

SUMMARY
Crohn’s disease (CD) is a chronic gastrointestinal disease, increasing in prevalence worldwide. CD is multifactorial, involving the complex interplay of genetic, immune, and environmental factors, necessitating a systems-level understanding of its etiology. To characterize cell type-specific transcriptional heterogeneity in active CD, we profiled 720,633 cells from terminal ileum and colon of 71 donors with varying inflammation status. Our integrated datasets revealed organ and compartment-specific responses to acute and chronic inflammation; most immune changes were in cell composition while transcriptional changes dominated among epithelial and stromal cells. These changes correlated with endoscopic inflammation, but small and large intestines exhibited distinct responses, particularly apparent when focusing on IBD risk genes. Finally, we mapped markers of disease-associated myofibroblast activation, and identified CHMP1A, TBX3, and RNF168 as regulators of fibrotic complications. Altogether, our results provide a roadmap for understanding cell type- and organ-specific differences in CD and potential directions for therapeutic development.
Keywords: inflammatory bowel disease (IBD), inflammation, single-cell RNA sequencing, high-dimensional profiling, myofibroblasts
Graphical Abstract
eTOC Blurb
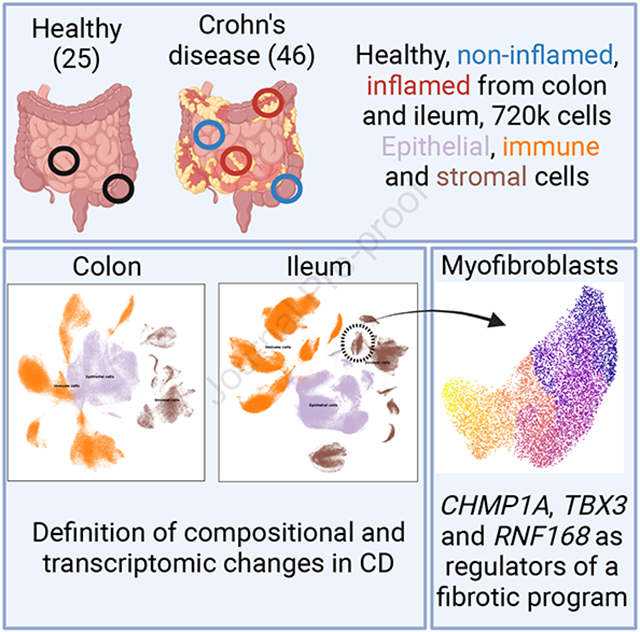
Crohn’s disease (CD) is a heterogenous condition impacting the ileum and colon in unique ways. Here, Kong et al. define the unique epithelial, stromal, and immune characteristics of CD by generated a single-cell transcriptomic atlas of the ileum and colon, and uncover novel regulators of collagen production in disease-associated fibroblasts.
INTRODUCTION
The inflammatory bowel diseases (IBD), comprising ulcerative colitis (UC) and Crohn’s disease (CD), are immune-mediated relapsing-remitting chronic disorders affecting millions of people worldwide. The prevalence of IBD is increasing1,2, and the treatment burden in the United States alone is estimated at $14.6–31.6 billion3. CD and UC are both characterized by a dysfunctional and hyperactive immune response resulting in uncontrolled inflammation4. In CD, this inflammation and resulting damage affects all layers of the gut, whereas in UC, this is limited to the colonic epithelium. Unlike UC, CD is generally characterized by discontinuous inflammation that can occur in distinct segments of the intestinal tract. Additionally, analyses of colonic mucosal samples of CD and UC have shown marked separations between the two diseases, in particular among T cell subsets5. CD primarily affects the ileum and colon, and recent work has suggested that ileal-dominant and colonic CD should be considered separate disease subtypes6,7. This highlights the importance of understanding whether and how the cellular processes underlying colonic and ileal inflammation differ.
Genome-wide association studies8–10 and exome sequencing studies11 define a broad set of risk genes related to epithelial barrier function, microbe sensing and restriction, and adaptive immunity12. This is perhaps not surprising given the fact that the proper function of the intestine is also characterized by a complex set of interactions amongst multiple host cell types– including epithelial, stromal and immune cells– and environmental factors– including dietary chemicals and microbes. Understanding the complex cellular networks that characterize health and IBD pathogenesis requires high-resolution system-level measurements such as single-cell RNA sequencing. For example, in UC, both compartment-specific13–15 and tissue-wide16 single-cell analyses illustrate changes in epithelial cell subsets, adaptive immune cells, and stromal compartments. These are associated with disease but also importantly with treatment outcomes. For example, an oncostatin M (OSM) circuit in inflammatory monocytes and fibroblasts is associated with resistance to anti-TNF therapy. These single-cell datasets also help functionalize genetic risk loci by mapping gene expression to specific cell types. In the context of CD, high-resolution studies correlate genetic and cellular modules in the ileum with disease outcomes and altered T cell subset distributions in inflammation17. The cellular module highlighted in that study demonstrates an enrichment of cytokine-cytokine receptor and chemokine-chemokine receptor pairs, but also an increase in OSM, suggesting some commonalities between anti-TNF resistance mechanisms in CD and UC. Single-cell technologies have also been used to identify CD-associated expression changes linked with the reactivation of developmental programs in a pediatric cohort18, and to map immune cell programs which are adopted by IBD to recruit and retain immune cells in inflammation19.
Because CD can occur across both the small and large bowel, which are characterized by distinct cellular networks in health, a cross-organ analysis would be a key resource to understand the mechanistic commonalities and differences between ileal, ileocolonic and colonic CD. To address this, we collected tissue from a total of 71 CD patients and non-IBD donors from inflamed and non-inflamed regions of the terminal ileum and colon, and used single-cell sequencing to elucidate the cell type- and location-specific changes that occur in CD. Our work identified a complex network of changes associated with disease. We observed broad compositional changes across immune and stromal cell subsets, while transcriptional reprogramming was more pronounced across epithelial cells, highlighting the different factors that participate in rewiring during disease. We demonstrated that some of these changes were restricted to either the colon or the ileum suggesting distinct tissue-specific responses. We also combined these transcriptomic analyses with existing genetic datasets to map risk gene expression. Finally, we mapped disease-associated changes in fibroblast gene expression and validated three regulators of fibroblast collagen induction that may represent novel targets for the management of fibrotic complications, thus demonstrating the applicability of the dataset. Altogether, this work offers a comprehensive view of the common cellular and transcriptomic changes associated with Crohn’s disease and can also serve as a foundational resource to explore the impact of disease progression and therapeutic strategies.
RESULTS
Terminal ileum and colon biopsies show both region- and disease-associated shifts in cell type composition
We collected data from 136 samples from 46 CD and 25 non-IBD patients at Massachusetts General Hospital. These include 24 samples from 12 non-IBD donors published previously16,20. In the majority of samples, we separated and independently processed the epithelial (E) and lamina propria (L) fractions (for a total of 89 epithelial channels and 100 L channels), while 36 samples were processed without separation, altogether resulting in 720,633 high quality single-cell transcriptomes in 225 channels (Fig. 1A, Table S1, Methods). Samples were obtained from three segments of the GI tract: 289,730 cells from colon (CO), 77,554 cells across the small bowel (SB) and 353,349 cells specifically from terminal ileum (TI). Due to the small number of SB samples, these were combined with TI samples for all analyses.
Figure 1. Single-cell measurement of healthy, non-inflamed and inflamed terminal ileum (TI) and colon (CO), biopsies in Crohn’s Disease.

(A) Workflow of biopsy collection and scRNA-seq measurements. *Note that some patients contributed samples to multiple locations and/or inflammation statuses. (B-C) UMAP visualization of all cells in TI and CO, colored by cell type compartment (B) and detailed cell types within compartments (C). (D) Principal coordinates analysis (PCoA) of Bray-Curtis dissimilarities from sample-level cell type composition profiles. Top: Colored by layer information: E: epithelium, L: lamina propria, N: not separated. Middle: Colored by location. Bottom: Colored by disease status (Healthy, Non-inflamed or Inflamed). (E) Barplots show significant differences in cell type frequency for non-inflamed (blue) and inflamed (red) samples relative to healthy (green) samples in immune, stromal and epithelial compartments in TI (top-row) and CO (bottom-row). (*adjusted p<0.05, **adjusted p<0.01, red* = overrepresented in inflamed or non-inflamed samples vs. healthy, blue* = underrepresented). The total number of cells contributing to each bar is also shown. In TI, the sample size for Healthy/Non-inflamed/Inflamed was 13/49/16 for the stromal and immune compartments, and 11/45/14 for epithelial. In CO, these were 32/20/6 and 32/18/5 respectively.
Because of the segmental nature of inflammation in CD, we specifically aimed to compare inflamed and non-inflamed regions. As part of clinical care, patients were scored using a simple endoscopic score for Crohn’s disease (SES-CD), which was summed across all the segments evaluated21. When active disease was present (SES-CD ≥ 3 across the whole intestine), we aimed to collect samples from both visibly inflamed and non-inflamed regions (i.e. regions that have a segmental score of 0). In inactive disease (SES-CD ≤ 2), we only collected non-inflamed samples (see Methods).
Annotation by cell type markers (Methods, Table S2) first broadly classified cells into three major cell type compartments (numbers listed for colon and TI, respectively): 97,788 and 154,136 epithelial cells, 39,433 and 75,695 stromal cells, and 152,509 and 201,072 immune cells (Fig. 1B). We evaluated standard quality metrics (number of genes and UMI per cell, percentage of mitochondrial reads) within each type of sample processing (Fig. S1A). The number of genes and UMI per cell was generally lower in the non-separated sample, potentially reflecting a lower recovery. The fraction of mitochondrial genes generally showed more limited differences between the different sample fractions, but as expected the percentage of mitochondrial reads was higher in epithelial cells compared to immune or stromal cells, as epithelial cells are prone to death by anoikis during tissue dissociation. To account for these technical factors, we adjusted for layer and the number of genes detected in applicable downstream analyses.
