

Abstract
A series of 24 new 1H-1,2,3-triazole hybrids of 3-O-acetyl-11-keto-β-boswellic acid (β-AKBA (1)) and 11-keto-β-boswellic acid (β-KBA (2)) was designed and synthesized by employing “click” chemistry in a highly efficient manner. The 1,3-dipolar cycloaddition reaction between β-AKBA-propargyl ester intermediate 3 or β-KBA-propargyl ester intermediate 4 with substituted aromatic azides 5a–5k in the presence of copper iodide (CuI) and Hünig’s base furnished the desired products—1H-1,2,3-triazole hybrids of β-AKBA (6a–6k) and β-KBA (7a–7k)—in high yields. All new synthesized compounds were characterized by 1H-, 13C-NMR spectroscopy, and HR-ESI-MS spectrometry. Furthermore, their α-glucosidase-inhibitory activity was evaluated in vitro. Interestingly, the results obtained from the α-glucosidase-inhibitory assay revealed that all the synthesized derivatives are highly potent inhibitors, with IC50 values ranging from 0.22 to 5.32 µM. Among all the compounds, 6f, 7h, 6j, 6h, 6g, 6c, 6k, 7g, and 7k exhibited exceptional inhibitory potency and were found to be several times more potent than the parent compounds 1 and 2, as well as standard acarbose. Kinetic studies of compounds 6g and 7h exhibited competitive and mixed types of inhibition, with ki values of 0.84 ± 0.007 and 1.18 ± 0.0012 µM, respectively. Molecular docking was carried out to investigate the binding modes of these compounds with α-glucosidase. The molecular docking interactions indicated that that all compounds are well fitted in the active site of α-glucosidase, where His280, Gln279, Asp215, His351, Arg442, and Arg315 mainly stabilize the binding of these compounds. The current study demonstrates the usefulness of incorporating a 1H-1,2,3-triazole moiety into the medicinally fascinating boswellic acids skeleton.
Keywords: 3-O-acetyl-11-keto-β-boswellic acid (AKBA); 11-keto-β-boswellic acid (KBA); 1H-1,2,3-triazole hybrids; click chemistry; α-glucosidase inhibitors; molecular docking studies
1. Introduction
Frankincense (oleo-gum exudates of Boswellia ssp.) is the most precious resin religiously, economically, socially, and medicinally. The distribution of the Boswellia trees ranges from South Arabia to India to the horn of Africa [1]. Frankincense exists in traditional Chinese medicine to treat ulcers, rheumatoid arthritis, dysmenorrhea, swelling, osteoarthritis, pain from injuries, and amenorrhea [2,3,4]. B. sacra is endemic to Oman, and the resin (olibanum or frankincense) of B. serrata is known for its superb anti-inflammatory and pro-apoptotic activity [5,6]. Frankincense and triterpenoids from B. sacra and other species of the same genus have been used in traditional medicine since ancient times to treat obesity and lipid disorders [4,7].
The oleo-gum resin of Boswellia contains an abundant quantity of pentacyclic triterpenoid acids and boswellic acids (BAs), and is reported to have multiple beneficial bioactivities: particularly, anticancer [8], hepatoprotective [9,10,11], anti-inflammatory [5], 5-lipoxygenase [12], neuroprotective [13], apoptosis [14], antiproliferative [15], antidepressant [1], anti-arthritic, and anti-tumor [7,9] bioactivities. Due to their known biological activities, boswellic acids are of special interest [16,17,18]. Due to the superb anti-inflammatory activity of β-AKBA (1) and β-KBA (2) (Figure 1), we selected them as precursors in the current study [15,19].
Figure 1.

Chemical structures of β-AKBA (1) and β-KBA (2).
In addition, the importance of 1H-1,2,3-triazole molecules in pharmaceuticals and agrochemicals has increased [20]. Triazole compounds play a key role in organic chemistry due to their multiple and broad range of applications in bio-medicinal, biochemical, and material sciences [21]. The interest of the compounds containing the triazole moiety underwent a substantial growth over the past few decades. Furthermore, heterocycles containing 1,2,3-triazole scaffolds are known to have biological activities including anti-microbial [22], anti-bacterial [23], anti-viral [24], anti-HIV [25], anti-inflammatory [26], and anti-cancer [27] activities. Our group have recently reported the synthetic modifications of the β-AKBA and β-ABA hybrid triazole moieties as potent carbonic anhydrase-II [28].
In the drug discovery process, the incorporation of an active pharmacophoric moiety into a core bioactive natural product is a key step in exploring a wide range of new chemical entities with enhanced biological activities. With this objective in our mind, and in continuation of our research interest involving the chemical transformation of compounds 1 and 2 through the molecular hybridization approach, we herein describe the synthesis of novel β-AKBA (1)- and β-KBA (2)-based 1H-1,2,3-triazole hybrids and molecular docking studies with respect to their α-glucosidase activities.
Hyperglycemia is one of the most serious diabetes outcomes, leading to long-term health issues such as cardiovascular diseases, metastatic cancer, retinopathy, and nephropathy [29]. According to a worldwide projection, factors such as population growth, aging, urbanization, and lifestyle may cause the global prevalence rate of diabetes to rise to 54 percent by 2030 [30]. α-glucosidase is a hydrolase enzyme which is present on the brush border of the small intestine and catalyzes carbohydrates digestion into glucose molecules, which are then absorbed into blood. The use of α-glucosidase inhibitors has been shown to be a reliable approach for drug development and to overcome hyperglycemia. As a result, α-glucosidase inhibitors (AGIs) have received a lot of interest due to their therapeutic usage in the treatment of nocturnal hypoglycemia [31]. A fundamental treatment method used to overcome hyperglycemia is to reduce carbohydrate digestion and absorption, which provides an insight into the important role of α-glucosidase. As a result, novel AGIs with fewer side effects are urgently needed, in contrast to the currently existing clinical AGIs, such as acarbose, voglibose, and miglitol [32,33]. Recently, it was reported in the literature that both 1H-1,2,3-triazole and BAs are active pharmacophores of α-glucosidase inhibitors [34,35,36]. In the current study, we conjugated the 1H-1,2,3-triazole with BAs to discover their hybrid behavior against α-glucosidase.
Keeping in mind the importance of boswellic acids, triazole moieties, and AGIs, we planned to synthesize their hybrids between BAs and triazoles, and screened them against the α-glucosidase enzyme. Furthermore, this is the first report, to the best of authors’ knowledge, on the synthetic hybrids of 1H-1,2,3-triazole with β-AKBA (1) and β-KBA (2). In addition, here, we report the crystal structure of compound 4 for the first time. Additionally, some structure–activity relationships, molecular docking, and kinetic studies of the active analogs are also discussed.
2. Results and Discussion
2.1. Chemistry
The compounds β-AKBA (1) (900 mg) and β-KBA (2) (840 mg) were employed as starting materials, which were obtained as white crystals from the oleo-gum resin of Boswellia sacra [37]. As the concentration of β-AKBA is low in the resin of B. sacra compared to other boswellic acids (BA, KBA, and ABA), in order to prepare (1) and (2) in large quantities, we transformed a cluster of BAs (20 gm) into β-AKBA and purified them through column chromatography (70–230 mesh) using the increasing polarity of a 20–30% n-hexane/EtOAc solvent system as a mobile phase to obtain pure β-AKBA (3.2 gm). In the second step, β-AKBA (1.6 gm) was deacetylated and purified through column chromatography (CC) using 30–40% n-hexane/EtOAc as a mobile phase to afford pure β-KBA (1.4 gm) (Scheme 1). The structures of the compounds were confirmed through 1D (1H- and 13C) NMR spectroscopy and mass spectrometry (HR-ESI-MS) [7,15,37].
Scheme 1.

Large-scale production of β-AKBA (1) and β-KBA (2). Reagents and conditions: (a) Ac2O/Py/DMAP, CH2Cl2, room temperature, 6 h; (b) NBS/CaCO3/H2O/hν, dioxane, room temperature, 10 h; (c) 0.5 N KOH in iPrOH, reflux, 10 h.
The synthetic scheme of 1H-1,2,3-triazole hybrids of β-AKBA (1) and β-KBA (2) is depicted in Scheme 2. In the present study, a series of new analogues of β-AKBA and β-KBA containing the 1H-1,2,3-triazole moiety were designed and synthesized. The desired 1H-1,2,3-triazole hybrids (6a–6k and 7a–7k) were achieved using a two-step protocol with propargylation followed by Huisgen’s 1,3-dipolar cycloaddition reaction. Thereby, in the initial step, compounds 1 and 2 were treated in DMF with propargyl bromide in the presence of potassium carbonate at room temperature to afford the β-AKBA-propargyl ester intermediate 3 (yield 96%) and β-KBA-propargyl ester intermediate 4 [38,39,40,41].
Scheme 2.

Reagents and conditions: (a) propargyl bromide, K2CO3, DMF, room temperature, 18 h, 94–96%; (b) R1-N3 (different substituted aromatic azides, 5a–k), CuI, Et3N, CH3CN, room temperature, 3 h, 6a–6k (87–96%), 7a–7k (86–96%).
Step 2 was carried out using “click” chemistry. In step 2, a 1,3-dipolar cycloaddition reaction was conducted between β-AKBA-propargyl ester intermediate 3 and aromatic azides 5a–5k, in the presence of copper iodide (CuI) and Hünig’s base in MeCN, to obtain the desired products—1H-1,2,3-triazole hybrids of β-AKBA derivatives 6a–6k—in very good yields (87–96%) [40,41]. Under similar conditions, the 1H-1,2,3-triazole hybrids of β-KBA derivatives 7a–7k in excellent yields of 86–96% were obtained from β-KBA-propargyl ester intermediate 4 (Table 1).
Table 1.
Synthesis of 1H-1,2,3-triazole hybrids of β-AKBA a β-KBA.
| Compounds | Azides (5a-k) R1-Group |
1H-1,2,3-Triazole Products (6a–k and 7a–k) |
Yields of Products % a |
|---|---|---|---|
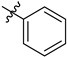
| 3 | Ph | 6a | 90 |
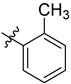
| 3 | 2-MeC6H4 | 6b | 87 |
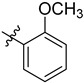
| 3 | 2-MeOC6H4 | 6c | 89 |
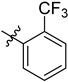
| 3 | 2-F3CC6H4 | 6d | 96 |
| 3 | 4-MeOC6H4 | 6e | 88 |
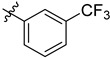
| 3 | 3-F3CC6H4 | 6f | 95 |
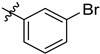
| 3 | 3-BrC6H4 | 6g | 92 |
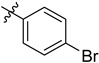
| 3 | 4-BrC6H4 | 6h | 91 |
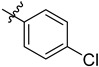
| 3 | 4-ClC6H4 | 6i | 93 |
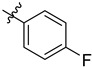
| 3 | 4-FC6H4 | 6j | 94 |
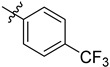
| 3 | 4-F3CC6H4 | 6k | 96 |
| 4 | Ph | 7a | 89 |
| 4 | 2-MeC6H4 | 7b | 86 |
| 4 | 2-MeOC6H4 | 7c | 88 |
| 4 | 2-F3CC6H4 | 7d | 96 |
| 4 | 4-MeOC6H4 | 7e | 87 |
| 4 | 3-F3CC6H4 | 7f | 94 |
| 4 | 3-BrC6H4 | 7g | 91 |
| 4 | 4-BrC6H4 | 7h | 93 |
| 4 | 4-ClC6H4 | 7i | 92 |
| 4 | 4-FC6H4 | 7j | 94 |
| 4 | 4-F3CC6H4 | 7k | 96 |
a Yields refer to pure isolated products.
