

Abstract
Advances in high-throughput and digital technologies have required the adoption of big data for handling complex tasks in life sciences. However, the drift to big data led researchers to face technical and infrastructural challenges for storing, sharing, and analysing them. In fact, this kind of tasks requires distributed computing systems and algorithms able to ensure efficient processing. Cutting edge distributed programming frameworks allow to implement flexible algorithms able to adapt the computation to the data over on-premise HPC clusters or cloud architectures. In this context, Apache Spark is a very powerful HPC engine for large-scale data processing on clusters. Also thanks to specialised libraries for working with structured and relational data, it allows to support machine learning, graph-based computation, and stream processing. This review article is aimed at helping life sciences researchers to ascertain the features of Apache Spark and to assess whether it can be successfully used in their research activities.
MSC: 00-01, 99-00
Keywords: Apache Spark, Big data, Parallel computing, HPC
1. Introduction
The continuous technological progress in data acquisition is generating a huge amount of data in several research fields. Among the manifold sources of data that are causing the drift toward big data, let us recall in particular i) the studies on the human genome and phenome, ii) microscopy and imaging tasks, and iii) the digitisation of medical records and clinical exams.
Studies on the human genome and phenome – An increase of about 50% in human genome and phenome data deposition has been pointed out by the EMBL-EBI 2020 annual report [1]. Among other studies, let us recall phenome-wide association studies [2], which is an emerging method to identify cross-phenotype associations. To this end, an extensive use of electronic health records is made, along with data aggregation from traditional epidemiological studies.
Microscopy and imaging tasks – Rapid advances in microscopy and medical imaging have brought an ever increasing production of data. Just to give an idea, the EMBL-EBI 2020 annual report highlights an increase of 120% for imaging data and of 164% for electron cryo-microscopy data. Medical imaging devices are another important source of data. Also thanks to the improvements related to the advances on artificial intelligence, diagnostic imaging techniques (e.g., computed tomography scans, x-rays, magnetic resonance imaging) are increasingly used in medical practice.
Digitisation of medical records and clinical exams – The advances in these fields are generating an ever-increasing amount of recorded data. The interested reader may consult the review article of Atasoy et al. [3], which provides an interdisciplinary overview and synthesis of patient care digitisation, also pointing to the relationships with computer science, medicine, management information systems, and economics.
Summarising, the common feature of all cited fields is big data, a technology that is radically changing the research landscape and offers unprecedented opportunities for further scientific insights. These opportunities are mainly related to the wide use of machine learning (ML) and data mining (DM) algorithms, which allow to make accurate predictions and to uncover fine-grained patterns [4]. Beyond the aspects related to data analysis, big data has also given a major support to stakeholders and researchers for dealing with technical and infrastructural challenges, which include i) devising proper policies and techniques for ensuring security and privacy while storing and sharing data, as well as ii) implementing proper computing systems tailored to analyse big data. Big data refers to large and complex datasets, which require powerful and scalable1 computing systems able to efficiently analyse them.
In Europe, data security is regulated by the General Data Protection Regulation (GDPR) [5], which defines specific rules to protect sensitive personal data used for research purposes [6]. Hence, data storing and data sharing must be carried out in accordance with all indications aimed at protecting data. Furthermore, data sharing also poses problems related to the transfer of large volumes of data. In particular, proper strategies are required to ensure that only data relevant to the current analysis task is moved. As for the need to rely on secure, efficient and scalable distributed computing systems, different initiatives are supporting researchers in the task of dealing with these challenges. Without claiming to be exhaustive, let us recall some relevant initiatives. The Global Alliance for Genomics and Health (GA4GH) [7] community implements frameworks and standards for secure sharing of genomic data. Horizon Europe, the European programme for research and innovation, funded the development of European research infrastructures aimed at implementing facilities to provide resources and services for the research communities. As for health, let us mention BBMRI-ERIC [8] the research infrastructure for biobanking and biomolecular resources, ELIXIR [9] the research infrastructure for life-science resources, and Euro-Bioimaging the research infrastructure for imaging technologies in biological and biomedical sciences. The cited research infrastructures implement specific services devised for their communities, which include the ability of storing, sharing, and analysing big data.
More generally, big data is known to target high-performance computing (HPC) systems, as distributing the computation across multiple nodes ensures that the data analysis is completed on time. Distributed computing is a paradigm in which the components of a software system are shared among multiple computing nodes and run as a single system. A major benefit of distributed computing is the capability of increasing the overall computing power by simply adding further nodes on need.
Among the tools and infrastructures devised to perform big data analysis, Apache Spark [10] (Spark hereinafter) is a prime choice, thanks to its ability to provide an advanced programming model and specific libraries aimed at supporting various kinds of computations. Although Spark is starting to be widely adopted in several application and research fields, it is not yet widely known and used in the life sciences, where Apache Hadoop is most known. In fact, different features make Spark and Hadoop complement each other.
This article is aimed at helping researchers to understand the main features of the Spark framework and to show its applicability to tasks generically attributable to life sciences. With this goal in mind, a literature review of relevant Spark applications is made, regarding the specific challenges that may hold for the corresponding research fields. To the best of our knowledge, only one review article with a similar target has been published by Guo et al. [11] in 2018, focusing on bioinformatics applications based on Spark. In this work, the scope of interest extends beyond bioinformatics to the life sciences.
The remainder of this article is organized as follows. First, the Spark framework is described and then a review of Spark-based applications is provided. A discussion on how Spark can be integrated with other technologies follows, to help researchers taking a decision about adopting it to solve relevant problems. Finally, conclusions are drawn.
2. Apache Spark
Spark is a unified computing engine and a set of specialized libraries for large-scale data processing on computer clusters. Originally developed at the UC Berkeley AMPLab in 2009, in 2013 Spark was donated to the Apache Software Foundation.
From an abstract perspective, Spark is a distributed data processing engine, whose components work and communicate on a cluster of computers. The highest-level units of computation are Spark applications, each being a computational resource that runs a user program in a distributed environment. Any Spark application puts into practice a master-slave policy, which consists of two processes –i.e., driver program (the master) and a set of executors (the workers). Depending on the execution mode, driver program and executors are distributed over the cluster by means of a cluster manager. With full support to various programming languages (i.e., Scala, Java, Python, and R), Spark implements ad-hoc libraries for SQL, ML, stream processing and graph computing.2
Spark also supports three data abstraction types (i.e., RDDs, DataFrames, and Datasets) which consist of a logical structure of the data distributed across the nodes of a cluster (see the Appendix). As for storage management, by default Spark adopts the local file system of the running host, with particular emphasis on the Hadoop file system (HDFS), HBase, and various cloud-based file systems –including Amazon S3, Google Cloud and Microsoft Azure Storage. At the time of writing, Spark is the most actively developed distributed programming framework, taking worldwide contributions from hundreds of organizations.
This section is organised as follows: first, the basic information for understanding what is a Spark application is given. Then a few words are spent to briefly describe the Spark ecosystem and to compare Spark with Hadoop [12].
2.1. Spark applications
From an abstract perspective, Spark is a distributed data processing engine, whose components work and communicate on a cluster of machines. In Spark, the highest-level units of computation are Spark applications, each being a distributed computational resource that runs a user program. Any Spark application puts into practice a master-slave policy, which consists of two processes –i.e., driver program (the master) and a set of executors (the workers). Depending on the execution mode, driver program and executors are distributed over the cluster by means of a cluster manager (see Fig. 1).
Figure 1.
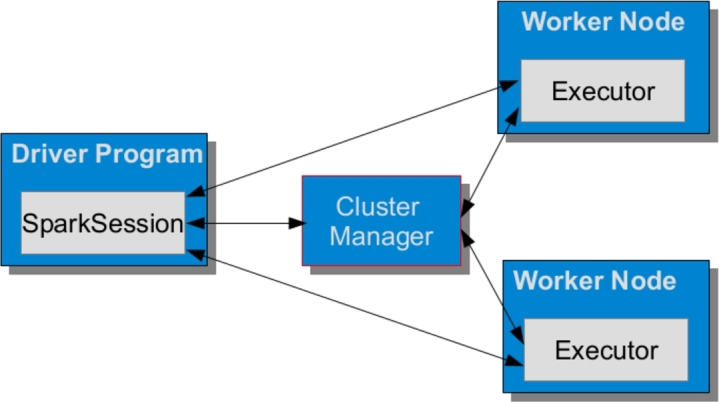
Snapshot of a Spark application.
Driver program – The main responsibilities of the driver program are i) to maintain all relevant information during the lifetime of the application; ii) to interact with the user program; and iii) to analyse, distribute, and schedule the computation across the executors. A SparkSession is a unified solution devised to make available all Spark operations on data. Initially, the SparkSession asks the cluster manager to find proper locations for all executors on the cluster and then sends them the application code, together with the tasks to be run.
Executors – A Spark executor runs on each worker node in the cluster for the entire lifetime of an application. Executors are entrusted with handling application tasks and with reporting the state of the computation and the results back to the SparkSession. Depending on the settings that characterise the corresponding Spark application, executors can be statically or dynamically allocated.
Cluster manager – The main responsibilities of the cluster manager are to start executor processes over the cluster and to partition resources across applications. Spark supports various cluster managers, i.e., the default standalone cluster manager, Apache Hadoop YARN [13], Apache Mesos [14], and Kubernetes [15]. The cluster manager may be involved in different ways while running a Spark application, according to the adopted execution mode. The supported execution modes are local mode, client mode, and cluster mode. The simplest setting occurs in the first case (i.e., when a Spark application is run on a local machine) and is typically used to test the application during its development. In client mode, the cluster manager is only responsible for taking care of the executors, whereas the client machine is responsible for maintaining the driver program. The most general execution model is the last one, in which the cluster manager is responsible for maintaining both driver program and executors.
