

SUMMARY
Synaptic plasticity depends on rapid experience-dependent changes in the number of neurotransmitter receptors. Previously, we demonstrated that motor-mediated transport of AMPA receptors (AMPARs) to and from synapses is a critical determinant of synaptic strength. Here, we describe two convergent signaling pathways that coordinate the loading of synaptic AMPARs onto scaffolds, and scaffolds onto motors, thus providing a mechanism for experience-dependent changes in synaptic strength. We find that an evolutionarily conserved JIP-protein scaffold complex and two classes of mitogen-activated protein kinase (MAPK) proteins mediate AMPAR transport by kinesin-1 motors. Genetic analysis combined with in vivo, real-time imaging in Caenorhabditis elegans revealed that CaMKII is required for loading AMPARs onto the scaffold, and MAPK signaling is required for loading the scaffold complex onto motors. Our data support a model where CaMKII signaling and a MAPK-signaling pathway cooperate to facilitate the rapid exchange of AMPARs required for early stages of synaptic plasticity.
In brief
Regulated trafficking of AMPA receptors (AMPARs) contributes to glutamate-mediated neurotransmission and synaptic plasticity. Hoerndli et al. demonstrate that MAPK-associated scaffold proteins and MAPK signaling are essential for kinesin-mediated delivery and removal of synaptic AMPARs. Coordinated MAPK and CaMKII signaling regulates a mobile complex that contributes to the rapid exchange of synaptic AMPARs.
Graphical Abstract
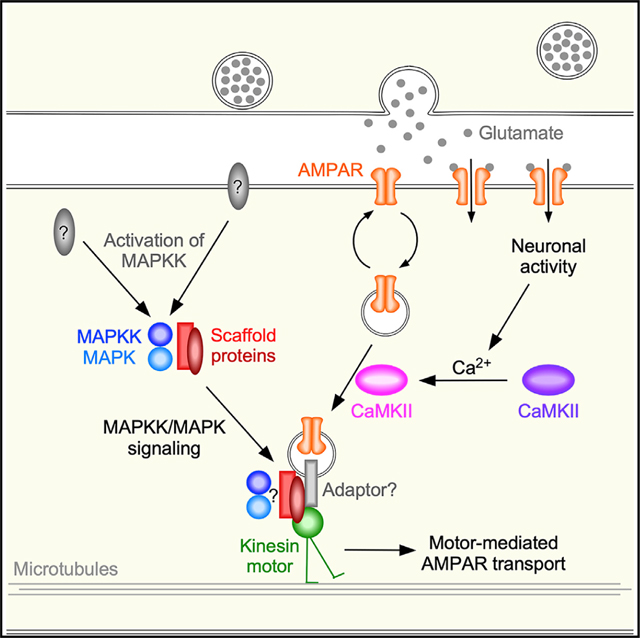
INTRODUCTION
Synapses are plastic, enabling changes in the strength of synaptic signaling. A major determinant of synaptic strength is the number of postsynaptic neurotransmitter receptors (Diering and Huganir, 2018; Huganir and Nicoll, 2013; Nicoll, 2017). Distinct patterns of synaptic signaling can cause lasting changes in receptor number, thus providing a mechanism for experience-dependent plasticity of the nervous system. In cellular models of learning, such as long-term potentiation (LTP) or long-term depression (LTD) in the hippocampus, synaptic plasticity is dependent on activity-dependent changes in the number of postsynaptic AMPA receptors (AMPARs), a subtype of the ionotropic glutamate receptor (iGluR) family (Herring and Nicoll, 2016; Huganir and Nicoll, 2013; Luscher and Malenka, 2012; Nabavi et al., 2014).
Calcium/calmodulin-dependent kinase II (CaMKII) has firmly established roles in the transduction of synaptic activity and in regulating the number of postsynaptic AMPARs (Bayer and Schulman, 2019; Lisman, 2017; Murakoshi et al., 2017; Nicoll and Roche, 2013). In previous work, we discovered that kinesin-1 motors mediated bidirectional transport of AMPARs along mixed-polarity microtubules (Hoerndli et al., 2013), and that UNC-43 (the Caenorhabditis elegans homolog of CaMKII) was required for the motor-mediated delivery and removal of synaptic AMPARs (Hoerndli et al., 2013, 2015a, 2015b). In unc-43 loss-of-function (lf) mutants, the number of AMPAR transport vesicles in neuronal processes was reduced to levels comparable with those observed in unc-116(lf) (kinesin-1) motor mutants. In previous studies, we demonstrated that the peak glutamate-gated current is primarily dependent on the GLR-1 AMPAR subunit (Brockie et al., 2001; Mellem et al., 2002; Zheng et al., 2004). Additionally, we found that the magnitude of the glutamate-gated current was greatly reduced in unc-43(lf) mutants (Hoerndli et al., 2015b), a finding that explained the altered synaptic function in these mutants. Finally, we demonstrated that altered AMPAR transport was associated with defective synaptic plasticity (Hoerndli et al., 2015b). These findings raised the question of whether CaMKII and kinesin-1 were sufficient for transport of AMPARs. Here, we sought to address two important questions: what cargo adaptor(s) carry the AMPAR cargo on kinesin-1 motors, and what regulates the transport of cargo?
While kinesin-1 can directly bind certain cargo proteins, indirect binding of cargo via adaptor or linker proteins can increase the diversity of transported cargo and contribute to the regulation of transport (Akhmanova and Hammer, 2010; Karcher et al., 2002; Schnapp, 2003; Verhey and Rapoport, 2001). Certain scaffold proteins can act as adaptors for motor-mediated transport of distinct cargo proteins (Fu and Holzbaur, 2014; Hirokawa and Noda, 2008) and provide a mechanism for the organization of signaling modules. An early study of the role of kinesin-1 in the transport of AMPARs identified the glutamate receptor interacting protein (GRIP1 scaffolding protein) as important for the steering of kinesin-1 to dendrites (Setou et al., 2002). However, the precise role(s) of GRIP1 with respect to transport remains to be determined. A conditional knockout of GRIP1 did not disrupt the steady-state trafficking of AMPARs, but did modify receptor recycling (Mao et al., 2010).
Scaffold proteins can act as adaptors and also bind to members of the mitogen-activated protein family of kinases (MAPKs) (Dhanasekaran et al., 2007). For example, the MAPK-associated scaffold protein, JIP3/Sunday Driver, binds to kinesin-1 and has an evolutionarily conserved role in the axonal transport of synapse-directed vesicles (Cockburn et al., 2018; Duncan and Goldstein, 2006). Similarly, the scaffold protein JIP1 can bind to kinesin-1 (Fu and Holzbaur, 2014), and also to the GluA4 AMPAR subunit (Vieira et al., 2010); however, a role for JIP1 in AMPAR transport has not been established. Notably, MAPKs have been implicated in synaptic plasticity, but the mechanism is not well understood (Mao and Wang, 2015; Shetty and Sajikumar, 2017; Zamora Chimal and De Schutter, 2018; Zhang et al., 2012).
In mammals, four genes encode JIP proteins: JIP1 and JIP2 are closely related by sequence, whereas JIP3 and JIP4 are a separate family (Whitmarsh, 2006; Zeke et al., 2016). In C. elegans, jip-1 and unc-16 encode homologs of JIP1 and JIP3, respectively (Byrd et al., 2001; Sakaguchi et al., 2004; van den Berg et al., 2013). unc-16(lf) mutants have mislocalized presynaptic vesicles and altered distribution of GLR-1 AMPARs along neuronal processes (Byrd et al., 2001). We noted that the size and spacing of GLR-1 accumulations in unc-16(lf) mutants were reminiscent of the altered distribution of GLR-1 observed in unc-116(lf) (kinesin heavy chain) and klc-2(lf) (kinesin light chain) kinesin-1 motor protein mutants (Hoerndli et al., 2013), and in unc-43(lf) (CaMKII) mutants (Hoerndli et al., 2015b). Additionally, UNC-16 (JIP3) physically interacts with JKK and JNK kinases (Byrd et al., 2001). These similarities motivated us to address whether the JIP scaffold proteins participate in kine-sin-1-mediated transport of AMPARs, and whether transport was dependent on MAPK-signaling molecules.
Here, in vivo imaging and electrophysiology studies combined with genetic analysis demonstrate that UNC-16 and JIP-1 scaffold proteins link kinesin-1 motors to GLR-1 (AMPAR) cargo. Transport of GLR-1 was severely diminished in unc-16(lf) and jip-1(lf) mutants, as was synaptic function. jip-1; unc-16 double mutants did not show an additive phenotype with respect to altered GLR-1 transport or GLR-1-mediated currents, which is consistent with these proteins forming a heteromeric scaffold-protein complex. Further, our data suggest that MAPK signaling is required for the association of the UNC-16/JIP-1 scaffold complex with kinesin-1 motors. We found that transport of GLR-1 and the scaffold proteins was defective in MAPK mutants. Additionally, double-mutant epistasis experiments indicated that MAPK loss-of-function mutants suppressed the increased AMPAR transport observed in CaMKII gain-of-function mutants.
Together, our data support a model where MAPK signaling and CaMKII signaling converge at the scaffold complex, leading to the coordinated loading of synaptic AMPAR cargo onto the scaffold, and of the scaffold onto kinesin-1 motors. We anticipate that this mechanism is conserved and might function to provide rapid exchange of AMPARs at synapses in response to experience-dependent changes in synaptic activity.
RESULTS
AMPAR transport to synapses is dependent on a heteromeric UNC-16/JIP-1 scaffold complex
We previously found that disrupted motor-mediated transport of the GLR-1 AMPAR subunit resulted in GLR-1 accumulation at synapses and, paradoxically, diminished AMPAR-mediated currents (Hoerndli et al., 2013, 2015b). In the absence of kinesin-1-mediated transport, GLR-1 reaches synapses via diffusion where it accumulates because removal from synapses is also dependent on kinesin-1. In the absence of kinesin-1, only monomeric GLR-1 AMPARs populate synapses and thus peak currents are diminished compared with wild-type controls (Hoerndli et al., 2013, 2015b).
The apparent accumulation of GLR-1 at synaptic puncta in unc-6(lf) mutants (Byrd et al., 2001) prompted us to investigate whether this scaffold protein was required for GLR-1 transport. We measured in vivo real-time transport of a functional GLR-1::GFP fusion protein that we selectively expressed in the pair of AVA interneurons that normally express GLR-1 (Hoerndli et al., 2013, 2015b). Compared with controls, the number of GLR-1::GFP transport vesicles (flux) was dramatically reduced in unc-16(lf) mutants (Figures 1A and 1B). The diminished flux was comparable with that observed in unc-116(lf) and klc-2(lf) kinesin-1 mutants (Hoerndli et al., 2013), consistent with UNC-16 (JIP3) contributing to kinesin-1-mediated transport of GLR-1 (Figures S1A and S1B). Cell-specific RNAi knockdown of UNC-16 in AVA decreased GLR-1 flux, indicating a cell-autonomous role for UNC-16 with respect to GLR-1 transport (Figures S1C and S1D).
Figure 1. GLR-1 transport is reduced in unc-16 and jip-1 scaffold-protein mutants.

(A) Kymographs of GLR-1::GFP transport in the AVA processes of transgenic control worms, unc-16(e109) and jip-1(km18) single and double loss-of-function (lf) mutants, and transgenic mutants that expressed either mCherry::UNC-16, mCherry::JIP-1, or mCherry::JIP3a in AVA.
