

Abstract
The epithelium of the urinary bladder must maintain a highly impermeable barrier despite large variations in urine volume during bladder filling and voiding. To study how the epithelium accommodates these volume changes, we mounted bladder tissue in modified Ussing chambers and subjected the tissue to mechanical stretch. Stretching the tissue for 5 h resulted in a 50% increase in lumenal surface area (from ∼2900 to 4300 μm2), exocytosis of a population of discoidal vesicles located in the apical cytoplasm of the superficial umbrella cells, and release of secretory proteins. Surprisingly, stretch also induced endocytosis of apical membrane and 100% of biotin-labeled membrane was internalized within 5 min after stretch. The endocytosed membrane was delivered to lysosomes and degraded by a leupeptin-sensitive pathway. Last, we show that the exocytic events were mediated, in part, by a cyclic adenosine monophosphate, protein kinase A-dependent process. Our results indicate that stretch modulates mucosal surface area by coordinating both exocytosis and endocytosis at the apical membrane of umbrella cells and provide insight into the mechanism of how mechanical forces regulate membrane traffic in nonexcitable cells.
INTRODUCTION
The plasma membrane forms the interface between the cell and its extracellular environment and in response to external stimuli coordinates changes in cellular function. The size and function of this organelle are regulated by the processes of endocytosis and exocytosis. Exocytosis is involved in the cell surface delivery and secretion of newly synthesized and recycled molecules, whereas endocytosis is required for the internalization and recovery of exocytosed membrane. In spite of the growing body of literature describing the effects of mechanical force on exocytosis (Lang et al., 1985; Dietz, 1988; Wirtz and Dobbs, 1990; Sadoshima and Izumo, 1993; Sadoshima et al., 1993; Xu et al., 1996; Steers et al., 1998; Clemow et al., 2000), much less is known about how mechanical stimuli affect endocytosis, although membrane tension may play an important role in this process (Fink and Cooper, 1996; Dai et al., 1997; Homann, 1998; Raucher and Sheetz, 1999; Apodaca, 2002; Morris and Homann, 2001).
One cell type that may provide additional insight into these phenomena is the specialized umbrella cell that lines the mucosal surface of the bladder and forms the barrier between the urine and the underlying tissue. This barrier must be maintained in spite of dramatic changes in lumenal urine volume during filling and voiding. The bladder accommodates these changes in large part by macroscopic and microscopic folding and unfolding of the mucosal surface. Furthermore, it is hypothesized that during filling the umbrella cells recruit a subapical pool of cytoplasmic vesicles (which are either fusiform or discoidal shaped) to fuse with the apical membrane, thereby increasing the mucosal surface area (Hicks, 1966; Porter et al., 1967). The evidence for vesicle insertion is largely based on two observations. First, a morphometric analysis showed that umbrella cells from full bladders contained significantly fewer vesicles than those from contracted bladders (Minsky and Chlapowski, 1978); and second, osmotic or mechanical stretch resulted in increased tissue capacitance, a measure of membrane surface area (Lewis and de Moura, 1982). However, a study that correlates stretch-activated changes in apical and/or basolateral surface area with changes in vesicle dynamics has yet to be performed. In addition, the underlying mechanism that regulates exocytosis in these cells is largely unexplored, although the cytoskeleton is likely to play a role in this process (Lewis and de Moura, 1982).
On voiding, the extra surface membrane is believed to be endocytosed, which replenishes the population of cytoplasmic vesicles, followed by a refolding of the umbrella cell membrane and mucosal surface in preparation for the next filling cycle (Porter et al., 1967; Minsky and Chlapowski, 1978). The actual role of endocytosis in these cells is unclear. The increases in capacitance observed after osmotic or hydrostatic stretch were partially reversed by removal of the stretch (Lewis and de Moura, 1984). In addition, some studies showed that when fluid phase markers were added to the bladder lumen, the markers were detected in vesicles after voiding (Hicks, 1966; Porter et al., 1967; Chang et al., 1994), consistent with reinternalization of membrane. However, the uptake of marker was modest, indicating that much of the vesicle membrane may not be derived from the apical surface. Other investigators failed to demonstrate uptake of horseradish peroxidase (HRP) or HRP-labeled lectin into vesicles during bladder contraction but instead observed marker in multivesicular bodies and lysosomes (Amano et al., 1991). Thus, the role that endocytosis plays in apical surface modulation of umbrella cells is currently not understood. Moreover, the effects of mechanical stimuli on endocytosis remain poorly characterized in this or other cell types (Apodaca, 2002).
Based on our results, we propose an alternative model of vesicle trafficking and surface area modulation in umbrella cells. We observed that initially upon stretch, there was little change in the apical or basolateral surface area. However, continued stretching induced large increases in apical surface area in combination with a decrease in vesicle surface area. The increase in apical surface area coincided with a significant increase in surface expression of uroplakin III, a membrane/vesicle protein, and secretion of 35S-labeled proteins. In contrast to the current model, we observed that stretch-induced vesicle fusion was accompanied by a simultaneous apical membrane endocytosis and that the internalized membrane was delivered to lysosomes. Last, we determined that the exocytic, but not the endocytic events were modulated, in part, by a cAMP, protein kinase A (PKA)-dependent mechanism. These data indicate that in addition to activating exocytosis, mechanical stimuli also induce endocytosis and the balance of these events governs the size of the umbrella cell apical plasma membrane.
MATERIALS AND METHODS
Materials
Unless specified otherwise, all chemicals were of reagent quality or better and were obtained from Sigma (St. Louis, MO).
Mechanical Stretch of Uroepithelium
Animal experiments were performed in accordance with the Animal Use and Care Committee of the University of Pittsburgh, Pittsburgh, PA. Urinary bladders were obtained from female New Zealand White rabbits (3–4 kg). Rabbits were euthanized with 300 mg of sodium pentobarbital, the bladder was exposed, the ends of the bladder were clamped with hemostats, an incision was made lengthwise along the bladder, and the opened bladder was surgically excised and washed in Krebs' solution (110 mM NaCl, 5.8 mM KCl, 25 mM NaHCO3, 1.2 mM KH2PO4, 2.0 mM CaCl2, 1.2 mM MgSO4, 11.1 mM glucose, pH 7.4). The bladder was trimmed of excess fat and placed on a rack (see Figure 1b in Lewis and Hanrahan, 1990), mucosal side down, in the same solution at 37°C bubbled with 95% air, 5% CO2 gas. The smooth muscle layers were removed by dissection with scissors and forceps. A plastic ring (containing a 2-cm2 opening) with sharp pins around the edges was coated with high vacuum grease (Dow Corning, Midland, MI), placed underneath the tissue, lifted up, and the tissue was pressed onto the pins with forceps to secure the piece onto the ring. Excess tissue surrounding the ring was trimmed with scissors and the ring (now with the tissue attached) was mounted between two halves of a modified Ussing chamber (Figure 1). Each hemichamber (apical and basolateral) was filled with 12.5 ml of Krebs' solution, the basolateral hemichamber was bubbled with gas, and the tissue was equilibrated for 30–60 min. Normally, each bladder yielded three rings of mounted tissue. Tissue capacitance and transepithelial resistance (TER) were monitored throughout the equilibration and only those preparations that exhibited a starting capacitance of ∼1.8–2.1 μF and a TER of >8000 cm2 were used.
Figure 1.

Modified Ussing chambers used to stretch bladder tissue. Electrode ports (A), Luer ports (B), plastic ring containing excised tissue (C), basal (serosal) chamber (D), apical (mucosal) chamber (E), and water jacket ports (F).
After equilibration, to induce stretch, additional Krebs' solution was added to the apical hemichamber to a volume of 14 ml (hemichamber capacity), and an additional 0.5 ml of Krebs' solution was injected by syringe to increase the back pressure in the apical hemichamber to 8 cm H2O as measured by a force transducer (AD Instruments, Mountain View, CA). In rabbit, 8 cm H2O is representative of a pressure normally observed during the extended storage phase of a bladder filling (Levin and Wein, 1982). When we compared this rapid filling with slower filling (0.1 ml/min), we observed no discernible difference in capacitance changes and therefore used the rapid filling technique to simplify our experiments. Capacitance and TER were measured throughout the experiment.
Capacitance Measurement
Capacitance was measured as a means of estimating surface area (where 1 μF ≅ 1 cm2 of actual membrane area) as described previously (Lewis and Diamond, 1976; Lewis and de Moura, 1982). Because the apical membrane of the umbrella cell is the major site of resistance, changes in capacitance primarily reflect changes in apical cell surface area (Lewis and Diamond, 1976; Lewis and de Moura, 1982). To measure capacitance, a square current pulse of 1 μA, generated by a MacLab 8s A/D convertor (AD Instruments, Victoria, Australia) interfaced with a 400-MHz PowerPC G3 Macintosh computer (Apple, Cupertino, CA) and a VCC MC6 current/voltage clamp (Physiological Instruments, San Diego, CA), was applied across the tissue for 200 ms following a 25-ms delay. The voltage response of the tissue was digitized and then recorded every 100 μs by using the Scope program (AD Instruments). An average of eight sweeps was recorded for each current pulse. The time constant, τ, was determined by calculating the length of time required to reach 63% of the steady-state voltage by using a data transformation routine that included curve fitting the voltage response to a single exponential. The R values for curve fitting were >0.99 under all conditions. The capacitance was determined using the formula C = τ/R, where C is the capacitance and R is the resistance. Resistance was determined by dividing the amplitude of the steady-state voltage response by the amplitude of the square current pulse. Because the area of the tissue mounted on the ring is 2 cm2, a capacitance of ∼2 μF ensured that the tissue was fairly smooth and free of large folds. This was confirmed by transmission electron microscopy (TEM) and scanning electron microscopy.