2.2. The Spark ecosystem
As highlighted by Fig. 2, the Spark Ecosystem is built upon the Spark Core, which is responsible for providing all basic I/O functionalities, scheduling and monitoring the jobs on Spark clusters, task dispatching, networking with different storage systems, fault recovery, and efficient memory management. As pointed out, the Core API also provides support for interacting with five programming languages, namely Scala, Java, Python, R, and SQL. Beyond the Spark Core, the Spark Ecosystem encompasses specific modules devised to support i) SQL with a distributed query engine, ii) the processing of live data streams, iii) machine learning and data mining tasks, iv) graph and graph-parallel computations. In recent distributions, a specific subsystem for dealing with the R language (i.e., SparkR) has also been made available.3
Figure 2.

Spark Ecosystem.
2.3. Spark vs Hadoop
Hadoop is a well known framework for distributed processing of large data sets across clusters of computers. It is widely used in different fields, including life sciences. Spark and Hadoop come with features that give support to researchers in the task of deciding which one to use for their purpose. Spark is a prime choice when the processing time is a significant constraint. In particular, applications that involve real time processing and/or ML tasks should be dealt with using the Spark framework –which can be up to 100x faster than Hadoop, thanks to its capability of performing in-memory processing. As for Hadoop, it is a primary choice for batch processing tasks, provided there no strict time constraints hold. Due to their complementary abilities, one may conclude that Spark and Hadoop complement each other. Nevertheless, nothing prevents from using them together. In fact, Spark is often used in conjunction with Hadoop to take advantage of its HDFS, for its ability of storing large volumes of data.4
3. Apache Spark in life sciences
The search for relevant articles has been constrained by the decision of reporting only recent ones (i.e., from 2018 on), to avoid any overlapping with the cited review article of Guo et al. [11]. The search for articles has been mainly conducted on PubMed looking for the keywords “Apache Spark” in all search fields. In total, 33 articles have been selected, whose common denominator is need of adopting Spark to deal with the computational challenges in various fields, including genomics, metagenomics, epigenomics, biomedical imaging, biomedical signal processing, gene-disease associations and drug discovery.
Table 1 reports a summary of the main features of the tools proposed in these articles. The table shows that apart from some tools that reports tests only on a multi-core workstation ([16], [17], [18], [19]), Spark has been widely used to implement tools aimed at parallelizing the computation on a distributed computing environment. Most of these tools have been specifically devised for, or tested on, a cloud environment ([20], [21], [22], [23], [24], [25], [26], [27], [28] [29], [30], [31], [32] [33], [34], [35], [36], [37]). Being the increasing availability of IaaS (Infrastructure as a Service) cloud computing services, it is desirable that the released tools are commonly designed to be supported also by such infrastructures.
Table 1.
Main features of surveyed Spark-based applications in the fields of life sciences. The table shows information about i) the main task performed ii) the Spark libraries that have been used, iii) if multi-node computing is supported iv) if multi-gpu computing is supported, v) if cloud support is declared, vi) if Apache Hadoop file system (HDFS) is used, vii) if external cluster manager (CMAN) is used, and xiii) the use of other external tools/frameworks (if any).
| Task | Spark libraries | Multi-node | Multi-GPU | Cloud | HDFS | Ext. CMAN | Ext. tools/frameworks | |
|---|---|---|---|---|---|---|---|---|
| Genomics | ||||||||
| genome assembly | ||||||||
| SORA [20] | de novo genome assembly | GraphX | ✓ | - | ✓ | - | - | - |
| variant calling | ||||||||
| DECA [21] | copy number variantion discovery | MLlib | ✓ | - | ✓ | - | - | ADAM |
| ADS-HCSpark [48] | SNPs and indels calling | - | ✓ | - | - | - | - | - |
| SparkGA2 [22] | variant calling | - | ✓ | - | ✓ | ✓ | - | - |
| SparkRA [49] | GATK best-practices pipeline | - | ✓ | - | - | - | - | - |
| DeepVariant on Spark [23] | SNPs and indels calling | - | ✓ | ✓ | ✓ | ✓ | - | Apache Parquet |
| VC@Scale [24] | SNPs and indels calling | - | ✓ | ✓ | ✓ | - | - | Apache Arrow |
| Halvade Somatic [25] | somatic variant calling | - | ✓ | - | ✓ | - | - | - |
| alignment | ||||||||
| IMOS [38] | long reads alignment | - | ✓ | - | - | - | - | - |
| SPARK-MSNA [56] | multiple sequence alignment | Streaming | ✓ | - | - | - | - | - |
| DHPGIndex [26] | indexing of compressed pan-genomes | - | ✓ | - | ✓ | ✓ | - | - |
| K-mer statistics | ||||||||
| FastKmer [27] | k-mer counting | - | ✓ | - | ✓ | ✓ | - | - |
| Discount [28] | k-mer counting | - | ✓ | - | ✓ | - | - | - |
| analysis of genomic datasets | ||||||||
| Sparkhit [29] | integrates a series of analytical methods | MLlib | ✓ | - | ✓ | ✓ | - | Open to docker containers |
| PyGMQL [30] | manipulation of region-based genomic files | - | ✓ | - | ✓ | ✓ | YARN | - |
| Mango [31] | a genomics visualization platform | SQL | ✓ | - | ✓ | ✓ | YARN | - |
| SeQuiLa [43] | querying and processing of genomic intervals | SQL | ✓ | - | - | - | YARN | - |
| PyBDA [61] | command line tool for statistical and ML-based analysis | MLLib | ✓ | - | - | - | - | Snakemake |
| DNA error correction | ||||||||
| SparkEC [41] | correction of sequencing data | - | ✓ | - | - | ✓ | - | Hadoop Sequence Parser (HSP) |
| genome compression | ||||||||
| SparkGC [42] | compression for large collections of genomes | - | ✓ | - | - | ✓ | - | - |
| Metagenomics | ||||||||
| Flint [32] | a metagenomics profiling pipeline | Streaming | ✓ | - | ✓ | - | - | - |
| GATK PathSeq [33] | discovery and identification of microbial sequences | - | ✓ | - | ✓ | - | - | - |
| MetaGO [16] | a pipeline to detect group-specific sequences for microbial communities | - | - | - | - | ✓ | - | - |
| Epigenomics | ||||||||
| BiSpark [39] | bisulfite sequencing aligner | - | ✓ | - | - | ✓ | - | - |
| Biomedical Imaging | ||||||||
| Name N/A [35] | fMRI big data analytics | Tensorflow on Spark (third-party package) | ✓ | ✓ | ✓ | ✓ | - | - |
| Orbit [36] | whole slide imaging | - | ✓ | ✓ | ✓ | - | - | - |
| Biomedical Signal Processing | ||||||||
| Name N/A [65] | real-time heart arrhythmia detection | Structured Streaming | ✓ | - | - | - | - | - |
| Name N/A [37] | systolic blood pressure prediction | Streaming | ✓ | - | ✓ | - | - | Apache Kafka |
| Gene-Disease Association | ||||||||
| PRSoS [40] | polygenic risk scores | - | ✓ | - | - | ✓ | - | - |
| Name N/A [17] | breast cancer prediction | MLlib | - | - | - | - | - | - |
| VIGLA-M [18] | visual analysis tool for gene expression levels in melanoma patients | - | - | - | - | - | - | - |
| RWRHN-FF [19] | high-throughput network inference | - | - | - | - | - | - | - |
| Drug Discovery | ||||||||
| Name N/A [34] | virtual screening | MLlib | ✓ | - | ✓ | ✓ | - | - |
It should be pointed out that some applications in the fields of genomics ([23], [24]) and biomedicine ([35], [36]) have also been designed to distribute the computation on multiple GPUs on multiple nodes. GPU technology is widely used in life science. The massive parallelization offered by many-core GPU devices is allowing to deal with different computational challenges. In particular, in the field of omics-sciences different GPU-based tools have been proposed which are commonly used by researhers. Typically, these tools are designed to be run on a single GPU or on a computing node equipped with more devices. The availability of highly scalable computing infrastructures opens to the possibility of run these tools on distributed multiple nodes equipped with multiple GPUs. Spark could be effectively used to adapt existing tools as well as to implement new ones to scale the GPUs computation on such infrastructures.
Similarly, the increasing use of DL in multiple fields of life sciences (e.g., pharmacogenomics, proteomics, biomedicine) is encouraging the implementation of scalable multi-GPU applications. In fact, building complex models consisting of millions of trainable parameters on very large datasets is challenging. Therefore, Spark could be used to implement highly scalable DL-based applications.
The Table 1 also highlights that Spark is also used with other frameworks. In particular, it is often used in conjunction with Hadoop to take advantange of its file system (i.e., HDFS) ([16], [22], [23], [26], [27], [30], [31], [34], [35], [38], [39], [40], [41],[42]) and of its cluster manager (i.e., YARN) ([30], [31], [43]).
In the following of this section the surveyed applications are described in more details. They have been logically grouped by sub-areas and then according to the main task performed.
3.1. Genomics
This category accounts for articles broadly framed within the field of genomics. The articles are grouped hereinafter into five categories, according to the main task they perform: i) de novo genome assembly, ii) variant calling, iii) sequence alignment, iv) k-mer analysis, v) analysis of genomic datasets, vi) DNA error correction, and vii) genome compression.
