

Abstract
Nitrogen mustard (NM) is a cytotoxic vesicant known to cause acute lung injury which progresses to fibrosis; this is associated with a sequential accumulation of pro- and anti-inflammatory macrophages in the lung which have been implicated in NM toxicity. Farnesoid X receptor (FXR) is a nuclear receptor involved in regulating lipid homeostasis and inflammation. In these studies, we analyzed the role of FXR in inflammatory macrophage activation, lung injury and oxidative stress following NM exposure. Wild-type (WT) and FXR−/− mice were treated intratracheally with PBS (control) or NM (0.08 mg/kg). Bronchoalveolar lavage fluid (BAL) and lung tissue were collected 3, 14 and 28 d later. NM caused progressive histopathologic alterations in the lung including inflammatory cell infiltration and alveolar wall thickening and increases in protein and cells in BAL; oxidative stress was also noted, as reflected by upregulation of heme oxygenase-1. These changes were more prominent in male FXR−/− mice. Flow cytometric analysis revealed that loss of FXR resulted in increases in proinflammatory macrophages at 3 d post NM; this correlated with upregulation of COX-2 and ARL11, markers of macrophage activation. Markers of anti-inflammatory macrophage activation, CD163 and STAT6, were also upregulated after NM; this response was exacerbated in FXR−/− mice at 14 d post-NM. These findings demonstrate that FXR plays a role in limiting macrophage inflammatory responses important in lung injury and oxidative stress. Maintaining or enhancing FXR function may represent a useful strategy in the development of countermeasures to treat mustard lung toxicity.
Keywords: FXR, Mustard vesicants, Lung toxicity, Macrophages, Oxidative stress
1. Introduction
Mustard vesicants including sulfur mustard (SM) and nitrogen mustard (NM) are cytotoxic alkylating agents. Originally synthesized as a chemical warfare agent, NM was subsequently developed for use in cancer chemotherapy. Both SM and NM are known to cause extensive damage to the respiratory tract, which can progress to fibrosis and other chronic diseases, and they remain high priority chemical threat agents. Tissue injury induced by mustards is associated with a macrophage-dominant inflammatory response (Malaviya et al., 2016; Sunil et al., 2020; Venosa et al., 2016). Macrophages are key in both the initiation and resolution of inflammatory responses to tissue injury and in the wound-healing process (Laskin et al., 2019). This activity is mediated by distinct macrophage subsets broadly classified as proinflammatory M1 and anti-inflammatory M2 which develop in response to molecules they encounter in the tissue microenvironment including cytokines, growth factors, lipids, and epigenetic modifiers (Homer et al., 2011; Laskin et al., 2019; Li et al., 2018). Under homeostatic conditions, the activity of inflammatory macrophages is tightly regulated. This is important as uncontrolled activation of M1 macrophages exacerbates tissue injury, while excessive M2 macrophage activity promotes the development of chronic disease (Laskin et al., 2011; Lech and Anders, 2013; Wynn and Vannella, 2016).
Accumulating evidence suggests that lung lipids play an important role in regulating macrophage inflammatory responses to inhaled toxicants (Francis et al., 2020; Madison et al., 2020; Morissette et al., 2015; Venosa et al., 2019). In this context, we previously reported that mustard exposure leads to disruption in lung lipids, characterized by increases in total lung phospholipids and alterations in the expression of surfactant proteins (Sunil et al., 2020; Sunil et al., 2018). Importantly, this correlated with derangements in macrophage inflammatory activity and impaired pulmonary functioning (Sunil et al., 2020; Sunil et al., 2018; Sunil et al., 2014; Venosa et al., 2019).
Farnesoid X receptor (FXR) is a ligand-activated nuclear receptor that functions to regulate lipid and bile acid homeostasis; it also has anti-inflammatory activity and is thought to be important in linking lipid metabolism and inflammation (Armstrong and Guo, 2017; Gai et al., 2018; Shaik et al., 2015). Mainly expressed in the liver and intestines, FXR has also been identified in alveolar epithelial cells and macrophages, pulmonary endothelial cells, and lung fibroblasts (Chen et al., 2016; Francis et al., 2020; Hendrick et al., 2014; Venosa et al., 2019). In earlier studies, we reported that FXR activity is down-regulated in the lung following exposure of rats to NM, a response associated with an accumulation of oxidized lipids in lung macrophages, the appearance of macrophage foam cells and the development of pulmonary fibrosis (Venosa et al., 2019). To analyze the role of FXR in these responses, we used genetically modified mice with a targeted deletion of the Fxr gene (Guo et al., 2006). We found that FXR deficiency plays a key role in regulating both pro- and anti-inflammatory macrophage activation following NM-induced lung injury and oxidative stress. These results provide insight into mechanisms mediating inflammation and fibrogenesis following NM-induced lung damage which may be useful in the development of effective countermeasures against pulmonary injury.
2. Methods
2.1. Animals and treatments
Male and female C57BL/6 J wild-type (WT) mice were purchased from The Jackson Laboratories (Bar Harbor, ME). FXR−/− mice (129-Fxrtm1Gonz) were bred at Rutgers University; mice were backcrossed to C57BL/6 J mice for 10 generations and genotyped by PCR as previously described (Guo et al., 2006). Mice were housed in filter-top microisolation cages in an AALAC approved animal care facility and provided food and water ad libitum. Animals received humane care in compliance with the institution’s guidelines outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Mice (12–15 wk) were exposed intratracheally to phosphate buffered saline (PBS) control or freshly prepared NM (0.08 mg/kg; mechlorethamine hydrochloride, Sigma Aldrich, St. Louis, MO) as previously described (Sunil et al., 2018). At this age, the phenotype of FXR−/− mice resembled WT mice.
2.2. Sample collection
Mice were euthanized 3 d, 14 d and 28 d after exposure by intraperitoneal injection of ketamine (135 mg/kg) and xylazine (30 mg/kg). Bronchoalveolar lavage (BAL) fluid was collected by slowly instilling and withdrawing 1 ml of ice-cold PBS into the lungs five times through a 20-gauge cannula in the trachea. Immediately following BAL collection, the left lobe was ligated, inflated via the trachea with 3% paraformaldehyde and removed. After 24 h at 4°C, the tissue was transferred to 50% ethanol and paraffin embedded. Sections (5 μm) were prepared and stained with hematoxylin and eosin and Masson’s trichrome. Images were scanned using an Olympus VS-120 Virtual Microscopy System and viewed using OlyVIA version 2.6 software (Olympus Corporation, Center Valley, PA). Lung sections were scored for epithelial hyperplasia, perivascular inflammation, perivascular edema, neutrophils, foamy alveolar macrophages, alveolar wall thickening, and fibrosis by a boardcertified pathologist (M. Goedken, Rutgers University).
2.3. BAL analysis
BAL fluid was centrifuged (300 x g, 8 min, 4°C), supernatants collected, aliquoted and stored at −80C until analysis. Cell pellets were resuspended in PBS and viable cells counted on a hemocytometer using trypan blue dye exclusion. Total protein was quantified in cell free BAL using a BCA Protein Assay kit (Pierce Biotechnologies Inc., Rockford, IL) with bovine serum albumin as the standard. Samples were analyzed in triplicate using a SpectraMAX M3 microplate reader at 560 nm (Molecular Devices, San Jose, CA).
2.4. Flow cytometry
BAL cells were centrifuged (400 x g for 6 min) and resuspended in 700 μl of red blood cell (RBC) lysis buffer (Sigma Aldrich) for 5 min at room temperature. Cells were then centrifuged (400 x g, 6 min) and resuspended in 100 μl of staining buffer (PBS containing 2% FBS and 0.02% sodium azide). This was followed by incubation for 10 min at 4°C with anti-mouse CD16/32 (1:100; BioLegend, San Diego, CA) to block non-specific binding, and then with FITC-conjugated anti-mouse CD11b (1:200; BioLegend), PE-CF594-conjugated anti-mouse Ly6C (1:200; BD Biosciences, San Jose, CA), PerCP-Cy5.5-conjugated anti-mouse Siglec F (1:200; BD Biosciences), PECy7-conjugated anti-mouse F4/80 (1:200; BioLegend), AlexaFluor647-conjugated anti-mouse Ly6G (1:200; BioLegend), AlexaFluor700-conjugated anti-mouse CD45 (1:200; BioLegend), Brilliant violet 421-conjugated anti-mouse CD11c (1:200; BioLegend) antibodies or appropriate isotypic controls for 30 min, followed by eFluor780-conjugated fixable viability dye (eBioscience, San Diego, CA) for 30 min. Cells were washed twice with staining buffer, fixed with 3% paraformaldehyde, and analyzed on a Gallios flow cytometer (Beckman Coulter Inc., Brea, CA). Data were analyzed using Beckman Coulter Kaluza version 2.1 software. Viable cells (10,000) were assessed for expression of CD45, followed by CD11b, Ly6G, Ly6C, F4/80, CD11c, and Siglec F.
