

Abstract
An efficient, eco-compatible, and very cheap method for the construction of fully substituted pyrazoles (Pzs) via eliminative nitrilimine-alkene 1,3-dipolar cycloaddition (ENAC) reaction was developed in excellent yield and high regioselectivity. Enaminones and nitrilimines generated in situ were selected as dipolarophiles and dipoles, respectively. A deep screening of the employed base, solvent, and temperature was carried out to optimize reaction conditions. Recycling tests of ionic liquid were performed, furnishing efficient performance until six cycles. Finally, a plausible mechanism of cycloaddition was proposed. Then, the effect of three different structures of Pzs was evaluated on the F1FO-ATPase activity and mitochondrial permeability transition pore (mPTP) opening. The Pz derivatives’ titration curves of 6a, 6h, and 6o on the F1FO-ATPase showed a reduced activity of 86%, 35%, and 31%, respectively. Enzyme inhibition analysis depicted an uncompetitive mechanism with the typical formation of the tertiary complex enzyme-substrate-inhibitor (ESI). The dissociation constant of the ESI complex (Ki’) in the presence of the 6a had a lower order of magnitude than other Pzs. The pyrazole core might set the specific mechanism of inhibition with the F1FO-ATPase, whereas specific functional groups of Pzs might modulate the binding affinity. The mPTP opening decreased in Pz-treated mitochondria and the Pzs’ inhibitory effect on the mPTP was concentration-dependent with 6a and 6o. Indeed, the mPTP was more efficiently blocked with 0.1 mM 6a than with 1 mM 6a. On the contrary, 1 mM 6o had stronger desensitization of mPTP formation than 0.1 mM 6o. The F1FO-ATPase is a target of Pzs blocking mPTP formation.
Keywords: pyrazoles; 1,3-dipolar cycloaddition; hydrazonyl chlorides; mitochondrial permeability transition pore; F1FO-ATPase
1. Introduction
Pyrazoles (Pzs) are aromatic five-membered heterocyclic compounds characterized by two adjacent nitrogen atoms and three carbons in the ring. They can exist in three tautomeric forms due to the π-electrons shifting into the heterocycle ring [1]. Pzs show a large number of applications due to the aromatic character of the heterocycle core that makes them resistant to oxidation, reduction, and acid or basic hydrolysis [2]. For this reason, Pz derivatives are useful as agrochemicals, such as insecticides, fungicides, and herbicides [3,4]; or as dyes [5], sunscreen materials [6], analytical reagents [7], and powerful ligands in coordination chemistry [8]. In addition, polyaromatic pyrazoles have important photophysical, optical, and electronic properties [5,9]. Pzs and their derivatives surely play a significant role in medicinal chemistry towards the development of novel and more efficient drugs, considering their active participation in hydrogen bond formation, π-stacking, and dipole–dipole interactions with different biological targets [10]. Among others, Pzs present significant antibacterial [11], antimicrobic, anticancer, anti-inflammatory, anti-diabetic, and anti-degenerative activity [12]. In Figure 1, Betazole [13], CDPPB [14], Difenamizole [15], Fezolamine [16], Rimonabant [17], and Ruxolitinib [18,19] are examples of Pz-containing drugs already available on the market (Figure 1).
Figure 1.

Clinical uses of drugs containing a pyrazole core.
Especially, their potential bioactive properties have made this class of heterocycles attractive targets for organic synthesis and in particular, in the past decade, many methods to synthesize tetrasubstituted Pzs have been developed [20]. Among them, one of the first realized procedures was the cyclocondensation of substituted hydrazines with 1,3-diketones, known as the Knorr reaction that, however, turned out to be a low-performant reaction for the obtaining of a regioisomer mixture (Scheme 1a) [21]. An approach that surely solved the problem of regioselectivity was the multicomponent synthesis illustrated in Scheme 1b [22], but the method has limited applications due to the difficult preparation of reagents or use of closed reactors [23]. The 1,3-dipolar cycloaddition reaction between alkynes and nitrilimines generated in situ from hydrazonyl halides (Scheme 1c) provided tetrasubstituted Pzs in good yields without, however, the overcoming of very low regioselectivity [24,25].
Scheme 1.

Conventional approaches and our proposal for synthesis of tetrasubstituted pyrazoles.
With the aim to develop a novel and higher-performing synthesis of 1,3,4,5-tetrasubstituted Pzs, and in continuity with our experience in the construction of biologically active heterocycle compounds [26,27,28,29], especially by eliminative 1,3-dipolar cycloadditions [30,31], herein, we present our highly regioselective method founded on a base-promoted eliminative nitrilimine-alkene cycloaddition (ENAC). The reactions, performed between various enaminones and several nitrilimines generated in situ by hydrazonyl chlorides in ionic liquid as an eco-friendly solvent (Scheme 1d) are, to our knowledge, the first attempt of ENAC with enaminones as a dipolarophile.
In general, in 1,3-dipolar cycloadditions, olefins are a valid alternative to alkynes because of their easy availability and low-cost preparation [32]. In detail, the ENAC reaction proceeds through the formation of an unstable intermediate pyrazoline, which is easily transformed into the corresponding stable Pz by an eliminative step, as reported in several works of literature [33,34,35,36,37,38,39]. Moreover, enaminones are excellent and versatile dipolarophiles to promote the quick formation of N-containing heterocycles by eliminative cycloaddition [40] because they can easily lose a primary amine [41].
In this work, after an in-depth examination of the best reaction conditions by varying base, solvent, and temperature, we extended the procedure to several enaminones and nitrilimines, obtaining a series of 1,3,4,5-tetrasubstituted Pzs in very high yield and regioselective manner. We selected the [mPy]OTf as a non-conventional solvent by considering the ionic liquid typical properties (i.e., low vapor pressure, solvent recover/recycle, and so on) [42], the acclaimed strong stabilization of reaction intermediates [43], and its easy one-step preparation through a halide-free direct procedure [44]. Hence, recovery procedures to reuse the [mPy]OTf were conducted, observing an elevated performance until six cycles.
Finally, we proposed a probable mechanism of base-promoted eliminative nitrilimine-alkene cycloaddition, starting from enaminones and nitrilimines. Furthermore, we evaluated the effect of new Pzs on mitochondrial F1FO-ATPase and the mitochondrial permeability transition pore (mPTP) event, considering analogous previous studies conducted on triazole substrates. The F1FO-ATPase is evolutionarily specialized in ATP synthesis by using the protonmotive force across the inner mitochondrial membrane, and it is also the main candidate responsible for the mPTP formation [45,46,47]. The F1FO-ATPase as a bifunctional membrane-bound molecular machine transduces the energy of the transmembrane proton motive force (Δp) to the chemical energy in the form of ATP via torque rotation. Vice versa, working as an H+ pump reenergizes the inner mitochondrial membranes (IMM) building the Δp [48]. The drop of Δp increases mitochondrial ROS production and Ca2+ accumulation. These are molecular events that open the mPTP [49,50]. In several diseases, the mPTP is involved in triggering different forms of regulated cell death [51,52], and the F1FO-ATPase is considered the outstanding biological target in drug discovery to block this phenomenon [53,54,55,56].
2. Materials and Methods
Commercial starting materials were purchased from Merck (Milano, Italy) or Alfa Aesar (Karlsruhe, Germany) and were used without further purification. Reactions were monitored by TLC using silica plates 60-F264, commercially available from Merck (Milano, Italy). Mono and bidimensional 1H and 13C NMR experiments were recorded at 300, 500, and 125.7 MHz, respectively, in CDCl3 and DMSO-d6 as solvent using tetramethylsilane (TMS) as an internal standard (Bruker ACP 300 MHz and Bruker Avance 500 MHz with a 5 mm TBO probe, Rheinstetten, Germany). Chemical shifts are given in parts per million and coupling constants in Hertz. Regiochemistry was established by the NMR technique. High-resolution mass spectra (HRMS) were recorded with a Bruker Compact QTOF instrument (Bruker, Billerica, MA, USA). HRMS spectra were acquired in positive ion mode, with a mass resolution of 30,000. Mass calibration was performed with a solution of sodium formate clusters and processed in HPC mode. Spectra acquisition was performed in flow injection, with a full scan mode in the range of 50 to 500 m/z. N2 was the source of dry gas (V = 4 L/min, T = 180 °C). The ion formula of each compound was calculated with the Smart Formula tool of the Bruker software platform, analyzing the isotopic pattern ratio with 4 mDa mass confidence. All samples were dissolved in MeOH. The final substrates were further purified before biological assays by recrystallization. Synthesis and characterization of variously substituted benzoyl phenylhydrazines 3a–3g and hydrazonyl chlorides 4a–4g are realized by modified literature procedures [57,58], respectively] and reported in the Supplementary Materials. Enaminones 5a–5c were prepared according to procedures in the literature [59,60]. [mPy]OTf was prepared according to procedures in the literature [44] (see the Supplementary Materials for the synthetic procedure).