(B) Total (anterograde and retrograde) GLR-1::GFP transport events in control worms (n = 15), unc-16(lf) (n = 12), jip-1(lf) (n = 14), jip-1(lf); unc-16(lf) (n = 9), unc-16(lf); mCherry::UNC-16 (n = 14), unc-16(lf); mCherry::JIP3a (n = 7), and jip-1(lf); mCherry::JIP-1 (n = 10) (normalized to control). ***Significantly different from control, p < 0.001. Filled star, significantly different from unc-16(lf), p < 0.001. Open star, significantly different from jip-1(lf), p < 0.001. Horizontal bar indicates unc-16(lf) and jip-1(lf) were not significantly different from each other or from jip-1(lf); unc-16(lf) double mutants.
(C) Kymographs of GLR-1::GFP transport in transgenic control worms (without Phsp::jip-1 or Phsp::unc-16), and transgenic jip-1(lf) and unc-16(lf) that expressed either Phsp::jip-1 or Phsp::unc-16, respectively, and either with or without 1 h of heat shock (H.S.) at 32°C. Phsp represents the hsp16–2p heat-shock promoter.
(D) Total GLR-1::GFP transport events in control worms without (n = 16) or with (n = 5) heat shock, jip-1(lf); Phsp::jip-1 without (n = 30) or with (n = 26) heat shock, and unc-16(lf); Phsp::unc-16 without (n = 28) or with (n = 27) heat shock (normalized to control without heat-shock treatment). ****p < 0.0001. n.s., not significant. All statistical comparison used a Kruskal-Wallis test with Dunn’s multiple comparison.
All strains carried the glr-1(ky176) deletion mutation (see STAR Methods). Scale bars, 5 μm. Error bars represent standard error of the mean (SEM). See STAR Methods for a description of statistical analysis. See also Figures S1–S3, Tables S1, and S2.
We next asked whether additional JIP-family scaffold proteins participate in kinesin-1-mediated transport of GLR-1. The vertebrate scaffold protein JIP1 interacts with kinesin motors and JIP3 (Satake et al., 2013). jip-1 encodes the C. elegans homolog of JIP1 (Sakaguchi et al., 2004). We found that GLR-1 flux was also significantly reduced in jip-1(lf) mutants (Figures 1A and 1B). The transport defect of jip-1; unc-16 double mutants was indistinguishable (no additive effect) from that of unc-16 single mutants, consistent with jip-1 and unc-16 functioning in the same pathway (Figures 1A and 1B). Both UNC-16 and JIP-1 scaffold proteins function cell autonomously, as demonstrated by cell-specific rescue of the transport defects in the AVA interneurons of transgenic unc-16(lf) and jip-1(lf) mutants, respectively (Figures 1A and 1B). Importantly, we also found that cell-specific expression of mammalian JIP3a in the AVA neurons of transgenic unc-16(lf) mutants rescued the unc-16 mutant defects, indicating evolutionary conservation of protein function (Figures 1A and 1B). Although the number of GLR-1 transport vesicles was considerably reduced in the mutants (note: we observed a reduction in both anterograde and retrograde transport; Figure S2A), we did not find appreciable differences in their instantaneous velocity (Figure S2B), indicating that this aspect of kinesin-1 function was not appreciably dependent on UNC-16 or JIP-1.
To address whether the defective AMPAR transport observed in the scaffold-protein mutants was secondary to altered development of the nervous system, we used a heat-shock promoter (hsp16–2p) to induce expression of a wild-type unc-16 or jip-1 transgene at the adult stage. Heat shock of transgenic worms with a hsp16–2p::mCherry::jip-1 transgene was sufficient to drive expression of mCherry::JIP-1 as evaluated by mCherry fluorescence (Figure S3). Using the same protocol, we drove the expression of hsp16–2p::unc-16 or hsp16–2p::jip-1 in transgenic unc-16 or jip-1 mutants, respectively, and found that heat shock at the adult stage restored the number of GLR-1::GFP transport events to near wild-type levels (Figures 1C and 1D). These data demonstrate that the altered transport of GLR-1 in unc-16 and jip-1 mutants was not caused by a developmental abnormality and provide additional evidence for the importance of these scaffold proteins for GLR-1 transport in the adult nervous system. Because the mammalian homologs of UNC-16 and JIP-1 (JIP3 and JIP1, respectively) are known to interact (Satake et al., 2013), our data are consistent with the hypothesis that a heteromeric UNC-16/JIP-1 scaffold complex functions to regulate AMPAR trafficking.
The distribution and function of synaptic AMPARs are disrupted in unc-16 and jip-1 scaffold-protein mutants
We next asked whether the disrupted AMPAR transport found in the unc-16(lf) and jip-1(lf) scaffold-protein mutants modified the steady-state number of receptors at individual synapses. Previously, we found that the number of AMPARs at synapses was dependent on kinesin-1-mediated receptor transport. Thus, in unc-116(lf) kinesin-1 mutants, the balance of delivery and removal of AMPARs was altered, leading to an accumulation of AMPARs at synapses (Figures 2A and 2B) (Hoerndli et al., 2013, 2015b). We found similar accumulations of AMPARs in unc-16(lf) and jip-1(lf) scaffold-protein mutants (Figures 2A and 2B). The GLR-1::GFP puncta in jip-1; unc-16 double mutants did not significantly differ from that of the single mutants, providing additional evidence that JIP-1 and UNC-16 form a functional complex.
Figure 2. UNC-16 and JIP-1 are required for GLR-1 synaptic levels and function.

(A) Confocal images of GLR-1::GFP puncta in the proximal region of the AVA processes in transgenic control and mutant worms.
(B) GLR-1::GFP fluorescence intensity in the proximal AVA processes in transgenic control (n = 40), unc-116(lf) (n = 20), unc-16(lf) (n = 21), jip-1(lf) (n = 20), and jip-1(lf); unc-16(lf) (n = 20) (normalized to control). **Significantly different from control, p < 0.01. Horizontal bar indicates unc-116(lf), unc-16(lf), and jip-1(lf) single mutants were not significantly different from each other or from the jip-1(lf); unc-16(lf) double mutant (Kruskal-Wallis and Dunn’s multiple testing).
(C) Confocal images of GLR-1::GFP in the proximal AVA processes in transgenic control worms (without Phsp::jip-1 or Phsp::unc-16), and transgenic jip-1(lf) and unc-16(lf) that expressed either Phsp::jip-1 or Phsp::unc-16, respectively and either with or without heat-shock treatment (see STAR Methods).
(D) GLR-1::GFP fluorescence intensity in control worms without (n = 38) or with (n = 29) heat shock, jip-1(lf); Phsp::jip-1 without (n = 30) or with (n = 29) heat shock, and unc-16(lf); Phsp::unc-16 without (n = 28) or with (n = 24) heat shock (normalized to control without heat-shock treatment).*p < 0.05; **p < 0.01; n.s., not significant (Kruskal-Wallis and Dunn’s multiple testing).
(E) Currents measured in AVA neurons in response to glutamate application (black bars) in transgenic control and mutant worms. Cells were held at −60 mV.
(F) Peak glutamate-gated current in control (n = 12), unc-16(lf) (n = 8), jip-1(lf) (n = 7), jip-1(lf); unc-16(lf) (n = 3), and transgenic unc-16(lf); mCherry::UNC-16 mutants (n = 4). ****Significantly different from control, p < 0.0001. **Significantly different from unc-16(lf), p < 0.01. Horizontal bar indicates unc-16(lf) and jip-1(lf) single mutants were not significantly different from each other or from the jip-1(lf); unc-16(lf) double mutant (one-way ANOVA with Bonferroni correction).
Scale bars, 5 μm. Error bars represent SEM. See STAR Methods for a description of statistical analysis. See also Figure S3, Tables S1, and S2.
Consistent with the normalization of AMPAR transport events following heat-shock-induced expression of unc-16 or jip-1 in transgenic mutants (Figures 1C and 1D), we also found that the increased synaptic GLR-1::GFP fluorescence observed in unc-16 and jip-1 mutants was restored to near wild-type levels following heat shock of the transgenic mutants (Figures 2C and 2D).
Mutations that disrupt the motor-mediated transport of GLR-1 are associated with reduced glutamate-gated current (Hoerndli et al., 2013, 2015b). We therefore asked whether current was similarly reduced in scaffold-protein mutants. As described previously (Hoerndli et al., 2013, 2015b), we used in vivo patch-clamp techniques to record from AVA neurons, and found that glutamate-gated current was markedly reduced in the unc-16(lf) and jip-1(lf) single mutants (Figures 2E and 2F). The jip-1 and unc-16 single mutants were not statistically different from each other or from the jip-1; unc-16 double mutant, providing additional support of our hypothesis that an UNC-16/JIP-1 scaffold complex mediates AMPAR transport. Furthermore, the transgenic expression of mCherry::UNC-16 in AVA, which rescued GLR-1 transport in unc-16 mutant (Figures 1A and 1B), also restored glutamate-gated current (Figures 2E and 2F), providing additional support for a cell-specific role of the UNC-16 scaffold protein in AMPAR transport and synaptic function.
Delivery and removal of synaptic AMPARs are dependent on UNC-16 and JIP-1 scaffold proteins
An independent test of the necessity of scaffold proteins for AMAR delivery is provided by monitoring the rate of fluorescence recovery after photobleaching (FRAP) of fluorescently tagged AMPARs at synaptic sites (fluorescent puncta along neural processes) (Hoerndli et al., 2013). We found that fluorescence recovery of GLR-1::GFP 2 h after photobleaching was reduced in unc-16(lf) and jip-1(lf) mutants compared with controls (Figures 3A–3C; see Figure S4 for FRAP experiments on a shorter timescale). In these experiments, we bleached a segment (~500 μm in length) of the neuronal processes extending from the vulva to the distal end of the AVA processes and evaluated FRAP of GLR-1::GFP puncta in the distal processes (Figure 3A). This procedure eliminated recovery mediated by simple diffusion and allowed us to selectively measure recovery driven by active motor-mediated transport, as we have previously described (Hoerndli et al., 2013). The data were consistent with the reduced flux of GLR-1 transport vesicles observed in the kymographs (Figure 1). Given that puncta were, on average, larger in the mutants compared with control worms, we also quantified FRAP at puncta of comparable total intensity (within 10% of each other). Again, we found that fluorescence recovery was significantly slower in unc-16(lf) (7.6% ± 1.7%, n = 8, p < 0.0001), and in jip-1(lf) (14% ± 1.3%, n = 8, p < 0.01) compared with control worms (93% ± 17%, n = 8).
Figure 3. The UNC-16 and JIP-1 scaffold proteins are necessary for the delivery and removal of synaptic AMPARs.

(A) Schematic representing the distal region of the AVA processes where GLR-1::GFP puncta were photobleached (dashed box, ~500 μm) and imaged (boxed region) for quantification of GFP fluorescence recovery.
(B) Confocal images before, immediately after (bleach), and 2 h after photobleaching GLR-1::GFP. Scale bar, 5 μm.
(C) GLR-1::GFP fluorescence recovery 2 h after photobleaching in transgenic control worms (n = 9), unc-16(lf) (n = 9), and jip-1(lf) (n = 5) mutants as a percentage of the fluorescent signal before photobleaching.