2.5. Immunohistochemistry
Tissue sections were deparaffinized with xylene (4 min, 2×) followed by decreasing concentrations of ethanol (100%–50%) and then water. Antigen retrieval was performed using citrate buffer (10.2 mM sodium citrate, pH 6.0). Endogenous peroxidase was quenched with 3% H2O2 for 10 min. Tissue sections were incubated for 2 h at room temperature with 25–50% goat serum to block nonspecific binding, and then overnight at 4°C with rabbit antibody to heme oxygenase-1 (HO-1;1:500; Enzo Life Sciences, Famingdale, NY), cyclooxygenase-2 (COX-2; 1:1000; Abcam, Cambridge, MA), ADP-ribosylation factor-like protein 11 (ARL11; 1:100; Bioss, Woburn, MA), signal transducer and activator of transcription 6 (STAT6, 1:200; Abcam, Cambridge, MA), CD163 (1:100; Biorbyt, St. Louis, MO) or appropriate IgG controls. Sections were then incubated with biotinylated secondary antibody (Vector Labs, Burlingame, CA) for 30 min at RT. Binding was visualized using a peroxidase diaminobenzidine (DAB) Substrate Kit (Vector Labs). Semiquantitative analysis of macrophage staining was performed on fifteen random lung sections from each animal.
2.6. miRNA analysis
cDNA was generated from lung tissue samples using a TaqMan Advanced miRNA cDNA Synthesis Kit (Applied Biosystems, Waltham, MA). RT-qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a Quant Studio Flex 6 PCR machine (Applied Biosystems). Ct values were converted to ΔΔCt values and normalized to the geometric mean of endogenous controls, miR-16-5p and miR-221-3p, which were selected because of their stability (Donati et al., 2019; Pamedytyte et al., 2020).
2.7. Statistical analysis
Data were analyzed using two-way ANOVA, followed by Tukey’s post hoc test. A p value of ≤ 0.05 was considered statistically significant.
3. Results
3.1. Effects of loss of FXR on NM-induced lung injury, inflammation, and oxidative stress
In initial studies, we analyzed the effects of loss of FXR on BAL cell number and protein content, markers of alveolar epithelial barrier injury (Bhalla, 1999). In male, but not female WT and FXR−/− mice, BAL cell number and protein content were significantly increased relative to control mice at 3 d and 14 d post-NM (Fig. 1). No significant differences were observed between the genotypes. Whereas at 28 d post-NM, BAL cell and protein content were increased in both male and female WT mice, in FXR−/− mice they were only increased in male mice.
Fig. 1. Effects of loss of FXR on BAL protein and cell content.
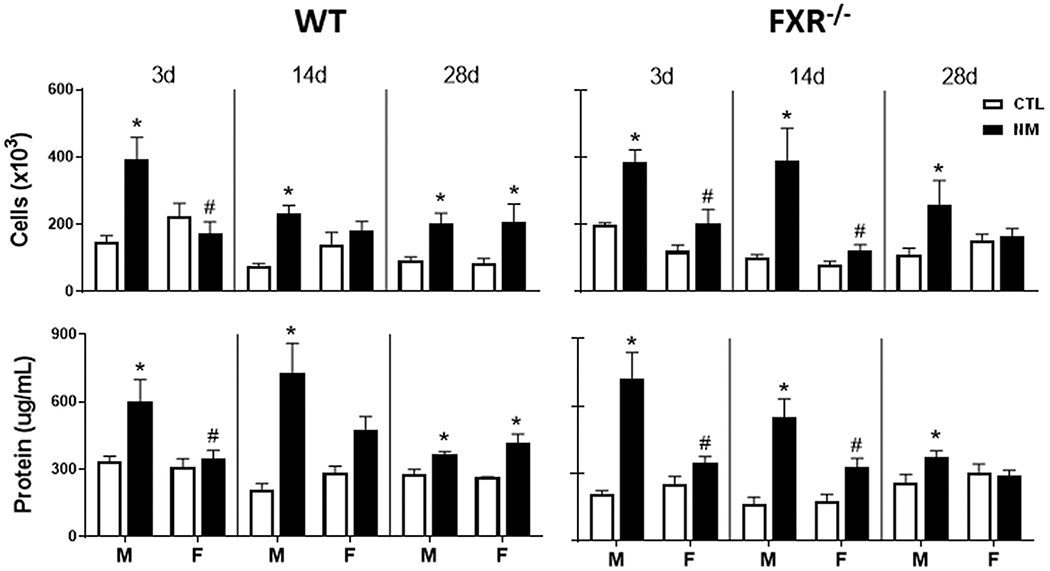
BAL was collected 3 d, 14 d, and 28 d after administration of PBS (CTL) or NM to male (M) and female (F) wild-type (WT) and FXR−/− mice. Upper panels: Viable cells were enumerated by trypan blue exclusion dye. Lower panels: Cell-free supernatants were analyzed in triplicate for protein using a BCA kit. Bars, mean ± SE (n = 4–13 mice/treatment group). *Significantly different (p ≤ 0.05) from CTL treated mice. #Significantly different (p ≤ 0.05) from male mice.
We next assessed the impact of the loss of FXR on NM-induced histopathological changes in the lungs. In male WT mice, NM caused increases in perivascular inflammation, edema and alveolar neutrophils, which were observed 3 d and 14 d post-exposure (Fig. 2 and Table 1). At 14 d post-NM, foamy macrophages were also noted, along with some alveolar wall thickening and fibrosis, as determined by Masson’s trichrome staining; these responses persisted for at least 28 d (Fig. 3 and Table 1). Loss of FXR was associated with significant increases in epithelial hyperplasia and edema, alveolar wall thickening and fibrosis at 14 d after NM administration, and perivascular edema at 14 d and 28 d. In male WT mice, NM also caused oxidative stress, as reflected by upregulation of HO-1 in lung macrophages (Fig. 4 and Table 2). Greater numbers of macrophages expressing HO-1 were observed in lungs of FXR−/− mice when compared to WT mice (Table 2). In contrast, in female WT mice, NM had no significant effects on histopathology or oxidative stress, a response not altered by loss of FXR (not shown). These data indicate that male mice were more sensitive to NM-induced toxicity than female mice. Based on these findings, in subsequent studies we focused only on male mice.
Fig. 2. Effects of loss of FXR on lung histology.
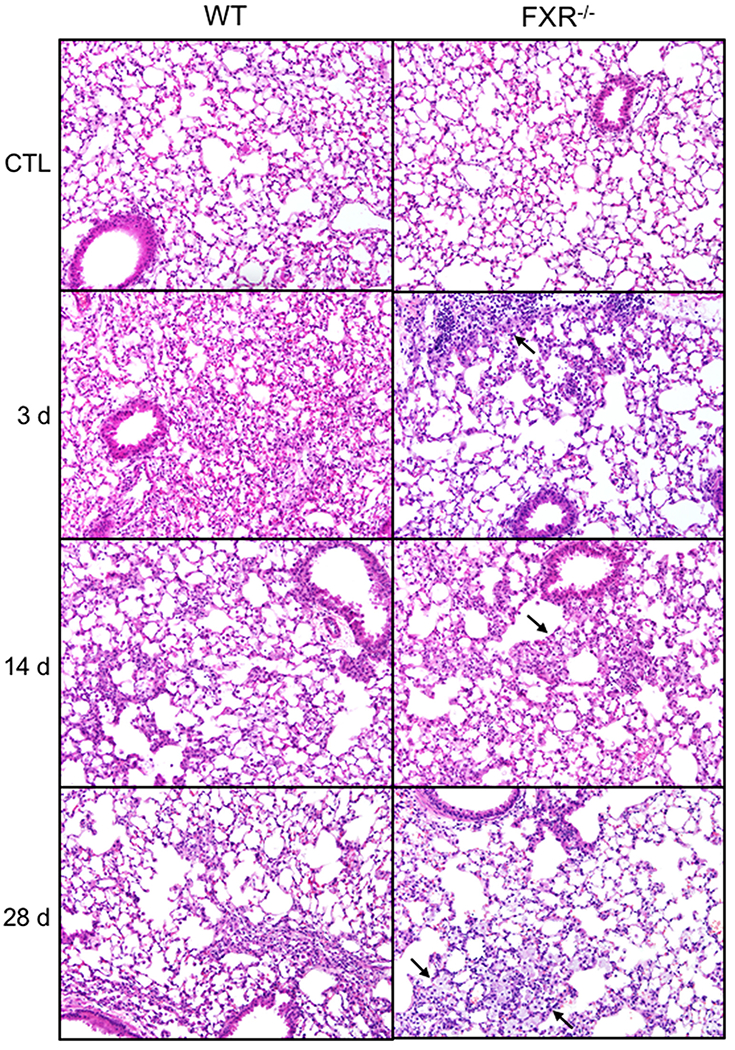
Tissue sections were prepared 3 d, 14 d and 28 d after administration of PBS (CTL) or nitrogen mustard (NM) to male wild-type (WT) and FXR−/− mice and stained with H&E. Representative sections from 4—5 mice/treatment group are shown. Original magnification, 20×. Arrows indicate differences observed between WT and FXR−/− mice.
