malformation. Cell Mol Life Sci. 2022;79:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anceriz N, Vandal K, Tessier PA. S100A9 mediates neutrophil adhesion to fibronectin through activation of beta2 integrins. Biochem Biophys Res Commun. 2007;354:84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim HY, Skokos EA, Myer DJ, Agaba P, Gonzalez AL. alphaVbeta3 integrin regulation of respiratory burst in fibrinogen adherent human neutrophils. Cell Mol Bioeng. 2014;7:231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nair KS, Zingde SM. Adhesion of neutrophils to fibronectin: role of the cd66 antigens. Cell Immunol. 2001;208:96–106. [DOI] [PubMed] [Google Scholar]
- 71.van den Berg JM, Mul FP, Schippers E, Weening JJ, Roos D, Kuijpers TW. Beta1 integrin activation on human neutrophils promotes beta2 integrin-mediated adhesion to fibronectin. Eur J Immunol. 2001;31:276–84. [DOI] [PubMed] [Google Scholar]
- 72.Bos JL, de Bruyn K, Enserink J, Kuiperij B, Rangarajan S, Rehmann H, et al. The role of Rap1 in integrin-mediated cell adhesion. Biochem Soc Trans. 2003;31:83–6. [DOI] [PubMed] [Google Scholar]
- 73.Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol. 2006;16:1796–806. [DOI] [PubMed] [Google Scholar]
- 74.Lagarrigue F, Kim C, Ginsberg MH. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood. 2016;128:479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmid MC, Franco I, Kang SW, Hirsch E, Quilliam LA, Varner JA. PI3-kinase gamma promotes Rap1amediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS ONE. 2013;8:e60226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lagarrigue F, Paul DS, Gingras AR, Valadez AJ, Sun H, Lin J, et al. Talin-1 is the principal platelet Rap1 effector of integrin activation. Blood. 2020;136:1180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kinashi T, Katagiri K. Regulation of lymphocyte adhesion and migration by the small GTPase Rap1 and its effector molecule, RAPL. Immunol Lett. 2004;93:1–5. [DOI] [PubMed] [Google Scholar]
- 78.Miertzschke M, Stanley P, Bunney TD, Rodrigues-Lima F, Hogg N, Katan M. Characterization of interactions of adapter protein RAPL/Nore1B with RAP GTPases and their role in T cell migration. J Biol Chem. 2007;282:30629–42. [DOI] [PubMed] [Google Scholar]
- 79.Galan JA, Avruch J. MST1/MST2 protein kinases: regulation and physiologic roles. Biochemistry. 2016;55:5507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dusaban SS, Kunkel MT, Smrcka AV, Brown JH. Thrombin promotes sustained signaling and inflammatory gene expression through the CDC25 and Ras-associating domains of phospholipase C. J Biol Chem. 2015;290:26776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barry DM, Koo Y, Norden PR, Wylie LA, Xu K, Wichaidit C, et al. Rasip1-mediated rho GTPase signaling regulates blood vessel tubulogenesis via nonmuscle myosin II. Circ Res. 2016;119:810–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Norden PR, Kim DJ, Barry DM, Cleaver OB, Davis GE. Cdc42 and k-Ras control endothelial tubulogenesis through apical membrane and cytoskeletal polarization: novel stimulatory roles for GTPase effectors, the small GTPases, Rac2 and Rap1b, and inhibitory influence of Arhgap31 and Rasa1. PLoS ONE. 2016;11:e0147758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu L, Aerbajinai W, Ahmed SM, Rodgers GP, Angers S, Parent CA. Radil controls neutrophil adhesion and motility through beta2-integrin activation. Mol Biol Cell. 2012;23:4751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.To JY, Smrcka AV. Activated heterotrimeric G protein alphai subunits inhibit Rap-dependent cell adhesion and promote cell migration. J Biol Chem. 2018;293:1570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Swamy H, Glading AJ. Contribution of protein-protein interactions to the endothelial-barrier-stabilizing function of KRIT1. J Cell Sci. 2022;135:jcs258816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Draheim KM, Huet-Calderwood C, Simon B, Calderwood DA. Nuclear localization of integrin cytoplasmic domain-associated protein-1 (ICAP1) influences beta1 integrin activation and recruits Krev/interaction trapped-1 (KRIT1) to the nucleus. J Biol Chem. 2017;292:1884–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Millon-Fremillon A, Bouvard D, Grichine A, Manet-Dupe S, Block MR, Albiges-Rizo C. Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent beta1-integrin affinity. J Cell Biol. 2008;180:427–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lord JM, Butcher S, Killampali V, Lascelles D, Salmon M. Neutrophil ageing and immunesenescence. Mech Ageing Dev. 2001;122:1521–35. [DOI] [PubMed] [Google Scholar]
- 89.Simmons SR, Bhalla M, Herring SE, Tchalla EYI, Bou Ghanem EN. Older but Not Wiser: the age-driven changes in neutrophil responses during pulmonary infections. Infect Immun. 2021;89:e00653–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jackson SJ, Andrews N, Ball D, Bellantuono I, Gray J, Hachoumi L, et al. Does age matter? The impact of rodent age on study outcomes. Lab Anim. 2017;51:160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carnieli DS, Yoshioka E, Silva LF, Lancas T, Arantes FM, Perini A, et al. Inflammation and remodeling in infantile, juvenile, and adult allergic sensitized mice. Pediatr Pulmonol. 2011;46:650–65. [DOI] [PubMed] [Google Scholar]
- 92.Lim K, Hyun YM, Lambert-Emo K, Topham DJ, Kim M. Visualization of integrin Mac-1 in vivo. J Immunol Methods. 2015;426:120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Silveira AAA, Dominical VM, Almeida CB, Chweih H, Ferreira WA Jr, Vicente CP, et al. TNF induces neutrophil adhesion via formin-dependent cytoskeletal reorganization and activation of beta-integrin function. J Leukoc Biol. 2018;103:87–98. [DOI] [PubMed] [Google Scholar]
- 94.Hendey B, Zhu CL, Greenstein S. Fas activation opposes PMA-stimulated changes in the localization of PKCdelta: a mechanism for reducing neutrophil adhesion to endothelial cells. J Leukoc Biol. 2002;71:863–70. [PubMed] [Google Scholar]
- 95.Kamoshida G, Kikuchi-Ueda T, Nishida S, Tansho-Nagakawa S, Ubagai T, Ono Y. Pathogenic bacterium Acinetobacter baumannii inhibits the formation of neutrophil extracellular traps by suppressing neutrophil adhesion. Front Immunol. 2018;9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pellegatta F, Radaelli A, Heltai S, Yan L, Chierchia SL, Folli F. Evidence for the involvement of phosphatidylinositol 3-kinase in fMLP-stimulated neutrophil adhesion to ICAM-1-transfected cells. J Cardiovasc Pharmacol. 2001;37:751–61. [DOI] [PubMed] [Google Scholar]
- 97.Itou T, Collins LV, Thoren FB, Dahlgren C, Karlsson A. Changes in activation states of murine polymorphonuclear leukocytes (PMN) during inflammation: a comparison of bone marrow and peritoneal exudate PMN. Clin Vaccine Immunol. 2006;13:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maruyama N, Tansho-Nagakawa S, Miyazaki C, Shimomura K, Ono Y, Abe S. Inhibition of neutrophil adhesion and antimicrobial activity by diluted hydrosol prepared from Rosa damascena. Biol Pharm Bull. 2017;40:161–8. [DOI] [PubMed] [Google Scholar]
- 99.Young JC, Chehoud C, Bittinger K, Bailey A, Diamond JM, Cantu E, et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am J Transplant. 2015;15:200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Konstandin MH, Sester U, Klemke M, Weschenfelder T, Wabnitz GH, Samstag Y. A novel flow-cytometry-based assay for quantification of affinity and avidity changes of integrins. J Immunol Methods. 2006;310:67–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and materials from this work are available upon reasonable request.