

Abstract
Accuracy of warfarin dose prediction algorithms may be improved by including data from diverse populations in genetic studies of dose variability. Here, we surveyed single nucleotide polymorphisms in vitamin K-related genes for association with warfarin dose requirements in two admixed Latino populations in standard-principal component adjusted and contemporary-local ancestry adjusted regression models. A total of 5 variants from vitamin K-related genes/pathways were associated with warfarin dose in both cohorts (p<0.0125) in standard models. Local ancestry-adjusted analysis unveiled 35 associated variants with absolute effects ranging from β = 9.04 (±2.23) to 39.18 (±10.89) per ancestral allele in the discovery cohort and β = 6.47 (±2.02) to 17.82 (±6.83) in the replication cohort. Importantly, we demonstrate the technical validity of the Tractor model in cohorts with admixed ancestry from three founder populations and bring attention to the technical hurdles obstructing the inclusion of diverse, especially admixed, populations in pharmacogenomic research.
Keywords: precision medicine, latino/a/e/x, local ancestry, GWAS
Introduction
The anticoagulant drug, warfarin, although commonly used worldwide, continues to result in a high frequency of adverse effects. To better predict a warfarin user’s therapeutic window, dose prediction algorithms have been developed based on genetic, demographic, and clinical variables. The more potent S-isomer of warfarin is primarily metabolized by CYP2C9 and genetic variants that reduce CYP2C9 function are known to result in reduced warfarin dose requirements. Similarly, warfarin’s mechanism of action is inhibition of vitamin K epoxide reductase (VKORC1) and genetic variants that increase abundance of VKORC1 protein result in reduced warfarin dose requirements. Derivation of pharmacogenomic dose prediction algorithms, which currently predict no more than 60% of the variability in warfarin stable dose; even in homogenous, well-represented-in-research populations1; may benefit from the inclusion of previously overlooked populations, such as many Latino populations2. Pharmacogenetics (PGx) research suffers from biases created by exclusion of non-European populations3,4. Furthermore, recent studies have shown increased representation of ancestrally diverse populations is beneficial for discovering novel loci and addressing health disparities5–7. This inclusion bias has the potential to widen already existing health disparities during downstream algorithm creation and clinical implementation. It is therefore critically important to undertake genetic studies in underrepresented populations, including Latino populations, so that benefits of optimal pharmacogenomic dosing in historically excluded individuals can be appropriately examined. Ultimately this work encourages equal opportunity to benefit from warfarin PGx dosing.
Beyond a staggering underrepresentation in genomics studies at large, a statistical hurdle prevents increased representation even when Latino participants enroll to a genomics study8. Study participants that do not match closely with widely used reference genomes are often excluded prior to downstream statistical analysis to decrease risk of false positive associations with population rather than phenotype9. Although it is possible that persons identifying as Latino are largely of European descent, due to the unique colonialist history of Latin America, individuals identifying as Latino likely have admixed ancestry of European and Native American or African ancestors. Diverse populations increase power to detect SNP associations on a surface level by increasing the pool of possible participants in research studies. Diverse populations also allow researchers to glean information based on the varying relatedness of individual genomes. Traditional mechanisms to account for population level genetic variation during statistical testing include adjusting for principal components (i.e. measures of dimensionality) in the regression model, however, this method is statistically expensive and largely an artifact of the data used to generate the components10. Innovative, methods, such as Tractor, that leverage local ancestry, or chromosome-level ancestry, are being developed to encourage the inclusion of diverse populations in genomic analyses11–13. Leveraging individual level ancestries of mosaic populations, we incorporate local ancestry information into our statistical model to facilitate the inclusion of admixed participants of a diverse population in our association analysis with consideration for population stratification.
Warfarin anticoagulation functions by inhibiting the vitamin K cycle of the host to reduce blood clotting ability. The required vitamin K hydroquinone is then lacking for carboxylation and consequent activation of vitamin K-dependent proteins within the clotting cycle. Because of this pathway, vitamin K is also commonly used as a reversal agent in the event of over anticoagulation with warfarin14. Additionally, warfarin initiation may come with education regarding the need for consistent intake of green leafy vegetables15. Given the close relationship of warfarin with vitamin K, we focused our genome wide analysis specifically on single nucleotide polymorphisms (SNPs) in genes within vitamin K pathways.
Here, we survey single nucleotide variant associations with warfarin dose in two Latino populations. Further, we examine genomic signals in vitamin K genes that may be relevant to warfarin dosing when considering local ancestry throughout the genome via the Tractor regression model11. With this workflow, we attempt to address the systematic exclusion of admixed participants in warfarin genetic research and validate an alternative method to adjustment with principal components which may be increasingly underpowered in smaller research populations.
Methods
Study Populations
Discovery Cohort
DNA specimens were collected from participants in an observational cohort study (ClinicalTrial.gov Identifier NCT02972385)16. Warfarin users (n = 154) with a stable dose were recruited during their usual warfarin management appointments at Banner University Medical Center and El Rio Community Clinics in Tucson, Arizona, USA between October 2016 and March 2020. Participants were over 18 years of age, self-reported Hispanic or Latino ethnicity and were on a stable dose of warfarin. Stable warfarin dose was defined as consistent dose for at least two consecutive study visits. Inability to give informed consent was the only exclusion criteria. This study included a mouthwash sample collected at the time of enrollment and a retrospective electronic medical record review, and thus no patients were withdrawn or lost to follow-up. All research activities involving human participants in this study were approved by the Institutional Review Boards at the University of Arizona (#1608762767) in August 2016. Written informed consent was obtained from all participants prior to study enrollment.
Replication Cohort
This is a follow-up analysis of DNA specimens that were collected from participants in an observational cohort study (ClinicalTrial.gov Identifier NCT01318057). A full description of the cohort and original study have been previously described17. Briefly, warfarin treated participants (n = 255) were recruited from the Veteran’s Affairs Caribbean Healthcare system-affiliated anticoagulation clinic in San Juan, Puerto Rico. Participants self-identified as Caribbean Hispanic Puerto Ricans, were over 21 years old and on a stable warfarin dose defined as consistent dose for at least three consecutive study visits. The portion of the cohort with full genome-wide genetic results (Infinium™ Human Omni Xpress BeadChip by Illumina, San Diego, CA, USA) and clinical data available (n = 96) was analyzed here.
Genetic Assays and Quality Control of Genomic Data
Discovery Cohort
The participants in the discovery cohort provided a mouthwash sample for genotyping. Mouthwash samples were processed for genomic DNA extraction using a Puregene Blood Core Kit (QIAGEN Inc., Venlo, Limburg) according to manufacturer’s instructions. DNA samples were genotyped using an Illumina (Illumina, San Diego, CA, USA) MEGAEX array which contains 2 036 060 SNPs, at VANTAGE (Vanderbilt Technology for Advanced Genomics, Nashville, TN).