Table 1. Summary of effects of loss of FXR on NM-induced lung histopathology.
Sections were prepared 3 d, 14 d and 28 d after administration of PBS (CTL) or nitrogen mustard (NM) to male wild-type (WT) and FXR−/− mice. Histopathologic changes were quantified in H&E and Masson’s trichrome stained sections as described in the Materials and Methods. Values are mean ± SE, n = 4–5 mice/treatment/time point.
| Histopathology | WT |
FXR−/− |
|||
|---|---|---|---|---|---|
| CTL | NM | CTL | NM | ||
| 3 d | Epithelial hyperplasia | – | – | – | – |
| Perivascular inflammation | – | 1.4 ± 0.2 | – | 1.75 ± 0.3 | |
| Perivascular edema | – | 1.4 ± 0.2 | – | 2.0 ± 0.0# | |
| Neutrophils (alveolar) | – | 1.0 ± 0.3 | – | 1.5 ± 0.3 | |
| Foamy alveolar macrophages | – | – | – | – | |
| Alveolar wall thickening | – | – | – | – | |
| Fibrosis | – | 0.4 ± 0.2 | – | 0.5 ± 0.3 | |
| 14 d | Epithelial hyperplasia | – | 0.2 ± 0.2 | – | 0.8 ± 0.2# |
| Perivascular inflammation | – | 0.8 ± 0.2 | – | 1.2 ± 0.4 | |
| Perivascular edema | – | – | – | 0.8 ± 0.4 | |
| Neutrophils (alveolar) | – | 0.4 ± 0.2 | – | 0.8 ± 0.2 | |
| Foamy alveolar macrophages | – | 1.4 ± 0.2 | – | 1.8 ± 0.2 | |
| Alveolar wall thickening | – | 1.6 ± 0.7 | – | 3.8 ± 0.7# | |
| Fibrosis | 0.2 ± 0.2 | 1.0 ± 0.3 | 0.2 ± 0.2 | 2.2 ± 0.4*# | |
| 28 d | Epithelial hyperplasia | – | – | – | – |
| Perivascular inflammation | – | 1.0 ± 0.0 | 0.2 ± 0.0 | 1.0 ± 0.0 | |
| Perivascular edema | – | – | – | – | |
| Neutrophils (alveolar) | – | – | – | – | |
| Foamy alveolar macrophages | – | 1.0 ± 0.0 | – | 1.0 ± 0.0 | |
| Alveolar wall thickening | – | 1.0 ± 0.0 | – | 1.0 ± 0.0 | |
| Fibrosis | – | 1.0 ± 0.0 | 0.4 ± 0.2 | 0.8 ± 0.2 | |
Significantly different (p ≤ 0.05) from CTL mice.
Significantly different (p ≤ 0.05) from WT mice.
Fig. 3. Effects of loss of FXR on the development of fibrosis.
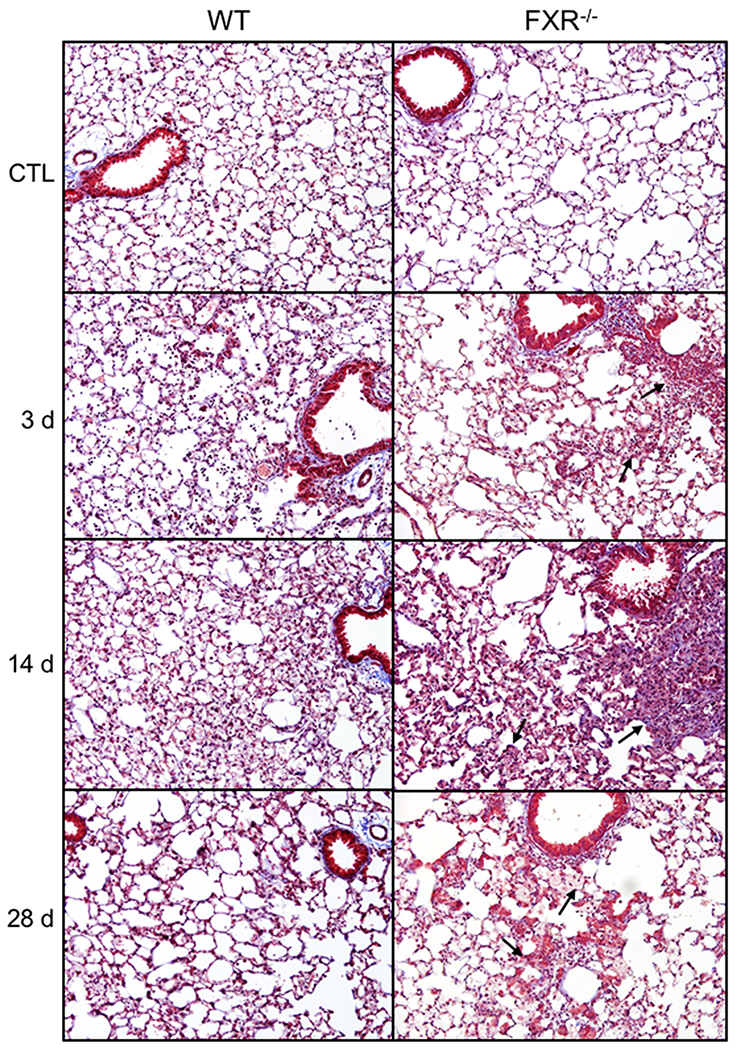
Tissue sections were prepared 3 d, 14 d, and 28 d after administration of PBS (CTL) or nitrogen mustard (NM) to male wild-type (WT) and FXR−/− mice and stained with Masson’s trichrome. Representative sections from 4–5 mice/treatment group are shown. Original magnification, 20×. Arrows indicate differences observed between WT and FXR−/− mice.
Fig. 4. Effects of loss of FXR on NM-induced expression of HO-1.