2.1. General Procedure for Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles 6a–6g
In a 50 mL two-necked round-bottom flask, equipped with a bubble condenser and magnetic stir bar, enaminone 5a–5c (0.5 g, 1 eq), anhydrous K2CO3 (2.5 eq), and hydrazonyl chloride 4a–4g (1.3 eq) were mixed with 12 mL of [mPy](OTf)/H2O (9:1 v:v). The reaction was heated at 50 °C for the appropriate time. The mixture was extracted with dichloromethane (3 × 10 mL) and the combined organic layer was washed with a saturated aqueous NH4Cl solution (3 × 10 mL), dried with anhydrous Na2SO4, filtered, and evaporated under vacuum. The crude was purified by flash chromatography (hexane/ethyl acetate 8:2 v:v), isolating product 6a–6g.
The regiochemistry was attributed by comparison with data in the literature for compound 6a [61]. Furthermore, the regioisomeric ratio was calculated by 1H NMR of the crude of reaction, by comparison of the CH3 singlet on the pyrazole ring for both regioisomers (see the Supplementary Materials).
2.2. Recovery and Recycling Procedure of Ionic Liquid [mPy](OTf)
The [mPy](OTf)/H2O mixture, recovered as residue insoluble in the extraction phase, was washed with diethyl ether (2 × 5 mL) and dried at 70 °C under vacuum conditions. Successive runs were performed in the recycled ionic liquid after the addition of water and fresh reagents.
1-(5-Methyl-1,3-diphenyl-1H-pyrazol-4-yl)ethanone (6a). White solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.13 (s, 3H, CH3), 2.56 (s, 3H, CH3), 7.41–7.48 (m, 4H, Ar), 7.48–7.50 (m, 3H, Ar), 7.50–7.53 (m, 1H, Ar), 7.53–7.59 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.82, 30.83, 120.56, 125.76, 128.41, 128.70, 128.73, 129.25, 129.44, 133.71, 138.56, 143.70, 153.32, 195.94. ESI(+)-MS: m/z [M + H]+ calcd for [C18H17N2O]+, 277.1335, found 277.1331.
1-(3-(4-Methoxyphenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone (6b). White solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.15 (s, 3H, CH3), 2.55 (s, 3H, CH3), 3.85 (s, 3H, CH3),6.95–7.01 (m, 2H, Ar), 7.40–7.55 (m, 7H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.78, 30.73, 55.28, 113.83, 120.49, 125.72, 125.95, 128.61, 129.19, 130.61, 138.59, 143.57, 153.04, 160.02, 196.01. ESI(+)-MS: m/z [M + H]+ calcd for [C19H19N2O2]+, 307.1441, found 307.1436.
1-(5-Methyl-1-phenyl-3-(p-tolyl)-1H-pyrazol-4-yl)ethanone (6c). Pale-yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.14 (s, 3H, CH3), 2.41 (s, 3H, CH3), 2.55 (s, 3H, CH3),7.22–7.29 (m, 2H, Ar), 7.39–7.54 (m, 7H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.75, 21.29, 30.78, 120.53, 125.72, 128.61, 129.05, 129.18, 129.25, 130.68, 138.50, 138.59, 143.55, 153.32, 196.05. ESI(+)-MS: m/z [M + H]+ calcd for [C19H19N2O]+, 291.1492, found 291.1492.
1-(5-Methyl-3-(4-nitrophenyl)-1-phenyl-1H-pyrazol-4-yl)ethanone (6d). Yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.24 (s, 3H, CH3), 2.57 (s, 3H, CH3), 7.44–7.60 (m, 5H, Ar), 7.74–7.82 (m, 2H, Ar), 8.28–8.35 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.89, 31.03, 120.82, 123.58, 125.78, 129.20, 129.46, 130.30, 138.27, 140.21, 143.97, 147.96, 150.78, 194.76. ESI(+)-MS: m/z [M + H]+ calcd for [C18H16N3O3]+, 322.1186, found 322.1186.
1-(3-(2-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone (6e). Yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.06 (s, 3H, CH3), 2.62 (s, 3H, CH3), 7.31–7.41 (m, 2H, Ar), 7.42–7.54 (m, 7H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 13.01, 29.61, 120.83, 125.70, 126.87, 128.73, 129.20, 129.65, 130.23, 131.64, 133.42, 134.30, 138.40, 143.79, 150.55, 194.70. ESI(+)-MS: m/z [M + H]+ calcd for [C18H16ClN2O]+, 311.0946, found 311.0941.
1-(3-(3-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone (6f). Yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.17 (s, 3H, CH3), 2.55 (s, 3H, CH3), 7.34–7.43 (m, 3H, Ar), 7.44–7.56 (m, 5H, Ar), 7.57–7.61 (m, 1H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.78, 30.87, 120.52, 125.72, 127.70, 128.80, 128.87, 129.30, 129.42, 129.59, 134.36, 135.45, 138.40, 143.83, 151.75, 195.34. ESI(+)-MS: m/z [M + H]+ calcd for [C18H16ClN2O]+, 311.0946, found 311.0952.
1-(3-(4-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone (6g). Yellow solid. 1H-NMR (CDCl3, 300 MHz): δ (ppm) 2.16 (s, 3H, CH3), 2.55 (s, 3H, CH3), 7.37–7.43 (m, 1H, Ar), 7.43–7.46 (m, 2H, Ar), 7.47–7.51 (m, 4H, Ar), 7.51–7.57 (m, 2H, Ar). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 12.79, 30.86, 120.54, 125.72, 128.62, 128.84, 129.29, 130.71, 132.15, 134.82, 138.43, 143.79, 151.98, 195.41. ESI(+)-MS: m/z [M + H]+ calcd for [C18H16ClN2O]+, 311.0946, found 311.0948.
(5-Methyl-1,3-diphenyl-1H-pyrazol-4-yl)(phenyl)methanone (6h). White solid. 1H-NMR (CDCl3, 300 MHz): δ (ppm) 2.45 (s, 3H, CH3), 7.11–7.19 (m, 3H, Ar), 7.19–7.32 (m, 3H, Ar), 7.36–7.46 (m, 3H, Ar), 7.50–7.61 (m, 4H, Ar), 7.69–7.77 (m, 2H, Ar). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 12.26, 118.86, 125.61, 127.92, 128.00, 128.04, 128.61, 128.70, 129.31, 129.77, 132.45, 132.53, 138.43, 138.92, 143.33, 152.52, 192.79. ESI(+)-MS: m/z [M + H]+ calcd for [C23H19N2O]+, 339.1492, found 339.1492.
(3-(4-Methoxyphenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (6i). White solid. 1H-NMR (CDCl3, 300 MHz): δ (ppm) 2.42 (s, 3H, CH3), 3.72 (s, 3H, CH3), 6.64–6.74 (m, 2H, Ar), 7.22–7.30 (m, 3H, Ar), 7.33–7.42 (m, 3H, Ar), 7.51–7.59 (m, 4H, Ar), 7.71–7.79 (m, 2H, Ar). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 12.24, 55.14, 113.44, 118.58, 124.98, 125.54, 128.05, 128.47, 129.23, 129.73, 129.85, 132.50, 138.40, 138.91, 143.10, 152.15, 159.34, 192.86. ESI(+)-MS: m/z [M + H]+ calcd for [C24H21N2O2]+, 369.1598, found 369.1597.
(5-Methyl-1-phenyl-3-(p-tolyl)-1H-pyrazol-4-yl)(phenyl)methanone (6j). White solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.07 (s, 3H, CH3), 2.41 (s, 3H, CH3), 7.22–7.29 (m, 6H, Ar), 7.29–7.34 (m, 3H, Ar), 7.34–7.41 (m, 3H, Ar), 7.56–7.63 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 21.37, 31.30, 121.88, 125.44, 127.82, 128.49, 128.81, 128.96, 129.07, 129.32, 129.56, 129.90, 130.42, 138.50, 139.15, 144.64, 152.46, 196.07. ESI(+)-MS: m/z [M + H]+ calcd for [C24H21N2O]+, 353.1648, found 353.1645.
