

Abstract
α3β4 Nicotinic acetylcholine receptor (nAChR) has been recognized as an emerging biomarker for the early detection of drug addiction. Herein, α3β4 nAChR ligands were designed and synthesized to improve the binding affinity and selectivity of two lead compounds, (S)-QND8 and (S)-T2, for the development of an α3β4 nAChR tracer. The structural modification was achieved by retaining the key features and expanding the molecular structure with a benzyloxy group to increase the lipophilicity for blood-brain barrier penetration and to extend the ligand-receptor interaction. The preserved key features are a fluorine atom for radiotracer development and a p-hydroxyl motif for ligand-receptor binding affinity. Four (R)- and (S)-quinuclidine-triazole (AK1-AK4) were synthesized and the binding affinity, together with selectivity to α3β4 nAChR subtype, were determined by competitive radioligand binding assay using [3H]epibatidine as a radioligand. Among all modified compounds, AK3 showed the highest binding affinity and selectivity to α3β4 nAChR with a Ki value of 3.18 nM, comparable to (S)-QND8 and (S)-T2 and 3069-fold higher affinity to α3β4 nAChR in comparison to α7 nAChR. The α3β4 nAChR selectivity of AK3 was considerably higher than those of (S)-QND8 (11.8-fold) and (S)-T2 (294-fold). AK3 was shown to be a promising α3β4 nAChR tracer for further development as a radiotracer for drug addiction.
Keywords: α3β4 Nicotinic acetylcholine receptor, quinuclidine, triazole, drug-seeking behavior monitoring, drug addiction
1. Introduction
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channel receptors in the Cys-loop superfamily, which are expressed in the central nervous system (CNS) and in the peripheral nervous system (PNS) [1,2,3]. The subunits of nAChRs can be alpha (α2–α10) and beta (β2–β4) subunits surrounding an ion pore. The nAChR subtypes can be divided into two classes, according to subunit assembly: homopentameric receptors such as α7 and α9 and heteropentameric receptors such as α3β4 and α4β2 [3]. The different subunit combinations bring about the distinct pharmacological profile of nAChR [1]. For example, the α7 and α4β2 nAChR subtypes, which are expressed at high levels in the brain, are involved in neurological disorders (Alzheimer’s disease, schizophrenia, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), and inflammation [4,5,6,7,8,9,10]), whereas the α3β4 nAChR is involved in depression and drug addiction [11,12,13].
Drug addiction, particularly nicotine addiction, is a worldwide epidemic that affects the lives of both smokers and non-smokers. The reward and reinforcement effects, through the mesolimbic pathway, and the aversive effects of withdrawal, via the medial habenula-interpeduncular (MHb-IPN) pathway, are involved in maintaining nicotine use [14,15,16]. Examples of subunits in nAChR subtypes that are mainly expressed in the mesolimbic pathway are α4, α6, α7, and β2 subunits [17], whereas α3, α5, and β4 subunits are in the MHb-IPN circuit [13,18,19]. Human genetic studies showed that gene clusters expressing α3, α5, and β4 subunits were associated with tobacco dependence and higher numbers of cigarettes smoked per day [20,21,22]. The expression of α3, α5, and β4 nAChR subunits in the MHb-IPN circuit has drawn attention to the study of both α3β4* and α5* nAChRs in nicotine aversion. However, some evidence supports the role of α4β2α5* nAChR in regulating glutamate transmission [23] and that of α3β4* nAChR in regulating acetylcholine release in the MHb-IPN pathway [24,25,26].
The α3β4 nAChR expressed in the MHb-IPN circuit [14,27,28,29] has been reported to play a role in drug or psychostimulant-seeking behavior including nicotine, morphine, methamphetamine, and alcohol [30]. The α3β4 nAChR is not only expressed in the interpeduncular nucleus (IPN) and the medial habenula (MHb), but also in the pineal gland, locus coeruleus, and hippocampus [31]. Several compounds are reported to bind with α3β4 nAChR, including 18-methoxycoronaridine (18-MC) [32], dextromethorphan [33], mecamylamine [34], SR16584 [35], (S)-T1 [36] and AT-1001 [37] (Figure 1). However, all of them except (S)-T1 and AT-1001 are non-selective ligands, which can bind with other subtypes or other receptors. For example, 18-MC can bind to an opioid receptor [38] and a muscle AChR [39], mecamylamine can antagonize several nAChR subtypes [40] and dextromethorphan can block N-methyl-D-aspartate (NMDA) receptor [41]. The selective α3β4 nAChR (S)-T1 and AT-1001 with Ki below 10 nanomolar were further developed to be α3β4 nAChR radiotracers by labeling with a radionuclide yielding [125I]AT-1012 [42] and (S)-[18F]T1 [36], respectively. [125I]AT-1012 labeled with 125I, a gamma-emitter, is a single-photon emission computed tomography (SPECT) tracer, while (S)-[18F]T1 containing 18F, a positron emitter, is a positron emission tomography (PET) imaging agent. The selective α3β4 nAChR radiotracer is a tool for studying psychostimulant and drug-seeking behavior and drug discovery.
Figure 1.

Structures of (a) α3β4 nAChR ligands and (b) radio-imaging agents.
Nowadays, the combined use of a radiotracer and a PET scanner is the most precise imaging technique to detect early-stage of diseases at a molecular level [43]. The scanner quantitatively measures biochemical and physiological processes by using a suitable radiotracer. The radiotracer comprises two parts: a specific tracer to localize a target or disease biomarker and a positron-emitting radionuclide, such as fluorine-18 (18F) or carbon-11 (11C), to produce gamma radiation via the process of positron annihilation [44]. The development of a radiotracer has three main steps: (i) design and synthesis of non-radioactive reference compound (formerly named cold standard), where the radionuclide in a radiotracer is a non-radioactive isotope; (ii) synthesis of a precursor for radiolabeling; and (iii) radiosynthesis or radiolabeling of a precursor to yield a radiotracer [45]. The starting point for the development of a new radiotracer is the design of a new tracer or search for available non-radioactive reference compounds with good binding affinity at a nanomolar level and high selectivity profile to a target (disease biomarker).
In this study, four compounds (AK1-AK4) were designed to increase the binding affinity and selectivity to α3β4 nAChR. The designed structures were synthesized by copper-catalyzed azide-alkyne cycloaddition (CuAAC) and evaluated in silico and in vitro for binding affinity and selectivity. The compound showing the highest affinity and selectivity to α3β4 nAChR will be selected as a non-radioactive reference compound for further development as an α3β4 nAChR PET imaging agent for drug addiction.