Quality control of the genome-wide variants in the discovery cohort was performed using PLINK 1.918 and the plinkQC package (version 0.3.4)19 in R (version 4.0.2)20. PlinkQC acts as a wrapper for the PLINK program allowing for interactive quality control in the RStudio environment. Due to the cohort being case-only and having admixed ancestries, departure from Hardy-Weinberg equilibrium was estimated under the null hypothesis of the predictable segregation ratio of specific matching genotypes with a liberal alpha value (p>1×10−10). In admixed populations especially, an inflation of heterozygote SNPs could be due to recent population substructure21 instead of genotyping error which is typically the reason these variants are filtered22. Monomorphic sites were removed. Per-marker and per-individual missingness were set to 10% to allow for maximum data prior to imputation, as it has been shown that filtering prior to imputation has little effect on imputation of common variants23. Samples were also removed due to relatedness, failed sex check, or outlying weekly warfarin doses. Finally, scripts to check the quality of the data and to prepare for imputation were implemented. The checkVCF.py (https://github.com/zhanxw/checkVCF) and McCarthy Group Tool for 1000G Imputation (https://www.well.ox.ac.uk/~wrayner/tools/) scripts were implemented prior to imputation with the Trans-Omics for Precision Medicine (TOPMed) imputation server. Non-SNPs, SNPs that did not align to the reference genome, and duplicate sites were removed. A total of 867 125 variants was subsequently used to impute additional variants using the TOPMed freeze8 reference panel in the TOPMed imputation server (https://imputation.biodatacatalyst.nhlbi.nih.gov/#!). Pre-phasing was performed using Eagle v2.424 and imputation via minimac425 within the TOPMED server. Imputed variants with quality R2 = 0.01 (the most accurately imputed 1% of variants) were kept for further analysis, giving a final number of 12 046 918 variants before filtering based on vitamin K pathway-related genes, and 103 277 after filtering.
Replication Cohort
The participants in the replication cohort provided a blood sample for genotyping. Processing for genomic DNA extraction using a QIAamp DNA Blood Maxi Kit (QIAGEN Inc., Venlo, Limburg) according to manufacturer’s instructions. DNA samples were genotyped using an Illumina (Illumina, San Diego, CA, USA) Infinium™ Human OmniExpress-24 v1.2 BeadChip which contains 713 599 SNPs.
Quality control of the genome-wide variants in the replication cohort was performed using PLINK 1.9. Samples and individuals with missingness greater than 10% were removed in addition to those with a minor allele frequency lower than 1%. A total of 649 437 variants was subsequently used to impute additional variants in the TOPMed imputation server in the same manner as in the discovery cohort. A final number of 4 593 221 before filtering based on vitamin K pathway-related genes, and 31 505 after filtering, was used in further analyses.
Local and Global Ancestry Estimation
We used default parameters with one iteration in RFMIX v2 (https://github.com/slowkoni/rfmix) to estimate local ancestry per chromosome in the discovery and replication cohorts26. Native American (Karitiana, Maya, Pima, Surui; n = 46) retrieved from the Human Genome Diversity Project (HGDP)27, and European (IBS; n = 107) and African (YRI; n = 108) populations retrieved from 1000 Genomes28 were merged to serve as reference. Global ancestry estimates were derived from an updated lai_global.py script from https://github.com/armartin/ancestry_pipeline29. We then surveyed local ancestry throughout the genome and at important warfarin pharmacogenes including Cytochrome P450 family 2 subfamily C member 9 (CYP2C9), Vitamin K epoxide Reductase Complex subunit 1 (VKORC1), Cytochrome P450 Family 4 Subfamily F Member 2 (CYP4F2), NAD(P)H Quinone Dehydrogenase 1 (NQO1), and 𝛾-glutamyl carboxylase (GGCX). Differences in proportions of the cohort with a specific ancestry at each pharmacogene were descriptively tested for statistical significance using the tableby function from the arsenal package in R30. In these analyses, individuals whose local ancestry was assigned to more than one population within the gene of interest were excluded. Finally, principal components were visualized to increase confidence in ancestry estimates using ggplot2 in R31.
Statistical Analyses
Patient baseline characteristics were summarized as counts and percentages or mean and standard deviation (sd) for categorical and continuous variables, respectively. Hypotheses were tested with chi-square goodness of fit test for equal counts for categorical variables using chisq.test and t-test for continuous variables with oneway.test using the tableone package in R32.
Given low sample sizes in both the discovery and replication cohorts, we limited our genome-wide scan to variants in genes in vitamin K pathways. We performed a KEGG (Kyoto Encyclopedia of Genes and Genomes) search for the terms “menaquinone” and “phylloquinone” to pull all genes from four pathways: Ubiquinone and other terpenoid-quinone biosynthesis, Vitamin digestion and absorption, Biosynthesis of cofactors (hsa01240), and Metabolic pathways (hsa01100)33. Uniprot was then used to translate KEGG gene nomenclature to ensembl34. Finally, biomart (version 2.44.4) was employed to retrieve a list of gene start and end positions for use in sub-setting the genome-wide data35. A full list can be found in Table S1.
In our primary analysis, linear regression models were adjusted with the top three principal components as covariates to account for population stratification, without controlling for local ancestry. Tractor was then used to calculate ancestral minor allele dosages at each SNP position and inform associations with warfarin dose in a series of secondary analyses11. The Tractor model employed tests each variant for an association with following model:
where x1 and x2 (0,1,2) represent the count of haplotypes of the first and second ancestries at the locus in question for each individual, x3-x5 (0,1,2) represent the count of risk alleles coming from each ancestry, and x6-xk are other covariates such as age, body size, an estimate of global ancestry, and so on. The model presented here is for a 3-way admixed population. Each regression was principal component-adjusted and performed with PLINK1.918 --linear or local ancestry-adjusted with a Tractor model and performed in Hail (https://github.com/hail-is/hail)36. Minor allele frequency cutoffs of 5% and a Hardy Weinberg cutoff of P < 1×10−6 were implemented in every regression, and analyses included only SNPs in vitamin K genes (n = 1589).
A SNP was further explored when it reached P < 0.0125 (0.05/4 KEGG pathways) in both cohorts with effects on warfarin dose in the same direction. P-values from tests of Hardy Weinberg equilibrium were extracted from PLINK1.9 using --hardy using an exact test37. These liberal replications were then examined for their association with warfarin dose after adjustment for variables described in the International Warfarin Pharmacogenetics Consortium (IWPC) model: age, height, weight, genotypes at VKORC1 and CYP2C9, amiodarone use and enzyme inducer use. Enzyme inducers surveyed here were phenytoin, carbamazepine, rifampin, and rifampicin38. Genotypes were derived from rs9923231 for VKORC1 and from rs1799853 and rs1057910 for CYP2C9*2 and *3, respectively. IWPC adjusted linear models were run with lm in R. All code for this analysis has been made available at github.com/karneslab/warf_latinx_local_vitk.
Results
Characterization of the study populations
After removing seven participants to greater than 10% missing data, two to cryptic relatedness, three to sex/gender mismatches, and one with therapeutic dose greater than four standard deviations above the mean suggesting non-compliance, our analysis included a discovery cohort of 141 self-reported Latino individuals recruited from Tucson, Arizona, USA and a replication cohort of 96 individuals recruited from San Juan, Puerto Rico. Baseline characteristics of each cohort are reported in Table 1. The discovery cohort contained more women (43% versus 1%, P < 0.001) and included a younger population, with more variation in age, (62.8 [standard deviation (sd) 16.8] years versus 69.2 [sd 10.0] years in Tucson and San Juan, respectively, P = 0.001). Body surface area was lower with more variation in the discovery cohort (1.9 [sd 0.3] versus 2.0 [sd 0.2] m2 in Tucson and San Juan, respectively, P = 0.004). A minority of the participants had variant allele copies for either CYP2C9*2 or CYP2C9*3 (18.8% and 20.8% in Tucson and San Juan, respectively, P = 0.83). Notably, none of the participants could be classified as “CYP2C9 Poor Metabolizers” by the system derived via the Clinical Pharmacogenetics Implementation Consortium (CPIC)39. VKORC1-1639G>A A allele carriage was variable between cohorts, with 53.9% (AA: 27.7%, GA: 52.5%) in the discovery cohort and 35.2% (AA: 13.5%, GA: 46.9%) in the replication.
Table 1.