3.1.1. De novo genome assembly
De novo genome assembly refers to a process aimed at building a genome by finding overlaps and merging DNA fragments. Most de novo genome assemblers implement an approach based on the overlap-layout-consensus (OLC) algorithm or on the de Bruijin graph. Paul et al. (2019) [20] present SORA (Scalable Overlap-graph Reduction Algorithm), a novel algorithm for de novo genome assembly based on the OLC approach. The algorithm is designed to deal with the extensive memory and processing time required by OLC. SORA leverages Spark and its GraphX library to accelerate the graph reductions for genome assemblies by compacting repetitive information of sequence overlaps in the cloud, in a local cluster, or using a stand-alone workstation. In particular, SORA is implemented and designed to leverage an assortment of computational operations in GraphX for construction, graph reading, transformation, and computation.
3.1.2. Variant calling
Variant calling is a main task of genomic analysis and represents a well known challenge in computational genomics. The literature search reported seven articles describing tools for variant calling using Spark. The reported works focus on implementing strategies aimed at efficiently porting existing algorithms to Spark (i.e., XHMM [44], GATK [45], DeepVariant [46]). These works implement multi-core, multi-node and multi-gpu strategies to optimize the computing performance.
Linderman et al. (2019) [21] propose DECA, a horizontally scalable implementation of the XHMM algorithm –which is aimed at discovering copy-number variants (CNVs) of the exome. DECA is based on Spark and ADAM [47], the latter being an in-memory distributed computing framework for genome analysis built on Spark. Compared to XHMM, DECA achieves a 35-fold speedup for calling CNVs in 2535 sample 1000 Genomes exome cohort.
ADS-HCSpark [48] implements a multi-core and multi-node parallelization of the GATK HaplotypeCaller algorithm. It should be pointed out that variant calling can result in some long tail tasks. In a distributed environment, this aspect need to be taken into account to avoid unbalanced workload among the workers that prevent scalability. In fact, an unbalanced workload of a single worker may cause a delay on the entire cluster. To deal with this potential issue, ADS-HCSpark implements an adaptive data segmentation that analyse the input files to predict the time-consuming blocks of data and dividing them into multiple new blocks whereas other blocks are keep in the original partitions.
Mushtaq et al. [22] (2019) propose SparkGA2 a framework built on Spark designed to efficiently run the GATK pipelines on highly scalable computing clusters. The framework has been designed with the aim to improve the performance with respect to the cutting-edge tools while optimizing the hardware resources utilization. If on the one hand, SparkGA2 exploits the in-memory computation to improve the performance, on the other hand it also implements specific strategies aimed at optimizing the amount of memory required to perform the computation. Specifically, its performance is optimized exploiting the efficient in-memory computation, whereas it uses an in-house implemented on-the-fly compression method to optimize the memory consumption. Experiments shown that SparkGA2 resulted faster than GATK best-practices pipeline on both a large cluster consisting of 67 nodes and a smart cluster of 6 nodes up to 22% and 9% respectively.
Al-Ars et al. (2020) [49] propose SparkRA to efficiently scale up the GATK RNA-seq variant calling pipeline on multi-core and multi-node platforms. The parallelization is achieved dividing the input data into a number of chunks that are processed by multiple instances of the GATK pipeline tools. In so doing, a static and a dynamic load balancing strategy is implemented, with the goal of limiting the time imbalance that occurs while dealing with different data chunks. Experimental results show that SparkRA is effective in utilizing CPU resources. CPU of all nodes is keep active at close to 100% utilization most of the time.
Huang et al. (2020) [23] propose DeepVariant-on-Spark, a Spark-based implementation of the deep-learning (DL) based variant caller DeepVariant. Devised to overcome the limitation of single GPU processing of DeepVariant, the algorithm leverages Spark to run multiple DeepVariant processes in parallel, thus addressing the scalability problem of variant calling. It enables a multi-node and multi-GPU parallelization, aimed at optimising resource allocation while accelerating the processing of the DeepVariant pipeline. DeepVariant-on-Spark has been deployed on the Google Cloud Platform (GCP), with the goal of making it more accessible to everyone.
Ahmad et al. (2021) [24] propose VC@Scale, another tool aimed at scaling DeepVariant. VC@Scale is a scalable and high-performance variant-calling workflow for cluster-scaled environments based on Spark and Apache Arrow [50]. The latter is a cross-language development platform for in-memory analytics, which enables data processing and data mobility by means of a standard columnar memory format. Integrated with Spark, Apache Arrow has been used for DNA data-preprocessing tasks –such as alignment, sorting, and duplicate removal. Then, DeepVariant was integrated into VC@Scale to create an open-source DeepVariant workflow on Spark for both CPU-only and CPU plus GPU clusters. Experiments, performed on the Dutch national supercomputer shown that VC@Scale was at least two times faster than other state-of-the-art implementations in the pre-processing stages.
Detection of somatic variants is another time consuming task. In fact, detecting low-frequency variants requires a high sequencing depth that generates large volume of data requiring high computing time. Halvade Somatic, a framework to efficiently performs somatic variant calling is presented in [25]. Halvade Somatic supports variant calling from both whole-genome sequencing and whole-exome sequencing. Its pipeline is implemented according to the GATK best practices recommendations. Spark is used to scale the computation and to manage data streams that are processed on different CPU cores in parallel. Halvade Somatic has been devised to scale the computation on multi-node and/or multi-core compute platforms. On a single node equipped with 36 cores it achieved a speedup of 4.3x with respect to the original pipeline. An additional speedup of 14.4x was achieved running it on 16 nodes.
3.1.3. Alignment
Alignment is a fundamental task in many analysis pipelines. Different solutions aimed at implementing fast and accurate sequence alignment tools have been proposed in the literature. Some implementations based on Spark are also available. The surveyed articles report three tools to deal with indexing of large pan-genomes, long-read alignment, and multiple sequence alignment.
Maarala et al. (2021) proposed DHPGIndex [26], a scalable distributed compression and indexing tool for a massive number of assembled genomes, with read alignment and sequence matching support. Built on top of Spark, DHPGIndex implements a distributed version of the compressed hybrid indexing [51] method, based on the Relative Lempel-Ziv (RLZ) algorithm [52]. Experiments shown that DHPGIndex performs compression and indexing of large pan-genomes very efficiently in terms of time. DHPGIndex can be used with the short read aligners BWA [53], with Bowtie [54], and with the sequence search tool BLAST [54].
A solution to deal with the task of long reads alignment is presented in [38]. In particular, an aligner devised for noisy PacBio reads –called IMOS– is proposed. IMOS is based on Meta-Aligner [55], an accurate aligner with uncompetitive performance in terms of computation time. Its implementation on Spark made it possible to overcome the problem related to its performance. IMOS distributes the long reads among the processing nodes using HDFS. Then, it uses Spark to call the instances of IMOS worker on the nodes. Finally, the workers run Meta-Aligner to align the long reads. Specific strategies have also been adopted to avoid unbalanced workloads and minimize storage usage. The implemented load balancer works on top of HDFS. It analyses a FastQ file to divide it in chunks with similar characteristics. To minimize storage usage, the reference files remains allocated into the main memory at runtime. This approach avoids storage-to-memory communication overheads.
Multiple sequence alignment (MSA) is a common task required by most genomic analyses. To deal with the computational complexity of this task, various heuristics have been proposed in the literature. SPARK-MSNA, is an MSA algorithm on Spark proposed by Vineetha et al. (2019) [56]. The algorithm uses both suffix tree and a modified Needleman-Wunsch algorithm. Suffix tree is used to identify common substrings, whereas Needleman-Wunsch is used for pairwise alignments. With the aim of improving the efficiency of pairwise alignments, a knowledge base is also created and supervised learning is used to guide the alignment. Spark has been used to implement the parallel computation of the suffix tree construction and of the pairwise alignment. Specifically, the suffix tree is partitioned vertically and each partition is built independently. Pairwise alignment is performed on the computing nodes using the modified pairwise algorithm. The adoption of Spark also reduces the network overhead thanks to the data locality concept of MapReduce.
3.1.4. K-mer analysis
Processing of k-mers is a task required by many sequence processing algorithms, such as genome assembly and genome size estimation. Brief summaries on two tools designed to tackle the k-mer counting task are reported hereinafter.
Petrillo et al. (2019) [27] propose FastKmer, an algorithm implemented on Spark aimed at extracting k-mer statistics from large collection of biological sequences, which extends the Hadoop-based tool KHC [57]. FastKmer consists of two main stages. In the first stage superkmers are extracted and sorted according to their signature, so that each group will be contained in a specific bin. Bin contents can vary, which typically ends up to anunbalanced partitioning over bins. To deal with this issue, FastKmer adopts the Longest Processing Time (LPT) algorithm. Initially, LPT sorts jobs by processing time and each job is assigned to the machine with the lowest load. In the second stage all statistics on k-mers are computed and collected.
Nystrom et al. [28] propose Discount, a further distributed k-mer counting tool on Spark, which introduces the universal frequency ordering. This ordering is a new combination of frequency-sampled minimizers and universal k-mer hitting sets. The universal frequency ordering yields both evenly distributed binning and small bin sizes. Experimental results show that Discount is the most efficient k-mer counting tool using about 1/8 of the memory with respect to comparable approaches.
3.1.5. Analysis of genomic datasets
The challenge in analysing genomic datasets goes along two major dimensions: algorithmic time complexity and data size, meaning that efficient algorithms and scalable computing platforms are required to analyse large-scale genomic data. As major tool for contrasting these issues, Spark has been used to implement various tools, aimed at performing analytical tasks, genomics visualization, fast querying and processing of genomic intervals, and customizable ML analyses. These tools are briefly reported hereinafter.
