

Summary
CTCF is a DNA-binding protein which plays critical roles in chromatin structure organization and transcriptional regulation; however, little is known about the functional determinants of different CTCF-binding sites (CBS). Using a conditional mouse model, we have identified one set of CBSs that are lost upon CTCF depletion (lost CBSs) and another set that persists (retained CBSs). Retained CBSs are more similar to the consensus CTCF-binding sequence and usually span tandem CTCF peaks. Lost CBSs are enriched at enhancers and promoters and associate with active chromatin marks and higher transcriptional activity. In contrast, retained CBSs are enriched at TAD and loop boundaries. Integration of ChIP-seq and RNA-seq data has revealed that retained CBSs are located at the boundaries between distinct chromatin states, acting as chromatin barriers. Our results provide evidence that transient, lost CBSs are involved in transcriptional regulation, whereas retained CBSs are critical for establishing higher-order chromatin architecture.
Subject areas: Molecular Structure, Molecular mechanism of gene regulation, Components of the immune system
Graphical abstract
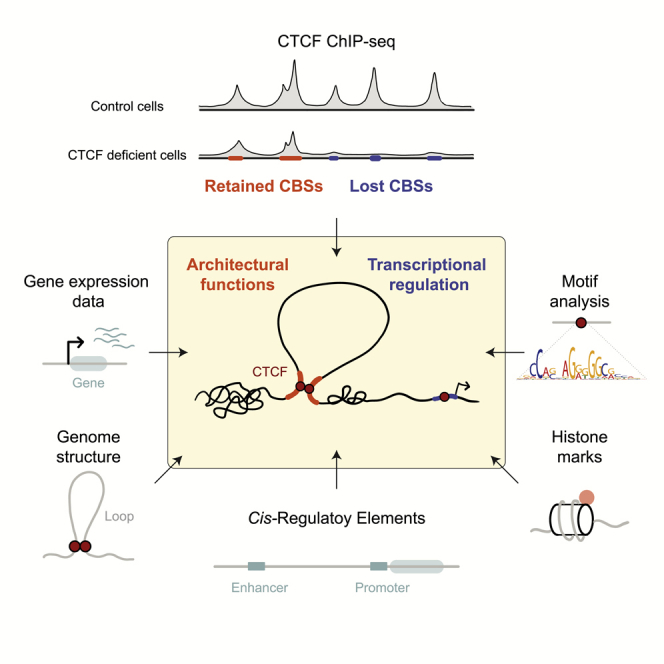
Highlights
-
•
We have defined sets of low-affinity and high-affinity CTCF-binding sites (CBSs)
-
•
CTCF-binding affinity effectively segregates distinct CTCF functions
-
•
Low-affinity CBSs are involved in transcriptional regulation through CREs
-
•
High-affinity CBSs are architectural and separate distinct chromatin states
Molecular Structure; Molecular mechanism of gene regulation; Components of the immune system.
Introduction
The 3D organization of chromatin is an essential factor in the regulation of all cellular and molecular processes, including the spatial and temporal regulation of gene expression.1,2 The mammalian genome is organized into multiple hierarchical layers that intimately integrate structure and function. These layers include chromosome territories, chromatin compartments A and B, topological associating domains (TADs), and chromatin loops.3 TADs are contiguous chromosomal regions ranging in size from 40 kb to 3 Mb (median size 185 kb) and characterized by high intradomain interaction.4 Functional genome features related to TAD organization include epigenetic profiles and transcriptional status,5 and TADs are considered gene regulatory domains in which gene expression is coordinated.6 Moreover, TAD boundaries can act as insulators that restrict enhancer action to promoters within the same TAD and thus prevent aberrant gene expression.7,8 Physical interaction between genes and cis-regulatory elements within the same TAD is achieved via intra-TAD loops.9,10
The zinc-finger DNA-binding protein CTCF plays an important role in chromatin organization at multiple levels.11 CTCF defines and stabilizes chromatin loops by binding to two CTCF-binding sites (CBSs) with convergent orientations coupled with cohesin-driven loop extrusion.12,13,14 CTCF is enriched at TAD boundaries,4 where it can establish chromatin barriers and block cross-domain interactions, thus acting as an insulator.15 Indeed, CTCF helps to prevent heterochromatin spreading at repressive chromatin domain boundaries.16,17,18 CTCF can also form intra-TAD loops,19 for instance through interaction with gene promoters or enhancers, and play more direct roles in transcriptional regulation.20,21,22,23 CTCF is thus critical for both architectural and direct gene regulation functions.
CTCF-depletion studies have revealed two classes of CBSs that presumably have differential CTCF affinities.19,21,22,24,25 The underlying cause of this differential affinity is poorly understood, but could be related to motif sequence24 or to the genomic location of the CBS, such as its distance from promoters.19,25 Higher affinity CBSs are enriched in TAD boundaries and long-range interactions, suggesting that high-affinity CBSs play an important role in constitutive chromatin architecture.9,22,25,26 However, no relationship has been established between CBS affinity and other specific CTCF functions, such as the promotion of enhancer interactions or the prevention of heterochromatin spreading. More comprehensive genome-wide analyses are thus needed to clarify the features of CBSs with different affinities and the CTCF functions associated with them, particularly for low-affinity CBSs, which lack an ascribed specific CTCF function.
Here, we explored the features that define CTCF affinity for CBSs and the functional consequences of this differential binding affinity. We characterize the predominant functions of high- and low-affinity CBSs, focusing on gene expression regulation via enhancer–promoter, TAD and intra-TAD loop formation, insulator function, and the establishment of boundaries between regions with different chromatin status. Our results define two functionally distinct sets of CBSs; one set of low-affinity CBSs that are predominantly involved in transcriptional regulation and a set of CBSs more resistant to CTCF depletion involved in loop formation and chromatin barriers.
