

Abstract
Intact glycopeptide analysis has been of great interest because it can elucidate glycosylation site information and glycan structural composition at the same time. However, mass spectrometry (MS)-based glycoproteomic analysis is hindered by the low abundance and poor ionization efficiency of glycopeptides. Relatively large amounts of starting materials are needed for the enrichment, which makes the identification and quantification of intact glycopeptides from samples with limited quantity more challenging. To overcome these limitations, we developed an improved isobaric labeling strategy with an additional boosting channel to enhance N,N-dimethyl leucine (DiLeu) tagging-based quantitative glycoproteomic analysis, termed as Boost-DiLeu. With the integration of one-tube sample processing workflow and high pH fractionation, 3514 quantifiable N-glycopeptides were identified from 30 μg HeLa cell tryptic digests with reliable quantification performance. Furthermore, this strategy was applied to human cerebrospinal fluid (CSF) samples to differentiate N-glycosylation profiles between Alzheimer’s disease (AD) patients and non-AD donors. The results revealed processes and pathways affected by dysregulated N-glycosylation in AD, including platelet degranulation, cell adhesion, and extracellular matrix, which highlighted the involvement of N-glycosylation aberrations in AD pathogenesis. Moreover, weighted gene co-expression network analysis (WGCNA) showed 9 modules of glycopeptides, two of which were associated with AD phenotype. Our results demonstrated the feasibility of using this strategy for in-depth glycoproteomic analysis of size-limited clinical samples. Taken together, we developed and optimized a strategy for enhanced comprehensive quantitative intact glycopeptide analysis with DiLeu labeling, showing significant promise for identifying novel therapeutic targets or biomarkers in biological systems with limited sample quantity.
Graphical Abstract
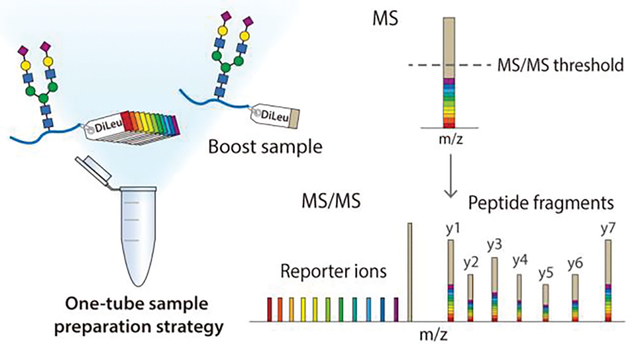
Introduction
Glycosylation is one of the most common post-translational modifications (PTMs), which is involved in many physiological processes including cell signaling, host-pathogen interaction, and immune response.1–3 Studies have shown that aberrant glycosylation plays a key role in the pathological processes during disease progressions, such as neurodegenerative diseases, diabetes, and cancers.4–7 Intact glycopeptide analysis has been of great interest recently because it retains information on glycosylation sites while elucidating peptide sequences and glycan structures, which enables the investigation of functional effects of heterogeneity across glycoproteome.8 However, akin to many other PTMs, mass-spectrometry (MS)-based glycoproteomic analysis has been hindered by the low abundance of glycopeptides among complex protein digests and their poor ionization efficiency.9,10
To overcome these limitations, various enrichment strategies have been developed to separate glycopeptides from other non-glycosylated peptides in biological samples before MS analysis.4 For example, hydrophilic interaction liquid chromatography (HILIC) has been extensively used to enrich glycopeptides based on their increased hydrophilicity introduced by glycan moieties.11–13 However, even utilizing these highly efficient enrichment methods, an in-depth glycoproteomic analysis still requires a relatively large amount of starting sample. This drawback particularly limits glycoproteomic analysis of precious clinical samples such as pathological tissues and cerebrospinal fluid (CSF), which cannot be expanded in vitro and are often limited in quantities for research.14
On the other hand, insufficient MS signal intensity from low abundance peptides usually leads to low proteome coverage in LC-MS/MS analysis, because the poor-quality MS/MS spectra hardly generate confident peptide identifications. Isobaric labeling can be used to enhance MS detection sensitivity in data-dependent acquisition (DDA) mode analysis since the signal intensities of precursor ions in the full scan are combined from all labeling channels of the same species.13 Abundant peptide backbone fragments and enhanced signal intensities in MS/MS facilitate identification while achieving multiplex quantification at the same time. Recent studies have taken advantage of isobaric labeling and introduced the concept of a “boosting” (or “carrier”) channel, in which a large amount of content-relevant samples is labeled by one channel of the isobaric tags and combined with those size-limited samples labeled by the remaining channels of the multiplex tags. The combined signal intensity of a given peptide can be greatly amplified and thus enhancing the detection of low-abundance peptides, as well as mitigating the impact of sample loss on low-volume samples during sample preparation.15,16 Such strategy has been applied to single-cell proteomics,17,18 quantification of phosphopeptides,15 phosphotyrosine-containing peptides,16 deglycosylated peptides of secreted glycoproteins,19 stable isotope labeling using amino acids in cell culture (SILAC)-labeled peptides,20 and low-abundance proteins in thermal proteome profiling (TPP).21 These studies showed promising features of the boosting approach to increase the proteome coverage and enable quantification of low-abundance proteins/peptides simultaneously, which would be especially useful for the PTM analysis of size-limited samples. Nonetheless, such strategy has not been applied to intact glycopeptide analysis yet.
Our group previously developed N,N-Dimethyl Leucine (DiLeu) isobaric tags for quantitative proteomics analysis, where the amine-reactive group reacted with N-termini and lysine residues of peptides similar to tandem mass tags (TMT)22 and isobaric tags for relative and absolute quantification (iTRAQ)23. Compared to these commercially available tags, DiLeu is much more cost-effective and can be synthesized in-house readily with high yields.24 It was first designed as a 4-plex set25 and then has been expanded to 12- and 21-plex by incorporating neutron encoding (NeuCode) strategy, which utilized the differences in mass between each isotope of every element, arising from their minute differences in nuclear binding energy.24,26–28 In 12-plex DiLeu setting, reporter ions are grouped into four regions ranging from m/z 115 to m/z 118, and the mass difference of adjacent channel is as small as 6 mDa within each region.24 DiLeu tag is well-suited for developing a boosting strategy for intact glycopeptides since it has been successfully applied to intact glycoproteomic quantification using cell lines and tissue samples in our previous studies.11,29 More importantly, it is less likely to be affected by the isotopic impurity “leakage” problem reported for 10-plex TMT due to the use of NeuCode strategy.15
In this study, we developed a 12-plex DiLeu tag-based boosting strategy (Boost-DiLeu) to perform a comprehensive quantitative N-glycoproteomic analysis of size-limited samples (Figure 1). To further recover glycopeptides from samples of small quantity, we adapted a one-tube processing strategy14 for DiLeu labeling, leveraging the benefits of an acid-cleavable detergent, RapiGest SF, to circumvent the necessities of sample desalting. The strategy was well established by carefully optimizing the sample handling processes, boosting-to-study channel (B/S) ratios, and instrumental parameters for data acquisition including automatic gain control (AGC) and ion injection time. Coupling with HILIC enrichment and high-pH (HpH) reversed-phase liquid chromatography (RPLC) fractionation, large-scale global mapping of N-glycoproteome was achieved from a small amount of HeLa cell digests. To further demonstrate the feasibility of the Boost-DiLeu strategy for size-limited clinical sample analysis, we quantified alterations of the N-glycoproteomes in CSF between Alzheimer’s disease (AD) patients and non-AD donors. Taken together, the Boost-DiLeu strategy not only increased the glycoproteome coverage but also enabled accurate and robust quantification, which shed light on future applications to site-specific quantitative glycoproteomic studies involving samples of limited availability.
Figure 1.
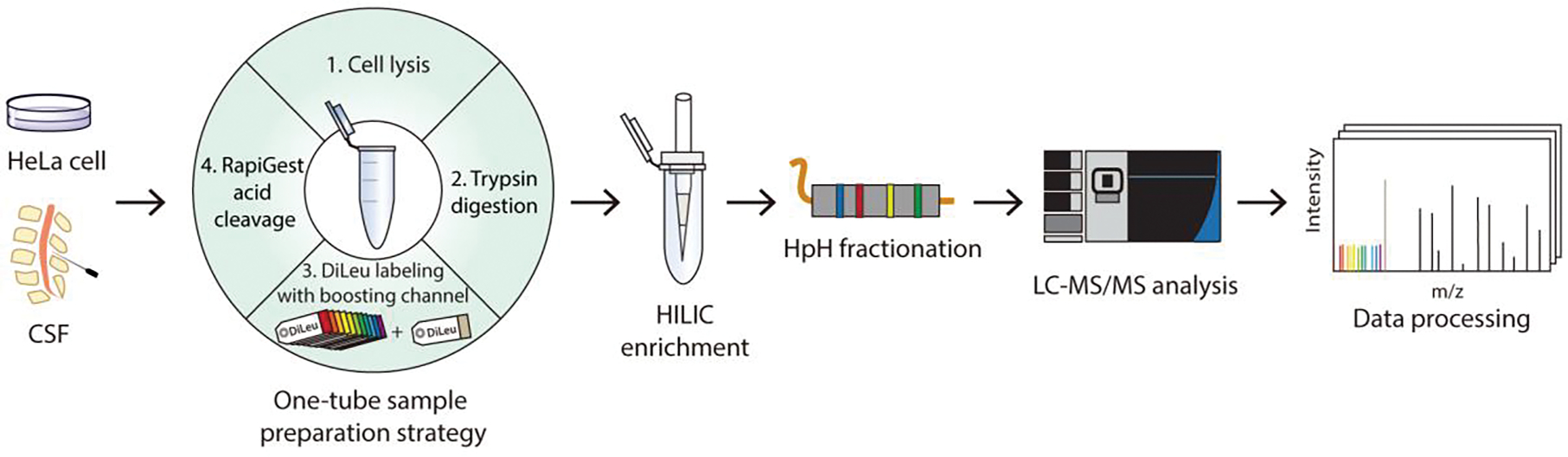
Workflow of Boost-DiLeu strategy. Proteins from biological samples were extracted, enzymatically digested, and labeled through one-tube sample preparation workflow. DiLeu 118d was used as a boosting channel. Samples were pooled after labeling for HILIC enrichment and HpH fractionation, followed by LC-MS/MS analysis.