Hung et al. (2018) [29] propose Sparkhit, a computational platform built on top of Spark and Hadoop for analysing large-scale genomic datasets on cluster and cloud platforms. Sparkhit integrates a series of analytical tools that perform a variety of genomics applications. In particular, it implements specific modules for metagenomic fragment recruitment (i.e., Sparkhit-recruiter) and short-read mapping (i.e., Sparkhit-mapper). Sparkhit also provides a wrapper tool (i.e., Sparkhit-piper) aimed at invoking and parallelizing third-party genomic analysis tools. It also extends the Spark MLlib for downstream data mining.
Nanni et al. [30] propose PyGMQL, a tool for manipulating region-based genomic files and their relative metadata. Built on top of the GMQL genomic big data management system [58], PyGMQL provides a set of functions for handling region data and their metadata, which can be scaled out to arbitrary clusters and implicitly apply to thousands of files, producing millions of regions.
Morrow et al. [31] propose Mango, a genomics visualization platform consisting of a genome browser and a Jupyter notebook that leverages the scalability of Spark by enabling interactive analysis over terabytes of sequencing data. Mango allows to manipulate and visualize datasets in a Jupyter notebook by providing access to genome track Jupyter widgets and summary visualizations. Using notebooks, it is also feasible to access any Spark SQL functions for filtering and querying genomic data. The genome browser provides traditional track views for variants and other features, while allowing users to remotely access datasets.
The SeQuiLa Spark package [43] has been devised and implemented to speed up analyses requiring intersection of genomic intervals. SeQuiLa represents genomic intervals as Spark/SQL tables, to facilitate the combined use of SQL interfaces and big data techniques while handling large-scale interval queries. The authors also implemented customized execution strategies to extend the SparkSQL Catalyst optimizer. A comparison with featureCounts [59] and the modified Spark SQL engine GenAp [60] pointed out that SeQuiLa was the fastest tool, achieving significant performance gain in genomic interval queries on single node and on cluster.
Dirmeier et al. [61] propose PyBDA, a Python-based ML command line tool for automated analysis of big biological data sets. PyBDA provides customizable ML pipelines to be run on high-performance clusters. It provides methods for dimension reduction, clustering, and supervised learning. PyDBA is built on Spark to automatically distribute data over the nodes of a cluster, with the goal of efficiently parallelizing expensive ML tasks. Snakemake is used to automatically schedule jobs to a computing cluster.
3.1.6. DNA error correction
Authors in [41] propose a DNA error correction tool built on Spark. The proposed tool is based on the multiple-sequence alignment tool CloudEC [62] built on Apache Hadoop. CloudEC performs very well in terms of accuracy whereas its performance in terms of computing time has significant room for improvement. SparkEC was specifically designed to improve the approach implemented in CloudEC in terms of computing time and usability. In particular, SparkEC exploited the efficient in memory data structures of Spark and a novel split-based processing strategy to improve the computing time with respect to CloudEC. Moreover, it was designed to avoid the heavy pre-processing steps required by CloudEC. In fact, CloudEC requires that the user convert the input datasets into an internal format without any parallel approach. The SparkEC implementation uses a specific library (i.e., Hadoop Sequence Parser) to avoid the pre-processing step. Experimental results carried-out on both short- and long-read datasets shown that SparkEC resulted up to 12x faster than CloudEC.
3.1.7. Genome compression
The continuous reduction of sequencing costs is at the basis of the enormous and constant growth of biological data. While this growth in available data offers new opportunities in the fields of the research and of the precision medicine, it also raises new challenges for implementing tools able to manage this massive amount of data efficiently and effectively. In this context the availability of suitable genome compression tools is required for efficient data storing and sharing. To deal with this challenge, authors in [42] propose SparkGC a Spark based genome compression for large collections of genomes. SparkGC was designed to be efficient in terms of both compression ratio and computing time. Efficiency in terms of compression ratio was achieved by optimizing the encoding scheme for the mapping results whereas the in-memory computation capabilities were exploited to optimize the compression time.
Compared with the best cutting-edge methods, SparkGC achieved a compression ratio that resulted better than 30%. As for the compression time, SparkGC runs on a single node resulted at least 3.8x fast than the cutting-edge tools while it scales well with an increasing number of nodes.
3.2. Metagenomics
The decrease in costs of high-throughput sequencing technologies has made it possible to measure the genomic composition of mixtures of microbes in their environment. Metagenomics studies enabled by these technologies made possible to investigate the microbial roles in the health. Contextually, these studies also raised computational challenges mainly related to the large volumes of generated data and to the analytical needs. In the following three specialized tools for metagenomics analysis are reported.
Valdes et al. (2019) [32] propose Flint, a pipeline built on top of Spark for fast real-time profiling of metagenomic samples against a large collection of reference genomes. In Flint, reference genomes are partitioned and each partition is assigned to a worker node on the Spark cluster. To avoid storage overhead the reads are streamed into the cluster. Then, Spark worker nodes (using Bowtie2 [63]) align reads to the distributed reference genomes according to a two-step MapReduce pipeline that repeatedly updates metagenomic profiles. Run on Amazon's Elastic MapReduce service of 64 machines, Flint was able to profile 1 million paired-end reads in 67 seconds –i.e., an order of magnitude faster than other state of the art algorithms.
Walker et al. (2018) [33] propose GATK PathSeq, an updated version of their computational pipeline PathSeq [64] for the discovery and identification of microbial sequences in genomic and transcriptomic libraries from eukaryotic hosts. This updated pipeline has been improved using faster computational approaches. Implemented using the GATK engine and Spark, it enables parallel data processing on local workstation, computing clusters and Google Cloud services. Specifically, Spark improves the performance of PathSeq in the steps aimed at removing low quality, low complexity, host-derived and duplicate reads. To this end, it is used to process batches of sequencing reads asynchronously in memory, thus maximizing resource utilization and minimizing hard-disk operations between pipeline stages.
Wang et al. (2018) [16] developed MetaGO, a new method to compare metagenomic samples. For different groups of microbial communities, information for potential biomarker discovery can be obtained by identifying group-specific sequences. The developed method uses long k-mers (≥30 bp) to identify group-specific sequences between two groups of metagenomic samples. MetaGO consists of three modules aimed at: i) creating a feature vector for each sample (i.e., the number of occurrences for each k-mer through all reads in one sample); ii) pre-processing the features; and finally iii) identifying group-specific features. In the second step, the feature vectors created at the previous step are integrated as a matrix. The time and memory required to perform this step increase drastically with the length of the k-mers and with the sample size. To deal with this computational issue, the pipeline has been parallelized using Spark. The analysis of each feature vector is partitioned on multiple workers using RDDs. MetaGO can be run on a local multi-core server or on a distributed cluster of machines that support HDFS.
3.3. Epigenomics
Epigenomics refers to the study of heritable phenotype changes that do not involve genotype alterations. DNA methylation is a common type of epigenetic change. Bisulfite treatment of DNA is acknowledged as the gold standard technique to study methylation. To detect the methylation levels, reads treated with the bisulfite must be aligned against a reference genome. Mapping these reads to a reference genome represents a significant computational challenge, mainly due to the increased search space and to the loss of information introduced by the treatment.
A highly scalable bisulfite aligner implemented on Spark (called BiSpark), devised to deal with the mapping of reads treated with bisulfite is proposed in [39]. Without going into unnecessary details, a common strategy to map bisulfite treated reads is based on a 3-letter nucleotide alphabet reduction algorithm. Some considerations are needed to apply this strategy in distributed environments. Two converted reads are generated from a single read producing four different alignment results. To find the best alignment, results should be moved among nodes. This data transfer is one of the most time-consuming parts of the distributed approach. To deal with this issue, BiSpark applies a specific feature function of Spark (i.e., context-level union) to ensure that all mappings of a read are merged in the same RDD. Moreover, the algorithm can give rise to an unbalanced data distribution, which typically contrasts the benefits of scalability. To deal this issue, BiSpark applies hash partitioning to get the data balanced over the computational nodes.
3.4. Biomedical imaging
Biomedical imaging refers to several different technologies that are used to acquire and analyse images of the human body in order to diagnose and/or monitor a disease. Analysis of these images may be time consuming and often a correct diagnosis is related to the experience of the physician. DL based approaches are implemented to automatically analyse and provide a diagnosis. DL techniques applied to this task require GPU devices and the training process may be very long –due to the data size and to the adopted algorithm. The possibility to distribute the training process on multiple GPUs helps to reduce this time.
An interesting work to scale DL on multiple GPUs using Spark is presented in 2019 by Makkie et al. [35]. The authors point to the main issues that occur in neuroimaging analysis using task-based functional magnetic resonance imaging (tfMRI). In fact, despite the wide use of this technology in neuroimaging, obtaining meaningful patterns from the complex structure of tfMRI is a challenging task that requires the adoption of proper technologies, like convolutional neural networks. The authors addressed the need of processing these very large datasets by implementing a distributed DL framework that allows to use TensorFlow on Spark. In so doing, the computation has been easily distributed on a cluster of GPUs. Experiments have been performed on data provided by the Human Connectome Project, and the corresponding results showed that the proposed system can be used on big data tfMRI modelling and analytics thanks to its efficiency and scalability. Spark was used on top of Hadoop to take advantage of the HDFS file system.
In 2020, Stritt et al. [36] proposed an application, called Orbit, for imaging analysis. The application domain is the analysis of whole slide images (WSIs). As this kind of images are often too large to be analysed with standard tools, Spark has been adopted to provide full-featured functionalities. In particular, Orbit is used i) to split images into smaller parts (to be further analysed with embedded ML algorithms), ii) to integrate third-party analysis tools based on DL, and iii) to support the analysis of huge amount of images on distributed computing environments. Orbit has been designed to be executed on both a multithreaded local computer as well as a multinode cluster. Summarizing, the system allows to apply existing algorithms designed to analyse smaller images towards very large images while it also allows the parallel processing of hundreds of WSI.