The 1H NMR spectrum of compound 3 showed characteristic signals at δ 2.46 and 4.71 corresponding to acetylenic (-C≡CH) and oxymethelene (-OCH2-) protons, respectively. The 13C NMR spectrum of compound 3 confirmed the above observations by exhibiting signals at δ 174.7, 76.8, 75.0, and 51.8, corresponding to the ester (-COOR), acetylene (C≡CH) and oxymethelene (-OCH2-) groups. Further confirmation of compound 3 was conducted by HRMS (ESI), which showed protonated molecular ion at (m/z) 551.3713 [M+H]+.
The 1H NMR spectrum of compound 4 showed prominent signals at δ 2.48 and 4.66, corresponding to acetylenic (-C≡CH) and oxymethelene (-OCH2-) protons, respectively. The 13C NMR spectrum of compound 4 confirmed the above observations by exhibiting signals at δ 167.1, 77.4, 74.7, and 51.5, corresponding to the ester (-COOR), acetylene (-C≡CH), and oxymethelene (-OCH2-) functionalities. Further confirmation of compound 4 was achieved by HRMS (ESI), which showed protonated molecular ion at (m/z) 509.3609 [M+H]+.
The structure of compound 4 was unambiguously confirmed using X-ray crystallography. The molecular structure of compound 4 is illustrated in Figure 2a, while the crystal data are depicted in Table S2 (Supporting Information). Compound 4 crystallizes in the orthorhombic space group P21P21P21. All cyclohexane rings of the pentacyclic backbone adopt chair conformations, with the exception of a ring containing sp2-hybdridized C-atoms (C12–C13). C9 and C11–C14 form an approximately planar region, with C8 out of the plane, resulting in a sofa or a half chair conformation of the ring. The double-bond character of C12–C13 was confirmed by their distance of 1.343 (4) Å. The bond distance between C3–O3 was 1.431 Å, which confirms their single-bond character. The bond distance between C11–O4 was 1.226 (3) Å, which suggests a double-bond character. The C32–C33 bond distance in the propargilate fragment was 1.164 (3), which confirms the triple-bond character. Compound 4 is stabilized by intramolecular hydrogen bonding. The O–H group forms a hydrogen bond with the oxygen (O4) of the neighboring molecule (dO3–H1⋯O4 = 2.08 (3) Å, ƟO3–H1⋯O4 = 172 (4)o), resulting in a hydrogen-bonded one-dimensional chain along c-axis (Figure 2b).
Figure 2.

X-ray crystal structure of compound 4 (a). Hydrogen bonding in 4; H-bonds are shown in blue (b).
The structures of all the synthesized compounds (6a–6k and 7a–7k) were characterized by spectral data analysis (1H and 13C NMR, and HRMS). 19F NMR spectroscopy was used for the compounds containing fluorine. The formation of 1H-1,2,3-triazole hybrids of β-AKBA (6a–6k) and β-KBA (7a–7k) derivatives was confirmed by the presence of a characteristic singlet observed in their corresponding 1H NMR spectra at δ 7.60–8.30, which can be attributed to the H-5′ proton of the triazole ring. Further, the 13C NMR spectra of the synthesized hybrids (6a–6k and 7a–7k) determined the diagnostic triazole ring carbon signals between δ 123~125 and 142~144. The structures of the synthesized 1H-1,2,3-triazole hybrids (6a–6k and 7a–7k) were further supported by HRMS.
2.2. α-Glucosidase Activity
The synthesized derivatives of boswellic acids were evaluated for their α-glucosidase-inhibitory potential. Consequently, all the compounds exhibited significant inhibitory activities in the range of 0.22–5.32 µM, at a level even superior to the standard inhibitor acarbose (IC50 = 942 ± 0.74 µM). Compounds 3 and 4, containing the same propargylic groups, displayed slight variations in their α-glucosidase-inhibitory activity after the addition of the triazole moiety with different substituents. Compound 3 with the propargylic group displayed a potent inhibitory activity, with an IC50 value of 4.72 ± 0.52 µM, while the addition of the triazole moiety in 6a was favorable in order to enhance the inhibitory activity (IC50 = 2.02 ± 0.031 µM). However, the substitution of the ortho-methyl group in 6b slightly decreased the inhibitory activity of 6b (IC50 = 2.25 ± 0.10 µM), as compared to 6a with no substitution. However, the substitution of the methoxy group at the ortho-position in 6c (IC50 = 1.23 µM) and 6e (IC50 = 2.01 µM) showed only a modest impact on the inhibition, as compared to substitution at the para-position. The substitution of the tri-fluoro methyl group at the meta-position of 6f (IC50 = 0.22 µM) remarkably increased the inhibitory activity, more so than substitutions at the ortho-and para-positions of compounds 6d (IC50 = 2.37 µM) and 6k (IC50 = 1.26 µM). Moreover, a minute variation was observed in the inhibitory potential by the substitution of the bromo phenyl group in compounds 6g and 6h, with IC50 values of 0.78 and 0.69 µM, respectively. In compound 6j, the addition of a highly electronegative fluorine atom at the meta-position displayed significant inhibitory potential, with an IC50 value of 0.58 µM, as compared to the replacement of the chlorine atom at the same position in 6i (IC50 = 3.06 µM) (Table 2).
Table 2.
In vitro α-glucosidase-inhibitory activities of compounds 6a–6k and 7a–7k.
| Compounds | IC50 ± µM | Compounds | IC50 ± µM | R1-Group |
|---|---|---|---|---|
| 6a | 2.02 ± 0.03 | 7a | 2.31 ± 0.17 |
|
| 6b | 2.25 ± 0.10 | 7b | 2.11 ± 0.10 |
|
| 6c | 1.23 ± 0.08 | 7c | 2.18 ± 0.01 |
|
| 6d | 2.37 ± 0.11 | 7d | 2.62 ± 0.06 |
|
| 6e | 2.01 ± 0.09 | 7e | 2.37 ± 0.07 |
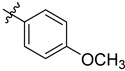
|
| 6f | 0.22 ± 0.08 | 7f | 2.52 ± 0.12 |
|
| 6g | 0.78 ± 0.01 | 7g | 1.34 ± 0.02 |
|
| 6h | 0.69 ± 0.04 | 7h | 0.43 ± 0.05 |
|
| 6i | 3.06 ± 0.22 | 7i | 3.18 ± 0.69 |
|
| 6j | 0.58 ± 0.04 | 7j | 2.64 ± 0.03 |
|
| 6k | 1.26 ± 0.05 | 7k | 1.39 ± 0.11 |
|
| Acarbose (Standard) | 942 ± 0.74 |
|
||
Compound 4 (IC50 value of 5.32 ± 0.15 µM) with the propargylic group displayed the least α-glucosidase-inhibitory potential among all the compounds. However, the addition of triazole group, along with various substitutions at different positions, improved the inhibitory potential of compound 4 derivatives. The phenyl-substituted derivative of 4 (7a) exhibited a higher level of in vitro inhibitory activity (IC50 = 2.31 µM) than compound 4. Similarly, 7b with the methyl substitution at the meta-position of the phenyl ring also displayed a potent inhibitory activity (IC50 = 2.11 µM), while the substitution of the tri-fluoro methyl group at the meta-and ortho-positions of 7d and 7f, respectively, produced only a slight difference in the inhibitory potential, with IC50 values of 2.62 and 2.52 µM, respectively, in contrast to the addition of the same group at the para-position in 7k, which displayed an increase in the inhibitory activity (IC50 = 1.39 µM). Compound 7c with a meta-methoxy-substituent displayed a slight increase in inhibitory activity (IC50 = 2.18 µM), as compared to 7e (IC50 = 2.37 µM) with a similar substituent at the para-position. Compound 7h with para-bromo substitution displayed more potent inhibitory activity (IC50 = 0.43 µM) than 7g (IC50 = 1.34 µM) with the same substitution at the ortho-position. In contrast, compound 7j with para-fluoro substitution exhibited more potent inhibitory activity (IC50 = 2.64 µM) than 7i with a chloro-substituent at the same para-position (IC50 = 3.18 µM). Comparing both series (6a–6k and 7a–7k), four compounds of AKBA derivatives (6c, 6f, 6g, and 6j) displayed a higher level of inhibition than KBA derivatives (7c, 7f, 7g, and 7j), compared to the standard, which showed the importance of the acetate group over hydroxyl, while the rest of the compounds determined an almost similar inhibition. Only one compound, 7h (KBA derivative), showed a higher level of inhibin than 6j (AKBA derivative), which could possibly be due to the substituted group attached to the triazoles moiety.
Kinetic Studies
To investigate their inhibitory mechanisms, the most potent compounds 6f and 7h were subjected to kinetic studies. Compound 6f exhibited a competitive type of inhibition, with a ki value of 0.84 ± 0.007 µM (Figure 3). In this type of inhibition, the inhibitor binds with the active site residue of enzyme and, therefore, Vmax remains the same, with an increase in the Km. The kinetic studies of compound 7h revealed a mixed type of inhibition, with a ki value of 1.18 ± 0.0012 µM (Figure 4). In this type of inhibition, both inhibitors—Vmax and Km—are affected by Vmax decreases and Km increases.
Figure 3.

Mode of inhibition of α-glucosidase by compound 6f: (A) Lineweaver–Burk plot of reciprocal of rate of reaction (velocities) vs. reciprocal of substrate p-nitro phenyl-α-D-glucopyranoside in the absence of (■), and in the presence of 1.50 (●), 0.75 µM (●), and 0.200 µM (■) of compound 6f. (B) Secondary replot of Lineweaver–Burk plot between the slopes of each Lineweaver–Burk plot vs. different concentrations of compound 6f. (C) Dixon plot of reciprocal of rate of reaction (velocities) vs. different concentrations of compound 6f.
Figure 4.

Mode of inhibition of α-glucosidase by compound 7h (A) Lineweaver–Burk plot of reciprocal of rate of reaction (velocities) vs. reciprocal of substrate p-nitro phenyl-α-D-glucopyranoside in the absence of (■), and in the presence of 3.00 (●), 1.50 µM (●), and 0.1875 µM (■) of compound 7h. (B) Secondary replot of Lineweaver–Burk plot between the slopes of each Lineweaver–Burk plot vs. different concentrations of compound 7h. (C) Dixon plot of reciprocal of rate of reaction (velocities) vs. different concentrations of compound 7h.
2.3. Molecular Docking Studies
Initially, the docking protocol was validated through the re-docking of the competitive inhibitor: maltose in the active site of the enzyme (PDB code: 3A4A). The re-docking results showed that the maltose was docked at its binding site with RMSD = 1.31Å (Figure S1, Supplementary Materials) and a docking score of −5.94 kcal/mol, which indicated that the docking method was reliable to use to predict the binding modes of our compounds.