Subject Characteristics.
| Tucson, AZ | San Juan, PR | P-valuea | |
|---|---|---|---|
| Participants (n) | 141 | 96 | |
| Warfarin dose (mg/week) (mean (SD)) | 33.84 (15.67) | 32.25 (11.64) | 0.397 |
| BSA (m2) | 1.90 (0.31) | 2.01 (0.22) | 0.004 |
| Age (years) | 62.77 (16.75) | 69.18 (10.00) | 0.001 |
| Male Gender (n (%)) | 81 (57.4) | 95 (99.0) | <0.001 |
| European Global Ancestry b | 0.60 (0.12) | 0.76 (0.11) | <0.001 |
| Amiodarone Users | 6 (4.3) | 1 (1.0) | 0.297 |
| Enzyme Inducer Users c | 5 (3.5) | 1 (1.0) | 0.433 |
| CYP2C9 Intermediate Metabolizer d | 26 (18.8) | 20 (20.8) | 0.834 |
| VKORC1e (%) | <0.001 | ||
| G/G | 28 (19.9) | 38 (39.6) | |
| G/A | 74 (52.5) | 45 (46.9) | |
| A/A | 39 (27.7) | 13 (13.5) |
Hypotheses were tested with chi-square goodness of fit test for equal counts for categorical variables using chisq.test() and t-test for continuous variables with oneway.test() using the tableone package in R
European ancestry was derived using individuals from the Iberian Peninsula as reference
Enzyme Inducers surveyed include: phenytoin, carbamazepine, rifampin and rifampicin
rs1799853 and rs1057910 were surveyed to proxy CYP2C9*2 and *3. Metabolizer status was derived following the current Clinical Pharmacogenetics Implementation Consortium Guideline for Warfarin. No participants in either cohort carried CYP2C9 variants on both alleles, thus there are no poor metabolizers to report.
VKORC1-1639G>A (rs9923231) was surveyed to proxy metabolizer status at VKORC1
AZ indicates Arizona; PR, Puerto Rico; mg, milligrams; SD, standard deviation; BSA, body surface area; m2, meters squared; CYP2C9, Cytochrome P450 family 2 subfamily C member 9; VKORC1, Vitamin K epoxide Reductase Complex subunit 1
Global ancestry estimates differed between cohorts, as shown in Figure 1. Both cohorts, however, were found to have appreciable European ancestry proportions (60.1% in Tucson and 75.9% in San Juan), as shown in Table 1. Local ancestry was visualized throughout the entire genome of each study participant. One example of this genome-wide local ancestry visualization from each cohort can be found in Figure S1. We additionally surveyed local ancestry at warfarin-impacting pharmacogenes VKORC1 and CYP2C9 (Table 2). Forty and 67 percent or more of the individuals in the Tucson and San Juan cohorts, respectively, were observed to have only European ancestry for the entire length of both CYP2C9 and VKORC1. Local ancestry estimates for warfarin pharmacogenes CYP4F2, GGCX, and NQO1 showed little similarity between cohorts (with the exception of GGCX) but were similar within each cohort (Table S2). Local ancestry at warfarin pharmacogenes, in general, was reflective of the slight increase in global European ancestry in the replication cohort. Results from principal components analyses corroborate global ancestry results, as displayed in Figure S2.
Figure 1.
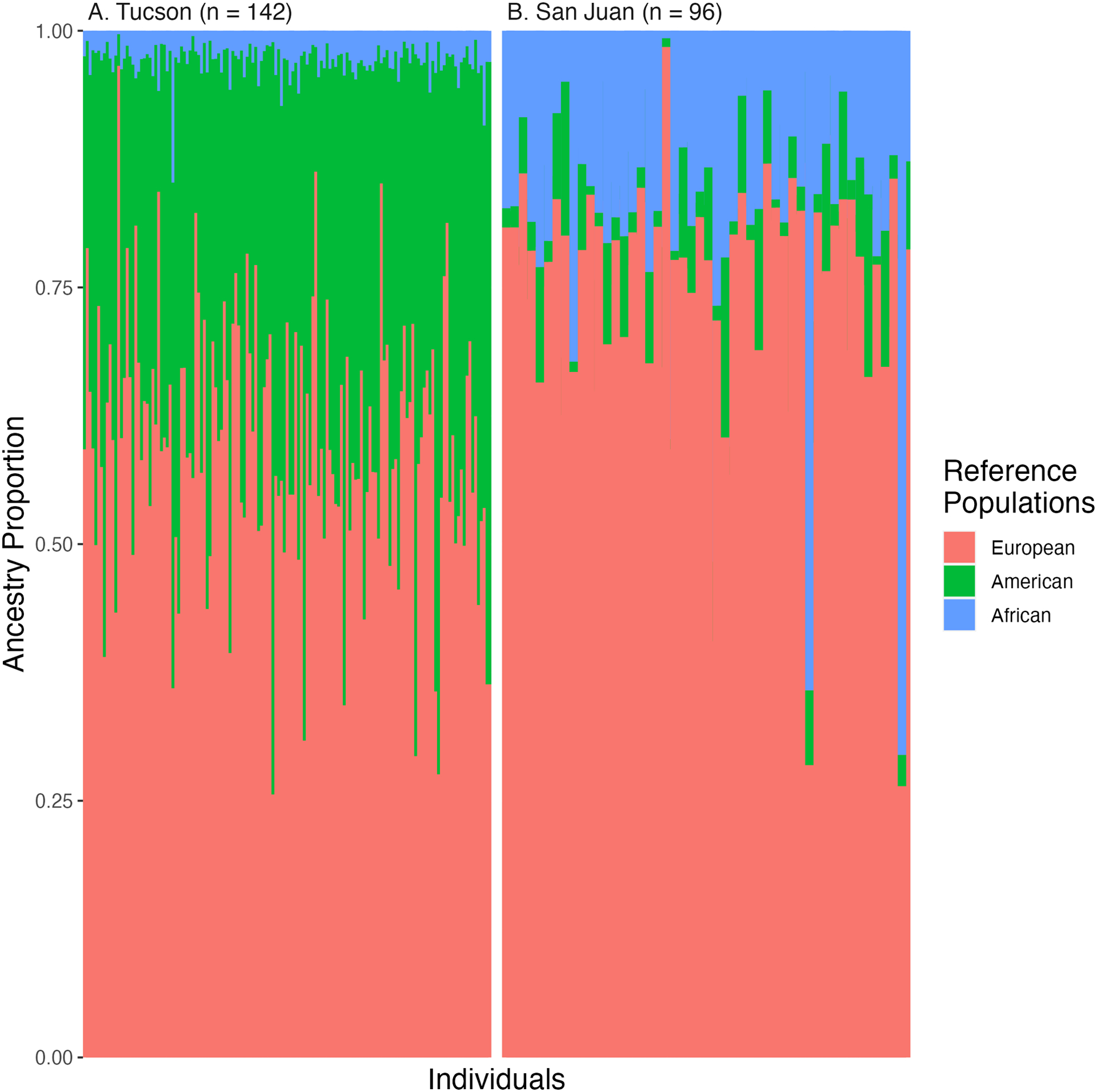
Global ancestry proportions per individual in the A) discovery and B) replication cohort as produced by RFMix_v2 when using references from Europeans of the Iberian Peninsula (Purple), Africans of Yorubaland (Yellow), and Indigenous Americas (from multiple ethnic groups, Green) as reference. The x-axis represents individuals while the y-axis represents proportions of each ancestry which are shown in contrasting colors.
Table 2.