Results
Differential CTCF binding distinguishes between high affinity, retained CBSs and low affinity, lost CBSs
To study the function of CTCF in the B cell genome, we used Ctcffl/fl; Cd19-Creki/+ mice to conditionally deplete CTCF from B cells27,28,29 (Figures S1A–S1C). Cd19-Cre-mediated depletion occurs gradually during bone marrow B cell differentiation and allows complete deletion in mature naive B cells.28,30 To assess the binding profile of CTCF in naive B cells, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) experiments in spleen B cells from Ctcffl/fl; Cd19-Creki/+ mice (CTCF-deficient, hereafter CTCFfl/fl) and from Ctcffl/+; Cd19-Creki/+ littermates (hereafter CTCFfl/+). CTCFfl/+ and CTCF+/+ B cells show no significant differences in activation or survival,31 and the CTCFfl/+ littermates were therefore used as controls. In this model, CTCFfl/fl naive B cells retain residual CTCF protein (Figure S1C).29
To identify CBS in CTCFfl/fl and CTCFfl/+ B cells, we performed peak calling on ChIP-seq data using MACS2. Principal component analysis showed that B cells of the same genotype clustered at PC1, which accounts for 95% of the variance, thus allowing separation of replicates according to genotype (Figure 1A). CTCF binding was markedly reduced but still detectable in CTCFfl/fl B cells (Figure S1D). Differential binding analysis revealed that 67.2% (29,172/43,388) of CBSs were lost in CTCFfl/fl B cells (lost CBSs), whereas 32.7% (14,213/43,388) of CBSs persisted (retained CBSs), and only 3 CBSs were gained in CTCFfl/fl B cells versus CTCFfl/+ cells (Figure 1B). Although retained CBSs generally had a higher binding signal than lost CBSs (Figure 1C), the classification into retained and lost CBSs does not reflect the signal ratio between CTCFfl/+ and CTCFfl/fl cells (Figure 1D), thus indicating that lost and retained CBSs are genuinely distinct CBS categories. In addition, we compared our CBSs groups with those previously described using an auxin-inducible degron (AID) system for CTCF degradation.25 We found our lost CBSs more enriched in the clusters with lower CTCF persistence (clusters 1 and 2) defined by the Blobel lab, while the highest overlap of our retained CBSs was with clusters with a higher CTCF persistence (5 and 6) (Figure S1E). This shows that our CBSs categories share similarities with those reported using other CTCF depletion models.
Figure 1.

Differential CTCF-binding affinity across the genome
(A) Principal component analysis (PCA) of CTCF ChIP-seq data. Each dot represents an individual mouse.
(B) Venn diagram showing the overlap between CTCFfl/+ and CTCFfl/fl binding sites. Numbers indicate the CBSs shared between CTCFfl/+ and CTCFfl/fl (retained CBSs, red shade), CTCFfl/+-specific (lost CBSs, blue shade) or CTCFfl/fl-specific (gained CBSs, gray shade).
(C) Profile plot (top) and heatmap (bottom) showing the average CTCF signal distribution at lost (left) and retained (right) CBSs from CTCFfl/fl and CTCFfl/+ cells. Regions spanning 1 kb upstream and downstream of the center of the CBS are shown.
(D) Volcano plot showing differential binding analysis (CTCFfl/+ vs CTCFfl/fl logFC and adjusted p value) of lost (blue) and retained (orange) CBSs.
Analysis of the size and distribution of retained and lost CBSs showed that retained CBSs were on average wider (Figure 2A) and very often harbored several summits (Figures 2B and 2C). In contrast, lost peaks were narrower and generally contained a single summit (Figures 2A–2C). Thus, retained CBSs more frequently contain tandem CTCF peaks, whereas lost CBSs are often composed of just one peak.
Figure 2.

CTCF lost and retained binding sites have distinct features
(A) Boxplot showing CBS size distribution of retained and lost CBSs. Statistical analysis was done with two-tailed unpaired Student’s t test (p value <2.2e-16).
(B) Frequency of the number of summits per lost CBS (blue) and retained CBS (orange) for each replicate. Data are represented as mean ± SD.
(C) Genome browser view of CTCF binding over a representative retained CBS and lost CBS for each of the CTCFfl/+ and CTCFfl/fl replicates.
(D) Percentage of CBSs containing a CTCF motif as defined by Homer. Statistical analysis was done with Fisher’s exact test (p value <0.0001).
(E) Boxplot showing CTCF motif scores in lost and retained CBSs, as defined by Homer. Statistical analysis was done with two-tailed unpaired Student’s t test (p value <2.2e-16).
(F) Overlap of lost and retained CBSs with those found in kidney, heart, lung, and liver CTCF ChIP-seq (ENCODE project datasets). Statistical analysis was done with Fisher’s exact test (p value <0.0001).
CTCF binds to a degenerate consensus DNA-binding motif composed of a ∼20-bp core,32 and CTCF-binding affinity can be estimated by similarity to this motif.33 HOMER motif analysis showed that 83.91% of retained CBSs contained CTCF motifs, whereas for lost CBSs this fraction was only 50.91% (Figure 2D). Moreover, retained CBSs contained more CTCF motifs and spanned a broader region than lost CBSs (Figures S2A and S2B). CTCF motifs in retained CBSs also had a higher consensus motif score than those in lost CBSs (Figure 2E), indicating a better fit with the CTCF consensus motif and supporting the idea that retained CBSs had higher CTCF affinity.
A secondary motif (U motif) located 5–6 bp upstream of the CTCF core motif (C motif) is believed to increase CTCF DNA-binding affinity34,35 (Figure S2C), indicating that the sequence context flanking the CTCF motif also plays a role in CTCF binding. A Spamo program analysis showed that the proportion of CBSs containing such U and C motifs was higher in retained than in lost CBSs (Figure S2D). This difference increased further with increasing consensus motif score (Figure S2E), indicating that retained CBSs had more consensus-like U and C motifs than lost CBSs, possibly contributing to their higher affinity.
An analysis of public databases showed that 94% of the retained CBSs identified in our study are widely conserved across different tissues, compared with only 42% of the lost CBSs (Figure 2F). Overall, these results indicate that lost CBSs tend to be of lower affinity and cell-type specific, whereas the retained CBSs have higher affinity for CTCF and are sites for constitutive CTCF binding.
Lost CTCF sites are involved in the direct regulation of gene expression
To determine whether lost and retained CBSs have distinct roles, we studied their distribution across different genomic regions. A positional enrichment plot showing the distribution of lost and retained CBSs at transcription start sites (TSS) revealed a genome-wide enrichment of lost CBSs at these loci (Figure 3A). Lost CBSs were also enriched near enhancer regions in splenic B cells, as defined in the enhancer Atlas 2.0 database (Figure 3B). Moreover, an analysis of ENCODE data on candidate cis regulatory regions in CH12 B cells showed that nearly 48% of the mapped lost CBSs overlapped with promoter or enhancer regions, compared with just 15% of retained CBSs (Figure 3C). Lost CBSs were also more enriched in the H3K4me3 and H3K27ac histone marks, which are associated with active promoters and enhancers, respectively (Figures 3D and S3A). Lost and retained CBSs also differed in the distribution of the H3K4me1 mark, which had a bimodal pattern in lost CBSs (Figures 3D and S3A) that could indicate active promoters.37
Figure 3.

Lost CBSs are associated with cis-regulatory elements
(A) Positional enrichment of lost or retained CBSs at TSS (data obtained from UCSC).
(B) Positional enrichment of lost or retained CBSs at enhancer regions from splenic B cells (data obtained from Enhancer Atlas 2.0).
(C) Percentage of lost or retained CBSs at different categories of cis-regulatory element from CH12 cell line (as defined in ENCODE encyclopedia).
(D) Histone ChIP-seq signal at lost or retained CBSs (±10 kb from the center of the binding site). Histone datasets were downloaded from GSE82144.36
(E) Boxplot showing gene expression (CPM) of genes with lost CBSs at their promoter region (-1 Kb to +1 Kb of TSS) and genes with retained CBSs at the promoter region. Statistical analysis was done with unpaired two-samples Wilcoxon test (p value <3.6e-10). Genes with CPM>150 are not shown in the plot to improve visualization, but they were considered for the statistical analysis.