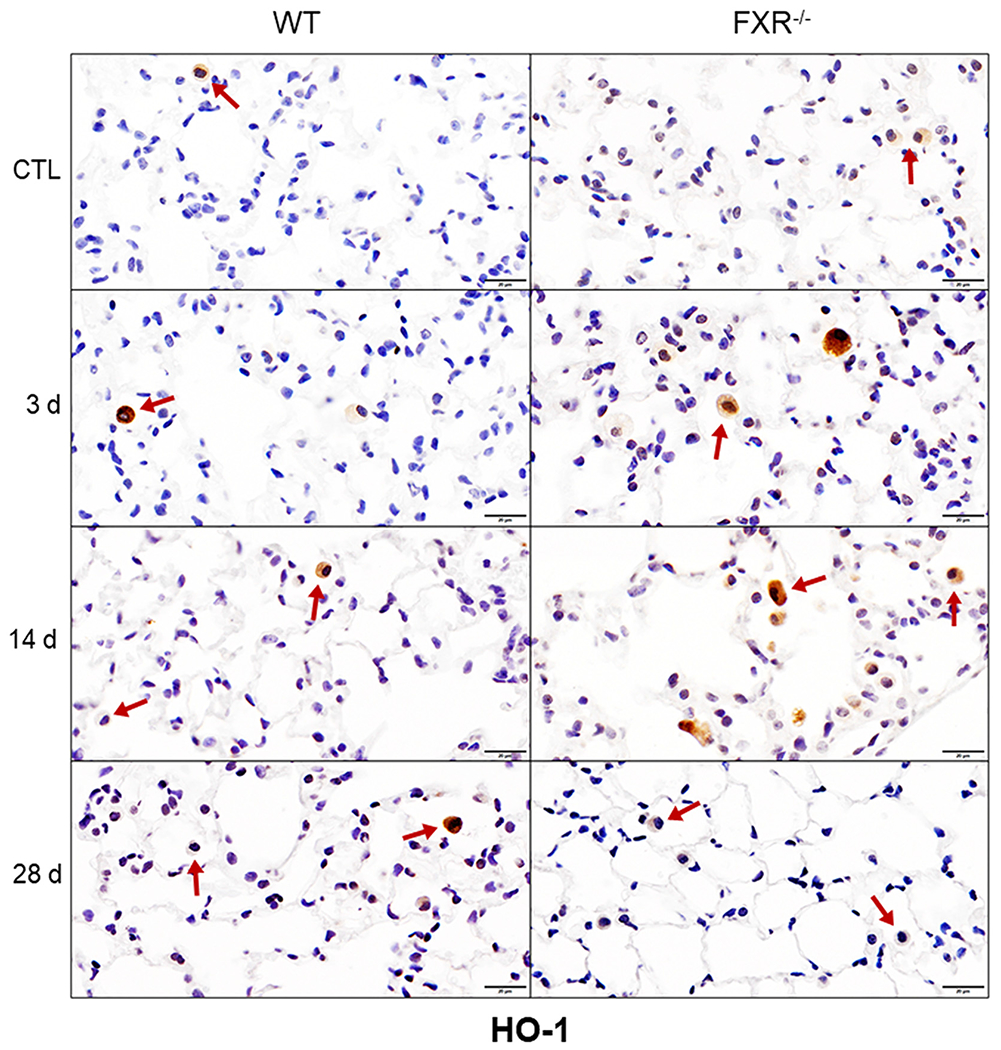
Lung sections prepared 3 d, 14 d and 28 d after administration of PBS (CTL) or NM to male wild-type (WT) and FXR−/− mice were immunostained with antibodies to HO-1. Binding was visualized using a peroxidase DAB substrate kit. Arrows indicate lung macrophages. Representative sections from 3– 5 mice/treatment group are shown. Original magnification, 40×.
Table 2. Semiquantitative analysis of immunohistochemistry in lung macrophages.
Lung sections prepared 3 d, 14 d and 28 d after administration of PBS (CTL) or nitrogen mustard (NM) to wild-type (WT) and FXR−/− mice were immunostained with antibodies to HO-1, COX-2, ARL11, STAT6, or CD163. Binding was visualized using a Vectastain kit. The number of macrophages positively staining for each antibody was quantified in 15 sections/mouse (original magnification 40×). Values are mean ± SE, n = 3–5 mice/treatment/time point. Data were analyzed by two-way ANOVA.
| Marker | WT |
FXR−/− |
|||
|---|---|---|---|---|---|
| CTL | NM | CTL | NM | ||
| 3 d | HO-1 | 21.0 ± 1.5 | 47.7 ± 0.7* | 61.3 ± 0.3# | 60.7 ± 2.9# |
| COX-2 | 5.0 ± 1.5 | 8.0 ± 2.1 | 7.7 ± 4.7 | 18.7 ± 1.8*,# | |
| ARL11 | 37.0 ± 2.5 | 38.7 ± 4.8 | 48.0 ± 3.2 | 44.0 ± 3.1 | |
| STAT6 | 45.7 ± 6.6 | 57.0 ± 8.0 | 48.0 ± 4.2 | 65.7 ± 11.6 | |
| CD163 | 29.0 ± 5.8 | 35.3 ± 4.8 | 26.0 ± 1.5 | 34.3 ± 1.8 | |
| 14 d | HO-1 | 8.0 ± 2.2 | 41.4 ± 6.5* | 21.4 ± 2.6 | 150.3 ± 6.5*,# |
| COX-2 | 13.8 ± 3.9 | 26.6 ± 6.4* | 22.2 ± 2.4 | 117.8 ± 6.1*,# | |
| ARL11 | 28.2 ± 4.4 | 59.4 ± 9.6* | 34.4 ± 1.7 | 102.4 ± 8.7*,# | |
| STAT6 | 31.0 ± 8.4 | 52.7 ± 9.0 | 40.0 ± 7.6 | 113.7 ± 18.8*,# | |
| CD163 | 4.6 ± 1.8 | 34.7 ± 4.9* | 14.2 ± 0.7 | 45.0 ± 13.5*,# | |
| 28 d | HO-1 | 24.0 ± 2.3 | 51.3 ± 2.2* | 13.0 ± 3.1# | 32.7 ± 2.0*,# |
| COX-2 | 30.0 ± 3.8 | 58.0 ± 6.4* | 20.7 ± 1.8 | 33.0 ± 5.2# | |
| ARL11 | 39.3 ± 2.8 | 54.3 ± 4.9 | 34.3 ± 5.4 | 54.7 ± 3.8* | |
| STAT6 | 58.7 ± 8.4 | 78.0 ± 12.7 | 42.0 ± 12.4 | 73.3 ± 3.8 | |
| CD163 | 38.3 ± 3.8 | 41.7 ± 9.4 | 20.3 ± 2.0 | 49.0 ± 3.2* | |
Significantly different (p ≤ 0.05) from CTL treated mice.
Significantly different (p ≤ 0.05) from WT mice.
3.2. Loss of FXR alters resident and inflammatory macrophage subsets responding to NM
Flow cytometric analysis of cells recovered from the lungs by BAL revealed a significant decrease in CD45+CD11b−Ly6G−Ly6C−F4/80+CD11c+ resident macrophages at 3 d, and to a lesser extent at 14 d post-NM in both WT and FXR−/− mice, with no effect at 28 d (Fig. 5). Conversely, significant increases in CD45+CD11b+Ly6G−Ly6C+F4/80+CD11c+ proinflammatory macrophages were noted in the lungs of both WT and FXR−/− mice following NM administration, most prominently at 3 d (Fig. 5). At this time, the response was greater in FXR−/− mice when compared to WT mice. Whereas CD45+CD11b+ Ly6G−Ly6C−F4/80+CD11c+ anti-inflammatory macrophages increased at all time points following NM exposure in WT and FXR−/− mice, there were no significant differences between the genotypes.
Fig. 5. Effects of loss of FXR on lung macrophages responding to NM.