Further detailed clustering and annotation resulted in 65 cell types/states (Fig. 1C, Fig. S1B), which we next used to perform an analysis of Bray-Curtis dissimilarities to identify the main drivers of cell type composition across samples. As expected, the processing of biopsies, either in a single digestion step or as separated epithelial and lamina propria fractions (Methods), was the main driver of cell type composition (Fig. 1D, top; PERMANOVA R2 = 0.32, p < 10−4). Location (colon vs terminal ileum; Fig. 1D, middle) also accounted for a large portion of the variability (R2 = 0.14, p < 10−4). We noted that the previously published16,20 control samples did not form an outgroup, and clustered with the rest of the colon samples from this study (Fig. S1C). Separation by disease status (healthy, non-inflamed or inflamed; Fig. 1D, bottom) was visible, with a less obvious role in the first two axes of variation compared to layer (PCoA plots for each layer can be found in Fig. S1D, where these differences are more apparent). These compositional differences were still statistically significant (R2 = 0.055, p < 10−4), prompting us to further evaluate the influence of disease and inflammation status in more detailed analyses.
CD and inflammation broadly restructure immune and stromal compartment composition
We first examined disease-related differences in cell type composition of each sample using Dirichlet regression (Methods, Fig. 1E, Fig. S1E–F). Consistent with their inflamed status, we found that numerous immune cell types were compositionally overrepresented in inflamed samples, after adjusting for layer differences (Methods). This compartment-level observation was replicated in both TI and colon, though individual cell types had different patterns. Overall, we observed greater remodeling of the immune and stromal compartments in both locations compared to epithelial cells, though this was predominantly among T cells in the colon and myeloid cells in TI. Specifically, in TI, we observed 10 of 27 (37%) immune cell groups were altered in disease (Inflamed vs Healthy) compared to 6/16 (38%) stromal cells and 3/17 (18%) epithelial cells. In the colon, 8 of 25 (32%) immune cell groups were altered in disease compared to 6/17 (35%) stromal cells and 0/13 (0%) epithelial cells, though several of these epithelial cell types were significantly reduced in non-inflamed samples. Plasma cells made up the largest fraction of immune cells in all conditions, and were underrepresented in the inflamed colon (Fig. S1F) (consistent with previous observations16). However, in TI, we found the opposite pattern, with plasma cells overrepresented in inflamed tissue. Some of these compositional changes in the immune compartment already existed in diseased non-inflamed samples (Fig. 1E, top), including several expanded myeloid cell subsets (DC2 in TI, mast cells in both TI and colon), an increase in some fibroblast subsets in TI, as well as the expansion of lymphatic endothelial cells. On the other hand, the expansion of some subsets such as S100A8 S100A9 monocytes was strongly associated with active inflammation. This is consistent with the presence of either underlying low-grade inflammation or permanent reshaping of cellular compartments even in regions of the intestine that do not appear inflamed by endoscopic examination.
With an opposite trend to plasma cells, several stromal cell subsets were enriched in inflammation in TI and depleted in CO. The abundance of several fibroblast subsets in particular was reduced in CO, including the SMOC2+ PTGIS+ and ADAMDEC+ Fibroblast clusters (Fig. 1E, bottom). A previously-identified IL11-producing inflammation-associated fibroblast16,20 was expanded in the inflamed colon, and not detected in TI. As fibroblasts are associated with resistance to anti-TNF therapy17, but also participate in the development of fibrotic lesions and strictures present in CD complications, these results suggest that there may be organ-specific processes that require further characterization. We further discuss some of the differentiation processes involved in these distinct fibroblast subsets in more detail below (Fig. 5). Pericytes were an exception to this trend, showing a large compositional enrichment in inflamed colon samples, which was absent in TI.
Figure 5. Molecular regulators of myofibroblast activation in inflamed tissues.

(A) UMAP of the myofibroblast sub-groups in TI (HHIP+ NPNT+ and GREM1+ GREM2+), overlaid with expression of myofibroblast markers and sub-group markers. (B) Myofibroblast sub-groups clustered into two distinct clusters. (C) Pseudotime analysis of TI myofibroblasts. (D) Expression profiles of 200 selected genes with significant expression changes with respect to pseudotime (Methods). Highlighted genes include sub-group marker genes from (A) for the myofibroblast sub-groups, as well as significant genes in (E). Expression (TP10K) was first smoothed with a cubic spline and standardized to highlight expression differences (Methods). (E) Volcano plots for differential expression of four collagen genes and four HHIP+ NPNT+ marker genes in siRNA knockdown of select genes. Collagen genes were selected based on their expression in this population of cells (COL5A3 and COL7A1), and expression in myofibroblasts in literature61. The top 5 genes by FDR are labeled. Dashed red line indicates FDR 0.05. (F) (Left) Fibroblasts were stained for COL7A1 expression (green) and counterstained with DAPI (blue) after knock-down of the indicated genes (left to right: control, TBX3, CHMP1A and RNF168). (Right) MFI of COL7A1 staining in the indicated cells with (pink) and without (gray) TGFβ treatment. Each dot represents the average of 30 images, n = 6 independent wells.
As expected, in samples where we collected both an epithelial fraction (stripping the epithelial layer with EDTA) and a lamina propria fraction (enzymatically digesting the underlying tissue), the epithelial fraction was mostly comprised of epithelial cells (mean 54% ± 24% sd), while the lamina propria fraction comprised mainly immune and stromal cells (mean 77% ± 19% sd). However, we also noted a significant representation of some immune cells in the epithelial fraction in TI, likely representing intraepithelial lymphocytes (IELs). As these cells can be ontogenically and functionally distinct from lamina propria lymphocytes22, we analyzed compositional changes for immune cells across epithelial fractions separately. In particular, we identified a population of ID3+ ENTPD1(CD39)+ IELs, consistent with previous reports 23 that was only detected in epithelial samples (Fig. S2A–B) and was compositionally underrepresented in non-inflamed disease samples (Fig. S2C). This suggested a remodeling of the IELs compartment that can persist in the absence of overt inflammation. We observed an enrichment of plasma cells, which was consistent with the general trend (Fig. 1E), but even more marked in the epithelial fraction as healthy epithelial samples did not contain any intra-epithelial plasma cells (Fig. S2C). Finally, we also noted an overrepresentation of other immune cell types, in particular CD4+ and CD8+ T, which was generally more marked than tissue-level trends where few differences were observed for these cell types. Overall, our results suggest a remodeling of the IEL compartment during disease, both in inflamed and non-inflamed tissue, that is characterized by a reduction in the frequency of bona fide ITGAE(CD103)+ ENTPD1(CD39)+ IELs and an increase in plasma cells and conventional T cells.
Inflammation-related differences in core IBD risk gene expression are site-specific
After analyzing differences in cell type composition, we focused on gene expression across these cell types. IBD has a significant genetic component and multiple GWAS as well as more recent exome studies have identified a large set of risk genes over the years, but the relevant cell types and mechanisms of action of some of these genes has remained relatively unclear. Using Gini coefficients as a measure of unequal expression distributions, we first quantified the cell subset specificity of a core set of IBD risk genes identified from fine mapping GWAS24. Most IBD risk genes were highly specific to certain cell types (mean Gini coefficient 0.55, Fig. 2A, Table 1). Expression specificity was largely consistent between TI and colon (Fig. 2A), with some exceptions including CARD9 and IL2RA. CARD9 is a key signaling protein involved in the innate immune system’s response to fungi and bacteria and is primarily expressed in the TI by a subset of macrophages, while it is expressed in colonic cells within a subset of dendritic cells. IL2RA, a receptor for interleukin 2, is specifically expressed in Tregs in the colon, and is expressed by a more diverse constellation of cell types in the TI, which includes Tregs but is dominated by PLA2G2D+ macrophages.
Figure 2. Expression profiles across cell types of core IBD risk genes.

(A) Scaled mean expression (Methods) of 20 core IBD risk genes in both TI and colon. (B) Fraction of cells from healthy, non-inflamed, and inflamed samples expressing core IBD genes for cell types with at least 200 cells available. Each point represents the fractions of cells within a cell type expressing that particular gene. Gene-cell type pairs with a difference in fraction of genes expressing greater than 0.3 (dashed lines) are labeled. Note that for readability, cell types are summarized to their category, causing some gene labels to be repeated for each specific cell type. Individually differentially expressed gene-cell type pairs (FDR<0.05) are highlighted (red).
Table 1. Gini coefficients for 20 core IBD risk genes in TI and colon.
Functional relevance to IBD from12.