To investigate the binding mode of the compounds in the active site of α-glucosidase, we carried out a molecular docking experiment. In the docking studies, it was observed that the propargyl-linked carbonyl moiety of 3 fit deep inside the active site and formed a hydrogen bond (H-bond) with the side chain of Arg442. In contrast, the carbonyl group of 4 did not interact with the surrounding residues; instead, its -OH group formed an H-bond with the side chain of Arg442. The boswellic acid skeleton of both of the compounds fit at the entrance of the active site and blocked the access to substrate in the active site. The binding modes of compounds 3 and 4 derivatives suggest that the addition of the triazole-substituted phenyl ring dragged the boswellic acid moieties of the compounds towards the entrance, while the substituted groups fit deep inside the active site.
The most active compound, 6f, mediated multiple H-bonds within the active site. The triazole-linked carbonyl oxygen of 6f formed an H-bond with His280, while the acetate moiety of 6f mediated a bidentate interaction with Arg315. Additionally, the side chain of Ser240 donated a H-bond to the carbonyl group in the boswellic acid. The binding modes of 7h and 6j reflect that the carbonyl and -OH moieties of 7h mediated H-bonding with Arg315 and Ser157, respectively, whereas the acetate and the carbonyl groups of 6j formed a bidentate interaction with Arg315 and Ser240, respectively. Similarly, the carbonyl moiety of 6h formed an H-bond with Arg315, while its triazole ring interacted with Arg442 through the H-bond. The carbonyl groups of 6g mediated H-bonds with His280 and Ser240. The side chains of Ser240 and Gln279 bound with the boswellic acid carbonyl oxygen and the phenyl-substituted methoxy group of 6c, respectively, while His280 bound with the triazole-substituted oxygen of 6c and carbonyl oxygen of 6k. Moreover, the -OH and the triazole-substituted oxygen of 7g formed H-bonds with the side chains of Glu411 and Gln279, respectively. The acetate moiety of 7k and 6e was linked with the side chain of Gln279.
Furthermore, the side chains of His351, Arg442, and Asp242 formed H-bonds with the triazole ring of 6a and the -OH of 7b and 7c, respectively. Meanwhile, 6b formed H-bonds with the side chain of Ser240 and His280 and -OH of 7a bind with Arg442. The carbonyl oxygen and fluorine atom of 6d formed an H-bond with the side chains of Gln279 and His351, respectively. Similarly, Ser240 provided an H-bond to the carbonyl oxygen of 7e. The binding modes of 7f and 7d showed that 7f formed H-bonds with the side chain of Gln279 and His112, while 7d only mediated an H-bond with the side chain of Arg213. The docked view of 7j suggested that the compound fit deep inside the active site and mediated multiple H-bonds with His351, Asp215, and Gln279. Similarly, the acetate, triazole, and carbonyl groups of 6i formed several H-bonds with Arg442, His351, Asn350, and Arg315. The docked conformation of 7i was found to be more surface-exposed, and formed only an H-bond with the Tyr158. Afterwards, compounds 2 and 1 exhibited the least inhibitory activities, compared to other compounds in this series.
The acetate and -OH groups of 2 mediated H-bonds with the side chains of Arg442 and Asp215, respectively. Meanwhile, the acetate moiety of 1 interacted with the side chains of Arg213 and His351. The binding modes of these compounds indicate that His280, Gln279, Asp215, His351, Arg442, and Arg315 are the important residues that provide H-bonds to these compounds, while Phe301 and Phe303 provide π–π interaction to most of the compound at the entrance of the active site. The binding mode of the most active hit is shown in Figure 5. The docking score of the compounds were in the range of −9.18 to −2.23 Kcal/mol. The atom-based protein–ligand interaction of each compound is tabulated in Table S1 (Supplementary Materials), along with their docking scores. The docking results support our experimental finding.
Figure 5.

The binding mode of the most active compound, 6f (shown in the purple stick model), in the active site of enzyme. The ligand-binding residues are presented in the orange stick model and H-bonds are shown in green lines.
3. Experimental Section
3.1. General
All experiments were carried out in dry reaction vessels under a dry nitrogen atmosphere. All reagents were purchased from Sigma-Aldrich, Germany. Solvents were purified and dried according to the standard procedures. The high-resolution electrospray ionisation mass spectra (HR-ESI-MS) were recorded on an Agilent 6530 LC Q-TOF LC/MS (Agilent, country of origin USA/EU, made in Singapore). The 1H and 13C NMR spectra were recorded on a nuclear magnetic resonance (NMR) spectrometer (Bruker, Zürich, Switzerland) operating at 600 MHz (150 MHz for 13C) using the solvent peak as an internal reference (CDCl3, δ H: 7.26; δ C: 77.0). The 19F NMR spectra were recorded at 564 MHz. Data were reported in the following order: chemical shift (δ) in ppm, multiplicities, and coupling constants (J) in Hertz (Hz). The column chromatography was carried out using silica gel of the selected particle size of 100–200 mesh. All reactions were monitored by thin layer chromatography (TLC) using silica gel F254 pre-coated plates (Merck, Darmstadt, Germany). Visualization was accomplished using UV-light and I2 stain. The solvents for column chromatography (EtOAc, n-hexane) were technical grade and distilled prior to use. The organic extracts were dried over anhydrous MgSO4. Single crystals of 4 were mounted on a MiTeGen loop with grease and examined on a Bruker D8 Venture APEX diffractometer equipped with a Photon 100 CCD area detector at 296 (2) K using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data were collected using the APEX-II software [42], integrated using SAINT [43], and corrected for absorption using a multi-scan approach (SADABS) [44]. The final cell constant was determined from full least squares refinement of all observed reflections. The structure was solved using intrinsic phasing (SHELXT) [45]. All non-H atoms were in subsequent difference maps and refined anisotropically. H-atoms were added at calculated positions and refined with a riding model. H atoms on O and N atoms were in a difference map and refined with constrained O—H and N—H distances. The structure of 4 was deposited with the CCDC (CCDC deposition number = 2,151,126).
3.2. Preparation of Boswellic Acids (BAs) Cluster
The air-dried ground material (100 g) of B. sacra resin was exhaustively extracted with 100% MeOH at room temperature (three times). The extract was evaporated to yield the yellowish residue (70 g). The MeOH extract was subjected to column chromatography (CC, SiO2 (70–230 mesh; Merck) using n-hexane/EtOAc up to 50% EtOAc, and then washed by pure EtOAc) to yielded eight fractions (BSF1–8). After taking comparative TLC with BA standards, three fractions (BSF3–5, 20 gm, 20–40% n-hexane/EtOAc) were combined and used for the large-scale production of β-AKBA.
3.3. General Procedure for Synthesis of 3 and 4
Anhydrous potassium carbonate (87.0 mg, 0.63 mmol) and propargyl bromide (75.0 mg, 0.63 mmol) were added successively to a solution of β-AKBA (1) or β-KBA (2) (200.0 mg, 0.42 mmol) in dry DMF (10 mL). The reaction mixture was stirred at room temperature for 18 h until the completion of the reaction (monitored by TLC analysis). After the completion of the reaction, it was extracted with ethyl acetate (3 times). The combined organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure on a rotary evaporator to obtain the crude product, which was purified by column chromatography on silica gel using n-hexane/ethyl acetate (95:5 v/v) as an eluent to furnish the pure compounds 3 (96%) and 4 (94%), respectively.
3.3.1. (Prop-2-yne-1-yl) 3α-acetyloxy-11-oxo-urs-12-en-24-oate (3)
White solid; yield = 96%; 1H NMR (600 MHz, chloroform-d) δ 5.51 (s, 1H), 5.30 (s, 1H), 4.70–4.62 (m, 2H), 2.50 (dt, J = 13.5, 3.5 Hz, 1H), 2.43 (d, J = 2.4 Hz, 1H), 2.38 (s, 1H), 2.18 (ddd, J = 14.6, 11.6, 3.3 Hz, 1H), 2.06 (s, 3H), 1.87 (dd, J = 13.5, 4.5 Hz, 2H), 1.81 (d, J = 7.6 Hz, 3H), 1.74–1.71 (m, 1H), 1.67–1.62 (m, 1H), 1.60–1.56 (m, 1H), 1.51 (d, J = 11.1 Hz, 1H), 1.47–1.41 (m, 3H), 1.39–1.35 (m, 2H), 1.31 (s, 3H), 1.23–1.19 (m, 2H), 1.17 (s, 3H), 1.15 (s, 3H), 1.04 (s, 3H), 1.02–0.98 (m, 1H), 0.91 (s, 3H), 0.84 (d, J = 14.1 Hz, 1H), 0.79 (s, 3H), 0.77 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 174.7, 170.1, 164.9, 130.4, 75.0, 73.0, 60.2, 59.0, 51.8, 50.4, 46.7, 45.0, 43.7, 40.9, 39.3, 37.2, 34.5, 33.9, 32.8, 30.8, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.7, 18.2, 17.4, 13.4; HRMS (ESI+): found [M+3H]+: 551.37139 (calculated for C36H55O4, 551.41004).
3.3.2. (Prop-2-yne-1-yl) 3α-hydroxy-11-oxo-urs-12-en-24-oate (4)
Light green-colour crystals; yield = 94%; 1H NMR (600 MHz, chloroform-d) δ 5.52 (s, 1H), 4.66 (t, J = 2.2 Hz, 2H), 4.09 (s, 1H), 2.48 (dd, J = 13.6, 3.6 Hz, 1H), 2.45–2.38 (m, 2H), 2.26 (t, J = 14.9 Hz, 1H), 2.07 (td, J = 13.7, 4.8 Hz, 1H), 1.87 (t, J = 13.2 Hz, 2H), 1.71 (d, J = 14.3 Hz, 1H), 1.65 (td, J = 13.0, 3.9 Hz, 1H), 1.52 (s, 3H), 1.46 (d, J = 12.7 Hz, 2H), 1.44–1.37 (m, 2H), 1.33 (d, J = 3.3 Hz, 1H), 1.31 (d, J = 3.1 Hz, 1H), 1.29 (s, 6H), 1.27–1.21 (m, 2H), 1.21–1.17 (m, 1H), 1.16 (s, 3H), 1.05 (s, 3H), 1.00 (dd, J = 12.7, 8.2 Hz, 1H), 0.92 (s, 3H), 0.90–0.83 (m, 1H), 0.80 (s, 3H), 0.77 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 175.8, 164.9, 156.1, 130.5, 77.4, 74.7, 70.5, 60.3, 59.0, 51.5, 48.8, 47.5, 45.0, 43.8, 40.9, 39.2, 37.4, 33.9, 33.8, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.0, 21.1, 20.5, 18.8, 18.2, 17.4, 13.3; HRMS (ESI+): found [M+H]+: 509.36097 (calculated for C33H49O4 required 509.36309).
3.4. General Procedure for Synthesis of 1H-1,2,3-Triazol Hybrids of 3-Acetyl-11-keto-β-boswellic Acid (6a–6k) and 11-Keto-β-boswellic Acid (7a–7k) Derivatives
CuI (20 mg, 0.104 mmol) and Et3N (0.022 mL, 0.156 mmol) at room temperature were added to a solution of β-AKBA-propargyl ester intermediate 3 (30 mg, 0.052 mmol) or β-KBA-propargyl ester intermediate 4 (30 mg, 0.051 mmol) and substituted aromatic azides 5a–k (0.062 mmol, 1.2 eq) in acetonitrile (10 mL), and the mixture was stirred for 3 h. The reaction mixture was diluted with EtOAc (30 mL), 20 mL of aqueous NH4Cl was added, and the aqueous layer was extracted with EtOAc (3 × 30 mL). Then, the combined organic layer was washed with brine (1 × 20 mL), dried over anhydrous MgSO4, filtered, and the filtrate was concentrated in vacuo. The crude residue was purified by flash column chromatography (silica gel, n-hexane/EtOAc, 85:15) to yield 6a–6k (87–96%) or 7a–7k (86–96%), respectively.