(D) Schematic representing GLR-1::Dendra2 puncta in the AVA processes before (top) and after photoconversion (dashed circles), and puncta imaged for quantification of red fluorescence remaining 2 h after photoconversion (boxed region).
(E) Confocal images of GLR-1::Dendra2 puncta (three examples per genotype) before, immediately after (converted), and 2 h after photoconverting GLR-1::Dendra2 from green to red fluorescence (red channel only). Scale bar, 1.3 μm.
(F) The percentage of red fluorescence remaining 2 h after photoconverting GLR-1::Dendra2 in transgenic control worms (n = 10), unc-16(lf) (n = 6), and jip-1(lf) (n = 8).
Significantly different from control, *p < 0.05 and ***p < 0.001 (Kruskal-Wallis with Dunn’s multiple testing). Error bars represent SEM. See also Figure S4, Tables S1, and S2.
To specifically address the question of receptor removal from synapses, we generated transgenic worms that expressed a functional fusion protein where GLR-1 was tagged with the photo-convertible fluorophore Dendra2 (Gurskaya et al., 2006; Hoerndli et al., 2015b). Removal of synaptic GLR-1::Dendra2 was measured by using UV light to photoconvert GLR-1::Dendra2 puncta from green to red fluorescence and then measuring the amount of red fluorescence remaining 2 h after photoconversion. Puncta of comparable intensities between all three genotypes were photoconverted. We found that the red-fluorescent signal in either unc-16 and jip-1 mutants was elevated compared with controls; i.e., GLR-1:Dendra2 was removed more slowly in the mutants (Figures 3D–3F). Together, these studies demonstrate essential roles for the UNC-16 and JIP-1 scaffold proteins in the delivery and removal of synaptic AMPARs.
UNC-16 is co-transported with JIP-1 and GLR-1
If UNC-16 is part of a scaffold complex mediating the transport of AMPARs, then we would expect co-transport of UNC-16 with GLR-1. Kymographic analysis revealed GLR-1::GFP transport events associated with mCherry::UNC-16 (Figure 4A; see Figure S5 demonstrating lack of bleed-through fluorescence). We found that GLR-1 transport was associated with UNC-16; however, UNC-16 was not exclusively associated with GLR-1, suggesting that this scaffold protein contributes to the transport of additional cargo. Similarly, we found that GFP::JIP-1 was co-transported with mCherry::UNC-16 (Figure 4B), further supporting the hypothesis that GLR-1 and the scaffold proteins, UNC-16 and JIP-1 are part of a transport complex. Importantly, the transport of UNC-16 was dependent on JIP-1 and on kinesin light chain (KLC-2) (Figures 4C and 4D), indicating that UNC-16 is immobile in the absence of JIP-1 and that the UNC-16/JIP-1 scaffold complex associates with kinesin-1 motors.
Figure 4. The UNC-16 and JIP-1 scaffold proteins are co-transported and are required for CaMKII-dependent AMPAR transport.

(A and B) Kymographs showing co-transport events (arrowheads) of mCherry::UNC-16 with either GLR-1::GFP (A), or GFP::JIP-1 (B) along the AVA processes. Scale bars, 2 μm.
(C) Kymographs of mCherry::UNC-16 transport in the AVA processes of transgenic control worms, and jip-1(lf) and klc-2(km11) lf mutants. All strains carried the unc-16(e109) mutation. Scale bar, 5 μm.
(D) Total mCherry::UNC-16 transport events in control (n = 9), jip-1(lf) (n = 11), and klc-2(lf) (n = 10). Significantly different from control, **p < 0.01 and ****p < 0.0001 (Kruskal-Wallis and Dunn’s multiple testing).
(E) Kymographs of GLR-1::GFP transport in the AVA processes of transgenic control worms, and various single and double mutants. Scale bars, 5 μm.
(F) Total GLR-1::GFP transport events in control worms (n = 9), unc-16(lf) (n = 9), jip-1(lf) (n = 14), and unc-43(n498sd) gain-of-function (gf) (n = 12) single mutants, and unc-43(gf); unc-16(lf) (n = 6) and jip-1(lf); unc-43(gf) (n = 6) double mutants (normalized to control without unc-43(gf)). Strains with a wild-type unc-43 allele or unc-43(gf) allele are indicated by white and gray bars, respectively. **p < 0.01; n.s., not significant using ANOVA with Kruskal-Wallis and Dunn’s multiple testing.
(G) Kymographs of mCherry::UNC-16 transport in transgenic control worms, unc-43(n498 n1186) lf, and unc-43(gf) mutants. All strains carried the unc-16(e109) mutation. Scale bars, 5 μm.
(H)Total mCherry::UNC-16 transport events in control (n = 13), unc-43(lf) (n = 9), and unc-43(gf) (n = 4). **Significantly different from control; p < 0.01; n.s., not significantly different from control (Mann-Whitney U test between samples acquired the same day).
Error bars represent SEM. See STAR Methods for a description of statistical analysis. See also Figure S5, Tables S1, and S2.
The UNC-16 and JIP-1 scaffold proteins act downstream of CaMKII signaling
We previously found that over 80% of GLR-1 transport is dependent on the kinase UNC-43 (CaMKII), which is critically important for the loading of GLR-1 cargo onto kinesin-1 motors (Hoerndli et al., 2015b). How might UNC-43 signaling interact with UNC-16 and JIP-1 scaffold proteins to regulate AMPAR transport? To address this question, we first examined GLR-1 transport in unc-43 gain-of-function (gf) mutants (Hoerndli et al., 2015b; Reiner et al., 1999), and asked if this was modified by either the unc-16(lf) or jip-1(lf) mutation.
As previously described, we found increased GLR-1 transport in unc-43(gf) mutants compared with controls (Figures 4E and 4F) (Hoerndli et al., 2015b). Transport in either unc-16(lf); unc-43(gf) or jip-1(lf); unc-43(gf) double mutants was comparable with that measured in unc-16(lf) or jip-1(lf) single mutants, respectively (Figures 4E and 4F), consistent with UNC-43 (CaMKII) signaling acting either on or upstream of the scaffold proteins. Thus, UNC-43-dependent transport of GLR-1 cargo via kinesin-1 motors is dependent on the presence of the scaffold proteins UNC-16 and JIP-1.
Is transport of the scaffold proteins dependent on UNC-43 (CaMKII)? If so, this could explain the observed decrease in GLR-1 transport in unc-43(lf) mutants (Hoerndli et al., 2015b). Although unc-43(lf) mutants showed a modest decrease in UNC-16 transport compared with controls (Figures 4G and 4H), the effect was insufficient to explain the dramatic decrease in GLR-1 transport observed in unc-43(lf) (Hoerndli et al., 2015b) (see also Figures 7C and 7D). We also asked whether the increased GLR-1 transport in unc-43(gf) mutants was secondary to an increase in the transport of UNC-16. In contrast to GLR-1 transport, UNC-16 transport was not increased in unc-43(gf) mutants compared with controls (Figures 4G and 4H). Thus, transport of the scaffold proteins themselves is mostly independent of UNC-43 (CaMKII), indicating that the changes in GLR-1 transport observed in unc-43 mutants (Figures 4E and 4F) are not secondary to changes in the scaffold complex. Together, these data suggest a model where CaMKII signaling facilitates loading of AMPARs onto a JIP-protein scaffold complex, which is then co-transported by kinesin-1 motors.
Figure 7. CaMKII and MAPK regulation of GLR-1 transport are interdependent.

(A) Kymographs of GLR-1::GFP transport in the AVA processes of transgenic control worms, and various single, double, and triple mutants.
(B) Total GLR-1::GFP transport events in unc-43(gf) (n = 12), unc-43(gf); jnk-1(lf) (n = 8), unc-43(gf); jkk-1(lf) (n = 15), and unc-43(gf); jnk-1(lf); jkk-1(lf) (n = 8) (normalized to same-day control without unc-43(gf)). Strains with a wild-type unc-43 allele or unc-43(gf) allele are indicated by white and gray bars, respectively. All data for strains without unc-43(gf) are the same as those shown in Figure 5B. *p < 0.05 and **p < 0.01(Kruskal-Wallis and Dunn’s multiple testing).
(C) Kymographs of GLR-1::GFP transport in control worms and unc-43(lf) mutants both with and without the JKK-1:JNK-1::TagRFP-T gain-of-function transgene (JKK-1::JNK-1(gf)).
(D) Total GLR-1::GFP transport events in control (n = 15), JKK-1:JNK-1(gf) (n = 8), unc-43(lf) (n = 8), and unc-43(lf); JKK-1:JNK-1(gf) (n = 10) (normalized to same-day control without JKK-1:JNK-1(gf)). ****p < 0.0001 (one-way ANOVA and Dunnett’s multiple testing).
(E) Kymographs of GLR-1::GFP transport in control worms, and various single and double mutants.
(F) Total GLR-1::GFP transport events in control worms (n = 13), mpk-1(oz140) (n = 7) and sek-1(km4) (n = 13) lf mutants, and mpk-1(lf); sek-1(lf) (n = 2) and jkk-1(lf) sek-1(lf) (n = 9) double mutants (normalized to same-day control). ****p < 0.0001 (one-way ANOVA and Dunnett’s multiple testing).
(G) Kymographs of GLR-1::GFP transport in control worms, and various single, double, and triple mutants.
(H) Total GLR-1::GFP transport events in control worms (n = 10), unc-43(gf) (n = 12), unc-43(gf); jkk-1(lf) (n = 15), unc-43(gf); sek-1(lf) (n = 15), and unc-43(gf); jkk-1(lf) sek-1(lf) (n = 10) (normalized to same-day control). **Significantly different from unc-43(gf); jkk-1(lf) sek-1(lf) triple mutant, p < 0.001. ***p < 0.001; n.s., not significant (one-way ANOVA and Tukey’s multiple testing).
(I) Total mCherry::UNC-16 transport events in transgenic control worms (n = 10), sek-1(lf) (n = 11), and jkk-1(lf) sek-1(lf) (n = 11) (normalized to same-day control). ***Significantly different from control and sek-1(lf), p < 0.0001; n.s., not significant using ANOVA with Kruskal-Wallis and Dunn’s multiple testing.
(J) Total mCherry::UNC-16 transport events in control worms (n = 12) and transgenic worms that expressed JKK-1::JNK-1::TagRFP-T gain-of-function (n = 14) (normalized to same-day control). ***p < 0.001 using Student’s t test.
(K) Model describing GLR-1 cargo loading onto kinesin-1 motors. Presumed neuronal activity leads to MAPK/MAPKK-dependent recruitment of the UNC-16/JIP-1 (JIPs) scaffold proteins, which enables CaMKII-dependent loading of GLR-1 cargo onto the kinesin-1 motor complex.
Strains carried either the glr-1(ky176) deletion mutation (A, C, E, and G) or the unc-16(e109) mutation (I and J) (see STAR Methods). Scale bars, 5 μm. Error bars represent SEM. See STAR Methods for a description of statistical analysis. See also Figures S6, S7, Tables S1, and S2.