To test whether the location of lost CBSs near promoters affected gene expression, we performed an RNA-seq experiment in CTCFfl/+ naive B cells. Genes harboring lost CBSs in the proximity of their promoter (-1 kb to +1 kb of the TSS) were more highly expressed than those harboring retained CBSs (Figure 3E). These results indicate that lost CBSs and retained CBSs are associated with distinct regulatory regions in chromatin and suggest that lost CBSs are preferentially involved in the direct regulation of gene expression through binding to promoter and enhancer regions.
Retained CTCF sites contribute to chromatin architecture and insulation
CTCF establishes TAD boundaries and intra-TAD loops in association with cohesin. To investigate whether lost and retained CBSs are differentially involved in loop formation, we analyzed the presence of the RAD21 cohesin subunit in CBSs, using public ChIP-seq data on RAD21 binding sites in naive B cells.38 This analysis showed greater cohesin deposition in retained CBSs than in lost CBSs (Figure 4A). We also conducted a positional enrichment analysis using previously reported loop boundaries detected in Hi-C and CTCF ChIA-PET experiments,36,38 finding a higher proportion of retained CBSs at loop boundaries (Figures 4B and S3B). Moreover, retained CBSs showed higher overlap with both TAD and intra-TAD loop anchors as defined by Matthews and Waxman9 (Figure S3C). These results support the idea that retained CBSs are principally involved in the genome architecture role of CTCF.
Figure 4.

Retained CBSs are associated with chromatin architecture
(A) RAD21 signal at lost or retained CBSs (±10 kb from the center of the binding site). Dataset were obtained from GSE98119.38
(B) Positional enrichment of lost or retained CBSs at CTCF-loop left and right boundaries, as defined by ChIA-PET (data were obtained from GSE98119).38
(C) Positional enrichment of lost or retained CBSs at strong (left) or weak (right) domain boundaries, as defined by Rao et al.4 (dataset downloaded from the 4dnucleome data portal, 4DNESK95HVFB).
(D) MNase-seq average signal at lost or retained CBSs (+-2 kb from the center of the binding site). Datasets were downloaded from GSE82144.36
We next studied the distribution of lost and retained CBSs at strong and weak TAD boundaries, as defined by Rao et al.4 At strong boundaries, retained CBSs were much more abundant than lost CBSs, whereas at weak boundaries both CBS types were present in similar amounts (Figure 4C). Nucleosome depletion near CTCF sites has been linked to CTCF binding strength and chromatin boundaries.39 Nucleosome density measured by MNase-seq36 was much lower in retained CBSs than in lost CBSs (Figure 4D), in line with our analysis thus far. These results suggest specific involvement of retained CBSs in establishing strong boundaries and preventing interaction between adjacent chromatin domains.
Retained CTCF sites act as barriers between distinct chromatin states
CTCF is enriched at the boundaries of H3K27me3 domains, which are associated with heterochromatin regions, suggesting a role for CTCF in maintaining the structure of repressed domains and preventing heterochromatin spreading.17,18,40 Moreover, CTCF promotes the removal of H3K27me3 marks, and the resulting H3K27me3-depleted state of CBSs can prevent the spread of histone modifications.41 To determine if this CTCF function is preferentially associated with retained CBSs, we plotted H3K27me3 signals at lost and retained CBSs (Figure 5A). We found high levels of H3K27me3 deposition at flanking regions of retained CBSs and a pronounced deletion of this mark at CBS centers, a distribution that was reversed in lost CBSs. A similar pattern of the H3K27me3 mark was observed in CBSs previously described by the Blobel lab using an AID system25 (Figure S3D). These results suggested preferential involvement of retained CBSs in delimiting H3K27me3 heterochromatin regions, most likely through the formation of loops (Figure 5B). To test this hypothesis, we analyzed the distribution of lost and retained CBSs and of H3K27me3 at chromatin loop boundaries. Making use of previously reported loop definition in naive B cells,36 we first performed a k-means clustering of loop boundaries based on H3K27me3 deposition. This analysis identified 4 types of boundary regions (Figure 5C): H3K27me3high/H3K27me3high (cluster 1), H3K27me3high/H3K27me3low (cluster 2), H3K27me3low/H3K27me3high (cluster 3), and H3K27me3low/H3K27me3low (cluster 4), where clusters 2 and 3 contain boundaries that separate active from inactive chromatin states. Overlap analysis showed that retained CBSs were more frequent than lost CBSs at boundaries, and this difference was especially marked at the boundaries within clusters 2 and 3 (Figure 5D). These results support the hypothesis that retained CBSs establish boundaries between regions with different chromatin states (Figure 5B).
Figure 5.

Retained CBSs insulate domains with different chromatin states
(A) H3K27me3 average signal at lost or retained CBSs (+-10 kb from the center of the binding site). Datasets were downloaded from GSE82144.36
(B) Model to propose a CTCF chromatin barrier function based on the distribution of H3K27me3 at retained CBSs. The cartoon at the top shows that CTCF can act as a chromatin barrier insulating euchromatin from H3K27me3 rich heterochromatin (1). We propose that CTCF at retained CBSs can establish insulator loops of repressed regions (left) or active regions (right) (2). The expected boundaries established by CTCF between active and repressed regions are depicted as blue boxes and those between repressed and active regions are depicted by pink boxes (3). Those transitions then would be expected to yield the H3K27me3 pattern shown in 4.
(C) Heatmap of H3K27me3 deposition observed in the loop boundaries defined by Hi-C. Datasets from GSE82144.36 Loop boundaries were clustered by k-means based on H3K27me3 distribution.
(D) Positional enrichment of CTCF lost or retained CBSs at loop boundaries segregated by clusters 2 and 3 (left) or clusters 1 and 4 (right).
To validate this idea, we first developed an algorithm to predict CTCF-mediated loops from our CTCF ChIP-seq data. Based on the known requirements for CTCF-mediated loop formation, we identified as potential loops regions <1 Mb that contained CBSs with convergent CTCF motifs at both anchors (Figure S4).42 The potential loops were then classified according to the types of CBS at their boundaries, as follows: (1) a potential loop flanked by two retained CBSs was considered retained, and (2) a potential loop flanked by two lost CBSs was considered lost. To determine if lost and retained loops correlated with gene activation or repression, we calculated the mean expression of the genes located within each loop and selected the 10% of loops with lower (repressed) or higher (active) mean expression (Figure S4). We next studied H3K27me3 deposition (Figures 6A and 6B) and gene expression (Figures 6C and 6D) in the two loop classes, identifying retained loops with repressed expression, retained loops with active expression, lost loops with repressed expression, and lost loops with active expression. Repressed loops had a stronger H3K27me3 signal than active loops, indicating that the classification of repressed and active loops according to gene expression effectively distinguishes inactive from active chromatin regions (Figures 6A and 6B). However, H3K27me3 signal was stronger in retained loops (both repressed and active) (Figure 6A), than in lost loops (Figure 6B), suggesting that chromatin activity is generally lower in retained than in lost loops. Moreover, boundary separation was much sharper in retained loops (Figure 6A, orange arrows) than in lost loops (Figure 6B, blue arrows). This is reinforced by the highly consistent transcriptional status of genes in retained loops (Figure 6C), whereas transcriptional activity in lost loops was relatively heterogeneous (Figure 6D).