3.5. Biomedical signal processing
Automatic biomedical signals processing is an important task aimed at identifying indicators of diseased states. Artificial Intelligence is used to manage and analyse large biomedical datasets leading to the clinical decisions and real-time applications. In the following, two works aimed at detecting real-time cardiac arrhythmias and predicting systolic blood pressure are reported.
In 2021, Ilbeigipour et al. [65] proposed a system aimed at detecting real-time cardiac arrhythmias. To this end, they implemented an online pipeline for running pre-processing stages of ECG data and arrhythmia detection on top of Spark. The pipeline encompasses data pre-processing and classification steps. These steps were deployed using Spark libraries and the Spark structured stream processing engine5 to classify ECG streaming data and real-time cardiac arrhythmia detection. The classification model was created using a RF and was pre-loaded into the pipeline. ECG test data was stored in files, the data being entered within a time interval of 5 seconds. The pipeline was evaluated in terms of classification performance and rate of delay in arrhythmia detection. While classification metrics are comparable with other methods, the proposed approach significantly reduces the latency, thanks to the speed up enforced by Spark.
In 2021 Saleh et al. [37] proposed a real-time system aimed at predicting systolic blood pressure (SBP), tailored to prevent and avoid problems that can arise from sudden changes in blood pressure. The system consists of an offline model and an online prediction pipeline. The first component is aimed at investigating different DL models, to identify the one with achieve best performance. The second component evaluates the model in real-time. Spark and Apache Kafka were used to deal with real-time constraints for streaming data. Streams of SBP time-series data are generated and then sent to an Apache Kafka topic.6 Subsequently, Spark streaming reads the data from the Kafka topic and forwards it to the DL model to predict the near future of SBP in real-time.
3.6. Gene-disease association
Disease gene prediction represents one of the main computational challenges of precision medicine. In the following, tools aimed at scoring polygenic risk, analysing gene expression levels, and analysing gene interaction networks are reported.
In 2018, Chen et al. [40] proposed PRSoS a novel approach based on Spark for generating polygenic risk scores. PRSoS has been designed to be implemented as a standalone version or on a cluster. The performance of PRSoS in terms of computing time was compared with that of PRSice [66] in the task of generating polygenic risk scores for major depressive disorder with different settings. Results shown that PRSoS outperforms PRSice when polygenic risk scores are generated for a large number of SNPs.
A study for breast cancer prediction using big data was reported by Alghunaim and Al-Baity [17] in 2019. This research work had a twofold goal. On the one hand, it was aimed at assessing the impact of gene expression and/or DNA methylation data in the prediction task. On the other hand, the benefits of using Spark and MLlib in place of WEKA [67] have been assessed. As for the second aspect, the goal was to assess their behaviour when dealing with large sets of data. Experiments performed using three classifiers, namely Support Vector Machines (SVM), Decision Trees (DT), and Random Forests (RF), shown that Spark outperformed WEKA in classifying the different types of data with respect to different measures.
In 2019, Navas et al. [18] proposed VIGLA-M, a visual analysis tool designed to support physicians and clinical researchers in the task of analysing gene expression levels in melanoma patients. VIGLA-M can be used to discover changes in gene expression levels of single patients and to discover clusters of patients tied by a relevant clinical correlation. The tool was tested on clinical datasets relating to metastatic melanoma patients.
In 2021, Joodaki et al. [19] proposed RWRHN-FF a novel algorithm aimed at finding genes involved in relevant phenotypes. Results obtained using gene interaction networks are often not accurate due to the high number of false positives. In this study, four gene-gene and four disease-disease networks are analysed to set up a more reliable data network. Experimental results shown that the networks integrated using a type-II fuzzy voter scheme outperformed the other methods used in a comparative benchmark in terms of AUC and precision. To limit the amount of time required to train RWRHN-FF on large heterogeneous networks, the authors have resorted to a parallel implementation on Spark. To assess the benefits of the Spark based implementation, it has been compared with a sequential version of the algorithm. Experiments carried out on heterogeneous networks of different sizes have shown significant improvements.
3.7. Drug discovery
In drug discovery virtual screening (VS) refers to a computational technique aimed at identifying the molecules that have the highest likelihood to bind to a drug target.
In [34] Ahmed et al. propose an iterative virtual screening method based on Spark. The time for docking can be notably reduced inferring in advance high-scoring ligands and avoiding those with low-score. The authors implemented a method that iteratively docks a set of ligands to form a training set, then a ligand-based model is trained on this set, and finally the ligands are predicted to exclude those with a low-score. These steps are repeated on another set of ligands until a given efficiency level is reached. Spark and MLlib are used to parallelize both the docking and the modelling on HPC clusters as well as on cloud resources. Although this method is aimed at shortening the virtual screening execution time, it also opens to opportunities for large-scale studies which may involve multiple target receptors and multiple large molecule libraries.
4. Discussion
4.1.
The Spark technology is increasingly adopted in life sciences. The previous section reports several research works that took advantage of Spark for its scalability, as well as its capability of supporting ML tasks and real-time analyses of streaming data. Some of these works also report the usage of Spark with other Apache frameworks, including Hadoop, Kafka, and Arrow. Moreover, only three works ([30], [31], [43]) refer the used cluster manager, although choosing the right one may be fundamental to optimize the use of computing resources.
Moreover, apart in [61], no articles focusing on workflow management have been found in recent literature. These aspects are discussed hereinafter.
4.1.1. Choosing a cluster manager
Although Spark provides a default standalone cluster manager, as previously noted also third-party cluster managers can be used –such as Apache Mesos, Hadoop YARN and Kubernetes. These cluster managers implement different strategies, meaning that they can be more or less suitable depending on the application at hand.
The default Spark cluster manager represents the easiest solution for cluster management when working with small clusters. However, working with big clusters may require the adoption of more sophisticated tools, such as YARN and Mesos. Being general resource managers, YARN and Mesos launch Spark as an application. However, this aspect has the positive effect of avoiding issues related to daemon overhead, which is a welcome feature on large scale clusters. The main difference between YARN and Mesos is that the former implements a request-based approach, whereas the latter an offer-based approach. In particular, YARN knows the available resources on the cluster and allocates them to run the application, whereas Mesos offers the available resources, leaving to Spark the right to decide which ones should be used.
Allocation policies are very important to optimize the use of resources. Each Spark application runs by default an executor on each cluster node and multiple applications are served according to a First-In First-Out policy. However, it is also feasible to statically partition the resources. For instance, one can bind an application to a specific set of nodes or can control the used memory. YARN provides a finer control of the resources, allowing to set the number of executors on the cluster and to set the memory and cores for executors. As for Mesos, static partitioning allows to limit the cores for each executor and to control the executor memory. Mesos also allows a dynamic sharing of the CPU cores. In particular, when an application is not running any task, the corresponding cores may be used to run tasks of other applications.
Recently, Spark added stable support for Kubernetes, which is a system for automated deployment, scaling, and management of containerized applications. Running Spark on Kubernetes brings benefits that are generating a growing interest. In particular, using the container technology for Spark applications makes easy to run them everywhere. Moreover, containers allow to isolate applications from each other, thus facilitating the run of different workloads on the same cluster. It should be pointed out that the container technology is widely used in bioinformatics, for it makes easier to install and run applications while enabling the reproducibility of analyses. Just to give an idea, at the time of writing, Biocontainers [68] amount to 10.3k tools on the Docker containers repository. Kubernetes also supports resource sharing among jobs, as well as priority assignment to containers. Summarising, Kubernetes is expected to make Spark more reliable and cost effective. The cons of using Kubernetes are that it requires expertize that researchers and/or organizations may need to acquire. However, some enterprise solutions already exist, such as RedHat OpenShift, which are able to simplify the deployment of Kubernetes clusters while providing features that make it more suitable for application in healthcare support.
4.1.2. Workflow management
Different solutions have been deployed to support researchers in the tasks of building, managing and sharing workflows –with the goal of making analyses reproducible. For instance, Common Workflow Language (CWL), Workflow Description Language (WDL), Guix Workflow Language (GWL), Nextflow [69], and Snakemake [70] are widely adopted solutions to describe and manage workflows. Best practices use workflow systems able to support the container technology, so that analyses on distributed computing systems can be easily orchestrated. However, workflow-oriented processing has some limitations when working with big data. Generally, these systems use a decoupled shared storage system for data synchronization and for storing intermediate results. Obviously, when working with big data, massive access to such storage systems may heavily affect the performance. Therefore, it is essential to rely on frameworks such as Spark to efficiently manage this kind of workflows.
In Spark, efficient workflow management can be supported using Apache Airflow [71]. This open-source platform allows to easily schedule and monitor workflows by means of a suitable web application. Airflow also provides many plug-and-play operators, ready to execute tasks on different platforms –such as Google Cloud Platform and Amazon Web Services. A workflow is represented here as a Directed Acyclic Graph (DAG), which contains individual pieces of work called Tasks. The DAG is able to run the underlying application in accordance with all dependencies set for the application itself. In particular, it allows to specify the dependencies between Tasks and the order for executing them. Currently, Airflow does not support any of the workflow languages used in biomedical research. A future support of these languages by Airflow would help researchers to easily run workflows on Spark. The cited paper of Kotliar et al. [72], presented at CWL-Airflow 2019, goes in this direction –being a package that adds support for CWL to Airflow.