2. Results and Discussion
Four quinuclidine-triazole compounds (AK1-AK4) were modified by merging the structures of two lead compounds, (S)-QND8 and (S)-T2 [46], followed by single point modification (Figure 2) to improve α3β4 nAChR binding affinity and selectivity profiles. Two pharmacophoric features, a chiral quinuclidine ring ((R) and (S)) acting as a cationic center and a triazole linker acting as a hydrogen bond acceptor [47], were kept in the modified structures. The key functional motifs in the hydrophobic part of (S)-QND8 and (S)-T2 were preserved: a hydroxyl group (-OH) for forming a hydrogen bond with the receptor [48] and a fluorine atom for further development as a positron-emitting radionuclide, fluorine-18. Besides retaining the fluorine atom and the hydroxyl group, the single-point modification was made by extending the hydrophobic part with a benzene ring at the hydroxyl function. The benefit of the added benzyloxy is that it can increase the lipophilicity of (S)-QND8 and (S)-T2 from log P of 1.87 and 2.03, respectively to log P of 4.03 and accordingly enhance brain penetration and subtype selectivity. The hydrophobic area has been reported to mediate nAChR subtype selectivity.
Figure 2.

The design strategy of α3β4 nAChR ligands.
The binding affinities (Ki) of AK1-AK4 and two lead compounds were presented in Table 1. All compounds exhibited binding affinity to α3β4 nAChR, α7 nAChR, and α4β2 nAChR in the range of 2.28–9760 nM. Comparing (R)- and (S)-enantiomers, the (S)-isomers of both AK1 and AK3 preferably bound to α3β4 nAChR, whereas their corresponding (R)-isomers (AK2 and AK4) selectivity bound to α7 nAChR. The stereoselective binding of (S)-enantiomers to α3β4 nAChR and (R)-enantiomers to α7 nAChR agrees with previous reports [46,48]. This might lead to some limitations of this study. The data discussed stereoselectivity came from one core structure, which is a quinuclidine ring.
Table 1.
In vitro binding affinity constants (Ki values) for binding of the quinuclidine-triazole derivatives towards human α3β4, α4β2, and α7 nAChRsa.
 R |
Binding Affinity Ki (nM) a |
Selectivity Ratios | ||||
|---|---|---|---|---|---|---|
| α3β4 b | α7 c | α4β2 c | α7/α3β4 | α4β2/α3β4 | α4β2/α7 | |
| Synthesized compounds | ||||||
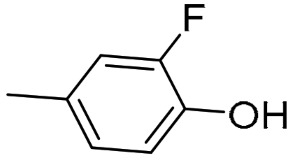 AK1 (S-isomer) |
2.28 (1.72; 2.85) |
26.7 $ (20.0; 33.4) |
504 (196; 813) |
11.7 | 221 | 18.9 |
 AK2 (R-isomer) |
601 #,$ (432; 770) |
4.49 $ (3.56; 5.43) |
5977 #,$ (4939; 7015) |
0.01 | 9.95 | 1331 |
 AK3 (S-isomer) |
3.18 (2.17; 4.18) |
9760 #,$ (9565; 9955) |
180 (160; 201) |
3069 | 56.6 | 0.02 |
 AK4 (R-isomer) |
112 (81.3; 142) |
53.6 $ (50.2; 57.1) |
4176 #,$ (4059; 4293) |
0.48 | 37.3 | 77.9 |
| Lead compounds | ||||||
 (S)-QND8 [46] |
2.48 ± 0.04 | 29.3 ± 0.18 $ | 461 ± 89 | 11.8 | 186 | 15.7 |
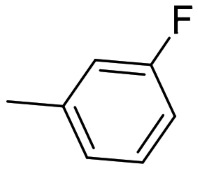 (S)-T2 [46] |
2.25 ± 0.42 | 660 ± 1.39 # | 519 ± 20 | 294 | 231 | 0.79 |
a Values represent the mean of two independent experiments performed in triplicates and reported in brackets. b Binding assays were performed with membrane preparations from HEK293 cells stably transfected with human α3β4 or α4β2 nAChR and the radiotracer [3H]epibatidine (working concentration ~ 0.5 nM; Kd = 0.025 nM for hα4β2 nAChR and Kd = 0.117 nM for hα3β4 nAChR). c Binding assays were performed with membrane preparations from SH-SY5Y cells stably transfected with human α7 nAChR and the radiotracer [3H]methyllycaconitine (working concentration ~ 0.5 nM; Kd = 2.0 nM). # p < 0.05 compared with (S)-QND8; $ p < 0.05 compared with (S)-T2.
The binding affinities of new (S)-enantiomers (AK1 and AK3) to α3β4 nAChR with Ki values of 2.28 and 3.18 nM are high, almost the same as those of the lead compounds, (S)-QND8, (S)-T2, and AT-1001 (Ki = 2.48, 2.25, and 2.60 nM, respectively) [46]. The selectivity of AK3 over the α7 subtype is the highest with a 3069-fold higher affinity to the α3β4 in comparison to the α7 subtype, whereas for all compounds the selectivity for the α4β2 subtype is much lower with a range of 10–220-fold. Molecular docking of AK1, AK3, and lead compounds to α3β4 and α7 nAChR homology models were performed to explain the remarkably high selectivity profile of AK3 to α3β4 over α7 nAChRs.
The molecular docking of new compounds and lead compounds to α3β4 nAChR homology model showed that all compounds aligned and formed hydrogen bonds and π-π interactions with amino acid residues in an aromatic cage located at the interface between the α3 subunit and β4 subunit. The protonated quinuclidine ring of all (S)-enantiomers pointed to Asp173, a key amino acid determinant to form a salt bridge and hydrogen bond interaction in slightly shorter distances than their (R)-counterparts, particularly of 1.74 Å of AK3 vs. 2.14 Å of AK4 (Figure 3, Table 2). In agreement with a previous report [48], the (S)-enantiomer of a quinuclidine ring allowed the docked pose to accommodate a salt bridge interaction with Asp173, the key determinant of the α3β4 nAChR binding. Other common key residues of α3β4 nAChR for binding are Trp149 and Tyr190 in the α3 subunit for which the interactions can be observed to Trp149 and/or Tyr190 in the modified compounds. For AK1, the protonated quinuclidine formed a salt bridge interaction with Asp173, and the phenolic −OH formed a hydrogen bond with Ser148. The additional interactions were cation-π interaction with Trp59, π-π interactions with Trp59, Trp149, Trp190, and Tyr197, and halogen bond with Trp149 (Figure 4A and Table 2). Even though the number of hydrogen bonds and π-π interaction of AK3 with key residues was less than those of AK1, the binding affinity of AK3 to α3β4 nAChR (Ki = 3.18 nM) was found to be comparable to AK1 (Ki = 2.28 nM). This result confirms that the extending benzyloxy group increased hydrophobic interactions as designed. These interactions from the expansion contributed to the similar binding affinity and ligand efficiency (LE), a parameter to assess the binding affinity of different molecular weights, of these two compounds (LE = −0.52 and −0.51 for AK1 and AK3) (Table 2).