TEM
Tissue was fixed by isovolumetric exchange of Krebs' buffer with 100 mM cacodylate buffer containing 1.5% (vol/vol) glutaraldehyde, 2% (wt/vol) paraformaldehyde, 1 mM CaCl2, 0.5 mM MgCl2 for 1–2 h at room temperature. When relevant, 0.01% (wt/vol) ruthenium red was included with the fixative. The samples were then osmicated 1–2 h with 1.5% (wt/vol) OsO4 in 100 mM cacodylate, washed several times with distilled water, and then block stained overnight in 0.5% (wt/vol) aqueous uranyl acetate. Cells were dehydrated in a graded series of ethanol, embedded in the epoxy resin LX-112 (Ladd, Burlington, VT), and sectioned with a diamond knife (Diatome, Fort Washington, PA). Sections, silver in color, were mounted on butvar-coated nickel grids (slotted grids for serial sectioning), contrasted with uranyl acetate and lead citrate, and viewed at 80 kV in a JEOL (Tokyo, Japan) 100 CX electron microscope.
Stereological Analysis
Epon blocks were sectioned perpendicular to the length of the epithelium to obtain vertical sections necessary to determine mean cell volume, apical and basolateral surface areas, and discoidal vesicle volume and surface area.
Mean Cell Volume.
The mean umbrella cell volume was estimated in either control or stretched cells by taking a minimum of 50 photographs at 1400× of randomly chosen umbrella cells, printing an 8- × 10-inch image of each cell, and overlaying a cycloid lattice grid on the image (Griffiths, 1993). When necessary, more than one photograph was taken to include the entire cell and the photographs were overlapped as a montage. The average volume fraction of the nucleus [V(nuc)] was determined by point counting and calculating the ratio of counts on the nucleus [P(nuc)] relative to counts on the cell [P(cell)] as described by the following formula (Griffiths, 1993):
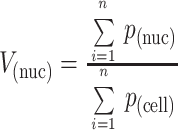 |
In a separate experiment, optical sections through the umbrella cell nuclei were taken by confocal microscopy, and the images were projected on to one another to determine the average diameter of the nucleus. The average nuclear volume was calculated using the formula 4/3πr3. Rabbit umbrella cells usually contain one nucleus per cell and the nuclei are generally spherical. The mean cell volume was calculated by dividing the volume of the nucleus by V(nuc) (Griffiths, 1993).
Membrane Surface Areas.
The apical and basolateral membrane surface areas of the umbrella cells were determined by overlaying a cycloid lattice grid as described above on the 8- × 10-inch images. Intersections of the cycloids with the apical or basolateral surface domains along with the number of points on the cell were counted. The surface density (Sv) was calculated using the following formula (Baddeley et al., 1986):
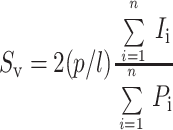 |
where (p/l) is the ratio of test points to test curve length and is a function of the cycloid grid, which in this case was 1/4.59 μm. For discoidal vesicle surface area, photographs were taken at 3600× and overlapping 8- × 10-inch images of the umbrella cells were used to determine Sv, which was calculated by counting the intersections of the cycloids with the vesicle membrane domains relative to the points on the cell. The cycloid grid used for this purpose had a p/l ratio of 1/0.87 μm. Surface area was determined by multiplying the membrane surface density by the volume of the cell (Baddeley et al., 1986).
Error Analysis.
The SE of the estimated values was determined by the bootstrap method (Efron and Tibshirani, 1993). Briefly, 50 individual geometric parameters (volume or surface area) were obtained for umbrella cells analyzed after point/intersection counting with the cycloid grid as described above. The values were recorded and copied into the Microsoft Excel computer software program (Microsoft, Redmond, WA) as a workbook. By using the Resampling Stats Excel add-in (available at www.resample.com), the data were resampled, with replacement, to obtain a pseudosample of data. The parameter was estimated from the pseudodata and the value was recorded in a separate column. This process was repeated 50 times to generate a column of 50 bootstrapped parameter estimates. A new mean (Xboot) and variance (s2boot) were determined for the bootstrapped values and the SE of the original estimated mean (Xorig) was determined by the following formula:
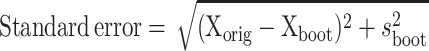 |
Statistical significance was determined by Student's t test, p < 0.05
Immunogold Labeling of Ultrathin Cryosections
Urinary bladders were isolated and stripped of muscle tissue as described above. After the experiment cells were fixed 45 min at room temperature with 0.1% (vol/vol) glutaraldehyde and 2% (wt/vol) paraformaldehyde in 100 mM cacodylate buffer containing 0.5 mM MgCl2 + 1 mM CaCl2, and cut into 0.5–1.0-mm2 cubes. The cubes were incubated 30 min at 37°C in 3% (wt/vol) gelatin and then overnight at 4°C in 1.3 M sucrose and 20% (wt/vol) polyvinylpyrrolidone (Mr 10,000). The cubes were mounted on cryo-stubs and frozen in liquid nitrogen. Cryosectioning was performed at −110°C in a Leica (Deerfield, IL) Ultracut E ultramicrotome with an FCS cryochamber attachment. The cryosections were labeled with culture supernatant from the mouse hybridoma cell line K8B12, which produces anti-uroplakin III antibody (Truschel et al., 1999) and subsequently with goat anti-mouse IgG Fc 5-nm gold (Amersham Biosciences, Piscataway, NJ) as described previously (Altschuler et al., 2000). Sections were viewed at 80–100 kV in a Jeol 100CX electron microscope (Peabody, MA).
Biotinylation of Apical Membrane Proteins
The tissue was mounted and equilibrated as described and then stretched or incubated in the chamber for the indicated amount of time. The tissue was placed on ice and the apical medium was replaced with 10 ml of ice-cold Krebs' solution containing 1 mg/ml sulfo-N-hydroxysuccinimide-disulfo-biotin (Pierce Chemical, Rockford, IL) and incubated for 15 min to biotinylate apical membrane proteins. After incubation, the biotin/Krebs solution was aspirated and fresh biotin in Krebs' was added for another 15 min on ice. The reaction was quenched by exchanging the apical solution with ice-cold minimal essential medium (MEM) (Invitrogen, Carlsbad, CA) + 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and washing 3 × 15 min.
Measurement of Uroplakin III at Cell Surface
After biotinylation, the chamber was disassembled and the epithelial cells were gently scraped and placed in 0.15 ml of 0.5% SDS-lysis buffer (100 mM NaCl, 50 mM tetraethylammonium, 5 mM EDTA, and 0.5% SDS) containing protease inhibitor cocktail (5 μg/ml leupeptin, 5 μg/ml antipain, 5 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride). The samples were boiled for 5 min and then shaken in a vortex mixer (Eppendorf - 543α mixer, Boulder, CO) for 15 min at 4°C. Protein lysate (25 μg) in 500 μl of mixed micelle buffer (150 mM NaCl, 20 mM triethanolamine, 5 mM EDTA, 110 mM sucrose, 1% Triton X-100, and 0.2% SDS) was incubated overnight at 4°C on a rotator with 80 μl of a 50% slurry of streptavidin beads (Pierce Chemical) washed before use three times with mixed micelle buffer and one time with final wash buffer (150 mM NaCl, 20 mM triethanolamine pH 8.1, and 5 mM EDTA). After overnight incubation, the beads were isolated by centrifugation at 14,000 rpm for 10 s, washed three times with mixed micelle buffer and once with final wash buffer. Biotinylated proteins bound to the streptavidin beads were separated by boiling for 5 min in Laemmli's sample buffer containing 100 mM dithiothreitol. An aliquot of the starting lysate (2.5 or 1.25 μg) was similarly boiled in sample buffer. The proteins were resolved on a 15% SDS-polyacrylamide gel and were transferred to Immobilon-P membranes (Millipore, Bedford, MA) for 75 min at 375-mA constant current. The membrane was blocked overnight in 5% (wt/vol) bovine serum albumin dissolved in phosphate-buffered saline (PBS). The blot was incubated for 2 h at room temperature with rotation with K8B12 supernatant diluted 1:1000 in PBS containing 1% dehydrated nonfat milk. The membrane was then washed with Tris-buffered saline-Tween (2.68 mM KCl, 0.5 M NaCl, 25 mM Tris-HCl pH 8.0, and 0.05% [vol/vol] Tween 20) 2 × 15 min and then 1 × 60 min and incubated in goat anti-mouse antibodies conjugated to HRP (Jackson Immunoresearch Laboratories, West Grove, PA) diluted 1:25,000 in PBS-milk for 1 h with rotation. The membrane was washed 2 × 15 min and 1 × 60 min in Tris-buffered saline-Tween, incubated for 1 min in Super Signal ECL reagent (Pierce Chemical), and exposed to XAR-5 film (Eastman Kodak, Rochester, NY). The bands were quantified by densitometry using the Molecular Analyst Program (Bio-Rad, Hercules, CA).