Local Ancestry at CYP2C9 and VKORC1
| Gene | Copiesa | Tucson, AZ (n = 141) | San Juan, PR (n = 96) | Total (n = 237) | P-valueb |
|---|---|---|---|---|---|
| CYP2C9 | EUR | < 0.001 | |||
| 0 | 28 (20.3%) | 8 (8.4%) | 36 (15.5%) | ||
| 1 | 55 (39.9%) | 23 (24.2%) | 78 (33.5%) | ||
| 2 | 55 (39.9%) | 64 (67.4%) | 119 (51.1%) | ||
| AMR | < 0.001 | ||||
| 0 | 58 (42.0%) | 87 (91.6%) | 145 (62.2%) | ||
| 1 | 56 (40.6%) | 6 (6.3%) | 62 (26.6%) | ||
| 2 | 24 (17.4%) | 2 (2.1%) | 26 (11.2%) | ||
| AFR | < 0.001 | ||||
| 0 | 131 (94.9%) | 70 (73.7%) | 201 (86.3%) | ||
| 1 | 7 (5.1%) | 21 (22.1%) | 28 (12.0%) | ||
| 2 | 0 (0.0%) | 4 (4.2%) | 4 (1.7%) | ||
| VKORC1 | EUR | < 0.001 | |||
| 0 | 23 (17.3%) | 4 (4.4%) | 27 (12.1%) | ||
| 1 | 54 (40.6%) | 21 (23.1%) | 75 (33.5%) | ||
| 2 | 56 (42.1%) | 66 (72.5%) | 122 (54.5%) | ||
| AMR | < 0.001 | ||||
| 0 | 59 (44.4%) | 88 (96.7%) | 147 (65.6%) | ||
| 1 | 52 (39.1%) | 3 (3.3%) | 55 (24.6%) | ||
| 2 | 22 (16.5%) | 0 (0.0%) | 22 (9.8%) | ||
| AFR | < 0.001 | ||||
| 0 | 129 (97.0%) | 69 (75.8%) | 198 (88.4%) | ||
| 1 | 4 (3.0%) | 18 (19.8%) | 22 (9.8%) | ||
| 2 | 0 (0.0%) | 4 (4.4%) | 4 (1.8%) |
Copies indicates the haploid count of ancestrally similar segments at a given gene and copies are denoted for each ancestral reference used
Hypotheses were tested with chi-square goodness of fit test for equal counts per cohort with tableby() from the arsenal package in R
AZ indicates Arizona; PR, Puerto Rico; CYP2C9, Cytochrome P450 family 2 subfamily C member 9; VKORC1, Vitamin K epoxide Reductase Complex subunit 1; EUR, Europeans of the Iberian Peninsula and Spain; AMR, Indigenous American (multiple ethnic groups); AFR, Africans of Yorubaland
Associations of vitamin K gene variants across the genome with stable warfarin dose
Results of the primary, principal components adjusted linear regression analyses are shown in Figure 2. A total of five variants from vitamin K genes in metabolic pathways, and cofactor and terpenoid-quinone synthesis pathways were associated with warfarin dose in both cohorts, with the same direction of effect, at a significance threshold of P < 0.0125 (Table 3). The variants showing the strongest significance overall were located in the intron of well-known warfarin impacting gene, VKORC1 (vitamin K epoxide reductase complex 1). Variants in VKORC1 showed negative associations with warfarin dose while all other associations were positive (Figure S3). After adjustment for known covariates, most replicated variant associations with warfarin dose were inconsistently statistically associated with warfarin dose between cohorts (Table 3).
Figure 2.
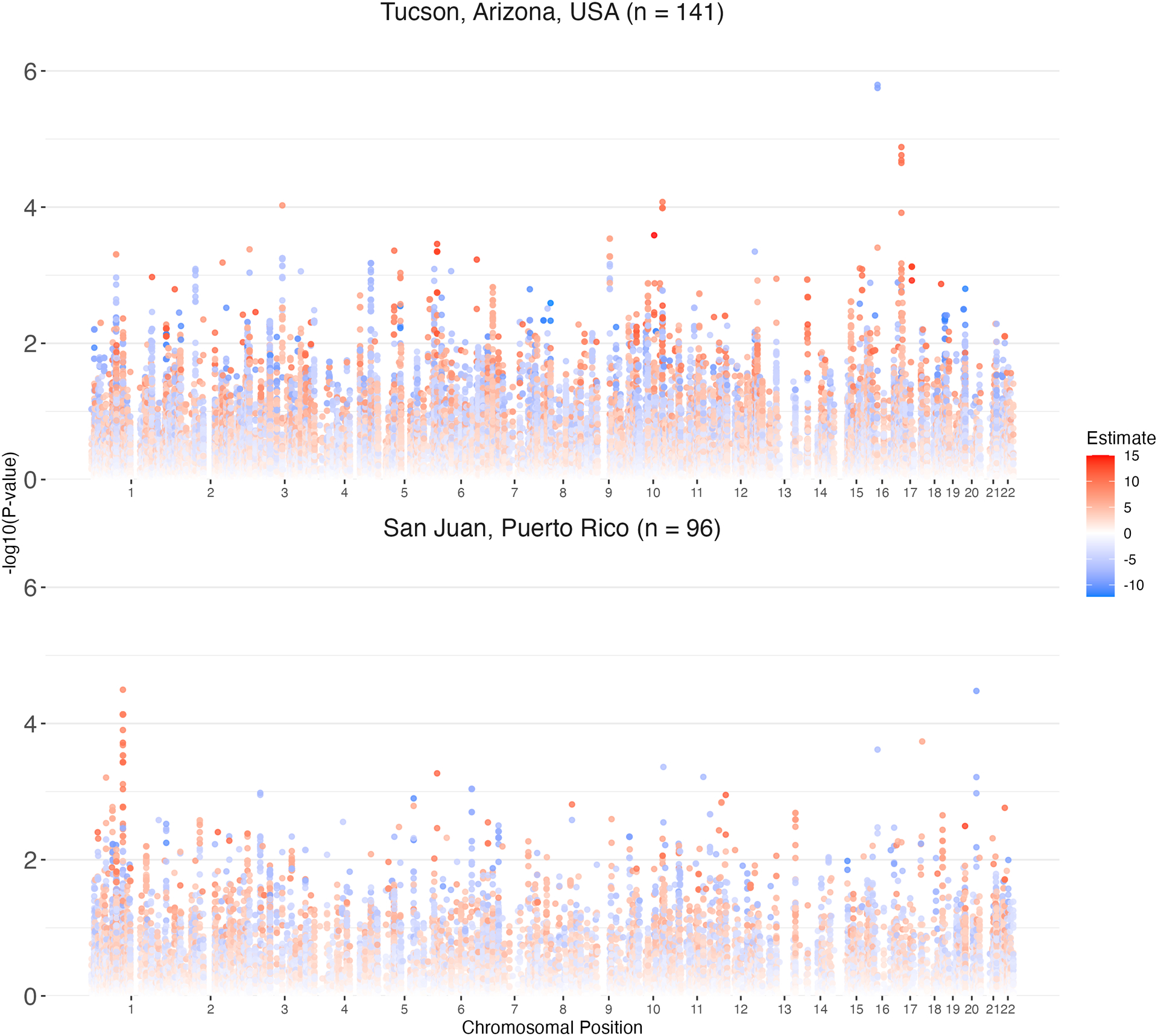
Manhattan plots of linear regression significance values from vitamin K-pathway related variants on warfarin dose in the A) discovery and B) replication cohort. The x-axis represents chromosomal location while the y-axis represents significance of the association (−log10[p value]). Each dot is a variant tested for an association with stable warfarin dose and the color of each dot represents the effect size of the association where blue and red colors are negative and positive effects, respectively.
Table 3.
