

Abstract
Animal models of tauopathy help in understanding the role of mutations in tau pathobiology. Here, we used adeno-associated viral (AAV) vectors to administer three tau genetic variants (tauwild-type, tauP301L, and tauR406W) intracranially into 12-month-old C57BL/6Nia mice and collected tissue at 16 months. Vectors designed to express green fluorescent protein controlled for surgical procedures and exogenous protein expression by AAV. The tau genetic variants produced considerably different phenotypes. Tauwild-type and tauP301L caused memory impairments. The tauP301L caused increased amounts of aggregated tau, measured both neurochemically and histologically. Tauwild-type produced elevated levels of soluble tau and phosphorylated tau by ELISA and increased staining for phosphorylated forms of tau histologically. However, only the tauwild-type caused localized atrophy of brain tissue at the sites near the injection. The tauR406W had low protein expression and produced no atrophy or memory impairments. This supports the potential use of AAV expressing tauwild-type in aged mice to examine events leading to neurodegeneration in Alzheimer’s disease pathology.
Keywords: tau, aging, mouse models, neurodegeneration, viral models
Introduction
Pathological tau is a defining feature in a variety of diseases referred to as tauopathies. Tauopathies include not only Alzheimer’s disease (AD), but frontotemporal dementia (FTD), progressive supranuclear palsy, corticobasal degeneration, and many others (Abbott et al., 2006; Bejanin et al., 2017; Gordon et al., 2018; Hanseeuw et al., 2019; Malpas et al., 2020). One of the differences between tauopathies is the regional progression of the disease and cell types affected. The MAPT gene encodes tau protein, and several mutations have been identified associated with FTD (Goedert, 2005). In addition, a rare tau variant, A152T increases the risk of developing AD 2–3-fold (Coppola, et al 2012; Labbe et al, 2015). The gap in translationally meaningful research models specific to AD needs to be addressed as treatments and therapeutic approaches have remained elusive for AD.
The identified tau mutations have been employed to create cell lines and transgenic animal models which have greatly contributed to the knowledge of the progression of pathological tau. Increased age is the greatest risk factor for an AD diagnosis and is an important consideration in modeling AD (Braak et al., 1996; Franceschi and Campisi, 2014). However, most transgenic animal models express the transgene for the entire lifespan and some models prove lethal before middle age. Viral vector models can be administered at different ages, permitting examination of the aging context on the time course and severity of tauopathy development. Additionally, viral vector models allow researchers to target selected structures in brain permitting regional comparisons. AAV serotype 9 (AAV9) is a variant that efficiently transduces neurons and can be used to induce tauopathy (Dayton et al., 2012; Mustroph et al., 2012; Vanderweyde et al., 2016).
Researchers have successfully used AAV to administer wild-type and mutated forms of tau that can induce pathology (Jaworski et al., 2009; Mustroph et al., 2012; Vanderweyde et al., 2016). It has been noted that tauwild-type does not aggregate in vitro as readily as mutated forms of tau (Hasegawa et al., 1998; Nacharaju et al., 1999). The P301L mutation has been used in the creation of transgenic mouse lines that develop pathological tau, behavioral deficits, an inflammatory response, and neural atrophy which are critical factors in modeling AD (Ramsden et al., 2005; Santacruz et al., 2005). In contrast to P301L, the R406W mutation has not been used in a viral method of delivery but has been used in cell work, and to a lesser extent, in transgenic lines. Clinically, the R406W mutation results in personality changes, memory loss, and mild parkinsonism (Heutink et al., 1997; Van Swieten et al., 1999). Individuals with R406W mutations tend to have a later age of onset (54.2–64.2 years of age) and an increase in the disease duration (up to 20 years), indicating that this mutation may exert less severe consequences (Van Swieten et al., 1999).
The goal of this study was to compare viral 2N4R tauwild-type, tauP301L, and tauR406W to determine if these isoforms resulted in distinct tauopathy phenotypes in middle-aged mice. To characterize these phenotypes, we tested mice for cognitive and motor deficits, examined tissues for differences in the expression, phosphorylation, and deposition of tau, and used multiple measures to assess if these variants induced neural atrophy. We found significant differences in the phenotypes of the tauopathy induced by each of these genetic variants of tau.
Materials and Methods
Animals and Experimental Design
Forty C57BL/6 mice were obtained from the National Institute of Aging colony and were balanced for sex. All animals were aged to one year and maintained on 12-hour light cycles with food and water provided ad libitum. At 12 months of age, all mice underwent bilateral intracranial stereotaxic surgeries to both hippocampi (coordinates ML:+/−2.7, AP:−2.7, DV:−3.0, relative to bregma) and anterior cortices (coordinates ML:+/−2.20, AP:2.2, DV:−3.0). Two μl of viral constructs (green fluorescent protein (GFP), tauwild-type, tauP301L, or tauR406W) were administered at each injection site at a flow rate of 0.5μl per minute using a pulled glass pipette needle that was attached to a 10μl Hamilton syringe (Hamilton, Reno, NV; cat. no. 80000) with heat shrink tubing. After the surgery, mice were placed on a heating pad to maintain body temperature and observed during postop recovery. At 3.5 months of viral incubation, behavioral assessments were conducted to ascertain if behavioral changes had developed. Brain tissues were collected two weeks after the behavioral assessments at 16 months of age. A flowchart of the procedures and numbers of mice are presented in Supplemental Figure 1.
Viral Constructs
Glycerol stocks of competent SURE2 cells containing 2N4R tauwild-type and GFP plasmids were donated by Dr. Kevin Nash from the University of South Florida. Gene fragments for 2N4R tauP301L and 2N4R tauR406W were ordered from Genewiz (Genewiz, South Plainfield, NJ, USA). Constructs were created containing the cytomegalovirus (CMV) enhancer with the chicken β- actin promoter (Gray et al., 2011), the three different tau isoform sequences (P301L, R406W, and wild-type), and the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE). WPRE has been reported to enhance the long-term expression of the viral cargo genes and increase the duration of viral expression (Loeb et al., 1999). The plasmid sequences were confirmed prior to virus production (P301L, R406W, and wild-type) (Genewiz, South Plainfield, NJ, USA). HEK293T cells were co-transfected with each tau plasmid along with two other plasmids that encode for AAV9 capsid proteins and adenovirus helper elements. Viral vector purification was conducted by iodixanol gradients and ultracentrifugation. The viral vectors were concentrated with Amicon Ultra-15 centrifugal filters (Sigma, Burlington, MA, USA; Cat. No. UFC910024) using Dulbecco’s phosphate buffered saline (DPBS; pH 7.4). The concentrated viral vector solutions were then aliquoted into 10μl volumes and stored at −80°C. Viral titers were measured by RT qPCR with primers targeted to the WPRE sequence. The viral vectors were diluted to 8.8×1011 vector genomes (vg)/ul. For each day of surgery, a fresh aliquot was thawed to minimize excess freeze-thaw cycles. To monitor the volume of injection, 8.8×1010vg/ml (a 1/10th dosage) of AAV9 GFP was added to each of the tau viral vectors.
Behavioral Assessments
After 3.5 months of viral vector incubation, behavioral assessments were conducted by an experimenter blind to the treatment group identity for each mouse. To first acclimate the mice to being routinely handled, and to measure motor activity, the open field task was conducted. Animals were placed into an open field enclosure (L 40mm/ W 40mm/H 40mm, BioSeb; Vitrolles, France) for 10 minutes and the total distance traveled was digitally recorded with AnyMaze software (Stoelting, Wood Dale, IL, USA; cat. no. 60000). Next, mice were tested for coordination and motor skill on an accelerating TSE rotating rod apparatus (TSE Systems, Chesterfield, MO, USA). A total of 8 trials were conducted over 2 days with 4 trials on each day. The trial duration was up to 5 minutes and the rotating rod continually increased in speed. The animals had at least a 20-minute inter-trial interval to minimize muscle fatigue. Fall latency was recorded for each trial.