3.4.1. (4-Phenyl-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6a)
Gummy white solid; yield = 90%; 1H NMR (600 MHz, chloroform-d) δ 8.01 (s, 1H), 7.69–7.64 (m, 2H), 7.49 (t, J = 7.7 Hz, 2H), 7.41 (t, J = 7.3 Hz, 1H), 5.48 (s, 1H), 5.29 (s, 3H), 2.46 (dt, J = 13.5, 3.5 Hz, 1H), 2.35 (s, 1H), 2.15 (t, J = 14.8 Hz, 1H), 2.04 (s, 3H), 2.00 (s, 1H), 1.87–1.80 (m, 3H), 1.80–1.74 (m, 1H), 1.70 (dd, J = 14.2, 3.1 Hz, 1H), 1.64–1.54 (m, 2H), 1.48 (s, 1H), 1.42 (dd, J = 12.8, 9.8 Hz, 2H), 1.35 (d, J = 11.5 Hz, 2H), 1.29 (s, 3H), 1.26 (d, J = 10.1 Hz, 1H), 1.22 (t, J = 7.1 Hz, 2H), 1.18 (d, J = 4.3 Hz, 1H), 1.14 (s, 3H), 1.09 (s, 3H), 0.99–0.96 (m, 1H), 0.90 (s, 3H), 0.86 (s, 3H), 0.77 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.6, 170.1, 165.0, 142.9, 136.8, 130.4, 129.8, 128.9, 122.3, 120.6, 73.1, 60.2, 59.0, 57.4, 50.4, 46.7, 45.0, 43.7, 40.8, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.4, 27.1, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; HRMS (ESI+): found [M+3H]+: 670.42317 (calculated for C42H60N3O4, 670.45838).
3.4.2. (4-(O-tolyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6b)
Gummy white solid; yield = 87%; 1H NMR (600 MHz, chloroform-d) δ 7.78 (s, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.32 (dd, J = 20.4, 7.6 Hz, 2H), 7.26 (s, 1H), 5.50 (s, 1H), 5.34 (d, J = 12.6 Hz, 1H), 5.32–5.28 (m, 2H), 2.47 (dd, J = 13.7, 3.6 Hz, 1H), 2.36 (s, 1H), 2.16 (s, 3H), 2.05 (s, 3H), 2.02 (s, 1H), 1.83 (ddd, J = 23.3, 14.1, 5.9 Hz, 2H), 1.73 (s, 1H), 1.68 (s, 3H), 1.66–1.54 (m, 3H), 1.50 (d, J = 11.0 Hz, 1H), 1.47–1.40 (m, 3H), 1.36 (d, J = 12.3 Hz, 2H), 1.31 (s, 3H), 1.23 (t, J = 5.4 Hz, 2H), 1.16 (s, 3H), 1.10 (s, 3H), 1.01–0.97 (m, 1H), 0.91 (s, 3H), 0.87 (s, 3H), 0.78 (s, 3H), 0.77 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.6, 170.1, 165.0, 142.1, 136.2, 133.6, 131.4, 130.4, 129.9, 126.9, 125.9, 125.6, 73.1, 60.2, 59.0, 57.4, 50.4, 46.7, 45.0, 43.7, 40.8, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.4, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.8, 17.4, 14.1, 13.2; HRMS (ESI+): found [M+H]+: 684.45097 (calculated for C42H58N3O5, 684.43765).
3.4.3. (4-(Ortho-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6c)
Gummy white solid; yield = 89%; 1H NMR (600 MHz, chloroform-d) δ 8.12 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.11–7.01 (m, 2H), 5.48 (s, 1H), 5.30 (d, J = 7.2 Hz, 3H), 3.85 (s, 3H), 2.45 (dt, J = 13.5, 3.5 Hz, 1H), 2.35 (s, 1H), 2.15 (t, J = 15.2 Hz, 1H), 2.04 (s, 3H), 2.01 (s, 1H), 1.89–1.75 (m, 3H), 1.73–1.69 (m, 1H), 1.69–1.51 (m, 3H), 1.49 (d, J = 10.9 Hz, 1H), 1.45–1.39 (m, 3H), 1.34 (d, J = 12.2 Hz, 2H), 1.29 (s, 3H), 1.22 (t, J = 7.1 Hz, 2H), 1.18 (s, 1H), 1.15 (s, 3H), 1.09 (s, 3H), 1.00–0.96 (m, 1H), 0.90 (s, 3H), 0.85 (s, 3H), 0.77 (s, 3H), 0.75 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.5, 170.1, 164.9, 151.1, 141.6, 130.4, 130.2, 126.3, 126.0, 125.4, 121.1, 112.2, 73.2, 60.2, 59.0, 57.5, 55.9, 50.4, 46.7, 45.0, 43.7, 40.8, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.5, 27.1, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 700.43333 (calculated for C42H58N3O6, 700.43256).
3.4.4. (4-(Ortho-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6d)
Gummy pale yellow-colour solid; yield = 96%; 1H NMR (600 MHz, Chloroform-d) δ 7.87 (s, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.66 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 5.51 (d, J = 8.4 Hz, 1H), 5.30 (d, J = 11.6 Hz, 3H), 2.47 (dt, J = 13.6, 3.6 Hz, 1H), 2.36 (s, 1H), 2.15 (ddd, J = 18.2, 9.5, 3.6 Hz, 1H), 2.05 (d, J = 5.0 Hz, 3H), 2.01 (s, 2H), 1.85 (ddd, J = 20.2, 12.8, 5.6 Hz, 2H), 1.78 (dd, J = 12.7, 3.1 Hz, 1H), 1.74–1.68 (m, 2H), 1.64 (dd, J = 13.0, 4.1 Hz, 1H), 1.60–1.56 (m, 1H), 1.50 (d, J = 11.0 Hz, 1H), 1.46–1.40 (m, 3H), 1.38–1.34 (m, 2H), 1.30 (s, 3H), 1.22 (t, J = 7.1 Hz, 3H), 1.14 (s, 3H), 1.11 (s, 3H), 0.91 (s, 3H), 0.89 (s, 3H), 0.78 (s, 3H), 0.76 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.5, 170.1, 165.0, 142.3, 134.5, 133.1, 130.5, 130.4, 128.9, 127.3, 126.8, 73.2, 60.2, 59.0, 57.2, 50.4, 46.7, 45.0, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.4, 27.1, 23.6, 23.5, 21.3, 21.1, 21.0, 20.5, 18.8, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −59.21; HRMS (ESI+): found [M+H]+: 738.40951 (calculated for C42H55F3N3O5, 738.40938).
3.4.5. (4-(Para-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6e)
Gummy pale yellow-colour solid; yield = 88%; 1H NMR (600 MHz, Chloroform-d) δ 7.92 (s, 1H), 7.56 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 5.48 (s, 1H), 5.28 (d, J = 5.7 Hz, 3H), 3.83 (s, 3H), 2.45 (dt, J = 13.5, 3.5 Hz, 1H), 2.35 (s, 1H), 2.19–2.11 (m, 1H), 2.04 (s, 3H), 2.01 (s, 2H), 1.86–1.75 (m, 3H), 1.70 (d, J = 13.7 Hz, 1H), 1.59 (ddd, J = 30.7, 14.3, 6.3 Hz, 2H), 1.49 (d, J = 11.1 Hz, 1H), 1.46–1.39 (m, 3H), 1.34 (d, J = 12.3 Hz, 2H), 1.29 (s, 3H), 1.22 (t, J = 7.1 Hz, 2H), 1.18 (d, J = 3.6 Hz, 1H), 1.14 (s, 3H), 1.09 (s, 3H), 1.00–0.96 (m, 1H), 0.90 (s, 3H), 0.85 (s, 3H), 0.77 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.6, 170.1, 164.9, 159.9, 142.7, 130.4, 130.2, 122.5, 122.3, 114.8, 73.1, 60.3, 60.2, 59.0, 57.4, 55.6, 50.5, 46.7, 45.0, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.4, 27.2, 23.7, 23.5, 21.2, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; HRMS (ESI+): found [M+Na]+: 722.41206 (calculated for C42H57N3NaO6, 722.41451).
3.4.6. (4-(Meta-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6f)
Gummy red-colour solid; yield = 95%; 1H NMR (600 MHz, Chloroform-d) δ 8.13 (s, 1H), 8.04 (s, 1H), 7.95 (d, J = 7.8 Hz, 1H), 7.72 (dt, J = 15.7, 7.8 Hz, 2H), 5.53 (s, 1H), 5.34 (s, 3H), 2.51 (dt, J = 13.7, 3.4 Hz, 1H), 2.40 (s, 1H), 2.22–2.16 (m, 1H), 2.09 (s, 3H), 2.06 (s, 1H), 1.93–1.82 (m, 2H), 1.76 (d, J = 13.7 Hz, 3H), 1.70–1.64 (m, 1H), 1.61 (dt, J = 15.4, 3.5 Hz, 1H), 1.54 (d, J = 11.1 Hz, 1H), 1.51–1.44 (m, 3H), 1.40 (d, J = 11.6 Hz, 2H), 1.34 (s, 3H), 1.27 (dd, J = 8.9, 5.1 Hz, 2H), 1.23 (d, J = 4.2 Hz, 1H), 1.19 (s, 3H), 1.15 (s, 3H), 1.05–1.00 (m, 1H), 0.95 (s, 3H), 0.91 (s, 3H), 0.82 (s, 3H), 0.80 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.1, 175.6, 170.1, 165.0, 143.4, 137.1, 132.6, 130.6, 130.4, 125.5, 124.1, 123.5, 122.3, 117.5, 73.0, 60.1, 59.0, 57.2, 50.5, 46.7, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −62.85; HRMS (ESI+): found [M+Na]+: 760.39146 (calculated for C42H54F3N3 NaO5, 760.39133).
3.4.7. (4-(Meta-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-Acetyloxy-11-oxo-urs-12-en-24-oate (6g)
Gummy orange-colour solid; yield = 92%; 1H NMR (600 MHz, Chloroform-d) δ 8.02 (s, 1H), 7.90 (d, J = 1.9 Hz, 1H), 7.63 (dd, J = 8.1, 2.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 5.49 (s, 1H), 5.28 (s, 3H), 2.46 (dt, J = 13.4, 3.4 Hz, 1H), 2.35 (s, 1H), 2.17–2.11 (m, 1H), 2.05 (s, 3H), 2.01 (d, J = 1.3 Hz, 2H), 1.85 (dt, J = 14.2, 7.1 Hz, 1H), 1.80–1.75 (m, 2H), 1.72–1.69 (m, 1H), 1.65–1.55 (m, 2H), 1.49 (d, J = 11.1 Hz, 1H), 1.45–1.41 (m, 2H), 1.36 (m, 1H), 1.35 (d, J = 11.9 Hz, 2H), 1.30 (s, 3H), 1.24–1.21 (m, 2H), 1.20–1.15 (m, 2H), 1.14 (s, 3H), 1.10 (s, 3H), 1.00–0.96 (m, 1H), 0.91 (s, 3H), 0.86 (s, 3H), 0.78 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.1, 175.6, 170.1, 164.9, 143.2, 137.7, 131.9, 131.1, 130.4, 123.7, 123.3, 122.2, 119.0, 73.1, 60.2, 59.0, 57.3, 50.4, 46.7, 45.0, 43.7, 40.8, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; HRMS (ESI+): found [M+Na]+: 770.31261 (calculated for C41H5479BrN3O5Na, 770.31445). found [M+Na]+: 772.31313 (calculated for C41H5481BrN3O5Na, 772.31342).