Figure 6.

Retained CBSs preferentially establish loops with distinct epigenetic and transcriptional status
(A and B) Heatmap showing the H3K27me3 deposition at retained (A) and lost (B) loops and their 50 kb flanking regions. Orange arrows depict retained CBSs (A). Blue arrows depict lost CBSs (B). Loop length was scaled for representation. H3K27me3 signal is normalized to the input signal.
(C and D) Heatmap showing gene expression levels at the retained (C) or lost (D) loops and their 50 kb flanking regions. Loops were scaled as in A and B.
These results confirm our hypothesis that retained CBSs contribute to the segregation of chromatin domains by blocking the spread of repressive histone marks. In addition, our data indicate that retained CBSs establish loops with stable chromatin boundaries, whereas loops flanked by lost CBSs are more transient, suggesting that they are more permissive to dynamic transcriptional regulation.
Discussion
In this study, we provide a detailed description of the features and functions of two groups of CBSs defined by differential CTCF binding: lost CBSs, which are more transient and involved in direct transcriptional regulation; and retained CBSs, which are more stable and implicated in higher-order chromatin structures. We assessed the effect of CTCF depletion on primary naive B cells using the conditional Ctcffl/fl; Cd19-Creki/+ mouse model, which is expected to promote complete CTCF depletion in mature naive B cells. However, we found significant residual CTCF binding to DNA in CTCFfl/fl cells, consistent with previous studies of CTCF depletion achieved both with conditional Cre models31,43,44 and with the AID system.19,21,22,25,45 In Cre models, this phenomenon may be partly due to incomplete deletion of the CTCF alleles.29 However, with the AID system there is well-established evidence that a subset of CBSs is more resistant to degron-mediated degradation, which has a major impact on the persistence of CTCF at these sites.25 We thus conclude that our model has allowed the identification of two sets of CBSs, retained and lost, most likely representing genomic regions with different CTCF affinities, as reported by others.19,24,25
The distinction between lost and retained CBSs is not random, and distinct features can be ascribed to each type of CBS. In particular, retained CBSs are longer and rich in tandemly bound CTCF. This reflects the preferential presence of tandem CTCF motifs within individual retained CBSs. Furthermore, CTCF motifs in retained CBSs are more similar to the consensus motif than are those in lost CBSs. In addition, a higher proportion of the retained CBSs contain the previously defined U-motif, which increases CTCF-binding affinity.34 CTCF binding can also be modulated by additional mechanisms such as those involving specific protein partners or long non-coding RNA;46,47 nevertheless, we believe that the nature of the CTCF motifs and the presence of the U-motif are likely to be major determinants of the divergent activities of lost and retained CBSs. Indeed, combined assessment of CTCF motif numbers, CTCF consensus motif score, and the presence of the U motif could be used to successfully predict the presence of retained CBSs based on DNA sequence alone.
A key finding of our study is that the distinction of CBSs into two classes has important functional implications. For example, lost CBSs are more often located at promoter and enhancer regions than are retained CBSs. Moreover, the epigenetic mark distribution and transcriptional data suggest that lost CBSs are more frequently associated with active chromatin and direct transcriptional regulation. CTCF likely regulates transcription through the formation of intra-TAD loops between enhancers and promoters.20,21 These enhancer-promoter loops are probably more transient than the structural loops formed by retained CBSs, and this transience likely makes enhancer-promoter loops refractory to Hi-C detection. This would explain why we did not detect lost CBS enrichment at intra-TAD loop anchors, whereas we did detect retained CBSs. Alternatively, CTCF may also function as a conventional transcription activator at lost CBSs, independently of its architectural role.15 Regardless of the specific mechanisms responsible for the transcriptional regulation by lost CBSs, this role in dynamic, fined-tuned gene regulation is consistent with the comparatively low conservation of lost CBSs between cell types.
In contrast, retained CBSs are predominantly involved in the regulation of chromatin structure. Previous depletion studies revealed the role of retained CBSs in establishing TAD boundaries.24,25 Our new findings show that retained CBSs are preferentially located at strong domain boundaries over weak boundaries. Moreover, the U-motif that we found to be enriched in retained CBSs was recently reported to be important for CTCF-mediated insulation.26 These results indicate that retained CBSs are involved in the isolation of chromatin domains from neighboring regions and suggest that they could be important for CTCF heterochromatin barrier function. In this regard, we found that retained CBSs are more abundant at boundaries separating regions with differing chromatin states than at boundaries separating chromatin with the same state. This function might depend on the nucleosome-depleted regions present in retained CBSs. The spreading of histone modifications requires the cooperative binding of enzymes to adjacent nucleosomes, so the lack of nucleosomes at locations within retained CBSs could arrest heterochromatin spreading at that point.39 The findings presented in this study reveal the multifunctional nature of CTCF and show how its distinct structural and regulatory functions are rooted in differential occupancy patterns.