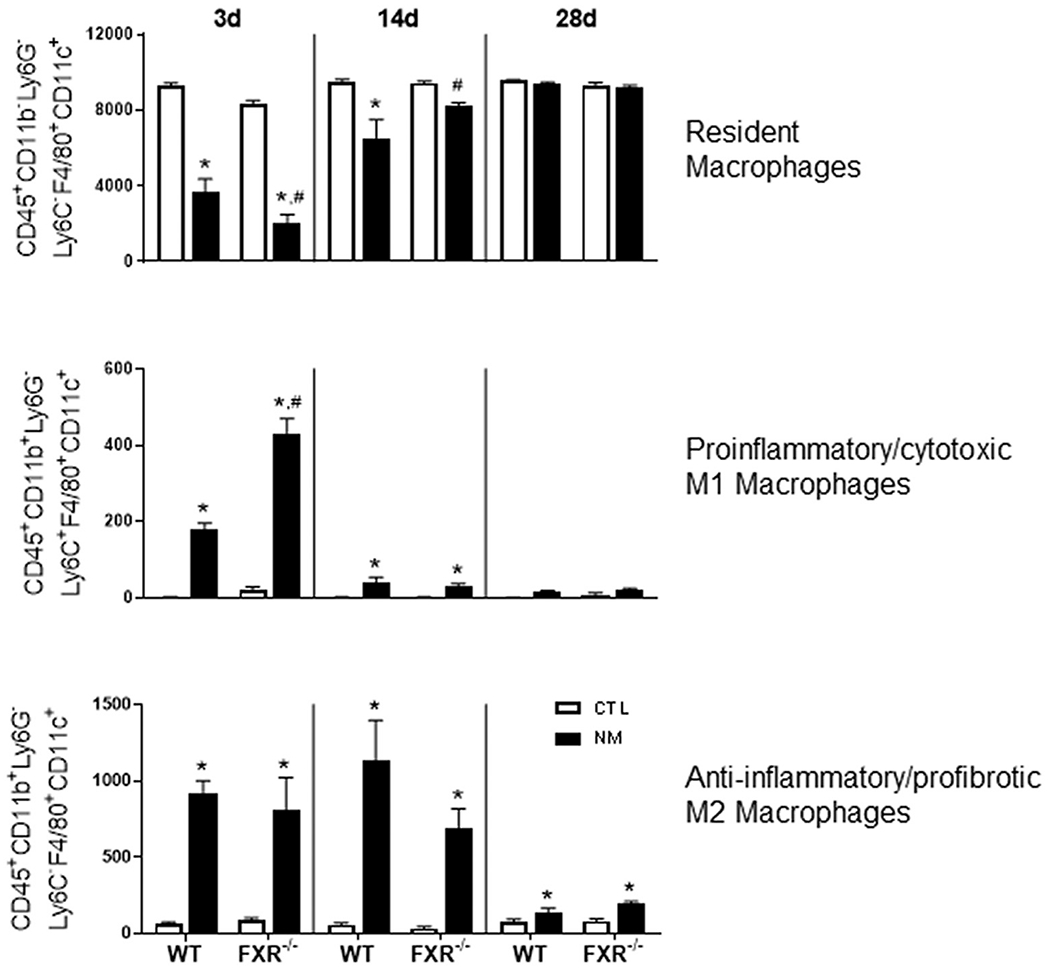
Lung macrophages were collected 3 d, 14 d, and 28 d after administration of PBS (CTL) or NM to male wild-type (WT) and FXR−/− mice. Cells were immunostained with antibodies to CD45, CD11b, Ly6C, Ly6G, F4/80, and CD11c or isotypic controls and analyzed by flow cytometry. Bars, mean ± SE (n = 5–9 mice/treatment group). Y-axis, macrophages/10,000 viable BAL cells. *Significantly different (p ≤ 0.05) from CTL treated mice. #Significantly different (p ≤ 0.05) from WT mice.
To determine if macrophages responding to NM were activated, we analyzed their expression of proinflammatory and anti-inflammatory proteins in histologic sections. ARL11 is an endogenously expressed protein that promotes proinflammatory activation of macrophages (Arya et al., 2018). Treatment of WT mice with NM resulted in a significant increase in numbers of macrophages staining for ARL11 at 14 d; this response was significantly greater in FXR−/− mice (Fig. 6 and Table 2). COX-2 expression was also upregulated in lung macrophages in WT mice following NM exposure; this was evident at all post exposure times (Fig. 6 and Table 2). Loss of FXR resulted in an exacerbated response to NM. Constitutive expression of COX-2 was also detected in alveolar epithelial type II cells in control WT and FXR−/− mice; expression increased after NM exposure (Fig. 6).
Fig. 6. Effects of loss of FXR on NM-induced increases in expression of COX-2 and ARL11.
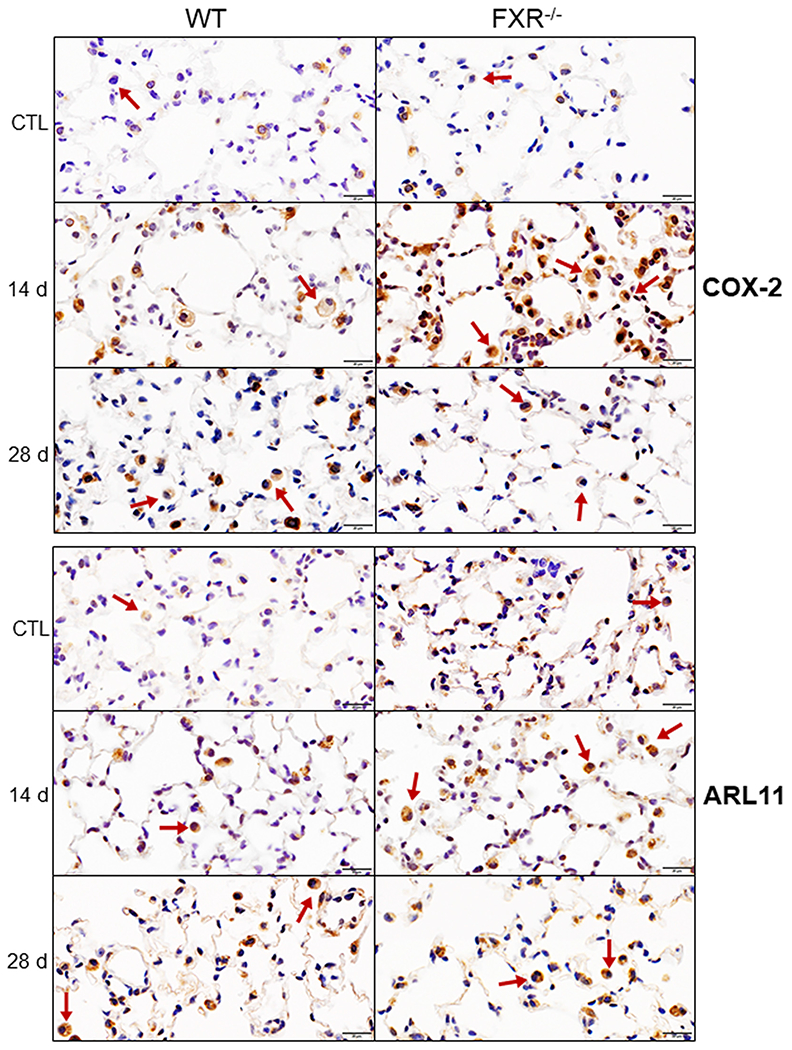
Lung sections prepared 14 d and 28 d after administration of PBS (CTL) or NM to male wild-type (WT) and FXR−/− mice were immunostained with antibodies to COX-2 and ARL11. Binding was visualized using a peroxidase DAB substrate kit. Arrows indicate lung macrophages. Representative sections from 3–5 mice/treatment group are shown. Original magnification, 40×.
We also found that markers of anti-inflammatory activation including CD163 and STAT6 were upregulated in lung macrophages in both WT and FXR−/− mice after NM (Fig. 7 and Table 2). This was most prominent at 14 d post NM exposure. At this time, the response of FXR−/− mice to NM was significantly greater than the response of WT mice. While in WT mice NM-induced increases in CD163 declined at 28 d, in FXR−/− mice they remained upregulated.
Fig. 7. Effects of loss of FXR on NM-induced increases in expression of STAT6 and CD163.
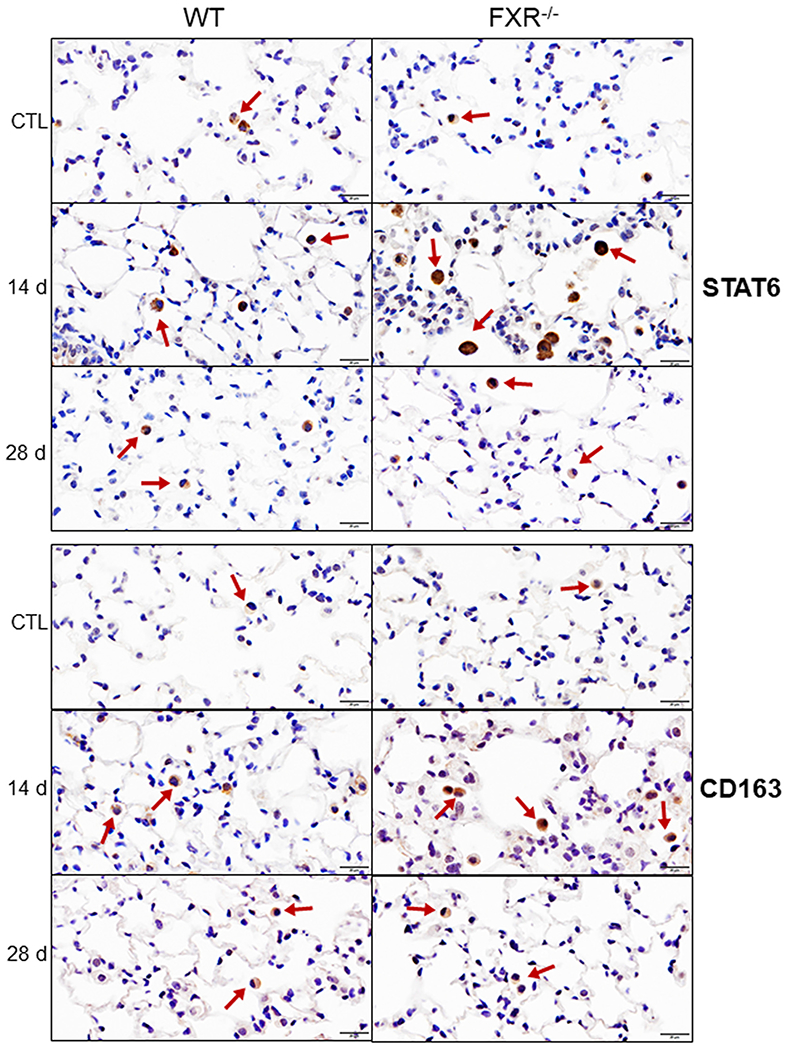
Lung sections prepared 14 d and 28 d after administration of PBS (CTL) or NM to male wild-type (WT) and FXR−/− mice were stained with antibodies to STAT6 and CD163. Binding was visualized using a peroxidase DAB substrate kit. Arrows indicate lung macrophages. Representative sections from 3–5 mice/treatment group are shown. Original magnification, 40×.