Apical Secretion
The tissue was mounted and equilibrated as described and then washed once with PBS (containing 2.0 mM Ca2+ and 1.0 mM Mg2+) and incubated for 30 min in cysteine/methionine-deficient MEM to starve the cells. The basolateral media were removed, the chamber was placed on its side (apical hemichamber down), and [35S]cysteine/methionine express labeling mix (taken from a 10-mCi/ml stock; PerkinElmer Life Sciences, Boston, MA) diluted in cysteine/methiomide deficient MEM to a final concentration of 0.25 mCi/ml in a volume of 267 μl was added to the basal side of the tissue for 30 min to metabolically label the cells. The tissue was washed three times with Krebs' solution and then chased for 60 min in Krebs' solution. After the chase, 500-μl aliquots were taken from the basolateral and apical media to determine background radioactivity; 500 μl of Krebs' solution was added back to each hemichamber; and then the tissue was stretched for 5, 15, 120, or 300 min or incubated without stretch as a control. At the indicated times, 500-μl aliquots were taken from the apical and basolateral hemichambers and spotted on 1.5-in2 pieces of filter paper (Whatman, Maidstone, England) and incubated at room temperature until dry. The pieces were placed in ice-cold 10% (vol/vol) trichloroacetic acid to precipitate secretory proteins, incubated with shaking at 4°C for 30 min, washed 3 × 5 min with ice-cold 70% (vol/vol) ethanol at 4°C, and then incubated at room temperature until dry. The pieces were placed in 6-ml scintillation vials, the vials were filled with 5 ml of Scintisafe scintillation fluid (Fisher Scientific, Pittsburgh, PA), and the radioactivity was determined by counting in a scintillation counter (Wallac, Turku, Finland).
Diffusion of Urea across Uroepithelium
The tissue was mounted and equilibrated as described and the basolateral or apical chamber was incubated with 167 μCi of [14C]urea (taken from a 5-mCi/ml stock diluted in H2O; American Radiolabeled Chemicals, St. Louis, MO). The tissue was then stretched or incubated without stretch as a control. Duplicate 30-μl aliquots were taken from the basolateral and apical hemichambers at 0, 15, 120, or 300 min and 60 μl of Krebs' solution was replaced to maintain a constant buffer volume. The aliquots were mixed with 10 ml of scintillation fluid and the samples were counted in a liquid scintillation counter. The amount of urea that crossed the uroepithelium was expressed as a percentage of the number of counts in the apical medium (or basolateral medium when added apically) divided by the total number of counts in the apical and basolateral media at each time point.
Measurement of Endocytosis
Tissue was mounted, equilibrated, and then biotinylated as described above. After biotinylation, the tissue was warmed to 37°C in Krebs' solution and stretched or incubated without stretch for the indicated amount of time to allow for internalization of the biotinylated proteins. After incubation, the chambers were placed on ice to stop all membrane trafficking. Cell-surface biotin was removed by incubating the tissue for 30 min at 4°C with 17 mg/ml 2-mercaptoethanesulfonic acid (MESNA) in 50 mM Tris pH 8.6, 100 mM NaCl, 0.2% bovine serum albumin, and 1 mM EDTA. The buffer was aspirated and the reaction was quenched for 10 min at 4°C with 25 mg/ml iodoacetic acid diluted in PBS. The iodoacetic acid was removed and the tissue was washed two times with ice-cold Krebs' buffer. The chamber was disassembled and the apical surface was gently scraped to remove the uroepithelium. The scraped cells were placed in 150 μl of 0.5% SDS-lysis buffer containing a protease inhibitor cocktail (described above). The samples were boiled for 5 min and then vortex shaken for 15 min at 4°C. Protein lysate (5 μg) was mixed with sample buffer lacking reducing agent and the proteins were resolved on 15% SDS-polyacrylamide gels and transferred to Immobilon-P as described above. To measure endocytosis, the biotinylated proteins were detected by incubating the blots with 0.1 μg/ml streptavidin-HRP (Jackson Immunoresearch Laboratories) in PBS containing 1% (wt/vol) dehydrated nonfat milk for 2 h at room temperature with rotation. The blots were washed and the bands were visualized and quantified by densitometry as described above. The amount of endocytosis was calculated as the quotient of the signal intensity of the MESNA-treated lane and the untreated lane for each time point.
Wheat Germ Agglutinin-Fluorescein Isothiocyanate (WGA-FITC) Internalization
The tissue was mounted and equilibrated as described and then cooled to 4°C. The apical compartment was incubated with 25 μg/ml WGA-FITC (Vector Laboratories, Burlingame, CA) in Krebs' solution, and the tissue was warmed and incubated for 15 min with or without stretch. The tissue was then placed on ice and the cell surface WGA-FITC was removed by washing 3 × 30 min with 50 mM N-acetyl glucosamine in Krebs' buffer on a shaker at 4°C. The tissue was then fixed, stained with 4,6-diamidino-2-phenylindole (Molecular Probes, Eugene, OR), and mounted as described (Apodaca et al., 1994). Samples were viewed in a TE300 Nikon (Melville, NY) microscope equipped for epifluorescence and outfitted with a Ludl (Electronic Products, Hawthorne, NY) Z-focus controller. The images were collected and processed by digital deconvolution with Open Lab software (Improvision, Coventry, England). Alternatively, the tissue was stretched for 2 or 5 h, the pressure was released, and WGA-FITC was added to the apical chamber. The tissue was then restretched for 15 min, cooled, and washed with N-acetyl glucosamine at 4°C followed by fixation and mounting as described above. To observe endocytosis in whole bladders, excised bladders were washed with Krebs' buffer as described above but otherwise left intact. The contracted bladders were incubated in Krebs' buffer (gassed with 95% air, 5% CO2) for 60 min and then attached to a 60-ml syringe by using ligatures. The bladders were filled with 30 ml of Krebs' buffer containing WGA-FITC and then incubated for 15 min at 37°C. The bladders were rapidly chilled to 4°C, surface-bound WGA-FITC was removed by treatment with N-acetyl glucosamine at 4°C, and the tissue was fixed as described above. Imaging for the latter two experiments was performed on a TCS confocal microscope equipped with krypton, argon, and helium-neon lasers (Leica). Images were acquired using a 40× plan-apochromat objective (zoom of 2) and the appropriate filter combination. The images (1024 × 1024 pixels) were saved in a tag information file format and the contrast levels of the images adjusted in the Photoshop program (Adobe Photoshop, Mountain View, CA) on a Power PC G-4 Macintosh (Apple). The contrast-corrected images were imported into Freehand (Macromedia, San Francisco, CA) and printed from a Kodak 8650PS dye sublimation printer.
Measurement of Intracellular Levels of cAMP
Bladder tissue was mounted and was incubated for 15–300 min in the presence or absence of stretch. After the designated time point, the tissue was removed from the chamber and the mucosal surface was gently scraped with a cell scraper (number 83.1830; Sarstedt, Newton, NC) to selectively remove the epithelial cells. Intracellular concentrations of cAMP in the epithelial cells were determined using a Biotrak kit (RPA 538; Amersham Biosciences) according to the manufacturer's instructions.
RESULTS
Stretch-regulated Changes in Umbrella Cell Apical Surface Area
It has been argued that umbrella cells accommodate increased urine volume by simply unfolding their surface membranes (Koss, 1969; Staehelin et al., 1972). Therefore, our first goal was to determine whether stretch resulted in changes in the apical surface area of umbrella cells or simply caused unfolding of the membrane. To mimic the extended storage phase of bladder filling we chose a method of stretch that exposed the uroepithelium to a constant physiological pressure over relatively long periods of time. In rabbits this phase lasts ∼5 h (Levin et al., 1993) and the lumenal pressure (∼8 cm H2O) remains relatively constant (Levin and Wein, 1982).
Rabbit uroepithelium, which is comprised of umbrella, intermediate, and basal cell layers was stripped of underlying muscle tissue and mounted in modified Ussing chambers. The muscle tissue was removed as previously described to prevent unwanted contraction and ease of tissue manipulation (Lewis and Hanrahan, 1990). After a period of equilibration, hydrostatic force was applied to the mucosal chamber until a pressure of 8 cm H2O was generated and then maintained throughout the duration of the experiment (see MATERIALS AND METHODS). To confirm that the tissue remained intact and retained its barrier function, TER was monitored throughout the experiment. The initial TER (11,396 ± 760 Ωcm2) dipped after stretch, but rapidly recovered and at the end of the 5-h stretch TER was 14,222 ± 1,062 Ωcm2. The initial fall in TER likely reflects increased ion conductance because the short-circuit current significantly increased upon stretch and then subsided (our unpublished data).
Changes in surface area were measured in real time by monitoring tissue capacitance. This electrophysiological parameter is directly proportional to surface area (1 μF ≅ 1 cm2) and in bladder tissue is thought to primarily measure changes in the surface area of the apical plasma membrane of the umbrella cells (Lewis and Diamond, 1976). If surface unfolding was sufficient to accommodate the stretch response, no changes in capacitance were expected. However, when tissue was stretched the capacitance rapidly rose ∼15% within the first 20 min followed by a slower increase to 50% after 5 h of stretch (Figure 2). This biphasic response in capacitance was similar to earlier studies that used osmotic stretch (Lewis and de Moura, 1982; Lewis and de Moura, 1984). Stretching the tissue for longer than 5 h did not significantly increase the capacitance above 50% (our unpublished data).
Figure 2.

Capacitance changes in response to stretch. Bladder tissue was mounted and equilibrated as described in MATERIALS AND METHODS. The change in capacitance of stretched cells (▪) or unstretched control cells (♦) is shown. Data is mean ± SEM (n = 5). The error bars are smaller than the symbols.
As a control, the tissue was incubated in the chamber without the addition of hydrostatic pressure. Throughout the 5-h period of incubation the capacitance of the control tissue was unchanged, confirming that incubation in the chamber was not sufficient to elicit changes in surface area (Figure 2). Tissue pretreated with cycloheximide exhibited an initial rise in capacitance of ∼25% that remained unchanged for the 5-h period of stretch (our unpublished data), indicating that protein synthesis was important in the later stages of stretch. In summary, the data indicated that our method of stretch was effective for increasing apical surface area of umbrella cells and were inconsistent with the hypothesis that surface unfolding is the only mechanism responsible for accommodating increased urine volume (Koss, 1969; Staehelin et al., 1972).