3.4.8. (4-(Para-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6h)
Gummy orange-colour solid; yield = 91%; 1H NMR (600 MHz, Chloroform-d) δ 8.00 (s, 1H), 7.63 (d, J = 8.3 Hz, 2H), 7.58 (d, J = 8.5 Hz, 2H), 5.49 (s, 1H), 5.29 (d, J = 4.5 Hz, 3H), 2.46 (dt, J = 13.5, 3.5 Hz, 1H), 2.35 (s, 1H), 2.14 (tt, J = 14.8, 3.3 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 1H), 1.85 (dt, J = 14.0, 7.0 Hz, 1H), 1.74 (td, J = 18.1, 17.0, 7.6 Hz, 3H), 1.59 (ddd, J = 32.7, 14.2, 6.3 Hz, 2H), 1.49 (s, 1H), 1.47–1.38 (m, 3H), 1.38–1.32 (m, 2H), 1.30 (s, 3H), 1.23 (t, J = 6.9 Hz, 2H), 1.20–1.15 (m, 2H), 1.14 (s, 3H), 1.09 (s, 3H), 1.00–0.96 (m, 1H), 0.91 (s, 3H), 0.85 (s, 3H), 0.78 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.1, 175.6, 171.1, 170.1, 164.9, 143.2, 135.7, 132.9, 130.4, 122.6, 122.2, 122.0, 73.0, 60.2, 59.0, 57.3, 50.5, 46.7, 45.0, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.5, 27.2, 23.7, 23.5, 21.2, 21.1, 20.5, 18.8, 18.2, 17.3, 14.1, 13.1; HRMS (ESI+): found [M+Na]+: 770.31480 (calculated for C41H5479BrN3O5Na, 770.31445). found [M+Na]+: 772.31427 (calculated for C41H5481BrN3O5Na, 772.31460).
3.4.9. 4-(Para-chloro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6i)
Gummy red-colour solid; yield = 93%; 1H NMR (600 MHz, Chloroform-d) δ 8.01 (s, 1H), 7.65 (dd, J = 8.8, 2.1 Hz, 2H), 7.53–7.44 (m, 2H), 5.50 (s, 1H), 5.30 (s, 3H), 2.47 (d, J = 13.3 Hz, 1H), 2.36 (s, 1H), 2.15 (t, J = 14.4 Hz, 1H), 2.06 (s, 3H), 2.02 (d, J = 2.0 Hz, 1H), 1.86 (dt, J = 14.0, 7.0 Hz, 1H), 1.81–1.70 (m, 3H), 1.66–1.56 (m, 2H), 1.50 (d, J = 11.1 Hz, 1H), 1.48–1.41 (m, 3H), 1.36 (d, J = 12.1 Hz, 2H), 1.31 (s, 3H), 1.26–1.22 (m, 2H), 1.18 (d, J = 15.6 Hz, 2H), 1.15 (s, 3H), 1.11 (s, 3H), 0.99 (dd, J = 14.8, 4.3 Hz, 1H), 0.92 (s, 3H), 0.86 (s, 3H), 0.79 (s, 3H), 0.76 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.6, 170.1, 165.0, 143.2, 135.3, 134.8, 130.4, 130.0, 121.8, 73.0, 60.2, 59.0, 57.3, 50.5, 46.7, 45.0, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.9, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.4, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 704.38440 (calculated for C41H55ClN3O5, 704.38302).
3.4.10. 4-(Para-fluoro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6j)
Gummy yellow-colour solid; yield = 94%; 1H NMR (600 MHz, Chloroform-d) δ 7.97 (s, 1H), 7.70–7.63 (m, 2H), 7.20 (t, J = 7.9 Hz, 2H), 5.50 (s, 1H), 5.30 (d, J = 3.6 Hz, 3H), 2.47 (d, J = 13.3 Hz, 1H), 2.36 (s, 1H), 2.18–2.12 (m, 1H), 2.05 (d, J = 1.8 Hz, 3H), 2.03 (s, 1H), 1.88–1.84 (m, 1H), 1.84–1.67 (m, 3H), 1.66–1.56 (m, 2H), 1.50 (d, J = 11.2 Hz, 1H), 1.47–1.40 (m, 3H), 1.35 (d, J = 12.2 Hz, 2H), 1.30 (s, 3H), 1.29–1.24 (m, 2H), 1.24–1.19 (m, 2H), 1.15 (s, 3H), 1.10 (s, 3H), 0.99 (dd, J = 14.3, 4.3 Hz, 1H), 0.91 (s, 3H), 0.86 (s, 3H), 0.78 (s, 3H), 0.76 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.2, 175.6, 170.1, 165.0, 163.3, 161.7, 143.0, 133.0, 130.4, 122.7, 122.6, 116.8, 116.7, 73.1, 60.2, 59.0, 57.3, 50.5, 46.7, 45.0, 43.7, 40.9, 39.2, 37.2, 34.5, 33.9, 32.7, 30.9, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.4, 14.2, 13.1; 19F NMR (564 MHz, chloroform-d) δ −111.78; HRMS (ESI+): found [M+H]+: 688.41281 (calculated for C41H55FN3O5, 688.41258).
3.4.11. 4-(Para-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6k)
Gummy orange-colour solid; yield = 96%; 1H NMR (600 MHz, Chloroform-d) δ 8.09 (s, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.3 Hz, 2H), 5.50 (s, 1H), 5.31 (s, 3H), 2.46 (dd, J = 10.4, 7.0 Hz, 1H), 2.36 (s, 1H), 2.18–2.12 (m, 1H), 2.06 (s, 3H), 1.87 (dd, J = 13.7, 5.0 Hz, 1H), 1.80 (d, J = 13.1 Hz, 1H), 1.63 (td, J = 13.0, 3.9 Hz, 3H), 1.59–1.49 (m, 2H), 1.47–1.39 (m, 3H), 1.36 (d, J = 11.7 Hz, 2H), 1.31 (s, 3H), 1.27 (s, 1H), 1.23 (q, J = 5.1, 2.8 Hz, 2H), 1.23–1.17 (m, 2H), 1.16 (s, 3H), 1.11 (s, 3H), 1.02–0.97 (m, 1H), 0.91 (s, 3H), 0.85 (s, 3H), 0.78 (s, 3H), 0.76 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.1, 175.6, 170.1, 165.0, 143.4, 139.1, 130.4, 127.1, 122.2, 120.5, 73.0, 60.1, 59.0, 57.2, 50.5, 46.7, 45.0, 43.7, 40.8, 39.3, 39.2, 37.2, 34.5, 33.9, 32.7, 30.8, 28.8, 27.5, 27.2, 23.7, 23.5, 21.3, 21.1, 20.5, 18.8, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −62.65; HRMS (ESI+): found [M+H]+: 738.41080 (calculated for C42H55F3N3O5, 738.40938).
3.4.12. (4-Phenyl-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7a)
Gummy pale yellow-colour solid; yield = 89%; 1H NMR (600 MHz, Chloroform-d) δ 8.01 (s, 1H), 7.70–7.63 (m, 2H), 7.53–7.47 (m, 2H), 7.45–7.40 (m, 1H), 5.48 (s, 1H), 5.34–5.24 (m, 2H), 2.43 (d, J = 13.6 Hz, 1H), 2.37 (s, 1H), 2.26–2.18 (m, 1H), 2.01 (d, J = 1.8 Hz, 2H), 1.85–1.76 (m, 3H), 1.69 (d, J = 14.1 Hz, 1H), 1.61 (dt, J = 13.2, 7.7 Hz, 1H), 1.53–1.47 (m, 2H), 1.46–1.34 (m, 5H), 1.34–1.28 (m, 2H), 1.28–1.26 (m, 6H), 1.24–1.21 (m, 2H), 1.18–1.13 (m, 1H), 1.09 (s, 3H), 1.00–0.94 (m, 1H), 0.90 (s, 3H), 0.86 (d, J = 1.9 Hz, 3H), 0.77 (d, J = 1.9 Hz, 3H), 0.75 (dd, J = 6.4, 1.8 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 165.0, 143.1, 136.8, 130.4, 129.8, 128.9, 122.2, 120.6, 70.5, 60.3, 59.0, 57.1, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.8, 32.8, 30.9, 28.8, 27.4, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 628.41011 (calculated for C39H54N3O4, 628.41143).
3.4.13. (4-(Ortho-tolyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7b)
Gummy pale yellow-colour solid; yield = 86%; 1H NMR (600 MHz, Chloroform-d) δ 7.77 (s, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.37–7.28 (m, 2H), 7.26 (d, J = 7.8 Hz, 1H), 5.49 (d, J = 1.8 Hz, 1H), 5.35–5.25 (m, 2H), 2.43 (d, J = 13.5 Hz, 1H), 2.38 (s, 1H), 2.25–2.18 (m, 1H), 2.16 (d, J = 1.9 Hz, 3H), 2.09–2.03 (m, 1H), 2.01 (d, J = 1.7 Hz, 1H), 1.87–1.75 (m, 3H), 1.70 (d, J = 14.2 Hz, 1H), 1.62 (dt, J = 13.2 Hz, 1H), 1.50 (t, J = 13.1 Hz, 2H), 1.47–1.35 (m, 5H), 1.31 (d, J = 14.6 Hz, 2H), 1.28–1.26 (m, 6H), 1.24–1.22 (m, 2H), 1.19–1.15 (m, 1H), 1.09 (s, 3H), 1.00–0.95 (m, 1H), 0.91 (s, 3H), 0.86 (d, J = 1.7 Hz, 3H), 0.77 (d, J = 1.8 Hz, 3H), 0.75 (dd, J = 6.5, 1.7 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.6, 136.2, 133.6, 131.4, 130.4, 129.9, 126.9, 125.9, 125.6, 70.5, 60.3, 59.0, 57.1, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.4, 33.9, 33.8, 32.8, 30.9, 28.8, 27.4, 27.1, 26.2, 24.2, 21.1, 20.5, 18.9, 18.2, 17.7, 17.4, 14.1, 13.2; HRMS (ESI+): found [M+H]+: 642.42489 (calculated for C40H56N3O4, 642.42708).