Experimental Section
Supplementary Experimental Procedures.
Details about chemicals and materials, cell culture, sample preparation with the conventional protocol, offline HpH fractionation, global proteomic analysis, and expanded data analysis are provided in the Supporting Information.
HeLa Cell Sample Preparation.
In one-tube sample processing workflow, HeLa cell pellets were solubilized in 0.1% (w/v) RapiGest prepared in 100 mM TEAB with 1% (v/v) protease inhibitor and phosphatase inhibitor. The mixture was then incubated at 60 °C for 30 min and sonicated at 4 °C for 1 min. Lysates were centrifuged at 21000 g at 4 °C for 5 min and the supernatant was collected. Protein BCA assay, reduction, alkylation, and trypsin digestion were performed in the same way as mentioned in the conventional method. After digestion, peptides were dried down in vacuo without acidification and desalting.
CSF Sample Preparation.
Detailed information on CSF samples is provided in Supporting Information and Supplemental Table S1. The study was approved by the regional ethics committee at the University of Gothenburg. After a BCA protein assay, 30 μg proteins were aliquoted for each study channel. 900 μg proteins pooled from both AD patients and non-AD donors CSF samples at a ratio of 3:5 were used as boosting channel. The aliquots were dried down in vacuo and treated with the one-tube sample preparation approach as described above.
DiLeu Labeling.
DiLeu tag synthesis and labeling were performed as previously reported.24,29 DiLeu tags were activated in anhydrous DMF with DMTMM and NMM at 0.6x molar ratios to tags. The mixture was vortexed at room temperature for 1h and the supernatant was then added to each sample for labeling. After vortexing at room temperature for 2 h, the reaction was quenched by adding 5% NH2OH to a final concentration of 0.25%. Each batch of labeled peptides was pooled together and dried down in vacuo.
SAX-HILIC Enrichment.
Enrichment of DiLeu-labeled glycopeptides was performed with in-house packed strong anion exchange (SAX)-HILIC SPE tips following previous publications with minor modification.29,30 Briefly, 3 mg of cotton wool was plugged into a 200 μL empty TopTip, which was placed on a 2 mL microcentrifuge tube with an adapter unit. After activation in 1% TFA for 15 min, the SAX material was transferred into the cotton-packed TopTip at a beads-to-peptide ratio of 30:1. The solvent was removed at 200 g for 2 min. The stationary phase was conditioned with 300 μL 1% TFA and loading buffer (80% ACN/1% TFA) three times. The DiLeu-labeled sample was dissolved in 300 μL loading buffer and loaded onto the SAX-cotton tip. The tip was centrifuged at 200 g for 2 min and flow-through was reloaded to the SAX-cotton four more times to ensure complete retention. The SAX-cotton tip was washed with 300 μL loading buffer six times, and then eluted with 150μL 0.1% FA solution three times. Samples were dried down in vacuo before direct MS analysis or HpH fractionation.
LC-MS/MS Analysis.
Lyophilized samples reconstituted in 0.1% FA were loaded onto a 15 cm length, 75 μm i.d. in-house packed Bridged Ethylene Hybrid C18 (1.7 μm, 130 Å, Waters) column, and analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) interfaced with a Dionex UltiMate 3000 UPLC system (Thermo Fisher Scientific, San Jose, CA). Two technical replicates were run for each fraction. For intact N-glycopeptides analysis, binary buffers (A, 0.1% FA; B, 90% ACN, 0.1%FA) were used in LC and the linear gradient was from 0 to 30% B for 80 min. MS survey scans of peptides were acquired from 400 to 2000 m/z at a resolution of 120 K, using an AGC target setting of 4E5 and a maximum injection time of 100 ms. For MS2 scan, a duty cycle of 3s was set in top speed mode. Only spectra with a charge state among 2–7 were selected for fragmentation by stepped higher-energy collision dissociation (HCD) with normalized collision energy (NCE) of 30 ± 3%. The MS2 spectra were acquired with a resolution of 60 K, a lower mass limit of m/z 110, and dynamic exclusion of 12 s with 10 ppm mass tolerance. Different AGC settings and maximum injection times were tested for method optimization. All data were acquired in profile mode.
Data Analysis.
Raw files were searched against UniProt Homo sapiens reviewed database (August 2020, 20311 sequences) using Byonic search engine (version 2.9.38, Protein Metrics Inc) embedded within Proteome Discoverer 2.1 (PD 2.1, Thermo Fisher Scientific). Trypsin was selected as the enzyme and two maximum missed cleavages were allowed. Searches were performed with a precursor mass tolerance of 15 ppm and a fragment mass tolerance of 0.03 Da. Static modifications were specified as carbamidomethylation (+57.02146 Da) on cysteine residues and 12-plex DiLeu (+145.12801 Da) on peptide N-terminus and lysine residues. Dynamic modifications consisted of oxidation of methionine residues (+15.99492 Da), deamidation (+0.984016 Da) of asparagine and glutamine residues, and N-glycosylation. Oxidation and deamidation were set as “rare” modification, and N-glycosylation was set as “common” modification. Glycan modifications were searched against a glycan database expanded from the Byonic embedded 182 human N-glycan database to include mannose-6-phosphate (M6P) glycans consisting of HexNAc (2–4) Hex (3–9) Phospho (1–2) modifications. N-glycopeptides were filtered at a 1% peptide FDR, Byonic score >150 and log|Prob| >1. Glycopeptides were exclusively categorized into six glycosylation type categories based on the glycan composition identified: (1) M6P (containing M6P glycan); (2) both sialylated and fucosylated; (3) sialylated only; (4) fucosylated only; (5) complex/hybrid (> 2 HexNAc); (6) oligomannose (2 HexNAc and ≥ 5 Hex), and (7) paucimannose (1–2 HexNAc and < 5 Hex). Quantification was performed in PD 2.1 with a reporter ion integration tolerance of 10 ppm for the most confident centroid. Reporter ion intensities were exported, and isotopic interference correction was performed with a Python script according to the previously described equations.24 Reporter ion intensity of each channel was normalized by median to correct systematic biases of 12-plex DiLeu tags via Perseus.31 Student’s t-test was performed, where significant change was defined by a p-value less than 0.05 and fold-change over 1.5.31 The MS data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the accession code PXD029269.32
Results and Discussion
One-tube Sample Processing Workflow for DiLeu Labeled Glycoproteomics.
Recently, Wu et al introduced a strategy termed Nanogram TMT Processing in One Tube (NanoTPOT) to achieve desalt-free isobaric labeling using TMT tags in a single tube.14 An acid-cleavable detergent, RapiGest SF, was utilized to eliminate the need for commonly used SDS lysis buffer and urea that are detrimental to MS and tag labeling. Here we adapted and optimized this strategy to conduct desalting-free DiLeu labeling. To assess the performance of this optimized one-tube DiLeu labeling sample processing, the same amounts of HeLa cell pellets were processed both in the conventional method and this method as described in the experimental section. SDS-PAGE result suggested that the trypsin digestion was complete in both methods (Figure S1). In terms of proteome coverage, both workflows identified comparable numbers of labeled peptides in global proteome analysis (Figure S2A), and 80.1% of proteins identified in one-tube processing workflow were also found in the conventional method (Figure S2B). For DiLeu labeled glycoproteomics, the one-tube sample processing method outperformed the conventional one in all aspects, including identification of glycopeptide-spectrum matches (GPSMs), unique glycopeptides and corresponding glycoproteins (Figure 2A). Among all identified glycoproteins, 57.1% were found in both methods, while the one-tube processing method recovered 13.8% (35 out of 254) more unique glycoproteins than the conventional one (Figure 2B, Table S2). The excellent performance of the one-tube sample processing method can be attributed to two factors: (1) RapiGest is a preferred reagent for handling membrane proteins and the protein precipitation step can be omitted since there is no need to remove RapiGest after extraction.33 (2) Avoid desalting before labeling effectively reduces sample loss, especially for hydrophilic glycopeptides. Taken together, the one-tube sample processing strategy is suitable for DiLeu labeled glycoproteomic analysis.