Morphological Changes in Stretched Uroepithelium
The morphological changes that accompanied stretch were also examined. Tissue was mounted in the chambers and stretched for either 20 min or 5 h, or was incubated without stretch as a control. The tissue was subsequently fixed and examined by TEM. In control tissue (unstretched), the umbrella cells were roughly cuboidal and often exhibited portions of cytoplasm that extended downward into the underlying intermediate cell layer (Figure 3A). This morphology is typical of umbrella cells observed in contracted bladders (Hicks, 1975). Both the apical and the basolateral surfaces exhibited some degree of folding, although there was considerable variation among the cells. When viewed at higher magnification, numerous discoidal vesicles were commonly observed throughout the cytoplasm but appeared to be concentrated in the apical region of the cell just beneath the plasma membrane (Figure 3B). There were no observable differences between cells incubated without stretch for 20 min or 5 h.
Figure 3.

Morphological analysis of control and stretched umbrella cells. Cells were mounted in chambers and incubated for 5 h without stretch (A and B), or stretched for 20 min (C and D) or 5 h (E and F), fixed, and processed for TEM. (A, C, and E) Low-magnification views of representative umbrella cells from each experimental condition. The arrows delineate the borders of the cells. (B, D, and F) Higher magnification views of the apical portion of each umbrella cell. Representative discoidal vesicles are marked by arrowheads.
When the cells were subjected to stretch for 20 min, we observed very few morphological changes relative to controls. The majority of the cells retained a cuboidal-like shape and exhibited a similar degree of apical and basolateral surface folding (Figure 3C). Similar to controls, higher magnification images of these cells revealed an abundance of discoidal vesicles present in the cytoplasm just below the apical membrane (Figure 3D), although the relative abundance of vesicles localized in the center of the cell appeared to be diminished. This change in distribution may represent a shuttling of vesicles to the apical membrane during the initial period of stretch to prime the vesicles for fusion.
In contrast to control cells, or to those stretched for 20 min, umbrella cells subjected to 5 h of stretch underwent dramatic changes in morphology (Figure 3E). The depth of the cell was drastically reduced with a concomitant increase in cell length. In addition to these changes, the apical and basal surfaces were flattened and retained only a small degree of membrane folding. Some cells exhibited an exaggerated folding of the lateral membrane (Figure 3F), which is also observed in expanded bladders (Minsky and Chlapowski, 1978). Another striking feature of the 5-h stretch samples was the paucity of discoidal vesicles in the cytoplasm (Figure 3F). This morphological change in the uroepithelium after 5 h of stretch was consistent with an earlier study that examined umbrella cells in expanded bladders (Minsky and Chlapowski, 1978). These data not only confirmed the physiological nature of our system but also demonstrated that the stretch-induced morphological transition in umbrella cells required a considerable amount of time to occur.
Quantification of Changes in Apical, Basolateral, and Vesicle Surface Area in Response to Stretch
Changes in apical surface area as determined by capacitance are only estimates and do not provide any information about the source of added membrane. Furthermore, some of these changes in capacitance may reflect alterations in basolateral surface area. Therefore, we sought an alternative experimental method to quantify changes in apical, basolateral, and vesicle surface area. To do this, we analyzed the tissue by stereology, a statistical analysis that calculates three-dimensional geometric parameters (volume and surface area) based on multiple two-dimensional images (tissue sections) (Griffiths, 1993).
First, it was necessary to demonstrate that a stereological analysis could accurately distinguish between the apical membrane and the vesicular membrane pools. Indeed, it has been argued that many of the vesicular structures observed by TEM are actually continuations of the plasma membrane that only appear to be separate structures based on the plane of the cross section (Koss, 1969). Obviously, this visual artifact would render our stereological analysis ineffective for measuring changes in surface area of either membrane pool. Therefore, to confirm that the vesicular structures observed by TEM are distinct from the plasma membrane, bladder tissue was incubated for 20 min without stretch and then fixed in the presence of ruthenium red, a membrane-impermeant stain used to distinguish between the plasma membrane (including its invaginations) and structures that are distinct from the plasma membrane (Hayat, 1989). In tissue that was incubated with ruthenium red, none of the vesicles acquired the stain (Figure 4A), including those vesicles that closely abutted the plasma membrane (arrows). These data clearly demonstrated that the vesicular structures represent a distinct pool of membrane that is not in continuity with the apical surface.
Figure 4.

Stereological analysis of umbrella cells and determination of vesicle/membrane continuity. (A) Mounted bladder tissue was fixed in the presence of 0.01% (wt/vol) ruthenium red and processed for TEM. Arrows point to discoidal vesicles that are in proximity, but not continuous with the apical membrane. (B–F) Serial sections of an umbrella cell reveal the discontinuity of vesicles with each other and the plasma membrane. Arrowheads depict individual discoidal vesicles as they appear and disappear within the sections. The dashed box surrounds an area of clustered discoidal vesicles that remain distinct from one another in all sections. A large plasma membrane-associated vesicle is marked with an asterisk. Uroepithelium was incubated in chambers for 20 min with stretch (░⃞) or without (▪) (G) or for 5 h with stretch (░⃞) or without stretch (▪) (H). The apical, basolateral, and vesicle surface areas were determined as described in MATERIALS AND METHODS. Shown is mean ± SEM (n = 50). Values that are significantly different (p < 0.05) from matched control values are marked with an asterisk.
It has also been proposed that the vesicles are not distinct from one another but instead form a large interconnected network of tubular canals (Koss, 1969). To confirm that the vesicles are individual and distinct intracellular structures, bladder tissue was mounted in the chambers for 20 min without stretch, fixed, and processed for TEM. The tissue was serial sectioned and the sections were ordered successively to enable us to track individual vesicles as they appeared and disappeared within the cytoplasm of a single cell. Figure 4, B–F, displays an example of five TEM micrographs taken from the apical portion of an umbrella cell. Each arrowhead tracks an individual vesicle as it appears and then fades from view. Analysis of many areas where vesicles were clustered together (dashed box) showed no fusion of individual vesicles. In addition, all vesicular structures remained separate from the plasma membrane in all sections examined with the exception of occasional infoldings of the plasma membrane that appeared as much larger vacuolar structures. An example of the latter is marked by the asterisks in Figure 4, B and C. Taken together, these results confirm that the vesicles are individual, intracellular structures that are distinct from the apical surface.
Next, we quantified the apical, basolateral, and vesicle surface areas of the umbrella cells in 20-min and 5-h control samples, or tissue that had been stretched for 20 min or 5 h. In 20-min control samples, the average apical cell surface area was ∼2700 μm2, the basolateral surface area was measured at ∼4600 μm2, and the vesicles occupied ∼8500 μm2 of surface area (Figure 4G). Thus, the surface area of the vesicles in control conditions was >3 times the surface area of the apical membrane. These results clearly indicated that the cells possess a tremendous reserve capacity to increase apical surface area via the fusion of vesicles with the apical membrane. Relative to controls, the samples stretched for 20 min exhibited no significant changes in any of the three parameters. These data were consistent with the morphological observations described above but were inconsistent with the capacitance data (see DISCUSSION).
After 5 h of stretch, the apical surface area was increased ∼50% over the matched control (from ∼2900 to 4300 μm2) with no significant change in basolateral surface area (Figure 4H). These results were consistent with the capacitance data and indicated that a considerable amount of membrane was added to the apical surface in response to stretch. However, when the changes in vesicle surface area were quantified after 5 h of stretch, it was found that the surface area of the vesicles decreased from ∼7200 μm2 to <1000 μm2 (Figure 4H). This revealed that the increase in apical surface area (∼1400 μm2) accounted for only a small fraction of the decrease in vesicle surface area (∼6200 μm2), indicating that an additional cellular event was occurring during the stretch response.
Stretch Increases Cell Surface Expression of Uroplakin III and Induces Apical Secretion of Metabolically Labeled Proteins
One prediction of the vesicle fusion model is that stretch will result in the delivery and incorporation of vesicle cargo into the apical membrane. A cargo molecule that can be used for this purpose is uroplakin III, a 47-kDa transmembrane component of both the apical membrane plaques and discoidal vesicles (Figure 5A) (Wu and Sun, 1993; Liang et al., 2001). We demonstrated that uroplakin III was associated with the plasma membrane and discoidal vesicles of rabbit tissue by immunogold labeling with a uroplakin III antibody (Figure 5A). Although uroplakin III was present on both the apical surface and the discoidal vesicles of umbrella cells, it was not found in significant amounts elsewhere in the epithelium (Wu and Sun, 1993; our unpublished data).
Figure 5.

Uroplakin III localization and stretch-induced increases in cell surface uroplakin III expression and metabolically labeled protein secretion. (A) Ultrathin cryosections of 5-h control tissue labeled with anti-uroplakin III antibody and secondary antibody conjugated to 5-nm gold. Asterisks denote examples of uroplakin III-positive vesicles. (B) Representative Western blots of total and cell surface uroplakin III expression in control or stretched samples. The bottom panel is exposed longer to allow the total cell signal in stretched samples to be visible. (C) Quantification of relative expression of cell-associated uroplakin III at the apical surface for each condition. Shown is mean ± SEM (n = 5). (D) Samples were stretched for 5 h and labeled with anti-uroplakin III antibody and secondary antibody conjugated to 5-nm gold. (E) Cells were pulsed with [35S]cysteine/methionine, chased for 1 h, and stretched or incubated without stretch for 5, 15, 120, or 300 min. Shown is average cpm present in apical medium at each time point ± SEM (n = 3). (F) Percentage of basally added [14C]urea that appeared in the apical medium after 0, 15, 120, or 300 min of stretch or incubation without stretch. Shown is mean ± SEM (n = 3).