To assess visuospatial navigation memory, the radial arm water maze (RAWM) was conducted as previously described (Alamed et al., 2006). RAWM consists of 4 tasks that were completed over 4 days. The RAWM apparatus has 6-arms radiating out from the center of the maze. The goal arm contained a platform that permits mice to stand without further swimming and acted as a reward. The goal arm remained the same arm for each trial, while the start arm for each trial was varied. Thus, to locate the platform, the mice learned to use extra-maze cues and a spatial hypothesis to solve the task. The goal arm was changed between animals to reduce possible use of olfactory clues. For each day, there were 15 trials that lasted for a duration of 1-minute. Day 1 was the training day and the platform alternated between being visible or hidden just beneath the water surface. If the animal was unable to complete the task within 1 minute, the animals were guided to the platform. Day 2 was the testing day, where only the hidden platform was used, and the animals were not guided to the platform if they failed to locate it within 1 minute. A reversal task was completed on day 3 where the hidden platform was placed in the arm 180° from the original goal arm for that individual mouse. The number of errors (entry of all four limbs into an incorrect arm) was recorded for days 1, 2, and 3. If the mouse failed to enter an arm within 15 seconds, this was also counted as an error. For each day, three trials were averaged together and resulted in 5 blocks of trials for each day of the assessment. The final task on day 4 was the open pool performance control which assessed the animal’s ability to see, swim to, and ascend the platform. In this task, the visible platform was placed in the center of the maze, and the external cues were removed. The latency to ascend to the platform was recorded, and mice failing to reach this goal within 20 seconds on the final trial block were excluded from the study due to performance impairment. One animal died prior to behavioral testing. In total, 12 animals were excluded from the RAWM data analysis. Two animals had visible ocular occlusions, 2 were unable to swim sufficiently well to be tested, 5 were noted to have seizures on one of the days of testing, and 3 animals were excluded due to failure to find and ascend the platform in the open pool task. The final sample size included GFP, n = 7; tauP301L, n = 5 tauR406W, n = 9; and tauwild-type, n = 6. The data presented only includes the mice that successfully completed the task and did not have seizures.
Tissue Collection
Brain tissues were collected after a 4-month viral vector incubation period. Mice were anesthetized with diluted Euthasol® solution (3.9 mg/kg of pentobarbital and 0.5mg/kg of phenytoin). Once anesthetized, animals were placed on a heating pad to maintain body temperature to minimize hypothermia-related tau phosphorylation (Planel et al., 2007). Animals were transcardially perfused with 25ml of 0.9% saline. Once perfused, the brains were immediately extracted, bisected sagitally and the right hippocampus (HPC) was dissected on a cold stage and frozen on dry ice. Samples were stored at −80°C until homogenized. The left hemispheres were immersed in 20ml of freshly prepared buffered 4% paraformaldehyde (pH 7.4) for 24 hours at 4°C. The left hemispheres were then cryoprotected using successive immersions in sucrose solutions (10%, 20%, and 30%) each for 24 hours at 4°C. The left hemispheres were then frozen and sectioned horizontally with a sliding microtome at a 25μm thickness except for every 10th section that was collected at 50μm thickness for hippocampal volumetric analysis. Tissue sections were stored in DPBS with 10 mM sodium azide solution (7.4 pH) at 4°C until staining protocols were conducted.
Histopathology
Sections were placed into free-floating multisample staining trays for each immunohistochemical marker and were conducted as previously described (Gordon et al., 2002). To eliminate endogenous peroxidase tissues were placed into a blocking solution (10% methanol, 3% H2O2 in phosphate buffered saline pH 7.4; PBS) and then washed 3 times with PBS. Next, tissues were placed in a permeabilization solution (0.2% lysine, 1%Trition X100, 4% normal goat serum in PBS) before being placed into the primary antibody overnight (GFP, Abcam ab13970 1:30k; total tau, HT7, Invitrogen MN1000B, 1:5k; phosphorylated Ser199/Thr205-tau, AT8, Invitrogen MN1020B, 1:10k; phosphorylated Ser396-tau, Anaspec AS 54977-025, 1:30k; MHCII, BD Pharmingen 556999, 1:5k). The following day, tissues were washed 3 times with PBS and incubated in the corresponding secondary antibody at a 1:3k dilution for 2 hours (goat anti-rabbit IgG, Vector Laboratories BA-1000; goat anti-rat IgG, Vector Laboratories BA-9400; goat anti-chicken IgY, Vector Laboratories BA-9010), unless the primary antibody was biotinylated. Tissues were then washed 3 times with PBS and placed in Vectastain® Elite® ABC kit solution (Vector Laboratories, Burlingame, CA; cat. no. PK-6100). After the 1-hour incubation, tissues were washed 2 times with PBS and the third wash with tris buffered saline pH 7.4 (TBS). Then tissues were placed into the color development solution (0.05% diaminobenzidine, 0.5% Ni2+, 0.03% H2O2 in TBS) for up to 5 minutes. Color development was stopped by removing tissues from the solution and washing with TBS for 1 wash, followed by 2 PBS washes. Once the stain was complete, tissues were mounted on microscope slides (Fisher Scientific, Waltham, MA; cat. no. 1255015), dehydrated, and cover slipped with DPX mounting medium (Electron Microscopy Sciences, Hatfield, PA; cat. no. 13512).
For the cresyl violet and Gallyas stains, tissue sections were first mounted on microscope slides and dried overnight. The cresyl violet staining visualizes the nissl bodies within the neuron, largely ribosomal RNA. This stain was conducted on 50μm tissue sections. On the day of the protocol, slides were rehydrated briefly and then placed in a 0.05% cresyl violet dye solution and destained in acetic acid solution (pH 3.5) to reduce background signaling. Slides were rinsed with water, dehydrated and coverslipped with DPX (Electronic Microscopy Sciences Cat. No. 13512).
Gallyas staining is a silver stain that allows for the visualization of silver positive neurofibrillary tau tangles (Uchihara, 2007). On the day of staining, the sections were briefly rehydrated and placed in a pretreatment solution of 5% periodic acid for 5 minutes. Then slides were washed prior to being immersed in a silver enhancing solution (10% potassium iodide, 4%NaOH, 0.035% silver nitrate) for one minute, then in a 0.5% acetic acid solution for 10 minutes. Slides were then immersed for 10 more minutes in a working developer solution (2.5% sodium carbonate, 0.1% ammonium nitrate, 0.1% silver nitrate, 0.5% tungstosilic acid, 0.2% formaldehyde). Upon development, slides were washed with 0.5% acetic acid followed by water washes. Next, slides were placed in gold toning solution (0.1% gold chloride solution) followed by an additional water wash. Finally, slides were placed in a solution to remove traces of unconjugated silver salts (0.1% sodium thiosulphate solution) followed by a water wash. Slides were immediately dehydrated and coverslipped.
Slides were digitally scanned at 200x magnification using ZEISS Axio Scan.Z1 and analyzed by NearCYTE tissue analysis software (NearCYTE, Pittsburgh, PA). Analyses were conducted by first segmenting immunopositive pixels by establishing threshold parameters applicable to the range of specimens analyzed. Then the ratio of immunopositive pixels to the total pixel area over the entire section was calculated. Using the entire section for analysis permitted evaluation of both the staining within injection region and the degree of spread outside the local injection area (which for some mice was subject to significant atrophy). The only exception to this analysis was the cresyl violet staining where stained slides were used to calculate the hippocampal volume using the method of Cavalieri using the equation, V = A × T, where A = sum of the hippocampal areas (μm2) on the sections analyzed, and T = section interval (number of sections) × t (section thickness (50μm). In addition, the cresyl violet stained sections were also used for measuring cortical thickness using the NearCYTE tissue analysis measure tool (μm). The cortical regions that were used for the cortical thickness were anterior to the injection site, at the injection site, and posterior to the injection site (see Figure 2 for locations).
Figure 2.