Figure 2.
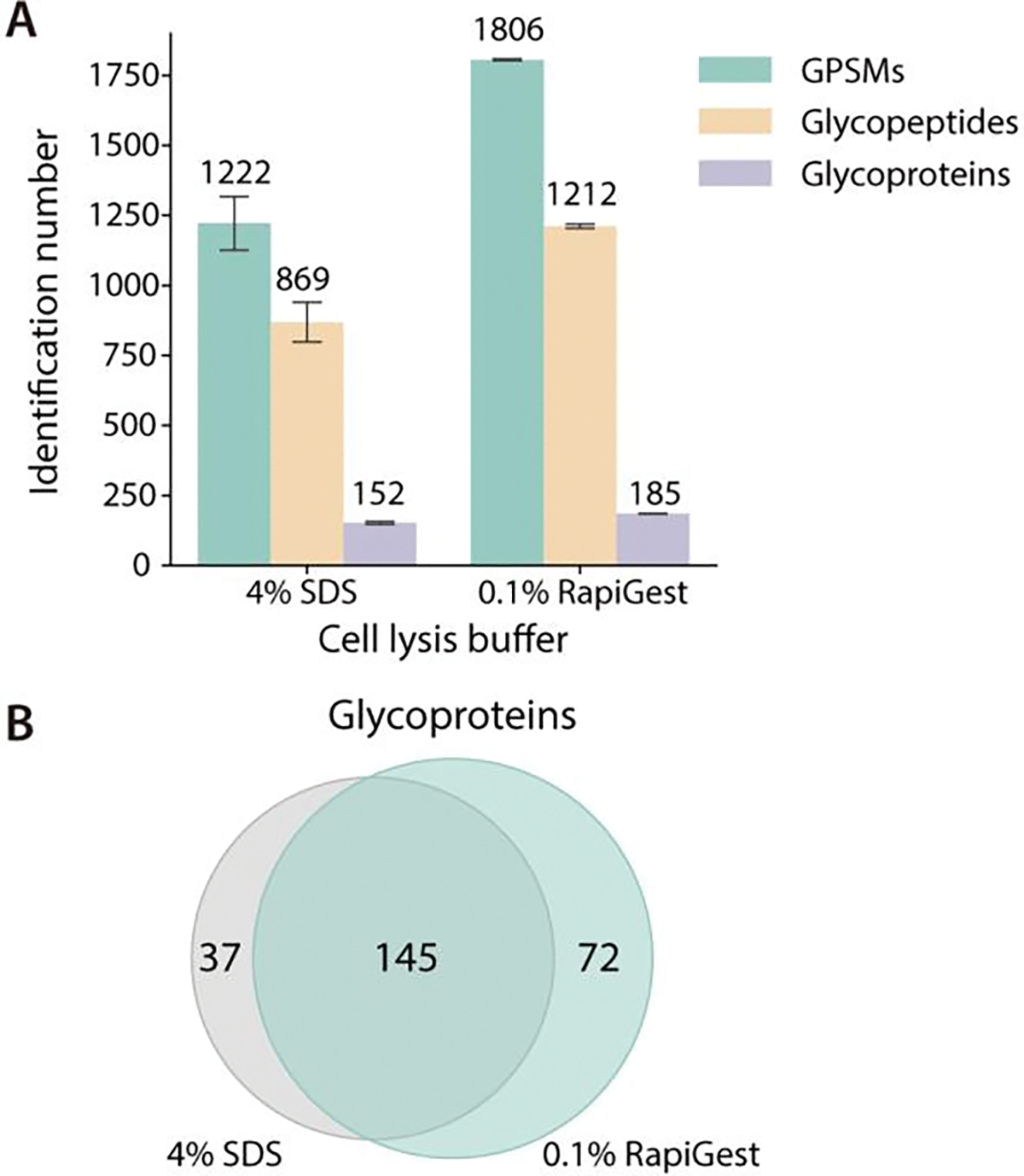
Comparison of sample preparation using conventional and one-tube sample processing workflow. (A) Identification number in glycoproteomic analysis. (B) Overlap of glycoproteins by the two methods as shown using Venn diagram.
Evaluation of the Boost-DiLeu strategy.
In the Boost-DiLeu strategy, the analysis of small-quantity samples labeled with study channels was enhanced by utilizing a boosting channel that contained a much larger quantity of labeled glycopeptides from analogous biological samples. After being pooled together, isobaric labeled glycopeptides from each channel appeared as a single precursor ion at the MS1 level, and the existence of boosting channel greatly increased the intensity of the precursor ions and facilitated triggering MS2 fragmentation. To validate the design of the Boost-DiLeu strategy and evaluate the effects of different boosting-channel-to-study-channel (B/S) ratios of sample amount on glycoproteomic coverage and quantification, a series of comparisons were performed using HeLa cell tryptic digests prepared with the one-tube sample processing workflow. The first three channels (115a,115b, and 116a) in 12-plex DiLeu were set as study channels and loaded with 10 μg tryptic peptides respectively. The last channel, DiLeu 118d, was selected as the boosting channel to generate different B/S ratios at 30x, 50x, and 100x (Figure 3A). A control group was prepared at the same time that only consisted of the first three study channels. Each group of samples was pooled together after labeling and enriched through HILIC before LC-MS/MS analysis.
Figure 3.
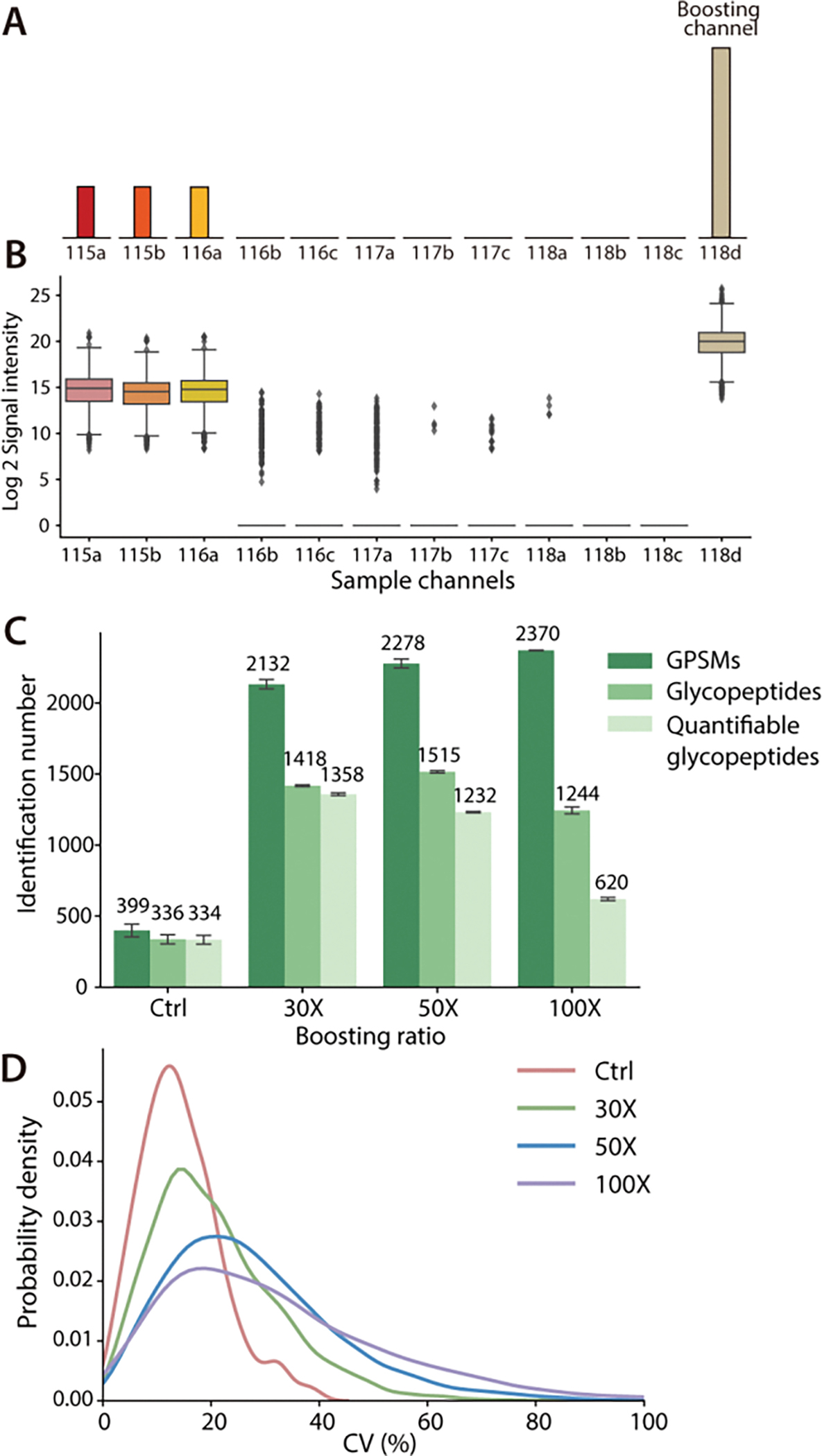
Comparison of different B/S ratios. (A) Experimental design: First three channels were labeled as study channels and DiLeu 118d channel was used as a boosting channel. (B) Reporter ion signal intensities at a 30x B/S ratio. (C) Identification number of GPSMs, total glycopeptides and quantifiable glycopeptides at different B/S ratios. (D) CV distribution of quantifiable glycopeptides.