(5-Methyl-3-(4-nitrophenyl)-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (6k). Yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.04 (s, 3H, CH3), 7.23–7.29 (m, 2H, Ar), 7.29–7.36 (m, 5H, Ar), 7.38–7.47 (m, 3H, Ar), 7.90–7.99 (m, 2H, Ar), 8.26–8.32 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 31.16, 122.03, 123.36, 125.42, 128.38, 128.90, 129.01, 129.21, 129.91, 130.05, 130.32, 138.77, 139.39, 145.72, 147.77, 150.18, 195.30. ESI(+)-MS: m/z [M + H]+ calcd for [C23H18N3O3]+, 384.1343, found 384.1352.
(3-(2-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (6l). Pale-yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.53 (s, 3H, CH3), 7.6–7.15 (m, 2H, Ar), 7.16–7.22 (m, 2H, Ar), 7.27–7.42 (m, 2H, Ar), 7.43–7.50 (m, 2H, Ar), 7.50–7.61 (m, 4H, Ar), 7.61–7.68 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.50, 120.10, 125.58, 126.46, 127.57, 128.68, 129.29, 129.35, 129.43, 129.50, 131.79, 131.98, 132.33, 133.44, 138.51, 138.74, 143.37, 150.60, 192.16. ESI(+)-MS: m/z [M + H]+ calcd for [C23H18ClN2O]+, 373.1102, found 373.1110.
(3-(3-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (6m). Pale-yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.44 (s, 3H, CH3), 7.01–7.10 (m, 1H, Ar), 7.10–7.16 (m, 1H, Ar), 7.20–7.31 (m, 4H, Ar), 7.36–7.43 (m, 1H, Ar), 7.48–7.52 (m, 1H, Ar), 7.52–7.58 (m, 4H, Ar), 7.68–7.75 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.28, 118.90, 125.58, 126.95, 127.98, 128.19, 128.57, 128.80, 129.17, 129.38, 129.62, 132.72, 133.96, 134.22, 138.41, 138.72, 143.56, 151.09, 192.49. ESI(+)-MS: m/z [M + H]+ calcd for [C23H18ClN2O]+, 373.1102, found 373.1117.
(3-(4-Chlorophenyl)-5-methyl-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (6n). Pale-yellow solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 2.41 (s, 3H, CH3), 7.10–7.17 (m, 2H, Ar), 7.27–7.32 (m, 2H, Ar), 7.35–7.40 (m, 2H, Ar), 7.42–7.48 (m, 2H, Ar), 7.49–7.58 (m, 4H, Ar), 7.68–7.76 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 12.29, 125.58, 128.23, 128.48, 128.76, 129.37, 129.72, 129.86, 130.36, 131.00, 132.81, 133.94, 138.33, 138.78, 143.41, 151.27, 192.57. ESI(+)-MS: m/z [M + H]+ calcd for [C23H18ClN2O]+, 373.1102, found 373.1114.
Phenyl(1,3,5-triphenyl-1H-pyrazol-4-yl)methanone (6o). White solid. 1H NMR (CDCl3, 300 MHz): δ (ppm) 7.08–7.21 (m, 2H, Ar), 7.30–7.40 (m, 4H, Ar), 7.42–7.58 (m, 6H, Ar), 7.61–7.69 (m, 3H, Ar), 7.82–7.90 (m, 3H, Ar), 7.91–8.03 (m, 2H, Ar). 13C NMR (CDCl3, 125 MHz): δ (ppm) 120.24, 123.21, 124.57, 127.03, 128.30, 128.37, 128.57, 128.71, 128.77, 129.09, 129.45, 129.86, 131.82, 135.00, 137.94, 139.41, 139.87, 161.54, 165.80, 189.71.ESI(+)-MS: m/z [M + H]+ calcd for [C28H21N2O]+, 401.1648, found 401.1647.
2.3. Preparation of Mitochondrial Fraction
Swine hearts were collected in a local abattoir and transported to the laboratory within 2 h on ice at 0–4 °C. After removing as much fat and blood clots as possible, approximately 30–40 g of heart tissue was rinsed in ice-cold Tris-HCl wash buffer (medium A) consisting of 0.25 M sucrose, 10 mM Tris (hydroxymethyl)-aminomethane (Tris), pH 7.4 and finely chopped into fine pieces with scissors. Subsequently, the tissues were gently dried on absorbent paper, weighed, and homogenized with Ultraturrax T25 in medium B (0.25 M of sucrose, 10 mM of Tris, 1 mM of EDTA (free acid), 0.5 mg/mL of BSA without fatty acid, pH 7.4 with HCl) in a ratio of 10 mL of medium B to 1 g of fresh tissue. The tissue was then carefully homogenized by a motorized Teflon pestle homogenizer (Braun Melsungen type 853202) at 650 rpm with 3 strokes up and down. The mitochondrial fraction was then obtained by gradual centrifugation (Sorvall RC2-B, SS34 rotor). The homogenate was centrifuged at 1000× g for 5 min, thus obtaining a supernatant and a pellet. The pellet was re-homogenized again under the same conditions as the first homogenization, and recentrifuged at 1000× g for 5 min. The supernatants collected from these two centrifugations, filtered through four cotton gauze layers, were centrifuged at 10,500× g for 10 min to produce the raw mitochondrial pellet. The raw pellet was resuspended in medium A and further centrifuged at 10,500× g for 10 min to obtain the final mitochondrial pellet. The latter was resuspended by gentle agitation using a Potter Elvejehm Teflon homogenizer in a small volume of medium A, thus obtaining a protein concentration of 30 mg/mL [62]. All steps were performed at 0–4 °C. Protein concentration was determined according to the Bradford colorimetric method using the Bio-Rad Protein Assay kit II with BSA as standard [63]. The mitochondrial preparations were then stored in liquid nitrogen.
2.4. Mitochondrial F-ATPase Activity Assay
After the thawing of the mitochondria from the liquid nitrogen, different mitochondrial preparations were used to evaluate F-ATPase activity. To measure the hydrolysis capacity of ATP by Mg2+-activated F1FO-ATPase, 1 mL of a reaction medium, consisting of 0.15 mg mitochondrial protein and 75 mM ethanolamine-HCl buffer at pH 9.0, was used in the presence of 6.0 mM Na2ATP and 2.0 mM MgCl2; while the same buffer was used to determine the activity of F1FO-ATPase activated with Ca2+ but at pH 8.8 in the presence of 3.0 mM Na2ATP and 2.0 mM CaCl2. The test involves a 5 min pre-incubation at 37 °C with the subsequent addition of the Na2ATP substrate to start the reaction. After 5 min, the reaction was stopped using 1 mL of an ice-cold aqueous solution of 15% (w/w) trichloroacetic acid (TCA). At this point, the samples were centrifuged for 15 min at 3500 rpm (Eppendorf Centrifuge 5202). The indirect determination of the F-ATPase activity was defined spectrophotometrically [64] by calculating the concentration of inorganic phosphate (Pi) hydrolyzed by known quantities of mitochondrial protein present in the supernatant. Therefore, before the start of the reaction, 1.0 µL of 3.0 mg/mL of oligomycin was added to the mixture, used in F-ATPase tests as it represents a specific inhibitor of F-ATPase capable of selectively blocking the FO subunit, solubilized in dimethyl sulfoxide (DMSO). For each series of experiments, at the same time as the conditions being tested, the total ATPase activity was calculated by evaluating the Pi in control tubes containing 1.0 μL of DMSO per mL of the reaction system. In the experiments we conducted, a 3.0 µg/mL dose of oligomycin gave the greatest inhibition of F-ATPase [65]. In each experiment, F1FO-ATPase activity was obtained as the difference between hydrolyzed Pi in the presence of oligomycin and hydrolyzed Pi by total ATPase activity and expressed as µmol Pi · mg protein−1 · min−1. The concentration of Pi hydrolyzed by known amounts of mitochondrial protein, which is an indirect measure of ATPase activity was evaluated spectrophotometrically according to Fiske and Subbarow [66].