To test the hypothesis that the discoidal vesicles were contributing to the increase in apical surface area, bladder tissue was left unstretched or was stretched for either 20 min or 5 h. The cell surface was biotinylated, the cells were lysed, and the biotinylated fraction was recovered with streptavidin beads. The amount of uroplakin III at the cell surface relative to the total cellular uroplakin III was determined by Western blot (Figure 5B). In control conditions, <5% of the total uroplakin III pool was present on the apical surface (Figure 5C). This is consistent with the large amount of surface area present in the vesicles relative to the apical membrane. This value remained unchanged after 20 min of stretch. In contrast, after 5 h of stretch, the fraction of uroplakin III at the cell surface increased to ∼32%, and the absolute amount at the surface increased ∼67 ± 18% above that observed in controls. This latter value is consistent with vesicle fusion and delivery of uroplakin III to the apical surface. In support of this, immunogold labeling demonstrated that uroplakin III was found at the plasma membrane in stretched tissue (Figure 5D). As noted in Figure 3F, there was a significant decrease in the number of discoidal vesicles.
Also evident from the Western blot analysis was that the total cell expression of uroplakin III was considerably lower after 5 h of stretch than in control. This effect was not simply due to a conformational change in the protein, because blotting for total cell surface proteins with streptavidin-HRP yielded a similar decrease in signal (Figure 6E). This result indicated that a significant portion of the apical membrane, including uroplakin III, was being degraded during stretch and is consistent with the decrease in vesicles observed both in this study and in that of Minsky and Chlapowski (1978).
Figure 6.

Effect of stretch on endocytosis. Umbrella cells were surface biotinylated and incubated for 0, 5, 15, 30, 60, or 120 min without stretch (A) or with stretch (B). MESNA-protected biotinylated proteins were isolated and Western blots were probed with streptavidin-HRP. Representative blot of control tissue (A) shows no visible endocytosis by the lack of signal in MESNA-treated lanes. Stretched tissue (B) exhibits large MESNA-protected signal, indicating endocytosis. (C) Quantification of endocytosis in control and stretched tissue. Shown is mean ± SEM (n = 5). Values that are significantly different (p < 0.05) from matched control values are marked with an asterisk. (D) WGA-FITC was internalized for 15 min in control unstretched (a) or stretched (b) tissue. Surface-bound WGA-FITC was removed by incubation at 4°C with N-acetyl glucosamine, and the samples were fixed, stained with 4,6-diamidino-2-phenylindole, and then examined byepifluorescence microscopy. A projection of digitally deconvoluted sections is shown. (Dc) Bladder cells were stretched for 2 h, WGA-FITC was added to the chamber and the samples stretched for an additional 15 min. Surface WGA-FITC was removed, the samples were fixed, and then examined in a confocal microscope. A projection of multiple XY sections is shown. (Dd) WGA-FITC was added to an excised but otherwise intact bladder for 15 min. Surface WGA-FITC was removed, the samples were fixed, and then examined in a confocal microscope. A projection of multiple XY sections is shown. (E) Apical surface proteins were biotinylated and the tissue was stretched for 0–300 min. The cells were lysed and biotinylated proteins detected by probing Western blots with streptavidin-HRP. Treatment with 40 μM leupeptin (Leup) prevented the degradation, demonstrating a lysosomal-mediated pathway, whereas treatment with lactacystin (Lact) had no effect. (F) Quantification of apical membrane protein degradation upon stretch. Values are relative to unstretched tissue biotinylated at t = 0. Shown is mean ± SEM (n = 4). Values that are significantly different (p < 0.5) from unstretched tissue at t = 0 are marked with an asterisk.
To confirm that exocytosis was occurring throughout the 5-h stretch period, we measured the secretion of metabolically labeled proteins over time. Unlike proteins anchored to the membrane, secreted proteins are not efficiently reinternalized after reaching the apical surface and thus provide an alternative way to detect exocytosis. Growth hormone, a secretory protein, has previously been localized to discoidal/fusiform vesicles (Kerr et al., 1997). Tissue was radiolabeled with [35S]cysteine/methionine for 30 min and chased for 1 h to label secretory proteins. The tissue was then stretched for 5, 15, 120, or 300 min or incubated without stretch as a control and the amount of radioactivity in the apical medium was determined. In control tissue, the radioactivity detected in the apical medium was <100 counts/minute (cpm) regardless of the duration of incubation, indicating that little secretion occurred under these conditions (Figure 5E). In contrast, within 5 min of stretch the apical cpm measured 7,980 ± 2,100 and this amount increased to 214,687 ± 40,252 cpm after 5 h. This secretion was completely blocked by adding 10 μg/ml brefeldin A (our unpublished data), demonstrating a bona fide membrane trafficking event and not nonspecific release.
Stretch also stimulated significant release of secretory proteins into the basolateral chamber (our unpublished data). To further ensure that the apical secretion we observed was not the result of paracellular diffusion of secretory proteins from the basolateral-to-apical chamber, [14C]urea was added to the basolateral chamber and the appearance of radioactivity in the apical chamber was determined in control or stretched tissue. In either case the amount of [14C]urea that crossed the cells was <0.4% after 5 h of stretch, demonstrating that the integrity of the umbrella cell layer was not compromised (Figure 5F). Apical-to-basolateral diffusion of [14C]urea was also determined and was not significantly different from basolateral-to-apical diffusion (our unpublished data). Taken together, these results confirmed that stretch induces a rapid and continual secretion of protein and that this secretion represents a genuine exocytic event.
Stretch Induces Endocytosis at the Apical Surface of Umbrella Cells
The stereological analysis revealed that after 5 h of stretch the increase in apical surface area accounted for only a small fraction of the loss in vesicle surface area. Furthermore, the biochemical data indicated that uroplakin III was degraded during the 5 h of stretch. Therefore, we hypothesized that membrane added to the apical surface during stretch was being endocytosed and degraded.
To test whether stretch induced endocytosis, the tissue was mounted in the modified Ussing chambers and the apical cell surface was biotinylated at 4°C with sulfo-N-hydroxysuccinimide-disulfo-biotin to label surface proteins. Several protein species were biotinylated (Figure 6A). The major protein species was a doublet that migrated with a molecular mass in the 67–70 kDa range. The identity of the doublet is not known. Several smaller molecular weight species were also detected, one of which was confirmed to be uroplakin III by probing blots of biotinylated proteins with anti-uroplakin III antibodies (our unpublished data). Exposing the blot for longer periods of time also revealed that other protein species were biotinylated, including proteins that migrated ∼15, 27, and 28 kDa (our unpublished data), which are the molecular masses of uroplakins II, Ia, and Ib, respectively (Wu et al., 1994). After biotinylation, the tissue was warmed and stretched for 0–120 min or incubated for the same amount of time in the absence of stretch as a control. The internalized biotin-labeled proteins were detected by treating cells with the membrane-impermeant reducing agent MESNA (see MATERIALS AND METHODS).
In control cells, the MESNA treatment removed essentially all of the biotin regardless of the duration of incubation (Figure 6A). This showed that little or no endocytosis occurred. In contrast, stretched cells exhibited a large MESNA-protected signal after 5 min, demonstrating that a significant amount of the surface membrane was rapidly internalized (Figure 6B). The quantification of this and other blots indicated that nearly 100% of the labeled membrane was endocytosed within 5 min of stretch (Figure 6C) and this value remained constant over a 2-h period of incubation. The latter data indicated that in the time frame examined, little recycling of biotinylated apical proteins occurred after endocytosis.
WGA-FITC, which binds to N-acetyl glucosamine-modified proteins and lipids on the apical surface of the umbrella cells (Fujiyama et al., 1995), was used as alternative marker of endocytosis. The lectin was included in the apical half of the Ussing chamber and the tissue was then stretched for 15 min or incubated without stretch for the same amount of time. After the incubation, surface-bound WGA-FITC was removed with N-acetyl glucosamine treatment at 4°C and the internalization of WGA-FITC was determined by fluorescence microscopy. No uptake was observed in control (unstretched) samples either after 15 min (Figure 6Da) or after 5 h (our unpublished data). Cells stretched for 15 min exhibited a significant amount of fluorescence in punctate structures, consistent with uptake into vesicular structures (Figure 6Db). Confocal microscopy verified that the endocytic structures were localized to the apical cytoplasm of the umbrella cells and that no staining was observed in the intermediate cell layer. To assess whether endocytosis continued throughout the stretch period, WGA-FITC was added to the apical chamber of the preparations 2 or 5 h after the initiation of stretch. Uptake of WGA-FITC was observed under both conditions (data for uptake after 2 h is shown in Figure 6Dc).
As final confirmation that stretch stimulates endocytosis in bladder umbrella cells we filled excised but otherwise intact bladders with buffer containing WGA-FITC for 15 min at 37°C. The mucosal surface was treated to remove cell surface lectin, fixed, and imaged. Again, significant uptake of lectin was observed in the umbrella cell population (Figure 6Dd). The latter observation is consistent with previous experiments that showed uptake of fluid-phase markers in bladders that were emptied and then filled with fluid-phase marker (Hicks, 1966; Porter et al., 1967). Although these original observations were taken as evidence of uptake after voiding, filling the bladder stretches the tissue, and the internalization previously observed is likely to be the stretch-regulated endocytosis described in this study.