Figure 3.

The overlay binding modes and molecular interactions of (S)- and (R)-enantiomers at the binding site of α3β4 nAChR: docked conformations of (a) AK1 (cyan) and AK2 (magenta) and (b) AK3 (yellow) and AK4 (violet). Orange and green indicated residues in α3-principal and β4-complementary subunits, respectively.
Table 2.
The amino acid residues involved in the binding interaction to α3β4 nAChRs.
| Cpds | α3β4 nAChRs | Binding Free Energy (ΔG, kcal/mol) | Ligand Efficiency * (LE) |
|||
|---|---|---|---|---|---|---|
| H-Bond (Distance in Å) | Cation-π | π-π | Halogen | |||
| AK1 | Ser148 (1.97), Asp173 (1.90) | Asp173, Trp59 |
Trp59, Trp149, Tyr190, Tyr197 | Trp149 | −10.94 | −0.52 |
| AK2 | Ser148 (1.94), Asp173 (1.91) |
Asp173 | Trp59, Tyr190, Tyr197 | Ser148, Trp149 |
−11.36 | −0.54 |
| AK3 | Asp173 (1.74) | Asp173 | Trp59, Trp149 | Trp149 | −14.21 | −0.51 |
| AK4 | Asp173 (2.14) | Asp173 | Trp59, Trp149 | Trp149 | −14.14 | −0.51 |
| (S)-QND8 | Trp149 (1.87), Asp173 (1.56) | Asp173 | Tyr190 | - | −13.28 | −0.66 |
| (S)-T2 | Asp173 (1.85) | Asp173 | Trp59, Trp149, Tyr190, Tyr197 | Ser148 | −10.42 | −0.52 |
The hydrogen bonds were analyzed and measured by AutoDock4.2, and the salt bridges, cation-π, and π-π interactions were analyzed by BIOVIA Discovery Studio Visualized. The interaction of amino acid residues with protonated quinuclidine is presented in bold. * LE is the ratio of Gibbs free energy (ΔG) to the number of non-hydrogen atoms of the ligand.
Figure 4.

The binding interactions of (S)-enantiomers (a) AK1 and (b) AK3 and (R)-enantiomers (c) AK2 and (d) AK4 to α3β4 nAChR. A and B indicated α3 and β4 subunits, respectively.
In terms of selectivity, AK3 possesses a 3069-fold selectivity to α3β4 nAChR over α7 nAChR, whereas the selectivity of AK1 and the lead compounds (S)-QND8 and (S)-T2 was much lower. The α7 nAChR binding poses of AK3 and (S)-QND8 showed that the quinuclidine ring of these two compounds is aligned in different directions leading to the higher number of hydrogen bond interactions of (S)-QND8 (Figure 5B). Three hydrogen bond interactions of (S)-QND8 to α7 nAChR are composed of the interaction of protonated quinuclidine to Asp164 and Ser166 and the interaction of -OH to Tyr93. Only one hydrogen bond interaction between protonated quinuclidine to Tyr93 was observed in AK3 resulting in less preference for the α7 subtype. Hence, the extension approach, via the added benzyloxy group in the AK3 structure, significantly enhanced the selectivity profile by the hydrophobic interactions with the residues in a β4-complementary subunit (Figure 3B). Therefore, the added benzyloxy group is the key contributor to the enhanced α3β4-selectivity of AK3 by providing the hydrophobic interactions with the β4 subunit.
Figure 5.

The overlay binding modes of AK3 and (S)-QND8 against (a) α3β4 nAChR and (b) α7 nAChR. Orange, green, and light pink indicated α3, β4, and α7 subunits, respectively.
MD simulation of AK3 to α3β4 and α7 nAChRs was performed to gain more insight into the high affinity and selectivity of AK3 to α3β4 nAChR (Ki = 3.18 nM, 3069-fold selectivity over α7 nAChR). The strong interaction from the salt bridge formation between protonated quinuclidine and the carboxylate of Asp173, which appeared to mediate high affinity and selectivity to α3β4 nAChR, was detected by MD simulation. Besides the salt bridge formation, the major interactions observed in the MD simulation of AK3 to α3β4 nAChR are four π-π interactions between both middle and terminal benzene rings of the hydrophobic part of AK3 and residues Tyr93, Trp149, Tyr190, and Tyr 197 (Figure 6a). When compared with molecular docking, most of the interacting amino acid residues were the same (Tyr93, Ser148, Trp149, Ser150, and Tyr197 in principal α3 subunit and Ala42, Trp59, Ile113, Leu123, Pro125, and Asp173 in complementary β4 subunit) but the binding interaction types were altered, due to the flexible and solvated receptor in MD simulation. For example, the halogen bond between a fluorine atom and Trp149 and π-π interactions of a triazole ring to Trp59 and Trp149 observed in molecular docking were replaced by additional π-π interactions of middle and terminal benzene rings to Tyr93, Tyr190, and Tyr 197 in MD simulation (Figure 6a). Although the number of main interactions in the complexes of AK3-α3β4 nAChR and AK3-α7 nAChR, were equal (four π-π interactions), the salt bridge interaction between the protonated quinuclidine and Asp173 in AK3-α3β4 nAChR complex (Figure 6b) was stronger than the hydrogen bond interaction to Ser166 of α7 nAChR. The results from both molecular docking and MD simulation supported the higher affinity and selectivity of AK3 to α3β4 nAChR than α7 nAChR.
Figure 6.

The binding modes of AK3 to (a) α3β4 nAChR and (b) α7 nAChR from MD simulation compared to molecular docking. A and B indicate principal and complementary subunits, respectively. Red circles indicate different interacting amino acid residues between MD simulation and molecular docking.