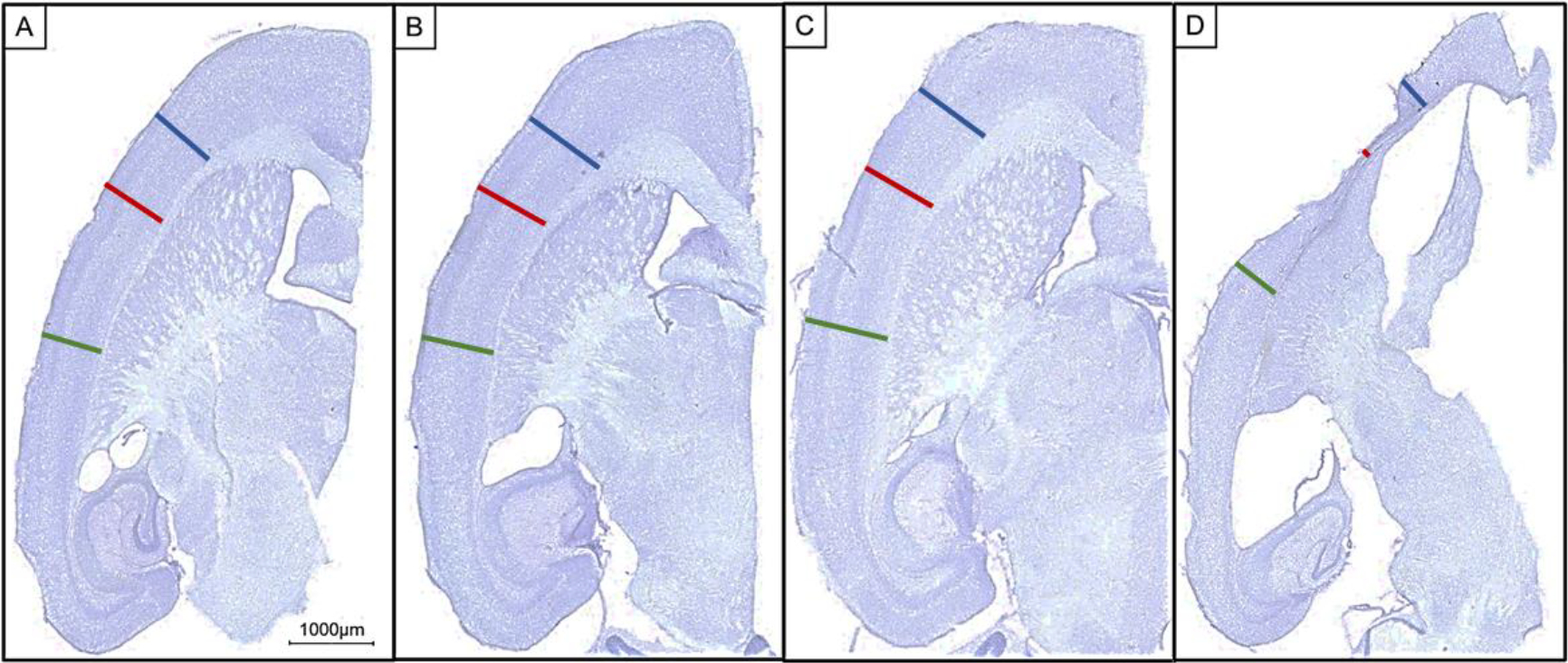
Micrograph representation of horizontal sections that reveal atrophy in the tauwild-type variant injection group compared to all other groups. Scale bar 1000μm. (A) GFP injected. (B) TauP301L injected. (C) TauR406W injected. (D) Tauwild-type injected. Blue bar = anterior to the injection site cortical measurement. Red bar = injection site cortical measurement. Green bar = posterior to the injection site cortical measurement.
Tissue Homogenization and Biochemical Analysis
The HPC was used for all biochemical analyses. The HPC was weighed and homogenized in 10 volume/weight of RIPA buffer (50mM Tris-Cl, 150nM NaCl,1% NP40, 0.1% SDS, 0.05% sodium deoxycholate) that contained protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO, USA; Cat. No. P8340), deacetylase inhibitor cocktail (MedChem Express, Monmouth Junction, NJ, USA; Cat. No. HY-K0030) phenylmethanesulfonyl fluoride (Sigma Aldrich, St. Louis, MO, USA; Cat. No. P7626), and phosphate inhibitor cocktails II and III (Sigma Aldrich, St. Louis, MO, USA; Cat. Nos. P5726 & P0044). Tissues were homogenized with a rotating mechanical pestle followed by sonication. Once homogenized, a 25ul aliquot was centrifuged at 50,000g at 4°C for 1 hour. The detergent-soluble fraction was obtained and stored at −80°C. The pellet was then resuspended in 70% formic acid with the mechanical pestle and incubated on a tube revolver for 30 minutes. One M tris-base (pH 7.6) was added to each sample. Each sample was individually adjusted to a pH of 7 with 15.1M NaOH. These detergent insoluble fractions were stored at −80°C. Pierce BCA protein assays were used to determine the protein concentrations for both detergent soluble and insoluble fractions.
ELISA assays were conducted to measure total tau ( Fisher Scientific, Cat. No. KHB0041), phosphorylated Ser199-tau (Fisher Scientific, Cat. No. KHB7041), and phosphorylated Ser396-tau (Fisher Scientific, Cat. No. KHB7031), both detergent soluble and insoluble fractions were assayed. Sample aliquots were first diluted (1:1000) in RIPA and then further diluted using the dilution buffer provided by each kit to a concentration empirically determined to be near the center on the standard curve for that analyte. Soluble oligomeric tau (using the TOC1 antibody) was conducted on the detergent soluble fraction using a sandwich ELISA protocol (Kanaan et al., 2016). The TOC1 ELISA required a 25x dilution. All concentrations derived from the ELISA assays were then normalized to the total protein concentration from BCA protein assays for each fraction.
In addition, the detergent soluble fraction was also used for western blotting analyses. Ten μg of sample protein was loaded on a 4–20% polyacrylamide Criterion™ TGX Stain-Free Gels (Bio-Rad, Hercules, Cali; Cat. No. 5678094) and electrophoresed with tris glycine running buffer (Bio-Rad, Hercules, Cali; Cat. No. 1610732). After electrophoresis, the gels were transferred to 0.2μm midi nitrocellulose membranes (Bio-Rad, Hercules, Cali; Cat. No.1704159) using Bio-Rad Trans-Turbo Transfer System (Bio-Rad, Hercules, Cali; Cat. No.12003154). Membranes were blocked in 5% nonfat dry milk for 45 minutes. The primary antibodies used for western blotting analyses included total tau (HT7; Invitrogen MN1000, 1:10k), phospho-tau Ser202 and Thr205 (AT8; Invitrogen MN1020, 1:10k), phospho-tau Ser396 (Anaspec; AS 54977-025, 1:2k), and actin (Sigma A2066). Once the membranes were blocked, they were incubated in the primary antibodies overnight at 4°C. The following morning membranes were washed with tris buffered saline (TBS, 50 mM Tris-Cl, pH 7.6; 150 mM NaCl, pH 7.6) plus 0.07% tween-20 and then incubated in the corresponding secondary antibody for 1hour (AntiRabbit IgG IR dye 680cw, LI-COR Biosciences #926-32211 or Anti-Mouse IgG IR Dye 800cw. LI-COR Biosciences # 926-32280) at a 1:10k dilution). Membranes were then washed again with TSB+0.07%tween-20 and then imaged using Bio-Rad ChemiDoc MP. The tau probes were normalized to actin through the Image Lab software (Bio-Rad, Hercules, Cali).
Statistical Analyses
All graphical figures were created with GraphPad Prism 8 (La Jolla, CA) and statistical analyses were conducted by using IBM SPSS Statistics 25 (Armonk, New York, USA). To access any group differences either one-way ANOVA or two-way repeated mixed measures ANOVAs were conducted. For the open field task, a one-way ANOVA was conducted to determine if there was a significant group difference (GFP, tauP301L, tauR406W, and tauwild-type) based on the tau variant injection group for the total distance traveled during a 10-minute trial. Because the rotarod encompasses 8 trials conducted over 2 consecutive days, a two-way repeated measures ANOVA was conducted to determine if there was any difference of the tau variant injection group for the amount of time the animals were able to remain on a rotating rod. For each day of the RAWM tasks, there were 5 blocks of trials and two-way repeated measures ANOVAs were conducted. Finally, assessments of each stain, biochemical analysis, change in body weight, and hippocampal weights were analyzed by one-way ANOVAs. To assess cortical thickness at the anterior cortical injection sites, a two-way repeated measures ANOVA was conducted. If the ANOVA indicated any significant between subject differences, the individual means were tested by a Tukey’s post hoc test to determine which groups were significantly different. For measurements of tau histology and tau ELISA values, only the three treatment groups that were injected with tau expressing viral vectors were included. The GFP group had no human tau expression to be detected at the antibody concentration used for optimal detection of injected tau (Supplementary Figure 2). Including these near zero values would have statistically masked key differences in expression between the tau variant groups.
Results
Behavioral Assessments
One of the goals of this study was to determine if any of the tau variants resulted in a behavioral phenotype. For the behavioral assessments, the GFP control group were used for comparison. For the open field task, two animals from the tauR406W injection group were excluded from the assessment due to a software failure to record performance during their testing. There was no evidence of group differences based on viral vector treatment group for the open field task, F(3,33) = .122, p = .946. Similarly, rotarod performance revealed no significant difference between the experimental groups (F(3,35) = .928, p = .437).
To assess cognitive performance based on treatment group, mice were tested over 4 days on the RAWM task. Two-way repeated measures ANOVAs were conducted for each day of the RAWM tasks. For the training day, there were no group differences, F(3,23) = .796, p =.508. However, when the testing day was examined, there were significant group differences, F(3,23) = 7.716, p = .001; Figure 1). Tukey’s post-hoc analysis detected that the tauP301L mice performed significantly more errors when compared to the GFP (p = .002) and the tauR406W (p <.001) variant injection groups. The tauwild-type injection group also performed significantly more errors when compared to GFP (p < .001) and tauR406W (p < .0001) but was not significantly different when compared to the tauP301L(p = .817). When the reversal task was assessed, there was no evidence of any group differences, F(3,23) = 1.572, p = .223). All animals were evaluated for any performance impairments using the open pool task. Three mice were designated as having performance impairments as they required longer than 20 seconds to find and ascend the visual platform and failed to improve over trials (48 seconds ± 18 for these 3 mice; for other mice on the final block the mean was 9.1 seconds). The data from these three mice were excluded from all radial arm water maze analysis. A repeated measures two-way ANOVA identified significant group differences among the remaining mice, F(3,23) = 8.468, p= .0006. Tukey’s post-hoc analysis identified that the tauwild-type group was the only tau variant injection group that took significantly longer to find and ascend the visual platform when compared to all other tau groups (p < .001).