Endocytosed Membrane Is Delivered to Lysosomes
Having established that stretch induced apical membrane endocytosis, we next examined the hypothesis that internalized membrane was being degraded. Accordingly, the kinetics of degradation was determined by biotinylating the cell surface at 4°C and subsequently stretching for 5–300 min. After each time point, the tissue was lysed; the cohort of biotinylated proteins was isolated; and the proteins were separated by SDS-PAGE, transferred to membranes, and probed with streptavidin-HRP (Figure 6E). The fraction of protein remaining after each time point relative to the starting amount was quantified (Figure 6F). The total amount of biotinylated proteins remained constant for up to 120 min of stretch. However, by 210 min there was a significant decrease in protein to ∼20% of the starting value and by 300 min the protein levels were reduced to ∼9% (Figure 6F). This result was consistent with our previous experiment with uroplakin III and indicated that the apical membrane proteins were being degraded.
Two mechanisms can account for degradation within cells; delivery to lysosomes or proteasomes. To discriminate between the two pathways, the apical cell surface was biotinylated at 4°C, and the tissue was stretched for 5 h in the presence of 40 μM leupeptin to block lysosomal degradation (Ihrke et al., 1998). Consistent with lysosomal delivery, incubation with leupeptin inhibited the degradation that was observed in cells stretched without the drug treatment (Figure 6, E and F). To rule out the possibility of proteasomal-mediated degradation, we treated the cells with 10 μM lactacystin to inhibit proteasome function (Fenteany et al., 1995). Lactacystin treatment had no effect on the degradation of apical membrane proteins (Figure 6F), confirming that degradation was via a lysosomal-mediated pathway. These data supported our hypothesis that the internalized membrane was delivered to lysosomes where it was degraded.
The Stretch Response Is a cAMP-mediated Event
In other mechanically sensitive tissues, mechanical stimulation is associated with elevated levels of the second messenger cAMP (Russo et al., 1989; Sandy et al., 1989; Watson et al., 1989). To ascertain the role of cAMP in the uroepithelium, we measured intracellular levels of cAMP in control and stretched tissue over time. Stretching the tissue caused the intracellular levels of cAMP to rise more than twofold within 15 min (Figure 7A). These levels remained elevated throughout the 5-h period of stretch, implicating cAMP in the stretch response. To test the effects of elevating cAMP in the umbrella cells, we incubated the tissue with 10 μM forskolin and 500 μM 3-isobutyl-1-methylxanthine (IBMX) to raise intracellular cAMP levels. Unexpectedly, we observed an unusually large increase of 120% in capacitance over a 5-h period, versus the 50% change observed with stretch alone (Figure 7B). Incubating the tissue with 8-bromo-cAMP, a membrane-permeant analog of cAMP, induced a similar effect as forskolin/IBMX (our unpublished data) and treatment with di-deoxy forskolin, a nonfunctional forskolin analog, had no effect on capacitance (our unpublished data). We also explored the effect on capacitance when forskolin was added to stretched tissue. The response was similar to control tissue treated with forskolin but the net rise in capacitance was slightly greater (∼140 vs. 120%). The above-mentioned data demonstrated that cAMP dramatically altered membrane trafficking at the apical surface of umbrella cells.
Figure 7.

Role of cAMP and PKA in the stretch response. (A) Intracellular levels of cAMP were measured in control cells (▪) and stretched cells (░⃞) after 15, 30, 120, or 300 min. Shown is mean ± SEM (n = 4). Values that are significantly different (p < 0.05) from matched control values are marked with an asterisk. (B) Effects of forskolin and H89 on capacitance. Cells were treated with 10 μM forskolin/500 μM IBMX without stretch (▪), treated with forskolin/IBMX and 10 μM H89 (▴), or stretched in the presence of 10 μM H89 alone (●). For comparison, the capacitance readings from Figure 1 of stretched (♦) and control (□) cells are shown. Shown is mean ± SEM (n = 6). (C) Effects of forskolin/IBMX on umbrella cell morphology. Tissue was treated with forskolin/IBMX without stretch for 5 h and processed for TEM. Notice the highly convoluted apical membrane. (D) Changes in surface area resulting from Forskolin/IBMX treatment. Tissue was treated with forskolin/IBMX (░⃞) and stereological analysis was performed as described. Control surface areas are shown for comparison (▪), n = 50. Values that are significantly different (p < 0.05) from matched control values are marked with an asterisk. (E) Endocytosis was determined in forskolin/IBMX-treated cells. Shown is representative Western blot after 0, 5, 15, 30, 60, or 120 min with or without MESNA treatment.
A likely effector of cAMP is PKA, the target of the H89 kinase inhibitor (Fujihara et al., 1993). In stretched tissue treated with H89, the capacitance initially rose ∼20% but failed to increase any further, implicating PKA as a possible effector of the stretch response (Figure 7B). Moreover, the rise in capacitance observed after incubating the tissue in forskolin/IBMX was also significantly inhibited when the tissue was pretreated with H89. These findings underscored the importance of cAMP and PKA in the regulation of mucosal surface area and indicated that cAMP modulated, in part, the response of the umbrella cell to mechanical stretch.
The significant changes observed in capacitance when the tissue was incubated with forskolin/IBMX prompted us to examine the morphology of the umbrella cells after forskolin treatment and to assess the effect of these drugs on endocytosis. Accordingly, we treated unstretched umbrella cells with forskolin/IBMX for 5 h and processed the tissue for TEM. Treatment with forskolin/IBMX (Figure 7C) elicited exaggerated convolutions in the apical cell membranes, supporting the large increases observed in capacitance. Stereological measurements confirmed the capacitance results (Figure 7D). The apical surface area of forskolin-treated cells increased to ∼6500 μm2, more than double the apical surface area of an unstretched umbrella cell. No significant changes in basolateral surface area were detected. There was a significant decrease in vesicle area that accompanied the forskolin/IBMX-induced fusion. However, unlike stretched tissue a significant amount of vesicle area remained. Last, we measured the effects of forskolin/IBMX on endocytosis in control unstretched tissue. The amount of MESNA-protected biotin remained constant over a 2-h time period, demonstrating that no endocytosis occurred (Figure 7E). This result indicated that forskolin may be modulating apical surface area by means of discoidal vesicle exocytosis without affecting apical membrane endocytosis.
DISCUSSION
Stretch-induced Changes in Apical Surface Area of Umbrella Cells
The ability of the bladder to retain urine requires a barrier epithelium that can accommodate large variations in urine volume. At the cellular level this is likely accomplished by changes in umbrella cell architecture and surface area. The latter is thought to be modulated by discoidal vesicle exocytosis (Minsky and Chlapowski, 1978; Lewis and de Moura, 1982, 1984). The underlying intermediate and basal cell layers also undergo morphological transitions, but much less is known about their cellular biology (Hicks, 1975).
Our data confirm that stretch increases the apical surface area of umbrella cells, and these changes are likely mediated by discoidal vesicles. Mechanical distention of the uroepithelium resulted in an ∼50% increase in capacitance after a 5-h period of stretch. This large change in capacitance is consistent with, but significantly greater than the previous observation that short-term mechanical bowing of the uroepithelium increased capacitance by ∼18% (Lewis and de Moura, 1982). Furthermore, a stereological analysis confirmed that the apical surface area of the umbrella cells increased by 50%, whereas there was no statistically significant change in basolateral surface area. The stereological analysis also revealed that stretch was accompanied by a fourfold decrease in vesicle surface area. These data concur with previous observations that the numerical density of discoidal vesicles was decreased in expanded versus contracted bladders (Minsky and Chlapowski, 1978). Biotinylation experiments demonstrated that both the relative and absolute amount of uroplakin III (a “cargo” molecule found in discoidal vesicles) at the cell surface significantly increased after 5 h of stretch, consistent with discoidal vesicle fusion. Last, secretion of metabolically labeled proteins confirmed a rapid and significant apical secretion in stretched tissue that increased over time. Although exocytosis was observed shortly after stretch, the majority of exocytosis occurred between the 2- and 5-h period. Although the above-mentioned results are consistent with the fusion of discoidal vesicles during stretch, we cannot rule out that some of the observed changes in surface area were the result of other membranous organelles fusing with the apical surface of the umbrella cells.
Release of metabolically labeled secretory proteins was observed within 5 min of stretch, indicating that stretch rapidly stimulated exocytosis in umbrella cells. Whether these early exocytic events result in a change in the apical surface area of these cells is unclear. We observed a small increase (∼15%) in capacitance within the first 20 min of stretch. However, a stereological analysis showed no significant change in apical, basolateral, or vesicle surface area. Furthermore, we observed that the amount of uroplakin III at the cell surface was unchanged after 20 min of stretch. Because stretch stimulates ion transport (our unpublished data), the small capacitance change we observe may be imaginary (Gillis, 1995). Alternatively, the area change may be too small to be detected by either stereology or biotinylation.
Mechanical Stretch Stimulates Apical Endocytosis in Umbrella Cells
In the classical model, vesicle endocytosis is proposed to occur only upon voiding (i.e., after release of stretch) (Hicks, 966; Porter et al., 1967). Our data are inconsistent with this aspect of the model. Most notably, we observed that essentially all biotinylated apical membrane proteins, general markers of the apical plasma membrane, were endocytosed within 5 min of mechanical stretch. However, we cannot rule out that only a subpopulation of apical membrane is biotinylated and that we are overestimating the amount of endocytosis. Moreover, we observed uptake of WGA-FITC in stretched preparations of isolated uroepithelium or upon filling excised but otherwise intact bladders.