Limitations of the study
In this study, we have associated certain molecular features with the loss or persistence of CTCF at a binding site. However, we cannot rule out that other factors such as specific protein partners or long non-coding RNA are involved. In addition, experiments of genetic modification of CBSs will be necessary to get further insights on the relationship between the molecular characteristics of CBSs and CTCF function.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-α-Tubulin (DM1A) | Sigma | Cat# T9026; RRID:AB_477593 |
| Rabbit polyclonal anti-CTCF | Millipore | Cat# 07-729; RRID:AB_441965 |
| Goat polyclonal Anti-Rabbit HRP | DAKO | P0448; RRID:AB_2617138 |
| Goat polyclonal Anti-Mouse HRP | DAKO | P0447; RRID:AB_2617137 |
| Rabbit polyclonal anti-CTCF | Diagenode | Cat# C15410210-50; RRID:AB_2753160 |
| Rat anti-CD43 MicroBeads | Miltenyi Biotec | Cat# 130-049-801; RRID:AB_2861373 |
| Chemicals, peptides, and recombinant proteins | ||
| ACK Lysing Buffer | Lonza | Cat# BP10-548E |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| iDeal ChIP-seq Kit for Transcription Factors | Diagenode | Cat# C01010055 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE207640 |
| ChIP-seq, ChIA-PET and Hi-C | Kieffer-Kwon et al.36 and Vian et al.38 | GEO: GSE82144 GEO: GSE98119 |
| Hi-C | Rao et al.4 | 4D nucleome: 4DNESK95HVFB |
| TAD and intra-TAD loop anchors | Matthews and Waxman9 | GEO: GSE102999 |
| CTCF binding sites | Luan et al.25 | GEO: GSE150415 |
| Experimental models: Organisms/strains | ||
| Mouse: CTCFfl/fl | Heath et al.27 | N/A |
| Mouse: B6.129P2(C)-Cd19tm1(cre)Cgn/J | Jackson laboratories | Cat# 006785; RRID:IMSR_JAX:006785 |
| Oligonucleotides | ||
| CTCF deletion Fwd GGGCATCAGATCTCATTAAGGA | Perez-Garcia et al.31 | N/A |
| CTCF deletion Rv ACTCCATCTCTAGCCTCTCTATT | Perez-Garcia et al.31 | N/A |
| Software and algorithms | ||
| Loop prediction algorithm | This paper, GitHub | https://github.com/ARRlab/Loop_prediction_algorithm |
| Prism – version 6 | GraphPad | https://www.graphpad.com/scientificsoftware/prism |
| R | RCoreTeam | http://www.R-project.org/ |
| MACS2 | Zhang et al.48 | https://github.com/macs3-project/MACS/ |
| BedTools | Quinlan and Hall49 | https://bedtools.readthedocs.io/en/latest/ |
| DESeq2 | Love et al.50 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| HOMER | Heinz et al.51 | http://homer.ucsd.edu/ |
| deeptools | Ramírez et al.52 | https://deeptools.readthedocs.io/en/develop/ |
| FastQC | Krueger et al.53 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Criscuolo and Brisse54 | http://code.google.com/p/cutadapt/ |
| BWA | Li and Durbin55 | http://bio-bwa.sourceforge.net/ |
| Picard | Broad Institute | http://broadinstitute.github.io/picard |
| bamtools | Barnett et al.56 | https://github.com/pezmaster31/bamtools/wiki |
| ChIPQC | Carroll et al.57 | https://doi.org/10.18129/B9.bioc.ChIPQC |
| DiffBind | Ross-Innes et al.58 | http://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| samtools | Li et al.59 | http://htslib.org/ |
| IGV | Robinson et al.60 | http://www.broadinstitute.org/igv/ |
| ChIPseeker | Yu et al.61 | https://bioconductor.org/packages/ChIPseeker/ |
| Spamo - MEMEsuite | Whitington et al.62 | http://meme-suite.org/ |
| Other | ||
| CTCF signal visualization | This paper, UCSC genome browser | https://genome.ucsc.edu/s/lab_arr/mm10 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Almudena R. Ramiro (aramiro@cnic.es).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Conditional CTCF-deficient mice (Ctcffl/fl; Cd19-Creki/+) were obtained by breeding Ctcffl/fl mice27 with Cd19-Creki/+ mice.28 Six- to thirteen-week-old healthy mice of both sexes were used for ChIP-seq and RNA-seq experiments, including three littermates per group. Mice were housed in pathogen-free conditions under a 12 h dark/light cycle with food ad libitum. All animal procedures were conducted in accordance with EU Directive 2010/63/UE, enforced in Spanish law under Real Decreto 53/2013. The procedures were reviewed by the Institutional Animal Care and Use Committee (IACUC) of the Centro Nacional de Investigaciones Cardiovasculares and were approved by the Consejería de Medio Ambiente, Administración Local y Ordenación del Territorio of the Comunidad de Madrid (Ref: PROEX 341/14).
Method details
B cell purification
Naïve B cells were isolated from the spleen. Spleens were meshed through 70 μm pore nylon cell strainers (BD Falcon) in complete RPMI medium supplemented with 10% FBS and penicillin (50 U/ml) and streptomycin (50 μg/ml). Erythrocytes were lysed by incubating the cell suspension in erythrocyte lysis buffer (ACK Lysing Buffer, BioWhittaker) for 4 minutes at room temperature. After washing with cold complete RPMI, B cells were isolated by immunomagnetic depletion using anti-CD43 beads (Miltenyi Biotec). B cell purity was >95%.
qPCR for CTCF genomic deletion
For analysis of CTCF depletion, genomic DNA was isolated from B cells using phenol:chloroform:isoamyl alcohol and ethanol precipitation. DNA was quantified by SYBR green assay (Applied Biosystems). The CTCF primers used were as follows: 5’ – GGG CAT CAG ATC TCA TTA AGG A -3′ (forward) and 5’ – ACT CCA TCT CTA GCC TCT CTA TT -3′ (reverse). PCR amplification was performed with 7900HT Fast Real-Time PCR System thermal cycler (Applied Biosystems).
Immunoblotting
B cells were incubated in RIPA lysis buffer in the presence of protease inhibitors (Roche) and phosphatase inhibitors (Roche), and lysates were cleared by centrifugation. Total protein was size-fractionated on SDS-PAGE 8% acrylamide-bisacrylamide gels and transferred to Protan nitrocellulose membrane (Whatman) in transfer buffer (0.19 M glycine, 25 mM Tris base, and 0.01% SDS) containing 20% methanol (90 min at 0.4 A). Membranes were probed with anti-mouse-CTCF (1/1,000, Millipore) and anti-mouse-tubulin (1/5,000, Sigma-Aldrich). Then, membranes were incubated with HRP-conjugated anti-rabbit (1/5,000, DAKO) and anti-mouse (1/10,000, DAKO) antibodies, respectively, and developed with ClarityTM Western ECL Substrate (Bio-Rad).
ChIP-seq
ChIP was performed according to the Diagenode protocol (iDeal ChIP-seq Kit for Transcription Factors C01010055). In brief, 5 million cells were crosslinked in 1% formaldehyde (Sigma) for 10 min at 37°C and quenched with 0.125 M cold glycine. Cell pellets were lysed in 1 mL RIPA buffer (10 mM Tris-HCl, 1 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, pH 8.0) at 4°C for 20 min and centrifuged at 2300 x g for 5 min at 4°C. Nuclei were suspended in 500 μL 0.5% SDS lysis buffer (0.5% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0) and sonicated using the Covaris system (shearing time 15 min, 10% duty cycle, 200 cycles per burst, and 175 W PIP). Of the sheared chromatin, 1% was set aside (input), and the rest was incubated overnight at 4°C with anti-CTCF antibody (Diagenode). Immunoprecipitated chromatin was eluted and decrosslinked for 8 hours. DNA was purified and quantified with an Invitrogen Qubit Fluorometer. Finally, 3–4 ng of DNA were used to prepare libraries using the Illumina NEBNext Ultra II DNA Library Prep Kit. ChIP-seq and input control libraries from three biological replicates per genotype were sequenced in a Illumina HiSeq 2500 next-generation sequencer.