5. Conclusions
In this work, the Spark framework has been described. Then, a literature review of Spark-based applications in life sciences has been made, showing how Spark can be adopted to manage and analyse big data in different fields related to life sciences. Although most of the applications fall in the field of genomics, it has been shown that Spark can also be adopted to address computational challenges in metagenomics, epigenomics, biomedical imaging, biomedical signal processing, gene-disease association and drug-discovery.
The reported research literature put into evidence that Spark could be integrated with other technologies, with specific focus on the capability of efficiently supporting containers and on workflow management. As a final note, the integration of Spark with Kubernetes and Airflow is expected to rapidly increase the usage of Spark within a large community of researchers involved in life sciences.
Funding statement
This work was supported by Ministero dell'Università e della Ricerca through the PNRR-IR0000031 project to AM (Strengthening of the Biobanking and Biomolecular Resources Research Infrastructure) and the BBMRI.it (Italian national node of BBMRI-ERIC) a research infrastructure financed by the Italian Government to MG.
CRediT authorship contribution statement
All authors listed have significantly contributed to the development and the writing of this article.
Declaration of Competing Interest
The authors declare no competing interests.
Footnotes
Scalability is the property of a system to increase or decrease the resources used for computation in response to changes in processing demands.
The ordering used to list out programming languages supported by Spark is not random. In fact, Spark has been developed in Scala, which is an extension of Java able to provide full support for the functional programming style. Python and R follow, due to their wide diffusion. Recall also that Scala and Java are strongly typed, whereas Python and R are untyped.
A more detailed description of the Spark Ecosystem is reported in the Appendix.
The interested reader can find a more detailed comparison between Spark and Hadoop in the Appendix.
Apache structured streaming is an advance of the standard streaming module. The difference stands in the use of DataFrames instead of DStreams, as found in the original streaming model.
Kafka's topics are implemented as distributed transaction logs, a transaction log being an abstract data type to which new data areonly ever appended (i.e., never overwritten).
Recall that in a lazy computation model an operation is performed only when strictly required.
Appendix A. Spark data abstractions
In a distributed system, data abstraction consists of a logical structure of the data distributed across the nodes of a cluster. Spark supports three core abstractions, namely RDDs, DataFrames, and Datasets. Table A.2 shows an overview of these data structures, whose characteristics are briefly summarized hereinafter.
Table A.2.
Main features of RDD, DataFrame and Dataset. Recall that the native language support is given by Scala and Java. Additional language support is reported in the table.
| RDD | DataFrame | Dataset | |
|---|---|---|---|
| Immutable | ✓ | ✓ | ✓ |
| Fault-tolerant | ✓ | ✓ | ✓ |
| Type-safe | ✓ | - | ✓ |
| Schema | - | ✓ | ✓ |
| Optimizer Engine | - | ✓ | ✓ |
| Additional languages | Python, R | Python, R | - |
An RDD (Resilient Distributed Dataset) [73] is an immutable and fault-tolerant distributed collection of data partitioned across a cluster, which can be operated in parallel. RDD is type safe and comes with low-level APIs that enable high control over the physical data structure. Although they are the primary data abstraction in Spark, RDDs do not integrate any optimization engine, meaning that the strategy aimed at optimising performance and memory management must be directly handled by programmers. RDD APIs are supported by Scala, Java, Python, and R.
A DataFrame is an immutable and distributed collection of data, defined on top of RDDs and organized into named columns. Conceptually, DataFrames are similar to the data frames of Python (through the Pandas library) and R. DataFrames are also strictly related to relational database tables, the main difference being that the former allow to define schemas on distributed collections of data –being distributed over the nodes of a cluster. Due to their strict relationship with RDDs, also DataFrames are fault-tolerant. Unlike RDDs, they come with a built-in optimization engine (i.e., Catalyst, based on Scala's functional programming constructs). DataFrames are not type safe and are supported by Scala, Java, Python, and R.
A Dataset is an immutable and distributed data structure, defined on top of DataFrame. In a way, Datasets combine the benefits of RDDs and DataFrames. In fact, they are type-safe and accept relational queries. Like DataFrames, also Datasets make use of the Catalyst engine for optimization. Fault-tolerance is extended also to Datasets, for they are defined on top of DataFrames. Being a collection of strongly-typed JVM objects, this data abstraction is only supported by Scala and Java.
Appendix B. Spark ecosystem
The Spark Ecosystem consists of the Spark Core and specific modules aimed at providing support to SQL (i.e., Spark/SQL), process live data streams (i.e., Streaming), machine learning (i.e., MLlib), graphs and graph-parallel computations (i.e., GraphX) and R package (i.e., Spark/R). A description of these components follows hereinafter.
Spark/SQL is a distributed SQL query engine, yielding the capability of performing SQL queries inside Spark programs. It can be accessed in several ways, including the standard SQL interface and the Dataset API, depending on the user needs and implementation choices. It can also run ETL functions on data expressed in various formats, such as Parquet, ORC, JSON, and JDBC. Sparl/SQL can also be used to read data from an existing Hive installation as well.
Streaming enables scalable, high-throughput, and fault-tolerant processing of live data streams. Beyond TCP sockets for handling incoming data, Streaming can also be used in synergy with various distributed log technologies, including Apache Kafka, Amazon Kinesis, Microsoft Azure Event Hubs and Google Pub/Sub. Data handled by Streaming are typically processed using standard or ad-hoc algorithms. However, MLlib and GraphX may also be involved, typically framed within data pathways defined to support real-time analysis. Results obtained from streamed data can be stored to filesystems, databases, and/or live dashboards.
MLlib is a ML library, aimed at providing off-the-shelf tools for ML and DM tasks on scalable and complex systems. Fully integrated with the data abstractions provided by Spark, this library supports most of the ML algorithms and techniques. Further support is given to feature handling (in particular to feature extraction and feature selection), as well as to the construction, evaluation, and tuning of ML pipelines. Data and algorithm persistence is also addressed thanks to facilities implemented for saving and loading algorithms, models, and pipelines. Further relevant utilities, such as linear algebra, statistics, and various data handling tools, are also available off-the-shelf.
GraphX[74] is a graph processing framework built on top of Spark. Designed to support graphs and graph-parallel computations, GraphX is a generalisation of MapReduce based on the Graph abstraction, which consists of a directed multigraph with user-defined objects attached to each vertex and edge. Some fundamental operators, like subgraph, joinVertices, and aggregateMessages, are provided to support Graph-based computations. With the goal of simplify graph analytics tasks, a growing collection of graph algorithms and builders (including an optimized variant of the Pregel API) is also provided.
Spark/R is a new entry in the Spark Ecosystem, devised as R package to provide a distributed data frame implementation and to support relevant operations like selection, filtering, and aggregation on large data-sets. The same functionalities are also available on Python through the pySpark interface.
Appendix C. Spark vs Hadoop
With the goal of keeping the comparison simple, Spark and Hadoop are compared according to few major features, i.e., performance, costs, fault-tolerance, scalability, data processing and support for ML. A summary of the comparison is given in Table C.3.
Table C.3.
Spark vs Hadoop: comparison in terms of performance, infrastructural costs, fault-tolerance, scalability, data processing and support for ML.
| Spark | Hadoop | |
|---|---|---|
| Performance | 3 ÷ 100 times faster | Fast |
| Infrastructural Costs | More expensive | Cheaper |
| Fault-tolerance | Yes | Yes |
| Scalability | Yes | Yes |
| Data processing | Real time processing | Batch processing |
| Support for ML | MLlib | Mahout |
Performance – In terms of computation efficiency, Spark outperforms Hadoop, being typically up to 3x faster for large workloads and up to 100x faster for small ones. This ability depends on the core strategies adopted by Hadoop and Spark for processing data. Hadoop's calculation engine is MapReduce, whose typical behaviour can be summarised as follows. First, input data are split into chunks, distributed across the cluster nodes and processed by map tasks. Afterwards, the output generated by the map tasks is shuffled and sorted, and finally, the reduce tasks operate to provide the result of the overall computation. Hadoop MapReduce tasks are disk-oriented, meaning that each operation takes its input from disk and writes the result back to disk. On the one hand, this strategy facilitates the handling of failures, as –when needed– the computation can be easily resumed from the intermediate results stored on disk. On the other hand, these frequent disk accesses negatively affect the performance in time. Unlike Hadoop, by default Spark processes input data and intermediate results in RAM. The difference between the extensive use of disk performed by Hadoop and the in-memory strategy adopted by Spark generates the speedup (i.e., up to 100x) observed on average between Spark and Hadoop.
Costs – Although both frameworks are open source, their implementations require different hardware constraints. On the one hand, Hadoop requires high disk storage, whose cost is low and ever decreasing. On the other hand, the in-memory strategy used by Spark requires large amounts of RAM memory, which is more expensive than storage. However, it is also true that the time required for computation is lower –thus positively affecting the productivity and the savings in terms of power consumption. The same considerations can be made for cloud-based deployments, as although the hourly cost of in-memory servers is higher than that based on disk storage, the reduced computation time translates into overall cost savings.
Fault-tolerance – One of the main components of the Hadoop ecosystem is HDFS, which is a fault-tolerant distributed file system. Files are split into blocks and replicated many times across many nodes. This strategy ensures that a failure occurring at a specific node does not negatively affect the computation, as an HDFS file can be easily rebuilt by retrieving the missing blocks from other nodes. Note that, in case of crash, Hadoop resumes the execution from the point at which the failure has occurred (as all data and intermediate results are stored on disk). As for Spark, fault-tolerance is ensured by the RDDs, for they are immutable collection of objects logically partitioned on the nodes of a cluster. Hence, if a node crashes, its RDD partition will be assigned to another node which will operate the same logical execution plan. Note that, when the crash of a node occurs, the processing must be restarted from the beginning (on another node), as Spark data is typically located in RAM memory.