Despite significant amounts of endocytosis, there was little apparent change in apical surface area during the first 20 min of stretch, thus indicating that the amount of endocytosis must have been balanced by roughly equivalent amounts of exocytosis. This rapid exocytosis could be a result of either recycled membrane or delivery of newly synthesized vesicle membrane. Because apical surface area increased over the longer time scale, the rate of exocytosis must ultimately outpace the rate of endocytosis. This is supported by the large increase in secretion between 2 and 5 h of stretch. Nonetheless, endocytosis continues throughout the entire period of stretch as evidenced by uptake of WGA-FITC at 2 and 5 h of stretch.
Although is may seem counterintuitive that exocytosis and endocytosis are occurring simultaneously in stretched umbrella cells, these two processes occur constitutively and simultaneously in all cells, even under resting conditions (Harter and Wieland, 1996; Mukherjee et al., 1997). Coupled exocytosis/endocytosis likely plays a key role in fine-tuning the changes in umbrella cell surface area as a result of stretch and/or other external stimuli (see discussion below). Furthermore, rapid endocytosis/exocytosis would provide the umbrella cells with a means of quickly replacing old or damaged surface plaques with new ones in preparation for imminent lumen expansion. In this regard, it has recently been shown that expression of uroplakin III, a major plaque constituent, is required for the maintenance of bladder barrier function (Hu et al., 2000). Finally, exocytosis decreases membrane tension (Morris and Homann, 2001), which is compensated for by endocytosis. Regardless of its function, endocytosis clearly represents a major pathway of membrane trafficking during stretch and its specific functions and regulations will be addressed in future studies.
In the present analysis, we have not directly examined the events that occur when stretch is released. Because stretch increases apical surface area, this added membrane must be endocytosed at some point after voiding. Minsky and Chlapowski (1978) observed that within 10 min after voiding, the number of discoidal vesicles in the umbrella cells increased by approximately twofold compared with the number of vesicles in distended bladders (Minsky and Chlapowski, 1978). This implies that some vesicles are replenished within a short period of time after stretch. The increased number of discoidal vesicles could reflect apical membrane internalization and/or rapid delivery of newly synthesized vesicles from the Golgi (Hicks, 1966). This assembly of vesicles in the Golgi is likely to be significant. Only a small fraction of discoidal/fusiform vesicles is labeled by apically endocytosed fluid or membrane markers, indicating that they are primarily involved in exocytic traffic (Hicks, 1966; Porter et al., 1967; Amano et al., 1991). Moreover, from our data it is evident that the amount of internalized membrane would be insufficient to replenish the surface area of the discoidal vesicles lost during stretch.
Endocytosed Membrane Is Delivered to Lysosomes
Discoidal vesicles or discoidal vesicle-derived membrane was previously observed in multivesicular bodies as well as lysosomes (Porter et al., 1967; Amano et al., 1991). However, the extent of apical membrane entry into this pathway and the role that stretch played in this process were previously unknown. We observed that upon internalization biotin-labeled apical surface proteins were eventually degraded. This proteolytic event was prevented by the lysosomal enzyme inhibitor leupeptin, but not by the proteasome inhibitor lactacystin. This finding is consistent with the decrease in vesicles observed in this study and that described previously (Minsky and Chlapowski, 1978). However, degradation was not apparent until some time after 2 h of stretch. In other cell types degradation can proceed as quickly as 15–20 min after internalization and delivery to lysosomes usually occurs within 30–45 min of endocytosis (Griffiths et al., 1989). It is possible that either the delivery of endocytosed membrane to late endosomes/lysosomes is slow in umbrella cells or that stretch up-regulates a pathway that results in enhanced degradation of discoidal vesicles. The latter possibility would explain the sudden change in protein signal after 120 min of stretch. Alternatively, there may be some recycling of vesicle membrane that escaped our detection.
Role of cAMP in Stretch-regulated Exocytosis
In addition to elucidating the dynamics of membrane trafficking in response to stretch, we have also gained insight into one of the signal transduction pathways that may regulate these trafficking events. Our data indicate that a cAMP-PKA pathway plays a role in the stretch response. Increased amounts of cAMP were observed in stretched tissue and elevating the intracellular concentration of cAMP in unstretched umbrella cells with forskolin/IBMX resulted in a >100% increase in apical cell surface area. These events were blocked by treatment with the PKA antagonist H89. Importantly, stretch-regulated changes in membrane capacitance were significantly decreased by H89, further confirming that the cAMP-PKA pathway is required for vesicle exocytosis. Our results are consistent with the recent observation that cultured uroepithelial cells or bladder explants secrete tissue-type plasminogen activator and urokinase in a cAMP-dependent manner (Deng et al., 2001), although the study did not examine the effects of stretch on secretion. The results of our experimentation have additional significance because they indicate that the surface area of umbrella cells can be regulated by external stimuli independent of stretch. These stimuli could come in the form of growth factors, hormones, or neurotransmitters present in the blood or released by the bladder-associated musculature and nerves. Interestingly, some afferent neuronal processes terminate within the uroepithelium and umbrella cells apparently express surface receptors for various neurotransmitters (Birder et al., 1998). As such, the apical surface area of umbrella cells may be regulated by mechanical as well as exocrine/paracrine stimuli.
The dramatic increases in umbrella cell surface area observed in unstretched tissue treated with agents that raise intracellular cAMP occurred in the apparent absence of endocytosis. This observation underscores the importance of endocytosis in regulating umbrella cell apical surface area. Although stretch stimulated endocytosis of the apical membrane, treatment with forskolin/IBMX without stretch had no effect on endocytosis but resulted in a greater increase in surface area. These observations indicate that the response to stretch may involve at least two levels of regulation, one that operates by modulating intracellular levels of cAMP and the other that uses another uncharacterized mechanism. Other cellular events rapidly triggered by mechanical stretch include stimulation of stretch-activated channels, activation of the integrin-associated focal adhesion kinase, phosphoinositide turnover, and autocrine release of growth factors (Vandenburgh, 1992). Some or all of these mechanosensory mechanisms could play an important role in signaling exocytosis/endocytosis in umbrella cells.
In summary, our findings further elucidate the poorly understood process of mucosal surface area regulation in the uroepithelium during stretch. The data not only shed light on the membrane trafficking that occurs in these cells but also provide insight into the intracellular signal cascade that is responsible for this trafficking. Moreover, we have uncovered an additional pathway for apical endocytosis that is modulated by mechanical force (other examples of stretch-regulated endocytosis are given in Apodaca, 2002). This discovery broadens our understanding of the effects of mechanical stimuli on membrane traffic, especially in regard to the pivotal function of endocytosis in balancing the exocytic response of the cell. The coordinated balance between these two plasma membrane events in umbrella cells is likely to play a similarly important role in other cell types that are responsive to mechanical forces.
ACKNOWLEDGMENTS
We thank Drs. Rebecca Hughey, Nick Johnson, Linton Traub, and Ora Weisz for insightful comments and critiques while preparing this manuscript. This work was supported by R01 grants from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health to G.A. (R01DK51970) and M.Z (R01DK48217). The Laboratory of Epithelial Cell Biology is supported in part by an equipment grant from Dialysis Clinic Inc.
Abbreviations used:
- HRP
horseradish peroxidase
- IBMX
3-isobutyl-1-methylxanthine
- MESNA
2-mercaptoethanesulfonic acid
- PKA
protein kinase A
- TEM
transmission electron microscopy
- TER
transepithelial resistance
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.01–09–0435. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.01–09–0435.