DiLeu 118d was selected as the boosting channel because the reporter ion in this channel had no interference with other channels. Its −1 isotopic peak (2H to 1H) caused by isotopic impurity was at m/z 117.14656, which was 3mDa away from the DiLeu 12-plex 117c reporter ion at m/z 117.14363. Such mass difference can be resolved using high-resolution capability during LC-MS/MS analysis. Figure 3B displayed the reporter ion signal intensity distribution at a 30x B/S ratio. Similar reporter ion intensity distribution in study channels were observed in all groups and none of the empty channels had unusually high reporter ion signal, indicating that 118d channel successfully served as a boosting channel and avoided the isotopic interference problem reported in 10-plex TMT tags,15 which ensured high quantification accuracy and maximum multiplexing capacity of DiLeu tags.
Figure 3C demonstrated the effectiveness of adding a boosting channel, as the quantifiable glycopeptides in study channels, which stand for glycopeptides without missing reporter ion intensities, increased three times at B/S 30x compared to the control group. As expected, the identified number of GPSMs went up from 2132 to 2370 as the B/S ratios increased. However, the unique glycopeptide number and the quantifiable glycopeptides dropped as the B/S ratios increased. Simultaneously, an increased median coefficient of variations (CVs) from 13.41% to 28.19% (Figure 3D) was observed, which showed a similar trend reported in other studies.15,18 Since the total number of ions entering the Orbitrap was controlled in each scan, ions from boosting channel were likely to occupy more space than ions in study channels. At a higher B/S ratio, the signal intensity of reporter ions from study channels showed a slightly decreasing trend (Figure S3), which could be a result of reporter ion suppression. This negatively affected the quantification performance, including more missing values found in 50x and 100x groups and increased CVs. The same comparison was also made at three different B/S ratios using samples prepared with the conventional method (Figure S4). Despite the lower identification number, a similar trend of fewer quantifiable glycopeptides and a higher median of CVs was observed with the increase of B/S ratios, suggesting that 30x boosting ratio was optimal in the aspect of identification and quantification accuracy.
Optimization of Instrument Parameters.
When performing MS/MS analysis, the number of precursor ions reaching to orbitrap analyzer is controlled by AGC setting and maximum injection time. These parameters are essential to balance the detection sensitivity and MS2 scan rate. For global proteomic analysis, AGC is usually set in the range of 5E4 to 1E5 to improve the overall coverage.18 However, such a setting may not be well-suited for PTM analysis, especially in the case where a large portion of boosting samples are mixed with study samples. Ions from boosting samples can fill in the analyzer quickly, thus impairing the detection of signals from study channels. To understand the influence of AGC settings on glycoproteome coverage and quantification quality, four different AGC settings including 5E3, 3E4, 5E4, and 5E5 were compared with DiLeu-labeled HeLa cell tryptic digests with a 30x boosting channel added. The maximum injection time was fixed at 200 ms. In general, a higher AGC setting allows more ions to accumulate for MS/MS analysis at expense of MS2 scan rate and longer duty cycle time, thus affecting proteome coverage.18,34 Such a trend was observed in Figure 4A, where AGC 3E4 achieved the highest identification numbers of GPSMs, glycopeptides, and quantifiable glycopeptides while higher AGC values led to decreasing identification numbers. As reflected in Figure 4B, the median injection time increased from 18 ms to 200 ms when increasing AGC. The lowest AGC setting 5E3 resulted in significantly poorer glycoproteome coverage, indicating the necessity of allowing enough ions to enter the orbitrap for high-quality MS/MS analysis. On the other hand, accumulating more ions improves quantification performance since more ions from study channels are allowed to enter the mass analyzer.18,35 Though the reporter ion intensity decreased slightly at higher AGC settings due to potential space charge effects36 or preference of fragmentation on glycosidic bonds (Figure 4D), the median of CV distribution decreased from 23.63% to 12.97% (Figure 4C).
Figure 4.
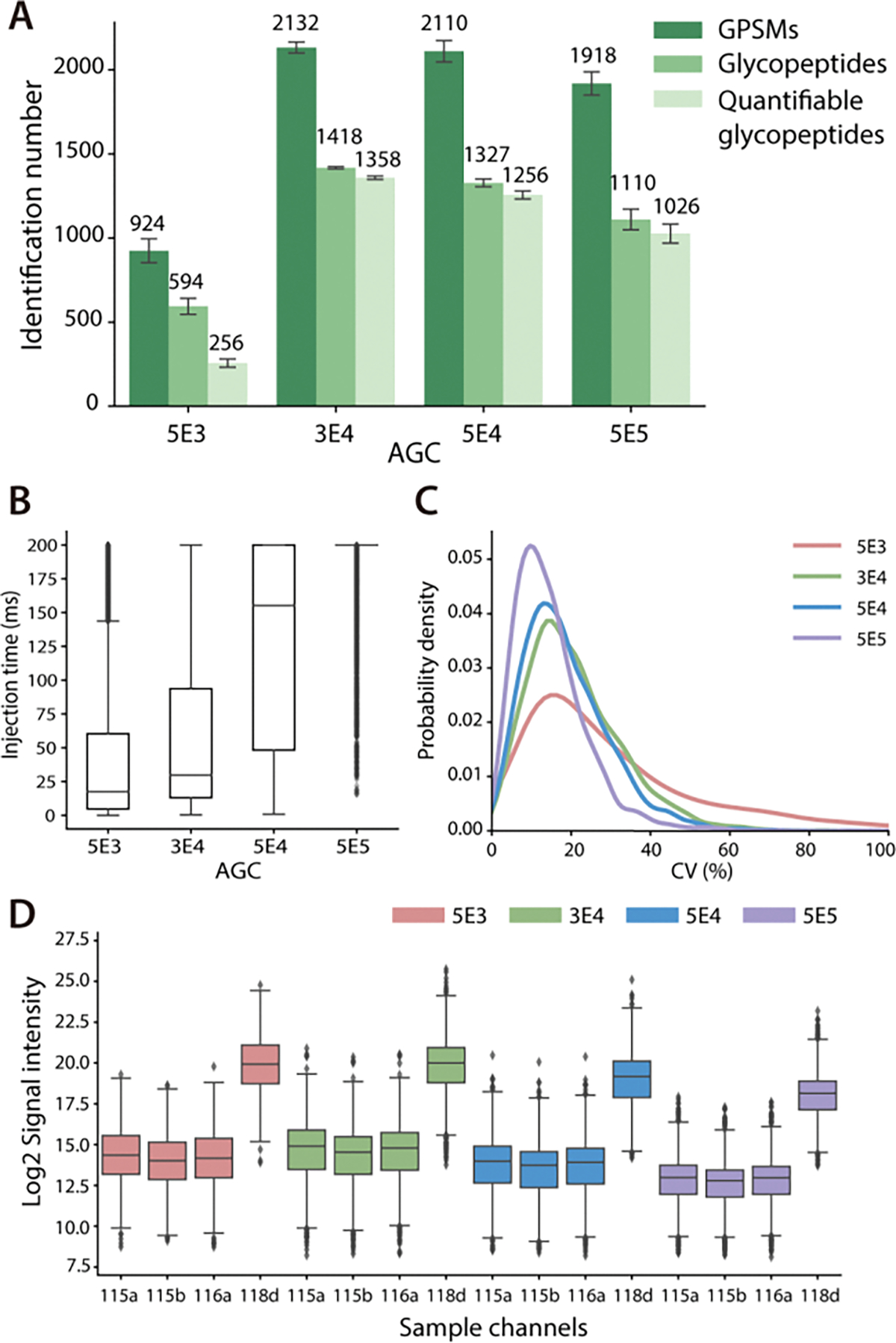
Comparison of different AGC settings. (A) Identification number of GPSMs, total glycopeptides and quantifiable glycopeptides with different AGC. (B) Distribution of actual ion injection time. (C) CV distribution of quantifiable glycopeptides. (D) Distribution of reporter ion signal intensities.
Besides the AGC settings, maximum injection time is another important parameter to control the number of ions for MS2 analysis. To determine an optimal setting, two different maximum injection time at 200 ms and 300 ms were evaluated with the same B/S ratio of 30x and AGC setting of 5E4. As shown in Figure S5A, increased injection time elongated duty cycle time, thus resulting in lower identification numbers. In terms of quantification performance, the distribution of reporter ion signal intensity (Figure S5B) and CVs (Figure S5C) were almost similar in both cases, despite that 200 ms injection time had a slightly lower median of CV of 16.61% than 17.71% of 300 ms injection time setting.