2.5. Kinetic Analysis
To calculate the values of IC50, i.e., the concentration of the inhibitor that causes half of the maximal inhibition of enzyme activity, the data on the enzymatic activity obtained in the absence of pyrazoles and the presence of increasing concentrations of pyrazoles were used to calculate the inhibition of the enzyme which, after background correction, have been adapted to a 3 parameter Equation (1), where the lower data limit (no enzyme inhibition) is 0. In Equation (1), the enzyme activity (y) is a function of the inhibitor concentration (x), “Range” is the uninhibited enzyme activity (in the absence of the inhibitor) and s is a slope factor. Since x is in the denominator, y falls at increasing x values.
| (1) |
The study of pyrazole inhibition mechanisms on Ca2+- or Mg2+-activated F1FO-ATPases was performed using Dixon and Cornish-Bowden diagrams [67]. Several experimental sets were designed in which the activity of F-ATPase was evaluated in the presence of increasing concentrations of pyrazoles at two concentrations of ATP, maintaining a constant concentration of the cofactor Mg2+ or Ca2+. The reciprocal of enzymatic activity, 1/v in the Dixon diagram or the s/v ratio in the Cornish-Bowden diagram, was plotted as a function of pyrazole concentration. In all plots, the specific activity of the enzyme was taken as the expression of v. The K’i values, which represent the dissociation constant of the ternary enzyme-substrate-inhibitor complex (ESI), were calculated as the abscissa (changed to positive) of the intercept of the lines obtained in the Cornish-Bowden graphs.
2.6. mPTP Assay
On freshly isolated mitochondrial fractions from swine heart, fresh mitochondrial suspensions (1 mg/mL) were energized in the assay buffer (130 mM KCl, 1 mM KH2PO4, 20 mM HEPES, pH 7.2 with TRIS), incubated at 37 °C with 1 μg/mL of rotenone and 5 mM of succinate. Selected doses of pyrazoles were added to the mitochondrial suspensions after the evaluation of the mPTP. The opening of mPTP was induced by the addition of Ca2+ 10 μM as a CaCl2 solution at fixed time intervals (1 min). The calcium retention capacity (CRC), the lowering of which indicates the opening of mPTP, was evaluated spectro-fluorophotometrically in the presence of 0.8 μM of Fura-FF. The probe has different spectral properties in the absence and presence of Ca2+; that is, it shows an excitation/emission spectrum of 365/514 nm in the absence of Ca2+ (Fura-FF low Ca2+) and shifts to 339/507 nm in the presence of high concentrations of Ca2+ (Fura-FF high Ca2+). An increase in the fluorescence intensity ratio (Fura-FF high Ca2+)/(Fura-FF low Ca2+), i.e., a decrease in CRC [68], indicates the opening of mPTP. All measurements were processed by LabSolutions RF software.
2.7. Statistical Analysis
The data represent the mean ± SD (shown as vertical bars in the figures) of the number of experiments reported in the figure captions. In each set of biochemical experiments, the analyses were carried out on at least three distinct mitochondrial preparations. The differences between the enzyme activity data in differently treated mitochondria were evaluated by one-way ANOVA followed by Dunnett’s test when the F values indicated a significance (P ≤ 0.05).
3. Results and Discussion
3.1. Synthesis
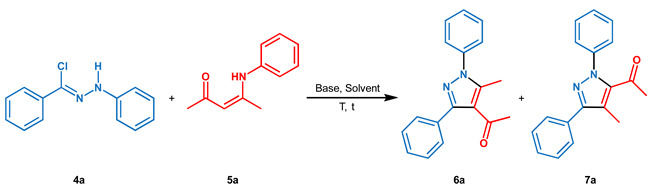
With the purpose of developing a highly regioselective synthesis of 1,3,4,5-tetrasubstituted pyrazoles by 1,3-dipolar cycloaddition reaction, a preventive preparation of dipoles and dipolarophiles was necessary. Firstly, hydrazonyl chlorides 4a–4g, a source of nitrilimines as 1,3-dipole, were synthesized from phenylhydrazine 1 and commercially available acyl chlorides 2a–2g, as described in Scheme 2.
Scheme 2.

Synthesis of hydrazonyl chlorides 4a–4g.
In detail, intermediate aroyl phenylhydrazines 3a–3g were obtained in very good yields and immediately transformed in the correspondent hydrazonyl chlorides 4a–4g, isolated in excellent yields (see the Supplementary Materials). In addition, three enaminones 5a–5c were prepared as dipolarophiles through procedures in the literature [59,60].
With the precursors in our hand, initial attempts to optimize the ENAC reaction were performed by choosing hydrazonyl chloride 4a and enaminone 5a as starting materials. The changes in solvent, base, and temperature are summarized in Table 1.
Table 1.
Optimization of reaction conditions.
| Entry a | Solvent b | Base | T (°C) | t (h) | Yield (%) c | 6a:7a e |
|---|---|---|---|---|---|---|
| 1 | [mPy](OTf)/H2O | Et3N | rt | 2 | 50 | 90:10 |
| 2 | [mPy](OTf)/H2O | Et3N | rt | 24 | 51 | 80:20 |
| 3 | [mPy](OTf)/H2O | Et3N | 50 | 2 | 67 | 96:4 |
| 4 | [mPy](OTf)/H2O | Et3N | 85 | 2 | 32 | 95:5 |
| 5 | [mPy](OTf)/H2O | DBU | 50 | 2 | 42 | 96:4 |
| 6 | [mPy](OTf)/H2O | DMAP | 50 | 2 | 38 | 96:4 |
| 7 | [mPy](OTf)/H2O | NaOH | 50 | 2 | traces | - |
| 8 | [mPy](OTf)/H2O | K2CO3 | rt | 2 | 72 | 96:4 |
| 9 | [mPy](OTf)/H2O | K2CO3 | 50 | 2 | 85 | 96:4 |
| 10 d | [mPy](OTf)/H2O | K2CO3 | 50 | 2 | 90 | 97:3 |
| 11 d | [Bmim]Cl/H2O | K2CO3 | 50 | 2 | 75 | 95:5 |
| 12 d | [Bmim][BF4]/H2O | K2CO3 | 50 | 2 | 78 | 96:4 |
| 13 d | DMF | K2CO3 | 50 | 2 | 52 | 76:24 |
| 14 d | DMSO | K2CO3 | 50 | 2 | 64 | 82:18 |
a Reaction Conditions: enaminone 5a (1 eq), base (2.5 eq), hydrazonyl chloride 4a (1 eq) in 12 mL of solvent mixture. b IL/H2O 9:1 v:v; c isolated yield. d reaction conditions: enaminone 5a (1 eq), base (2.5 eq), hydrazonyl chloride 4a (1.3 eq) in 12 mL of solvent mixture. e regioisomeric ratio was determined by 1H NMR analysis of the crude reaction mixture (see Supplementary Materials).
Initially, the reaction was tested in [mPy]OTf/H2O 9:1 v/v at room temperature with Et3N as the base, observing a low yield, also after the extension of the reaction time to 24 h (Table 1, entries 1 and 2). A temperature rise to 50 °C allowed an increase in yield in just 2 h (Table 1, entry 3). Unfortunately, the prolonging of the reaction times favored the degradation of reactants. A further increase in temperature until 85 °C drastically reduced reaction yields due to the decomposition of reagents (Table 1, entry 4). Subsequently, a screening of different bases was evaluated in the same reaction conditions. In more detail, DBU and DMAP were used as organic bases with results less satisfactory than triethylamine (Table 1, entries 5 and 6). Then, we decided to employ inorganic bases and we observed only traces of the final product in the presence of NaOH (Table 1, entry 7), while K2CO3 furnished 6a in high yield both at room temperature and at 50 °C (Table 1, entries 8 and 9). The slight increase of hydrazonyl chloride (1.3 eq) favored an elevated yield (90%, Table 1, entry 10). Finally, the change of ionic liquid as solvent did not show any yield improvement (Table 1, entries 11–12). In this context, a particular consideration concerns the presence of water that supports the solubility both of inorganic base and [mPy]OTf, which tends to solidify at a temperature between 25 and 60 °C. In addition, the reaction was conducted in common organic solvents (i.e., CH2Cl2, THF, AcOEt, and ACN) without observing any product (data not reported). On the contrary, the employment of DMF or DMSO furnished the final product in moderate yields and good regioisomeric ratios (Table 1, entries 13–14).