ChIP-seq analysis
ChIP-Seq sequencing reads were pre-processed by means of a pipeline that used FastQC53 to asses read quality, and Cutadapt54 to trim sequencing reads, eliminating Illumina adaptor remains, and to discard reads that were shorter than 30 bp. Resulting reads were then mapped against mouse genomic reference, GRCm38, with a pipeline that used bwa55 as aligner, Picard (http://broadinstitute.github.io/picard) to mark duplicate alignments, and bamtools56 to eliminate duplicate, chimeric and sub-optimally multi-mapped alignments, keeping only properly mapped reads. Alignments against the mitochondrial genome, chromosome Y and unplaced scaffolds were also removed. Once filtered alignments had been obtained, ChIP peaks were called with MACS2,48 using "-q 0.1" as the false discovery rate cut-off and removing those overlapping with mouse blacklisted regions. Using MAC2 we obtained a Narrowpeak file for each replicate where each identified peak is specified in a separate a row. When several of these peaks overlap between them, MACS2 assigns them the same peak identifier. Thus, we can know the number of small overlapping peaks (summits) contained in each peak.
Principal component analysis (PCA) was performed with the ChIPQC program.57 Next, filtered alignments and peaks, in BAM and BED formats, respectively, were processed with the R package DiffBind58 to define a consensus peak set including peaks found in at least 2 replicates (minOverlap = 0.66) and to define lost and retained CBSs through an occupancy analysis. DiffBind was also used to calculate and normalize peak coverage across samples, and to identify differential binding regions, using DESeq250 as analysis method. These data were used for volcanoplot representation, plotted with Graphpad Prism (version 6.01 for Windows, GraphPad Software, San Diego, CA, USA). CBS size was calculated based on the start and end coordinates present in the BED files. For density track visualization, BAM files were indexed with samtools,59 and BigWig files were then generated with bamCompare from the python tools deeptools suite.52 Using the computeMatrix and plotHeatmap functions in deeptools, the BigWig files were analyzed to yield a global evaluation of enrichment around the lost and retained CBS regions for each replicate.52 Both bigWig (signal) and BED (peak calls) files were visualised using Integrative Genomics Viewer (IGV).60 Additionaly, CBSs were annotated with ChIPseeker,61 using the genome annotation vM23 release from GENCODE. Overlapping regions were identified using the HOMER mergePeaks function with the ‘-d given’ option.51
To compare the number of summits within lost and retained CBSs, we generated a BED file containing the coordinates of regions from −200 to +200 bp around the summits detected for the 3 CTCF+/- group replicates. We then used the bedtools intersect function with the ‘-c’ option to count the number of summits per CBS in lost or retained groups.49 Finally, we represented the percentage of lost or retained CBSs with 1, 2, 3, 4 or more summits for each replicate.
CTCF motif analysis
Motif analysis was conducted with HOMER motif discovery software.51 Using the findMotifsGenome.pl function and the ‘-find’ option together with the HOMER CTCF-motif matrix, we extracted the matching score for every motif instance at each binding site (additional flags: -size given -mask). The motif distribution was determined using the annotatePealks.pl program with the ‘-size 1000 -hist 10’ options. The ‘-nmotifs’ option was used to obtain the number of motifs per CBS (additional flags: -size given).
Enrichment of the previously described CTCF upstream motif34 was assessed with Spamo,62 which permits simultaneous enrichment analysis of several motifs separated by variable sequence lengths. We considered only those cases in which the upstream motif was on the same strand as the core motif and positioned 5–6 bp upstream of it, as recommended by Nakahashi and collegues.34
Positional enrichment plot
We used positional enrichment plots to represent the distribution of lost and retained CBSs at specific genomic regions. A 10 or 100 kb region around a given genomic site (e.g. TSS regions) was divided into windows of 10 or 100 bp respectively. For each window, the percentage of overlapping lost or retained CBSs was calculated as the percentage of bp covered by CBSs within each window:
In cases where it was necessary to compare the values obtained in two plots (strong boundaries vs. weak boundaries), the overlap was additionally normalised to the number of genomic regions studied (e.g. number of strong boundaries). In these cases, the normalized overlap values were multiplied by 1000, so that the final number indicates the percentage of overlapping CBSs per 1000 regions.
CTCF loop boundary data were obtained from published ChIA-PET38 or Hi-C36 data, domain boundaries from published Hi-C data,4 TSSs in the mm10 genome from the UCSC database, and enhancer regions in Mus musculus splenic B cells from the enhancer Atlas 2.0 database.
CTCF binding site annotation
CBSs were classified into candidate cis-Regulatory Elements (cCREs) using the CH12-specific ENCODE encyclopedia. For each CBS, we selected a 400 bp window around the summit to avoid bias due to size difference between the lost and retained CBSs. The percentage of CBSs corresponding to each category was calculated using the HOMER mergePeaks function with the ‘-d given’ option.51 A similar analysis was performed for TAD anchors, intra- TAD anchors, lone CTCF sites, and CAC sites, as defined by Matthews BJ and Waxman DJ.9
Distribution of cohesin and histone modifications
BigWig files containing MNase-seq data and ChIP-seq data for RAD21, H3K4me1, H3K4me3, H3K27ac, and H3K27me336 were used to study the enrichment of these marks around lost and retained CBSs using the computeMatrix and plotProfile functions in deeptools.52
RNA-seq
RNA was extracted with the Qiagen RNeasy kit and was treated with DNase. Total RNA (500 ng) was used to generate libraries with the TruSeq RNA sample preparation kit v2 (Illumina). Briefly, poly-A RNA was purified using poly-T oligo- attached magnetic beads in two purification rounds, followed by fragmentation and first and second cDNA strand synthesis. cDNA 3′ ends were then adenylated, and the adapters were ligated followed by PCR library amplification. Finally, library size was checked with the Agilent 2100 Bioanalyzer DNA 1000 chip, and library concentrations were determined with a Qubi® fluorometer (Life Technologies). Libraries were sequenced in a HiSeq2500 next-generation sequencer (Illumina) to generate 60-base single reads.
RNA-seq analysis
RNA-Seq sequencing reads were pre-processed by means of a pipeline that used FastQC,53 to asses read quality, and Cutadapt54 to trim sequencing reads, eliminating Illumina adaptor remains, and to discard reads that were shorter than 30 bp. Resulting reads were mapped against reference transcriptome GRCm38.76 and quantified using RSEM.63 Expected expression counts calculated with RSEM were then processed with an analysis pipeline that used the Bioconductor package Limma64 for normalization (using TMM method), taking only into account those genes expressed with at least 1 count per million (CPM) in at least two samples. BigWig coverage files were produced with BamCoverage52 from RSEM-generated BAM files.
CTCF-mediated loop prediction algorithm
Lost and retained CBSs were annotated for the presence and orientation of CTCF motifs using the findMotifsGenome program in HOMER. When differently oriented motifs were identified in the same CBS, both orientations were annotated. A custom python code (https://www.python.org/) was then used to define potential loop regions. Pairs of CBSs were selected according to the following conditions. 1) Both CBSs must belong to the same group (lost or retained). 2) For the 5′ CBS, the CTCF motif must be in forward orientation, and for the 3′ CBS the CTCF motif must be in reverse orientation. 3) The two CBSs must be separated by less than 1 Mb.