For (R)-enantiomers, AK2 and AK4 showed lower binding affinities to α3β4 nAChR with Ki values of 601 and 112 nM, respectively than their (S)-counterparts and the lead compounds. From molecular docking results, the ligand-receptor interactions provided by quinuclidine and hydrophobic pharmacophores of AK2 and AK4 were not different from those of AK1 and AK3 (Figure 3 and Table 2). Only the triazole ring pointed to a different angle leading to the lower number of π-π interactions between the triazole ring of AK2 and AK4 with α3β4 nAChR compared to their (S)-counterparts (AK1 and AK3) showing the important role of this moiety for the binding affinity (Figure 4). However, the (R)-enantiomers AK2 and AK4 bound to α7 nAChR with Ki values of 4.49 and 53.6 nM, respectively, which are higher than the values of their (S)-counterparts (Table 1). The molecular docking to the α7 nAChR homology model showed that the quinuclidine ring interacted with Trp149 and Tyr93, key determinants of the α7 subtype leading to the high affinity of AK2 and AK4 (Figure 6). The key interactions of AK2 are strong cation-π to Trp149, hydrogen bonds to Tyr93 and Trp149, and π-π interactions to Trp55 and Trp149, whereas AK4 bound to the receptor with the hydrogen bond to Tyr93 and π-π interaction to Trp149 in addition to halogen bonds with Ser148 and Trp149 (Figure 7, Table 3). The absence of a strong cation-π interaction of the quinuclidine ring together with the lack of -OH to form additional hydrogen bonds resulted in the lower binding affinity of AK4 compared to AK2. In addition, the extended benzyloxy group present in AK4 might have caused steric hindrance to the receptor, leading to a decrease in affinity. The higher binding affinities of AK2 than AK4 (Ki of 53.6 and 4.49 nM) were found to agree with the ligand efficiency (LE) of these two compounds: AK4 bound to α7 nAChR weaker than AK2 (LE = −0.47 and −0.52, respectively) (Table 3).
Figure 7.

The binding interactions of (a) AK2 and (b) AK4 to α7 nAChR. A and B indicated principal and complementary subunits, respectively.
Table 3.
The amino acid residues involved in the binding interaction to α7 nAChRs.
| Cpds | α7 nAChRs | Binding Free Energy (ΔG, kcal/mol) | Ligand Efficiency * (LE) |
|||
|---|---|---|---|---|---|---|
| H-Bond (Distance in Å) | Cation-π | π-π | Halogen | |||
| AK1 |
Tyr93 (2.06, 2.03), Ser148 (2.66) |
Tyr195 | Trp55, Trp149, Tyr188 | - | −11.08 | −0.53 |
| AK2 | Tyr93 (2.13), Trp149 (1.86) |
Trp149 | Trp55, Trp149 | - | −10.98 | −0.52 |
| AK3 |
Tyr93 (2.20) |
- | Trp55, Trp149 | Trp149 | −13.10 | −0.47 |
| AK4 |
Tyr93 (2.15) |
- | Trp149 | Ser148, Trp149 | −13.20 | −0.47 |
| (S)-QND8 | Tyr93 (2.06), Asp164 (1.49), Ser166 (2.85) |
- | Trp55 | - | −11.18 | −0.56 |
| (S)-T2 | - | Tyr55 | Trp149, Tyr195 | Ser148, Trp149 | −9.99 | −0.50 |
The hydrogen bonds were analyzed and measured by AutoDock4.2, and the salt bridges, cation-π, and π-π interactions were analyzed by BIOVIA Discovery Studio Visualized. The amino acid residues’ interaction with protonated quinuclidine is presented in bold. * LE is the ratio of Gibbs free energy (ΔG) to the number of non-hydrogen atoms of the ligand.
In terms of the structure-activity relationship (SAR) of quinuclidine-triazole derivatives targeting α3β4 nAChR, three components are required for high affinity and selectivity: the (S)-enantiomer of a quinuclidine ring, a triazole ring and a large hydrophobic group (extended benzene ring). Among four synthesized compounds, AK3 showed the highest binding affinity and selectivity to α3β4 nAChR, qualifying this compound as a non-radioactive reference compound for α3β4 nAChR. Therefore, AK3 is a high potential candidate for further development as α3β4 nAChR PET tracer for monitoring drug addiction, after replacing the fluorine atom with the radioactive isotope, fluorine-18.
3. Materials and Methods
3.1. Synthesis
All chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA), Merck (Darmstadt, Germany), AK Scientific (Union City, CA, USA), Oakwood (Estill, SC, USA), and Fisher Scientific (Waltham, MA, USA) and used without further purification. The NMR spectroscopic data (1H, 13C, COSY, HSQC, HMBC) were recorded with a Varian Mercury-300. High-Resolution Mass Spectra (HRMS) were recorded on the FT-ICR APEX II spectrometer using electrospray ionization (ESI) in positive ion mode.
AK1-AK4 were synthesized by the previously described methods [47]. In brief, the terminal alkyne and quinuclidine azide were prepared first. For the alkyne building block, 2-fluoro-4-iodophenol reacted with trimethylsilyl acetylene in base condition using PdCl2(PPh3)2 and CuI as catalysts under nitrogen atmosphere overnight and purified by SiO2 column chromatography using 15%EtOAc in hexane as a mobile phase. The intermediate compound was desilylated with TBAF in THF at room temperature and purified by SiO2 column chromatography using 15%EtOAc in hexane as a mobile phase to get the terminal alkyne (Scheme 1) for AK1 and AK2. For terminal alkyne of AK3 and AK4, 2-fluoro-4-iodophenol first reacted with benzyl bromide in base condition at room temperature overnight (Scheme 2) before performing a Sonogashira cross-coupling reaction. The crude product was purified by SiO2 column chromatography using 5%Et3N, 10%MeOH in DCM as a mobile phase.
Scheme 1.

Sonogashira cross-coupling reaction and desilylation with TBAF in THF.
Scheme 2.

Nucleophilic substitution to prepare iodo-containing molecules.
For the preparation of (R)- and (S)-quinuclidine azides, the reaction of trifluoromethanesulfonic anhydride and sodium azide was run in the mixture of water and toluene (1:1.5) at 0 °C for 2 h and the reaction mixture was extracted with toluene to yield trifluoromethanesulfonyl azide (TfN3). The freshly prepared TfN3 in toluene was added to (R)- or (S)- of 3-aminoquinuclidine, K2CO3, and CuSO4.5H2O in the mixture of water and methanol (1:2) at room temperature overnight (Scheme 3). The prepared azide building blocks were then used without further purification.
Scheme 3.

Azido quinuclidine preparation. * Indicates (R)- or (S)-enantiomer.