It is remarkable that the best reaction conditions (Table 1, entry 10) also led to the highest regioisomeric ratio (6a/7a: 97/3).
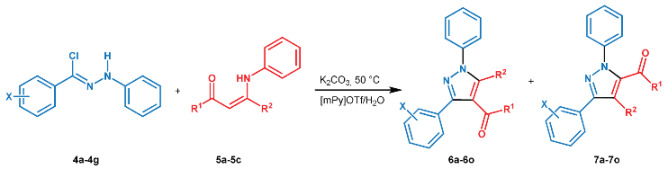
With the optimized reaction conditions in our hand, we extended the investigation on different enaminones 5a–5c and various hydrazonyl chlorides 4a–4g to synthesize a series of 1,3,4,5-tetrasubstituted pyrazoles 6a–6o (Table 2).
Table 2.
Synthesis of 1,3,4,5-tetrasubstituted pyrazoles by eliminative 1,3-dipolar cycloaddition.
| Entry a | Hydrazonyl Chloride |
X | Enaminone | R1 | R2 | Product | t (h) |
Yield 6a–6o (%) b |
6:7 c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4a | H | 5a | CH3 | CH3 | 6a | 2 | 90 | 97:3 |
| 2 | 4b | 4-OCH3 | 5a | CH3 | CH3 | 6b | 2 | 95 | 97:3 |
| 3 | 4c | 4-CH3 | 5a | CH3 | CH3 | 6c | 2 | 94 | 96:4 |
| 4 | 4d | 4-NO2 | 5a | CH3 | CH3 | 6d | 2 | 90 | 95:5 |
| 5 | 4e | 2-Cl | 5a | CH3 | CH3 | 6e | 2 | 92 | 96:4 |
| 6 | 4f | 3-Cl | 5a | CH3 | CH3 | 6f | 2 | 90 | 94:6 |
| 7 | 4g | 4-Cl | 5a | CH3 | CH3 | 6g | 2 | 92 | 98:2 |
| 8 | 4a | H | 5b | Ph | CH3 | 6h | 4 | 81 | 93:7 |
| 9 | 4b | 4-OCH3 | 5b | Ph | CH3 | 6i | 4 | 83 | 96:4 |
| 10 | 4c | 4-CH3 | 5b | Ph | CH3 | 6j | 4 | 82 | 96:4 |
| 11 | 4d | 4-NO2 | 5b | Ph | CH3 | 6k | 4 | 80 | 95:5 |
| 12 | 4e | 2-Cl | 5b | Ph | CH3 | 6l | 4 | 82 | 95:5 |
| 13 | 4f | 3-Cl | 5b | Ph | CH3 | 6m | 4 | 84 | 96:4 |
| 14 | 4g | 4-Cl | 5b | Ph | CH3 | 6n | 4 | 81 | 96:4 |
| 15 | 4a | H | 5c | Ph | Ph | 6o | 4 | 76 | 94:6 |
a Reaction conditions: enaminone 5a–5c (1 eq), K2CO3 (2.5 eq), hydrazonyl chloride 4a–4g (1.3 eq) in 12 mL of solvent mixture. b isolated yield. c regioisomeric ratio values were determined by 1H NMR analysis of the crude reaction mixture (see Supplementary Materials).
As you can see from the data collected in Table 2, the reactivity of the enaminones changed in function in the presence of aryl or alkyl groups as substituents. Especially, enaminone 5a was more reactive than 5b and 5c, probably due to steric hindrance effects that determine a longer reaction time and lower yields (Table 2, entries 8–15). On the contrary, the different nature of functional groups on the aromatic moiety of hydrazonyl chlorides 4a–4g did not seem to affect the reaction trend (Table 2, entries 2–7 and 9–14).
Finally, to our delight, a very high regioselectivity was confirmed for all reactions, as reported in Table 2.
3.2. Reaction Mechanism
At this point, we proposed a possible reaction mechanism, as illustrated in Scheme 3.
Scheme 3.

Proposed mechanism for regioselective synthesis of tetrasubstituted pyrazoles by eliminative nitrilimine-alkene 1,3-dipolar cycloaddition reaction.
Initially, the base produces both the enolate form (A) of the enaminone 5a and deprotonates the hydrazonyl chloride 4a, generating the nitrilimine C in situ [69]. The latter acts as a dipole with A through a regioselective 1,3-dipolar cycloaddition reaction. Generally, the accepted mechanism of 1,3-dipolar cycloaddition is of a concerted nature, providing for the alignment of the dipole and the dipolarophile on two parallel planes with a highly ordered system and a low degree of entropy. Regioselectivity can be explained through the frontier molecular orbital theory (FMO) and considering the inverse reactivity of cycloaddition [70,71]. Probably, the best orbital interaction is between the HOMO of the dipolarophile and the LUMO of the nitrilimine [72]. Furthermore, when the base deprotonates the enaminone, the HOMO energy tends to increase activating the dipolarophile and improving its reactivity. Moreover, the so-called ionic self-assembly (ISA) structure of ionic liquids due to noncovalent electrostatic interactions can give clusters that act as a support of dipole and dipolarophile, favoring their alignment and improving the orbital interaction [73,74]. Then, the trapped reagents lead to the formation of a pyrazoline D that is immediately transformed into the corresponding pyrazole 6a by elimination reaction with consequent loss of aniline. The heterocycle aromatization process corresponds to the fast stadium of the reaction because it was not possible to observe the pyrazoline intermedium D.
3.3. Recycling Ionic Liquid
The [mPy]OTf ionic liquid has been analyzed also with respect to recovery and reuse in the reaction of enaminone 5a with hydrazonyl chloride 4a and the results are shown in Figure 2.
Figure 2.

Recovery and reuse of [mPy]OTf until six cycles.
As shown in Figure 2, similar reaction yields were obtained, showing that the ionic liquid remains active until six cycles and that it can be recovered efficiently.
3.4. Pzs’ Effect on F1FO-ATPase
In previous studies, we investigated the F1FO-ATPase inhibition activity of triazole derivatives with interesting results. [75,76] Considering the nitrogen-based heterocyclic structure both of triazoles and pyrazoles, we decided to conduct a study at the mitochondrial level to evaluate whether the synthesized pyrazole compounds (Pzs) contribute to blocking the molecular events related to different forms of cell death.
To perform structure–activity relationship studies of fully-substituted pyrazoles on the molecular mechanism of F1FO-ATPase activity, uncoupled mitochondria obtained by freeze-thawing were used for kinetic analysis. We selected three pyrazole substrates (6a, 6h, and 6o) for the main difference related to the number of aryl groups linked to the azole core. Specifically, 6a has a two-aryl group, 6h has a three-aryl group, and 6o has a four-aryl group. The Pzs’ effect, in the range of 0.01–1.0 mM, was evaluated on the F1FO-ATPase activity. The substrate-depending inhibition potency of Pzs, estimated as IC50 values, was calculated as 0.25 ± 0.01 mM 6a, 1.62 ± 0.85 mM 6h, and 0.21 ± 0.15 mM 6o (Table 3). However, the maximal inhibition of 6a, 6h, and 6o on the ATP hydrolysis by F1FO-ATPase was 87%, 79%, and 43%, respectively (Figure 3). Thus, the 6a has the highest inhibiting power on F1FO-ATPase.
Table 3.
Kinetic parameters for the F1FO-ATPase inhibition by Pzs. IC50 and K’i values were graphically obtained from the Pzs’ titration curve and Cornish-Bowden plots of Figure 3 and Figure 4, respectively.
| Entry a | Compound | IC50 (mM) | K’i (mM) |
|---|---|---|---|
| 1 | 6a | 0.25 ± 0.01 | 0.21 ± 0.03 |
| 2 | 6h | 1.62 ± 0.85 | 1.27 ± 0.25 |
| 3 | 6o | 0.21 ± 0.15 | 1.86 ± 0.33 |
a Data are the mean ± SD of three independent experiments carried out on different mitochondrial preparations.
Figure 3.

Effect of pyrazole derivatives 6a (A), 6h (B), and 6o (C) on the activity of mitochondrial F1FO-ATPase. F-ATPase activity is plotted against concentrations of Pzs (logarithmic scale). Data represent the mean ± SD from three independent experiments carried out on different mitochondrial preparations.