| Functional relevance in IBD | Gene names | Gini (TI) | Highest mean expression cell type (TI) | Gini (CO) | Highest mean expression cell type (CO) | |
|---|---|---|---|---|---|---|
|
| ||||||
| IL23R | Adaptive immunity, Th17 | IL23R | 0.84 | ILCs | 0.85 | ILCs |
| NKX2–3 | Restitution | NKX2–3 NKX23 NKX2C | 0.81 | Endothelial cells CA4+ CD36+ | 0.76 | Endothelial cells CD36+ |
| CARD9 | Microbe-sensing | CARD9 | 0.77 | Macrophages PLA2G2D+ | 0.77 | DC2 CD1D− |
| EBF1 | B cells | EBF1 COE1 EBF | 0.72 | Pericytes HIGD1B+ STEAP4+ | 0.82 | Pericytes HIGD1B+ STEAP4+ |
| IL2RA | Adaptive immunity, Treg | IL2RA | 0.70 | Macrophages PLA2G2D+ | 0.74 | Tregs |
| NOD2 | Microbe-sensing | NOD2 CARD15 IBD1 | 0.71 | Monocytes S100A8+ S100A9+ | 0.73 | Monocytes S100A8+ S100A9+ |
| HNF4A | UPR, Healing | HNF4A HNF4 NR2A1 TCF14 | 0.68 | Epithelial cells HBB+ HBA+ | 0.75 | Enterocytes TMIGD1+ MEP1A+ |
| PTPN22 | Tolerance | PTPN22 PTPN8 | 0.61 | T cells OGT+ | 0.67 | T cells OGT+ |
| LRRK2 | Lysosome function | LRRK2 PARK8 | 0.59 | Neutrophils S100A8+ S100A9+ | 0.65 | Monocytes S100A8+ S100A9+ |
| IKZF1 | B cells, Treg | IKZF1 IK1 IKAROS LYF1 ZNFN1A1 | 0.51 | T cells OGT+ | 0.61 | T cells OGT+ |
| SLC22A5 | (carnitine transporter) | SLC22A5 OCTN2 | 0.54 | Epithelial cells METTL12+ MAFB+ | 0.49 | Enterocytes TMIGD1+ MEP1A+ |
| GPR35 | Epithelial barrier | GPR35 | 0.50 | Neutrophils S100A8+ S100A9+ | 0.52 | Enterocytes TMIGD1+ MEP1A+ |
| INPP5E | (phosphatase) | INPP5E | 0.39 | T cells OGT+ | 0.42 | Pericytes HIGD1B+ STEAP4+ |
| PRDM1 | B cells, Treg | PRDM1 BLIMP1 | 0.37 | Plasma cells | 0.43 | Macrophages LYVE1+ |
| JAK2 | Adaptive immunity, Cytokines | JAK2 | 0.34 | Neutrophils S100A8+ S100A9+ | 0.44 | Monocytes S100A8+ S100A9+ |
| SMAD3 | Treg | SMAD3 MADH3 | 0.35 | Neutrophils S100A8+ S100A9+ | 0.42 | T cells OGT+ |
| IFIH1 | Microbe-sensing | IFIH1 MDA5 RH116 | 0.29 | Neutrophils S100A8+ S100A9+ | 0.36 | Monocytes S100A8+ S100A9+ |
| TYK2 | Adaptive immunity, Cytokines | TYK2 | 0.29 | Monocytes S100A8+ S100A9+ | 0.32 | T cells OGT+ |
| NFKB1 | Immune signaling | NFKB1 | 0.23 | Neutrophils S100A8+ S100A9+ | 0.32 | Monocytes S100A8+ S100A9+ |
| EP300 | (transcriptional coactivator) | EP300 P300 | 0.22 | Tuft cells | 0.31 | T cells OGT+ |
Then, we examined how disease and inflammation status impact the expression of risk genes. Core IBD risk gene expression distributions between healthy and diseased samples show gene, cell type, and location differences. Myeloid cells in TI tend to have reduced expression of core IBD risk genes in diseased samples (Fig. 2B, left panels). This reduction is not visible for other cell types. NKX2–3 in particular is expressed in a higher fraction of healthy stromal cells compared to both inflamed and non-inflamed stromal cells (Fig. 2B, left panels). On the other hand, in the colon, numerous IBD risk genes were expressed in a higher fraction of both inflamed and non-inflamed cells compared to healthy cells (Fig. 2B, right panels). Differential expression analysis (Methods) further highlighted PRDM1 as differentially expressed (DE) in several cell types in colon (Fig. 2B, right panels). DE core IBD genes (FDR < 0.05, Fig S3A–B) were further biased towards being up-regulated in the colon (208 up-regulated gene-cell type pairs vs 7 down), while comparatively fewer were observed in TI (1 up-regulated gene-cell type pairs vs 13 down). These results suggest that even in the context of shared risk genes between different subtypes of inflammatory bowel disease (ileal and colonic Crohn’s but also for many of these genes ulcerative colitis, which was not included here), the changes in gene expression associated with inflammation are distinct across ileum and colon.
To contextualize these changes between sites, we additionally looked for baseline differences between the two sites among the healthy donors (Fig. S3C–D). We found that, in general, these IBD risk genes have lower expression in colon than in TI, in particular for immune-related cell types (Fig. S3E), showing the opposite trend from the DE results above in inflammation. This is particularly true of EP300, and PRDM1, the latter of which was highlighted above.
In addition to these well-characterized core-IBD genes, exome sequencing approaches recently identified five additional IBD-associated loci: IL10RA, DOK2, CCR7, PTAFR, and PDLIM511. IL10RA, DOK2, CCR7, and PTAFR were primarily expressed in immune cells, while PDLIM5 had more broadly-distributed expression (Fig. S4A). PDLIM5 showed the greatest expression differences in disease, frequently overexpressed in inflamed samples compared to healthy colon samples (Fig. S4B, Table S3). While further investigation of PDLIM5’s role in epithelial cells is warranted, these analyses demonstrate the potential of scRNAseq resources to identify relevant cell types for risk genes, and suggests that PDLIM5 coding variants could modulate the epithelial barrier.
Inflammation-associated transcriptional changes are largely site-specific and more pronounced in the colon
Differential expression in inflamed versus healthy tissue was quantified on a per location and per cell type basis (Methods, Fig. 3A–B, Table S3). There was some consistency between the differential expression profiles between the two sites (Fig. 3C), primarily observed in epithelial and stromal cells (Spearman rho = 0.25 and 0.34, respectively; P < 10−307), with immune cells showing the least correlation (rho = 0.21; P = 10−204). These weak though highly significant correlations indicate there is commonality in the genetic programs driving the inflammation signatures in the two sites, though the two sites still behave very differently. We therefore quantified the degree to which different cell types exhibited a more consistent set of DEGs in the two locations (Methods), and find that several myeloid cell types, DC2 CD1D−, Macrophages, and Mature DCs, exhibit the most consistent inflammatory signal (Fig. 3D). Consistent genes in this group highlight existing IBD-associated genes including STAT125, LSP126, and HIF1A27(Table S4). Our results also replicate and extend a previous finding that inflammation-related expression differences are highly correlated with the differences between non-inflamed vs healthy already present in individuals with IBD16, which we observe in both sites (Fig. 3E).
Figure 3. Location- and cell type-specific differential expression in active CD.

(A) Number of differentially-expressed genes between inflamed CD and healthy samples in TI, broken down by cell type (discrete component of a MAST model; FDR < 0.05). (B) Same as (A) but for colon. (C) Relationship between differential expression in TI and colon for each cell compartment. (D) Consistency score (Methods) of differential expression in inflammation for each common cell type between TI and CO. (E) Relationship between inflamed vs healthy and non-inflamed vs healthy samples in TI and colon within each cell compartment. (F) Fraction of cell types in which KEGG pathways are significantly enriched (FDR < 0.05; see Methods) within each compartment and location, split by enrichment direction (all pathway enrichment results in Table S5).
Differential expression was more pronounced in the colon compared to TI, in terms of the total numbers of differentially expressed genes (DEGs) detected (Fig. 3A–B). The transcriptional response to inflammation in the colon is therefore more marked than in TI. These differences were particularly strong among epithelial cell types, where some cell types in the colon showed thousands of DEGs, in particular among some enterocyte groups and goblet cells. This level of DEGs is roughly an order of magnitude larger than what we observed in immune cells. This is in stark contrast to the earlier observations at the compositional level, where epithelial cell types showed the least differences in inflammation (Fig. 1E). Some of these expression differences are already visible in non-inflamed tissue (Fig. S4C–D), particularly in a subset of goblet cells (with 1994 common DEGs, a 50% overlap), showing that these cells may already be primed for the inflammatory response in CD patients.
Pathways enriched in the epithelial compartment included antigen processing and presentation and cell adhesion molecules, as well as many disease-related pathways, which were broadly perturbed across numerous cell types (Fig. 3F; discussed in more detail in the following section). In the colon, numerous metabolic pathways were significantly down-regulated, largely due to down-regulation of the ketogenesis pathway (Fig. S4E), a common component of these enriched pathways. This suggests reduced potential for ketogenesis during inflammation. Treatment of IBD using ketogenic diets has been tested, with mixed results28,29. Ketogenesis is regulated in part by the PPAR signaling pathway, which also shows a consistent pattern of altered expression (Fig. 3F; Fig. S4E), in particular PPARG, suggesting that this is the key regulatory factor of ketogenesis in CD.
In contrast to DEGs in the epithelial compartment, we found that the majority of DEGs in the stromal cell types were consistently down-regulated across cell types in TI, while colon DEGs were more balanced (Fig. 3A–B). Despite this broad downward trend in TI DEGs, several pathways were positively enriched in this location, including oxidative phosphorylation and NOD-like receptor signaling pathways across numerous cell types. Other positively enriched pathways in TI included numerous disease-related pathways which were significantly enriched in particular in three cell types: HHIP+ NPNT+ Myofibroblasts, Glial cells, and Lymphatics (Table S5). These trends were not observed in the colon.
Despite expectations that cell types from the immune compartment would exhibit a greater inflammation-related DE signature, the immune compartment showed the least differential expression in inflammation (Fig. 3A–B), which is reflected in a reduced number of significantly enriched pathways (Fig. 3F). This effect was not explained by a difference in power linked to cell count differences, as the immune cells accounted for a plurality of cells in both TI and colon (46.7% and 52.6% respectively). Instead, this is consistent with the notion that compositional changes, for example caused by the infiltration of activated immune cells (Fig. 1E), are the main drivers of immune differences. Meanwhile, transcriptional changes in epithelial and to a lesser extent stromal cells appear to account for the majority of the response in that compartment. This effect is most pronounced in TI, where stromal and epithelial cell types both exhibited similar magnitude differences, while immune cells showed an order of magnitude fewer DEGs and have a similar bias towards down-regulation, also seen in stromal TI cells.
MHC class II genes drive distinct inflammatory signals in TI and the colon
Pathway enrichment analysis highlighted several immune- and disease-related pathways (Fig. 3F). These were largely driven by a core set of HLA genes common to all of these gene sets (Fig. S5A), primarily from MHC class II. Differential expression profiles of these genes revealed similar differential expression patterns in both TI and colon, with several notable exceptions. First, HLA genes as a group tended to be downregulated in inflammation in immune cells in the colon, particularly in dendritic cells, and this expression pattern was not observed in TI (Fig. S5A). Further, HLA-DRB5 was broadly overexpressed in inflammation in numerous epithelial cell types in TI yet it was mostly absent in the colon.