Noncoding microRNAs (miRs) have been identified as important regulators of inflammation and macrophage activation (Essandoh et al., 2016). In line with NM-induced proinflammatory macrophage activation, we found that proinflammatory miR-155 (Alivernini et al., 2018; Kurowska-Stolarska et al., 2011) was increased in the lungs of WT mice at 3 d post NM administration (Fig. 8). In FXR−/− mice, NM caused increases in proinflammatory miR-199a (Chen et al., 2008; Liu et al., 2018) and anti-inflammatory miR-146a and miR-223-3p (Boldin et al., 2011; Cheng et al., 2014; Yuan et al., 2021). Although NM had no significant effect on expression of miR-155 when compared to control in FXR−/− mice, there was a trend towards increased lung expression of this miRNA relative to WT mice. In contrast, NM had no effect on expression of miR-145a or miR-let-7d in either WT or FXR−/− mice.
Fig. 8. Effects of loss of FXR on miRNAs responding to NM.
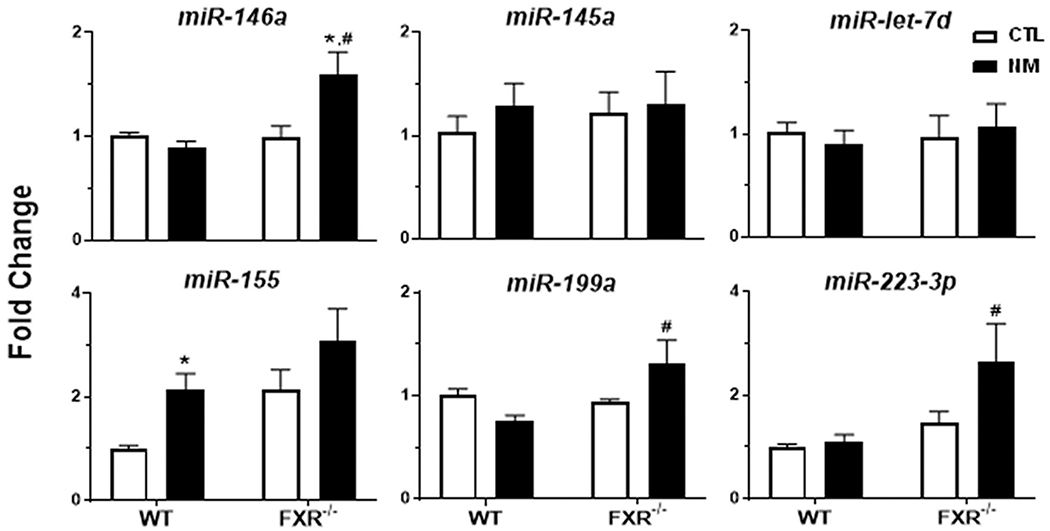
Lung tissue was collected 3 d after administration of PBS (CTL) or nitrogen mustard (NM) to male wild-type (WT) and FXR−/− mice. Data are presented as fold change relative to the geometric mean of miR-16-5p and miR-221-3p. Bars, mean ± SE (n = 5 mice/treatment group). *Significantly different (p ≤ 0.05) from CTL treated mice. #Significantly different (p ≤ 0.05) from WT mice.
4. Discussion
The present studies demonstrate that FXR plays a role in regulating lung macrophage responses to NM. This is based on our findings that loss of FXR results in increased numbers of proinflammatory/cytotoxic M1 macrophages in the lung responding to NM-induced injury. Moreover, after NM exposure, both M1 and anti-inflammatory/profibrotic M2 macrophages upregulate markers of activation. Conversely, resident macrophages, which are thought to be key in the resolution of inflammation and wound repair (Bain and MacDonald, 2022), are reduced in numbers. The fact that changes in macrophage subsets correlated with exaggerated histopathology, alveolar epithelial injury and oxidative stress provide additional support for the notion that these cells are important in the pathogenic response to mustard vesicants (Laskin et al., 2019; Venosa et al., 2016).
Consistent with previous studies (Malaviya et al., 2015; Sunil et al., 2020), NM was found to cause alveolar epithelial damage, as measured by increases in BAL cell and protein content. Notably, this response was greater in male FXR−/− mice relative to female mice, and accompanied by aggravated histopathologic alterations including perivascular edema, thickening of alveolar septal wall, inflammatory cell infiltrate, and collagen deposition. Oxidative stress was also evident in lungs of male, but not female, FXR−/− mice after NM. Epidemiological studies have shown that the prevalence of lung disease, including pulmonary fibrosis, acute respiratory distress syndrome and chronic obstructive disease (COPD) is greater in males than in females (Mannino et al., 1996; Moss and Mannino, 2002; Tam et al., 2011; Varkey, 2004). Similar sex differences in susceptibility of males have been noted in experimental models of acute lung injury induced by acid, lipopolysaccharide, hypoxia, and hyaluronan (Card et al., 2006; Erfinanda et al., 2021; Leary et al., 2019; Xie et al., 2021). Estrogen has been reported to protect rodents from lung damage induced by influenza, intestinal ischemia and reperfusion injury by altering pulmonary inflammatory responses (Breithaupt-Faloppa et al., 2014; Robinson et al., 2014). It remains to be determined if estrogen plays a role in protecting female FXR−/− mice from exacerbation of acute lung injury, oxidative stress, and fibrosis induced by NM and if this is due to altered macrophage responsiveness.
Following NM administration, we observed a significant increase in numbers of proinflammatory macrophages in the lung 3 d post NM exposure in FXR−/− mice relative to WT mice. This correlated with increases in expression of COX-2 and ARL11, demonstrating that proinflammatory macrophages responding to NM are activated. Loss of FXR also resulted in increases in miR-146a, miR-155, miR199a, and miR-223-3p following NM exposure. The anti-inflammatory actions of FXR have been reported to be due to its ability to suppress NF-κB (Fiorucci et al., 2011). Our findings of increased expression of miR-155 and miR199a, which are known to activate NF-κB (Chen et al., 2008; Mann et al., 2017), suggest a potential molecular mechanism underlying exacerbated proinflammatory macrophage activation in the lungs of FXR−/− mice. Of note, miR-146a and miR-223-3p which suppress NF-κB activity (Cheng et al., 2014; Zhou et al., 2018), were also upregulated in the lungs of FXR−/− mice following NM administration. miR-146a and miR-155 have been reported to work in tandem to fine-tune NF-κB activity (Mann et al., 2017). Increases in miR-146a as well as miR-223-3p may represent a compensatory attempt to blunt excessive NF-κB activity and promote wound repair.
We also found that COX-2 expression was upregulated in alveolar epithelial Type II cells in FXR−/− mice after NM administration. Type II cells are known to express FXR (Chen et al., 2016; Wang et al., 2016); it may be that loss of FXR leads to NF-κB activation in Type II cells either directly or indirectly via SHP-1, an atypical nuclear receptor that is a FXR target gene and functions as a transcriptional repressor (Fiorucci et al., 2018; Hoeke et al., 2014), and as a consequence increased expression of COX-2.