To understand the mechanism of inhibition by the pyrazole derivatives 6a, 6h, and 6o of the F1FO-ATPase, a kinetic inhibition analysis has been set up to define the features of the enzyme-inhibitor complex formation in relation to the ATP substrate present or absence. The building of Dixon and Cornish-Bowden plots, which complement one another [67], have been performed to identify the type of inhibition of Pzs. We always obtained parallel straight lines in Dixon plots (Figure 4A,C,E), whereas in Cornish-Bowden plots (Figure 4B,D,F) we depicted two straight lines intersecting above the x-axis.
Figure 4.

Mitochondrial F1FO-ATPase inhibition by Pzs. Dixon (A,C,E) and Cornish-Bowden (B,D,E) plots were obtained at two fixed ATP concentrations and designing the experiments as detailed in the “Kinetic analysis” section of Materials and Methods. All points represent the mean ± SD (vertical bars) of three independent experiments carried out on different mitochondrial preparations.
The uncompetitive mechanism of inhibition with respect to the ATP substrate was irrespective of the nature of Pzs. Therefore, Pzs can bind the F1FO-ATPase only when the enzyme-substrate (ES) complex was already formed, to yield the tertiary enzyme-substrate-inhibitor (ESI) complex. In addition, the Cornish-Bowden plots showed that the dissociation constant of the ESI complex (K’i) of the F1FO-ATPase inhibited with 6a was approximately six and nine times lower than 6h and 6o, respectively (Table 3). The results highlighted that in the presence of 6a the formation of the ternary complex was easier and stronger than in the presence of other Pzs.
The kinetic parameters corroborate that the 6a has a higher inhibition efficiency on F1FO-ATPase than other Pz derivatives tested, as shown by the lower IC50 value for 6a (Table 3, Entry 1). However, we can assert that the Pz core sets the specific mechanism of inhibition with the enzyme, whereas specific functional groups, in particular aryl groups in Pz derivatives, modulate the binding affinity with the F1FO-ATPase.
3.5. Pzs Effect on mPTP
Mitochondrial calcium retention is linked to IMM integrity. The IMM remains intact until a high channel conductance is not formed. Increasing Ca2+ concentration in mitochondria stimulates mPTP formation. The capability of intact mitochondria to accumulate Ca2+ is identified as CRC (Ca2+ retention capacity) and mPTP opening was measured when the Ca2+ pulses accumulated in the mitochondrial matrix were released. The Pzs were tested by adding 10 μM Ca2+ at subsequent steps of 1 min to succinate-energized freshly prepared mitochondrial suspensions. The CRC decreased in Pzs-treated mitochondria, which was revealed by an increase in fluorescence intensity, pointing out the Pzs ability to desensitize the mPTP opening (Figure 5A). In control mitochondria, the CRC decrease was revealed after 180 sec upon a two-train Ca2+ pulse, as shown by the rise in the Fura-FF ratio ((Fura-FF high Ca2+)/(Fura-FF low Ca2+)). Accordingly, the increase in CRC upon subsequent 10 μM Ca2+ additions at 1 min intervals indicated that the mPTP formation was triggered at a higher threshold value of Ca2+ concentration in the matrix with Pzs. In detail, the two concentrations of the Pzs tested (0.1 mM and 1 mM) had different desensitizing powers on the mPTP. In detail, 6a had no concentration-dependent difference in mPTP inhibition. The same Ca2+ pulses were required to form the pore. At 0.1 mM, 6h and 6o had a smaller inhibitor effect on mPTP than 1 mM 6a. Conversely, 1 mM 6h blocked the mPTP opening as well as 6a. Moreover, 6h attained a higher CRC value (low Fura-FF ratio) than other Pzs on the mPTP opening with both concentrations tested. On these bases, a higher CRC value would mirror a smaller mPTP size. F1FO-ATPase and the adenine nucleotide translocator, which support the high and low ion conductance, respectively, depict the different nature of the mPTP phenomenon related to the pore size and this physiological event could be affected by 6h [77]. Otherwise, in terms of mPTP desensitization to Ca2+, the effect on CRC was inversely proportional to the concentration of the 6o. This compound showed a typical profile with a delayed rise in the Fura-FF ratio increase that indicated a CRC decreased more marked at 1 mM than at 0.1 mM (Figure 5A).
Figure 5.

Evaluation of mPTP opening. Representative curves (A) of the calcium retention capacity (CRC) expressed as Fura-FF ratio, monitored in response to subsequent 10 μM CaCl2 pulses (shown by the triangles), as detailed in “mPTP assay” section of Materials and Methods, in untreated mitochondria (control) or in the presence of 0.1 or 1.0 mM Pzs. (B) Quantification of the mPTP is expressed as the ratio of the number of calcium pulses required to induce the mPTP in inhibited (CRCi) and treated (CRCo) mitochondria with different concentrations of Pzs. Data represent the mean ± SD from three independent experiments carried out on different mitochondrial preparations. * indicates significant differences with respect to the control (p ≤ 0.05). ** indicates significant differences (p ≤ 0.05) among treatments within the same Pzs.
Consistently, the PTP formation extent expressed as the ratio between inhibited and uninhibited CRC (CRCi/CRCo) [78] was significantly reduced by all Pzs. Moreover, in the presence of both the 6a concentrations tested, a half CRCi/CRCo value was produced. On the contrary, 6h inhibited the mPTP by 20% and 50% with 0.1 mM and 1.0 mM, respectively. A high overload of mitochondrial Ca2+ was required to open the mPTP with 1 mM 6o, highlighting a more pronounced protective effect on mPTP formation compared with the other different conditions tested in the presence of other Pzs. Indeed, the CRCi/CRCo values were decreased by 25% and 65% with 0.1 mM and 1.0 mM 6o, respectively (Figure 5B).
4. Conclusions
In conclusion, we have developed a new synthetic method to obtain 1,3,4,5-tetrasubstituted pyrazoles with complete regioselectivity, starting from hydrazonyl chlorides and enaminones in a very simple reaction medium involving K2CO3 and [mPy]OTf/H2O 9:1 v/v at 50 °C. The reaction consists of a typical Huisgen’s 1,3-dipolar cycloaddition to form a specific pyrazoline intermedium that is rapidly transformed in correspondent pyrazole by the elimination of aniline. The experimental observations revealed the special effects exerted by the ionic liquid in the reaction, due to the particular capacity to stabilize the reaction intermediates, allowed the formation of a pyrazole core. The proposed process provides an easy procedure, low reaction times, complete regioselectivity with no formation of by-products, and very good versatility of final fully substituted Pzs. In addition, it is evident that the ionic liquid can be easily recovered and reused for a few cycles without losing its efficiency.
The main difference between pyrazole structures is related to the number of aryl groups linked to the azole core. Specifically, 6a has a two-aryl group, 6h has a three-aryl group, and 6o has a four-aryl group. Pzs at the concentration of 0.1 mM were non-toxic on the F1FO-ATPase activity. Otherwise, at the high concentrations tested of 1 mM, only the 6h and 6o did not cause a strong percentage of inhibition of the F1FO-ATPase activity. Importantly, we have identified the F1FO-ATPase as the target of Pzs. The molecular mechanism of compounds on the enzyme arises from the core structure of Pzs and the aryl groups can lessen the inhibitory power of compounds. It is noteworthy that the data of structure–activity relationship analysis performed on the F1FO-ATPase conceive the link with the inhibition of mPTP opening. Thus, as the aryl groups increase, the inhibitory effect on F1FO-ATPase decreases, while the desensitization effect of the pore increases and the effect increases in a concentration-dependent manner with 6h or 6o. Moreover, the results corroborate the arcane nature of mPTP that relies on the structure and catalysis of F1FO-ATPase [79,80,81,82].
To sum up, future studies will aim to evaluate the time of half-life in the biological systems and stability of these molecules [83] to understand if the non-toxic action on F1FO-ATPase at low concentrations could maintain a potent mPTP inhibition.
Acknowledgments
Danilo Matteuzzi and Roberto Giusti (Department of Veterinary Medical Sciences, University of Bologna) are gratefully acknowledged for kindly conferring swine hearts from a local abattoir to Biochemistry laboratories.
Abbreviations
mPTP, mitochondrial permeability transition pore; Δp, proton motive force; IMM, inner mitochondrial membranes; Pzs, pyrazoles.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pharmaceutics15020498/s1, Scheme S1: synthesis of variously substituted benzoyl phenylhydrazines 3a–3g. Scheme S2: synthesis of variously substituted hydrazonyl chlorides 4a–4g. Characterization spectra of precursors and final products.