These results clearly demonstrated a trade-off between glycoproteome coverage and quantification performance in different AGC settings. Higher AGC improved quantification quality at the expense of the number of identified glycopeptides. Maximum injection time also had a great impact on glycoproteome coverage, while the influence on quantification performance was not as significant as AGC settings. To achieve a balance between glycoproteome coverage and quantification performance, AGC 3E4 and maximum injection time 200 ms were employed in the following experiments for glycoproteomics analysis.
Global Glycoproteome Mapping in HeLa Cell Line.
HpH provides an additional dimension of separation with conventional low-pH RPLC due to its high orthogonality and compatibility with direct MS analysis.37 Since the conventional HILIC fractionation based on decreasing ACN concentration showed less efficiency for glycopeptides fractionation, HpH fractionation was implemented to further boost the glycoproteome coverage in this study.38 Though HpH fractionation has been adopted in several PTM analyses to bolster the proteome coverage, additional separation requires a sufficient amount of sample to compensate for potential sample loss during the process, and this is usually unsuitable for samples of low quantity.15,39–41 With the Boost-DiLeu strategy, a larger amount of boosting sample can serve as a “carrier” sample that mitigate the impact of sample loss.42 After HILIC enrichment and HpH fractionation, a total of 4119 glycopeptides corresponding to 277 proteins were identified from 30 μg HeLa tryptic digest peptides labeled in the first three study channels, among which 3514 were quantifiable (Figure 5A, Table S3). Glycoproteome coverage was expanded by approximately 2.5-fold compared to the analyses without HpH fractionation. Figure 5B displayed the overlap of glycopeptides identified in four fractions, and 29.62%, 15.36%, 17.56%, and 23.64% of glycopeptides were unique in each fraction, indicating the high separation efficiency provided by HpH fractionation. Such comprehensive intact glycoproteome coverage also enabled global profiling of site-specific micro-heterogeneity of HeLa cell glycosylation, as visualized in Figure 5C. This glycoprotein-glycan network diagram mapped which glycans (outer nodes, 151 total) modified which proteins (inner bar, 277 total). The glycoproteins were sorted based on the different numbers of glycosites, and all nodes and edges were colored by the corresponding glycosylation type. Of note, more than half of the glycoproteins had only one glycosylation site, but 79.1% of glycosites have more than one glycan. In addition, several glycosylation patterns were observed, such as the prevalence of oligomannose glycosylation (44.11% of glycopeptides). Interestingly, 34 out of 151 glycans were found to be M6P glycans (a representative MS/MS spectrum shown in Figure S6). Due to the negatively charged phosphate group and extremely hydrophilic property, detection of M6P glycopeptides usually suffers from interference by other N-glycopeptides.43 With HpH fractionation, M6P glycopeptides were better separated from other glycopeptides so the detection sensitivity was enhanced. Along with improved glycoproteome coverage, the Boost-DiLeu strategy also presented good quantification performance after HpH fractionation. Figure 5D showed the median intensities of study channels were close to 1:1:1 without any isotopic interference from the DiLeu 118d channel. CV median of 16.75% (Figure 5E) and average Pearson correlation over 0.97 (Figure 5F) among three biological replicates demonstrated the robustness and accuracy of quantification of this strategy.
Figure 5.
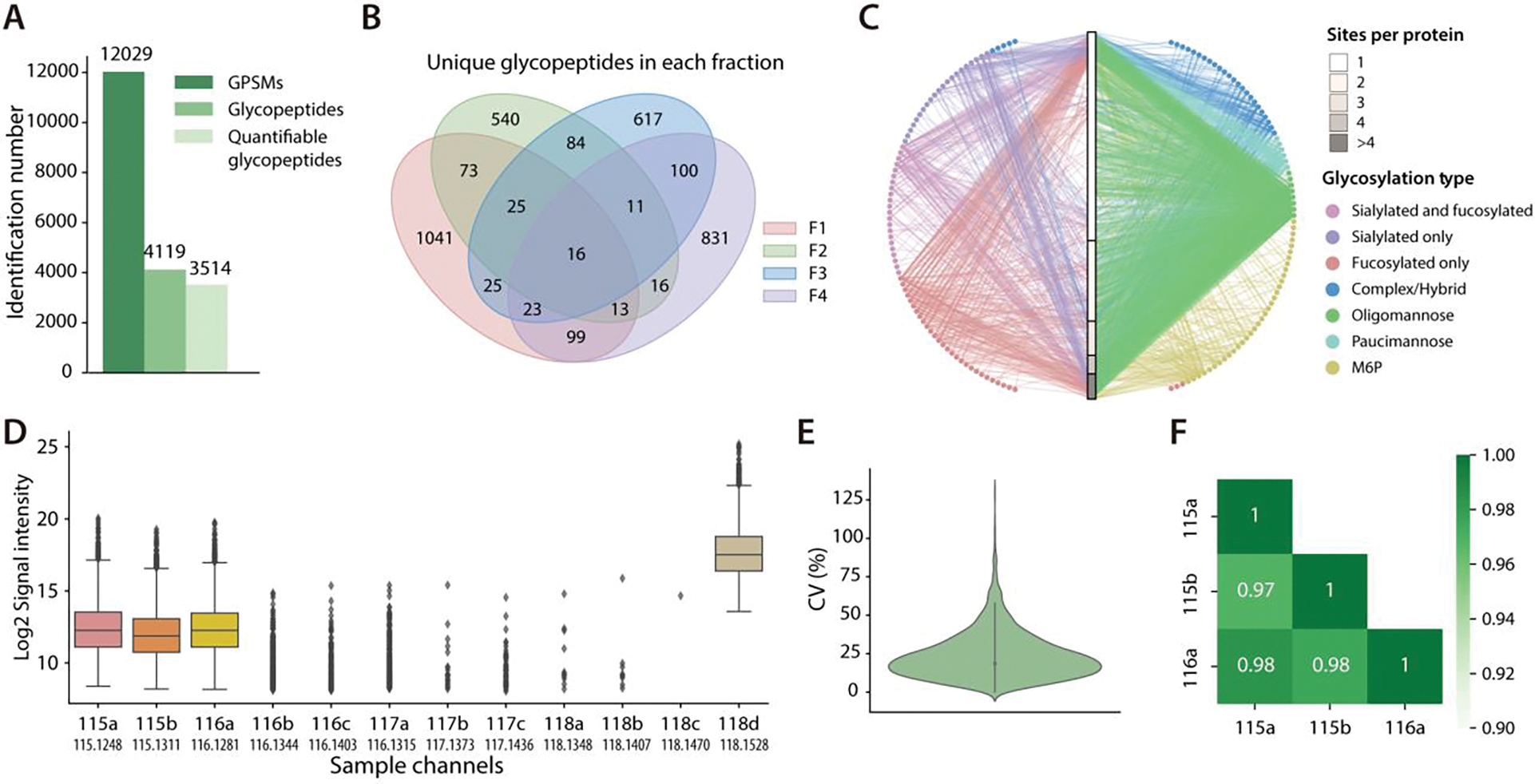
Global glycoproteome mapping in HeLa cell line. (A) Identification number of GPSMs, total glycopeptides and quantifiable glycopeptides. (B) Overlap of unique glycopeptides identified in each fraction. (C) A glycoprotein-glycan network diagram that maps which glycans (outer nodes) modify which proteins (inner bar). Glycoproteins are sorted by the number of glycosites. Glycan nodes and their linkage to the proteins are colored according to the glycosylation type. (D) Distribution of reporter ion signal intensities across 12 channels. (E) CV distribution of quantifiable glycopeptides. (F) Pearson correlation of reporter ion intensities between three study channels.
Site-specific Quantitative Glycoproteomic Analysis of Human CSF Samples.