3.4.14. (4-(Ortho-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7c)
Gummy pale yellow-colour solid; yield = 88%; 1H NMR (600 MHz, Chloroform-d) δ 8.11 (s, 1H), 7.71 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.07 (q, J = 8.0 Hz, 2H), 5.48 (d, J = 1.9 Hz, 1H), 5.34–5.27 (m, 2H), 3.86 (d, J = 1.8 Hz, 3H), 2.43 (d, J = 13.5 Hz, 1H), 2.37 (s, 1H), 2.23 (ddd, J = 16.5, 9.5, 3.2 Hz, 1H), 2.03 (dd, J = 18.1, 3.9 Hz, 3H), 1.82 (ddd, J = 17.5, 12.1, 4.2 Hz, 3H), 1.69 (d, J = 14.3 Hz, 1H), 1.65–1.59 (m, 1H), 1.50 (t, J = 14.4 Hz, 2H), 1.46–1.36 (m, 5H), 1.27 (t, J = 2.5 Hz, 6H), 1.23 (td, J = 7.2, 1.8 Hz, 3H), 1.18–1.15 (m, 1H), 1.09 (s, 3H), 0.99–0.95 (m, 1H), 0.90 (s, 3H), 0.86 (d, J = 1.9 Hz, 3H), 0.77 (d, J = 1.9 Hz, 3H), 0.76–0.74 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 199.5, 176.6, 165.0, 151.1, 141.8, 130.4, 130.2, 126.2, 125.5, 121.2, 112.2, 70.5, 60.3, 59.0, 57.2, 55.9, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.8, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.2, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.2; HRMS (ESI+): found [M+H]+: 658.42103 (calculated for C40H56N3O5, 658.42200).
3.4.15. (4-(Ortho-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7d)
Gummy white solid; yield = 96%; 1H NMR (600 MHz, Chloroform-d) δ 7.87 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 5.50 (d, J = 1.9 Hz, 1H), 5.30 (s, 2H), 2.44 (d, J = 13.6 Hz, 1H), 2.38 (s, 1H), 2.26–2.19 (m, 1H), 1.88–1.77 (m, 2H), 1.85–1.79 (m, 2H), 1.71–1.61 (m, 3H), 1.51 (t, J = 12.5 Hz, 2H), 1.47–1.36 (m, 5H), 1.31 (d, J = 14.9 Hz, 2H), 1.28–1.25 (m, 6H), 1.23 (td, J = 7.1, 1.9 Hz, 2H), 1.20–1.16 (m, 1H), 1.11 (s, 3H), 1.00–0.95 (m, 1H), 0.92–0.89 (m, 6H), 0.78 (d, J = 1.9 Hz, 3H), 0.76 (dd, J = 6.5, 1.8 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.6, 165.0, 142.5, 134.6, 133.1, 130.5, 130.4, 128.9, 127.2, 126.7, 126.7, 70.5, 60.3, 59.0, 57.0, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.4, 33.9, 33.8, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −59.20; HRMS (ESI+): found [M+H]+: 696.39734 (calculated for C40H53F3N3O4, 696.39882).
3.4.16. (4-(Para-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7e)
Gummy white solid; yield = 87%; 1H NMR (600 MHz, Chloroform-d) δ 7.96 (s, 1H), 7.57 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.2 Hz, 2H), 5.49 (s, 1H), 5.28 (s, 2H), 3.84 (d, J = 1.8 Hz, 3H), 2.44 (d, J = 13.7 Hz, 1H), 2.37 (s, 1H), 2.26–2.20 (m, 1H), 2.08–2.00 (m, 3H), 1.89–1.75 (m, 3H), 1.70 (s, 1H), 1.63 (d, J = 13.7 Hz, 1H), 1.49 (d, J = 11.8 Hz, 3H), 1.45–1.40 (m, 3H), 1.33 (d, J = 18.5 Hz, 2H), 1.27 (s, 6H), 1.24–1.22 (m, 2H), 1.18–1.15 (m, 1H), 1.10 (d, J = 6.4 Hz, 3H), 0.99–0.96 (m, 1H), 0.91 (s, 3H), 0.86 (s, 3H), 0.78 (s, 3H), 0.75 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.6, 165.0, 159.9, 130.4, 122.3, 114.8, 70.5, 60.3, 59.0, 57.1, 55.6, 48.9, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.8, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 658.42086 (calculated for C40H56N3O5, 658.42200)..4.17. (4-(meta-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7f)
Gummy pale yellow-colour solid; yield = 94%; 1H NMR (600 MHz, Chloroform-d) δ 8.09 (s, 1H), 7.99 (s, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.68 (dd, J = 17.2, 7.8 Hz, 2H), 5.49 (d, J = 1.9 Hz, 1H), 5.29 (s, 2H), 2.44 (d, J = 13.8 Hz, 1H), 2.38 (s, 1H), 2.26–2.19 (m, 1H), 2.03 (dd, J = 18.7, 3.5 Hz, 2H), 1.86–1.77 (m, 2H), 1.68 (dd, J = 24.7, 11.1 Hz, 3H), 1.52 (d, J = 18.9 Hz, 2H), 1.47–1.38 (m, 5H), 1.35–1.30 (m, 2H), 1.29–1.27 (m, 6H), 1.23 (dt, J = 7.1, 3.5 Hz, 2H), 1.17 (dd, J = 11.4, 3.0 Hz, 1H), 1.10 (s, 3H), 1.00–0.96 (m, 1H), 0.91 (s, 3H), 0.87 (d, J = 1.9 Hz, 3H), 0.78 (d, J = 1.9 Hz, 3H), 0.75 (dd, J = 6.5, 1.9 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 165.0, 137.1, 130.6, 130.4, 125.5, 123.5, 117.4, 70.5, 60.3, 60.3, 59.0, 56.9, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.7, 32.8, 30.9, 28.8, 27.4, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −62.85; HRMS (ESI+): found [M+H]+: 696.39788 (calculated for C40H53F3N3O4, 696.39882).
3.4.17. (4-(Meta-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7g)
Gummy white solid; yield = 91%; 1H NMR (600 MHz, Chloroform-d) δ 8.02 (s, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 1H), 7.38 (td, J = 8.1, 1.6 Hz, 1H), 5.49 (s, 1H), 5.28 (s, 2H), 2.44 (d, J = 13.7 Hz, 1H), 2.38 (s, 1H), 2.26–2.19 (m, 1H), 2.07–2.02 (m, 2H), 1.86–1.77 (m, 2H), 1.72–1.62 (m, 3H), 1.51 (dd, J = 17.6, 12.4 Hz, 2H), 1.47–1.37 (m, 5H), 1.31 (d, J = 14.6 Hz, 2H), 1.27 (d, J = 2.6 Hz, 6H), 1.24 (dd, J = 7.2, 1.6 Hz, 2H), 1.18–1.15 (m, 1H), 1.10 (s, 3H), 1.00–0.96 (m, 1H), 0.91 (s, 3H), 0.86 (s, 3H), 0.78 (s, 3H), 0.75 (dd, J = 6.5, 1.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 165.0, 131.9, 131.1, 130.4, 123.6, 123.3, 119.0, 70.5, 60.3, 60.3, 59.0, 57.0, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.8, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.3, 17.4, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 706.32360 (calculated for C39H5279BrN3O4, 706.32194). found [M+H]+: 708.32173 (calculated for C39H5281BrN3O4 required 708.32190).
3.4.18. (4-(Para-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7h)
Gummy white solid; yield = 93%; 1H NMR (600 MHz, Chloroform-d) δ 8.00 (s, 1H), 7.64 (dd, J = 8.7, 1.9 Hz, 2H), 7.61–7.54 (m, 2H), 5.49 (s, 1H), 5.28 (s, 2H), 2.43 (d, J = 13.4 Hz, 1H), 2.37 (s, 1H), 2.22 (ddd, J = 15.8, 9.1, 3.1 Hz, 1H), 2.02 (d, J = 1.7 Hz, 2H), 1.86–1.75 (m, 2H), 1.72–1.63 (m, 3H), 1.50 (d, J = 14.0 Hz, 2H), 1.47–1.35 (m, 5H), 1.34–1.29 (m, 2H), 1.26 (s, 6H), 1.25–1.22 (m, 2H), 1.17 (dd, J = 11.9, 3.5 Hz, 1H), 1.09 (s, 3H), 0.97 (dd, J = 14.5, 4.0 Hz, 1H), 0.91 (s, 3H), 0.84 (s, 3H), 0.78 (d, J = 1.8 Hz, 3H), 0.76–0.74 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 171.1, 165.0, 135.8, 132.9, 130.4, 122.6, 121.9, 70.5, 60.3, 59.0, 57.0, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.7, 32.8, 30.9, 28.8, 27.5, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; HRMS (ESI+): found (M+): 706.32121 (clad. for C39H5279BrN3O4, 706.32194). found [M+]: 708.32030 (calculated for C39H5281BrN3O4, 708.32060).
3.4.19. 4-(Para-chloro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7i)
Gummy orange-colour solid; yield = 92%; 1H NMR (600 MHz, Chloroform-d) δ 7.99 (s, 1H), 7.68–7.59 (m, 2H), 7.55–7.44 (m, 2H), 5.49 (s, 1H), 5.27 (s, 2H), 2.46–2.40 (m, 1H), 2.37 (s, 1H), 2.21 (d, J = 15.4, 14.9, 3.9 Hz, 1H), 2.05–2.01 (m, 2H), 1.82 (ddd, J = 32.8, 16.2, 8.9 Hz, 2H), 1.72–1.59 (m, 3H), 1.51 (d, J = 15.7 Hz, 2H), 1.47–1.36 (m, 5H), 1.30 (d, J = 13.6 Hz, 2H), 1.26 (s, 6H), 1.24–1.22 (m, 2H), 1.18–1.15 (m, 1H), 1.09 (s, 3H), 1.00–0.95 (m, 1H), 0.91 (s, 3H), 0.84 (s, 3H), 0.77 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 165.1, 135.3, 134.7, 130.4, 130.0, 122.2, 121.7, 70.5, 60.3, 60.3, 59.0, 57.0, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.7, 32.8, 30.8, 28.8, 27.4, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; HRMS (ESI+): found [M+H]+: 662.37150 (calculated for C39H53ClN3O4, 662.37246).
3.4.20. 4-(Para-fluoro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7j)
Gummy pale yellow-colour solid; yield = 94%; 1H NMR (600 MHz, Chloroform-d) δ 7.96 (d, J = 1.7 Hz, 1H), 7.65 (ddd, J = 8.9, 4.5, 1.7 Hz, 2H), 7.24–7.15 (m, 2H), 5.48 (s, 1H), 5.27 (dd, J = 4.8, 1.6 Hz, 2H), 2.45–2.39 (m, 1H), 2.37 (s, 1H), 2.26–2.17 (m, 1H), 2.01 (d, J = 1.6 Hz, 2H), 1.85–1.74 (m, 2H), 1.71–1.58 (m, 3H), 1.54–1.47 (m, 2H), 1.48–1.36 (m, 5H), 1.35–1.29 (m, 2H), 1.26 (s, 6H), 1.24–1.21 (m, 2H), 1.16 (dt, J = 13.9, 3.2 Hz, 1H), 1.09 (s, 3H), 0.99–0.95 (m, 1H), 0.90 (s, 3H), 0.84 (d, J = 1.7 Hz, 3H), 0.77 (d, J = 1.7 Hz, 3H), 0.75 (dd, J = 6.4, 1.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.5, 176.7, 165.1, 143.2, 133.0, 130.4, 122.6, 122.5, 122.4, 116.8, 116.7, 70.5, 60.3, 60.3, 59.0, 57.0, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.7, 32.8, 30.8, 28.8, 27.4, 27.1, 26.2, 24.2, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −111.79; HRMS (ESI+): found [M+H]+: 646.40098 (calculated for C39H53FN3O4 required 646.40201).