Replicated loci from primary analyses of vitamin K pathway-related variants
| Gene | SNP | HWEa | Cohort | Modelb | Effect Size (±SE) | P-valuec |
|---|---|---|---|---|---|---|
| ACSM6, Uncharacterized LOC107984257 | chr10:95205120:G:A (rs650243)d | 1.00 | Tucson | PC | 11.03 (±3.799) | 4.29×10−3 |
| 0.22 | San Juan | PC | 9.737 (±3.634) | 8.74×10−3 | ||
| Tucson | IWPC | 4.74 (±2.7) | 0.08 | |||
| San Juan | IWPC | 7.37 (±3.51) | 0.04 | |||
| COX10 | chr17:14207584:A:C (rs11078234) | 1.80×10−3 | Tucson | PC | 6.454 (±2.21) | 4.10×10−3 |
| 0.42 | San Juan | PC | 5.706 (±2.009) | 5.56×10−3 | ||
| Tucson | IWPC | 4.1 (±1.54) | 8.95×10−3 | |||
| San Juan | IWPC | 4.08 (±2.27) | 0.08 | |||
| VKORC1 | chr16:31093188:C:A (rs8050894) | 0.87 | Tucson | PC | −8.745 (±1.743) | 1.6×10−6 |
| 0.51 | San Juan | PC | −4.92 (±1.633) | 3.36×10−3 | ||
| Tucson | IWPC | −7.42 (±5.29) | 0.16 | |||
| San Juan | IWPC | −6.09 (±1.94) | 2.30×10−3 | |||
| VKORC1 | chr16:31093557:G:A (rs9934438) | 0.61 | Tucson | PC | −9.011 (±1.804) | 1.78×10−6 |
| 0.10 | San Juan | PC | −4.727 (±1.605) | 4.10×10−3 | ||
| Tucson | IWPC | - | - | |||
| San Juan | IWPC | - | - |
--hardy in plink1.9 tells the null hypothesis that observed genotype frequencies are not significantly different from those predicted for a population in equilibrium
Model refers to the regression that produced the outlined result, where PC stands for adjustment for the top three principal components, and IWPC stands for adjustment for the variables included in the IWPC dose prediction algorithm38 IWPC models were adjusted for age, height, weight, genotypes at VKORC1 and CYP2C9, amiodarone use and enzyme inducer use.
Hypotheses were tested with --linear of PLINK1.9, and with lm() from the stats package in R in IWPC adjusted models, a p-value less than 0.0125 is summarized as p < 0.0125
chr10:95197958:T:A (rs619297), in complete linkage, was also replicated
SNP indicates single nucleotide polymorphism; HWE, Hardy Weinberg Equilibrium; SE, standard error; ACSM6, Acyl-CoA Synthetase Medium Chain Family Member 6; COX10, Cytochrome C Oxidase Assembly Factor Heme A:Farnesyltransferase; VKORC1, Vitamin K epoxide reductase complex subunit 1; PC, principal components; and IWPC, international warfarin pharmacogenetics consortium
We then performed local ancestry adjusted regressions in both cohorts (Figure 3). Local ancestry adjusted regressions uncovered 35 variants in five different genes with relationships on warfarin dose with statistically significant absolute effect sizes ranging from β = 9.04 (±2.23) to 39.18 (±10.89) per ancestral allele in the discovery cohort and from β = 6.47 (±2.02) to 17.82 (±6.83) in the replication cohort, as summarized in Table 4. All statistically significant replications, except those in VKORC1, were pulled from African local ancestry estimates. Notably, Tractor models portions of the genome resembling different reference populations separately. Many variants meeting our replication criteria showed a statistically insignificant relationship with warfarin dose in the two populations where neither association nor replication were observed. For example, variation in the MGAT1 gene is statistically associated with warfarin dose only in the African local ancestry estimates. Plotting the associations in the entire cohort illustrates how traditional linear models may miss many of the observed signals (Figure S4). Our results suggest that comparing portions of the genome with high ancestral similarity uncovers variation that would be missed when analyzing all portions of the genome simultaneously. After adjustment for other known warfarin dose influencing variables (e.g. age, body size) using the IWPC prediction model, no variants outside of VKORC1 showed a statistically significant association with warfarin dose. A full data table of results can be found in Table S3.
Figure 3.
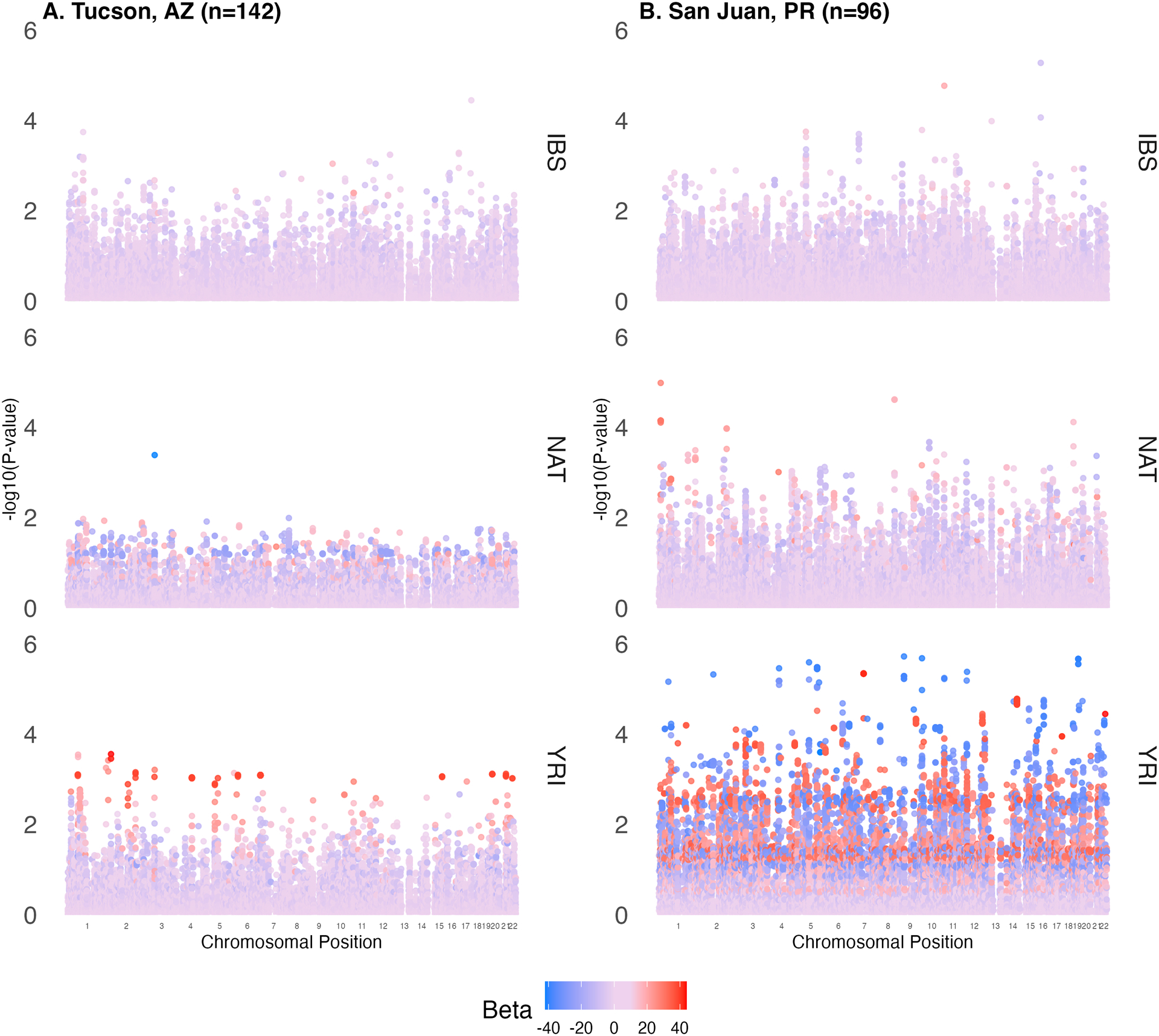
Manhattan plots of vitamin K pathway-related variants in linear regression associations for individuals with appreciable amounts of ancestry representing A) European people of the Iberian Peninsula, B) Africans of Yorubaland, and C) Indigenous Americans. Results from the discovery cohort are shown on the left and replication on the right for each ancestry. The x-axis represents chromosomal location while the y-axis represents significance of the association (−log10[p value]). Each dot is a variant that was tested for an association with stable warfarin dose and the color of that dot represents the effect size of the association where blue, purple and red colors represent negative, neutral, and positive effects, respectively.