Through direct interchanges with the extracellular fluid of central nervous system (CNS), CSF can directly reflect pathological changes in the CNS, providing the opportunity for biomarker discovery in neurological diseases.44–47 However, the analysis of CSF samples is challenging due to the limited availability and the relatively low total protein concentration (0.2 – 0.8 mg/mL).48 To address these challenges, the Boost-DiLeu strategy was adopted to profile glycoproteomic changes in CSF between AD patients and non-AD donors at B/S ratio of 30x. In total, 1321 intact N-glycopeptides were identified, and 1172 of them were quantifiable (with at least three valid reporter ion intensities in one group), mapping to 164 glycoproteins and 158 different glycans (Figure S7A, Table S4). Figure S7B shows the glycoprotein-glycan network of all quantifiable glycopeptides. Nearly 62% of the glycoproteins were observed with only one glycosite, yet 47% of the glycosites were modified with more than one glycan. In the meantime, 5% of glycoproteins were found to have more than 4 glycosylation sites, suggesting a high degree of heterogeneity in CSF glycosylation. Among all quantifiable glycopeptides, 49% and 22% of them were sialylated and fucosylated, and the prevalence of these two glycosylation types was also in line with our previously reported label-free study of N-glycopeptides in CSF samples.39
Compared to non-AD controls, 18 glycopeptides were found significantly changed, with 9 up-regulated and 9 down-regulated by student’s t-test (p-value<0.05) with a fold-change over 1.5 (Figure 6A, Figure S8A). The corresponding 14 glycoproteins were not observed to display alteration of abundances in parallel global proteomics analysis, suggesting that the dysregulation of glycopeptides likely occurred at the PTM level (Figure S8B). Among these glycopeptides, 13 of them were sialylated and 7 were fucosylated, which further emphasized the significance of the two types of glycosylation on the pathogenesis of AD.5,49 GO analysis showed that these dysregulated glycoproteins were significantly enriched in platelet degranulation, reverse cholesterol transport, cell adhesion, and complement activation, which have been reported to be associated with neuronal degeneration in AD (Figure S8D).50–53 Majority of dysregulated glycoproteins in AD were localized in the extracellular region, in accordance with the fact that CSF is in direct contact with the extracellular space of the brain and many N-glycoproteins are secreted proteins (Figure S8E).54,55 Protein-protein interaction network indicated that these proteins were mainly related to platelet degranulation and response to elevated platelet cytosolic Ca2+ (Figure S8C). Platelet degranulation was reported to play a pivotal role in platelet-mediated amyloid-β (Aβ) oligomerization in AD, which is believed to be greatly relevant to disease progression.56,57 Alterations in calcium homeostasis have been found to be critically implicated in brain aging and the neuropathology of AD, and the involvement of glycoproteins with response to elevated platelet cytosolic Ca2+ may provide deeper insight into the role of cytosolic calcium level during the progression of AD.58,59 Among these glycoproteins, four up-regulated sialyl-glycopeptides were observed at two sites of prostaglandin-H2 D-isomerase (PTHDS, Uniprot accession: P41222), a protein that is involved in multiple CNS functions and acts as a chaperone for preventing the formation of neurotoxic agents such as Aβ fibrils.60 This finding suggests that aberrant sialylation on this protein might play a specific role in AD progression through accelerated Aβ aggregation.61
Figure 6.
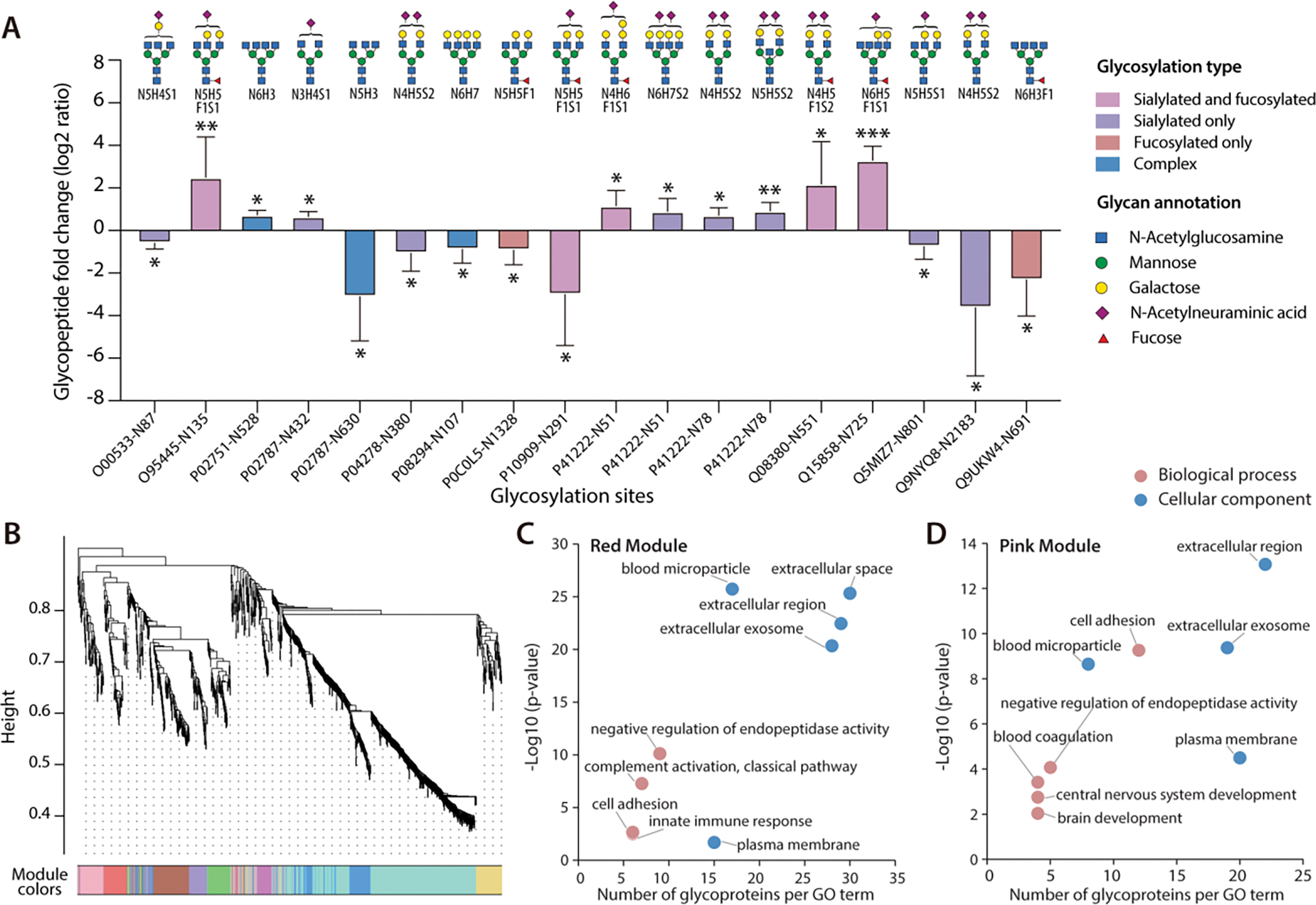
Site-specific quantitative glycoproteomic analysis of human CSF samples. (A) 18 aberrant N-glycopeptides in AD CSF samples with site-specific information (*p < 0.05, **p < 0.01, and ***p < 0.001). Each glycan structure only represents one possible glycan candidate due to the lack of structural information (N: HexNAc, H: Hex, F: Fucose, S: NeuAc). (B) WGCNA cluster dendrogram of 1172 glycopeptides. Nine modules with similar expression patterns were grouped via average linkage hierarchical clustering. (C) GO analysis of the red module glycopeptides clustered in WGCNA dendrogram. (D) GO analysis of the pink module glycopeptides clustered in WGCNA dendrogram.
To further study individual N-glycoproteome organization in CSF and its alterations in AD, WGCNA was conducted to construct an N-glycoproteomic network. Nine modules with similar expression patterns were grouped via average linkage hierarchical clustering (Figure 6B). Module-trait association analysis was performed to determine the correlation relationships between each module eigenglycopeptide and AD-relevant phenotypic traits and other clinical or sample characteristics (Figure S9A). Our analysis identified one positively correlated module (pink module) and one negatively correlated module (red module) that showed significant association with AD status, amyloid-β pathology, total-tau (T-tau), and phospho-tau 181 (P-tau 181) level. Subsequently, GO enrichment analysis was performed on the two disease-associated network modules (Figure 6C, 6D) which uncovered that these two modules were closely associated with brain development and extracellular function. In addition, the top 30 most connected hub glycopeptides in the two modules were plotted in Figure S9B and S9C.
Conclusions
Herein we successfully developed a robust and highly sensitive strategy, Boost-DiLeu for enhanced isobaric labeled quantitative intact glycoproteomic analysis. For the first time we incorporated the one-tube sample processing workflow into DiLeu labeling experiments, which not only simplified experimental steps, but also greatly reduced sample loss and increased proteome coverage. During labeling, we utilized a boosting channel consisting of relatively large amounts of biological samples analogous to samples of interest to enhance the MS signal, and this further increased glycoproteome coverage and ensured quantification performance. Boost/Study channel ratio of 30x was found to be optimal in both identification rate and quantification accuracy. MS2 parameters including AGC settings and maximum injection time were optimized, and a trade-off between glycoproteome coverage and quantification accuracy was observed. As a compromise, 3E4 AGC and 200 ms injection time were suggested as a starting point. Additional HpH fractionation after conventional HILIC enrichment provided complementary orthogonality in separation, which further boosted glycoproteome coverage. As a proof-of-principle experiment, the Boost-DiLeu strategy was applied to the site-specific N-glycoproteome analysis of human CSF samples from AD patients and non-AD donors. Overall, 1172 quantifiable intact glycopeptides were identified from 164 glycoproteins, and 18 glycopeptides were found to be dysregulated. WGCNA revealed two modules of glycopeptides significantly correlated with AD. These results demonstrated the feasibility of using this strategy for glycoproteomic analysis of size-limited clinical samples, which would be essential for future biomarker discovery and the development of personalized medicine. Notably, while we developed the Boost-DiLeu strategy with the 12-plex DiLeu tag set in this study, similar strategy may also be adapted to the 21-plex DiLeu tags for higher multiplex capacity without apparent isotopic interference from boosting channel.28 Future implementation of this strategy could also be extended to other DiLeu labeled PTM analyses of size-limited biological samples, such as phosphorylation and citrullination in clinical specimens.