MAPK signaling contributes to kinesin-1-mediated transport of AMPARs
Certain scaffold proteins can bind distinct classes of MAPKs that function in a signaling cascade. By localizing the kinases in close proximity to each other, scaffold proteins sharpen temporal response and enhance signaling sensitivity (Pan et al., 2012). To determine whether MAPKs or MAPK kinases (MAPKKs) contribute to AMPAR transport, we measured GLR-1 transport in two candidate MAPK-signaling mutants: jkk-1 (MAPKK of the JNK-signaling pathway) and jnk-1 (MAPK of the JNK-signaling pathway) (Ewbank, 2006; Kawasaki et al., 1999; Sagasti et al., 2001; Sakaguchi et al., 2004; Tanaka-Hino et al., 2002). Transport was reduced in both the jnk-1(lf) and jkk-1(lf) mutants, and defects of the single mutants were indistinguishable from those of the jnk-1(lf); jkk-1(lf) double mutants (Figures 5A and 5B). Both anterograde and retrograde transport were significantly reduced in jnk-1(lf); jkk-1(lf) compared with controls (Figure S6).
Figure 5. MAPK signaling pathways regulate AMPAR transport and synaptic accumulation.

(A) Kymographs of GLR-1::GFP transport in the AVA processes of transgenic control worms, jnk-1(gk7) and jkk-1(km2) lf mutants, and worms that expressed the JKK-1::JNK-1::TagRFP-T gain-of-function (gf) chimera. Scale bar, 5 μm.
(B) Total GLR-1::GFP transport events in control (n = 13), jnk-1(lf) (n = 8), jkk-1(lf) (n = 11), jnk-1(lf); jkk-1(lf) (n = 9), and JKK-1::JNK-1::TagRFP-T (n = 8) (normalized to control). Significantly different from control, **p < 0.01, and ****p < 0.0001. Horizontal bar indicates jnk-1(lf) and jkk-1(lf) single mutants were not significantly different from each other or from the jnk-1(lf); jkk-1(lf) double mutant (one-way ANOVA with Tukey’s multiple comparison).
(C) Confocal images of GLR-1::GFP in the distal AVA processes of transgenic control and mutant worms before, immediately after (bleach), and 2 h after photobleaching. Scale bar, 5 μm.
(D) GLR-1::GFP fluorescence recovery 2 h after photobleaching in control (n = 7), jnk-1(lf) (n = 8), and jkk-1(lf) (n = 8) as a percentage of the fluorescent signal before photobleaching.
(E) Confocal images of GLR-1::Dendra2 puncta (three examples per genotype) before, immediately after (converted), and 2 h after photoconverting GLR-1::Dendra2 from green to red fluorescence (red channel only). Scale bar, 1.3 μm.
(F) The percentage of red fluorescence remaining 2 h after photoconverting GLR-1::Dendra2 in control (n = 10), jnk-1(lf) (n = 9), and jkk-1(lf) (n = 10). ***Significantly different from same-day controls, p < 0.001.
All strains carried the glr-1(ky176) deletion mutation (see STAR Methods). Error bars represent SEM. See STAR Methods for a description of statistical analysis. See also Figure S6, Tables S1, and S2.
We next investigated the consequences of unregulated activation of the JNK-signaling pathway. We generated a fluorescently tagged (TagRFP-T) gain-of-function chimera as previously reported that links JKK-1 to JNK-1 (JKK-1::JNK-1) (Lei et al., 2002; Zheng et al., 1999). GLR-1::GFP flux in transgenic worms that expressed JKK-1::JNK-1(gf) in the AVA neurons was greatly increased compared with wild type (Figures 5A and 5B). These results are consistent with JNK signaling having a key regulatory role in motor-mediated AMPAR transport.
MAPK signaling is required for the delivery and removal of synaptic AMPARs
Loss of JNK-1 and JKK-1 function (Figure 5) partially phenocopied the defects in transport observed in the unc-16(lf) and jip-1(lf) scaffold-protein mutants (Figure 1). To further test the dependence of GLR-1 transport on the MAPK-signaling pathway, we either photobleached GLR-1::GFP or photoconverted GLR-1::Dendra2 at synapses, and measured changes in the fluorescent signal over time. GLR-1::GFP fluorescence recovery (a measure of GLR-1 delivery) was considerably reduced in jnk-1(lf) and jkk-1(lf) mutants (Figures 5C and 5D). The defect was also observed when examining GLR-1::GFP puncta of similar intensities (jnk-1(lf), 17% ± 3.1%, and jkk-1(lf), 18% ± 6.4%, n = 8; p < 0.01 compared with control, 86% ± 20%, n = 8). Similarly, loss of GLR-1::Dendra2 red fluorescence after photoconversion from green to red (a measure of GLR-1 removal) was significantly reduced in jnk-1 and jkk-1 mutants (puncta of similar fluorescence intensities were compared) (Figures 5E and 5F). Together, these results indicate that MAPK signaling is of critical importance for the delivery and removal of synaptic AMPARs.
MAPK signaling is required for loading of the scaffold proteins onto kinesin-1 motors
Does MAPK signaling require the presence of the UNC-16 and JIP-1 scaffold proteins? To address this, we expressed the JKK-1::JNK-1(gf) chimera specifically in AVA and measured transport of GLR-1. We found that the transgene-induced increase in GLR-1 transport was dependent on UNC-16 and JIP-1. Thus, expressing JKK-1::JNK-1(gf) in either unc-16(lf) or jip-1(lf) transgenic mutants did not rescue transport of GLR-1 (Figures 6A and 6B). These data indicate the MAPK signaling required for GLR-1 transport is dependent on context and requires the UNC-16 and JIP-1 scaffold proteins.
Figure 6. MAPK regulation of AMPAR transport requires the scaffold proteins.

(A) Kymographs of GLR-1::GFP transport in the AVA processes of transgenic control worms, unc-16(lf) and jip-1(lf), and transgenic control and mutant worms that expressed JKK-1::JNK-1::TagRFP-T gain-of-function chimera (JKK-1::JNK-1(gf)). All strains carried the glr-1(ky176) deletion mutation.
(B) Total GLR-1::GFP transport events in control without (n = 13) or with (n = 8) JKK-1:JNK-1(gf); unc-16(lf) without (n = 9) or with (n = 9) JKK-1:JNK-1(gf); and jip-1(lf) without (n = 9) or with (n = 5) JKK-1::JNK-1(gf). Data for control without JKK-1::JNK-1(gf) same as that shown in Figure 4F. ***p < 0.001.
(C) Kymographs of mCherry::UNC-16 transport in transgenic control worms, and jnk-1(lf) and jkk-1(lf) mutants. All strains carried the unc-16(e109) mutation.
(D) Total mCherry::UNC-16 transport events in control (n = 9), jnk-1(lf) (n = 10), and jkk-1(lf) (n = 10). Significantly different from control, *p < 0.05 and ***p < 0.001.
(E) Kymographs showing JKK-1::JNK-1::TagRFP-T and GLR-1::GFP co-transport events (arrowheads) in the AVA processes.
(F) Kymographs showing JKK-1::JNK-1::TagRFP-T transport in the AVA processes of transgenic control worms, and unc-16(lf) and jip-1(lf) mutants.
(G) Total JKK-1::JNK-1::TagRFP-T transport events in control (n = 10), unc-16(lf) (n = 10), and jip-1(lf) (n = 9). *Significantly different from control, p < 0.01; n.s., not significant.
(H) Three examples of single-plane confocal images of GLR-1::GFP, JKK-1::JNK-1::TagRFP-T, and the merge of both channels.
Scale bars, 5 μm. Error bars represent SEM. All statistics in this figure used a one-way ANOVA with Dunnett’s multiple testing as described in STAR Methods. See also Tables S1 and S2.
We reasoned that the altered transport of GLR-1 in MAPK mutants might be secondary to defective loading of the UNC-16/ JIP-1 scaffold complex onto motors. In support of this hypothesis, we found that the transport of UNC-16 was decreased in jnk1 and jkk-1 mutants compared with controls (Figures 6C and 6D). This suggests that the decreased AMPAR transport observed in MAPK mutants (Figures 5A and 5B) was secondary to reduced transport of the UNC-16 scaffold protein. We were unable to generate rescued transgenic lines with tagged variants of JKK-1 or JNK-1 for technical reasons associated with protein expression levels. However, we could image the JKK-1::JNK-1::TagRFP-T fusion protein, which we found was co-transported with GLR-1::GFP (Figure 6E). JKK-1::JNK-1::TagRFP-T transport was dependent on the UNC-16 scaffold protein (Figures 6F and 6G). Additionally, we found partial co-localization of GLR-1::GFP and JKK-1::JNK-1::TagRFP-T (Pearson’s correlation coefficient: 0.40 ± 0.11, p < 0.03, n = 5), consistent with a dependence on MAPK signaling for loading of GLR-1/UNC-16/ JIP-1 cargo (Figure 6H).
To address the interaction between MAPK and UNC-43 (CaMKII) signaling, we asked whether the increased GLR-1 transport observed in unc-43(gf) mutants was dependent on MAPK signaling. We found that GLR-1 transport observed in unc-43(gf) mutants was reduced by jkk-1(lf) or jnk-1(lf), and that transport in unc-43(gf) was similarly reduced when both jnk-1 and jkk-1 were mutated (Figures 7A and 7B, gray bars). In contrast, we observed complete suppression of unc-43(gf) in the scaffold-protein mutants, where GLR-1 transport in unc-16(lf); unc-43(gf) and jip-1(lf); unc-43(gf) double mutants was indistinguishable from unc-16(lf) or jip-1(lf) single mutants, respectively (Figures 4E and 4F). Importantly, GLR-1 transport in JKK-1::JNK-1(gf) mutants was entirely dependent on CaMKII, with transport in transgenic unc-43(lf) mutants that expressed JKK-1::JNK-1(gf) indistinguishable from unc-43(lf) single mutants (Figures 7C and 7D).
Our data demonstrate that transport of GLR-1 was dependent on an UNC-16/JIP-1 scaffold complex, and mutations that disrupt this scaffold (e.g., loss of UNC-16) could not be overcome by gain-of-function mutations in either UNC-43 (CaMKII) or the JNK-1/JKK-1 (MAPK) signaling pathway (Figures 4 and 6). In contrast, we found that increased transport of GLR-1 in unc-43(gf) was only partially suppressed by mutations in jnk-1(lf) or jkk-1(lf) (Figures 7A and 7B). Together, these results suggest that an additional signaling pathway(s) contributes to the loading of the UNC-16/JIP-1 scaffold onto kinesin-1 motors and motivated us to test whether additional MAPK-signaling proteins contribute to GLR-1 transport.
We found diminished transport in sek-1(lf) mutants (a MAPKK of the p38-signaling pathway) (Sagasti et al., 2001; Tanaka-Hino et al., 2002), and mpk-1(lf) mutants (a MAPK of an ERK-signaling pathway) (Lackner et al., 1994) (Figures 7E and 7F). Similar to jkk-1(lf), the sek-1(lf) mutation only partially suppressed the unc-43(gf) phenotype; however, almost complete suppression was observed when both jkk-1 and sek-1 were mutated (Figures 7G and 7H). This additive effect highlights the interactions of the MAPK signaling pathways (Fabian et al., 2021; Fey et al., 2012; Kim et al., 2004; Raman et al., 2007). We next examined the transport of mCherry::UNC-16 in the kinase mutants. We found that transport of UNC-16 was not significantly diminished in sek-1 mutants but was dramatically and significantly decreased in jkk-1 sek-1 double mutants (Figures 7I and S7A). In complementary experiments, we found that JKK-1::JNK-1(gf) increased mCherry::UNC-16 transport (Figures 7J and S7B), thus strengthening our conclusion that MAPK pathways control the loading of the JIP-1/UNC-16 scaffold complex onto kinesin motors.