Table 4.
Replicated loci from secondary genome-wide vitamin K analyses adjusting for local ancestry
| Gene | SNPa | HWEb | Cohort | Modelc | Ancestry | Frequencyd | Effect Size (±SE) | P-valuee |
|---|---|---|---|---|---|---|---|---|
| AK4 | chr1:65640280:G:Af | 0.12 | Tucson | Tractor | AFR | 0.88 | −3.44 (±2.53) | 0.18 |
| 0.21 | San Juan | Tractor | AFR | −4.21 (±2.94) | 0.16 | |||
| Tucson | Tractor | EUR | 0.95 | −4.81 (±4.47) | 0.28 | |||
| San Juan | Tractor | EUR | −21.62 (±11.42) | 0.06 | ||||
| Tucson | Tractor | NAT | 0.71 | 22.30 (±8.57) | 0.01 | |||
| San Juan | Tractor | NAT | 12.49 (±4.37) | 5.31×10−3 | ||||
| Tucson | IWPC | - | 0.51 (±1.46) | 0.73 | ||||
| San Juan | IWPC | - | −0.05 (±2.81) | 0.99 | ||||
| HK2 | chr2:75064362:T:Cg | 0.85 | Tucson | Tractor | AFR | 1.00 | −38.98 (±10.89) | 4.77×10−4 |
| 0.21 | San Juan | Tractor | AFR | −10.59 (±4.1) | 1.14×10−2 | |||
| Tucson | Tractor | EUR | 0.99 | −1.81 (±2.24) | 0.42 | |||
| San Juan | Tractor | EUR | −0.48 (±2.26) | 0.83 | ||||
| Tucson | Tractor | NAT | 0.99 | 1.3 (±3.63) | 0.72 | |||
| San Juan | Tractor | NAT | −2.89 (±10.4) | 0.78 | ||||
| Tucson | IWPC | - | −0.77 (±1.4) | 0.58 | ||||
| San Juan | IWPC | - | −1.22 (±2.15) | 0.57 | ||||
| MGAT1 | chr5:180226673:A:C | 0.12 | Tucson | Tractor | AFR | 0.37 | 31.31 (±8.2) | 2.04×10−4 |
| 0.13 | San Juan | Tractor | AFR | 15.63 (±4.46) | 7.26×10−4 | |||
| Tucson | Tractor | EUR | 0.89 | −5.17 (±2.81) | 6.82×10−2 | |||
| San Juan | Tractor | EUR | −4.36 (±2.61) | 9.87×10−2 | ||||
| Tucson | Tractor | NAT | 0.78 | −0.84 (±3.41) | 0.80 | |||
| San Juan | Tractor | NAT | 1.67 (±6.19) | 0.79 | ||||
| Tucson | IWPC | - | 0.2 (±1.53) | 0.90 | ||||
| San Juan | IWPC | - | 3.9 (±2.26) | 8.81×10−2 | ||||
| NEU3/OR2AT2P | chr11:74726828:C:T | 0.25 | Tucson | Tractor | AFR | 0.85 | 28.13 (±9.92) | 5.25×10−3 |
| 0.10 | San Juan | Tractor | AFR | 10.01 (±3.86) | 1.11×10−2 | |||
| Tucson | Tractor | EUR | 0.53 | 0.44 (±2.72) | 0.87 | |||
| San Juan | Tractor | EUR | 3.76 (±2.24) | 9.69×10−2 | ||||
| Tucson | Tractor | NAT | 0.73 | −3.48 (±3.27) | 0.29 | |||
| San Juan | Tractor | NAT | 3.36 (±6.83) | 0.62 | ||||
| Tucson | IWPC | - | 0.11 (±1.56) | 0.94 | ||||
| San Juan | IWPC | - | 5.3 (±2.08) | 1.25×10−2 | ||||
| ST8SIA5 | chr18:44291905:C:T | 1 | Tucson | Tractor | AFR | 0.89 | 35.31 (±11.69) | 3.02×10−3 |
| 0.65 | San Juan | Tractor | AFR | 17.82 (±6.83) | 1.07×10−2 | |||
| Tucson | Tractor | EUR | 0.67 | 0.31 (±2.67) | 0.90 | |||
| San Juan | Tractor | EUR | −1.83 (±2.92) | 0.53 | ||||
| Tucson | Tractor | NAT | 0.79 | −0.62 (±3.43) | 0.86 | |||
| San Juan | Tractor | NAT | −4.34 (±7.62) | 0.57 | ||||
| Tucson | IWPC | - | −0.16 (±1.47) | 0.91 | ||||
| San Juan | IWPC | - | 1.11 (±2.69) | 0.68 | ||||
| VKORC1 | chr16:31104509:C:Gh | 0.87 | Tucson | Tractor | AFR | 0.74 | −11.8 (±7.68) | 0.13 |
| 0.51 | San Juan | Tractor | AFR | −2.28 (±3.38) | 0.50 | |||
| Tucson | Tractor | EUR | 0.60 | −9.04 (±2.23) | 8.61×10−5 | |||
| San Juan | Tractor | EUR | −6.58 (±2) | 1.46×10−3 | ||||
| Tucson | Tractor | NAT | 0.56 | −7.52 (±3.12) | 1.73×10−2 | |||
| San Juan | Tractor | NAT | 6.96 (±6.21) | 0.27 | ||||
| Tucson | IWPC | - | −7.42 (±5.29) | 0.16 | ||||
| San Juan | IWPC | - | −6.09 (±1.94) | 2.30×10−3 |
Chromosomal position in GRCh37.p13
--hardy in plink1.9 tells the null hypothesis that observed genotype frequencies are not significantly different from those predicted for a population in equilibrium
Model refers to the regression that produced the outlined result, where IWPC stands for adjustment for the variables included in the IWPC dose prediction algorithm38 and Tractor stands for adjustment for local ancestry by the Tractor regression model11 IWPC models were adjusted for age, height, weight, genotypes at VKORC1 and CYP2C9, amiodarone use and enzyme inducer use. Tractor models were adjusted for local ancestry calls.