Figure 1.

Performance on the testing day of the radial arm water maze. AAV9 tauP301L and AAV9 tauwild-type performed significantly worse and made more errors on the second day of the radial arm water maze when compared to AAV9 tauR406W and AAV9 GFP. The number of errors made on the testing day of the radial arm water maze were grouped into five blocks of trials. Data are presented as mean (symbol) ±SEM (error bars). ** p<0.01 Tukey’s post-hoc analysis demonstrating a significant group differences of a one-way repeated measures ANOVA across the five block trials on the testing day. Mice found unable to perform the task (see methods) were excluded from the analysis and not shown here.
Measures of Atrophy
Visual inspection of the histological sections demonstrated that the tauwild-type injection group had atrophy at the injection sites (Figure 2). This was corroborated by several measures of atrophy that included hippocampal volume, hippocampal weight, cortical thickness, and changes in body weight between the day of injection and the day of tissue collection (Figure 3). When examining group differences in hippocampal volume, there was a main effect of tau variant injection group, (F(3,35) = 5.668, p = .003), and tauwild-type mice had a significant reduction in hippocampal volume when compared to all other treatment groups (p’s ≤ .04; Figure 3A). In addition, hippocampal weight measured from dissected samples used for neurochemistry indicated significant overall group differences, (F(3,35) = 5.388, p = .004; Figure 3B). The tauwild-type had a significant reduction in hippocampal weight compared to the tauR406W mice (p = .007). In addition, the P301L mice had a significant reduction in hippocampal weight when compared to the R406W group (p = 0.03). To quantify the group difference observed at the cortical injection site, three measurements of cortical thickness were taken, including anterior to the injection site, at the injection site, and posterior to the injection site. A two-way repeated measures ANOVA was conducted and there was a significant interaction of cortical thickness measurement site and treatment group (F(6,70) = 5.350, p = .0001; Figure 3C), where the tauwild-type had the greatest amount of cortical atrophy that was largest at the injection site. The main effect for cortical thickness was significant (F(2,70) = 17.00, p < .001), where the overall means for the cortical thickness was thinner at the injection site (M = 876.71, SEM = 28.86) compared to the cortex anterior (M = 1031.80 μm, SEM = 32.04) and posterior to the injection site (M = 994.29, SEM = 12.87). In addition, there was a main effect for injection group (F(3,35) = 10.07, p < .0001) where only the tauwild-type had a significant reduction in cortical thickness when compared to the GFP, tauP301L, and tauR406W groups (p < .001).
Figure 3.

Quantification of indicators of atrophy following intracranial AAV-tau injections. The tauwild-type intracranial injection exhibited the greatest amount of atrophy. (A, B, and D) scatterplots of individual animal’s expression values are plotted by injection groups for panels. Each dot represents one mouse. Data are presented as mean ±SEM (error bars). (A) Quantification of hippocampal volume estimated using 8 sections spanning the entire hippocampus by the method of Cavalieri indicated that the tau wild-type mice had a significant reduction of hippocampal volume compared to the other injections. (B) The hippocampal weights measured in milligrams demonstrated that tauP301L and tauwild-type mice had the lowest hippocampal weights. (C) The cortical thickness measured in micrometers anterior to the cortical injection site, the injection site, and posterior to the injection site confirmed the loss of cortical volume in the tauwild-type mice. (D) The change in body weight in grams prior to receiving intracranial injection compared to the weight at the time of tissue collection. Results from one-way ANOVA (A, B, and D) and one-way repeated mixed measures ANOVA (C). Results. * p<.05 * p<0.05, **p<0.01, *** p≤0.001 Tukey’s post-hoc analyses.
To examine any significant changes in body weight, a one-way ANOVA was conducted for the difference in weight from the time of surgery until the time of tissue collection. There was a significant effect of injection group (F(3,35) = 4.858, p =.006). Tukey’s posthoc analysis revealed a significant group difference between the GFP and tauwild-type injection groups (p =.004; Figure 3D).
Histological Measurements
To investigate if these treatment group differences in atrophy were due to tauopathy, immunohistochemical measurements of several tau isoforms were conducted to evaluate tauopathy (Figures 4 and 5). The following analyses for the histological measurements of tau were conducted across the tau variants only, as the GFP injection group did not have any positive signal for any tau measurements (Supplemental Figure 2). In mice injected with AAV tau variants, there were neurons labeled by antibodies staining for total tau (HT7); phospho-tau Ser199, Thr205 (AT8) and phospho-tau Ser396 (rabbit polyclonal serum; Figure 4A–C). These labeled neurons were found surrounding both the hippocampus and anterior cortical injection sites. In addition, in mice injected with tauP301L, there were occasional neurons that were labeled by the Gallyas silver method (Figure 4D). In other mice, there were very few Gallyas positive profiles.
Figure 4.
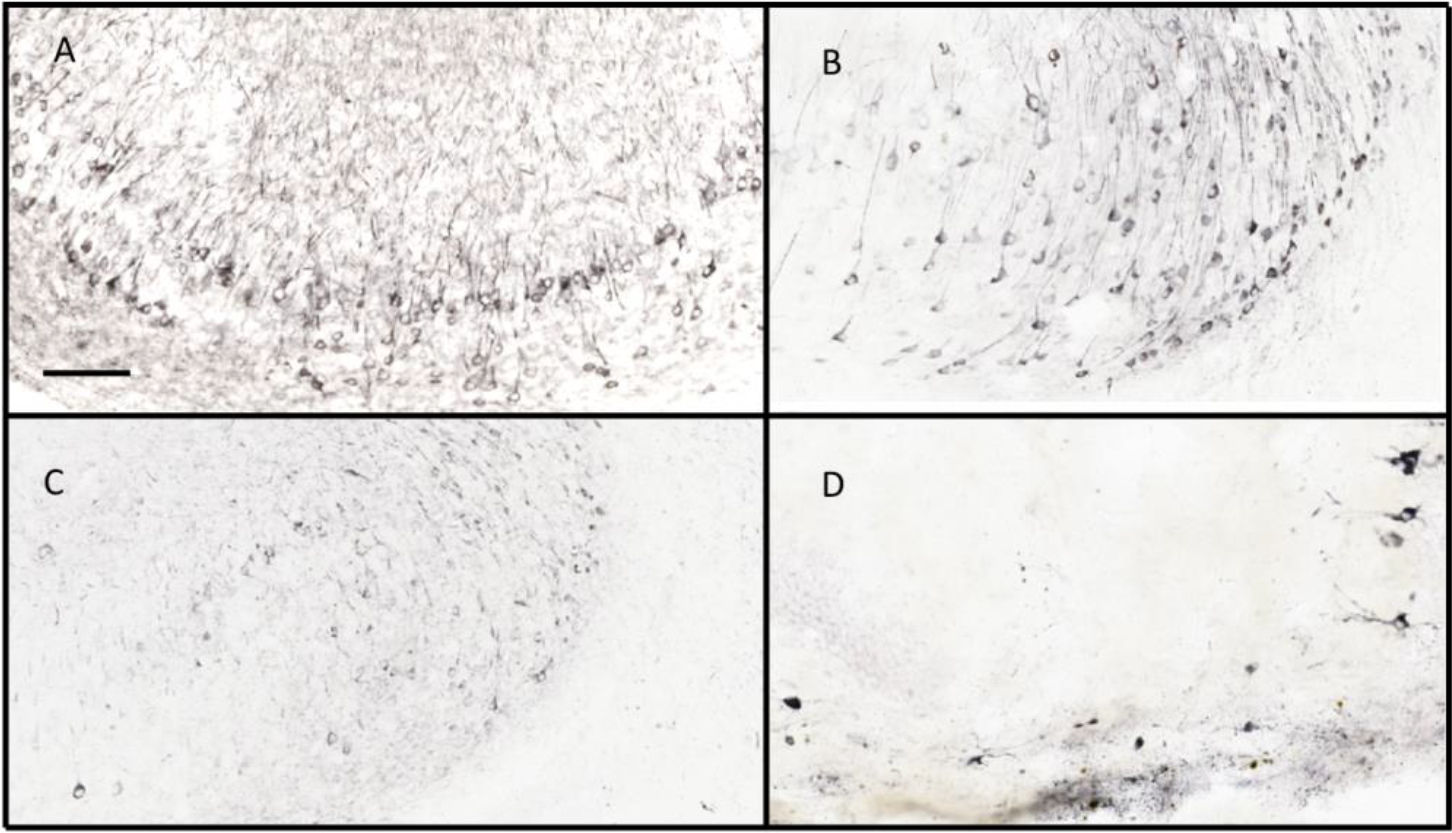
Immunostaining of mouse hippocampus for tau markers after intracranial injections of AAV-tau variants. Total tau was stained by antibody HT7 in the CA1/subiculum region of mouse hippocampus in a horizontal section. Positive stain can be detected in a large number of pyramidal cell bodies, as well as multiple dendritic profiles in the molecular layers above the cell bodies. (Figure 4A). Staining for phospho-tau Ser199-Thr205 by AT8 reveals a significantly smaller portion of pyramidal cells being labeled, as well as their proximal apical dendrites (Figure 4B). Figure 4C shows the staining pattern with a polyclonal antibody against phospho-tau Ser396. This staining is less intense in the cell bodies of pyramidal cells, and lightly stains a number of dendritic profiles in the molecular layer. Gallyas silver staining for neurofibrillary tangles stains very few neurons and primarily in the tauP301L group of mice. Several neurons can be seen along the right-hand side of the panel, including apical dendrites. Other more punctate staining is hard to determine if it is cellular but is largely absent from most mice in the study. Scale bar is 100 μm.