Our results indicate that at least two MAPK pathways can promote loading of the scaffold proteins onto kinesin-1 and contribute to GLR-1 transport. In conclusion, we provide a mechanistic model of AMPAR transport in which cooperative MAPK-and CaMKII-mediated signaling function at distinct steps to regulate loading of AMPARs onto kinesin-1 motors (Figure 7K).
DISCUSSION
Synapses have two major and interrelated roles: they provide a mechanism for rapid communication between neurons enabling the computing power of neuronal networks, and they facilitate plasticity of the nervous system. Each of the thousands of synapses in a given neuron is subject to experience-dependent changes in strength that reflect activity-dependent changes in the number of postsynaptic neurotransmitter receptors (Granger and Nicoll, 2014). How then are distinct synapses distributed along neuronal processes, and far from the cell body, supplied with the appropriate type and number of receptors? This is a classic logistics problem, which we have shown can be managed by the motor-mediated delivery of receptors to specific synapses, and by the motor-mediated removal from synapses for either redistribution (Hoerndli et al., 2013) or degradation (Kennedy and Ehlers, 2006). Rapid motor-mediated transport of AMPARs also occurs in vertebrate neurons (Hangen et al., 2018), and altered trafficking of AMPARs is associated with cognitive disorders (Volk et al., 2015).
We have discovered that motor-mediated receptor transport is dependent on two signaling pathways: a complex MAPK-signaling cascade required for loading the UNC-16/JIP-1 scaffold complex onto microtubule-based kinesin-1 motors, and CaMKII signaling, which mediates loading of synaptic AMPAR cargo onto the scaffold. We propose that this coordinated dual-signaling mechanism might contribute to the coupling of neuronal activity to the loading of AMPAR cargo onto kinesin-1 motors, thus providing a rapid and spatially confined mechanism to control the number of receptors in response to the activity of individual synapses (Figure 7K).
AMPAR transport is dependent on a kinesin-1-associated scaffold complex
We demonstrated that the MAPK-associated scaffold protein, UNC-16, co-transported with the JIP-1 scaffold protein and with the GLR-1 AMPAR subunit along the processes of the AVA neurons. Additionally, we found that UNC-16 and JIP-1 transport to synapses was co-dependent, consistent with the scaffold proteins forming a complex required for AMPAR transport by kinesin-1. This finding is also consistent with an earlier study that demonstrated that the mammalian homologs of UNC-16 and JIP-1 (JIP3a and JIP1, respectively) and UNC-116 (kinesin-1) form a ternary complex (Satake et al., 2013). Furthermore, we showed that vertebrate JIP3a rescued the AMPAR transport defects of unc-16(lf) mutants, indicating a conserved role in the delivery and removal of synaptic AMPARs.
The number of GLR-1 transport events was greatly decreased in unc-16 or jip-1 mutants. UNC-16 regulates the transport of a wide range of cargo (Byrd et al., 2001; Edwards et al., 2013, 2015; Sakamoto et al., 2005), and, consistent with this, only a subset of UNC-16 transport events was associated with GLR-1 and JIP-1. Glutamate-gated currents in the unc-16(lf) and jip-1(lf) scaffold-protein mutants were also significantly reduced, supporting defective receptor transport as the proximal cause of the reduced number of functional channels at synapses. We noted that deleting the scaffold proteins did not completely eliminate GLR-1 transport, indicating that transport vesicles might also directly associate with kinesin motors, or that additional scaffold proteins or compensatory mechanisms contribute to GLR-1 transport in the absence of JIP-1 and UNC-16.
MAPK signaling is required for transport of the UNC-16/ JIP-1 scaffold complex
The association of scaffold proteins with MAPKs allows for activation of signaling cascades in a localized environment (Pan et al., 2012). In certain cases, kinesin-mediated transport of specific vesicle subtypes is known to be dependent on MAPK-signaling pathways (Liang and Yang, 2019). Our data suggest that MAPK-signaling proteins, including JKK-1, JNK-1, SEK-1, and MPK-1, are required for association of the UNC-16/JIP-1 scaffold complex with kinesin-1 motors. Mutations that disrupted the JKK-1/JNK-1 pathway reduced UNC-16 transport by approximately 50% (Figure 6D), providing a mechanistic explanation for the approximate 50% decrease in GLR-1 transport in jnk-1 and jkk-1 mutants (Figure 5B). Double mutants that disrupted two separate MAPK-signaling pathways eliminated most UNC-16 transport (Figures 6D and 7I) and, correspondingly, GLR-1 transport (which was apparent in the presence of the unc-43(gf) mutation; Figure 7H).
CaMKII is required for the loading of AMPAR cargo onto the scaffold complex
Our genetic analysis indicates that UNC-43 (CaMKII) is required for the loading of AMPAR cargo onto the assembled scaffold proteins. Increased AMPAR flux in unc-43(gf) mutants was suppressed by mutating either unc-16 or jip-1 scaffold proteins (Figure 4F). Importantly, increased AMPAR flux in transgenic worms that expressed the JKK-1::JNK-1 gain-of-function variant was suppressed by a loss-of-function mutation in unc-43 (Figure 7D). These data suggest that, with respect to GLR-1 transport, the roles of MAPK and UNC-43 (CaMKII) signaling converge at the level of the scaffold proteins. We propose that CaMKII signaling promotes the loading of AMPAR cargo onto the UNC-16/JIP-1 scaffold, and that MAPK signaling is required for loading the scaffold complex onto kinesin-1 motors (Figures 7I–7K). Interestingly, CaMKII can activate MAPK signaling (Tanaka-Hino et al., 2002), and also phosphorylates the KLC-2 motor subunit (Hoerndli et al., 2015b), raising the intriguing possibility that activation of CaMKII can promote both the loading of AMPARs onto scaffold proteins and the loading of scaffold proteins onto kine-sin-1 motors (via possible CaMKII activation of MAPKs and/or direct activation of KLC-2), thus coordinating the delivery or removal of synaptic AMPARs with synaptic activity.
MAPK signaling: Implications for activity-dependent AMPAR delivery, coincidence detection, and synaptic plasticity
Our discovery that scaffold proteins and MAPKs have critical roles in kinesin-1-dependent delivery and removal of synaptic AMPARs has interesting implications for the control of synaptic strength. AMPARs are long lived (with an approximate lifetime of 10 days) (Fornasiero et al., 2018), and their numbers are relatively small (approximately 60 per synapse) (Richter et al., 2018). In contrast to protein-synthesis-dependent plasticity, which typically takes hours and might be reliant on local translation (Biever et al., 2019), classical early LTP is not dependent on protein synthesis (Abraham and Williams, 2008). Therefore, plastic changes in synaptic strength must rely on dynamic changes in the relative distribution of AMPARs at synapses. The speed of motor-mediated AMPAR transport is approximately 1.6 μm/s (Hoerndli et al., 2013, 2015b), allowing for rapid delivery and removal of receptors along neuronal processes. However, to accurately and efficiently regulate synaptic strength, the decision to deliver or remove cargo at specific synapses must also be sufficiently rapid to respond to synaptic activity.
We previously estimated that any given synapse is only a few seconds removed from a motor carrying AMPARs (Hoerndli et al., 2013). MAPK-signaling cascades can exhibit ultra-sensitivity, leading to switch-like changes in activity (Huang and Ferrell, 1996). Therefore, an AMPAR-associated signaling complex could facilitate rapid exchange of AMPARs by providing a decision-making switch (load or unload) for each motor as it passes a synapse. Interestingly, the estimated time constant of inactivation for synaptic CaMKII is approximately 6 s (Yasuda, 2017), which corresponds to the expected waiting time for kinesin-1-mediated AMPAR transport (Hoerndli et al., 2013).
The involvement of MAPK-signaling pathways in AMPAR transport has implications for current models of synaptic plasticity. LTP is a well-accepted cellular model of learning and memory that describes an enhancement of synaptic strength induced by patterned excitatory synaptic input. This phenomenon depends on activation of NMDA-type iGluRs, depolarization of the postsynaptic membrane, and activation of CaMKII (Nicoll and Roche, 2013). However, synaptic activity can also lead to the activation of MAPK-signaling cascades, which have been implicated in learning and memory (Gyurko et al., 2015; Ryu and Lee, 2016; Stornetta and Zhu, 2011; Tang and Yasuda, 2017). For example, dopamine signaling contributes to LTP in hippocampal CA1 neurons via a MAPK-signaling pathway (Shetty and Sajikumar, 2017). Additionally, MAPK signaling contributes to LTD in cerebellar Purkinje cells (Zamora Chimal and De Schutter, 2018), and LTP in the dentate gyrus (Wu et al., 2006). Importantly, MAPKs have been implicated in the synaptic removal of AMPARs (Myers et al., 2012), and may function in distinct synaptic microdomains to regulate the delivery and removal of synaptic AMPARs (Zhang et al., 2018; Zhu et al., 2002). We have now found that the regulation of AMPAR transport—delivery and removal—is dependent on both MAPK-and CaMKII-signaling cascades, suggesting that signaling by these pathways is central to the control of synaptic plasticity.
Considering the dependence of AMPAR transport on MAPK signaling, it is important to identify postsynaptic proteins required for this process. These proteins might contribute to the “stop” and “go” signals for motor-mediated delivery and removal of AMPARs, and thus contribute to synaptic plasticity. We anticipate that dysfunction of these signaling pathways might also be involved in the disordered synaptic signaling characteristic of neurodegenerative disorders such as Alzheimer disease (Huang and Mucke, 2012; Sala Frigerio and De Strooper, 2016). Interestingly, JIP3a (UNC-16 homolog) is an important regulator of axonal lysosomal transport, and regulates amyloid processing in a mouse model of Alzheimer disease (Gowrishankar et al., 2017). Our results demonstrate that scaffold proteins (UNC-16 and JIP-1), and associated MAPK-and MAPKK-signaling pathways (JNK-1 and JKK-1, and MPK-1 and SEK-1), are required for postsynaptic AMPAR transport. Thus, it is of fundamental importance to gain further mechanistic insight into how these proteins regulate the delivery and removal of AMPARs at synapses distributed along the vast dendritic processes of neurons. In light of earlier work relating kinesin-1 to Alzheimer disease (Brady and Morfini, 2017; Kamal et al., 2001), our work raises the intriguing possibility that disrupted dendritic transport of AMPARs might causally contribute to the synaptopathy associated with Alzheimer disease.
Limitations of the study
In this study, we used a genetic approach, combined with in vivo imaging and electrophysiology, to demonstrate that CaMKII-and MAPK-signaling pathways converge to regulate the motor-mediated transport of synaptic AMPARs. These signaling pathways are known to be involved in synaptic plasticity and the recruitment of AMPARs. Our study now provides evidence that these same signaling pathways contribute to the control of synaptic strength via regulation of motor-mediated transport of synaptic AMPARs. While our genetic approach has proved highly informative, it does not show direct biophysical interactions. Therefore, future studies using complementary biochemical approaches in suitable mammalian systems could more directly demonstrate the physical interactions between motors, scaffold proteins, and AMPARs. This would provide useful mechanistic insights into the loading and unloading of AMPAR cargo. Genetic and biochemical studies are also needed to identify additional molecules that are predicted to contribute to AMPAR transport, including co-effector proteins for the signaling pathways and adapter proteins linking AMPAR cargo to the scaffold.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andres V. Maricq (maricq@neuro.utah.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Microscopy and electrophysiology data will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains and growth conditions
Hermaphrodite nematodes, Caenorhabditis elegans (N2 Bristol strain, and mutant and transgenic strains derived from N2) were used in the study. All strains were raised at 20°C under standard conditions. Mutant alleles used are detailed in Table S1. Germline transformation to generate transgenic strains was achieved as described (Brockie et al., 2001). Transgenic worms were identified either by rescue of the lin-15(n765ts) mutant phenotype using a wild-type lin-15(+) transgene, or by expression of a soluble fluorescent marker, e.g., egl-20p::nls::DsRed.