Mucin and claudin expression changes highlight a site-specific rewiring of barrier functions
Cell surface mucins contribute to the protective mucosal barrier between the intestinal epithelium and the lumen. While this barrier protects against bacterial invasion30, it also modulates inflammatory signals31. Ectopic mucin expression may therefore contribute to an exaggerated immune response. We found that MUC1, a cell surface mucin typically expressed in the stomach32, was upregulated in non-inflamed TI samples in CD (Fig. S5B, right), and was further increased during inflammation across epithelial cell types in both TI and colon (Fig. S5B, left). In the colon specifically, we additionally observed broad up-regulation of several other mucins: MUC2, MUC4, MUC5B, and MUC12. These are more typical of colonic mucins with the exception of MUC5B, which is a salivary mucin.
We also observed differential expression for other constituents of the mucosal layer. In particular, TFF1, a trefoil peptide which stabilizes the mucosa33, followed a pattern consistent with MUC1 and was strongly up-regulated in the inflamed colon and weakly up-regulated in TI (Fig. S5B, left), with almost no changes in non-inflamed samples (Fig. S5B, right).
Claudins, on the other hand, serving as backbone of tight junctions, are involved in the establishment of barrier properties and help to maintain the specificity of tight junction permeability34,35. Increased permeability and remodeling of tight junctions has been seen in CD patients36. Altered expression of claudin 2 and occludin has also been observed prior to CD onset37. The overall expression patterns of detected Claudin family genes were consistent between TI and colon (Fig. S5C), but there were a few claudins that expressed differently between the sites (Fig. S5D). In particular, claudin 2 and 15, two pore-forming claudins, showed higher expression among many epithelial cell types in TI compared to colon, suggesting increased paracellular permeability in the TI epithelium. On the other hand, claudins 3,4 and 5, which are sealing or barrier-forming claudins, showed higher expression in several colon stromal cell types, indicating greater barrier function in colon. We then focused on the impact of disease on the expression of this family. Across sites, claudins overall showed consistent expression changes, but changes in the colon were more pronounced than in the TI. Among pore-forming claudins, claudin-2 was strongly upregulated, whereas claudin-15, which forms Na+ channels, was downregulated. Claudins 3, 4, 7 and 23 were broadly downregulated, and a number of these differences occurred in stem/cycling cells, indicating a potential interaction with epithelial proliferation or crypt biology. These disease-associated expression changes in claudins are broadly consistent with previous measurements35,36,38.
CD leads to metabolic changes in enteroendocrine cells
Enteroendocrine cells (EECs) sense microbial metabolites, and thus are key players in the initiation of the intestinal immune response39. Because of their rarity, EECs are hard to profile in single-cell studies, and studies have relied on ex vivo culture and enrichment of these cells40. Thus, their response to intestinal perturbations remains poorly characterized. Leveraging the size of our dataset, we focused on the TI epithelial compartment and detected 670 high-quality enteroendocrine cells (EEC), exhibiting high expression in markers CHGA and CHGB (Methods, Fig. 4A–C). These further clustered into 8 EEC subsets (Fig. 4A) based on established marker genes40,41. No donor or disease group was dominant in one of these subtypes, showing that this heterogeneity is not a donor-specific artifact (Fig. 4B). The two most common EEC subsets were both enterochromaffin (EC) cells, expressing TPH1 and REG4. N-cells and progenitors were the next largest EEC subsets, followed by several rarer EEC cell types: L-cells, D-cells, I-cells and K-cells.
Figure 4. Enteroendocrine Cells (EEC) in TI.

(A) UMAP of 670 EECs in TI, colored by subset. (B) Donor and disease composition among EEC subsets. (C) Expression of markers for major EEC subsets.
Given the limited number of EECs, only the two largest clusters, EC THP1+CES+ and EC REG4+NPW+, were used for differential expression analysis. In EC THP1+CES+ cells, DEGs suggested endoplasmic reticulum (ER) stress in CD, with UBA5, NCK1, SERINC3, CREB3L1, PDIA3, and TMEM33 showing altered expression in non-inflamed or inflamed tissue (FDR < 0.05; see Table S3). This was coupled with an overall increase in several respiratory genes, pointing to increased energy consumption by these EC cells. DEGs also included ATIC (FDR 0.001) and MTHFD1 (FDR 0.021), two genes involved in purine metabolism, which were both up-regulated in inflamed and non-inflamed samples (Table S3). Previous studies have found altered purine signaling in CD42, which may therefore be driven in part by these EC cells. DEGs in EC REG4+NPW+ cells were negatively enriched in oxidative phosphorylation (FDR 6.79 × 10−4) suggesting an inverse relationship between these two EC clusters.
In the colon, we also detected a similar, though smaller population of EECs with 164 cells in total (Fig. S6A–C). EC and progenitors were common subsets between TI and the colon. D/L/N-cells were less distinguishable in the colon, and I/K-cells were not detected. We also detected a unique subset among colon EECs (Fig. S6D) which was annotated as LEFTY1+. Marker genes from this subset were associated with colon homeostasis, tumor suppression, host defense against inflammation, and cytokine activity.
Pseudotime analysis identifies CHMP1A, TBX3, and RNF168 as regulators of collagen expression in myofibroblasts
As noted above, we observed that a population of myofibroblasts in the TI was expanded during ileal inflammation (denoted Myofibroblasts HHIP+ NPNT+; Fig. 1E, top). This myofibroblast population was enriched in genes involved in extracellular matrix deposition, such as COL18A1, and COL23A1 (Fig. 5A), which are implicated in beneficial wound healing responses43 but also associated with fibrotic strictures observed in CD44. This population of myofibroblasts clustered closely with the myofibroblast population denoted GREM1+ GREM2+ (Fig. 5B); however, we observed that GREM1+ GREM2+ myofibroblasts lacked collagen expression (Fig. 5A). To explore the regulatory network that drives collagen expression in CD myofibroblasts, we utilized pseudotime trajectory analysis (Methods) to organize cells starting from collagen-negative GREM1+ GREM2+ myofibroblasts to HHIP+ NPNT+ collagen-positive myofibroblasts (Fig. 5C).
This analysis revealed numerous genes with pseudotime-dependent expression in the transition between these myofibroblast groups (Fig. 5D). We selected a subset of these for follow-up based on gene annotation and expression levels, resulting in a set of 6 transcription factors and 10 other genes (Methods). We used 4 pooled siRNA oligos to knockdown (KD) each of these candidate genes in an arrayed approach in normal human intestinal fibroblasts. We assessed induction of COL4A1, COL4A2, COL5A3, and COL7A1, as well as four HHIP+ NPNT+ marker genes following stimulation with canonical collagen-inducer growth factor TGF-β. While some gene KDs had collagen-specific effects, we observed that KDs of CHMP1A, TBX3, and RNF168 significantly impaired production of several of these collagen genes and proteins (FDR < 0.05; Fig. 5E–F). In particular, the transcription factor TBX3 was strongly associated with TGF-β-driven collagen gene expression, and has previously been implicated in driving carcinomas and sarcomas45,46. TBX3 has also been reported in several clusters in a recent fibroblast cell atlas in mice47, including a cluster (characterized by Adamdec1) that is specifically associated with colitis in the perturbed-state dataset15. Both tumors and fibrotic scars are associated with enhanced deposition of extracellular matrix48, thus future efforts may investigate the in vivo role of TBX3 in driving tissue fibrosis in CD. We also observed a similar overall pattern in the colon, though fewer myofibroblast cells were sampled there (Fig. S6E–H). Other genes identified in the pseudotime analysis may therefore be of interest for further follow-up.
Finally, we applied the NicheNet algorithm49 to find putative ligands responsible for transitioning myofibroblasts between the two states. Since more differential expression was detected in the colon (Methods, Fig. 3B), we focused this analysis there. Ligands responsible for the induction of collagen genes more commonly interacted with GREM1+ GREM2+ myofibroblasts than with HHIP+ NPNT+ (Fig. S6I–J). In addition to the TGF-β signature, we also noticed activity by a related set of genes in these cells, BMP2/5/7, largely derived from other fibroblasts (Fig. S6I). BMP2/5/7 were all differentially expressed in at least one other fibroblast cell type in diseased samples, indicating that these may play a part in the miscommunication resulting in CD progression. Interestingly, these BMP ligands have been identified as markers of a mesenchymal niche in a previous study of the colonic mesenchyme, and CyTOF analysis based on a subset of markers suggests that these cells may be diminished in disease15.
DISCUSSION
In this study we describe the single-cell expression profiles of 720,633 cells from 71 patients, providing the largest single-cell resource to date to study CD. Our dataset covers the epithelial, immune and stromal compartments across multiple locations and multiple disease statuses, therefore allowing us to characterize the cell-type-specific differences along these important dimensions.
One striking difference observed across compartments was the nature of the response to disease and inflammation. The epithelium experienced the greatest changes in expression profiles, including a broad increase in expression of MHC class II genes, as found in Thomas et al50. Meanwhile, immune cell differences in gene expression were comparatively smaller (including decreased HLA expression), but their compositional changes were more marked. Stromal cells displayed both transcriptional and compositional changes, perhaps reflecting a joint reprogramming and tissue remodeling. In all three compartments, transcriptional changes in non-inflamed disease samples and inflamed samples were strongly correlated, a phenomenon that has also been reported in the case of ulcerative colitis both for broad cellular networks16 and more specifically among epithelial cells14. This may reflect ongoing disease processes even in the context of endoscopic remission. Specifically in CD, our results extend previous findings showing that cell type composition profiles poorly discriminate between inactive and active CD5. Understanding the pathways that are involved in the maintenance of this “inflamed-like” transcriptional network in endoscopically normal tissue might uncover key targets for disease-modifying therapies and ultimately curative approaches.
As CD can occur throughout the intestinal tract, we were also able to directly compare the inflammatory response in the colon and TI, and observed a notably stronger transcriptional response in the colon. Expression differences in TI and colon were correlated, though not strongly so, indicating that the transcriptional programs underlying the inflammatory response are largely different in the two sites. Among pathways specifically enriched in the colon, we found numerous metabolic pathways largely different due to a down-regulation of the ketogenesis sub-pathway, driven by PPARG51. Interestingly, ketogenic diets have been trialed with mixed success in treating CD28,29. Our results provide a novel resource to analyze the network associated with the ketogenesis pathway and may offer insights on the individuality of patient responses to ketogenic diets, which may also be driven by personalized factors such as the microbiome, as has been reported with epilepsy52. More broadly, this may be combined with recent developments in single-cell proteomics and metabolomics (reviewed by Islam, et al53), which provide the opportunity to directly explore the associations between transcriptional programs and metabolic networks.