To define loops with high and low transcriptional levels, we used RNA-seq data to calculate the average expression of all genes contained in each loop. Next, we calculated the probability density function (PDF) using the expression values of all genes detected in our RNA-seq data. PDF allows to calculate the probability for a given gene to have certain expression level. Then, we obtained the PDF for the expression of several genes by convolving as many PDFs as genes in the loop. Expression values among the top 10% or the bottom 10% of the distribution, were scored as active or repressed, respectively (Figure S4).
Quantification and statistical analysis
Statistical analyses were performed with GraphPad Prism (version 6.01 for Windows, GraphPad Software, San Diego, CA, USA) or R packages. Continuous data were analyzed by two-tailed Student t test. Non-normally distributed data were analyzed with the unpaired two-sample Wilcoxon test. Significant association between two categorical variables was determined with the Fisher test. Differences were considered statistically significant at P≤0.05.
Acknowledgments
We thank all the members of the B lymphocyte Biology lab for helpful suggestions, Sonia Mur for technical assistance, Ana Losada and Ana Cuadrado for helpful discussions on experimental design, Román Pérez Santalla for his help on algorithm design, Simon Bartlett for English editing, and the CNIC Genomics Unit for ChIP-seq and RNA-seq. We also thank Rafael Casellas and Erez Aiden for sharing HiC data with us and Jing Luan and Gerd Blobel for their help to access their datasets. This work was supported by grants from the Spanish Ministerio de Economía, Industria y Competitividad and ERDF, A way of making Europe (SAF2016-75511-R), the Spanish Ministerio de Ciencia e Innovación (PID2019-106773RB-I00/AEI/10.13039/501100011033) and the “la Caixa” Banking Foundation under the project code HR17-00247 to A.R.R. F.S.-C. received support from the Spanish Ministerio de Economía y Competitividad (RTI2018-102084-B-I00). E.M.-Z. and A.R.-R. are fellows of the research training program (FPI) funded by the Ministerio de Economía y Competitividad (BES-2014-069525) and Ministerio de Ciencia e Innovación (PRE2020-091873). M.J.G., F.S.-C., and A.R.R. are supported by CNIC. The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the Ministerio de Ciencia e Innovación (MCIN) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence, CEX2020-001041-S funded by MICIN/AEI/10.13039/501100011033.
Author contributions
E.M.-Z. conducted ChIP-seq and RNA-seq experiments; M.J.G. performed mapping and peak calling for ChIP-Seq data and RNA-seq analysis; E.M.-Z. and A.R.-R. performed downstream data analysis and wrote the manuscript; F.S.-C. provided support for bioinformatic analyses; A.R.R. supervised the study and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects.
We support inclusive, diverse, and equitable conduct of research.
Published: February 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106106.
Supplemental information
Data and code availability
-
•
RNA-seq and ChIP-seq data from this study have been deposited at GEO and are publicly available under accession code GSE207640. This paper analyzes existing, publicly available data. These accession numbers are listed in the key resources table.
-
•
Algorithm code is deposited at GitHub and is publicly available (https://github.com/ARRlab/Loop_prediction_algorithm).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.de Laat W., Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 2.Gibcus J.H., Dekker J. The hierarchy of the 3D genome. Mol. Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng H., Xie W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019;20:535–550. doi: 10.1038/s41580-019-0132-4. [DOI] [PubMed] [Google Scholar]
- 4.Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon J.R., Gorkin D.U., Ren B. Chromatin domains: the unit of chromosome organization. Mol. Cell. 2016;62:668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flavahan W.A., Drier Y., Liau B.B., Gillespie S.M., Venteicher A.S., Stemmer-Rachamimov A.O., Suvà M.L., Bernstein B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matthews B.J., Waxman D.J. Computational prediction of CTCF/cohesin-based intra-TAD loops that insulate chromatin contacts and gene expression in mouse liver. Elife. 2018;7:e34077. doi: 10.7554/eLife.34077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavalheiro G.R., Pollex T., Furlong E.E. To loop or not to loop: what is the role of TADs in enhancer function and gene regulation? Curr. Opin. Genet. Dev. 2021;67:119–129. doi: 10.1016/j.gde.2020.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Ong C.T., Corces V.G. CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y., Zhang X., Dai H.Q., Hu H., Alt F.W. The role of chromatin loop extrusion in antibody diversification. Nat. Rev. Immunol. 2022;22:550–566. doi: 10.1038/s41577-022-00679-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merkenschlager M., Nora E.P. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genom. Hum. Genet. 2016;17:17–43. doi: 10.1146/annurev-genom-083115-022339. [DOI] [PubMed] [Google Scholar]
- 14.Davidson I.F., Peters J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021;22:445–464. doi: 10.1038/s41580-021-00349-7. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H., Lam J., Zhang D., Lan Y., Vermunt M.W., Keller C.A., Giardine B., Hardison R.C., Blobel G.A. CTCF and transcription influence chromatin structure re-configuration after mitosis. Nat. Commun. 2021;12:5157. doi: 10.1038/s41467-021-25418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang J., Kim Y.W., Park S., Kang Y., Kim A. Multiple CTCF sites cooperate with each other to maintain a TAD for enhancer-promoter interaction in the β-globin locus. FASEB J. 2021;35:e21768. doi: 10.1096/fj.202100105RR. [DOI] [PubMed] [Google Scholar]
- 17.Cuddapah S., Jothi R., Schones D.E., Roh T.Y., Cui K., Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narendra V., Rocha P.P., An D., Raviram R., Skok J.A., Mazzoni E.O., Reinberg D. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347:1017–1021. doi: 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyle J., Zhang Y., Wright S., Xu B., Shao Y., Easton J., Tian L., Feng R., Xu P., Li C. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer-promoter looping. Nucleic Acids Res. 2019;47:6699–6713. doi: 10.1093/nar/gkz462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanssen L.L.P., Kassouf M.T., Oudelaar A.M., Biggs D., Preece C., Downes D.J., Gosden M., Sharpe J.A., Sloane-Stanley J.A., Hughes J.R., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 2017;19:952–961. doi: 10.1038/ncb3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubo N., Ishii H., Xiong X., Bianco S., Meitinger F., Hu R., Hocker J.D., Conte M., Gorkin D., Yu M., et al. Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol. 2021;28:152–161. doi: 10.1038/s41594-020-00539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz-Velasco M., Kumar M., Lai M.C., Bhat P., Solis-Pinson A.B., Reyes A., Kleinsorg S., Noh K.M., Gibson T.J., Zaugg J.B. CTCF-mediated chromatin loops between promoter and gene body regulate alternative splicing across individuals. Cell Syst. 2017;5:628–637.e6. doi: 10.1016/j.cels.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Khoury A., Achinger-Kawecka J., Bert S.A., Smith G.C., French H.J., Luu P.L., Peters T.J., Du Q., Parry A.J., Valdes-Mora F., et al. Constitutively bound CTCF sites maintain 3D chromatin architecture and long-range epigenetically regulated domains. Nat. Commun. 