Author Contributions
Conceptualization, V.A., A.D.N., L.M. and S.N.; methodology, V.A., C.A. and P.C.; validation, V.A., C.A., P.C., F.T. and S.N.; formal analysis, V.A., C.A., L.M. and G.F.; investigation, V.A., C.A., A.J., F.O. and M.A.T.; resources, P.C., A.D.N., L.M. and S.N.; data curation, V.A., C.A., P.C., A.J., F.O. and M.A.T.; writing—original draft preparation, V.A., C.A., A.D.N., L.M. and S.N.; writing—review and editing, V.A., A.D.N., L.M. and S.N.; supervision, A.D.N., L.M. and S.N.; funding acquisition, P.C., A.D.N., L.M. and S.N. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was financed by the University of Bologna, Italy, the RFO grant to SN, and an Alma Idea 2022 grant to SN. This research was also funded by the University of Calabria and Calabria Region (PAC CA-LABRIA 2014–2020-Asse Prioritario 12, Azione B 10.5.12 CUP: H28D19000040006). We thank the Italian Ministry of University and Research (MUR) for two doctoral grants.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Gomes P.M.O., Ouro P.M.S., Silva A.M.S., Silva V.L.M. Styrylpyrazoles: Properties, synthesis and transformations. Molecules. 2020;25:5886. doi: 10.3390/molecules25245886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar H., Bansal K.K., Goyal A. Synthetic methods and antimicrobial perspective of pyrazole derivatives: An insight. Anti-Infect. Agents. 2020;18:207–223. doi: 10.2174/2211352517666191022103831. [DOI] [Google Scholar]
- 3.Giornal F., Pazenok S., Rodefeld L., Lui N., Vors J.-P., Leroux F.R. Synthesis of diversely fluorinated pyrazoles as novel active agrochemical ingredients. J. Fluorine Chem. 2013;152:2–11. doi: 10.1016/j.jfluchem.2012.11.008. [DOI] [Google Scholar]
- 4.Judge N.R., Chacktas G., Ma L., Schink A., Buckpesch R., Schmutzler D., Machettira A.B., Dietrich H., Asmus E., Bierer D., et al. Flexible synthesis and herbicidal activity of fully substituted 3-hydroxypyrazoles. Eur. J. Org. Chem. 2021;2021:5677–5684. doi: 10.1002/ejoc.202100468. [DOI] [Google Scholar]
- 5.Catalan J., Fabero F., Claramunt R.M., Santa Maria M.D., Foces-Foces M.C., Cano F.H., Martinez-Ripoll M., Elguero J., Sastre R. New ultraviolet stabilizers: 3- and 5-(20 -hydroxyphenyl)pyrazoles. J. Am. Chem. Soc. 1992;114:5039–5048. doi: 10.1021/ja00039a014. [DOI] [Google Scholar]
- 6.Garcia H., Iborra S., Miranda M.A., Morera I.M., Primo J. Pyrazoles and isoxazoles derived from 2-hydroxyaryl phenylethynyl ketones: Synthesis and spectrophotometric evaluation of their potential applicability as sunscreens. Heterocycles. 1991;32:1745–1748. doi: 10.1002/chin.199217164. [DOI] [Google Scholar]
- 7.Busev A.I., Akimov V.K., Gusev S.I. Pyrazolone Derivatives as Analytical Reagents. Russ. Chem. Rev. 1965;34:237. doi: 10.1070/RC1965v034n03ABEH001426. [DOI] [Google Scholar]
- 8.Trofimenko S. Coordination chemistry of pyrazole-derived ligands. Chem. Rev. 1972;72:497–509. [Google Scholar]
- 9.Maeda H., Ito Y., Kusunose Y., Nakanishi T. Dipyrrolylpyrazoles: Anion receptors in protonated form and efficient building blocks for organized structures. Chem. Commun. 2007:1136–1138. doi: 10.1039/B615787D. [DOI] [PubMed] [Google Scholar]
- 10.Massah M., Balmeh N., Goodarzi K., Fard N.A. Molecular, docking analysis of H1 and H2 antihistamines groups with L-asparaginase II for reducing allergenicity; an in silico approach. Inform. Med. Unlocked. 2022;28:100865. doi: 10.1016/j.imu.2022.100865. [DOI] [Google Scholar]
- 11.Alam M.A. Antibacterial Pyrazoles: Tackling resistant bacteria. Future Med. Chem. 2022;14:343–362. doi: 10.4155/fmc-2021-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karrouchi K., Radi S., Ramli Y., Taoufik J., Mabkhot Y.N., Al-aizari F., Ansar M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules. 2018;23:134. doi: 10.3390/molecules23010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verma K., Lahariya A.K., Dubey S., Verma A.K., Das A., Schneider K.A., Bharti P.K. An integrated virtual screening and drug repurposing strategy for the discovery of new antimalarial drugs against plasmodium falciparum phosphatidylinositol 3-kinase. J. Cell. Biochem. 2021;122:1326–1336. doi: 10.1002/jcb.29954. [DOI] [PubMed] [Google Scholar]
- 14.Yang S.-T., Wang M., Galvin V., Yang Y., Arnsten A.F.T. Effects of blocking mGluR5 on primate dorsolateral prefrontal cortical neuronal filing and working memory performance. Psychopharmacology. 2021;238:97–106. doi: 10.1007/s00213-020-05661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennani F.E., Doudach L., Cherrah Y., Ramli Y., Karrouchi K., Ansar M., Faouzi M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorganic Chem. 2020;97:103470. doi: 10.1016/j.bioorg.2019.103470. [DOI] [PubMed] [Google Scholar]
- 16.Chuang H., Huang L.-C.S., Kapoor M., Liao Y.-J., Yang C.-L., Chang C.-C., Wu C.-Y., Hwu J.R., Huang T.-J., Hsu M.-H. Design and synthesis of pyridine-pyrazole-sulfonate derivatives as potential anti-HBV agents. Med. Chem. Commun. 2016;7:832–836. doi: 10.1039/C6MD00008H. [DOI] [Google Scholar]
- 17.Priya D., Gopinath P., Dhivya L.S., Vijaybabu A., Haritha M., Palaniappan S., Kathiravan M.K. Structural insights into pyrazoles as agents against anti-inflammatory and related disorders. Chem. Sel. 2022;7:e202104429. doi: 10.1002/slct.202104429. [DOI] [Google Scholar]
- 18.Wang Y.M., Teusink-Cross A., Elborai Y., Krupski M.C., Nelson A.S., Grimley M.S., Flannery A., Mehta P.A., Bleesing J.J., Chandra S., et al. Ruxolitinib for the treatment of chronic GVHD and overlap syndrome in children and young adults. Transplantation. 2022;106:412–419. doi: 10.1097/TP.0000000000003768. [DOI] [PubMed] [Google Scholar]
- 19.Niknam Z., Jafari A., Golchin A., Pouya F.D., Nemati M., Rezaei-Tavirani M., Rasmi Y. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 2022;27:6. doi: 10.1186/s40001-021-00626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dadiboyena S., Nefzi A. Synthesis of functionalized tetrasubstituted pyrazolyl heterocycles—A review. Eur. J. Med. Chem. 2011;46:5258–5275. doi: 10.1016/j.ejmech.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Naoum F., Kasiotis K.M., Magiatis P., Haroutounian S.A. Synthesis of novel nitro-substituted triaryl pyrazole derivatives as potential estrogen receptor ligands. Molecules. 2007;12:1259–1273. doi: 10.3390/12071259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng X., Mani N.S. Reaction of N-Monosubstituted hydrazones with nitroolefins: A novel regioselective pyrazole synthesis. Org. Lett. 2006;8:3505–3508. doi: 10.1021/ol061226v. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed M.S.M., Kobayashi K., Mori A. One-pot construction of pyrazoles and isoxazoles with palladium-catalyzed four-component coupling. Org. Lett. 2005;7:4487–4489. doi: 10.1002/chin.200606123. [DOI] [PubMed] [Google Scholar]