In addition to the analysis of these large scale, cross-compartment changes, the scale of our study also allowed us to focus on rarer cell subsets. For example, we identified a sizable and transcriptionally distinct subset of immune cells in our epithelial fractions (i.e. cells detached from the tissue with EDTA + DTT disruption of junctions, in the absence of enzymatic digestion). We confirmed that these cells are intraepithelial lymphocytes, consistent with a previously-described ID3+ ENTPD1(CD39)+ IEL group23. Importantly, we observed an overall depletion of these cells in diseased samples, reminiscent of the remodeling of the IEL compartment that has been described in celiac disease54. However, it is important to note that IELs can be comprised of both conventional and non-conventional T cells, and the latter express a restricted set of TCRs that can recognize a range of self and non-self molecules55. These cells have for example been shown to regulate nutrient sensing56 as well as inflammation23. An important follow-up to this work will be to understand the repertoire of IELs in health and disease, for example using V(D)J sequencing approaches.
We were also able to characterize in detail the response of rare cells such as enteroendocrine cells in the context of inflammation. Previous studies have used ex vivo expansion to characterize the transcriptomic profile of the different enteroendocrine cell subsets40, but such approaches cannot capture the changes that may occur in disease in these cells. Our work shows an enrichment of ER stress signatures in the context of disease. This finding is particularly relevant given the role of ER stress and the unfolded protein response in the genetic risk for Crohn’s disease12,57,58. The role of ER stress in secretory cells in the intestine, such as Paneth cells or goblet cells, has been long recognized59 and the enrichment of ER stress in EECs during disease suggests that the secretory function of these cells may also be affected in Crohn’s potentially modulating their function and interactions with the nervous system.
Beyond the study of IBD-associated risk genes, single-cell atlases offer unprecedented opportunities to map the emergence of disease associated cell states. Here, we focused specifically on fibroblasts, as these cells have been associated with pathology and therapy resistance60 but remain poorly characterized in IBD compared to immune or epithelial subsets. For this, we leveraged our transcriptomic data to identify a subset of genes linked to the transition between two myofibroblast subsets with differential abundance in disease and with differential collagen production characteristics. From this, we validated a set of genes, TBX3, RNF168, CHMP1A, which impact collagen production in these cells. These genes may therefore be involved in CD-related fibrotic strictures and suggest novel therapeutic hypotheses for the management of this complication. Altogether, we demonstrate approaches that can leverage single-cell data to both facilitate variant to function assignment and the identification of disease pathway specific targets. We expect that similar future studies focused on other risk genes and compartments could broadly extend our understanding of the functional regulators of CD.
In conclusion, in this study we described the transcriptional perturbations in active CD at an unprecedented level of detail. The resulting analysis and dataset provide a framework for further investigation into the complex dysregulation of the gastrointestinal immune response in CD, and a testing ground for cell type specific differences in this disease.
LIMITATIONS OF THE STUDY
In this study, we report on single-cell analyses performed across 46 Crohn’s disease subjects and 25 non-IBD controls in a single center. This number enabled us to deeply characterize the differences associated with both active inflammation and non-inflamed intestinal tissue in Crohn’s disease. However, it is important to note that there are many layers of heterogeneity that we were not powered to address here and that would require more targeted collections. For example, understanding the impact of biologics on tissue state will likely require focusing enrollment on a limited number of therapeutic agents and obtaining longitudinal samples pre- and post-treatment. Additionally, it will be critical to enroll participants from a diverse range of ancestries and risk genes to understand the impact of genetics on disease phenotype.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ramnik J. Xavier (xavier@molbio.mgh.harvard.edu)
Materials Availability:
This study did not generate any novel reagents, all materials are commercially available as listed in the key resource table.
Data and Code Availability:
The datasets generated during the current study are available for download from the controlled-access data repository, Broad DUOS (Accession DUOS-000146 CD_Atlas_2021_GIDER; DUOS-000145 CD_Atlas_2021_PRISM). The analyzed data reported in this paper is available at the Broad Single Cell Portal (SCP1884).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and tissue sample collection
Subjects were enrolled in either the PRISM (Prospective Registry in IBD Study at MGH, protocol 2004P001067, used for all CD patients and some controls) or the GIDER (GI disease and endoscopy registry, protocol 2015P000275, used for the remaining controls) study at Massachusetts General Hospital (MGH). Informed consent was obtained from all patients in accordance with the respective protocol and sequencing and data storage and publication plans were approved by the MGH IRB and the Office for Research Subject Protection at the Broad Institute. Clinical information and metadata for the samples in this study were provided in Table S1. Healthy controls were recruited at the time of routine colonoscopy. Healthy controls were individuals without a history of inflammatory bowel disease (IBD), a 1st degree relative with IBD, histories of autoimmune disease, immune mediated conditions, infectious colitis, and colon cancer, or a family history of colon cancer, and who were overall healthy with no other disease history. CD patients were included based on having a clinical diagnosis of Crohn’s disease, and observed to have active disease via macroscopic assessment from a physician during an endoscopy as part of routine clinical care. Biopsies were obtained during endoscopy, using biopsy forceps that were used in standard of care. The presence or absence of inflammation was visually evaluated by the endoscopist at the time of collection. To ensure this evaluation was consistent across endoscopists, we used the simple endoscopic score for Crohn’s disease (SES-CD)21. This score consists of segmental scores that are then summed to obtain an overall indicator of disease activity. At the patient level, an SES-CD of 0–2 indicates remission/inactive disease while an SES-CD score ≥ 3 indicates active inflammation, criteria that are consistent with previous studies62,63. Of note, this score has been used in clinical trials64 and was shown to have limited variability across raters65. In patients with active disease, we aimed to collect both inflamed biopsies (segmental score > 0 and visible inflammation) and non-inflamed biopsies (segmental score = 0). Biopsy bites were immediately placed into cryovials containing Advanced DMEM F-12 and placed on wet ice for transport.
METHODS DETAILS
Epithelial Layer Dissociation
On arrival, biopsy bites were washed 2x in cold PBS and in 3x in cold PBS/10mM EDTA. The tissue was then added to 25 mL PBS/10mM EDTA and placed in a rotating incubator at 37°C for 15 minutes. Following incubation, the tissue rested on ice for 10 minutes and was then shaken vigorously for 10–15 seconds. The supernatant was collected as fraction 1 and additional fractions were collected until the supernatant had visible crypts when viewed under the microscope. Tissue was kept on ice in a small amount of PBS/10mM EDTA for further lamina propria digestion, and fraction(s) with visible crypts were combined and spun down at 330g for 3 minutes. Supernatant was removed and the pellet was resuspended in 1mL pre-warmed TrypLE express (Thermo Fisher) for 1 minute. 1mL PBS was added to quench the reaction followed by another 4 mL PBS and the single cell suspension was spun down at 330g for 3 minutes. The pellet was resuspended in 1mL PBS and transferred to a 1.5 mL microcentrifuge tube, spun again at 300g for 3 minutes and resuspended in 50–200μl 0.4% BSA-PBS for 10X single cell loading.
Lamina Propria Layer Dissociation
Tissue saved on ice from epithelial layer digestion was moved into a 5mL snap-cap centrifuge tube with 5mL RPMI 1640 (Gibco, cat no. 11875093) supplemented with 2% FBS, 200μl Liberase TM (2.5 mg/ml, Roche, cat. no. 5401119001, reconstituted in injection-quality sterile water) and 50μl DNase I (10 mg/ml, Roche, cat. no. 10104159001, reconstituted in injection-quality sterile water). Tissue was incubated in a rotating incubator 37°C for 45 minutes. Following incubation, 0.5 mL FBS was added directly to the snap-cap tube which was then vortexed for 20 seconds. Tissue and media were poured over a 70μm filter into a falcon tube and 2% FBS-RPMI was added over the filter up to 30 mL. Sample was then spun down at 450g for 3 minutes. Supernatant was removed and pellet was resuspended in 1 mL 0.4% BSA-PBS and transferred to a microcentrifuge tube. Cell suspension was spun down at 300g for 3 minutes, supernatant was removed and pellet was resuspended in 1 mL ACK Lysing Buffer (Gibco, cat. no. A1049201) and incubated at room temperature for 1 minute. Cell suspension was spun down again at 300g for 3 minutes, washed two additional times in 0.4% BSA-PBS and resuspended in a final dilution of 50–200μl 0.4% BSA-PBS.
Single-Cell Profiling
Epithelial and lamina propria single cell suspensions were counted and, if necessary, diluted to a concentration of 200–2000 cells per μl. 10,000 cells from each sample were then loaded on a Chromium controller (10X Genomics). Samples were processed either with v2 or single-indexed v3.1 chemistry as described below, and chemistry type for each sample is included in Table S1.
For v2 samples, cells were loaded on a Chromium Single Cell A Chip (PN-120236) with gel beads from the Chromium Single Cell 3’ Library & Gel Bead Kit v2 (PN-120237) and indexed according to the Chromium i7 Multiplex Kit (PN-120262) instructions. Libraries were sequenced on either a NextSeq or a HiSeq X (both from Illumina), according to manufacturer’s instructions (Read 1, Cell barcode and UMI, 26bp, i7 index : 8bp, i5 index : none, Read 2, insert, 98bp).
For single-index v3.1 samples, cells were loaded on a Chromium Next GEM Chip G Single Cell Kit (PN-1000120) with GEMs from the Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 (PN-1000121) and indexed according to the Single Index Kit T Set A (PN-1000213) instructions. Libraries were sequenced on either a NextSeq or a HiSeq X (both from Illumina), according to manufacturer’s instructions (Read 1, Cell barcode and UMI, 26bp, i7 index : 8bp, i5 index : none, Read 2, insert, 91 or 96bp).
siRNA KD experiments in myofibroblasts
Normal colon-derived intestinal human fibroblasts (CCD-18Co) were obtained from the American Type Culture Collection (CRL-1459). Fibroblasts were maintained in DMEM containing GlutaMAX (Thermo Fisher, Catalog #10566016), supplemented with 10% (vol/vol) heat-inactivated FBS, NEAA (Gibco), penicillin/streptomycin (Corning). Cells were cultured at 37 °C with 5% CO2.