Scalability – There are two types of scalability in Hadoop: vertical and horizontal (the former is also referred as “scale-up”, whereas the latter as “scale-out”). Vertical scaling addresses the scalability problem by increasing the hardware capacity of individual machines (e.g., adding RAM). As for horizontal scaling, it is typically performed by adding more machines to the cluster. As new nodes can be added according to a “plug-and-play” policy, there is no downtime or green zone while scaling out. In Hadoop, HDFS enables high scalability, for it has been designed under the specific constraint of supporting scaling out. In particular, while Hadoop is running, one can easily add nodes to accommodate larger workloads. As for Spark, it does not have its own distributed file system; however, large amounts of data can be dealt with by using HDFS.
Data processing – Hadoop is more suitable for batch processing, whereas Spark for in-memory computations. Hence, real-time processing is supported only by Spark. Note that Spark computations are lazy7 on transformations (e.g., map, filter, groupByKey) and eager on actions (e.g., reduce, collect, take). This policy is adopted for Spark RDDs and Datasets. In so doing, Spark optimizes the usage of the network bandwidth.
Support for ML – Both frameworks provide support for ML, with Spark resulting faster than Hadoop. Hadoop provides support for ML with Apache Mahout [75], whereas Spark provides MLlib [76] which performs iterative in-memory ML computations. The difference in performance (in favour of Spark) depends on the fact that Hadoop splits data into chunks that may be too large for ML algorithms.
Data availability
No data was used for the research described in the article.
References
- 1.https://www.embl.org/files/wp-content/uploads/EMBL-EBI_Annual-Report_2020.pdf Embl-ebi annual report 2020. URL.
- 2.Unravelling the Human Genome–Phenome Relationship Using Phenome-Wide Association StudiesNat. Rev. Genet. 2016;17:129–145. doi: 10.1038/nrg.2015.36. [DOI] [PubMed] [Google Scholar]
- 3.Atasoy H., Greenwood B.N., McCullough J.S. 2019. The digitization of patient care: a review of the effects of electronic health records on health care quality and utilization. Tech. Rep. [DOI] [PubMed] [Google Scholar]
- 4.Zhou L., Pan S., Wang J., Vasilakos A. Machine learning on big data: opportunities and challenges. Neurocomputing. 2017;237:350–361. doi: 10.1016/j.neucom.2017.01.026. [DOI] [Google Scholar]
- 5.Parliament, Eurpean Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation) Off. J. Eur. Union. 2016;119(1) [Google Scholar]
- 6.Mondschein C., Monda C. Fundamentals of Clinical Data Science. 2019. The EU's general data protection regulation (GDPR) in a research context; pp. 55–71. [PubMed] [Google Scholar]
- 7.https://www.ga4gh.org Global alliance for genomics and health, URL. [DOI] [PubMed]
- 8.Litton J.-E. Launch of an infrastructure for health research: BBMRI-ERIC. Biopreserv. Biobank. 2018;16(3):233–241. doi: 10.1089/bio.2018.0027. [DOI] [PubMed] [Google Scholar]
- 9.Durinx C., McEntyre J., Appel R., Apweiler R., Barlow M., Blomberg N., Cook C., Gasteiger E., Kim J.-H., Lopez R., Redaschi N., Stockinger H., Teixeira D., Valencia A. Identifying ELIXIR core data resources. F1000Res. 2017:5. doi: 10.12688/f1000research.9656.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaharia M., Chowdhury M., Franklin M., Shenker S., Stoica I. 2nd USENIX Workshop on Hot Topics in Cloud Computing. vol. 2010. 2010. Spark: cluster computing with working sets; p. 95. (HotCloud). [Google Scholar]
- 11.Guo R., Zhao Y., Zou Q., Fang X., Peng S. Bioinformatics applications on apache spark. GigaScience. 2018;7(8) doi: 10.1093/gigascience/giy098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apache hadoop. https://hadoop.apache.org
- 13.Vavilapalli V., Murthy A., Douglas C., Agarwal S., Konar M., Evans R., Graves T., Lowe J., Shah H., Seth S., Saha B., Curino C., O'Malley O., Radia S., Reed B., Baldeschwieler E. Apache hadoop YARN: yet another resource negotiator. Proceedings of the 4th Annual Symposium on Cloud Computing; SoCC 2013; 2013. pp. 1–16. [DOI] [Google Scholar]
- 14.Hindman B., Konwinski A., Zaharia M., Ghodsi A., Joseph A., Katz R., Shenker S., Stoica I. Proceedings of NSDI 2011: 8th USENIX Symposium on Networked Systems Design and Implementation. USENIX Association; Boston, MA: 2011. Mesos: a platform for fine-grained resource sharing in the data center; pp. 295–308.https://www.usenix.org/conference/nsdi11/mesos-platform-fine-grained-resource-sharing-data-center [Google Scholar]
- 15.Kubernetes. https://kubernetes.io
- 16.Wang Y., Fu L., Ren J., Yu Z., Chen T., Sun F. Identifying group-specific sequences for microbial communities using long k-mer sequence signatures. Front. Microbiol. 2018;9:872. doi: 10.3389/fmicb.2018.00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alghunaim S., Al-Baity H. On the scalability of machine-learning algorithms for breast cancer prediction in big data context. IEEE Access. 2019;7:91535–91546. doi: 10.1109/ACCESS.2019.2927080. [DOI] [Google Scholar]
- 18.Navas-Delgado I., García-Nieto J., López-Camacho E., Rybinski M., Lavado R., Guerrero M., Aldana-Montes J. VIGLA-M: visual gene expression data analytics. BMC Bioinform. 2019;20(4):1–11. doi: 10.1186/s12859-019-2695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joodaki M., Ghadiri N., Maleki Z., Lotfi Shahreza M. A scalable random walk with restart on heterogeneous networks with apache spark for ranking disease-related genes through type-ii fuzzy data fusion. J. Biomed. Inform. 2021;115 doi: 10.1016/j.jbi.2021.103688. [DOI] [PubMed] [Google Scholar]
- 20.Paul A., Lawrence D., Song M., Lim S.-H., Pan C., Ahn T.-H. Using apache spark on genome assembly for scalable overlap-graph reduction. Hum. Genomics. 2019;13(1):48. doi: 10.1186/s40246-019-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linderman M., Chia D., Wallace F., Nothaft F. DECA: scalable XHMM exome copy-number variant calling with ADAM and Apache Spark. BMC Bioinform. 2019;20(1):1–8. doi: 10.1186/s12859-019-3108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mushtaq H., Ahmed N., Al-Ars Z. SparkGA2: production-quality memory-efficient Apache Spark based genome analysis framework. PLoS ONE. 2019;14(12) doi: 10.1371/journal.pone.0224784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang P.-J., Chang J.-H., Lin H.-H., Li Y.-X., Lee C.-C., Su C.-T., Li Y.-L., Chang M.-T., Weng S., Cheng W.-H., Chiu C.-H., Tang P. DeepVariant-on-Spark: small-scale genome analysis using a cloud-based computing framework. Comput. Math. Methods Med. 2020;2020 doi: 10.1155/2020/7231205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad T., Al Ars Z., Hofstee H. Vc@Scale: scalable and high-performance variant calling on cluster environments. GigaScience. 2021;10(9) doi: 10.1093/gigascience/giab057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decap D., De Schaetzen Van Brienen L., Larmuseau M., Costanza P., Herzeel C., Wuyts R., Marchal K., Fostier J. Halvade somatic: somatic variant calling with apache spark. GigaScience. 2022;11 doi: 10.1093/gigascience/giab094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maarala A., Arasalo O., Valenzuela D., Makinen V., Heljanko K. Distributed hybrid-indexing of compressed pan-genomes for scalable and fast sequence alignment. PLoS ONE. 2021;16(8) doi: 10.1371/journal.pone.0255260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferraro Petrillo U., Sorella M., Cattaneo G., Giancarlo R., Rombo S. Analyzing big datasets of genomic sequences: fast and scalable collection of k-mer statistics. BMC Bioinform. 2019;20(4):1–14. doi: 10.1186/s12859-019-2694-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nyström-Persson J., Keeble-Gagnère G., Zawad N. Compact and evenly distributed k-mer binning for genomic sequences. Bioinformatics. 2021;37(17):2563–2569. doi: 10.1093/bioinformatics/btab156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang L., Krüger J., Sczyrba A. Analyzing large scale genomic data on the cloud with sparkhit. Bioinformatics. 2018;34(9):1457–1465. doi: 10.1093/bioinformatics/btx808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nanni L., Pinoli P., Canakoglu A., Ceri S. PyGMQL: scalable data extraction and analysis for heterogeneous genomic datasets. BMC Bioinform. 2019;20(1):1–11. doi: 10.1186/s12859-019-3159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrow A., He G., Nothaft F., Tu E., Paschall J., Yosef N., Joseph A. Mango: exploratory data analysis for large-scale sequencing datasets. Cell Syst. 2019;9(6):609–613. doi: 10.1016/j.cels.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Valdes C., Stebliankin V., Narasimhan G. Large scale microbiome profiling in the cloud. Bioinformatics. 2019;35(14) doi: 10.1093/bioinformatics/btz356. https://doi.org/10.1093/bioinformatics/btz356 i13–i22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker M., Pedamallu C., Ojesina A., Bullman S., Sharpe T., Whelan C., Meyerson M. GATK PathSeq: a customizable computational tool for the discovery and identification of microbial sequences in libraries from eukaryotic hosts. Bioinformatics. 2018;34(24):4287–4289. doi: 10.1093/bioinformatics/bty501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed L., Georgiev V., Capuccini M., Toor S., Schaal W., Laure E., Spjuth O. Efficient iterative virtual screening with apache spark and conformal prediction. J. Cheminform. 2018;10(1):1–8. doi: 10.1186/s13321-018-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makkie M., Huang H., Zhao Y., Vasilakos A., Liu T. Fast and scalable distributed deep convolutional autoencoder for fMRI big data analytics. Neurocomputing. 2019;325:20–30. doi: 10.1016/j.neucom.2018.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stritt M., Stalder A., Vezzali E. Orbit image analysis: an open-source whole slide image analysis tool. PLoS Comput. Biol. 2020;16(2) doi: 10.1371/journal.pcbi.1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saleh H., Younis E., Sahal R., Ali A. Predicting systolic blood pressure in real-time using streaming data and deep learning. Mob. Netw. Appl. 2021;26(1):326–335. doi: 10.1007/s11036-020-01645-w. [DOI] [Google Scholar]