3.4.21. 4-(Para-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7k)
Gummy white solid; yield = 96%; 1H NMR (600 MHz, Chloroform-d) δ 8.08 (s, 1H), 7.85 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 5.49 (s, 1H), 5.35–5.23 (m, 2H), 2.45–2.40 (m, 1H), 2.37 (s, 1H), 2.25–2.18 (m, 1H), 2.03 (dd, J = 19.6, 3.2 Hz, 2H), 1.86–1.76 (m, 2H), 1.72–1.59 (m, 3H), 1.54–1.48 (m, 2H), 1.47–1.35 (m, 5H), 1.35–1.29 (m, 2H), 1.27 (s, 6H), 1.23 (td, J = 7.1, 1.5 Hz, 2H), 1.19–1.15 (m, 1H), 1.09 (s, 3H), 0.99–0.95 (m, 1H), 0.91 (s, 3H), 0.84 (s, 3H), 0.77 (s, 3H), 0.75 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 199.4, 176.7, 171.1, 165.1, 143.6, 139.1, 130.4, 127.2, 127.1, 122.1, 120.5, 70.5, 60.2, 59.0, 56.9, 48.8, 47.5, 45.0, 43.7, 40.9, 39.2, 37.3, 33.9, 33.7, 32.8, 30.8, 28.8, 27.4, 27.1, 26.2, 24.1, 21.1, 20.5, 18.9, 18.2, 17.4, 14.1, 13.1; 19F NMR (564 MHz, chloroform-d) δ −62.66; HRMS (ESI+): found [M+H]+: 696.40558 (calculated for C40H53F3N3O3, 696.39882).
3.5. α-Glucosidase-Inhibitory Assay
The α-glucosidase (EC 3.2.1.20; Saccharomyces cerevisiae) assay was carried out using 50 mM of phosphate buffer (pH = 6.8). The test samples were dissolved in DMSO. The enzyme (1U/2mL) dissolved in phosphate buffer: 20 µL/well of enzyme, 20 µL/well of test samples, and 135 µL/well of phosphate buffer were employed in the 96-well plate. After adding all of these into the 96-well plate, followed by 15 min incubation at 37 °C, 25 µL/well of substrate para-nitro phenyl-α-D-glucopyranoside (0.7 mM) was added to the 96-well plate, and measurements were taken at 400 nm for 30 min using a spectrophotometer (xMarkTM Microplate, Bio-Rad, Hercules, CA, USA). As a control, 7% DMSO was employed. Acarbose was used as a standard inhibitor [35]. For mechanistic studies, a similar procedure was employed, with the addition of four different substrate concentrations: 0.1, 0.2, 0.4, and 0.8 µM. The percentage of inhibition was estimated using the following formula:
3.6. In Silico Docking Studies
In this study, we conducted a docking experiment using Molecular Operating Environment (MOE version 2020.09) [46]. The X-ray crystal structure of α-glucosidase from Saccharomyces cerevisiae in a complex with ligand (α-D-glucopyranose) was taken from the Protein databank with PDB code 3A4A [47]. This structure was selected because of its high resolution, i.e., 1.6 Å. The protein file was prepared using the QuickPrep module of MOE, which adds missing hydrogen atoms to the protein structure, calculates the charges of each atom of protein (AMBER 10: EHT force field), and refines the structure after a short minimization, with a gradient of 0.1 kcal/mol/Å. The ligands files were prepared for docking by Chem Draw and converted into 3D-form by the MOE WASH module in the MOE database. WASH adds hydrogen atoms on the ligand and calculates the partial charges (AMBER 10: EHT force field) on each atom of the ligand. Moreover, in the database, WASG minimizes the structure of each ligand with the default parameter, i.e., gradient = 0.1 kcal/mol/Å. After preparing the protein file and ligand database, docking was performed using the Triangle Matcher placement method and London dG scoring function. Thirty docked conformations of each ligand were saved and, using the conformational sampling method, the best pose of each ligand was selected according to the docking score and ligand’s interactions with the enzyme.
4. Conclusions
A series of 24 novel 1H-1,2,3-triazole hybrids of β-AKBA (6a–6k) and β-KBA (7a–7k) was designed and synthesized by employing “click” chemistry in a highly efficient manner. The study of α-glucosidase inhibition determined that all the synthesized derivatives are highly potent inhibitors, with IC50 values ranging from 0.22 to 5.32 µM. Among all the synthesized compounds, nine compounds (6f, 7h, 6j, 6h, 6g, 6c, 6k, 7g, and 7k) exhibited the highest inhibition and were found to be several times more potent than the standard (acarbose). Furthermore, molecular docking was performed to discover the binding modes of these compounds with the α-glucosidase enzyme. The results showed that all compounds are accommodated well in the active site of α-glucosidase, where His280, Gln279, Asp215, His351, Arg442, and Arg315 are mainly stabilized the binding of these compounds. These encouraging results enable us to deeply explore the diabetic potential of boswellic acids hybrid triazoles in the field of medicinal chemistry. Furthermore, here, the α-glucosidase activity of all the active compounds is reported for the first time for these BA derivatives. The pharmacophoric features of substituted triazoles at the C-24 position of boswellic acids might be an interesting lead for the further development of new diabetic-like drug molecules.
Acknowledgments
Financial support from the Research Council through the Research Grant Programs (BFP/RGP/EBR/21/005) and (BFP/RGP/CBS/21/006) is gratefully acknowledged. The authors would like to thank the University of Nizwa for their generous support of this project. We also thank the analytical and technical staff of the University of Nizwa for assistance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph16020229/s1. Figures S1–S80: 1H, 13C-NMR and HRMS spectra of the synthesized compounds (3,4 and 6a–6k, and 7a–7k); Figure S81: The re-docked conformation (green stick model) of maltose is superimposed on the X-ray conformation (grey stick model) present in the PDB code 3A4A; Table S1. The docking results of all the compounds; Table S2. Crystal data of the compound 4.
Author Contributions
N.U.R., T.K.M., T.A. and S.K.A. isolated and synthesized all the derivatives of AKBA and KBA. S.U. and A.K. screened the compounds against α-glucosidase-inhibitory activity. N.U.R. and S.K.A. performed structural elucidation and wrote the original draft of the manuscript. S.A.H. performed the docking study of the compounds, while M.U.A. conducted the X-ray diffraction study of the crystal structure. R.C. and A.A.-H. supervised the project, assisted in writing, reviewing, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The spectroscopic data presented in this study are available in the supporting information.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
Funding will be provided by The Research Council through the Research Grant Program (BFP/RGP/EBR/21/005).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Al-Harrasi A., Csuk R., Khan A., Hussain J. Distribution of the Anti-Inflammatory and Anti-Depressant Compounds: Incensole and Incensole Acetate in Genus Boswellia. Phytochemistry. 2019;161:28–40. doi: 10.1016/j.phytochem.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Asteggiano A., Curatolo L., Schiavo V., Occhipinti A., Medana C. Development, Validation, and Application of a Simple and Rugged HPLC Method for Boswellic Acids for a Comparative Study of Their Abundance in Different Species of Boswellia Gum Resins. Appl. Sci. 2023;13:1254. doi: 10.3390/app13031254. [DOI] [Google Scholar]
- 3.Liu Y.M. Harmonograph of Uighur, Part One. Xinjiang Sci. Technol. Hyg. Publ. House, Urumqi. 1999:222. [Google Scholar]
- 4.The Chinese Pharmacopeia Pharmacopia. China Medical Sciences Press; Beijing, China: 2015. p. 223. CotC 1. [Google Scholar]
- 5.Singh S., Khajuria A., Taneja S.C., Johri R.K., Singh J., Qazi G.N. Boswellic Acids: A Leukotriene Inhibitor Also Effective through Topical Application in Inflammatory Disorders. Phytomedicine. 2008;15:400–407. doi: 10.1016/j.phymed.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Bertocchi M., Isani G., Medici F., Andreani G., Tubon Usca I., Roncada P., Forni M., Bernardini C. Anti-inflammatory activity of Boswellia serrata extracts: An in vitro study on porcine aortic endothelial cells. Oxid. Med. Cell. Longev. 2018;2018:2504305. doi: 10.1155/2018/2504305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehrzadi S., Tavakolifar B., Huseini H.F., Mosavat S.H., Heydari M. The effects of Boswellia serrata gum resin on the blood glucose and lipid profile of diabetic patients: A double-blind randomized placebo-controlled clinical trial. J. Evid. Based Integr. Med. 2018;13:2515690X18772728. doi: 10.1177/2515690X18772728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Y., Zhang Q., Sun L. Five Terpenoids from the Gum Resin of Boswellia Carterii and Their Cytotoxicity. Fitoterapia. 2021;154:105017. doi: 10.1016/j.fitote.2021.105017. [DOI] [PubMed] [Google Scholar]
- 9.Barakat B.M., Ahmed H.I., Bahr H.I., Elbahaie A.M. Protective effect of boswellic acids against doxorubicin-induced hepatotoxicity: Impact on Nrf2/HO-1 defense pathway. Oxid. Med. Cell. Longev. 2018;2018:8296451. doi: 10.1155/2018/8296451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y.G., Ren J., Wang A.G., Yang J.B., Ji T.F., Ma Q.G., Tian J., Su Y.L. Hepatoprotective prenylaromadendrane-type diterpenes from the gum resin of Boswellia carterii. J. Nat. Prod. 2013;76:2074–2079. doi: 10.1021/np400526b. [DOI] [PubMed] [Google Scholar]
- 11.Mahesh B.U., Shrivastava S., Pragada R.R., Naidu V.G., Sistla R. Antioxidant and hepatoprotective effects of Boswellia ovalifoliolata bark extracts. Chin. J. Nat. Med. 2014;12:663–671. doi: 10.1016/S1875-5364(14)60101-1. [DOI] [PubMed] [Google Scholar]
- 12.Safayhi H., Mack T., Sabieraj J., Anazodo M.I., Subramanian L.R., Ammon H.P. Boswellic Acids: Novel, Specific, Nonredox Inhibitors of 5-Lipoxygenase. J. Pharmacol. Exp. Ther. 1992;261:1143–1146. [PubMed] [Google Scholar]