The prepared terminal alkynes and quinuclidine azides reacted via copper-catalyzed azide-alkyne cycloaddition (CuAAC) or click chemistry using 20 mol% of sodium ascorbate and 5 mol% of copper sulfate as catalysts to yield AK1-AK4 as quinuclidine triazole compounds (Scheme 4).
Scheme 4.

Copper-catalyzed azide-alkyne cycloaddition (CuAAC). * Indicates (R)- or (S)-enantiomer.
2-Fluoro-4-(1-((1R,3S,4R)-quinuclidin-3-yl)-1H-1,2,3-triazol-4-yl)phenol [AK1]
(S)-3-Azidoquinuclidine (0.135 g, 0.91 mmol) reacted with 4-ethynyl-2-fluorophenol (0.170 g, 1.29 mmol) using 20 mol% of sodium ascorbate and 5 mol% of copper sulfate as catalysts to yield a white solid (70.90 mg, 71.43%). Rf = 0.25, MP: 258 °C (dec); FTIR (ATR) (cm−1): 3384 (O-H stretching), 2943, 2878 (aliphatic C-H stretching), 1619, 1566 (aromatic C=C stretching), 1459 (aliphatic C-H bending), 1293 (aromatic C-N stretching), 1200 (aliphatic C-N stretching), 1049 (aliphatic C-O stretching), 874, 781, (C=C bending); 1H NMR (300 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.62 (dd, J = 12.4, 1.9 Hz, 1H), 7.52 (d, J = 7.90 Hz, 1H), 7.02 (m, 1H), 4.75 (m, 1H), 3.35 (m, 2H), 2.97 (m, 1H), 2.80 (m, 3H), 2.17 (m, 1H), 1.73 (m, 2H), 1.41 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 153.24, 150.23, 145.95, 145.01, 122.04, 120.61, 118.58, 113.35, 58.22, 52.10, 46.94, 46.78, 27.96, 25.30, 19.82; HRMS (ESI) calculated (C15H18FN4O, MH+): 289.1459, found 289.1455.
2-Fluoro-4-(1-((1S,3R,4S)-quinuclidin-3-yl)-1H-1,2,3-triazol-4-yl)phenol [AK2]
(R)-3-Azidoquinuclidine (0.275 g, 1.81 mmol) reacted with 4-ethynyl-2-fluorophenol (0.351 g, 2.58 mmol) using 20 mol% of sodium ascorbate and 5 mol% of copper sulfate as catalysts to yield a white solid (143.2 mg, 27.62%). Rf = 0.25, MP: 275 °C (dec); FTIR (ATR) (cm−1): 3128 (O-H stretching), 2948, 2870 (aliphatic C-H stretching), 1619, 1563 (aromatic C=C stretching), 1462 (aliphatic C-H bending), 1293 (aromatic C-N stretching), 1200 (aliphatic C-N stretching), 1035 (aliphatic C-O stretching), 883, 781, (C=C bending); 1H NMR (300 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.61 (dd, J = 12.4, 1.9 Hz, 1H), 7.53 (d, J = 7.90 Hz, 1H), 7.01 (m, 1H), 4.72 (m, 1H), 3.31 (m, 2H), 2.96 (m, 1H), 2.76 (m, 3H), 2.15 (m, 1H), 1.71 (m, 2H), 1.38 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 153.25, 150.07, 145.95, 145.20, 121.95, 120.56, 118.61, 113.55, 57.81, 52.35, 47.03, 46.79, 28.12, 25.79, 20.08; HRMS (ESI) calculated (C15H18FN4O, MH+): 289.1459, found: 289.1457.
(1R,3S,4R)-3-(4-(4-(Benzyloxy)-3-fluorophenyl)-1H-1,2,3-triazol-1-yl)quinuclidine [AK3]
(S)-3-Azidoquinuclidine (0.090 g, 0.60 mmol) reacted with 1-(benzyloxy)-4-ethynyl-2-fluorobenzene (0.120 g, 0.53 mmol) using 20 mol% of sodium ascorbate and 5 mol% of copper sulfate as catalysts to yield a white solid (271.5 mg, 62.07%). Rf = 0.625, MP:173–175 °C; FTIR (ATR) (cm−1): 3100, 3020 (aromatic C-H stretching), 2934, 2867 (aliphatic C-H stretching), 1630, 1585, 1510 (aromatic C=C stretching), 1451 (aliphatic C-H bending), 1380 (aromatic C-N stretching), 1274 (aliphatic C-N stretching), 1133, 1026 (aliphatic C-O stretching), 880, 793 (C=C bending); 1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.72 (dd, J = 12.6, 1.5 Hz, 1H), 7.65 (d, J = 8.6 Hz, 1H), 7.47 (m, 1H), 7.47 (m, 1H), 7.42 (m, 2H), 7.35 (m, 2H), 5.22 (s, 1H), 4.76 (m, 1H), 3.61 (m, 2H), 2.94 (m, 2H), 2.21 (m, 1H), 1.77 (m, 2H), 1.44 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 153.63, 150.40, 145.78, 145.64, 136.48, 128.54, 128.14, 127.93, 121.34, 120.61, 115.91, 112.73, 70.31, 57.40, 51.95, 46.60, 27.65, 25.31, 19.60; HRMS (ESI) calculated (C22H23FN4O, MH+): 379.1929, found: 379.1925.
(1S,3R,4S)-3-(4-(4-(Benzyloxy)-3-fluorophenyl)-1H-1,2,3-triazol-1-yl)quinuclidine [AK4]
(R)-3-Azidoquinuclidine (0.234 g, 1.54 mmol) reacted with 1-(benzyloxy)-4-ethynyl-2-fluorobenzene (0.349 g, 1.54 mmol) using 20 mol% of sodium ascorbate and 5 mol% of copper sulfate as catalysts to yield a white solid (285.1 mg, 73.53%). Rf = 0.625, MP:180–181 °C; FTIR (ATR) (cm−1): 3100, 3040 (aromatic C-H stretching), 2937, 2864 (aliphatic C-H stretching), 1630, 1512 (aromatic C=C stretching), 1456 (aliphatic C-H bending), 1380 (aromatic C-N stretching), 1276, 1223 (aliphatic C-N stretching), 1127, 1018 (aliphatic C-O stretching), 883, 740 (C=C bending); 1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.71 (dd, J = 12.8, 1.9 Hz, 1H), 7.65 (d, J = 8.65, 1H), 7.49 (m, 1H), 7.47 (m, 1H), 7.42 (m, 2H), 7.35 (m, 2H) 5.22 (s, 2H), 4.76 (1H), 3.61 (m, 2H), 2.94 (m, 4H), 2.21 (m, 1H), 1.77 (m, 2H), 1.44 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 153.63, 150.40, 145.78, 145.11, 136.48, 128.54, 128.13, 121.96, 120.99, 112.99, 112.73, 70.31, 57.44, 51.97, 46.61, 27.67, 25.35, 19.63; HRMS (ESI) calculated (C22H23FN4O, MH+): 379.1929, found: 379.1927.