- 38.Hadadian Nejad Yousefi M., Goudarzi M., Motahari S. IMOS: improved Meta-aligner and Minimap2 on Spark. BMC Bioinform. 2019;20(1):1–14. doi: 10.1186/s12859-018-2592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soe S., Park Y., Chae H. BiSpark: a Spark-based highly scalable aligner for bisulfite sequencing data. BMC Bioinform. 2018;19(1):1–9. doi: 10.1186/s12859-018-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L., Yao N., Garg E., Zhu Y., Nguyen T., Pokhvisneva I., Hari Dass S., Unternaehrer E., Gaudreau H., Forest M., McEwen L., MacIsaac J., Kobor M., Greenwood C., Silveira P., Meaney M., O'Donnell K. PRS-on-Spark (PRSoS): a novel, efficient and flexible approach for generating polygenic risk scores. BMC Bioinform. 2018;19(1):1–9. doi: 10.1186/s12859-018-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Expósito R., Martínez-Sánchez M., Touriño J. SparkEC: speeding up alignment-based DNA error correction tools. BMC Bioinform. 2022;23(1):1–17. doi: 10.1186/s12859-022-05013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao H., Hu G., Liu S., Fang H., Ji Y. SparkGC: Spark based genome compression for large collections of genomes. BMC Bioinform. 2022;23(1):1–21. doi: 10.1186/s12859-022-04825-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiewiórka M., Leśniewska A., Szmurło A., Stepień K., Borowiak M., Okoniewski M., Gambin T. SeQuila: an elastic, fast and scalable SQL-oriented solution for processing and querying genomic intervals. Bioinformatics. 2019;35(12):2156–2158. doi: 10.1093/bioinformatics/bty940. [DOI] [PubMed] [Google Scholar]
- 44.Fromer M., Moran J., Chambert K., Banks E., Bergen S., Ruderfer D., Handsaker R., McCarroll S., O'Donovan M., Owen M., Kirov G., Sullivan P., Hultman C., Sklar P., Purcell S. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012;91(4):597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Auwera G., Carneiro M., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., Banks E., Garimella K., Altshuler D., Gabriel S., DePristo M. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013;43(1) doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poplin R., Chang P.-C., Alexander D., Schwartz S., Colthurst T., Ku A., Newburger D., Dijamco J., Nguyen N., Afshar P., Gross S., Dorfman L., McLean C., Depristo M. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018;36(10):983–987. doi: 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 47.Nothaft F., Massie M., Danford T., Zhang Z., Laserson U., Yeksigian C., Kottalam J., Ahuja A., Hammerbacher J., Linderman M., Franklin M., Joseph A., Patterson D. Proceedings of the ACM SIGMOD International Conference on Management of Data. 2015. Rethinking data-intensive science using scalable analytics systems; pp. 631–646. [DOI] [Google Scholar]
- 48.Xiao A., Wu Z., Dong S. ADS-HCSpark: a scalable HaplotypeCaller leveraging adaptive data segmentation to accelerate variant calling on Spark. BMC Bioinform. 2019;20(1):1–13. doi: 10.1186/s12859-019-2665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Ars Z., Wang S., Mushtaq H. SparkRA: enabling big data scalability for the GATK RNA-seq pipeline with Apache Spark. Genes. 2020;11(1):53. doi: 10.3390/genes11010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.https://apache.arrow.org Apache arrow. URL.
- 51.Ferrada H., Gagie T., Hirvola T., Puglisi S. Hybrid indexes for repetitive datasets. Philos. Trans. - Royal Soc. A, Math. Phys. Eng. Sci. 2014;372(2016) doi: 10.1098/rsta.2013.0137. [DOI] [PubMed] [Google Scholar]
- 52.Hoobin C., Puglisi S., Zobel J. Relative Lempel-Ziv factorization for efficient storage and retrieval of web collections. Proc. VLDB Endow. 2011;5:265–273. doi: 10.14778/2078331.2078341. [DOI] [Google Scholar]
- 53.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B., Trapnell C., Pop M., Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):1–10. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nashta-ali D., Aliyari A., Moghadam A., Edrisi M., Motahari S., Khalaj B. Meta-aligner: long-read alignment based on genome statistics. BMC Bioinform. 2017;18(1):1–9. doi: 10.1186/s12859-017-1518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vineetha V., Biji C., Nair A. SPARK-MSNA: efficient algorithm on Apache Spark for aligning multiple similar DNA/RNA sequences with supervised learning. Sci. Rep. 2019;9(1):1–11. doi: 10.1038/s41598-019-42966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferraro Petrillo U., Roscigno G., Cattaneo G., Giancarlo R. Informational and linguistic analysis of large genomic sequence collections via efficient hadoop cluster algorithms. Bioinformatics. 2018;34(11):1826–1833. doi: 10.1093/bioinformatics/bty018. [DOI] [PubMed] [Google Scholar]
- 58.Masseroli M., Pinoli P., Venco F., Kaitoua A., Jalili V., Palluzzi F., Muller H., Ceri S. Genometric query language: a novel approach to large-scale genomic data management. Bioinformatics. 2015;31(12):1881–1888. doi: 10.1093/bioinformatics/btv048. [DOI] [PubMed] [Google Scholar]
- 59.Liao Y., Smyth G., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 60.Kozanitis C., Patterson D. GenAp: a distributed SQL interface for genomic data. BMC Bioinform. 2016;17(1):1–8. doi: 10.1186/s12859-016-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dirmeier S., Emmenlauer M., Dehio C., Beerenwinkel N. PyBDa: a command line tool for automated analysis of big biological data sets. BMC Bioinform. 2019;20(1):1–6. doi: 10.1186/s12859-019-3087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung W.-C., Ho J.-M., Lin C.-Y., Lee D. 2017 IEEE International Conference on Big Data (Big Data) IEEE; 2017. CloudEC: a mapreduce-based algorithm for correcting errors in next-generation sequencing big data; pp. 2836–2842. [Google Scholar]
- 63.Langmead B., Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kostic A., Ojesina A., Pedamallu C., Jung J., Verhaak R., Getz G., Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 2011;29(5):393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ilbeigipour S., Albadvi A., Noughabi E. Akhondzadeh. Real-time heart arrhythmia detection using apache spark structured streaming. J. Healthc. Eng. 2021;2021 doi: 10.1155/2021/6624829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Euesden J., Lewis C., O'Reilly P. PRSice: polygenic risk score software. Bioinformatics. 2015;31(9):1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Witten I.H., Frank E. 2nd edition. Morgan Kaufmann; San Francisco: 2005. Data Mining: Practical Machine Learning Tools and Techniques. [Google Scholar]
- 68.da Veiga Leprevost F., Grüning B., Alves Aflitos S., Röst H., Uszkoreit J., Barsnes H., Vaudel M., Moreno P., Gatto L., Weber J., Bai M., Jimenez R., Sachsenberg T., Pfeuffer J., Vera Alvarez R., Griss J., Nesvizhskii A., Perez-Riverol Y. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017;33(16):2580–2582. doi: 10.1093/bioinformatics/btx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Tommaso P., Chatzou M., Floden E., Barja P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35(4):316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 70.Köster J., Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28(19):2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 71.Apache airflow. https://airflow.apache.org
- 72.Kotliar M., Kartashov A., Barski A. CWL-airflow: a lightweight pipeline manager supporting common workflow language. GigaScience. 2019;8(7) doi: 10.1093/gigascience/giz084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaharia M., Chowdhury M., Das T., Dave A., Ma J., McCauley M., Franklin M., Shenker S., Stoica I. Proceedings of NSDI 2012: 9th USENIX Symposium on Networked Systems Design and Implementation. 2012. Resilient distributed datasets: a fault-tolerant abstraction for in-memory cluster computing; pp. 15–28. [Google Scholar]
- 74.Gonzalez J., Xin R., Dave A., Crankshaw D., Franklin M., Stoica I. GraphX: graph processing in a distributed dataflow framework. Proceedings of the 11th USENIX Symposium on Operating Systems Design and Implementation; OSDI 2014; 2014. pp. 599–613. [Google Scholar]
- 75.Apache mahout. https://mahout.apache.org
- 76.Meng X., Bradley J., Yavuz B., Sparks E., Venkataraman S., Liu D., Freeman J., Tsai D., Amde M., Owen S., Xin D., Xin R., Franklin M., Zadeh R., Zaharia M., Talwalkar A. MLlib: machine learning in Apache Spark. J. Mach. Learn. Res. 2016;17(1):1235–1241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.