- 13.Moussaieff A., Shein N.A., Tsenter J., Grigoriadis S., Simeonidou C., Alexandrovich A.G., Trembovler V., Ben-Neriah Y., Schmitz M.L., Fiebich B.L., et al. Incensole Acetate: A Novel Neuroprotective Agent Isolated from Boswellia Carterii. J. Cereb. Blood Flow Metab. 2008;28:1341–1352. doi: 10.1038/jcbfm.2008.28. [DOI] [PubMed] [Google Scholar]
- 14.Liu J.J., Nilsson Å., Oredsson S., Badmaev V., Zhao W.Z., Duan R.D. Boswellic Acids Trigger Apoptosis via a Pathway Dependent on Caspase-8 Activation but Independent on Fas/Fas Ligand Interaction in Colon Cancer HT-29 Cells. Carcinogenesis. 2002;23:2087–2093. doi: 10.1093/carcin/23.12.2087. [DOI] [PubMed] [Google Scholar]
- 15.Solanki N., Mehta M., Chellappan D.K., Gupta G., Hansbro N.G., Tambuwala M.M., AA Aljabali A., Paudel K.R., Liu G., Satija S., et al. Antiproliferative Effects of Boswellic Acid-Loaded Chitosan Nanoparticles on Human Lung Cancer Cell Line A549. Future Med. Chem. 2020;12:2019–2034. doi: 10.4155/fmc-2020-0083. [DOI] [PubMed] [Google Scholar]
- 16.Shah B.A., Qazi G.N., Taneja S.C. Boswellic Acids: A Group of Medicinally Important Compounds. Nat. Prod. Rep. 2009;26:72–89. doi: 10.1039/B809437N. [DOI] [PubMed] [Google Scholar]
- 17.Hussain H., Ali I., Wang D., Hakkim F.L., Westermann B., Rashan L., Ahmed I., Green I.R. Boswellic Acids: Privileged Structures to Develop Lead Compounds for Anticancer Drug Discovery. Expert Opin. Drug Discov. 2021;16:851–867. doi: 10.1080/17460441.2021.1892640. [DOI] [PubMed] [Google Scholar]
- 18.Iram F., Khan S.A., Husain A. Phytochemistry and Potential Therapeutic Actions of Boswellic Acids: A Mini-Review. Asian Pac. J. Trop. Biomed. 2017;7:513–523. doi: 10.1016/j.apjtb.2017.05.001. [DOI] [Google Scholar]
- 19.Li C., He Q., Xu Y., Lou H., Fan P. Synthesis of 3-O-Acetyl-11-Keto-β-Boswellic Acid (AKBA)-Derived Amides and Their Mitochondria-Targeted Antitumor Activities. ACS Omega. 2022;7:9853–9866. doi: 10.1021/acsomega.2c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma K.N., Mondal D., Bera S. Pharmacological and Cellular Significance of Triazole-Surrogated Compounds. Curr. Org. Chem. 2019;23:2505–2572. doi: 10.2174/1385272823666191021114906. [DOI] [Google Scholar]
- 21.Lauria A., Delisi R., Mingoia F., Terenzi A., Martorana A., Barone G., Almerico A.M. 1,2,3-Triazole in Heterocyclic Compounds, Endowed with Biological Activity, through 1,3-Dipolar Cycloadditions. Eur. J. Org. Chem. 2014;2014:3289–3306. doi: 10.1002/ejoc.201301695. [DOI] [Google Scholar]
- 22.Nehra N., Tittal R.K., Ghule V.D. 1,2,3-Triazoles of 8-Hydroxyquinoline and HBT: Synthesis and Studies (DNA Binding, Antimicrobial, Molecular Docking, ADME, and DFT) ACS Omega. 2021;6:27089–27100. doi: 10.1021/acsomega.1c03668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunn G.L., Hoover J.R., Berges D.A., Taggart J.J., Davis L.D., Dietz E.M., Jakas D.R., Yim N., Actor P., Uri J.V., et al. Orally active 7-phenylglycyl cephalosporins. Structure-activity studies related to cefatrizine (SK&F 60771) J. Antibiotics. 1976;29:65–80. doi: 10.7164/antibiotics.29.65. [DOI] [PubMed] [Google Scholar]
- 24.El-Sayed W.A., Khalaf H.S., Mohamed S.F., Hussien H.A., Kutkat O.M., Amr A.E. Synthesis and Antiviral Activity of 1,2,3-Triazole Glycosides Based Substituted Pyridine via Click Cycloaddition. Russ. J. Gen. Chem. 2017;87:2444–2453. doi: 10.1134/S1070363217100279. [DOI] [Google Scholar]
- 25.Tian Y., Liu Z., Liu J., Huang B., Kang D., Zhang H., De Clercq E., Daelemans D., Pannecouque C., Lee K.-H., et al. Targeting the Entrance Channel of NNIBP: Discovery of Diarylnicotinamide 1,4-Disubstituted 1,2,3-Triazoles as Novel HIV-1 NNRTIs with High Potency against Wild-Type and E138K Mutant Virus. Eur. J. Med. Chem. 2018;151:339–350. doi: 10.1016/j.ejmech.2018.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angajala K.K., Vianala S., Macha R., Raghavender M., Thupurani M.K., Pathi P.J. Synthesis, Anti-Inflammatory, Bactericidal Activities and Docking Studies of Novel 1,2,3-Triazoles Derived from Ibuprofen Using Click Chemistry. Springerplus. 2016;5:423. doi: 10.1186/s40064-016-2052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Bourakadi K., Mekhzoum M.E.M., Saby C., Morjani H., Chakchak H., Merghoub N., el kacem Qaiss A., Bouhfid R. Synthesis, Characterization and in Vitro Anticancer Activity of Thiabendazole-Derived 1,2,3-Triazole Derivatives. New J. Chem. 2020;44:12099–12106. doi: 10.1039/C9NJ05685H. [DOI] [Google Scholar]
- 28.Avula S.K., Rehman N.U., Khan M., Halim S.A., Khan A., Rafiq K., Csuk R., Das B., Al-Harrasi A. New Synthetic 1H-1,2,3-Triazole Derivatives of 3-O-Acetyl-β-Boswellic Acid and 3-O-Acetyl-11-Keto-β-Boswellic Acid from Boswellia Sacra Inhibit Carbonic Anhydrase II in Vitro. Med. Chem. Res. 2021;30:1185–1198. doi: 10.1007/s00044-021-02723-8. [DOI] [Google Scholar]
- 29.Kausar N., Ullah S., Khan M.A., Zafar H., Atia-tul-Wahab, Choudhary M.I., Yousuf S. Celebrex Derivatives: Synthesis, α-Glucosidase Inhibition, Crystal Structures and Molecular Docking Studies. Bioorg. Chem. 2021;106:104499. doi: 10.1016/j.bioorg.2020.104499. [DOI] [PubMed] [Google Scholar]
- 30.Shaw J.E., Sicree R.A., Zimmet P.Z. Global Estimates of the Prevalence of Diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 31.McCulloch D.K., Kurtz A.B., Tattersall R.B. A new approach to the treatment of nocturnal hypoglycemia using alpha-glucosidase inhibition. Diabetes Care. 1983;6:483–487. doi: 10.2337/diacare.6.5.483. [DOI] [PubMed] [Google Scholar]
- 32.Umm-E-Farwa, Ullah S., Khan M.A., Zafar H., Atia-tul-Wahab, Younus M., Choudhary M.I., Basha F.Z. Dibenzazepine-Linked Isoxazoles: New and Potent Class of α-Glucosidase Inhibitors. Bioorg. Med. Chem. Lett. 2021;40:127979. doi: 10.1016/j.bmcl.2021.127979. [DOI] [PubMed] [Google Scholar]
- 33.Akhter S., Ullah S., Yousuf S., Atia-tul-Wahab, Siddiqui H., Choudhary M.I. Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Benzamide Derivatives of Thiourea as Potent Inhibitors of α-Glucosidase in-Vitro. Bioorg. Chem. 2021;107:104531. doi: 10.1016/j.bioorg.2020.104531. [DOI] [PubMed] [Google Scholar]
- 34.Maurya A.K., Mulpuru V., Mishra N. Discovery of Novel Coumarin Analogs against the α-Glucosidase Protein Target of Diabetes Mellitus: Pharmacophore-Based QSAR, Docking, and Molecular Dynamics Simulation Studies. ACS Omega. 2020;5:32234–32249. doi: 10.1021/acsomega.0c03871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rehman N.U., Khan A., Al-Harrasi A., Hussain H., Wadood A., Riaz M., Al-Abri Z. New α-Glucosidase Inhibitors from the Resins of Boswellia Species with Structure–Glucosidase Activity and Molecular Docking Studies. Bioorg. Chem. 2018;79:27–33. doi: 10.1016/j.bioorg.2018.04.020. [DOI] [PubMed] [Google Scholar]
- 36.Ye G.-J., Lan T., Huang Z.-X., Cheng X.-N., Cai C.-Y., Ding S.-M., Xie M.-L., Wang B. Design and Synthesis of Novel Xanthone-Triazole Derivatives as Potential Antidiabetic Agents: α-Glucosidase Inhibition and Glucose Uptake Promotion. Eur. J. Med. Chem. 2019;177:362–373. doi: 10.1016/j.ejmech.2019.05.045. [DOI] [PubMed] [Google Scholar]
- 37.Jauch J., Bergmann J. An Efficient Method for the Large-Scale Preparation of 3-O-Acetyl-11-Oxo-β-Boswellic Acid and Other Boswellic Acids. Eur. J. Org. Chem. 2003;2003:4752–4756. doi: 10.1002/ejoc.200300386. [DOI] [Google Scholar]
- 38.Spivak A.Y., Gubaidullin R.R., Galimshina Z.R., Nedopekina D.A., Odinokov V.N. Effective Synthesis of Novel C(2)-Propargyl Derivatives of Betulinic and Ursolic Acids and Their Conjugation with β-d-Glucopyranoside Azides via Click Chemistry. Tetrahedron. 2016;72:1249–1256. doi: 10.1016/j.tet.2016.01.024. [DOI] [Google Scholar]
- 39.Carvalho I., Andrade P., Campo V.L., Guedes P.M.M., Sesti-Costa R., Silva J.S., Schenkman S., Dedola S., Hill L., Rejzek M., et al. ‘Click Chemistry’ Synthesis of a Library of 1,2,3-Triazole-Substituted Galactose Derivatives and Their Evaluation against Trypanosoma Cruzi and Its Cell Surface Trans-Sialidase. Bioorg. Med. Chem. 2010;18:2412–2427. doi: 10.1016/j.bmc.2010.02.053. [DOI] [PubMed] [Google Scholar]
- 40.Das R., Mukhopadhyay B. Use of ‘Click Chemistry’ for the Synthesis of Carbohydrate-Porphyrin Dendrimers and Their Multivalent Approach toward Lectin Sensing. Tetrahedron Lett. 2016;57:1775–1781. doi: 10.1016/j.tetlet.2016.03.031. [DOI] [Google Scholar]
- 41.Artyushin O.I., Sharova E.V., Vinogradova N.M., Genkina G.K., Moiseeva A.A., Klemenkova Z.S., Orshanskaya I.R., Shtro A.A., Kadyrova R.A., Zarubaev V.V., et al. Synthesis of Camphecene Derivatives Using Click Chemistry Methodology and Study of Their Antiviral Activity. Bioorg. Med. Chem. Lett. 2017;27:2181–2184. doi: 10.1016/j.bmcl.2017.03.051. [DOI] [PubMed] [Google Scholar]
- 42.APEX-II, Bruker AXS, Madison, WI, USA
- 43.SADABS, Bruker AXS, Madison, WI, USA
- 44.Bruker SAINT. AXS, Madison, WI, USA
- 45.Sheldrick G.M. Acta Crystallographica Section A. Found. Adv. 2015;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chemical Computing Group ULC Molecular Operating Environment, 1010 Sherbooke St. West, Suite 910, Montr. QC, Canada, 2014
- 47.Yamamoto K., Miyake H., Kusunoki M., Osaki S. Crystal Structures of Isomaltase from Saccharomyces Cerevisiae and in Complex with Its Competitive Inhibitor Maltose. FEBS J. 2010;277:4205–4214. doi: 10.1111/j.1742-4658.2010.07810.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The spectroscopic data presented in this study are available in the supporting information.