Figure 5.
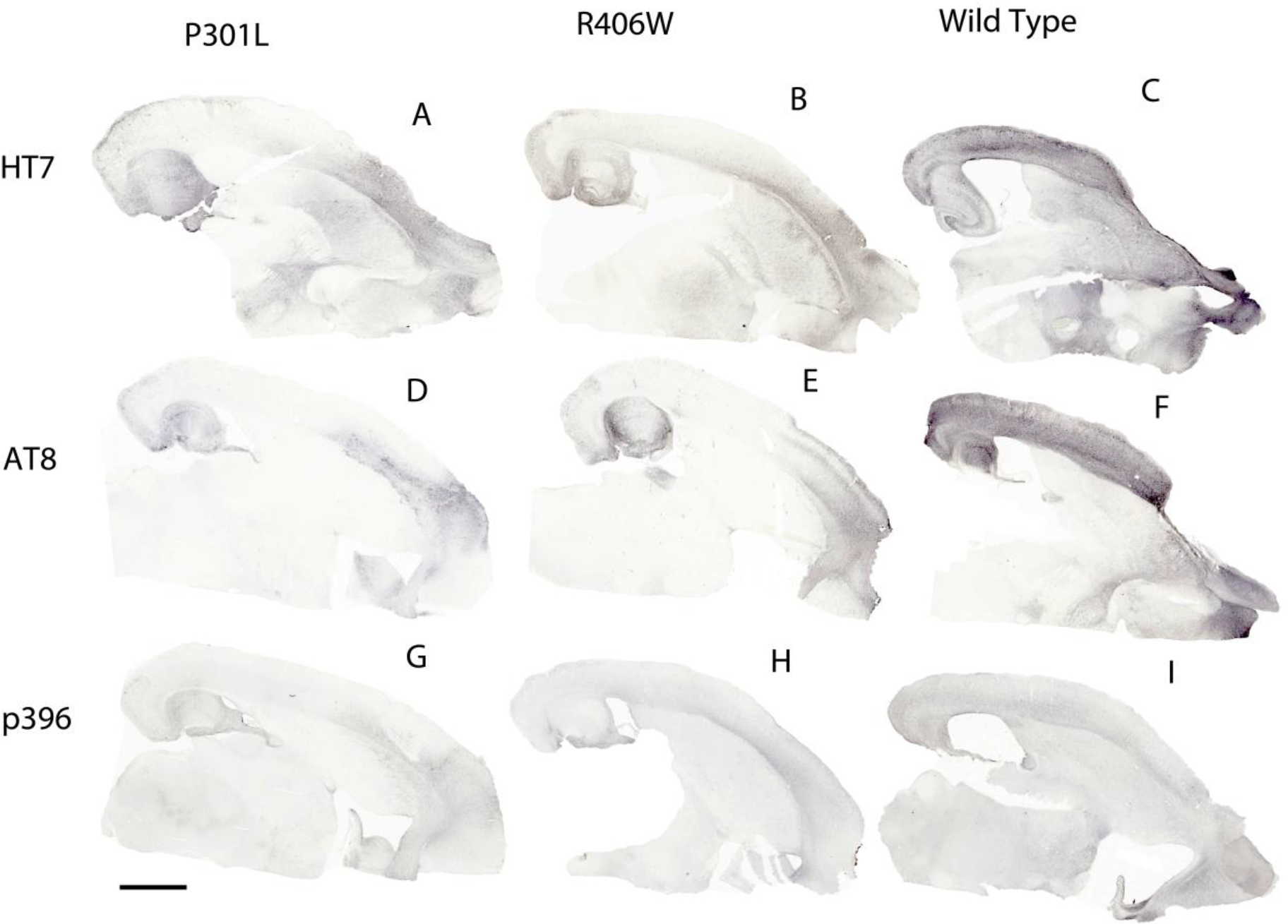
Immunohistochemistry for tau and phosphorylated tau epitopes in mice injected with AAV9 viral vectors expressing tauwild-type, tauP310L or tauR406W. Horizontal sections were stained for HT7 (total tau; panels A-C), AT8 (labeling tau phosphorylated at residues 199 and 202; Panels D-F) and an antibody recognizing tau phosphorylated at residue 396 (panels G-I). Scale bar = 1 mm.
Widespread expression of tau was observed throughout the hippocampus and cortex after AAV9-tau viral delivery (Figure 5). Strong expression was observed in frontal, insular, somatosensory, and entorhinal cortex areas. Staining for phosphorylated epitopes was more restricted to areas of injection in the frontal/insular cortex and hippocampus. When histological total tau (HT7) was quantified for the different tau variant groups, each of the tau viruses expressed tau but there were significant group differences (Figure 6A; F(2,28) = 34.696, p <.001). The tauwild-type injection group had a significantly greater amount of HT7 staining than the other tau injection groups (Figure 5A–C; Figure 6A; p’s < .001). To further evaluate tauopathy, phospho-tau Ser199/threonine 205 (AT8) and phospho-tau Ser396 were quantified. There were significant group differences for AT8 staining (F(2,28) = 22.510, p <.001; Figure 5D–F; Figure 6B). The tauwild-type had significantly greater amount of AT8 staining when compared to tauP301L and tauR406W (p’s <.001). Phospho-tau Ser396 also had significant group differences (F(2,28) = 5.720, p = .009) and tauwild-type had a significantly greater amount of staining compared to the tauR406W injection group (p = .006; Figure 5G–I; Figure 6C). In addition to examining tau immunohistochemistry, the Gallyas silver method was applied to stain for NFTs. Although overall little staining was apparent (Figure 4D), there were significant group differences (F(2,28) = 17.989, p <.001). In contrast to the phospho-tau markers, the tauP301L variant injection group had significantly greater amount of silver staining compared to tauR406Wand tauwild-type (p’s < .001; Figure 6D).
Figure 6.

Quantification of histological tau staining from Figure 5. The amount of staining was quantified per hemisphere due to atrophy of some regions of interest in some conditions. The tauwild-type injected group had the greatest amount of histological total and phosphorylated tau, whereas the tauP301L injected group had the greatest amount of silver positive neurofibrillary tangles. Scatterplots of individual animal’s expression values are plotted by injection and age groups. (A) Quantification of positive fractional area for total tau (HT7) staining. (B) Quantification of positive fractional area for phosphorylated tau Ser199/Thr205 (AT8) staining. (C) Quantification of positive fractional area for phosphorylated tau Ser396 staining. (D) Quantification of positive fractional area for silver positive Gallyas neurofibrillary tau tangle staining. Data are presented as mean ±SEM (error bars). Each dot represents one mouse. **p<0.01, *** p <0.001 Tukey’s post-hoc analyses.
In addition to histological tau measurements, innate immune activation was examined by MHCII staining of microglia (Supplemental Figure 3) where the GFP injection group was included. There were significant group differences (F(3,35) = 14.478, p <.001), where tauR406W (p = .03), and tauwild-type (p <.001) injection groups had a greater amount of MHCII expression when compared to the GFP group. Furthermore, the tauwild-type had the greatest amount of MHCII when compared to tauP301L (p <.001), and tauR406W (p = .01).
Soluble Tau
There were significant differences in total tau (F(2,26) = 15.192, p <.001) biochemically measured by ELISA. The tauR406W mice had significantly lower amounts of total tau when compared to tauP301L (p < .001) or tauwild-type mice (p = .002; Figure 7A). The soluble phospho-tau Ser199 demonstrated significant group differences (F(2,26)=15.366, p <.001; Figure 7B), and the post hoc analyses revealed that the tauR406W group had significantly lower amount of phospho-tau Ser199 compared to the tauP301L (p = .009) and the tauwild-type (p <.001) variant injection groups. Like the phospho-tau Ser199, phospho-tau Ser396 revealed significant group effects (F(2,26) = 17.300, p <.001; Figure 6C). The tauwild-type group had significantly greater amounts of phospho-tau Ser396 compared to tauP301L (p = .005) and tauR406W (p < .001) groups. In addition, the amount of tau oligomers measured with the TOC1 antibody exhibited significant group differences (F(2,26) = 10.92, p < .001). Again, the tauR406W mice had a significantly lower concentration of tau oligomers when compared to tauP301L mice (p < .001),Figure 6D).