METHOD DETAILS
Transgenic arrays
Transgenic animals expressed the following transgenic arrays: akIs141, rig-3p::HA::glr-1::gfp; akIs154, rig-3p::HA::glr-1::Dendra2; akEx4426, flp-18p::mCherry::unc-16 + egl-20p::nls::DsRed; akEx4395, flp-18p::mCherry::JIP3a + egl-20p::nls::DsRed; akEx4866, flp-18p::mCherry::jip-1 + egl-20p::nls::DsRed; akEx4489, flp-18p::gfp::jip-1 + flp-18p::mCherry::unc-16; akEx4490, flp-18p::gfp::jip-1 + flp-18p::mCherry::unc-16; akEx4824, flp-18p::jkk-1::jnk-1::TagRFP-T + egl-20p::nls::DsRed; akEx3831, flp-18p::unc-16(RNAi) + rig-3p::mCherry; akEx3832, flp-18p::unc-16(RNAi) + rig-3p::mCherry; csfEx7, rig-3p::mCherry + hsp16–2p::jip-1; csfEx12, rig-3p::mCherry+ hsp16–2p::unc-16; csfEx20, rig-3p:GLR-1:::mCherry + rig-3p:SEP::GLR-2; csfEx172, flp-18p::mCherry::UNC-16+ flp-18p::JKK-1:: JNK-1; csfEx176, flp-18p::mCherry::UNC-16.
DNA plasmids
pDM1494, rig-3p::mCherry; pDM1437, rig-3p::HA::glr-1::gfp; pDM1550, rig-3p::HA::glr-1::Dendra2; pDM2438, flp-18p::mCherry:: jip-1; pDM2389, flp-18p::mCherry::unc-16; pDM2370, flp-18p::mCherry::JIP3a; pDM2355, flp-18p::gfp::jip-1; pDM2271, flp-18p::jkk-1::jnk-1::TagRFP-T; pDM3006, flp-18p::jkk-1::jnk-1; pDM2296, hsp16–2p::unc-16; pDM2665, hsp16–2p::jip-1; pDM2118, rig-3p::SEP::GLR-2; pCT61, egl-20p::nls::DsRed; pJM23, lin-15p::lin-15.
Plasmid descriptions are also given in the Key resources table. jip-1 fusion clones (pDM2438 and pDM2355) were assembled via PCR amplification off of EST clones (yk1389g01, yk1193c03 and yk1057b08; gifts from Y. Kohara, National Institute of Genetics, Japan) and confirmed by sequencing. The unc-16 mini-gene was generated by PCR amplification from pCZ350 (Byrd et al., 2001). The JIP3a coding sequence was amplified off of pcdna3 T7 JIP3a (gift from Roger Davis, Addgene plasmid #53457). To generate pDM2271 and pDM3206 (flp-18p::jkk-1::jnk-1::TagRFP-T and flp-18p::jkk-1::jnk-1), jkk-1 and jnk-1 were both cloned from first strand cDNA generated from mixed-stage C. elegans RNA. Clones were then fused via PCR and included the flexible 12 amino acid linker VRVGGSGGTGGS (Zheng et al., 1999).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Muscimol | Sigma | Cat#: M1523 |
| Polybead Microsphere 0.1 μm | Polysciences | Cat#: 00876–15 |
| TetraSpek Fluorescent Microspheres | ThermoFisher Scientific | Cat#: T14792 |
|
| ||
| Critical commercial assays | ||
|
| ||
| QIAprep Spin Miniprep (250) | QIAGEN | Cat#: 27106 |
| HiSpeed Plasmid Midi Kit (25) | QIAGEN | Cat#: 12643 |
| PureLink HQ Mini Plasmid DNA Purification Kit | Invitrogen | Cat#: K210002 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C. elegans: unc-16(e109) | CGC | CB109 |
| C. elegans: jip-1(km18) | Eric Jorgensen | EJ10713 |
| C. elegans: unc-43(n498sd) | CGC | MT1092 |
| C. elegans: unc-43(n498n1186) | CGC | MT2605 |
| C. elegans: jnk-1(gk7) | CGC | VC8 |
| C. elegans: jkk-1(km2) | CGC | KU2 |
| C. elegans: sek-1(km4) | CGC | KU4 |
| C. elegans: klc-2(km11) | Yishi Jin | VM5950 |
| C. elegans: mpk-1(oz140)/dpy-17(e164) unc-79(e1068) | CGC | MT8186 |
| C. elegans: lin-15(n765ts) | CGC | MT8189 |
| C. elegans: akIs141; glr-1(ky176) | Hoerndli et al., 2013 | VM6552 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109) | This study | VM10878 |
| C. elegans: jip-1(km18) akIs141; glr-1(ky176) | This study | VM11282 |
| C. elegans: jip-1(km18) akIs141; glr-1(ky176) unc-16(e109) | This study | VM11987 |
| C. elegans: akIs141; unc-116(rh24) | This study | VM7851 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); akEx4426 | This study | VM12250 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); akEx4395 | This study | VM12182 |
| C. elegans: glr-1(ky176); akIs154 | Hoerndli et al., 2013 | VM7989 |
| C. elegans: glr-1(ky176) unc-16(e109); akIs154 | This study | VM11316 |
| C. elegans: jip-1(km18); glr-1(ky176); akIs154 | This study | VM12326 |
| C. elegans: lin-15(n765ts); akEx4489 | This study | VM12344 |
| C. elegans: lin-15(n765ts); akEx4490 | This study | VM12345 |
| C. elegans: jip-1(km18) akIs141; glr-1(ky176); akEx4871 | This study | VM12967 |
| C. elegans: jip-1(km18) akIs141; glr-1(ky176); akEx4866 | This study | FJH158 |
| C. elegans: jip-1(km18); akIs141; glr-1(ky176) unc-16(e109); akEx4426 | This study | VM12919 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); klc-(km11); akEx4426 | This study | FJH119 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); unc-43(n498sd) | This study | VM11000 |
| C. elegans: akIs141 jip-1(km18); glr-1(ky176); unc-43(n498sd) | This study | VM11982 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); unc-43(n498n1186); akEx4426 | This study | FJH96 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); unc-43(n498sd); akEx4426 | This study | VM13722 |
| C. elegans: akIs141; glr-1(ky176); jnk-1(gk7) | This study | VM11129 |
| C. elegans: akIs141; glr-1(ky176); jkk-1(km2) | This study | VM11128 |
| C. elegans: akIs141; glr-1(ky176); jnk-1(gk7); jkk-1(km2) | This study | VM11127 |
| C. elegans: akIs141; glr-1(ky176); akEx4824 | This study | VM12850 |
| C. elegans: jnk-1(gk7); glr-1(ky176); akIs154 | This study | VM10793 |
| C. elegans: jkk-1(km2); glr-1(ky176); akIs154 | This study | VM11447 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); akEx4824 | This study | VM12851 |
| C. elegans: akIs141 jip-1(km18); glr-1(ky176); akEx4824 | This study | VM12849 |
| C. elegans: akIs141; glr-1(ky176); jnk-1(gk7); akEx4426 | This study | VM12917 |
| C. elegans: jkk-1(km2); glr-1(ky176); akIs141; akEx4426 | This study | VM13045 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498sd) | Hoerndli et al., 2015a, 2015b | VM7295 |
| C. elegans: akIs141; glr-1(ky176); jnk-1(gk7) unc-43(n498sd) | This study | VM11444 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498sd); jkk-1(km2) | This study | VM11468 |
| C. elegans: akIs141; glr-1(ky176); jnk-1(gk7) unc-43(n498sd); jkk-1(km2) | This study | VM11443 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498n1186) | Hoerndli et al., 2015a, 2015b | VM8199 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498n1186); akEx4824 | This study | VM12529 |
| C. elegans: akIs141; mpk-1(oz140)/dpy-17(e164) unc-79(e1068) | This study | VM12947 |
| C. elegans: akIs141; glr-1(ky176); sek-1(km4) | This study | VM12930 |
| C. elegans: akIs141; mpk-1(oz140)/ dpy-17(e164) unc-79(e1068); sek-1(km4) | This study | VM14138 |
| C. elegans: akIs141; glr-1(ky176); jkk-1(km2) sek-1(km4) | This study | VM12524 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498sd); jkk-1(km2) | This study | VM11468 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498sd); jkk-1(km2) sek-1(km4); | This study | VM12969 |
| C. elegans: akIs141; glr-1(ky176); unc-43(n498sd); sek-1(km4) | This study | VM12932 |
| C. elegans: akIs141 II; glr-1(ky176) unc-16(e109) III; sek-1(km4); akEx4426 | This study | VM13005 |
| C. elegans: akIs141 II; glr-1(ky176) unc-16(e109) III; jkk-1(km2) sek-1(km4) X; akEx4426 | This study | VM13047 |
| C. elegans: akIs141; glr-1(ky176); akEx3831 | This study | VM11292 |
| C. elegans: akIs141; glr-1(ky176); akEx3832 | This study | VM11293 |
| C. elegans: akIs141 jip-1(km18); glr-1(ky176); csfEx7 | This study | FJH97 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); csfEx12 | This study | FJH102 |
| C. elegans: glr-2(ak10) glr-1(ky176); lin-15(n765ts); csfEx20 | This study | FJH127 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); csfEx172 | This study | FJH420 |
| C. elegans: akIs141; glr-1(ky176) unc-16(e109); csfEx176 | This study | FJH424 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S2 | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: pCZ350 | Yishi Jin | NA |
| Plasmid: pCDNA3 T7 JIP3a | Rogers Davis | Addgene plasmid #53457 |
| EST clones for sek-1 | Yuki Kohara | yk1489b11 |
| EST clones for jip-1 | Yuki Kohara | yk1389g01,yk1193c03 and yk1057b08 |
| rig-3p::mCherry | Hoerndli et al., 2013 | pDM1494 |
| rig-3p::HA::glr-1::gfp | Hoerndli et al., 2013 | pDM1437 |
| rig-3p::HA::glr-1::Dendra2 | Hoerndli et al., 2013 | pDM1550 |
| flp-18p::mCherry::jip-1 | This study | pDM2438 |
| flp-18p::mCherry::unc-16 | This study | pDM2389 |
| flp-18p::mCherry::JIP3a | This study | pDM2370 |
| flp-18p::gfp::jip-1 | This study | pDM2355 |
| flp-18p::jkk-1::jnk-1::TagRFP-T | This study | pDM2271 |
| flp-18p::jkk-1::jnk-1 | This study | pDM3006 |
| hsp16–2p::unc-16 | This study | pDM2296 |
| hsp16–2p::jip-1 | This study | pDM2665 |
| egl-20p::nls::dsRed | (Brockie et al., 2013) | pCT61 |
| lin-15p::lin-15 | (Huang et al., 1994) | pJM23 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| KymoAnalyzer ImageJ Plugin | Neumann et al., 2017 | N/A |
| Igor pro 6.3 | WaveMetrics | https://www.wavemetrics.com/products/igorpro/igorpro.htm |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html?s_tid=hp_products_matlab |
| Adobe Creative Cloud, Photoshop and Illustrator | https://www.adobe.com/creativecloud | |
Double stranded RNA interference (RNAi): The flp-18p promoter sequence was based on that published in (Feinberg et al., 2008). unc-16(RNAi) was expressed in the AVA neurons using the flp-18p promoter sequence and generated using published protocols (Esposito et al., 2007). Primer sequences are listed in Table S2.