Pre-designed pooled duplexes of siRNA oligomers were purchased from Sigma-Aldrich and re-suspended in nuclease-free water at 20μM. Seeded CCD-18Co fibroblasts were transfected with 20nmol siRNA complexed with Lipofectamine RNAiMAX (Thermo Fisher) in Opti-MEM media (Thermo Fisher). 24 hours later, cells were washed with PBS, and replenished with fresh media with or without the addition of 10ng/ml of human TGF-β (Invivogen) for 24 hours. Cells were washed in PBS, and resuspended in TRIzol reagent (Thermo Fisher) for RNA isolation.
RNA was extracted from fibroblasts in TRIzol reagent following the manufacturer’s protocol (Thermo Fisher). Equal amounts of RNA were used to synthesize cDNA with the iScript cDNA synthesis kit (Bio-Rad Laboratories). iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories) was used for qRT-PCR on the C1000 Touch Thermal Cycler (Bio-Rad Laboratories). Gene expression was calculated with the ΔΔCt calculation with Hprt as the reference housekeeping gene. Oligos used for qRT-PCR can be found in Table S6.
Collagen Immunofluorescence
siRNA-transfected CCD18-Co fibroblasts were seeded in a 96-well CellCarrier-96 Ultra microplate (PerkinElmer, #6055302) overnight. The next day, the cells were treated with or without the addition of 10ng/ml of human TGF-β (Invivogen, rcyc-htgfb1) for 24 hours.
Cells were then fixed in 2% PFA (Electron Microscopy Services, #15710-S) followed by permeabilization with 0.2% Triton X-100. The cells were then washed with PBS and blocked with 4% BSA-PBS. Following blocking, cells were incubated with 5μg/mL anti-Col7a1 (ThermoFisher, #MA5-41570) in 4% BSA-PBS for one hour at room temperature. Cells were then washed with PBS and incubated with 1:500 dilution AF488 (ThermoFisher, #A-21202), 1:5000 dilution of HCS CellMask Red (ThermoFisher, #H32712) and 1:5000 dilution of Hoechst 33342 (ThermoFisher, #H3570) in 4% BSA-PBS for one hour. Cells were then washed with PBS and imaged.
For imaging, the Opera Phenix High-Content/High-Throughput imaging system (Perkin Elmer) was used. 31 different fields were imaged at 6 replicates per sample at 20x water immersion in the confocal setting. Image analysis was performed with the Harmony software (Perkin Elmer). Cell nuclei were identified with Hoechst staining, and each cell boundary demarcated by HCS CellMask Red. Median fluorescence intensity of AF488-labeled Col7a1 was quantified in each individual cell and the median values per sample well obtained followed by subtraction of background fluorescence.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-cell data processing
After sequencing, BCL files were demultiplexed with Cell Ranger v3.0.2, then fastq files were aligned to the human genome (hg19). CellBender v2-alpha66 was used to remove systematic biases and background noise (learning_rate= 2e-5), and Scrublet v0.2.167 was used to identify doublets and remove low quality cells (with default settings). Cloud-based Cumulus v1.068 was then used to perform the batch correction (using the Harmony algorithm) on the aggregated gene-count matrices, this was done separately for TI and colon samples. Unless specified otherwise, gene expression was quantified by the default logarithmic expression values in Cumulus, specifically ln(TP100k + 1), where TP100k = 105 * NUMI / CellNUMI, NUMI is the number of UMIs detected for that gene in that cell, and CellNUMI is the total number of UMIs detected in the cell.
To help balance the comparison between inflamed and healthy groups in the colon, we included data from 12 non-IBD patients (24 samples total, which were further layer-separated into 48 total channels) from16. For this, we processed these samples together with the rest of the 8 non-IBD samples from the colon location, using the same bioinformatics pipeline, which included Harmony for batch correction. Resulting samples clustered with existing samples from colon.
Scaled mean expression
Expression values presented in Figs. 2A, S4A, and S5D were obtained by scaling the ln(TP100K + 1) expression value by the root mean squared expression to produce an “expression z-score”.
Single-cell clustering and ordination
Clustering and UMAP visualization were done by following the Cumulus default settings. Principal coordinates analysis (PCoA) of cell type compositions (Fig. 1D and Fig. S1C–D) was performed using the pco function of the labdsv R package from Bray-Curtis dissimilarities between the compositional profiles.
Cell type identification and signatures
Cell clusters for each location were first manually classified into three compartments based on expression of known marker genes: Epithelial (EPCAM, KRT8, and KRT18), Stromal (CDH5, COL1A1, COL1A2, COL6A2, and VWF), and Immune (CD45/PTPRC, CD3D, CD3G, CD3E, CD79A, CD79B, CD14, CD16, CD68, CD83, CSF1R, FCER1G).
Each compartment was then re-clustered individually per location, and fine-grained cell types were identified using a combination of an automatic cell type annotation step in Cumulus (function infer_cell_types with markers = ‘human_immune’), and manual inspection and adjustment based on previously identified markers19. Briefly, Epithelial cells were clustered into Enterocytes (RBP2, ANPEP, FABP2), Stem cells (LGR5, ASCL2, SMOC2, RGMB, OLFM4), Goblets (CLCA1, SPDEF, FCGBP, ZG16, MUC2), Paneth cells (DEFA5, DEFA6, REG3A), Tuft cells (LRMP, SH2D6), Enteroendocrine cells (CHGA, CHGB, NEUROD1) and Cycling cells (UBE2C, TOP2A, MKI67, HMGB2). Stromal cells were clustered into Fibroblasts (ADAMDEC1, PDGFRA, BMP4), Myofibroblasts (TAGLN, ACTG2), Lymphatics (CCL21, TFF3), Endothelial cells (CD36, DARC/ACKR169), Pericytes (NOTCH3, MCAM/CD146, RGS5) and Glial cells (FOXD3, MPZ, CDH19, PLP1, SOX10, S100B, ERBB3). Immune cells were first clustered into T cells (CD3D, CD3G, CD3E), B cells (CD79A, MS4A1/CD20, CD19), and Myeloid cells (CD14, CD16, HLA-DR). T cells were further sub-clustered into CD8 T cells (CD8A, CD8B), CD4 T cells (CD4), ILCs (RORC, IL1R1, IL23R, KIT, TNFSF4, PCDH9), NK cells (EOMES, PRF1, NKG7). B cells were sub-clustered into Plasma cells (SDC1, MZB1, SSR4, XBP1), B cells (BANK, MS4A1/CD30, ADAM28, VPREB3) and Germinal Center (GC) B cells (LRMP, GPT2, PAG1)70. Myeloid cells were sub-clustered into Mast cells (GATA2, CPA3, HPGDS), classical Macrophages (CD163, C1QB, C1QC), classical Monocytes (FCN1, S100A4, S100A6), DC1 (CLEC9A, XCR1) and DC2 (CLEC10A, FCER1A). Some cell clusters were further subdivided if there was evidence of heterogeneity in the UMAP. These are identified by one or two genes whose expression distinguishes these clusters, for example T cells CD4+ IL17A+. A summary of markers used and expression across clusters can be found in Table S2.
Cell type compositional analysis
Cell type composition PCoAs in Fig. 1D were generated using Bray-Curtis dissimilarities between the cell type composition profiles for each channel. PERMANOVA analysis was done using the adonis function in the R package vegan using the same dissimilarity matrix, using 9999 permutations.
Differential cell type abundances were determined as previously described16 using Dirichlet regression with R package DirichletReg, to account for the compositional nature of cell type counts within a sample. For epithelial cell types, only samples from the epithelial layer or non-separated samples were used, while for stromal and immune cell types, lamina propria and non-separated samples were used. Sample layer separation (separated vs non-separated) was regressed out by testing the formula “Normalized counts ~ Layer separation + Disease status”.
IBD risk genes selection
Core IBD risk genes were obtained from Table 1 in Huang, et al24, and only genes associated with IBD or CD and which have nonzero expression in at least 3% of cells in at least one cell type were included (Table 1).
Differential expression analysis
Differential expression analysis was performed using MAST71. DE analysis was only run for cell types for which there were at least 10 cells in each disease group (healthy, non-inflamed, inflamed). For each cell type and location, low-expression genes were first filtered out (minimum 10% cells with non-zero expression in at least one of the disease groups). To speed up the tests, for cell types with more than 10000 cells, we first sub-sampled the cells using a hierarchical even subsampling algorithm: at each level of the hierarchy (disease group > donor > sample), an even number of cells were sampled from each possible pool at the lower level, such that 10000 cells were sampled in total. This ensured that disease groups, donors, and samples with few total cells were still adequately represented in the sampled dataset. To further speed up the tests, gene expression for each gene was first fitted with an anti-conservative fixed effect model in MAST, with formula “Expression ~ NGenes + Layer + DiseaseGroup”. Genes with no disease-related difference were filtered out (nominal P > 0.05 in the discrete and continuous components for both the Non-Inflamed - Healthy and Inflamed - Healthy contrasts, from likelihood ratio tests). Remaining genes were fit with a mixed-effect model in MAST using formula “Expression ~ NGenes + Layer + DiseaseGroup + (1 | Donor) + (1 | Channel)”, to account for additional correlations between cells from the same donor and from the same samples. P-values were obtained from likelihood ratio tests. FDR-corrected p-values were calculated from all tested genes (from all cell types and locations), using the P-value from the mixed effect model if available, or from the fixed effect model if not (to avoid selection bias from the fixed effect pre-filter). Unless otherwise specified, all reported coefficients and FDR values are from the discrete component of the MAST model16.