Figure 7.

Quantification of soluble tau by ELISA. AAV9 tauP301L and tauwild-type exhibit early and late phosphorylated soluble fraction markers of tauopathy. (A) The total tau concentration measured by ELISA assays on the detergent soluble hippocampal fraction normalized to total protein. (B) The Ser199 concentration measured by ELISA assays on the detergent insoluble hippocampal fraction normalized to total protein. (C) Quantification of the positive fractional area of histological phospho-tau Ser396. (D) The oligomeric tau concentration measured by ELISA assays on the detergent soluble hippocampal fraction normalized to total protein. Data are presented as mean ±SEM (error bars). Each symbol represents one mouse. * p<0.05, ** p<0.01, *** p <0.001 Tukey’s post-hoc analyses.
We further analyzed the ELISA data by examining the ratios of phospho-tau to total tau for each mouse (Table 1). These data indicate a significant increase in the phospho-tau/total tau ratio in the tauwild-type group for both the phospho-tau Ser199 and phospho-tau Ser396 by 2–3-fold. There were no differences in the ratio of TOC-1 positive tau to total tau, or insoluble tau isoforms.
Table 1.
Phospho-Tau ELISA Values as a Percentage of Total Tau
| TauP301L | TauR406W | TauWild-type | One-way | ANOVA | |
|---|---|---|---|---|---|
|
| |||||
| M (SEM) | M(SEM) | M(SEM) | F | p | |
|
| |||||
| Soluble pSer199/total tau | 1.50(0.20) | 2.08(0.35) | 3.30(0.17)** | 17.14 | <.001 |
| Soluble pSer396/total tau | 0.66(0.15) | 0.13(0.20) | 2.17(0.28)** | 23.67 | <.001 |
| Soluble TOC1/total tau | 0.17(0.03) | 0.17(0.04) | 0.11(0.01) | 1.20 | 0.32 |
| Insoluble pSer199/total tau | 1.2(0.42) | 1.04(3.55) | 2.51(0.77) | 0.69 | 0.51 |
| Insoluble pSer396/total tau | 5.06(1.43) | 3.67(1.84) | 4.10(1.74) | 0.17 | 0.85 |
Tauwild-type > tauP301L and tauR406W P < 0.001 by Tukey’s posthoc test
Means and standard error of the mean are presented as percentages.
In addition to the ELISA assays, western blots were conducted for the detergent soluble fraction and analyzed for tau markers. For HT7 there was significant group differences (F(2,25)=8.107, p =.002; Supplemental Figure 4A) where tauR406W had a significant reduction in total tau concentration compared to tauP301L (p =.002) and tauwild-type (p =.02). When examining AT8, there was a significant F test, F(2,25) = 3.560, p =0.04. However, when Tukey’s post hoc analysis was conducted there were no statistically significant differences, but it followed a similar trend as the ELISA measurement for phosphorylated Ser199 (Supplemental Figure 4C). Furthermore phospho-tau Ser396 had significant group differences (F(2,26)=6.810, p= .004; Supplemental Figure 4E) that was detected as an increase in the tauwild-type compared to tauR406W (p=.003). Unsurprisingly, the ELISA measurements for total tau, phospho-tau Ser199, and phospho-tau Ser396 were significantly correlated to the western blot analyses for total tau (HT7; r=,70, p<.0001), AT8 (phospho-tau 202/thr205; r=.58, p=.0001), and phospho-tau Ser396 (r=.58, p =.0001), respectively.
Insoluble Tau
The detergent insoluble fraction was also examined for amount of total tau, phospho-tau Ser199, and phospho-tau Ser396 (Figure 8). Three samples were unable to be processed due to inadequate sample volume. The following insoluble tau analyses were conducted across the tau variants as the GFP injection group did not have any positive signal for any tau measurements. The insoluble total tau demonstrated significant group differences (F(2,23) = 14.581, p <.001), where the tauP301L had significantly greater amounts compared to tauR406W (p < .001) and tauwild-type (p = .005). The phospho-tau Ser199 also varied significantly by group, (F(2,23) = 7.244, p = .004) with significantly greater amounts in tauP301L when compared to the tauR406W variant injection group (p = .003). Finally, the amount of the insoluble fraction for phospho-tau Ser396 also exhibited significant differences (F(2,23) = 10.647, p < .001) where the tauP301L group had greater amounts compared to both the tauR406W ( p <.001) and the tauwild-type (p = .005) variant injection groups.
Figure 8.

Quantification of insoluble tau by ELISA. AAV9 tauP301L and tauwild-type exhibit early and late phosphorylated indicators of insoluble markers of tauopathy. (A). Total tau levels measured by ELISA assays with the detergent insoluble hippocampal fraction normalized to total protein. (B). The Ser199 concentration measured by ELISA assays on the detergent insoluble hippocampal fraction normalized to total protein. (C). The Ser396 concentration measured by ELISA assays on the detergent insoluble hippocampal fraction normalized to total protein. Data are presented as mean ±SEM (error bars). Each symbol represents one mouse. * p<0.05, ** p<0.01, *** p ≤0.001 Tukey’s post-hoc analyses.
Discussion
The purpose of the study was to compare three different tau genetic variants for differences among the phenotypes that emerged following direct administration into the brains of middle-aged mice. We found considerable differences among the phenotypes with the three tau genetic variants. Behaviorally, both the tauP301L and tauwild-type injection groups demonstrated declines in a spatial navigation memory task when compared to the GFP control and tauR406W injection groups. In terms of atrophy, the tauwild-type injection group had consistent reductions in histological hippocampal volume, dissected hippocampal weight, histological cortical thickness, and body weight loss over the duration of the study. The tauP301L injected mice had some reduction in hippocampal weight (compared to tauR406W mice, but not GFP injected mice), but no change in the other measures. There were no indications of atrophic changes in the mice administered AAV expressing tauR406W. These data suggest that the tauwild-type overexpression selectively resulted in significant neurodegeneration relative to the other treatment groups.
When examining measures of tau, two patterns emerged. The tauwild-type group had elevated levels of soluble phosphorylated tau isoforms measured by ELISA (presumably monomeric tau). This was consistent with the increased immunostaining for total tau and two phosphorylated tau isoforms. Additionally, when expressed as a percentage of total tau, the phosphorylated isoforms were 2–3 times more prevalent in the tauwild-type than the other two treatment groups. Conversely, the tauP301L group had larger amounts of what are presumed to be aggregated forms of tau, total and phosphorylated tau isoforms in the detergent insoluble fraction, the appearance of Gallyas silver-stained fibrils, and elevated oligomeric isoforms measured by the TOC1 antibody. Thus, in general, the tauP301L injected mice had a greater degree of tau aggregation.
Unexpectedly, the mice with administered viral vectors expressing tauR406W had a minimal phenotype. They were unaffected behaviorally, minimally deposited tau into insoluble fractions and had low levels of tau detected histologically. There have been several tauR406W transgenic mice developed previously which expressed a phenotype resembling that of P301L mice (reviewed in Denk and Wade-Martins (2009). The R406W mutation is outside the microtubule binding domain and, unlike the other tau mutations, produces tau fibrils that are both 3R and 4Rtau immunopositive (like Alzheimer’s; (de Silva et al., 2006), and formed paired helical filaments ultrastructurally (like Alzheimer’s; (Reed et al., 1997). Also, like Alzheimer’s, but not other FTD cases, the R406W mutation carriers have elevated tau phosphorylation of residue 217 in CSF (Sato et al., 2021). Thus, it was hoped this might form a more Alzheimer-like tau phenotype. However, it appears the expression of this mutation was considerably lower than the other two tau variants, despite the same promoter/enhancer elements and the same tau sequence in the constructs, other than residue 406.