3.2. Binding Affinity
The binding affinities of quinuclidine triazole compounds AK1-AK4 to nAChRs were determined by radioligand displacement assays [46]. SH-SY5Y cells stably transfected with human α7 nAChR and HEK293 cells stably transfected with human α4β2 nAChR or α3β4 nAChR were used in the experiments. Cells were collected, sedimented (800 rpm, 3 min), diluted with 50 mM TRIS-HCl, pH 7.4, and stored at −25 °C until use. Frozen cell suspensions were thawed and homogenized by a 27-gauge needle and diluted with incubation buffer (50 mM TRIS-HCl, pH 7.4, 120 mM NaCl, and 5 mM KCl). The membrane suspension was incubated with (±)-[3H]epibatidine (0.3 to 0.6 nM final concentration; molar activity 2.22 GBq/mmol). Nonspecific binding was determined by co-incubation with 300 µM (-)-nicotine tartrate. The incubation was performed at room temperature for 120 min and terminated by rapid filtration using Whatman GF/B glass-fiber filters presoaked in 0.3% polyethyleneimine and a 48-channel harvester (Biomedical Research and Development Laboratories, Gaithersburg, MD, USA) followed by 4 times washing with ice-cold 50 mM TRIS-HCl, pH 7.4. Filter-bound radioactivity was quantified by liquid scintillation counting. The 50% inhibition concentrations (IC50) were estimated from the competition curves by nonlinear regression using GraphPad Prism software and the Ki values were calculated according to the Cheng-Prusoff equation [49].
3.3. Molecular Docking
The homology models of α3β4 and α7 nAChRs were prepared as described in our previous study [48]. Briefly, the human amino acid sequences of α3β4 and α7 nAChRs were downloaded from UniProt for searching a proper protein template from Protein Data Bank (PDB) by Blast protein in Chimera 1.10.2 (Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, CA, USA). The amino acid sequences of α3β4 nAChR or α7 nAChR and acetylcholine binding protein (AChBP) (PDB ID 5AFH) used as a template were aligned by Clustal Omega (Conway Institute, University College Dublin, Dublin, Ireland) and the homology model of α3β4 nAChR and α7 nAChR was generated by Modeller 9.15 (University of California San Francisco, San Francisco, CA, USA). Several parameters i.e., the Discrete Optimized Protein Energy (DOPE) score, the GA341 score, and the Ramachandran plot were used to evaluate model quality. The structures of AK1–AK4 were drawn as protonated forms by Chem Draw Ultra 12.0 program (PerkinElmer, Waltham, MA, USA). The parameters for the molecular docking study with AutoDock4.2 included: 100 GA runs, a population size of 150, a maximum of 10,000,000 evaluations, and a maximum of 27,000 generations. The similar 3D conformations orientation within 2.0 Å were grouped as conformation clusters. The docked poses in the highest cluster as well as free binding energies (∆G binding) and ligand efficiency (LE) were analyzed. The binding interactions between ligand and target protein were visually analyzed by AutoDock4.2 in addition to BIOVIA Discovery Studio Visualized (Biovia, San Diego, CA, USA).
3.4. Molecular Dynamics (MD) Simulation
The molecular docking complexes of AK3 to a homology model of α3β4 and α7 nAChRs were optimized by MD simulation using NAMD software (University of Illinois at Urbana-Champaign, Urbana, IL, USA) [50] with CHARMM force field [51]. The complexes were solvated in the TIP3P model water box. The charge of the system was neutralized with an appropriate number of counter ions. Initially, the water box was minimized by the conjugate gradient method. Before the MD simulation, the system was equilibrated for 200 ps using an NPT ensemble at 310 K and 1 atm which was controlled by the Nosé-Hoover Langevin piston method [52] with 2 fs time steps and SHAKE algorithm. Periodic boundary conditions (PBC) and Particle Mesh Ewald (PME) method [52] were used for calculation. In the production steps, 100 ns of MD simulations were performed with trajectories saving every 2 ns for analysis. The complexes’ stability was evaluated using root mean square deviation (RMSD) (Supplementary Figure S1). Finally, the complexes were analyzed by BIOVIA Discovery Studio 2020 (Biovia, San Diego, CA, USA) [53].
3.5. Statistical Analysis
Data are represented as mean derived from two independent experiments performed in triplicate. The mean differences were statistically analyzed by one-way ANOVA followed by Tukey’s multiple comparison test using GraphPad Prism software.
4. Conclusions
The structural modification of α3β4 nAChR ligands for the development of a PET tracer has been achieved. The newly designed quinuclidine-triazole derivative AK3 showed good binding affinity (Ki = 3.18 nM) and a significantly enhanced selectivity α7 nAChR (3069-fold). The structural features contributing to the significant improvement of affinity and selectivity profiles of AK3 are the (S)-enantiomer of the quinuclidine ring, the triazole linker, and the extended hydrophobic part for interaction with the β4 subunit. The presence of a fluorine atom in AK3 provides the opportunity to develop AK3 as an α3β4 nAChR-targeted PET tracer. Therefore, AK3 is a promising compound for further development as a drug-seeking behavior monitoring agent.
Acknowledgments
The authors acknowledge the National e-Science Infrastructure Consortium for providing computing resources that have contributed to the research results reported within this paper. In addition, the authors thank Tina Spalholz, and Helmholtz-Zentrum Dresden Rossendorf (HZDR) for the technical support with the radioligand binding assays.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24043614/s1.