REFERENCES
- Altschuler Y, Kinlough C, Poland P, Apodaca G, Weisz O, Hughey R. Clathrin-mediated endocytosis of MUC1 is regulated by glycosylation. Mol Biol Cell. 2000;11:819–831. doi: 10.1091/mbc.11.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano O, Kataoka S, Yamamoto T. Turnover of asymmetric unit membranes in the transitional epithelial superficial cells of the rat urinary bladder. Anat Rec. 1991;229:9–15. doi: 10.1002/ar.1092290103. [DOI] [PubMed] [Google Scholar]
- Apodaca G, Katz L, Mostov K. Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. J Cell Biol. 1994;125:67–86. doi: 10.1083/jcb.125.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apodaca G. Modulation of membrane traffic by mechanical stimuli. Am J Physiol. 2002;282:F179–F190. doi: 10.1152/ajprenal.2002.282.2.F179. [DOI] [PubMed] [Google Scholar]
- Baddeley A, Gunderson H, Cruz-Orive L. Estimation of surface area from vertical sections. J Microsc. 1986;142:259–276. doi: 10.1111/j.1365-2818.1986.tb04282.x. [DOI] [PubMed] [Google Scholar]
- Birder L, Apodaca G, De Groat W, Kanai A. Adrenergic- and capsaicin-evoked nitric oxide release from urothelium and afferent nerves in urinary bladder. Am J Physiol. 1998;275:F226–F229. doi: 10.1152/ajprenal.1998.275.2.F226. [DOI] [PubMed] [Google Scholar]
- Chang A, Hammond T, Sun T, Zeidel M. Permeability properties of the mammalian bladder apical membrane. Am J Physiol. 1994;267:C1483–C1492. doi: 10.1152/ajpcell.1994.267.5.C1483. [DOI] [PubMed] [Google Scholar]
- Clemow D, Steers W, Tuttle J. Stretch-activated signaling of nerve growth factor secretion in bladder, and vascular smooth muscle cells from hypertensive, and hyperactive rats. J Cell Physiol. 2000;183:289–300. doi: 10.1002/(SICI)1097-4652(200006)183:3<289::AID-JCP1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Dai J, Ting-Beall P, Sheetz M. The Secretion-coupled endocytosis correlates with membrane tension changes in RBL 2H3 cell. J Gen Physiol. 1997;110:1–10. doi: 10.1085/jgp.110.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng F, Ding M, Lavker R, Sun T-T. Urothelial function reconsidered: a role in urinary protein secretion. Proc Natl Acad Sci USA. 2001;98:154–159. doi: 10.1073/pnas.98.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz J. Release of atrial natriuretic factor from heart-lung preparation by atrial distension. Am J Physiol. 1988;247:R1093–R1096. doi: 10.1152/ajpregu.1984.247.6.R1093. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ. Introduction to the Bootstrap. Boca Raton, FL: CRC Press; 1993. [Google Scholar]
- Fenteany G, Standaert R, Lane W, Choi S, Corey E, Schreiber S. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Fink R, Cooper M. Apical membrane turnover is accelerated near cell-cell contacts in an embryonic epithelium. Dev Biol. 1996;174:180–189. doi: 10.1006/dbio.1996.0064. [DOI] [PubMed] [Google Scholar]
- Fujihara M, Muroi M, Muroi Y, Ito N, Suzuki T. Mechanism of lipopolysaccharide-triggered junB activation in a mouse macrophage-like cell line. J Biol Chem. 1993;268:14898–14905. [PubMed] [Google Scholar]
- Fujiyama C, Masake Z, Sugihara H. Reconstruction of the urinary bladder mucosa in three-dimensional collagen gel culture: fibroblast-extracellular matrix interactions on the differentiation of transitional epithelial cells. J Urol. 1995;153:2060–2067. [PubMed] [Google Scholar]
- Gillis K. Techniques for membrane capacitance measurements. In: Sakman B, Neher E, editors. Single Channel Recording. New York: Plenum Press; 1995. pp. 155–198. [Google Scholar]
- Griffiths G. Fine Structure Immuno-cytochemistry. Berlin: Springer-Verlag; 1993. [Google Scholar]
- Griffiths G, Back R, Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J Cell Biol. 1989;109:2703–2720. doi: 10.1083/jcb.109.6.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter C, Wieland F. The secretory pathway: mechanisms of protein sorting and transport. Biochim Biophys Acta. 1996;1286:75–93. doi: 10.1016/0304-4157(96)00003-2. [DOI] [PubMed] [Google Scholar]
- Hayat M. Principles and Techniques of Electron Microscopy. Boca Raton, FL: CRC Press; 1989. [Google Scholar]
- Hicks R. The function of the Golgi complex in transitional epithelium. J Cell Biol. 1966;30:623–644. doi: 10.1083/jcb.30.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks M. The mammalian urinary bladder: an accommodating organ. Biol Rev. 1975;50:215–246. doi: 10.1111/j.1469-185x.1975.tb01057.x. [DOI] [PubMed] [Google Scholar]
- Homann U. Fusion and fission of plasma-membrane material accommodates for osmotically induced changes in the surface area of guard-cell protoplasts. Planta. 1998;206:329–333. [Google Scholar]
- Hu P, Deng F-M, Liang F-X, Hu C-M, Auerbach A, Shapiro E, Wu X-R, Kachlar B, Sun T-T. Ablation of uroplakin III gene results in small urothelial plaques, urothelial leakage, and vesicoureteral reflux. J Cell Biol. 2000;151:961–971. doi: 10.1083/jcb.151.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrke G, Martin G, Shanks M, Schrader M, Schroer T, Hubbard A. Apical plasma membrane proteins and endolyn-78 travel through a subapical compartment in polarized WIF-B hepatocytes. J Cell Biol. 1998;141:115–133. doi: 10.1083/jcb.141.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr D, Liang F, Bondioli K, Zhao H, Kreibich G, Wall R, Sun T-T. The bladder as a bioreactor: urothelium production and secretion of growth hormone into urine. Nat Biotech. 1997;16:75–79. doi: 10.1038/nbt0198-75. [DOI] [PubMed] [Google Scholar]
- Koss L. The asymmetric unit membranes of the epithelium of the urinary bladder of the rat. Lab Invest. 1969;21:154–168. [PubMed] [Google Scholar]
- Lang R, Tholken H, Ganten F, Luft F, Ruskoaho H, Unger T. Atrial natriuretic factor-a circulating hormone stimulated by volume loading. Nature. 1985;314:264–266. doi: 10.1038/314264a0. [DOI] [PubMed] [Google Scholar]
- Levin R, Longhurst P, Monson F, Haugaard N, Wein A. Experimental studies on bladder outlet obstruction. In: Lepor H, Lawson R K, editors. Prostate Diseases. Philadelphia: WB Saunders; 1993. pp. 119–130. [Google Scholar]
- Levin R, Wein A. Response of the in vitro whole bladder (rabbit) preparation to autonomic agonists. J Urol. 1982;128:1087–1090. doi: 10.1016/s0022-5347(17)53350-9. [DOI] [PubMed] [Google Scholar]
- Lewis S, Diamond J. Na+transport by rabbit urinary bladder, a tight epithelium. J Membr Biol. 1976;28:1–40. doi: 10.1007/BF01869689. [DOI] [PubMed] [Google Scholar]
- Lewis S, Hanrahan J. Physiological approaches for studying mammalian urinary bladder epithelium. Methods Enzymol. 1990;192:632–650. doi: 10.1016/0076-6879(90)92100-r. [DOI] [PubMed] [Google Scholar]
- Lewis S, de Moura J. Incorporation of cytoplasmic vesicles into apical membrane of mammalian urinary bladder epithelium. Nature. 1982;297:685–688. doi: 10.1038/297685a0. [DOI] [PubMed] [Google Scholar]
- Lewis S, de Moura J. Apical membrane area of rabbit urinary bladder increases by fusion of intracellular vesicle: an electrophysiological study. J Membr Biol. 1984;82:123–136. doi: 10.1007/BF01868937. [DOI] [PubMed] [Google Scholar]
- Liang F-X, Riedel I, Deng F-M, Zhou G, Xu C, Wu X-R, Kong X-P, Moll R, Sun T-T. Organization of uroplakin subunits: transmembrane topology, pair formation and plaque composition. Biochem J. 2001;355:13–18. doi: 10.1042/0264-6021:3550013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minsky B, Chlapowski F. Morphometric analysis of the translocation of lumenal membrane between cytoplasm and cell surface of transitional epithelial cells during the expansion-contraction cycles of mammalian urinary bladder. J Cell Biol. 1978;77:685–697. doi: 10.1083/jcb.77.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C, Homann U. Cell surface area regulation, and membrane tension. J Membr Biol. 2001;179:79–102. doi: 10.1007/s002320010040. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Ghosh R, Maxfield F. Endocytosis. Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Porter K, Kenyon K, Badenhausen S. Specializations of the unit membrane. Protoplasma. 1967;63:262–274. [PubMed] [Google Scholar]
- Raucher D, Sheetz M. Membrane expansion increases endocytosis rate during mitosis. J Cell Biol. 1999;144:497–506. doi: 10.1083/jcb.144.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo L, Rannels S, Laslow K, Rannels D. Stretch-related changes in lung cAMP after partial pneumonectomy. Am J Physiol. 1989;257:E261–E268. doi: 10.1152/ajpendo.1989.257.2.E261. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993;12:1681–1692. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy J, Meghji S, Farndale R, Meikle M. Dual elevation of cyclic AMP and inositol phosphates in response to mechanical deformation of murine osteoblasts. Biochim Biophys Acta. 1989;1010:265–269. doi: 10.1016/0167-4889(89)90171-7. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter H, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- Staehelin L, Chlapowski F, Bonneville M. Lumenal plasma membrane of the urinary bladder I. Three-dimensional reconstruction from freeze-etch images. J Cell Biol. 1972;53:73–91. doi: 10.1083/jcb.53.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steers W, Broder S, Persson K, Bruns D, Ferguson II J, Bruns M, Tuttle J. Mechanical stretch increases secretion of parathyroid hormone-related protein by cultured bladder smooth mucsle cells. J Urol. 1998;160:908–912. doi: 10.1016/S0022-5347(01)62831-3. [DOI] [PubMed] [Google Scholar]
- Truschel S, Ruiz W, Shulman T, Pilewski J, Sun T-T, Zeidel M, Apodaca G. Primary uroepithelial cultures: a model system to analyze umbrella cell barrier function. J Biol Chem. 1999;274:15020–15029. doi: 10.1074/jbc.274.21.15020. [DOI] [PubMed] [Google Scholar]
- Vandenburgh H. Mechanical forces and their second messengers in stimulating cell growth in vitro. Am J Physiol. 1992;262:R350–R355. doi: 10.1152/ajpregu.1992.262.3.R350. [DOI] [PubMed] [Google Scholar]
- Watson P, Haneda T, Morgan H. Effect of higher aortic pressure on ribosome formation and cAMP content in rat heart. Am J Physiol. 1989;256:C1257–C1261. doi: 10.1152/ajpcell.1989.256.6.C1257. [DOI] [PubMed] [Google Scholar]
- Wirtz H, Dobbs L. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 1990;250:1266–1269. doi: 10.1126/science.2173861. [DOI] [PubMed] [Google Scholar]
- Wu X-R, Lin J-H, Walz T, Haner M, Yu J, Aebi U, Sun T-T. Mammalian uroplakins. J Biol Chem. 1994;269:13716–13724. [PubMed] [Google Scholar]
- Wu X-R, Sun T-T. Molecular cloning of a 47 kDa tissue-specific and differentiation-dependent urothelial cell surface glycoprotein. J Cell Sci. 1993;106:31–43. doi: 10.1242/jcs.106.1.31. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu M, Liu J, Caniggia I, Post M. Mechanical strain induces constitutive and regulated secretion of glycosaminoglycans and proteoglycans in fetal lung cells. J Cell Sci. 1996;109:1605–1613. doi: 10.1242/jcs.109.6.1605. [DOI] [PubMed] [Google Scholar]