In light of our results, it is important to note the differences that have been observed previously with diverse forms of tau. Mutreja et al. (2018) demonstrated that tauP301L produced a greater reduction of microtubule assembly and stabilization with an increase in the polymerization of tau aggregates when compared to tauwild-type and tauR406W. Furthermore, the electron micrographs revealed that tauP301L generated the longest filaments. Similarly, our results suggest that tauP301L generated an increased amount of NFTs by Gallyas staining and insoluble tau by ELISA. Furthermore, Maeda et al. (2018) reported differences in the amount of oligomeric tau produced by tauP301L, tauwild-type, and tauR406W. This study found that mutated forms of tau resulted in an increased number of oligomeric granules when compared to tauwild-type. Interestingly that is consistent with our results showing that AAV tauP301L yielded a greater amount of tau oligomers by ELISA compared to the tauR406W injection groups. In a study with transgenic models expressing comparable amounts of wild-type and P301L tau, Kimura et al (2010) demonstrated that mice expressing wild-type tau had memory impairments and synaptic loss. Conversely, the P301L transgenics had 10–20% neuron loss, but no memory impairment. They further found that the fraction of tau that was phosphorylated was higher in the wild type than P301L mice and that only the P301L mice had insoluble forms of tau. These latter two observations are fully consistent with our results using AAV injections of these variants.
Somewhat surprising is that the aggregated forms of tau did not correlate with the degree of atrophy across the three tau treatment groups. For some time, it was presumed that the insoluble fibrillary forms of tau were associated with neurotoxicity (Iba et al, 2013; Narasimhan et al, 2017). Pathologically, the strongest AD histopathological correlate with cognitive function was neurofibrillary tangles (Nelson et al, 2012). The tauP301L injected mice had increased aggregation, but minimal atrophy. The tauwild type injected mice had seemingly higher levels of soluble forms of tau and phospho-tau, and this was associated with considerable atrophy in the brain. Although often studies in single models link aggregation and toxicity together, some studies find this is not the case. For example, in the rTg4510 mice which develop both neuron loss and aggregation, the suppression of the transgene with doxycycline resulted in rescue of the memory deficit and arrest of the atrophy yet continued increases in the amounts of aggregated tau (silver positive tangles; (Santacruz et al., 2005). Additional work in the rTg4510 line noted a regional mismatch between neurofibrillary pathology and neuron loss (Spires et al., 2006). There were regions in which neuron loss preceded the formation of neurofibrillary pathology, and regions with abundant tangles that had no neuron loss. The latter reference also confirmed that suppressing the tau transgene with doxycycline halted the neuron loss but failed to reduce the neurofibrillary pathology. Furthermore, Andorfer et al (2003) found no linkage between tau tangles in aged hTau mice and neuron loss. Perhaps most relevant is a study by d’Orange et al. (2018). These authors injected rats with AAV2 constructs containing either wild-type tau, or wild-type tau plus a ΔK280 pro-aggregate peptide. They reported that the wild-type construct alone produced neuron loss in the hippocampus without formation of Gallyas positive fibrils. However, the wild-type plus the pro-aggregate peptide induced Gallyas positive tangles but lacked neuron loss. They further noted that the AT8 immunostaining in the wild type only mice exceeded that for the wild-type plus pro-aggregate peptide mice. This is consistent with our finding of increased AT8 immunostaining in the wild type versus P301L mice. The Kayed group has argued that it is the smaller aggregates of tau, oligomers and protofibrils, that are the most toxic tau variants (Ghag et al 2018). In our work, the measure of oligomers by TOC1 was not informative with respect to the distinctions between the tauwild-type and tauP301L mice.
We selected mice between 12 and 16 months for this study to begin tau exposure at middle-age when some physiological functions are starting to decline and ending the exposure at an age where spontaneous memory impairment is rare in the absence of tauopathy (at 20 months, some C57BL/6 mice have spontaneous memory impairments; (Yanai and Endo, 2021); our unpublished data). In humans living with non-familial AD, pathology can begin to accumulate in middle age (50–65 years of age). Given the success with the intracranial AAV method of inducing tauopathy, we will perform a more detailed assessment of age as a variable in mice younger and older than 12 months at the time of surgery to assess whether the aged brain context has an influence on the development of tauopathy.
No study is without limitation. This study only included one viral vector incubation period of 4 months. It would be advantageous to evaluate the tauwild-type at earlier and later time points in comparison to the 4-month viral incubation period to see when atrophic changes start to emerge and whether longer exposures might lead to more aggregation and neurofibrillary pathology. Another limitation to this approach is the intraparenchymal injection procedure. In addition to producing local tissue damage due to craniotomy and needle insertion, the distribution of the viral vector forms a gradient from the center of the injection out to the fringes. Thus, some neurons see much higher expression levels than others, and other portions of the brain are not impacted. It is uncertain if a more uniform administration of these vectors might lead to different outcomes.
In conclusion both the tauP301L and tauwild-type induced models of tauopathy yet had considerably different phenotypes. These viral vectors induced memory deficits but apparently did so in different ways. TauP301L demonstrated a greater amount of aggregated tau including some NFTs. These might lead to disruption of synaptic circuits, interfering with normal brain function. Tauwild-type injections demonstrated greater amounts of soluble tau, phosphorylated tau and gross tissue atrophy. Thus, it would seem likely that the cognitive impairments associated with the wild-type tau are secondary to loss of brain structures critical for establishment of new memories. In modeling AD tauopathy, one of the criticisms, especially in transgenic models, is that the mutations typically used are specific to FTD (and result in different structures for the tau aggregates than in AD (Scheres et al., 2020). The use of tauwild-type can bypass the use of a mutated form of tau and the viral vector modeling allows for targeting the areas impacted in the progression of AD pathology. The findings that injections of AAV9 tauwild-type into middle-aged mice can produce pathological tau, a reduction in spatial navigational memory, and immune activation justifies further investigation of this model for AD-related tauopathy.
Supplementary Material
Supplemental Figure 1. Experimental design and flowchart with sample sizes for the comparison of intracranial viral GFP, tauP301L, tauR406W, and tauwild-type injections. Red boxes indicate attrition due to mortality, health concerns, or inability to swim. GFP= green florescent protein (control virus).
Supplemental Figure 2. Immunohistochemistry for tau and phosphorylated tau epitopes in mice injected with AAV9 GFP. (A) Section stained with HT7 (total tau). (B) Section stained with AT8 (labeling tau phosphorylated at residues 199 and 202). (C) Section staining with antibody recognizing tau phosphorylated at residue 396.
Supplemental Figure 3. Quantification of histological MHCII staining. AAV9 tauwild-type had the greatest immune response measured by MHCII. Quantification of positive area ratio of MHCII expression microglia by MHCII staining. Data are presented as mean ±SEM (error bars). Each dot represents one mouse. ** p<0.01, *** p <0.001 Tukey’s post-hoc analyses.
Supplemental Figure 4. Quantification of soluble tau by western blot normalized to actin. (A-B). Total tau levels measured by western blot with the detergent soluble hippocampal fraction normalized to actin. (C-D). AT8 measured by western blot with the detergent soluble hippocampal fraction normalized to total actin. (E-F). The Ser396 measured by western blot with the detergent soluble hippocampal fraction normalized to actin. Data are presented as mean ±SEM (error bars). Each symbol represents one mouse. * p<0.05, ** p<0.01 Tukey’s post-hoc analyses. AT8 measurement (panel C) indicated significant ANOVA (P < 0.05), but no significant means comparison by Tukey’s post hoc analysis.
Highlights.
Intracranial AAV9 tau mutation variants have different phenotypes
AAV9 tauwild-type injected mice had significant atrophy and increased soluble tau
AAV9 tauP301L injected mice had more aggregated tau
AAV9 tauR406W group failed to induce pathological changes
Both wild-type and P301L injected mice had impaired spatial memory
Acknowledgements
This work was supported by AG 051500 to DM and AG 062217 to MNG. We thank Dr. Nicholas Kanaan (Michigan State University) for generous provision of the TOC1 antibody and instructions on the ELISA method using this antibody. In addition, we thank Dr. Kevin Nash (University of South Florida) for generous provision of glycerol stocks of competent SURE2 cell that contained 2N4R tauwild-type and GFP plasmids for the viral preparation of this study.