Author Contributions
Conceptualization, J.S., O.V., and K.A.; Formal analysis, A.K., W.D.-C., S.C., O.V. and K.A.; Funding acquisition, A.K., J.S., O.V. and K.A.; Investigation, A.K., W.D.-C., S.C. and K.A.; Writing—original draft, A.K. and K.A.; Writing—review & editing, A.K., W.D.-C., S.C., J.S., C.C., O.V. and K.A. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data underlying the results are available as part of the article and no additional source data are required.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research is supported by National Cyclotron and PET Centre, Chulabhorn Hospital, Chulabhorn Royal Academy to A.K. and funded by the National Research Council of Thailand under Molecular Probes for Imaging Research Network (NRCT: N10A650046) to J.S., O.V., and K.A.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Dani J.A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015;124:3–19. doi: 10.1016/bs.irn.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dineley K.T., Pandya A.A., Yakel J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015;36:96–108. doi: 10.1016/j.tips.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gotti C., Clementi F., Fornari A., Gaimarri A., Guiducci S., Manfredi I., Moretti M., Pedrazzi P., Pucci L., Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 4.Roberts J.P., Stokoe S.A., Sathler M.F., Nichols R.A., Kim S. Selective coactivation of α7- and α4β2-nicotinic acetylcholine receptors reverses beta-amyloid-induced synaptic dysfunction. J. Biol. Chem. 2021;296:100402. doi: 10.1016/j.jbc.2021.100402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Letsinger A.C., Gu Z., Yakel J.L. α7 Nicotinic acetylcholine receptors in the hippocampal circuit: Taming complexity. Trends Neurosci. 2022;45:145–157. doi: 10.1016/j.tins.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Posadas I., López-Hernández B., Fau-Ceña V., Ceña V. Nicotinic receptors in neurodegeneration. Curr. Neuropharmacol. 2013;11:298–314. doi: 10.2174/1570159X11311030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyohara J., Hashimoto K. α7 Nicotinic receptor agonists: Potential therapeutic drugs for treatment of cognitive impairments in Schizophrenia and Alzheimer’s Disease. Open Med. Chem. J. 2010;4:37–56. doi: 10.2174/1874104501004010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dani J.A., Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 9.Wilens T.E., Decker M.W. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: Focus on cognition. Biochem. Pharmacol. 2007;74:1212–1223. doi: 10.1016/j.bcp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haydar N.S., Dunlop J. Neuronal nicotinic acetylcholine receptors—Targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top. Med. Chem. 2010;10:144–152. doi: 10.2174/156802610790410983. [DOI] [PubMed] [Google Scholar]
- 11.Glick S.D., Maisonneuve I.M., Dickinson H.A. 18-MC reduces methamphetamine and nicotine self-administration in rats. NeuroReport. 2000;11:2013–2015. doi: 10.1097/00001756-200006260-00041. [DOI] [PubMed] [Google Scholar]
- 12.Taraschenko O.D., Panchal V., Maisonneuve I.M., Glick S.D. Is antagonism of α3β4 nicotinic receptors a strategy to reduce morphine dependence? Eur. J. Pharmacol. 2005;513:207–218. doi: 10.1016/j.ejphar.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Wills L., Kenny P.J. Addiction-related neuroadaptations following chronic nicotine exposure. J. Neurochem. 2021;157:1652–1673. doi: 10.1111/jnc.15356. [DOI] [PubMed] [Google Scholar]
- 14.Wittenberg R.E., Wolfman S.L., De Biasi M., Dani J.A. Nicotinic acetylcholine receptors and nicotine addiction: A brief introduction. Neuropharmacology. 2020;177:108256. doi: 10.1016/j.neuropharm.2020.108256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenny P.J., Markou A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol. Biochem. Behav. 2001;70:531–549. doi: 10.1016/S0091-3057(01)00651-7. [DOI] [PubMed] [Google Scholar]
- 16.Picciotto M.R., Kenny P.J. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold. Spring. Harb. Perspect. Med. 2013;3:a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Castro-Neto A.G., Rameh-de-Albuquerque R.C., de Medeiros P.F.P., Uchôa R., Santos B.S. Chapter 49—Neuroscience of tobacco and crack cocaine use: Metabolism, effects, and symptomatology. In: Preedy V.R., editor. Neuroscience of Alcohol: Mechanisms and Treatment. Academic Press; London, UK: 2019. pp. 403–410. [Google Scholar]
- 18.Fowler C.D., Lu Q., Johnson P.M., Marks M.J., Kenny P.J. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elayouby K.S., Ishikawa M., Dukes A.J., Smith A.C.W., Lu Q., Fowler C.D., Kenny P.J. α3* Nicotinic acetylcholine receptors in the habenula-interpeduncular nucleus circuit regulate nicotine intake. J. Neurosci. 2021;41:1779–1787. doi: 10.1523/JNEUROSCI.0127-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saccone N.L., Wang J.C., Breslau N., Johnson E.O., Hatsukami D., Saccone S.F., Grucza R.A., Sun L., Duan W., Budde J., et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res. 2009;69:6848–6856. doi: 10.1158/0008-5472.CAN-09-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ware J.J., van den Bree M.B., Munafo M.R. Association of the CHRNA5-A3-B4 gene cluster with heaviness of smoking: A meta-analysis. Nicotine. Tob. Res. 2011;13:1167–1175. doi: 10.1093/ntr/ntr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spitz M.R., Amos C.I., Dong Q., Lin J., Wu X. The CHRNA5-A3 region on chromosome 15q24-25.1 is a risk factor both for nicotine dependence and for lung cancer. J. Natl. Cancer Instig. 2008;100:1552–1556. doi: 10.1093/jnci/djn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholze P., Huck S. The α5 nicotinic acetylcholine receptor subunit differentially modulates α4β2* and α3β4* receptors. Front. Synaptic. Neurosci. 2020;12:607959. doi: 10.3389/fnsyn.2020.607959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grady S.R., Moretti M., Zoli M., Marks M.J., Zanardi A., Pucci L., Clementi F., Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J. Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain R.J., Taraschenko O.D., Glick S.D. Effects of nicotine, methamphetamine and cocaine on extracellular levels of acetylcholine in the interpeduncular nucleus of rats. Neurosci. Lett. 2008;440:270–274. doi: 10.1016/j.neulet.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khroyan T.V., Yasuda D., Toll L., Polgar W.E., Zaveri N.T. High affinity α3β4 nicotinic acetylcholine receptor ligands AT-1001 and AT-1012 attenuate cocaine-induced conditioned place preference and behavioral sensitization in mice. Biochem. Pharmacol. 