Confocal microscopy
One-day old adult worms were mounted on 10% agarose pads with 1.6 μL of 30 mM muscimol. Unless otherwise specified, fluorescently tagged GLR-1, UNC-16, or JIP-1 were expressed in a glr-1(ky176), unc-16(e109) or jip-1(km18) mutant background, respectively.
Streaming imaging and quantification of fluorescence were performed as previously described (Hoerndli et al., 2013). Confocal images were acquired using a Nikon Ti Eclipse microscope, or an Olympus IX83 microscope, equipped with a WaveFX-X1 spinning disk confocal system (Quorum Technologies) with 491 nm and 561 nm excitation lasers (Coherent). Images were captured using Cascade II 1024 EMCCD cameras (Photometrics) and a Nikon 10031.49 NA TIRF objective, or using iXon 512 ultra EMCCD cameras (Andor) and an Olympus 10031.4 NA objective. Photobleaching was achieved using a Mosaic digital mirror device (Andor) combined with 488 nm light from either a mixed-gas Argon laser from Coherent, or a 3 W Coherent solid-state laser (Genesis MZ MTM). Both lasers used a total output of 0.5 W with a pulse time of 1 s for photobleaching. Image acquisition and device control were enabled by Metamorph 7.7.7 (Molecular Devices). Transport analysis was performed using the ImageJ plugin KymoAnalyzer (Neumann et al., 2017). Individual transport events were included in the analysis if the fluorescent signal remained in the focal plane as it passed through the field of view. At least 10 kymographs per experimental group were used for the stop and velocity analyses.
Simultaneous dual-channel streaming imaging was acquired using an Olympus IX83 microscope, with 2 Andor iXon 512 and a WaveFx-X1 spinning disk modified by Applied Spectral with a (DAPI-GFP/Cy3-Cy7) beam splitter. Prior to every imaging session, dual-channel pixel alignment was performed using a TetraSpek Fluorescent Microspheres slide with sphere sizes ranging from 0.1–4 μm in different regions (Thermo Fisher Scientific T14792). Live streaming images were typically acquired using a 200 ms exposure and a total of 250 frames. Photobleaching prior to imaging was performed using a 1 s pulse of 0.5 W from a 488 nm Coherent solid-state laser. For dual imaging experiments, we verified lack of signal bleed-through (Figure S5).
FRAP and photoconversion
Photobleaching of GLR-1::GFP fluorescence in the AVA processes and FRAP analysis were performed as previously described (Hoerndli et al., 2013). Briefly, confocal stacks of GLR-1::GFP at the distal end of AVA were acquired before and immediately after photobleaching of the entire region of AVA distal to the vulva. Worms were then taken off the microscopy pad and transferred to NGM (nematode growth media) plates with food (E. coli strain OP50), where they were allowed to recover and feed for 2 h. Worms were then put back on slides for confocal imaging of the photobleached region. Because the worms were remounted, there was some variability in alignment of the photobleached region; however, this did not affect our analysis, which examined the average recovery of fluorescence. For photoconversion of GLR-1::Dendra2, 2–3 synaptic puncta in the proximal region of AVA (of comparable total intensity between genotypes analyzed) were converted sequentially using a 750 ms pulse of 35 mW/mm2 from a 405 nm laser. Immobilization and animal recovery was performed as previously described (Hoerndli et al., 2013). Similar to FRAP of GFP, animals were taken off the microscopy pad for 2 h to feed and recover. Quantification of the GLR-1::Dendra2 signal was performed on maximum projections of confocal stacks that were generated using Meta-Morph’s stack arithmetic function. Background fluorescence was subtracted from the average fluorescence of the imaged region. FRAP recovery curves were fit in Prism (version 9.3) using the non-linear regression hyperbola model. For FRAP of GLR-1::mCherry (Figure S4), approximately 25 μm on either side of the region of interest (ROI) was photobleached before fluorescence recovery was measured in the ROI. All genotypes at 16 minutes post-photobleaching were compared using a one-way ANOVA, with Bartlett’s multiple comparison test establishing that unc-16(lf) and jip-1(lf) were statistically different from wild-type control animals (p < 0.01).
Analysis of co-localization
The degree of co-localization between GLR-1::GFP and JKK-1::JNK-1::TagRFP-T was determined as previously described (Lei et al., 2017). Briefly, after subtracting background fluorescence, a region of interest was drawn along the ventral nerve cord. Pearson’s correlation coefficient (PCC) was then calculated using the Coloc-2 analysis function in Fiji-ImageJ (https://imagej.net/Fiji). Statistical significance between the calculated PCC and that expected for no co-localization (a PCC of 0) was determined using a one-sample, one-tailed Student’s t-test (McDonald and Dunn, 2013).
Heat-shock induced gene expression
To maximize heat-shock induced expression of the hsp16–2p::jip-1 and hsp16–2p::unc-16 transgenes, all animals were maintained at 15°C for one generation prior to heat-shock treatment. For analysis of GLR-1::GFP transport, young adult animals were heat shocked for 1 h at 32°C in a water bath. Animals were allowed to recover for 20 min at 20°C before imaging as described. For analysis of GLR-1::GFP puncta fluorescence, animals were subjected to two heat-shock treatments. First, L4 animals were heat shocked at 32°C for 1 h and then transferred to 20°C overnight (12 h) followed by a second heat shock at 32°C for 1 h. Animals were left to recover for 1 h at 20°C before imaging as described. All imaging was completed within 30 min following heat shock/recovery. Confocal images of mCherry::JIP-1 (Figure S3) were acquired in transgenic worms that expressed hsp16–2p::mCherry::jip-1 and soluble flp-18p::mCherry. Worms were heat shocked for 1 h at 32°C followed by 20 min at room temperature prior to imaging.
Electrophysiology
Electrophysiological recordings were performed using previously described patch-clamp techniques (Mellem et al., 2002). Currents were evoked by pressure application of 3 mM glutamate via a Picospritzer II (General Valve Corp.). Cells were held at −60 mV. Data was acquired using a HEKA EPC9 amplifier and analyzed using IGOR Pro. All worms expressed GLR-1::GFP in AVA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Replication and randomization
In vivo microscopy experiments were performed using adult animals with controls included each day. Imaging was reproduced by quantifying multiple animals per genotype (as stated in the figure legends). Electrophysiological recordings from AVA neurons in vivo were performed over multiple days with control animals included each day. Recordings were reproduced by measuring currents in multiple dissected animals per genotype (as stated in the figure legends). Sample sizes used in this study were in accordance with previously published data.
Statistical tests
A non-parametric Mann-Whitney U test was used for all datasets with less than or equal to three groups. Datasets with non-gaussian, unequal standard deviations were analyzed using a conservative non-parametric Kruskal-Wallis with Dunn’s multiple testing corrections. Datasets with gaussian distribution and equal standard deviations were analyzed using a one-way ANOVA with correction for multiple testing. Data is presented as mean ± standard error of the mean (SEM). Groups were considered statistically different from one another if p < 0.05. Sample sizes are listed in the figure legends and no statistical methods were used to predetermine sample size. Prism 9.0 was used for all statistical analyses.
Supplementary Material
Highlights.
Mobile MAPK-associated scaffold proteins are required for AMPAR transport
MAPKKs and MAPKs are required for loading scaffold proteins onto kinesin motors
CaMKII is required for loading AMPARs onto scaffold proteins
CaMKII and MAPK signaling contribute to the rapid exchange of synaptic AMPARs
ACKNOWLEDGMENTS
We thank members of the Maricq laboratory for comments on the manuscript, and the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440) for providing worm strains. We thank Dr. Dayton Goodell and Dr. Monica Vetter for comments on the manuscript, Dr. Yuji Kohara for providing multiple ESTs, Dr. Erik Jorgensen for the jip-1(km18) mutant, Dr. Yishi Jin for the unc-16 rescue plasmid (pCZ350), and Dr. Roger Davis for the pcdna3 T7 JIP3a (Addgene #53457). This research was made possible by support from a grant from the Human Frontier Science Program (A.V.M.), NIH grants NS35812 and DA035080 (A.V.M.), and by postdoctoral fellowships from the Swiss National Science Foundation, Switzerland (F.J.H.) and NIH grant NS115947 (F.J.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110577.