Differential expression consistency in CO and TI
To quantify the degree of consistency between DEGs in CO and in TI (Fig. 3D and Table S3), we calculated the expected overlap between the DEG lists, and quantified the “consistency score” of a pair of lists as the ratio between the observed overlap compared to expected. Specifically, for two DEG lists A and B (corresponding to DEG lists in CO and TI) and a total number of genes N, the expected overlap (ignoring direction) was first estimated as E = |A| × |B| / N. A DEG was only considered “consistent” if its direction was the same in the two lists. We therefore first split A into A+ and A− (and likewise for B) for DEGs with positive and negative directions. The consistency score was defined as 2(|A+ ∩ B+| + |A− ∩ B−|) / E. A p-value was obtained (Table S4) by an upper-tailed Poisson test for the number of consistent DEG pairs, |A+ ∩ B+| + |A− ∩ B−|, with λ = E/2. P-values were adjusted using Benjamini-Hochberg FDR correction.
Pathway enrichment analysis
KEGG pathway enrichment analyses were performed by using R package fgsea72, fast preranked gene set enrichment analysis (GSEA): minSize=3, maxSize=500, nperm=100,000. Gene sets “c2.cp.kegg.v7.0.symbols.gmt” was in used the analysis, and these gene sets were obtained from the MSigDB73 collections: https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp. The default fgsea multiple hypothesis correction was used (Benjamini-Hochberg). Fig. 3F contains the 34 pathways that were significant (FDR <0.05) in at least 10% of all cell types per compartment. All pathway enrichment results in Table S5.
Pseudotime analysis in myofibroblasts
Pseudotime trajectory was calculated with Monocle 374 in the TI myofibroblasts. Clustering was performed with the louvain method with k = 500. Genes that have expression changed significantly over pseudotime were extracted using Moran’s I test for spatial correlation (q-value < 0.1). The top 200 genes by fraction of myofibroblast cells expressing them were selected from among this significant set and are presented in Fig. 5D. Expression in this heatmap was smoothed using a cubic spline using the smooth.spline function in R with smoothing parameter spar = 1.5. Genes were ordered by the pseudotime of maximum expression. Genes were selected for follow-up by prioritizing genes annotated as transcription factors, or which are DNA or RNA-binding. The following genes were selected for follow up: RNF168, GREM1, ZNF451, ZNF263, EDNRB, PTCH1, TBX3, CYP1B1, CHMP1A, GREM2, APOE, HOPX, RGMA, PKNOX1.
Ligand activity analysis
Ligand activity analysis was performed using nichenetr (An open source R implementation of NicheNet: https://github.com/saeyslab/nichenetr). The function nichenet_seuratobj_aggregate was used to predict ligand-receptor activity in different cell types. Default parameters were used with the exception of expression_pct which was set to 0.05. Top ligands were selected with a Pearson score higher than 0.08.
Supplementary Material
Figure S1, related to Figure 1. QC Metrics and cell type distribution by disease and inflammation status.
Figure S2, related to Figure 1. Intraepithelial lymphocytes (IELs) in TI.
Figure S3, related to Figure 2. Expression of IBD-related genes in disease in TI and CO.
Figure S4, related to Figure 3. Expression of five IBD-associated loci, non-inflamed vs healthy differential expression, and the Ketogenesis and PPAR signaling pathways in CD.
Figure S5, related to Figure 2–3. Differential expression in HLA, mucin and TFF genes in TI and CO.
Figure S6, related to Figure 4–5. Enteroendocrine Cells (EEC) and Myofibroblasts in the colon.
Table S1, related to Figure 1: Clinical metadata and sample information. Description of each individual and sample profiled in the study.
Table S2, related to Figure 1: Marker genes for fine-grained cell type identification.
Table S3: Differential expression results. Tab1, related to Figure 2–3: Differentially expressed genes (DEGs) for cell types, lineages during disease and locations. DEGs in this table were genes with the adjusted p value for the discrete coefficient <0.1 (i.e. Discrete FDR<0.1). Tab2, related to Figure 4: Differentially expressed genes (DEGs) for Enteroendocrine cells (EECs).
Table S4, related to Figure 3: Consistency score of differential expression in inflammation for each common cell type between TI and CO.
Table S5, related to Figure 3: KEGG enrichment results.
Table S6, related to Figure 5: Oligos used for qRT-PCR in siRNA KD experiments in myofibroblasts.
Highlights.
scRNAseq atlas of 720k ileal and colonic cells in Crohn’s disease (CD) and controls
Compositional and transcriptomic changes across immune, epithelial and stromal cells
Colonic tissues show stronger transcriptomic changes in inflammation and disease
CHMP1A, TBX3, and RNF168 may regulate a CD-associated program in fibroblasts
ACKNOWLEDGEMENTS
The authors thank participating patients and research staff at the Center for the Study of Inflammatory Bowel Disease. The authors also thank Luke Besse, Allison Higgins and Eric Chen for project and data management, Sean Kim for sample management and metadata collection, and the Broad Genomics Platform for help with sequencing data generation. The authors are also grateful to Heather Kang for editorial assistance with the manuscript and figures, and Dr. Huajun Han for discussions on gut barrier integrity in CD. This study was supported by funding from the National Institutes of Health (RC2 DK114784 and P30 DK43351 to R.J.X.), The Helmsley Charitable Trust, and The Crohn’s and Colitis Foundation. The authors gratefully acknowledge the use of the Opera Phenix High-Content/High-Throughput imaging system at the Broad Institute, funded by the S10 Grant NIH OD-026839-01.
Footnotes
DECLARATION OF INTERESTS
R.J.X. is a co-founder of Celsius Therapeutics and Jnana Therapeutics
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. (2012). Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 2.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. (2017). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778. [DOI] [PubMed] [Google Scholar]
- 3. [No title] https://www.crohnscolitisfoundation.org/sites/default/files/2019-02/Updated%20IBD%20Factbook.pdf.
- 4.Khor B, Gardet A, and Xavier RJ (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, Spencer D, Field M, McCourt C, Toothaker J, et al. (2020). Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology 159, 591–608.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dulai PS, Singh S, Vande Casteele N, Boland BS, Rivera-Nieves J, Ernst PB, Eckmann L, Barrett KE, Chang JT, and Sandborn WJ (2019). Should We Divide Crohn’s Disease Into Ileum-Dominant and Isolated Colonic Diseases? Clin. Gastroenterol. Hepatol. 17, 2634–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, et al. (2016). Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 387, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGovern DPB, Gardet A, Törkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RTH, Lagacé C, Li C, et al. (2010). Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 42, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji S-G, et al. (2017). Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sazonovs A, Stevens CR, Venkataraman GR, Yuan K, Avila B, Abreu MT, Ahmad T, Allez M, Ananthakrishnan AN, Atzmon G, et al. (2021). Sequencing of over 100,000 individuals identifies multiple genes and rare variants associated with Crohns disease susceptibility. medRxiv, 2021.June.15.21258641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham DB, and Xavier RJ (2020). Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, Kim ES, Limary AE, Jin W, Milner JJ, et al. (2020). Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol 5. 10.1126/sciimmunol.abb4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, Davis S, Kinchen J, Chen HH, Alham NK, et al. (2019). Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567, 49–55. [DOI] [PubMed] [Google Scholar]
- 15.Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, Ashley N, Cubitt L, Mellado-Gomez E, Attar M, et al. (2018). Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175, 372–386.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, Gettler K, Chuang L-S, Nayar S, Greenstein AJ, et al. (2019). Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elmentaite R, Ross ADB, Roberts K, James KR, Ortmann D, Gomes T, Nayak K, Tuck L, Pritchard S, Bayraktar OA, et al. (2020). Single-Cell Sequencing of Developing Human Gut Reveals Transcriptional Links to Childhood Crohn’s Disease. Dev. Cell 55, 771–783.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, Kleshchevnikov V, Dabrowska M, Pritchard S, Bolt L, et al. (2021). Cells of the human intestinal tract mapped across space and time. Nature 597, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jasso GJ, Jaiswal A, Varma M, Laszewski T, Grauel A, Omar A, Silva N, Dranoff G, Porter JA, Mansfield K, et al. (2022). Colon stroma mediates an inflammation-driven fibroblastic response controlling matrix remodeling and healing. PLoS Biol. 20, e3001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daperno M, D’Haens G, Van Assche G, Baert F, Bulois P, Maunoury V, Sostegni R, Rocca R, Pera A, Gevers A, et al. (2004). Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: the SES-CD. Gastrointest. Endosc. 60, 505–512. [DOI] [PubMed] [Google Scholar]
- 22.Mayassi T, and Jabri B (2018). Human intraepithelial lymphocytes. Mucosal Immunol. 11, 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S, Rosadini CV, Esaulova E, Di Luccia B, Kinnett B, et al. (2021). Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 12, 1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, Andersen V, Cleynen I, Cortes A, Crins F, et al. (2017). Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber S, Rosenstiel P, Hampe J, Nikolaus S, Groessner B, Schottelius A, Kühbacher T, Hämling J, Fölsch UR, and Seegert D (2002). Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 51, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, and Haase VH (2004). Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest. 114, 1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenz-Meyer H, Bauer P, Nicolay C, Schulz B, Purrmann J, Fleig WE, Scheurlen C, Koop I, Pudel V, and Carr L (1996). Omega-3 fatty acids and low carbohydrate diet for maintenance of remission in Crohn’s disease. A randomized controlled multicenter trial. Study Group Members (German Crohn’s Disease Study Group). Scand. J. Gastroenterol. 31, 778–785. [DOI] [PubMed] [Google Scholar]