2015;97:531–541. doi: 10.1016/j.bcp.2015.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pang X., Liu L., Ngolab J., Zhao-Shea R., McIntosh J.M., Gardner P.D., Tapper A.R. Habenula cholinergic neurons regulate anxiety during nicotine withdrawal via nicotinic acetylcholine receptors. Neuropharmacology. 2016;107:294–304. doi: 10.1016/j.neuropharm.2016.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLaughlin I., Dani J.A., De Biasi M. Nicotine withdrawal. Curr. Top. Behav. Neurosci. 2015;24:99–123. doi: 10.1007/978-3-319-13482-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaughlin I., Dani J.A., De Biasi M. The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J. Neurochem. 2017;142((Suppl. S2)):130–143. doi: 10.1111/jnc.14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaveri N., Jiang F., Olsen C., Polgar W., Toll L. Novel α3β4 nicotinic acetylcholine receptor-selective ligands. Discovery, structure-activity studies, and pharmacological evaluation. J. Med. Chem. 2010;53:8187–8191. doi: 10.1021/jm1006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millar N.S., Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 32.Glick S., Sell E., Maisonneuve I. Brain regions mediating α3β4 nicotinic antagonist effects of 18-MC on methamphetamine and sucrose self-administration. Eur. J. Pharmacol. 2008;599:91–95. doi: 10.1016/j.ejphar.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez S., Bertolino M., Xiao Y., Pringle K.E., Caruso F., Kellar K. Dextromethorphan and its metabolite dextrorphan block α3β4 neuronal nicotinic receptors. J. Pharmacol. Exp. Ther. 2000;293:962–967. [PubMed] [Google Scholar]
- 34.Nickell J.R., Grinevich V.P., Siripurapu K.B., Smith A.M., Dwoskin L.P. Potential therapeutic uses of mecamylamine and its stereoisomers. Pharmacol. Biochem. Behav. 2013;108:28–43. doi: 10.1016/j.pbb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki S., Cohen S.M., Arnold L.L., Pennington K.L., Kato H., Naiki T., Naiki-Ito A., Yamashita Y., Takahashi S. Cotinine, a major nicotine metabolite, induces cell proliferation on urothelium in vitro and in vivo. Toxicol. 2020;429:152325. doi: 10.1016/j.tox.2019.152325. [DOI] [PubMed] [Google Scholar]
- 36.Sarasamkan J., Fischer S., Deuther-Conrad W., Ludwig F.A., Scheunemann M., Arunrungvichian K., Vajragupta O., Brust P. Radiosynthesis of (S)-[18F]T1: The first PET radioligand for molecular imaging of α3β4 nicotinic acetylcholine receptors. Appl. Radiat. Isot. 2017;124:106–113. doi: 10.1016/j.apradiso.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Cippitelli A., Wu J., Gaiolini K.A., Mercatelli D., Schoch J., Gorman M., Ramirez A., Ciccocioppo R., Khroyan T.V., Yasuda D., et al. AT-1001: A high-affinity α3β4 nAChR ligand with novel nicotine-suppressive pharmacology. Br. J. Pharmacol. 2015;172:1834–1845. doi: 10.1111/bph.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonio T., Childers S.R., Rothman R.B., Dersch C.M., King C., Kuehne M., Bornmann W.G., Eshleman A.J., Janowsky A., Simon E.R., et al. Effect of Iboga alkaloids on micro-opioid receptor-coupled G protein activation. PLoS. ONE. 2013;8:e77262. doi: 10.1371/journal.pone.0077262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arias H.R., Rosenberg A., Feuerbach D., Targowska-Duda K.M., Maciejewski R., Jozwiak K., Moaddel R., Glick S.D., Wainer I.W. Interaction of 18-methoxycoronaridine with nicotinic acetylcholine receptors in different conformational states. Biochim. Biophys. Acta. 2010;1798:1153–1163. doi: 10.1016/j.bbamem.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham C.S., Moerke M.J., McMahon L.R. Discriminative stimulus effects of mecamylamine and nicotine in rhesus monkeys: Central and peripheral mechanisms. Pharmacol. Biochem. Behav. 2019;179:27–33. doi: 10.1016/j.pbb.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stahl S.M. Dextromethorphan/Bupropion: A novel oral NMDA (N-methyl-d-aspartate) receptor antagonist with multimodal activity. CNS Spectr. 2019;24:461–466. doi: 10.1017/S1092852919001470. [DOI] [PubMed] [Google Scholar]
- 42.Wu J., Perry D.C., Bupp J.E., Jiang F., Polgar W.E., Toll L., Zaveri N.T. [125I]AT-1012, a new high affinity radioligand for the α3β4 nicotinic acetylcholine receptors. Neuropharmacology. 2014;77:193–199. doi: 10.1016/j.neuropharm.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Townsend D.W. Positron emission tomography/computed tomography. Semin. Nucl. Med. 2008;38:152–166. doi: 10.1053/j.semnuclmed.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Cherry S.R. Fundamentals of positron emission tomography and applications in preclinical drug development. J. Clin. Pharmacol. 2001;41:482–491. doi: 10.1177/00912700122010357. [DOI] [PubMed] [Google Scholar]
- 45.Korat S., Bidesi N.S.R., Bonanno F., Di Nanni A., Hoang A.N.N., Herfert K., Maurer A., Battisti U.M., Bowden G.D., Thonon D., et al. Alpha-synuclein PET tracer development-an overview about current efforts. Pharmaceuticals. 2021;14:847. doi: 10.3390/ph14090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarasamkan J., Scheunemann M., Apaijai N., Palee S., Parichatikanond W., Arunrungvichian K., Fischer S., Chattipakorn S., Deuther-Conrad W., Schuurmann G., et al. Varying chirality across nicotinic acetylcholine receptor subtypes: Selective binding of quinuclidine triazole compounds. ACS. Med. Chem. Lett. 2016;7:890–895. doi: 10.1021/acsmedchemlett.6b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arunrungvichian K., Fokin V.V., Vajragupta O., Taylor P. Selectivity optimization of substituted 1,2,3-triazoles as α7 nicotinic acetylcholine receptor agonists. ACS. Chem. Neurosci. 2015;6:1317–1330. doi: 10.1021/acschemneuro.5b00058. [DOI] [PubMed] [Google Scholar]
- 48.Arunrungvichian K., Chongruchiroj S., Sarasamkan J., Schuurmann G., Brust P., Vajragupta O. In silico finding of key interaction mediated α3β4 and α7 nicotinic acetylcholine receptor ligand selectivity of quinuclidine-triazole chemotype. Int. J. Mol. Sci. 2020;21:6189. doi: 10.3390/ijms21176189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newton P., Harrison P., Clulow S. A novel method for determination of the affinity of protein: Protein interactions in homogeneous assays. J. Biomol. Screen. 2008;13:674–682. doi: 10.1177/1087057108321086. [DOI] [PubMed] [Google Scholar]
- 50.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 53.BIOVIA. Dassault Systèmes . BIOVIA Discovery Studio. Dassault Systèmes; San Diego, CA, USA: 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data underlying the results are available as part of the article and no additional source data are required.