Allele frequency of reference allele in 1000 Genomes cohort, except chr5:180226673:A:C which is summarized from gnomAD cohort, from African, European, and American reference populations
Hypotheses were tested with hl.agg.linreg() from Hail in Tractor models, and with lm() from the stats package in R in IWPC adjusted models, a p-value less than 0.05 is summarized as p < 0.05
Additional variants (n=23) in linkage also replicated. See Table S2 for the full table of replicated variant associations
Additional variants (n=4) in linkage also replicated. See Table S2 for the full table of replicated variant associations
One additional variant in linkage also replicated. See Table S2 for the full table of replicated variant associations
SNP indicates single nucleotide polymorphism; SE, standard error; AK4, Adenylate Kinase 4; HK2, Hexokinase 2; MGAT1, Mannosyl (α−1,3-)-glycoprotein β−1,2-N-acetylglucosaminyltransferase; NEU3, Neuraminidase 3; OR2AT2P, olfactory receptor, family 2, subfamily AT, member 2 pseudogene; ST8SIA5, ST8 Alpha-N-Acetyl-Neuraminide Alpha-2,8-Sialyltransferase 5; VKORC1, Vitamin K epoxide reductase complex subunit 1; IWPC, international warfarin pharmacogenetics consortium; EUR, Europeans of the Iberian Peninsula and Spain; AMR, Indigenous Americans (multiple ethnic groups); AFR, Africans of Yorubalandsd
Discussion
Two geographically, ethnically, and ancestrally distinct Latino warfarin-taking cohorts were investigated for association between SNPs in vitamin K-related genes and warfarin dose requirements. Our analysis highlights a promising, and novel, method for identifying phenotypically-related variants in ancestrally diverse cohorts, both in terms of current genomes and recent historical admixture. We identified variants in the VKORC1 gene in both our primary and secondary analyses, highlighting the validity of the Tractor model for future research on genetic predictors in non-European patients.
Little is known regarding the impact of genomic variants on warfarin dose requirements for populations that may be described as Latino. A single genome-wide association study of warfarin stable dose in a Brazilian cohort corroborated warfarin associations in variants of CYP2C9 and VKORC140. While a Brazilian population may, or may not, be described at Latino, the cohort surveyed exhibits ancestral admixture from three founder populations with a majority of the genome representing European reference genomes (>75% in Parra et. al., and >68% here, on average). Given the ancestral admixture of the Brazilian population and other Latino populations, principal components were employed to control for global ancestral estimates in previous analyses. Our comparable analysis provides further evidence for the association of VKORC1 polymorphisms on warfarin dose. Intronic VKORC1 variants rs9934438 and rs8050894 were significantly associated in both of our tested cohorts, regardless of allele frequencies differences. Principal components, however, can dramatically decrease the power of an analysis and may have led to filtering out important relationships with strict statistical thresholds.
Tractor has yet to be applied to a 3-way admixed (with significant ancestral portions from three founder populations) cohort, nor has it been used in pharmacogenomic research, to date. It is imperative to note the need for comparable ancestry proportions when assessing more than one admixed population. As shown in Figure 1, the only appreciable ancestry proportion that was comparable between the replication and discovery cohort was the section that aligned with European ancestry. Thus, although including a seemingly comparable 3-way admixed replication cohort, we had low power to interrogate portions of the ancestry that aligned with American or African ancestry. Unrealistic effect sizes and statistical artifacts can be seen throughout our Tractor analyses (Figure 3), specifically in lower proportion ancestral segments (i.e. African ancestral proportions), highlighting the need for larger cohorts overall to balance the ever-growing number of SNPs and ancestral identities. The only vitamin K pathway variants that were implicated by both our primary and secondary analyses are located inVKORC1. Although the associations of warfarin dose with genes presented in this analysis are preliminary, Tractor was able to identify known warfarin associated VKORC1 variants while retaining all study participants, regardless of admixture, and gaining information from the haplotypes of the cohort.
The disparate ancestries of the cohorts draws attention to a key importance of how we in the U.S. categorize individuals as Latino, especially in warfarin dose prediction research. Latin American populations, as mentioned previously, have a unique history of European colonization, which has led to the admixture of many Indigenous American people’s genomes with those of the colonizing Europeans and enslaved Africans they brought to the Americas. This admixture highlights challenges for the current Clinical Pharmacogenetics Implementation Consortium guideline for warfarin dosing which discriminates between patients with [West] African ancestry and non-African ancestry41. For example, should any amount of African ancestry require an individual to obtain additional genotyping prior to benefiting from PGx dosing? Further, does the patient’s African ancestry need to be recent to be relevant, i.e., do Latino patients with colonial admixture events from the 1500s require additional genetic testing or will knowledge of recent African ancestry be a proxy for African ancestry.
More broadly, the classification of individuals as Latino for prediction of phenotypic outcomes is largely uninformative. Individuals identifying as Latino may be as diverse as humans themselves given that the definition of Latino employs an expansive geographical region, Latin America. The region of the Americas where colonizers spoke Latin-derived languages encompasses an expansive land mass where a person of nearly any ancestral background may reside. The definition of Latino is not interchangeable with Hispanic, since Spain has colonized many countries worldwide in its imperialist history, apart from countries in the Americas, and this would imply that the language spoken by the person is biologically meaningful in some way.
Additional limitations of this analysis, apart from those discussed above, include the low sample sizes in both surveyed cohorts. Even with restriction to vitamin K-related genes our analyses were highly underpowered to make statistically-guided decisions in the face of multiple comparisons adjustments. Some false negative observations are thereby likely in our results due to these low sample sizes. However, future studies using this approach in admixed populations with larger sample sizes have the potential to discover novel variation influencing warfarin dose variability. Ancestry estimates, both local and global, are inherently biased due to the reference genomes employed for their creation being European centered42. The outcome variable, weekly warfarin dose, is also vulnerable to collection biases due to the potential for patient non-adherence, which was not addressed in this study. Finally, our manuscript did not include data regarding the use of novel oral anticoagulants (NOACs), which may be preferred in many clinical scenarios. NOACs may be an appropriate alternative to warfarin considering observations of high variability in warfarin dose43 and risk for warfarin-related intracranial hemorrhage44. Given the long track record of warfarin use in clinical practice, its affordable cost, and limited clinical utility of NOACs in special populations (children, renal impairment, elderly, mechanical heart valves), warfarin is likely to continue to be preferentially used over NOACs in a substantial proportion of patients.
Conclusions
We surveyed the vitamin K-related genome for single nucleotide variants associated with stable warfarin dose in two Latino populations. To address population stratification, we compared Tractor, which takes the local ancestry at each variant into consideration while modeling, to standard principal components adjusted models. Variants in the intron of VKORC1 were shown to be in association with warfarin stable dose in both analyses, demonstrating the statistical validity of the Tractor model. However, further results of this study are highly descriptive due to low sample sizes. Our analysis highlights the disparate ancestral proportions spanning populations that are both socially classified as Latino and the care that should be taken when making demographic classifications for pharmacogenetic testing.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
Pharmacogenetic variants affect warfarin stable dose without doubt. Studies have shown increased representation of ancestrally diverse populations is beneficial for discovering novel loci and addressing health disparities.
What question did this study address?
This study aimed to survey the genomes of two populations of patients who identify as Hispanic or Latino with the intention of addressing a lack of warfarin pharmacogenomic research in this population.
What does this study add to our knowledge?
This study validates the local ancestry adjusted regression model, Tractor, in a 3-way admixed population, thereby lowering the barrier to inclusion in warfarin pharmacogenomic research. We confirm the association of known warfarin pharmacogene, VKORC1, using Tractor and highlight the novel statistical methodology of including patients with admixed genomes with minimal loss of statistical power.
How might this change clinical pharmacology or translational science?
Our study challenges the current clinical recommendation to treat differentially based on West African Ancestry to consider the impact of this guideline on patients who identify as Hispanic or Latino and consequentially have a high probability of genomic signatures that resemble multiple reference ancestral populations.
Acknowledgements
We would like to formally thank and acknowledge all the participants in the discovery and replication cohorts for their participation in the study and trust in our research teams. We also acknowledge the participants who contributed to the Human Genome Diversity Project (HGDP) in the past. Thank you. This work would not be possible without the generous donations of participants to the greater good of science. We would also like to direct attention to the literature which highlights prior ethical issues with collection of HGDP data in indigenous peoples45,46, and encourage future researchers to consult the CARE Principles for Indigenous Data Governance (Collective Benefit, Authority to Control, Responsibility, and Ethics) when making data decisions regarding Indigenous populations47.