Supplementary Material
Acknowledgements
This work was supported, in part, by the National Institutes of Health Grants RF1AG052324, U01CA231081, and R01 DK071801 (to L.L.). The Orbitrap instruments were purchased through the support of an NIH Shared Instrument Grant (NIH-NCRR S10RR029531 to L.L.) and the University of Wisconsin-Madison, Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. L.L. would like to acknowledge NIH grants R21AG065728, and S10OD025084, as well as funding support from a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy. H.Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL.
Footnotes
H.Z. has served on scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). The other authors declare no conflict of interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website: Experimental Section; data availability; SDS-PAGE result; Proteomic-level comparison of two sample preparation methods; Distribution of reporter ion intensities at different B/S ratios; Comparison of different B/S ratios with conventional method; Different maximum injection time setting comparison; Representative M6P GPSM; Site-specific quantitative glycoproteomic analysis of human CSF samples. Table of CSF subject information; table of glycoproteins from two sample preparation methods; table of quantifiable glycopeptides in HeLa cell; table of quantifiable glycopeptides in CSF; table of GO analysis result of WGCNA modules.
Reference
- (1).Ohtsubo K; Marth JD Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [DOI] [PubMed] [Google Scholar]
- (2).Reily C; Stewart TJ; Renfrow MB; Novak J Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15 (6), 346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lin B; Qing X; Liao J; Zhuo K Role of Protein Glycosylation in Host-Pathogen Interaction. Cells 2020, 9 (4), 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chen Z; Huang J; Li L Recent Advances in Mass Spectrometry (MS)-Based Glycoproteomics in Complex Biological Samples. Trends Analyt Chem. 2018, 118 (Cell 126 2006), 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fang P; Xie J; Sang S; Zhang L; Liu M; Yang L; Xu Y; Yan G; Yao J; Gao X; Qian W; Wang Z; Zhang Y; Yang P; Shen H Multilayered N-Glycoproteome Profiling Reveals Highly Heterogeneous and Dysregulated Protein N-Glycosylation Related to Alzheimer’s Disease. Anal. Chem. 2019, 92 (1), 867–874. [DOI] [PubMed] [Google Scholar]
- (6).Hu Y; Pan J; Shah P; Ao M; Thomas SN; Liu Y; Chen L; Schnaubelt M; Clark DJ; Rodriguez H; Boja ES; Hiltke T; Kinsinger CR; Rodland KD; Li QK; Qian J; Zhang Z; Chan DW; Zhang H; Consortium CPTA; Pandey A; Paulovich A; Hoofnagle A; Zhang B; Mani DR; Liebler DC; Ransohoff DF; Fenyo D; Tabb DL; Levine DA; Kuhn E; White FM; Whiteley GA; Zhu H; Shih I-M; Bavarva J; McDermott JE; Whiteaker J; Ketchum KA; Clauser KR; Ruggles K; Elburn K; Ding L; Hannick L; Zimmerman LJ; Watson M; Thiagarajan M; Ellis MJC; Oberti M; Mesri M; Sanders ME; Borucki M; Gillette MA; Snyder M; Edwards NJ; Vatanian N; Rudnick PA; McGarvey PB; Mertins P; Townsend RR; Thangudu RR; Smith RD; Rivers RC; Slebos RJC; Payne SH; Davies SR; Cai S; Stein SE; Carr SA; Skates SJ; Madhavan S; Liu T; Chen X; Zhao Y; Wang Y; Shi Z Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 2020, 33 (3), 108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tang L; Chen X; Zhang X; Guo Y; Su J; Zhang J; Peng C; Chen X N-Glycosylation in Progression of Skin Cancer. Med. Oncol. 2019, 36 (6), 50. [DOI] [PubMed] [Google Scholar]
- (8).Riley NM; Hebert AS; Westphall MS; Coon JJ Capturing Site-Specific Heterogeneity with Large-Scale N-Glycoproteome Analysis. Nat. Commun. 2019, 10 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nwosu CC; Strum JS; An HJ; Lebrilla CB Enhanced Detection and Identification of Glycopeptides in Negative Ion Mode Mass Spectrometry. Anal. Chem. 2010, 82 (23), 9654–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hart-Smith G; Raftery MJ Detection and Characterization of Low Abundance Glycopeptides Via Higher-Energy C-Trap Dissociation and Orbitrap Mass Analysis. J. Am. Soc. Mass Spectrom. 2012, 23 (1), 124–140. [DOI] [PubMed] [Google Scholar]
- (11).Chen Z; Yu Q; Hao L; Liu F; Johnson J; Tian Z; Kao WJ; Xu W; Li L Site-Specific Characterization and Quantitation of N -Glycopeptides in PKM2 Knockout Breast Cancer Cells Using DiLeu Isobaric Tags Enabled by Electron-Transfer/Higher-Energy Collision Dissociation (EThcD). Analyst 2018, 143 (11), 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mysling S; Palmisano G; Højrup P; Thaysen-Andersen M Utilizing Ion-Pairing Hydrophilic Interaction Chromatography Solid Phase Extraction for Efficient Glycopeptide Enrichment in Glycoproteomics. Anal. Chem. 2010, 82 (13), 5598–5609. [DOI] [PubMed] [Google Scholar]
- (13).Qing G; Yan J; He X; Li X; Liang X Recent Advances in Hydrophilic Interaction Liquid Interaction Chromatography Materials for Glycopeptide Enrichment and Glycan Separation. Trends Anal. Chem 2020, 124, 115570. [Google Scholar]
- (14).Wu R; Pai A; Liu L; Xing S; Lu Y NanoTPOT: Enhanced Sample Preparation for Quantitative Nanoproteomic Analysis. Anal. Chem. 2020, 92 (9), 6235–6240. [DOI] [PubMed] [Google Scholar]
- (15).Yi L; Tsai C-F; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian W-J Boosting to Amplify Signal with Isobaric Labeling (BASIL) Strategy for Comprehensive Quantitative Phosphoproteomic Characterization of Small Populations of Cells. Anal. Chem. 2019, 91 (9), 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Slavov N Single-Cell Protein Analysis by Mass Spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tsai C-F; Zhao R; Williams SM; Moore RJ; Schultz K; Chrisler WB; Pasa-Tolic L; Rodland KD; Smith RD; Shi T; Zhu Y; Liu T An Improved Boosting to Amplify Signal with Isobaric Labeling (IBASIL) Strategy for Precise Quantitative Single-Cell Proteomics. Mol. Cell Proteomics 2020, 19 (5), 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Suttapitugsakul S; Tong M; Sun F; Wu R Enhancing Comprehensive Analysis of Secreted Glycoproteins from Cultured Cells without Serum Starvation. Anal. Chem. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Klann K; Tascher G; Münch C Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by MTORC1 and EIF2α. Mol. Cell 2020, 77 (4), 913–925.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Justice SAP; McCracken NA; Victorino JF; Qi GD; Wijeratne AB; Mosley AL Boosting Detection of Low-Abundance Proteins in Thermal Proteome Profiling Experiments by Addition of an Isobaric Trigger Channel to TMT Multiplexes. Anal. Chem. 2021, 93 (18), 7000–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Mohammed A; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (23).Ross P; Huang Y; Marchese J; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin D Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3 (12), 1154–1169. [DOI] [PubMed] [Google Scholar]
- (24).Frost DC; Greer T; Li L High-Resolution Enabled 12-Plex DiLeu Isobaric Tags for Quantitative Proteomics. Anal. Chem. 2015, 87 (3), 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Xiang F; Ye H; Chen R; Fu Q; Li LN,N-Dimethyl Leucines as Novel Isobaric Tandem Mass Tags for Quantitative Proteomics and Peptidomics. Anal. Chem. 2010, 82 (7), 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sleno L The Use of Mass Defect in Modern Mass Spectrometry. J. Mass Spectrom. 2012, 47 (2), 226–236. [DOI] [PubMed] [Google Scholar]
- (27).Hebert AS; Merrill AE; Bailey DJ; Still AJ; Westphall MS; Strieter ER; Pagliarini DJ; Coon JJ Neutron-Encoded Mass Signatures for Multiplexed Proteome Quantification. Nat. Methods 2013, 10 (4), 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Frost DC; Feng Y; Li L 21-Plex DiLeu Isobaric Tags for High-Throughput Quantitative Proteomics. Anal. Chem. 2020, 92 (12), 8228–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dieterich IA; Cui Y; Braun MM; Lawton AJ; Robinson NH; Peotter JL; Yu Q; Casler JC; Glick BS; Audhya A; Denu JM; Li L; Puglielli L Acetyl-CoA Flux from the Cytosol to the ER Regulates Engagement and Quality of the Secretory Pathway. Sci. Rep. 2021, 11 (1), 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cui Y; Yang K; Tabang DN; Huang J; Tang W; Li L Finding the Sweet Spot in ERLIC Mobile Phase for Simultaneous Enrichment of N-Glyco and Phosphopeptides. J. Am. Soc. Mass Spectrom. 2019, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13 (9), 731–740. [DOI] [PubMed] [Google Scholar]