In contrast to the effects of loss of FXR on numbers of proinflammatory macrophages in the lung, anti-inflammatory macrophages remained unchanged following NM administration. However, increases in expression of the anti-inflammatory activation markers, STAT6 and CD163, were noted in lung macrophages 14 d post NM exposure, a response that coincided with the appearance of foamy macrophages and fibrosis. These data suggest a transition of lung macrophages from an anti-inflammatory to a profibrotic M2 phenotype. In earlier studies, we demonstrated that FXR activity is suppressed following NM exposure; thus, expression of target genes that regulate lipid uptake including Cd36 were increased following NM exposure, while lipid efflux transporters, Abca1 and Abcg1, were downregulated; this was linked to increases in macrophage lipid content and foam cell formation (Venosa et al., 2019). Macrophage foam cells have been shown to play a key role in pulmonary fibrosis induced by bleomycin (Romero et al., 2015) and we speculate that they play a similar role in NM-induced pulmonary fibrosis. In “nonclassical” bile acid target organs such as the lungs and airways, FXR also exerts prominent anti-inflammatory and antifibrotic actions (Wu et al., 2020). This is thought to be due to its ability to suppress the activity of inflammatory cells either directly via inhibition of NF-κ or indirectly via activation of SHP-1 (Armstrong and Guo, 2017). FXR also downregulates expression of TGF-β1/SNAI1 and SNAI2 to suppress endothelial-to-mesenchymal transition and regulates the proportion of MMPs/TIMPs to blunt fibrogenesis (Comeglio et al., 2017; Fiorucci et al., 2018). Studies are in process to assess mechanisms underlying anti-inflammatory and anti-fibrotic actions of FXR in the lung following NM exposure.
Resident alveolar macrophages function as immune sentinels, protecting the lung from inhaled xenobiotics and initiating inflammatory responses (Byrne et al., 2015; Laskin et al., 2019). They also play a central role in the resolution of inflammation (Hussell and Bell, 2014; Kopf et al., 2015). Treatment of mice with NM resulted in a rapid (within 3 d) and transient decrease in resident macrophages in the lung; this response was exacerbated in mice lacking FXR. Decreases in resident macrophages early in the progression of NM-induced lung injury may contribute to excessive activation of proinflammatory macrophages. Subsequent recovery of resident macrophages by 14 d is likely due to proliferation of remaining subpopulations (Wynn and Vannella, 2016).
Accumulating evidence suggests that FXR plays an important role in both physiological and pathological responses of the respiratory system. Importantly, FXR is involved in protecting against the development of chronic diseases such as COPD, asthma, and fibrosis, pathologies observed in humans after mustard vesicant exposure (Comeglio et al., 2017; Fiorucci et al., 2018; Wu et al., 2020). Our findings that FXR is involved in limiting acute injury, oxidative stress and fibrosis induced by NM and that this response involves alterations in the behavior of resident and inflammatory macrophages are intriguing as they suggest a new target for the development of efficacious medical countermeasures. In this context, recent studies have shown that treatment of animals with the FXR agonist, obeticholic acid (OCA), improves pulmonary function and markers of inflammation and fibrosis in models of lung toxicity induced by bleomycin, lipopolysaccharide, and monocrotaline (Comeglio et al., 2019; Fei et al., 2019; Vignozzi et al., 2017). Studies are in progress to determine if OCA similarly reduces lung toxicity and fibrosis induced by NM.
Funding
This work was supported by the National Institutes of Health [AR055073, GM135258, ES004738, ES033698, ES005022, ES030984, S10OD026876, and T32ES007148].
Footnotes
CRediT authorship contribution statement
Alexa Murray: Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Visualization. Tanvi Banota: Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – review & editing, Visualization. Grace L. Guo: Conceptualization, Resources, Supervision, Writing – review & editing, Funding acquisition. Ley Cody Smith: Methodology, Validation, Investigation, Writing – review & editing. Jaclynn A. Meshanni: Methodology, Validation, Investigation, Writing – review & editing. Jordan Lee: Methodology, Validation, Investigation, Writing – review & editing. Bo Kong: Methodology, Validation, Investigation. Elena V. Abramova: Methodology, Validation, Investigation. Michael Goedken: Conceptualization, Supervision, Writing – review & editing, Funding acquisition. Andrew J. Gow: Conceptualization, Supervision, Writing – review & editing. Jeffrey D. Laskin: Conceptualization, Supervision, Writing – review & editing, Funding acquisition. Debra L. Laskin: Conceptualization, Supervision, Validation, Writing – review & editing, Visualization, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
No data was used for the research described in the article.
References
- Alivernini S, Gremese E, McSharry C, et al. , 2018. MicroRNA-155—at the critical interface of innate and adaptive immunity in arthritis. Front. Immunol 8 10.3389/fimmu.2017.01932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong LE, Guo GL, 2017. Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep 3 (2), 92–100. 10.1007/S40495-017-0085-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya SB, Kumar G, Kaur H, et al. , 2018. ARL11 regulates lipopolysaccharide-stimulated macrophage activation by promoting mitogen-activated protein kinase (MAPK) signaling. J. Biol. Chem 293 (25), 9892–9909. 10.1074/jbc.RAl17.000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, MacDonald AS, 2022. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol. 15 (2), 223–234. 10.1038/s41385-021-00480-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla DK, 1999. Ozone-induced lung inflammation and mucosal barrier disruption: toxicology, mechanisms, and implications. J. Toxicol. Environ. Health B 2 (1), 31–86. 10.1080/109374099281232. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Taganov KD, Rao DS, et al. , 2011. MiR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med 208 (6), 1189–1201. 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breithaupt-Faloppa AC, Thais Fantozzi E, Romero DC, et al. , 2014. Acute effects of estradiol on lung inflammation due to intestinal ischemic insult in male rats. Shock. 41 (3), 208–213. 10.1097/shk.0000000000000092. [DOI] [PubMed] [Google Scholar]
- Byrne AJ, Mathie SA, Gregory LG, et al. , 2015. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 70 (12), 1189–1196. 10.1136/thoraxjnl-2015-207020. [DOI] [PubMed] [Google Scholar]
- Card JW, Carey MA, Bradbury JA, et al. , 2006. Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J. Immunol 177 (1), 621. 10.4049/jimmunol.177.l.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Alvero AB, Silasi DA, et al. , 2008. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 27 (34), 4712–4723. 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Cai H-R, Xue S, et al. , 2016. Bile acids induce activation of alveolar epithelial cells and lung fibroblasts through farnesoid X receptor-dependent and independent pathways. Respirology. 21 (6), 1075–1080. 10.1111/resp.12815. [DOI] [PubMed] [Google Scholar]
- Cheng HS, Njock M-S, Khyzha N, et al. , 2014. Noncoding RNAs regulate NF-κB signaling to modulate blood vessel inflammation. Front. Genet 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeglio P, Filippi S, Sarchielli E, et al. , 2017. Anti-fibrotic effects of chronic treatment with the selective FXR agonist obeticholic acid in the bleomycin-induced rat model of pulmonary fibrosis. J. Steroid Biochem. Mol. Biol 168, 26–37. 10.1016/j.jsbmb.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Comeglio P, Filippi S, Sarchielli E, et al. , 2019. Therapeutic effects of obeticholic acid (OCA) treatment in a bleomycin-induced pulmonary fibrosis rat model. J. Endocrinol. Investig 42 (3), 283–294. 10.1007/s40618-018-0913-1. [DOI] [PubMed] [Google Scholar]
- Donati S, Ciuffi S, Brandi ML, 2019. Human circulating miRNAs real-time qRT-PCR-based analysis: an overview of endogenous reference genes used for data normalization. Int. J. Mol. Sci 20 (18) 10.3390/ijms20184353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erfinanda L, Ravindran K, Kohse F, et al. , 2021. Oestrogen-mediated upregulation of the mas receptor contributes to sex differences in acute lung injury and lung vascular barrier regulation. Eur. Respir. J 57 (1), 2000921. 10.1183/13993003.00921-2020. [DOI] [PubMed] [Google Scholar]