REFERENCES
- Abraham WC, and Williams JM (2008). LTP maintenance and its protein synthesis-dependence. Neurobiol. Learn. Mem 89, 260–268. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, and Hammer JA 3rd. (2010). Linking molecular motors to membrane cargo. Curr. Opin. Cell Biol 22, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, and Schulman H. (2019). CaM kinase: still inspiring at 40. Neuron 103, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biever A, Donlin-Asp PG, and Schuman EM (2019). Local translation in neuronal processes. Curr. Opin. Neurobiol 57, 141–148. [DOI] [PubMed] [Google Scholar]
- Brady ST, and Morfini GA (2017). Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol. Dis 105, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Jensen M, Mellem JE, Jensen E, Yamasaki T, Wang R, Maxfield D, Thacker C, Hoerndli F, Dunn PJ, Tomita S, et al. (2013). Cornichons control ER export of AMPA receptors to regulate synaptic excitability. Neuron 80, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Mellem JE, Hills T, Madsen DM, and Maricq AV (2001). The C. elegans glutamate receptor subunit NMR-1 is required for slow NMDA-activated currents that regulate reversal frequency during locomotion. Neuron 31, 617–630. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, and Jin Y. (2001). UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 32, 787–800. [DOI] [PubMed] [Google Scholar]
- Cockburn JJB, Hesketh SJ, Mulhair P, Thomsen M, O’Connell MJ, and Way M. (2018). Insights into kinesin-1 activation from the crystal structure of KLC2 bound to JIP3. Structure 26, 1486–1498.e1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, and Reddy EP (2007). Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202. [DOI] [PubMed] [Google Scholar]
- Diering GH, and Huganir RL (2018). The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JE, and Goldstein LS (2006). The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Yorks RM, Morrison LM, Hoover CM, and Miller KG (2015). Synapse-assembly proteins maintain synaptic vesicle cluster stability and regulate synaptic vesicle transport in Caenorhabditis elegans. Genetics 201, 91–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Yu SC, Hoover CM, Phillips BC, Richmond JE, and Miller KG (2013). An organelle gatekeeper function for Caenorhabditis elegans UNC-16 (JIP3) at the axon initial segment. Genetics 194, 143–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Di Schiavi E, Bergamasco C, and Bazzicalupo P. (2007). Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395, 170–176. [DOI] [PubMed] [Google Scholar]
- Ewbank JJ (2006). Signaling in the immune response. WormBook, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian DK, Fuentealba M, Donertas HM, Partridge L, and Thornton JM (2021). Functional conservation in genes and pathways linking ageing and immunity. Immun. Ageing 18, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, and Bargmann CI (2008). GFP Reconstitution across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353–363. [DOI] [PubMed] [Google Scholar]
- Fey D, Croucher DR, Kolch W, and Kholodenko BN (2012). Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front. Physiol 3, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornasiero EF, Mandad S, Wildhagen H, Alevra M, Rammner B, Keihani S, Opazo F, Urban I, Ischebeck T, Sakib MS, et al. (2018). Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun 9, 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MM, and Holzbaur EL (2014). Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends Cell Biol. 24, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Wu Y, and Ferguson SM (2017). Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol 216, 3291–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, and Nicoll RA (2014). Expression mechanisms underlying long-term potentiation: a postsynaptic view, 10 years on. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, and Lukyanov KA (2006). Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol 24, 461–465. [DOI] [PubMed] [Google Scholar]
- Gyurko MD, Csermely P, Soti C, and Stetak A. (2015). Distinct roles of the RasGAP family proteins in C. elegans associative learning and memory. Sci. Rep 5, 15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangen E, Cordelieres FP, Petersen JD, Choquet D, and Coussen F. (2018). Neuronal activity and intracellular calcium levels regulate intracellular transport of newly synthesized AMPAR. Cell Rep. 24, 1001–1012.e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, and Nicoll RA (2016). Long-term potentiation: from CaMKII to AMPA receptor trafficking. Annu. Rev. Physiol 78, 351–365. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, and Noda Y. (2008). Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol. Rev 88, 1089–1118. [DOI] [PubMed] [Google Scholar]
- Hoerndli FJ, Kallarackal AJ, and Maricq AV (2015a). Mobile AMPARs are required for synaptic plasticity. Channels (Austin) 9, 230–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerndli FJ, Maxfield DA, Brockie PJ, Mellem JE, Jensen E, Wang R, Madsen DM, and Maricq AV (2013). Kinesin-1 regulates synaptic strength by mediating the delivery, removal, and redistribution of AMPA receptors. Neuron 80, 1421–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerndli FJ, Wang R, Mellem JE, Kallarackal A, Brockie PJ, Thacker C, Madsen DM, and Maricq AV (2015b). Neuronal activity and CaMKII regulate kinesin-mediated transport of synaptic AMPARs. Neuron 86, 457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, and Ferrell JE Jr. (1996). Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U S A 93, 10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, and Mucke L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LS, Tzou P, and Sternberg PW (1994). The lin-15 locus encodes two negative regulators of Caenorhabditis elegans vulval development. Mol. Biol. Cell 5, 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, and Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, and Goldstein LS (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414, 643–648. [DOI] [PubMed] [Google Scholar]
- Karcher RL, Deacon SW, and Gelfand VI (2002). Motor-cargo interactions: the key to transport specificity. Trends Cell Biol. 12, 21–27. [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, and Matsumoto K. (1999). A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 18, 3604–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, and Ehlers MD (2006). Organelles and trafficking machinery for postsynaptic plasticity. Annu. Rev. Neurosci 29, 325–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, and Ausubel FM (2004). Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. U S A 101, 10990–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Kornfeld K, Miller LM, Horvitz HR, and Kim SK (1994). A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev. 8, 160–173. [DOI] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, and Davis RJ (2002). The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol 22, 4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei N, Mellem JE, Brockie PJ, Madsen DM, and Maricq AV (2017). NRAP-1 is a presynaptically released NMDA receptor auxiliary protein that modifies synaptic strength. Neuron 96, 1303–1316.e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YJ, and Yang WX (2019). Kinesins in MAPK cascade: how kinesin motors are involved in the MAPK pathway? Gene 684, 1–9. [DOI] [PubMed] [Google Scholar]
- Lisman J. (2017). Glutamatergic synapses are structurally and biochemically complex because of multiple plasticity processes: long-term potentiation, long-term depression, short-term potentiation and scaling. Philos. Trans. R. Soc. Lond. B Biol. Sci 372, 20160260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, and Malenka RC (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspect. Biol 4, a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Takamiya K, Thomas G, Lin DT, and Huganir RL (2010). GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocystcomplex interactions. Proc. Natl. Acad. Sci. U S A 107, 19038–19043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao LM, and Wang JQ (2015). Synaptically localized mitogen-activated protein kinases: local substrates and regulation. Mol. Neurobiol 53, 6309–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, and Dunn KW (2013). Statistical tests for measures of co-localization in biological microscopy. J. Microsc 252, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellem JE, Brockie PJ, Zheng Y, Madsen DM, and Maricq AV (2002). Decoding of polymodal sensory stimuli by postsynaptic glutamate receptors in C. elegans. Neuron 36, 933–944. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Shin ME, Parra-Bueno P, Szatmari EM, Shibata ACE, and Yasuda R. (2017). Kinetics of endogenous CaMKII required for synaptic plasticity revealed by optogenetic kinase inhibitor. Neuron 94, 690. [DOI] [PubMed] [Google Scholar]
- Myers KR, Wang G, Sheng Y, Conger KK, Casanova JE, and Zhu JJ (2012). Arf6-GEF BRAG1 regulates JNK-mediated synaptic removal of GluA1-containing AMPA receptors: a new mechanism for nonsyndromic X-linked mental disorder. J. Neurosci 32, 11716–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, and Malinow R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Chassefeyre R, Campbell GE, and Encalada SE (2017). KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA (2017). A brief history of long-term potentiation. Neuron 93, 281–290. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, and Roche KW (2013). Long-term potentiation: peeling the on- ion. Neuropharmacology 74, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CQ, Sudol M, Sheetz M, and Low BC (2012). Modularity and functional plasticity of scaffold proteins as p(l)acemakers in cell signaling. Cell Signal 24, 2143–2165. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, and Cobb MH (2007). Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112. [DOI] [PubMed] [Google Scholar]
- Reiner DJ, Newton EM, Tian H, and Thomas JH (1999). Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM ki- nase II. Nature 402, 199–203. [DOI] [PubMed] [Google Scholar]
- Richter KN, Wildhagen H, Helm MS, Ussling JE, Schikorski T, and Rizzoli SO (2018). Comparative synaptosome imaging: a semi-quantitative method to obtain copy numbers for synaptic and neuronal proteins. Sci. Rep 8, 14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HH, and Lee YS (2016). Cell type-specific roles of RAS-MAPK signaling in learning and memory: implications in neurodevelopmental disorders. Neurobiol. Learn. Mem 135, 13–21. [DOI] [PubMed] [Google Scholar]
- Sagasti A, Hisamoto N, Hyodo J, Tanaka-Hino M, Matsumoto K, and Bargmann CI (2001). The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell 105, 221–232. [DOI] [PubMed] [Google Scholar]
- Sakaguchi A, Matsumoto K, and Hisamoto N. (2004). Roles of MAP kinase cascades in Caenorhabditis elegans. J. Biochem 136, 7–11. [DOI] [PubMed] [Google Scholar]
- Sakamoto R, Byrd DT, Brown HM, Hisamoto N, Matsumoto K, and Jin Y. (2005). The Caenorhabditis elegans UNC-14 RUN domain protein binds to the kinesin-1 and UNC-16 complex and regulates synaptic vesicle localization. Mol. Biol. Cell 16, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, and De Strooper B. (2016). Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu. Rev. Neurosci 39, 57–79. [DOI] [PubMed] [Google Scholar]
- Satake T, Otsuki K, Banba Y, Suenaga J, Hirano H, Yamanaka Y, Ohno S, and Hirai S. (2013). The interaction of Kinesin-1 with its adaptor protein JIP1 can be regulated via proteins binding to the JIP1-PTB domain. BMC Cell Biol. 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnapp BJ (2003). Trafficking of signaling modules by kinesin motors. J. Cel. Sci 116, 2125–2135. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, and Hirokawa N. (2002). Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417, 83–87. [DOI] [PubMed] [Google Scholar]
- Shetty MS, and Sajikumar S. (2017). Differential involvement of Ca(2+)/ calmodulin-dependent protein kinases and mitogen-activated protein kinases in the dopamine D1/D5 receptor-mediated potentiation in hippocampal CA1 pyramidal neurons. Neurobiol. Learn. Mem 138, 111–120. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, and Zhu JJ (2011). Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist : a Rev. J. bringing Neurobiol. Neurol. Psychiatry 17, 54–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Hino M, Sagasti A, Hisamoto N, Kawasaki M, Nakano S, Ninomiya-Tsuji J, Bargmann CI, and Matsumoto K. (2002). SEK-1 MAPKK mediates Ca2+ signaling to determine neuronal asymmetric development in Caenorhabditis elegans. EMBO Rep. 3, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, and Yasuda R. (2017). Imaging ERK and PKA activation in single dendritic spines during structural plasticity. Neuron 93, 1315–1324.e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg MC, van Gogh IJ, Smits AM, van Triest M, Dansen TB, Visscher M, Polderman PE, Vliem MJ, Rehmann H, and Burgering BM (2013). The small GTPase RALA controls c-Jun N-terminal kinase-mediated FOXO activation by regulation of a JIP1 scaffold complex. J. Biol. Chem 288, 21729–21741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, and Rapoport TA (2001). Kinesin carries the signal. Trends Biochem. Sci 26, 545–550. [DOI] [PubMed] [Google Scholar]
- Vieira M, Fernandes J, Burgeiro A, Thomas GM, Huganir RL, Duarte CB, Carvalho AL, and Santos AE (2010). Excitotoxicity through Ca2+-permeable AMPA receptors requires Ca2+-dependent JNK activation. Neurobiol. Dis 40, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk L, Chiu SL, Sharma K, and Huganir RL (2015). Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci 38, 127–149. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ (2006). The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans 34, 828–832. [DOI] [PubMed] [Google Scholar]
- Wu J, Rowan MJ, and Anwyl R. (2006). Long-term potentiation is mediated by multiple kinase cascades involving CaMKII or either PKA or p42/44 MAPK in the adult rat dentate gyrus in vitro. J. Neurophysiol 95, 3519–3527. [DOI] [PubMed] [Google Scholar]
- Yasuda R. (2017). Biophysics of biochemical signaling in dendritic spines: implications in synaptic plasticity. Biophys. J 113, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora Chimal CG, and De Schutter E. (2018). Ca(2+) requirements for long-term depression are frequency sensitive in Purkinje cells. Front. Mol. Neurosci 11, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeke A, Misheva M, Remenyi A, and Bogoyevitch MA (2016). JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev 80, 793–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GR, Zhao H, Choi EM, Svestka M, Wang X, Cook RG, and Geller AI (2012). CaMKII, MAPK, and CREB are coactivated in identified neurons in a neocortical circuit required for performing visual shape discriminations. Hippocampus 22, 2276–2289. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang P, Wang G, Zhang H, Zhang Y, Yu Y, Zhang M, Xiao J, Crespo P, Hell JW, et al. (2018). Ras and rap signal bidirectional synaptic plasticity via distinct subcellular microdomains. Neuron 98, 783–800.e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Xiang J, Hunter T, and Lin A. (1999). The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J. Biol. Chem 274, 28966–28971. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Mellem JE, Brockie PJ, Madsen DM, and Maricq AV (2004). SOL-1 is a CUB-domain protein required for GLR-1 glutamate receptor function in C. elegans. Nature 427, 451–457. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, and Malinow R. (2002). Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110, 443–455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microscopy and electrophysiology data will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.