- (32).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Pérez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yılmaz Ş; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaíno JA The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47 (Database issue), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wu F; Sun D; Wang N; Gong Y; Li L Comparison of Surfactant-Assisted Shotgun Methods Using Acid-Labile Surfactants and Sodium Dodecyl Sulfate for Membrane Proteome Analysis. Anal. Chim. Acta 2011, 698 (1–2), 36–43. [DOI] [PubMed] [Google Scholar]
- (34).Randall SM; Cardasis HL; Muddiman DC Factorial Experimental Designs Elucidate Significant Variables Affecting Data Acquisition on a Quadrupole Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2013, 24 (10), 1501–1512. [DOI] [PubMed] [Google Scholar]
- (35).Specht H; Slavov N Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers. J. Proteome Res. 2020, 20 (1), 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dabrowski R; Ripa R; Latza C; Annibal A; Antebi A Optimization of Mass Spectrometry Settings for Steroidomic Analysis in Young and Old Killifish. Anal. Bioanal. Chem. 2020, 412 (17), 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yang F; Shen Y; Camp DG; Smith RD High-PH Reversed-Phase Chromatography with Fraction Concatenation for 2D Proteomic Analysis. Expert Rev. Proteom 2014, 9 (2), 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Huang J; Liu X; Wang D; Cui Y; Shi X; Dong J; Ye M; Li L Dual-Functional Ti(IV)-IMAC Material Enables Simultaneous Enrichment and Separation of Diverse Glycopeptides and Phosphopeptides. Anal. Chem. 2021, 93 (24), 8568–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chen Z; Yu Q; Yu Q; Johnson J; Shipman R; Zhong X; Huang J; Asthana S; Carlsson C; Okonkwo O; Li L In-Depth Site-Specific Analysis of N-Glycoproteome in Human Cerebrospinal Fluid and Glycosylation Landscape Changes in Alzheimer’s Disease. Mol. Cell Proteomics 2021, 20, 100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fang P; Ji Y; Silbern I; Doebele C; Ninov M; Lenz C; Oellerich T; Pan K-T; Urlaub H A Streamlined Pipeline for Multiplexed Quantitative Site-Specific N-Glycoproteomics. Nat. Commun. 2020, 11 (1), 5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Song C; Ye M; Han G; Jiang X; Wang F; Yu Z; Chen R; Zou H Reversed-Phase-Reversed-Phase Liquid Chromatography Approach with High Orthogonality for Multidimensional Separation of Phosphopeptides. Anal. Chem. 2010, 82 (1), 53–56. [DOI] [PubMed] [Google Scholar]
- (42).Cheung TK; Lee C-Y; Bayer FP; McCoy A; Kuster B; Rose CM Defining the Carrier Proteome Limit for Single-Cell Proteomics. Nat. Methods 2020, 1–8. [DOI] [PubMed] [Google Scholar]
- (43).Huang J; Dong J; Shi X; Chen Z; Cui Y; Liu X; Ye M; Li L Dual-Functional Titanium(IV) Immobilized Metal Affinity Chromatography Approach for Enabling Large-Scale Profiling of Protein Mannose-6-Phosphate Glycosylation and Revealing Its Predominant Substrates. Anal. Chem. 2019, 91 (18), 11589–11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Segal MB Extracellular and Cerebrospinal Fluids. J. Inherit. Metab. Dis. 1993, 16 (4), 617–638. [DOI] [PubMed] [Google Scholar]
- (45).Abdi F; Quinn JF; Jankovic J; McIntosh M; Leverenz JB; Peskind E; Nixon R; Nutt J; Chung K; Zabetian C; Samii A; Lin M; Hattan S; Pan C; Wang Y; Jin J; Zhu D; Li GJ; Liu Y; Waichunas D; Montine TJ; Zhang J Detection of Biomarkers with a Multiplex Quantitative Proteomic Platform in Cerebrospinal Fluid of Patients with Neurodegenerative Disorders. J. Alzheimer’s Dis. 2006, 9 (3), 293–348. [DOI] [PubMed] [Google Scholar]
- (46).Zhang J Proteomics of Human Cerebrospinal Fluid – the Good, the Bad, and the Ugly. Proteom. - Clin. Appl 2007, 1 (8), 805–819. [DOI] [PubMed] [Google Scholar]
- (47).Chen Z; Wang D; Yu Q; Johnson J; Shipman R; Zhong X; Huang J; Yu Q; Zetterberg H; Asthana S; Carlsson C; Okonkwo O; Li L. In-Depth Site-Specific O-Glycosylation Analysis of Glycoproteins and Endogenous Peptides in Cerebrospinal Fluid (CSF) from Healthy Individuals, Mild Cognitive Impairment (MCI), and Alzheimer’s Disease (AD) Patients. Acs Chem Biol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shores KS; Knapp DR Assessment Approach for Evaluating High Abundance Protein Depletion Methods for Cerebrospinal Fluid (CSF) Proteomic Analysis. J. Proteome Res. 2007, 6 (9), 3739–3751. [DOI] [PubMed] [Google Scholar]
- (49).Yang K; Yang Z; Chen X; Li W The Significance of Sialylation on the Pathogenesis of Alzheimer’s Disease. Brain Res. Bull. 2021, 173, 116–123. [DOI] [PubMed] [Google Scholar]
- (50).Evin G; Li Q-X Platelets and Alzheimer’s Disease: Potential of APP as a Biomarker. World J. Psychiatry 2012, 2 (6), 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Knebl J; DeFazio P; Clearfield MB; Little L; McConathy WJ; Pherson RM; Lacko AG Plasma Lipids and Cholesterol Esterification in Alzheimer’s Disease. Mech. Ageing Dev. 1994, 73 (1), 69–77. [DOI] [PubMed] [Google Scholar]
- (52).Zhong X; Wang J; Carlsson C; Okonkwo O; Zetterberg H; Li L A Strategy for Discovery and Verification of Candidate Biomarkers in Cerebrospinal Fluid of Preclinical Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 11, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).McGeer PL; McGeer EG The Possible Role of Complement Activation in Alzheimer Disease. Trends Mol. Med. 2002, 8 (11), 519–523. [DOI] [PubMed] [Google Scholar]
- (54).Roth J Protein N-Glycosylation along the Secretory Pathway: Relationship to Organelle Topography and Function, Protein Quality Control, and Cell Interactions. Chem. Rev. 2002, 102 (2), 285–304. [DOI] [PubMed] [Google Scholar]
- (55).Zhong X; Yu Q; Ma F; Frost DC; Lu L; Chen Z; Zetterberg H; Carlsson C; Okonkwo O; Li L HOTMAQ: A Multiplexed Absolute Quantification Method for Targeted Proteomics. Anal. Chem. 2019, 91 (3), 2112–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Donner L; Fälker K; Gremer L; Klinker S; Pagani G; Ljungberg LU; Lothmann K; Rizzi F; Schaller M; Gohlke H; Willbold D; Grenegard M; Elvers M Platelets Contribute to Amyloid-β Aggregation in Cerebral Vessels through Integrin ΑIIbβ3–Induced Outside-in Signaling and Clusterin Release. Sci. Signal. 2016, 9 (429), ra52–ra52. [DOI] [PubMed] [Google Scholar]
- (57).Sackmann C; Hallbeck M Oligomeric Amyloid-β Induces Early and Widespread Changes to the Proteome in Human IPSC-Derived Neurons. Sci. Rep. 2020, 10 (1), 6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).PASCALE A; ETCHEBERRIGARAY R CALCIUM ALTERATIONS IN ALZHEIMER’S DISEASE: PATHOPHYSIOLOGY, MODELS AND THERAPEUTIC OPPORTUNITIES. Pharmacol. Res. 1999, 39 (2), 81–88. [DOI] [PubMed] [Google Scholar]
- (59).Řípová D; Platilová V; Strunecká A; Jirák R; Höschl C. Cytosolic Calcium Alterations in Platelets of Patients with Early Stages of Alzheimer’s Disease. Neurobiol. Aging 2000, 21 (5), 729–734. [DOI] [PubMed] [Google Scholar]
- (60).Chen CPC; Huang Y-C; Chang C-N; Chen J-L; Hsu C-C; Lin W-Y Changes of Cerebrospinal Fluid Protein Concentrations and Gait Patterns in Geriatric Normal Pressure Hydrocephalus Patients after Ventriculoperitoneal Shunting Surgery. Exp. Gerontol. 2018, 106, 109–115. [DOI] [PubMed] [Google Scholar]
- (61).Domenico FD; Pupo G; Giraldo E; Badìa M-C; Monllor P; Lloret A; Schininà ME; Giorgi A; Cini C; Tramutola A; Butterfield DA; Viña J; Perluigi M Oxidative Signature of Cerebrospinal Fluid from Mild Cognitive Impairment and Alzheimer Disease Patients. Free Radic. Biol. Med 2016, 91, 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.