2020;11:54. doi: 10.1038/s41467-019-13753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luan J., Xiang G., Gómez-García P.A., Tome J.M., Zhang Z., Vermunt M.W., Zhang H., Huang A., Keller C.A., Giardine B.M., et al. Distinct properties and functions of CTCF revealed by a rapidly inducible degron system. Cell Rep. 2021;34:108783. doi: 10.1016/j.celrep.2021.108783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H., Zhu Q., Jussila A., Han Y., Bintu B., Kern C., Conte M., Zhang Y., Bianco S., Chiariello A.M., et al. CTCF mediates dosage- and sequence-context-dependent transcriptional insulation by forming local chromatin domains. Nat. Genet. 2021;53:1064–1074. doi: 10.1038/s41588-021-00863-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath H., Ribeiro de Almeida C., Sleutels F., Dingjan G., van de Nobelen S., Jonkers I., Ling K.W., Gribnau J., Renkawitz R., Grosveld F., et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008;27:2839–2850. doi: 10.1038/emboj.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rickert R.C., Roes J., Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marina-Zárate E., Pérez-García A., Ramiro A.R. CCCTC-binding factor locks premature IgH germline transcription and restrains class switch recombination. Front. Immunol. 2017;8:1076. doi: 10.3389/fimmu.2017.01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hobeika E., Thiemann S., Storch B., Jumaa H., Nielsen P.J., Pelanda R., Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pérez-García A., Marina-Zárate E., Álvarez-Prado Á.F., Ligos J.M., Galjart N., Ramiro A.R. CTCF orchestrates the germinal centre transcriptional program and prevents premature plasma cell differentiation. Nat. Commun. 2017;8:16067. doi: 10.1038/ncomms16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim T.H., Abdullaev Z.K., Smith A.D., Ching K.A., Loukinov D.I., Green R.D., Zhang M.Q., Lobanenkov V.V., Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plasschaert R.N., Vigneau S., Tempera I., Gupta R., Maksimoska J., Everett L., Davuluri R., Mamorstein R., Lieberman P.M., Schultz D., et al. CTCF binding site sequence differences are associated with unique regulatory and functional trends during embryonic stem cell differentiation. Nucleic Acids Res. 2014;42:774–789. doi: 10.1093/nar/gkt910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakahashi H., Kieffer Kwon K.R., Resch W., Vian L., Dose M., Stavreva D., Hakim O., Pruett N., Nelson S., Yamane A., et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013;3:1678–1689. doi: 10.1016/j.celrep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soochit W., Sleutels F., Stik G., Bartkuhn M., Basu S., Hernandez S.C., Merzouk S., Vidal E., Boers R., Boers J., et al. CTCF chromatin residence time controls three-dimensional genome organization, gene expression and DNA methylation in pluripotent cells. Nat. Cell Biol. 2021;23:881–893. doi: 10.1038/s41556-021-00722-w. [DOI] [PubMed] [Google Scholar]
- 36.Kieffer-Kwon K.R., Nimura K., Rao S.S.P., Xu J., Jung S., Pekowska A., Dose M., Stevens E., Mathe E., Dong P., et al. Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Mol. Cell. 2017;67:566–578.e10. doi: 10.1016/j.molcel.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bae S., Lesch B.J. H3K4me1 distribution predicts transcription state and poising at promoters. Front. Cell Dev. Biol. 2020;8:289. doi: 10.3389/fcell.2020.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vian L., Pękowska A., Rao S.S.P., Kieffer-Kwon K.-R., Jung S., Baranello L., Huang S.-C., El Khattabi L., Dose M., Pruett N., et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;173:1165–1178.e20. doi: 10.1016/j.cell.2018.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarkson C.T., Deeks E.A., Samarista R., Mamayusupova H., Zhurkin V.B., Teif V.B. CTCF-dependent chromatin boundaries formed by asymmetric nucleosome arrays with decreased linker length. Nucleic Acids Res. 2019;47:11181–11196. doi: 10.1093/nar/gkz908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Bortle K., Ramos E., Takenaka N., Yang J., Wahi J.E., Corces V.G. Drosophila CTCF tandemly aligns with other insulator proteins at the borders of H3K27me3 domains. Genome Res. 2012;22:2176–2187. doi: 10.1101/gr.136788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weth O., Paprotka C., Günther K., Schulte A., Baierl M., Leers J., Galjart N., Renkawitz R. CTCF induces histone variant incorporation, erases the H3K27me3 histone mark and opens chromatin. Nucleic Acids Res. 2014;42:11941–11951. doi: 10.1093/nar/gku937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanborn A.L., Rao S.S.P., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA. 2015;112:E6456–E6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watson L.A., Wang X., Elbert A., Kernohan K.D., Galjart N., Bérubé N.G. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J. Neurosci. 2014;34:2860–2870. doi: 10.1523/jneurosci.3769-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soshnikova N., Montavon T., Leleu M., Galjart N., Duboule D. Functional analysis of CTCF during mammalian limb development. Dev. Cell. 2010;19:819–830. doi: 10.1016/j.devcel.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Stik G., Vidal E., Barrero M., Cuartero S., Vila-Casadesús M., Mendieta-Esteban J., Tian T.V., Choi J., Berenguer C., Abad A., et al. CTCF is dispensable for immune cell transdifferentiation but facilitates an acute inflammatory response. Nat. Genet. 2020;52:655–661. doi: 10.1038/s41588-020-0643-0. [DOI] [PubMed] [Google Scholar]
- 46.Saldaña-Meyer R., Rodriguez-Hernaez J., Escobar T., Nishana M., Jácome-López K., Nora E.P., Bruneau B.G., Tsirigos A., Furlan-Magaril M., Skok J., Reinberg D. RNA interactions are essential for CTCF-mediated genome organization. Mol. Cell. 2019;76:412–422.e5. doi: 10.1016/j.molcel.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gavrilov A.A., Sultanov R.I., Magnitov M.D., Galitsyna A.A., Dashinimaev E.B., Lieberman Aiden E., Razin S.V. RedChIP identifies noncoding RNAs associated with genomic sites occupied by Polycomb and CTCF proteins. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2116222119. e2116222119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krueger F., Andrews S.R., Osborne C.S. Large scale loss of data in low-diversity illumina sequencing libraries can be recovered by deferred cluster calling. PLoS One. 2011;6:e16607. doi: 10.1371/journal.pone.0016607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Criscuolo A., Brisse S. AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front. Genet. 2014;5:130. doi: 10.3389/fgene.2014.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barnett D.W., Garrison E.K., Quinlan A.R., Strömberg M.P., Marth G.T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carroll T.S., Liang Z., Salama R., Stark R., de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 2014;5:75. doi: 10.3389/fgene.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R., et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 62.Whitington T., Frith M.C., Johnson J., Bailey T.L. Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res. 2011;39:e98. doi: 10.1093/nar/gkr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq and ChIP-seq data from this study have been deposited at GEO and are publicly available under accession code GSE207640. This paper analyzes existing, publicly available data. These accession numbers are listed in the key resources table.
-
•
Algorithm code is deposited at GitHub and is publicly available (https://github.com/ARRlab/Loop_prediction_algorithm).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.