- 29.Kong C, Yan X, Liu Y, Huang L, Zhu Y, He J, Gao R, Kalady MF, Goel A, Qin H, et al. (2021). Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct Target Ther 6, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moncada DM, Kammanadiminti SJ, and Chadee K (2003). Mucin and Toll-like receptors in host defense against intestinal parasites. Trends Parasitol. 19, 305–311. [DOI] [PubMed] [Google Scholar]
- 31.McGuckin MA, Linden SK, Sutton P, and Florin TH (2011). Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9, 265–278. [DOI] [PubMed] [Google Scholar]
- 32.Gendler SJ (2001). MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia 6, 339–353. [DOI] [PubMed] [Google Scholar]
- 33.Aihara E, Engevik KA, and Montrose MH (2017). Trefoil Factor Peptides and Gastrointestinal Function. Annu. Rev. Physiol. 79, 357–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overgaard CE, Daugherty BL, Mitchell LA, and Koval M (2011). Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid. Redox Signal. 15, 1179–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, Ciclitira PJ, and Al-Hassi HO (2016). Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 22, 3117–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, and Schulzke J-D (2007). Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iliopoulou L, and Kollias G (2021). Harnessing murine models of Crohn’s disease ileitis to advance concepts of pathophysiology and treatment. Mucosal Immunol. 10.1038/s41385-021-00433-3. [DOI] [PubMed] [Google Scholar]
- 38.Zhu L, Han J, Li L, Wang Y, Li Y, and Zhang S (2019). Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front. Immunol. 10, 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Worthington JJ, Reimann F, and Gribble FM (2018). Enteroendocrine cell-ssensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. 11, 3–20. [DOI] [PubMed] [Google Scholar]
- 40.Beumer J, Puschhof J, Bauzá-Martinez J, Martínez-Silgado A, Elmentaite R, James KR, Ross A, Hendriks D, Artegiani B, Busslinger GA, et al. (2020). High-Resolution mRNA and Secretome Atlas of Human Enteroendocrine Cells. Cell 181, 1291–1306.e19. [DOI] [PubMed] [Google Scholar]
- 41.Gehart H, van Es JH, Hamer K, Beumer J, Kretzschmar K, Dekkers JF, Rios A, and Clevers H (2019). Identification of Enteroendocrine Regulators by Real-Time Single-Cell Differentiation Mapping. Cell 176, 1158–1173.e16. [DOI] [PubMed] [Google Scholar]
- 42.Longhi MS, Moss A, Jiang ZG, and Robson SC (2017). Purinergic signaling during intestinal inflammation. J. Mol. Med. 95, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurtner GC, Werner S, Barrandon Y, and Longaker MT (2008). Wound repair and regeneration. Nature 453, 314–321. [DOI] [PubMed] [Google Scholar]
- 44.Henderson NC, Rieder F, and Wynn TA (2020). Fibrosis: from mechanisms to medicines. Nature 587, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renard C-A, Labalette C, Armengol C, Cougot D, Wei Y, Cairo S, Pineau P, Neuveut C, de Reyniès A, Dejean A, et al. (2007). Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 67, 901–910. [DOI] [PubMed] [Google Scholar]
- 46.Willmer T, Cooper A, Sims D, Govender D, and Prince S (2016). The T-box transcription factor 3 is a promising biomarker and a key regulator of the oncogenic phenotype of a diverse range of sarcoma subtypes. Oncogenesis 5, e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, et al. (2021). Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579. [DOI] [PubMed] [Google Scholar]
- 48.MacCarthy-Morrogh L, and Martin P (2020). The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. 13. 10.1126/scisignal.aay8690. [DOI] [PubMed] [Google Scholar]
- 49.Browaeys R, Saelens W, and Saeys Y (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. [DOI] [PubMed] [Google Scholar]
- 50.Thomas MF, Slowikowski K, Manakongtreecheep K, Sen P, Tantivit J, Nasrallah M, Smith NP, Ramesh S, Zubiri L, Tirard A, et al. (2021). Altered interactions between circulating and tissue-resident CD8 T cells with the colonic mucosa define colitis associated with immune checkpoint inhibitors. bioRxiv, 2021.September.17.460868. 10.1101/2021.09.17.460868. [DOI] [Google Scholar]
- 51.Dubuquoy L, Rousseaux C, Thuru X, Peyrin-Biroulet L, Romano O, Chavatte P, Chamaillard M, and Desreumaux P (2006). PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 55, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, and Hsiao EY (2018). The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 173, 1728–1741.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Islam M, Chen B, Spraggins JM, Kelly RT, and Lau KS (2020). Use of Single-Cell-Omic Technologies to Study the Gastrointestinal Tract and Diseases, From Single Cell Identities to Patient Features. Gastroenterology 159, 453–466.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayassi T, Ladell K, Gudjonson H, McLaren JE, Shaw DG, Tran MT, Rokicka JJ, Lawrence I, Grenier J-C, van Unen V, et al. (2019). Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell 176, 967–981.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayassi T, Barreiro LB, Rossjohn J, and Jabri B (2021). A multilayered immune system through the lens of unconventional T cells. Nature 595, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sullivan ZA, Khoury-Hanold W, Lim J, Smillie C, Biton M, Reis BS, Zwick RK, Pope SD, Israni-Winger K, Parsa R, et al. (2021). γδ T cells regulate the intestinal response to nutrient sensing. Science 371. 10.1126/science.aba8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You K, Wang L, Chou C-H, Liu K, Nakata T, Jaiswal A, Yao J, Lefkovith A, Omar A, Perrigoue JG, et al. (2021). QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science 371. 10.1126/science.abb6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graham DB, Lefkovith A, Deelen P, de Klein N, Varma M, Boroughs A, Desch AN, Ng ACY, Guzman G, Schenone M, et al. (2016). TMEM258 Is a Component of the Oligosaccharyltransferase Complex Controlling ER Stress and Intestinal Inflammation. Cell Rep. 17, 2955–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaser A, Lee A-H, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EES, Higgins DE, Schreiber S, Glimcher LH, et al. (2008). XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, Christoforidou Z, Sathananthan D, Thomas T, Ravindran R, et al. (2021). IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 27, 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urushiyama H, Terasaki Y, Nagasaka S, Terasaki M, Kunugi S, Nagase T, Fukuda Y, and Shimizu A (2015). Role of α1 and α2 chains of type IV collagen in early fibrotic lesions of idiopathic interstitial pneumonias and migration of lung fibroblasts. Lab. Invest. 95, 872–885. [DOI] [PubMed] [Google Scholar]
- 62.Sipponen T, Nuutinen H, Turunen U, and Färkkilä M (2010). Endoscopic evaluation of Crohn’s disease activity: comparison of the CDEIS and the SES-CD. Inflamm. Bowel Dis. 16, 2131–2136. [DOI] [PubMed] [Google Scholar]
- 63.Sipponen T, Björkesten C-GAF, Färkkilä M, Nuutinen H, Savilahti E, and Kolho K-L (2010). Faecal calprotectin and lactoferrin are reliable surrogate markers of endoscopic response during Crohn’s disease treatment. Scand. J. Gastroenterol. 45, 325–331. [DOI] [PubMed] [Google Scholar]
- 64.Danese S, Sandborn WJ, Colombel J-F, Vermeire S, Glover SC, Rimola J, Siegelman J, Jones S, Bornstein JD, and Feagan BG (2019). Endoscopic, Radiologic, and Histologic Healing With Vedolizumab in Patients With Active Crohn’s Disease. Gastroenterology 157, 1007–1018.e7. [DOI] [PubMed] [Google Scholar]
- 65.Khanna R, Zou G, D’Haens G, Rutgeerts P, McDonald JWD, Daperno M, Feagan BG, Sandborn WJ, Dubcenco E, Stitt L, et al. (2016). Reliability among central readers in the evaluation of endoscopic findings from patients with Crohn’s disease. Gut 65, 1119–1125. [DOI] [PubMed] [Google Scholar]
- 66.Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv, 791699. 10.1101/791699. [DOI] [Google Scholar]
- 67.Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, Rosen Y, Slyper M, Kowalczyk MS, Villani A-C, et al. (2020). Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 17, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thiriot A, Perdomo C, Cheng G, Novitzky-Basso I, McArdle S, Kishimoto JK, Barreiro O, Mazo I, Triboulet R, Ley K, et al. (2017). Differential DARC/ACKR1 expression distinguishes venular from non-venular endothelial cells in murine tissues. BMC Biol. 15, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tedoldi S, Paterson JC, Cordell J, Tan S-Y, Jones M, Manek S, Dei Tos AP, Roberton H, Masir N, Natkunam Y, et al. (2006). Jaw1/LRMP, a germinal centre-associated marker for the immunohistological study of B-cell lymphomas. J. Pathol. 209, 454–463. [DOI] [PubMed] [Google Scholar]
- 71.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korotkevich G, Sukhov V, and Sergushichev A (2019). Fast gene set enrichment analysis. Cold Spring Harbor Laboratory, 060012. 10.1101/060012. [DOI] [Google Scholar]
- 73.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 1. QC Metrics and cell type distribution by disease and inflammation status.
Figure S2, related to Figure 1. Intraepithelial lymphocytes (IELs) in TI.
Figure S3, related to Figure 2. Expression of IBD-related genes in disease in TI and CO.
Figure S4, related to Figure 3. Expression of five IBD-associated loci, non-inflamed vs healthy differential expression, and the Ketogenesis and PPAR signaling pathways in CD.
Figure S5, related to Figure 2–3. Differential expression in HLA, mucin and TFF genes in TI and CO.
Figure S6, related to Figure 4–5. Enteroendocrine Cells (EEC) and Myofibroblasts in the colon.
Table S1, related to Figure 1: Clinical metadata and sample information. Description of each individual and sample profiled in the study.
Table S2, related to Figure 1: Marker genes for fine-grained cell type identification.
Table S3: Differential expression results. Tab1, related to Figure 2–3: Differentially expressed genes (DEGs) for cell types, lineages during disease and locations. DEGs in this table were genes with the adjusted p value for the discrete coefficient <0.1 (i.e. Discrete FDR<0.1). Tab2, related to Figure 4: Differentially expressed genes (DEGs) for Enteroendocrine cells (EECs).
Table S4, related to Figure 3: Consistency score of differential expression in inflammation for each common cell type between TI and CO.
Table S5, related to Figure 3: KEGG enrichment results.
Table S6, related to Figure 5: Oligos used for qRT-PCR in siRNA KD experiments in myofibroblasts.
Data Availability Statement
The datasets generated during the current study are available for download from the controlled-access data repository, Broad DUOS (Accession DUOS-000146 CD_Atlas_2021_GIDER; DUOS-000145 CD_Atlas_2021_PRISM). The analyzed data reported in this paper is available at the Broad Single Cell Portal (SCP1884).