We would like to thank members of the team who helped to recruit participants for the discovery cohort in this analysis: Juanita Gonzales, RN and Echo Fallon, PharmD. We thank the team at Vanderbilt Technologies for Advanced Genomics (VANTAGE) who provided genotyping services for these data. Finally, we would like to thank Dr. Elizabeth Atkinson and Mike Wilson for help troubleshooting preprocessing data for use with Tractor.
Funding
Funding for this analysis included support from the National Institutes of Health’s National Heart, Lung, and Blood Institute (NIH NHLBI Awards R01 HL158686 [JHK, LHC, MAP], R01 HL156993 [JHK], and K01 HL143137 [JHK]), an institutional career development award from the University of Arizona Health Science Center (JHK), and a Seed Grant to Promote Translational Research in Precision Medicine from the Flinn Foundation (JHK). HES is supported by the Finley and Florence Brown Endowed Research Award from the University of Arizona Sarver Heart Center. JBG is supported by the National Institute of Environmental Health Sciences (T32 ES007091). JD, KCC and ARL are supported by Research Centers in Minority Institutions (RCMI) grant U54 MD007600 (National Institute on Minority Health and Health Disparities) from the National Institutes of Health.
Footnotes
Conflict of Interest Statement:
The authors declared no competing interests for this work.
SUPPORTING INFORMATION
Supplementary information accompanies this paper on the Clinical Pharmacology & Therapeutics website (www.cpt-journal.com).
Data Availability Statement
Genome-wide data and phenotype data used for the discovery cohort have been provided on database of Genotypes and Phenotypes (dbGaP accession number: Pending [University of Arizona] and phs001496 [University of Puerto Rico])).
References
- 1.Steiner HE et al. Machine Learning for Prediction of Stable Warfarin Dose in US Latinos and Latin Americans. Front. Pharmacol 12, 749786 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaye JB et al. WARFARIN PHARMACOGENOMICS IN DIVERSE POPULATIONS. Pharmacotherapy 37, 1150–1163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McInnes G, Yee SW, Pershad Y & Altman RB Genomewide Association Studies in Pharmacogenomics. Clin. Pharmacol. Ther 110, 637–648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Popejoy AB & Fullerton SM Genomics is failing on diversity. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duncan L et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun 10, 3328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindorff LA et al. Prioritizing diversity in human genomics research. Nat. Rev. Genet 19, 175–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mills MC & Rahal C A scientometric review of genome-wide association studies. Commun. Biol 2, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner S et al. Quality Control Procedures for Genome-Wide Association Studies. Curr. Protoc. Hum. Genet 68, 1.19.1–1.19.18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elhaik E Why most Principal Component Analyses (PCA) in population genetic studies are wrong. bioRxiv Preprint. 2021.04.11.439381 (2021).doi: 10.1101/2021.04.11.439381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atkinson EG et al. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet 53, 195–204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yorgov D, Edwards KL & Santorico SA Use of admixture and association for detection of quantitative trait loci in the Type 2 Diabetes Genetic Exploration by Next-Generation Sequencing in Ethnic Samples (T2D-GENES) study. BMC Proc. 8, S6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasaniuc B et al. Enhanced Statistical Tests for GWAS in Admixed Populations: Assessment using African Americans from CARe and a Breast Cancer Consortium. PLOS Genet. 7, e1001371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanley JP Warfarin reversal. J. Clin. Pathol 57, 1132–1139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheah GM & Martens KH Coumadin knowledge deficits: do recently hospitalized patients know how to safely manage the medication? Home Healthc. Nurse 21, 94–100; quiz 101 (2003). [DOI] [PubMed] [Google Scholar]
- 16.El Rouby N et al. Multi-site Investigation of Genetic Determinants of Warfarin Dose Variability in Latinos. Clin. Transl. Sci (2020).doi: 10.1111/cts.12854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duconge J et al. A Novel Admixture-Based Pharmacogenetic Approach to Refine Warfarin Dosing in Caribbean Hispanics. PloS One 11, e0145480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hannah V Meyer meyer-lab-cshl/plinkQC: plinkQC version 0.3.4 (2021).doi: 10.5281/zenodo.5106689 [DOI] [Google Scholar]
- 20.R Core Team R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, Vienna, Austria. (2020) https://www.R-project.org/ [Google Scholar]
- 21.Adhikari K, Mendoza-Revilla J, Chacón-Duque JC, Fuentes-Guajardo M & Ruiz-Linares A Admixture in Latin America. Curr. Opin. Genet. Dev 41, 106–114 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Price AL, Zaitlen NA, Reich D & Patterson N New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet 11, 459–463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charon C, Allodji R, Meyer V & Deleuze J-F Impact of pre- and post-variant filtration strategies on imputation. Sci. Rep 11, 6214 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maples BK, Gravel S, Kenny EE & Bustamante CD RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet 93, 278–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavalli-Sforza LL The Human Genome Diversity Project: past, present and future. Nat. Rev. Genet 6, 333–340 (2005). [DOI] [PubMed] [Google Scholar]
- 28.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin AR et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet 100, 635–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinzen E, Sinnwell J, Atkinson E, Gunderson T & Dougherty G arsenal: An Arsenal of ‘R’ Functions for Large-Scale Statistical Summaries. (2021) URL https://CRAN.R-project.org/package=arsenal
- 31.Wickham Hadley, Navarro Danielle, and Thomas Lin Pedersen ggplot2: Elegant Graphics for Data Analysis. (2020) URL https://ggplot2-book.org/
- 32.Panos A & Mavridis D TableOne: an online web application and R package for summarising and visualising data. Evid. Based Ment. Health 23, 127–130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Furumichi M, Tanabe M, Sato Y & Morishima K KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Apweiler R et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smedley D et al. BioMart – biological queries made easy. BMC Genomics 10, 22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Hail team Hail 0.2 (2018) URL https://github.com/hail-is/hail
- 37.Wigginton JE, Cutler DJ & Abecasis GR A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet 76, 887–893 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.International Warfarin Pharmacogenetics Consortium et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med 360, 753–764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Relling MV & Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parra EJ et al. Genome-wide association study of warfarin maintenance dose in a Brazilian sample. Pharmacogenomics 16, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson J et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Pharmacogenetics-Guided Warfarin Dosing: 2017 Update. Clin. Pharmacol. Ther 102, 397–404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levy-Sakin M et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun 10, 1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gage BF, Fihn SD & White RH Management and dosing of warfarin therapy. Am. J. Med 109, 481–488 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Jiang H, Jiang Y, Ma H, Zeng H & Lv J Effects of rivaroxaban and warfarin on the risk of gastrointestinal bleeding and intracranial hemorrhage in patients with atrial fibrillation: Systematic review and meta-analysis. Clin. Cardiol 44, 1208–1215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dodson M & Williamson R Indigenous peoples and the morality of the Human Genome Diversity Project. J. Med. Ethics 25, 204–208 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ilkilic I & Paul NW Ethical aspects of genome diversity research: genome research into cultural diversity or cultural diversity in genome research? Med. Health Care Philos 12, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Carroll SR et al. The CARE Principles for Indigenous Data Governance. Data Sci. J 19, 43 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome-wide data and phenotype data used for the discovery cohort have been provided on database of Genotypes and Phenotypes (dbGaP accession number: Pending [University of Arizona] and phs001496 [University of Puerto Rico])).