- Essandoh K, Li Y, Huo J, et al. , 2016. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. 46 (2), 122–131. 10.1097/SHK.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Fu L, Hu B, et al. , 2019. Obeticholic acid alleviate lipopolysaccharide-induced acute lung injury via its anti-inflammatory effects in mice. Int. Immunopharmacol 66, 177–184. 10.1016/j.intimp.2018.ll.005. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Mencarelli A, Cipriani S, et al. , 2011. Activation of the farnesoid X receptor protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs in mice. Br. J. Pharmacol 164 (8), 1929–1938. 10.11111/j.1476-5381.2011.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Biagioli M, Zampella A, et al. , 2018. Bile acids activated receptors regulate innate immunity. Front. Immunol. 9 10.3389/fimmu.2018.01853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis M, Guo G, Kong B, et al. , 2020. Regulation of lung macrophage activation and oxidative stress following ozone exposure by farnesoid X receptor. Toxicol. Sci 177 (2), 441–453. 10.1093/toxsci/kfaalll. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai Z, Visentin M, Gui T, et al. , 2018. Effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-κB signaling, and hepatic inflammation. Mol. Pharmacol. 94 (2), 802. 10.1124/mol.117.111047. [DOI] [PubMed] [Google Scholar]
- Guo GL, Santamarina-Fojo S, Akiyama TE, et al. , 2006. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim. Biophys. Acta 1761 (12), 1401–1409. 10.1016/j.bbalip.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrick SM, Mroz MS, Greene CM, et al. , 2014. Bile acids stimulate chloride secretion through CFTR and calcium-activated Cl- channels in Calu-3 airway epithelial cells. Am. J. Phys. Lung Cell. Mol. Phys. 307 (5), L407–L418. 10.1152/ajplung.00352.2013. [DOI] [PubMed] [Google Scholar]
- Hoeke MO, Heegsma J, Hoekstra M, et al. , 2014. Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1. PLoS One 9 (2), e88011. 10.1371/journal.pone.0088011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer RJ, Elias JA, Lee CG, et al. , 2011. Modern concepts on the role of inflammation in pulmonary fibrosis. Arch. Pathol. Lab. Med. 135 (6), 780–788. 10.1043/2010-0296-ra.l. [DOI] [PubMed] [Google Scholar]
- Hussell T, Bell TJ, 2014. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14 (2), 81–93. 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- Kopf M, Schneider C, Nobs SP, 2015. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 16 (1), 36–44. 10.1038/ni.3052. [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Alivernini S, Ballantine Lucy E, et al. , 2011. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci 108 (27), 11193–11198. 10.1073/pnas.1019536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, et al. , 2011. Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288. 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin DL, Malaviya R, Laskin JD, 2019. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol. Sci. 168 (2), 287–301. 10.1093/toxsci/kfy309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary S, Das P, Ponnalagu D, et al. , 2019. Genetic strain and sex differences in a hyperoxia-induced mouse model of varying severity of bronchopulmonary dysplasia. Am. J. Pathol. 189 (5), 999–1014. 10.1016/j.ajpath.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lech M, Anders H-J, 2013. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta Mol. basis Dis 1832 (7), 989–997. 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Li H, Jiang T, Li M-Q, et al. , 2018. Transcriptional regulation of macrophages polarization by microRNAs. Front. Immunol. 9, 1175. 10.3389/fimmu.2018.01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Guan H, Zhang J-L, et al. , 2018. Acute downregulation of miR-199a attenuates sepsis-induced acute lung injury by targeting SIRT1. Am. J. Phys. Cell Phys. 314 (4), C449–C455. 10.1152/ajpcell.00173.2017. [DOI] [PubMed] [Google Scholar]
- Madison MC, Landers CT, Gu B-H, et al. , 2020. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J. Clin. Investig. 129 (10), 4290–4304. 10.1172/JCI128531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil VR, Venosa A, et al. , 2015. Attenuation of nitrogen mustard-induced pulmonary injury and fibrosis by anti-tumor necrosis factor-α antibody. Toxicol. Sci 148 (1), 71–88. 10.1093/toxsci/kiVl61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil VR, Venosa A, et al. , 2016. Macrophages and inflammatory mediators in pulmonary injury induced by mustard vesicants. Ann. N. Y. Acad. Sci 1374 (1), 168–175. 10.llll/nyas.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Mehta A, Zhao JL, et al. , 2017. An NF-κB-microRNA regulatory network tunes macrophage inflammatory responses. Nat. Commun 8 (1), 851. 10.1038/S41467-017-00972-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannino DM, Etzel RA, Parrish RG, 1996. Pulmonary fibrosis deaths in the United States, 1979-1991. An analysis of multipie-cause mortality data. Am. J. Respir. Crit. Care Med 153 (5), 1548–1552. 10.1164/ajrccm.153.5.8630600. [DOI] [PubMed] [Google Scholar]
- Morissette MC, Shen P, Thayaparan D, et al. , 2015. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur. Respir. J 46 (5), 1451. 10.1183/09031936.00216914. [DOI] [PubMed] [Google Scholar]
- Moss M, Mannino DM, 2002. Race and gender differences in acute respiratory distress syndrome deaths in the United States: an analysis of multiple-cause mortality data (1979–1996). Crit. Care Med 30 (8), 1679–1685. 10.1097/00003246-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Pamedytyte D, Leipute E, Zilaitiene B, et al. , 2020. Different stability of miRNAs and endogenous control genes in archival specimens of papillary thyroid carcinoma. Mol. Med 26 (1), 100. 10.1186/sl0020-020-00218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DP, Hall OJ, Nilles TL, et al. , 2014. 17β-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CDS T cell responses in the lungs. J. Virol 88 (9), 4711–4720. 10.1128/jvi.02081-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F, Shah D, Duong M, et al. , 2015. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol 53 (1), 74–86. 10.1165/rcmb.2014-03430C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik FB, Prasad DVR, Narala VR, 2015. Role of farnesoid X receptor in inflammation and resolution. Inflamm. Res 64 (1), 9–20. 10.1007/s00011-014-0780-y. [DOI] [PubMed] [Google Scholar]
- Sunil VR, Vayas KN, Cervelli JA, et al. , 2014. Pentoxifylline attenuates nitrogen mustard-induced acute lung injury, oxidative stress and inflammation. Exp. Mol. Pathol. 97 (1), 89–98. 10.1016/j.yexmp.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Vayas KN, Cervelli JA, et al. , 2018. Protective role of surfactant protein-D against lung injury and oxidative stress induced by nitrogen mustard. Toxicol. Sci 166 (1), 108–122. 10.1093/toxsci/kfyl88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Vayas KN, Abramova EV, et al. , 2020. Lung injury, oxidative stress and fibrosis in mice following exposure to nitrogen mustard. Toxicol. Appl. Pharmacol 387, 114798 10.1016/j.taap.2019.114798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, Morrish D, Wadsworth S, et al. , 2011. The role of female hormones on lung function in chronic lung diseases. BMC Womens Health 11, 24. 10.1186/1472-6874-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey AB, 2004. Chronic obstructive pulmonary disease in women: exploring gender differences. Curr. Opin. Pulm. Med 10 (2), 98–103. 10.1097/00063198-200403000-00003. [DOI] [PubMed] [Google Scholar]
- Venosa A, Malaviya R, Choi H, et al. , 2016. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 54 (3), 436–446. 10.1165/rcmb.2015-01200C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venosa A, Smith LC, Murray A, et al. , 2019. Regulation of macrophage foam cell formation during nitrogen mustard (NM)-induced pulmonary fibrosis by lung lipids. Toxicol. Sci 172 (2), 344–358. 10.1093/toxsci/kfzl87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignozzi L, Morelli A, Cellai I, et al. , 2017. Cardiopulmonary protective effects of the selective FXR agonist obeticholic acid in the rat model of monocrotaline-induced pulmonary hypertension. J. Steroid Biochem. Mol. Biol 165, 277–292. 10.1016/j.jsbmb.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Wang J, Huang Y, Hou X, et al. , 2016. Morphologic damage of rat alveolar epithelial type II cells induced by bile acids could be ameliorated by farnesoid X receptor inhibitor Z-guggulsterone in vitro. Biomed. Res. Int 2016, 9283204. 10.1155/2016/9283204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JN, Chen JR, Chen JL, 2020. Role of farnesoid X receptor in the pathogenesis of respiratory diseases. Can. Respir. J 2020, 9137251. 10.1155/2020/9137251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Vannella KM, 2016. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44 (3), 450–462. 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Xie D, Li B, et al. , 2021. Gender differences in low-molecular-mass-induced acute lung inflammation in mice. Int. J. Mol. Sci 22 (1), 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Wu Q, Wang Z, et al. , 2021. MiR-223: an immune regulator in infectious disorders. Front. Immunol 12 10.3389/fimmu.2021.781815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Pal AS, Hsu AY-H, et al. , 2018. MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. 22 (7), 1810–1823. 10.1016/j.celrep.2018.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.