Footnotes
Disclosures
DGM receives research support from Bright Minds Pharmaceuticals, and Hesperos Inc. He also has recently consulted with Abbvie and is on the Speakers Bureau for Biogen. No other authors have disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ, Rönnbäck L, Hansson E, 2006. Astrocyte–endothelial interactions at the blood–brain barrier. Nature Reviews Neuroscience 7(1), 41–53. [DOI] [PubMed] [Google Scholar]
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D, 2006. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nature protocols 1(4), 1671–1679. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P, 2003. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of Neurochemistry 86(3), 582–590 [DOI] [PubMed] [Google Scholar]
- Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, Ayakta N, Cantwell A, Janabi M, Lauriola M, 2017. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140(12), 3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, Bohl J, Reintjes R, 1996. Age, neurofibrillary changes, Aβ-amyloid and the onset of Alzheimer’s disease. Neuroscience letters 210(2), 87–90. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K, 2011. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. Journal of Neuropathology & Experimental Neurology 70(11), 960–969. [DOI] [PubMed] [Google Scholar]
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, Lane JR, Karydas AM, Kenet RO, Biernat J, Wang LS, Cotman CW, Decarli CS, Levey AI, Ringman JM, Mendez MF, Chui HC, Le Ber I, Brice A, Lupton MK, Preza E, Lovestone S, Powell J, Graff-Radford N, Petersen RC, Boeve BF, Lippa CF, Bigio EH, Mackenzie I, Finger E, Kertesz A, Caselli RJ, Gearing M, Juncos JL, Ghetti B, Spina S, Bordelon YM, Tourtellotte WW, Frosch MP, Vonsattel JP, Zarow C, Beach TG, Albin RL, Lieberman AP, Lee VM, Trojanowski JQ, Van Deerlin VM, Bird TD, Galasko DR, Masliah E, White CL, Troncoso JC, Hannequin D, Boxer AL, Geschwind MD, Kumar S, Mandelkow EM, Wszolek ZK, Uitti RJ, Dickson DW, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Alzheimer’s Disease Genetics C, Ross OA, Rademakers R, Schellenberg GD, Miller BL, Mandelkow E, Geschwind DH, 2012. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet 21(15), 3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Orange M, Aurégan G, Cheramy D, Gaudin-Guérif M, Lieger S, Guillermier M, Stimmer L, Joséphine C, Hérard A-S, Gaillard M-C, 2018. Potentiating tangle formation reduces acute toxicity of soluble tau species in the rat. Brain 141(2), 535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya S, Berns KI, 2008. Gene therapy using adeno-associated virus vectors. Clinical microbiology reviews 21(4), 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton RD, Wang DB, Klein RL, 2012. The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert opinion on biological therapy 12(6), 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva R, Lashley T, Strand C, Shiarli A-M, Shi J, Tian J, Bailey KL, Davies P, Bigio EH, Arima K, 2006. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R-and 4R-specific tau monoclonal antibodies. Acta neuropathologica 111(4), 329–340. [DOI] [PubMed] [Google Scholar]
- Denk F, Wade-Martins R, 2009. Knock-out and transgenic mouse models of tauopathies. Neurobiology of aging 30(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J, 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences 69(Suppl_1), S4–S9. [DOI] [PubMed] [Google Scholar]
- Ghag G, Bhatt N, Cantu DV, Guerrero-Munoz MJ, Ellsworth A, Sengupta U, Kayed R, 2018. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci 27(11), 1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, 2005. Tau gene mutations and their effects. Movement disorders: official journal of the Movement Disorder Society 20(S12), S45–S52. [DOI] [PubMed] [Google Scholar]
- Gordon BA, McCullough A, Mishra S, Blazey TM, Su Y, Christensen J, Dincer A, Jackson K, Hornbeck RC, Morris JC, 2018. Cross-sectional and longitudinal atrophy is preferentially associated with tau rather than amyloid β positron emission tomography pathology. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 10, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KL, Connor K, Melachrino JO, O’Callaghan JP, Morgan D, 2002. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol 173, 183–195. [DOI] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, 2019. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA neurology 76(8), 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M, 1998. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS letters 437(3), 207–210. [DOI] [PubMed] [Google Scholar]
- Heutink P, Stevens M, Rizzu P, Bakker E, Kros JM, Tibben A, Niermeijer MF, Van Duijn CM, Oostra BA, Van Swieten JC, 1997. Hereditary frontotemporal dementia is linked to chromosome 17q21—q22: A genetic and clinicopathological study of three dutch families. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 41(2), 150–159. [DOI] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM, 2013. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33(3), 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Lechat B, Croes S, Termont A, Demedts D, Borghgraef P, Devijver H, Filipkowski RK, Kaczmarek L, 2009. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PloS one 4(10), e7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC, 2016. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. Journal of Neuropathology & Experimental Neurology 75(1), 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A, 2010. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem 285(49), 38692–38699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe C, Ogaki K, Lorenzo-Betancor O, Soto-Ortolaza AI, Walton RL, Rayaprolu S, Fujioka S, Murray ME, Heckman MG, Puschmann A, McCarthy A, Lynch T, Siuda J, Opala G, Rudzinska M, Krygowska-Wajs A, Barcikowska M, Czyzewski K, Sanotsky Y, Rektorova I, McLean PJ, Rademakers R, Ertekin-Taner N, Hassan A, Ahlskog JE, Boeve BF, Petersen RC, Maraganore DM, Adler CH, Ferman TJ, Parisi JE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Dickson DW, Ross OA, 2015. Role for the microtubule-associated protein tau variant p.A152T in risk of alpha-synucleinopathies. Neurology 85(19), 1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ, 1999. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Human gene therapy 10(14), 2295–2305. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sato Y, Takashima A, 2018. Frontotemporal dementia with Parkinsonism linked to chromosome-17 mutations enhance tau oligomer formation. Neurobiology of aging 69, 26–32. [DOI] [PubMed] [Google Scholar]
- Malpas CB, Sharmin S, Kalincik T, 2020. The histopathological staging of tau, but not amyloid, corresponds to antemortem cognitive status, dementia stage, functional abilities and neuropsychiatric symptoms. International Journal of Neuroscience, 1–10. [DOI] [PubMed] [Google Scholar]
- Mustroph ML, King MA, Klein RL, Ramirez JJ, 2012. Adult-onset focal expression of mutated human tau in the hippocampus impairs spatial working memory of rats. Behavioural brain research 233(1), 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja Y, Combs B, Gamblin TC, 2018. FTDP-17 mutations alter the aggregation and microtubule stabilization propensity of tau in an isoform-specific fashion. Biochemistry 58(6), 742–754. [DOI] [PubMed] [Google Scholar]
- Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH, 1999. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS letters 447(2–3), 195–199. [DOI] [PubMed] [Google Scholar]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, 2017. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. Journal of Neuroscience 37(47), 11406–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG, 2012. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. Journal of neuropathology and experimental neurology 71(5), 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Richter KEG, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, 2007. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. Journal of Neuroscience 27(12), 3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, Santacruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH, 2005. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). Journal of Neuroscience 25(46), 10637–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LA, Schelper RL, Solodkin A, Van Hoesen GW, Solodkin A, Morris JC, Trojanowski JQ, Grabowski TJ, Talbot CJ, 1997. Autosomal dominant dementia with widespread neurofibrillary tangles. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 42(4), 564–572. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH, 2005. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733), 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C, Mallipeddi N, Ghoshal N, Wright BA, Day G.y.S., Davis AA, Kim AH, Zipfel GJ, Bateman RJ, Gabelle A, 2021. MAPT R406W increases tau T217 phosphorylation in absence of amyloid pathology. Annals of Clinical and Translational Neurology 8(9), 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Zhang W, Falcon B, Goedert M, 2020. Cryo-EM structures of tau filaments. Current Opinion in Structural Biology 64, 17–25. [DOI] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT, 2006. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. The American journal of pathology 168(5), 1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, 2007. Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta neuropathologica 113(5), 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, De Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer MF, 1999. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 46(4), 617–626. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, da Rocha EL, Jansen-West K, Frame AA, Citro A, Leszyk JD, 2016. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell reports 15(7), 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai S, Endo S, 2021. Functional Aging in Male C57BL/6J Mice Across the Life-Span: a Systematic Behavioral Analysis of Motor, Emotional, and Memory Function to Define an Aging Phenotype. Frontiers in Aging Neuroscience, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Experimental design and flowchart with sample sizes for the comparison of intracranial viral GFP, tauP301L, tauR406W, and tauwild-type injections. Red boxes indicate attrition due to mortality, health concerns, or inability to swim. GFP= green florescent protein (control virus).
Supplemental Figure 2. Immunohistochemistry for tau and phosphorylated tau epitopes in mice injected with AAV9 GFP. (A) Section stained with HT7 (total tau). (B) Section stained with AT8 (labeling tau phosphorylated at residues 199 and 202). (C) Section staining with antibody recognizing tau phosphorylated at residue 396.
Supplemental Figure 3. Quantification of histological MHCII staining. AAV9 tauwild-type had the greatest immune response measured by MHCII. Quantification of positive area ratio of MHCII expression microglia by MHCII staining. Data are presented as mean ±SEM (error bars). Each dot represents one mouse. ** p<0.01, *** p <0.001 Tukey’s post-hoc analyses.
Supplemental Figure 4. Quantification of soluble tau by western blot normalized to actin. (A-B). Total tau levels measured by western blot with the detergent soluble hippocampal fraction normalized to actin. (C-D). AT8 measured by western blot with the detergent soluble hippocampal fraction normalized to total actin. (E-F). The Ser396 measured by western blot with the detergent soluble hippocampal fraction normalized to actin. Data are presented as mean ±SEM (error bars). Each symbol represents one mouse. * p<0.05, ** p<0.01 Tukey’s post-hoc analyses. AT8 measurement (panel C) indicated significant ANOVA (P < 0.05), but no significant means comparison by Tukey’s post hoc analysis.