- 24.Bonini B.F., Franchini M.C., Gentili D., Locatelli E., Ricci A. 1,3-dipolar cycloaddition of nitrile imines with functionalized acetylenes: Regiocontrolled Sc(OTf)3-catalyzed synthesis of 4- and 5- substituted pyrazoles. Synlett. 2009;14:2328–2332. doi: 10.1002/chin.201003119. [DOI] [Google Scholar]
- 25.Conti P., Pinto A., Tamborini L., Rizzo V., De Micheli C. A regioselective route to 5-substituted pyrazole- and pyrazoline-3-phosphonic acids and esters. Tetrahedron. 2007;63:5554–5560. doi: 10.1016/j.tet.2007.04.027. [DOI] [Google Scholar]
- 26.Maiuolo L., Algieri V., Russo B., Tallarida M.A., Nardi M., Di Gioia M.L., Merchant Z., Merino P., Delso I., De Nino A. Synthesis, Biological and In Silico Evaluation of Pure Nucleobase-Containing Spiro (Indane-Isoxazolidine) Derivatives as Potential Inhibitors of MDM2-p53 Interaction. Molecules. 2019;24:2909. doi: 10.3390/molecules24162909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiuolo L., Feriotto G., Algieri V., Nardi M., Russo B., Di Gioia M.L., Furia E., Tallarida M.A., Mischiati C., De Nino A. Antiproliferative Activity of Novel Isatinyl/Indanyl Nitrones (INs) as Potential Spin Trapping of Free Radical Intermediates. MedChemComm. 2018;9:299–304. doi: 10.1039/C7MD00537G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiuolo L., De Nino A., Algieri V., Nardi M. Microwave-Assisted 1,3-Dipolar Cyclo-addition: Recent Advances In Synthesis of Isoxazolidines. Mini-Rev. Org. Chem. 2017;14:136–142. doi: 10.2174/1570193X14666170206123513. [DOI] [Google Scholar]
- 29.Costanzo P., Calandruccio C., Di Gioia M.L., Nardi M., Oliverio M., Procopio A. First multicomponent reaction exploiting glycerol carbonate synthesis. J. Clean. Prod. 2018;202:504–509. doi: 10.1016/j.jclepro.2018.08.120. [DOI] [Google Scholar]
- 30.De Nino A., Merino P., Algieri V., Nardi M., Di Gioia M.L., Russo B., Tallarida M.A., Maiuolo L. Synthesis of 1,5-functionalized 1,2,3-triazoles using ionic liquid/iron(III) chloride as an efficient and reusable homogeneous catalyst. Catalysts. 2018;8:364–376. doi: 10.3390/catal8090364. [DOI] [Google Scholar]
- 31.De Nino A., MAiuolo L., Costanzo P., Algieri V., Jiritano A., Olivito F., Tallarida M.A. Recent progress in catalytic synthesis of 1,2,3-triazoles. Catalysts. 2021;11:1120. doi: 10.3390/catal11091120. [DOI] [Google Scholar]
- 32.Shi C., Ma C., Ma H., Zhou X., Cao J., Fan Y., Huang G. Copper-catalyzed synthesis of 1,3,4-trisubstituted and 1,3,4,5-tetrasubstituted pyrazoles via [3+2] cycloadditions of hydrazones and nitroolefins. Tetrahedron. 2016;72:4055–4058. doi: 10.1016/j.tet.2016.05.034. [DOI] [Google Scholar]
- 33.Molteni G., Ponti A. The nitrilimine-alkene cycloaddition regioselectivity rationalized by density functional theory reactivity indices. Molecules. 2017;22:202–213. doi: 10.3390/molecules22020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowalczyk A., Utecht-Jarzyńska G., Mlostoń G., Jasiński M. Trifluoromethylated Pyrazoles via Sequential (3 + 2)-Cycloaddition of Fluorinated Nitrile Imines with Chalcones and Solvent-Dependent Deacylative Oxidation Reactions. Org. Lett. 2022;24:2499–2503. doi: 10.1021/acs.orglett.2c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y., Gao C.-F., Ma H., Nie J., Ma J.-A., Zhang F.-G. Quadruple Functionalized Pyrazole Pharmacophores by One-pot Regioselective [3+2] Cycloaddition of Fluorinated Nitrile Imines and Dicyanoalkenes. Chem. Asian J. 2022;17:e202200436. doi: 10.1002/asia.202200436. [DOI] [PubMed] [Google Scholar]
- 36.Tian Y.-C., Li J.-K., Zhang F.-G., Ma J.-A. Regioselective Decarboxylative Cycloaddition Route to Fully Substituted 3-CF3-Pyrazoles from Nitrilimines and Isoxazolidinediones. Adv. Synth. Catal. 2021;363:2093–2097. doi: 10.1002/adsc.202100091. [DOI] [Google Scholar]
- 37.Ghasempour L., Asghari S., Tajbakhsh M., Mohseni M. Preparation of New Spiropyrazole, Pyrazole and Hydantoin Derivatives and Investigation of Their Antioxidant and Antibacterial Activities. Chem. Biodivers. 2021;18:e21001. doi: 10.1002/cbdv.202100197. [DOI] [PubMed] [Google Scholar]
- 38.Yavari I., Taheri Z., Naeimabadi M., Bahemmat S., Halvag M.R. A Convenient Synthesis of Tetrasubstituted Pyrazoles from Nitrile Imines and 2-(Thioxothiazolidin-5-ylidene)acetates. Synlett. 2018;29:918–921. doi: 10.1055/s-0036-1591921. [DOI] [Google Scholar]
- 39.Filkina M.E., Baray D.N., Beloglazkina E.K., Grishin Y.K., Roznyatovsky V.A., Kukushkin M.E. Regioselective Cycloaddition of Nitrile Imines to 5-Methylidene-3-phenyl-hydantoin: Synthesis and DFT Calculations. Int. J. Mol. Sci. 2023;24:1289–1307. doi: 10.3390/ijms24021289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Nino A., Algieri V., Tallarida M.A., Costanzo P., Pedròn M., Tejero T., Merino P., Maiuolo L. Regioselective synthesis of 1,4,5,-trisubstituted-1,2,3-triazoles from ary azides and enaminones. Eur. J. Org. Chem. 2019;2019:5725–5731. doi: 10.1002/ejoc.201900889. [DOI] [Google Scholar]
- 41.Duan L., Zhou H., Gu Y., Gong P., Qin M. The use of enaminones and enamines as effective synthons for MSA-catalyzed regioselective synthesis of 1,3,4-tri- and 1,3,4,5-tetrasubstituted pyrazoles. New. J. Chem. 2019;43:16131–16137. doi: 10.1039/C9NJ03701B. [DOI] [Google Scholar]
- 42.Maiuolo L., Algieri V., Olivito F., De Nino A. Recent developments on 1,3-dipolar cycloaddition reactions by catalysis in green solvents. Catalysts. 2020;10:65–91. doi: 10.3390/catal10010065. [DOI] [Google Scholar]
- 43.Maiuolo L., Russo B., Algieri V., Nardi M., Di Gioia M.L., Tallarida M.A., De Nino A. Regioselective synthesis of 1,5-disubstituted 1,2,3-triazoles by 1,3-dipolar cycloaddition: Role of Er(OTf)3, ionic liquid and water. Tetrahedron Lett. 2019;60:672–674. doi: 10.1016/j.tetlet.2019.01.053. [DOI] [Google Scholar]
- 44.De Nino A., Maiuolo L., Merino P., Nardi M., Procopio A., Roca-López D., Russo B., Algieri V. Efficient Organocatalyst Supported on a Simple Ionic Liquid as a Recoverable System for the Asymmetric Diels-Alder Reaction in the Presence of Water. ChemCatChem. 2015;7:830–835. doi: 10.1002/cctc.201402973. [DOI] [Google Scholar]
- 45.Nesci S., Pagliarani A. Incoming News on the F-Type ATPase Structure and Functions in Mammalian Mitochondria. BBA Advances. 2021;1:100001–100003. doi: 10.1016/j.bbadva.2020.100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mnatsakanyan N., Llaguno M.C., Yang Y., Yan Y., Weber J., Sigworth F.J., Jonas E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019;10:5823–5833. doi: 10.1038/s41467-019-13766-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urbani A., Giorgio V., Carrer A., Franchin C., Arrigoni G., Jiko C., Abe K., Maeda S., Shinzawa-Itoh K., Bogers J.F.M., et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019;10:4341–4351. doi: 10.1038/s41467-019-12331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nesci S., Pagliarani A., Algieri C., Trombetti F. Mitochondrial F-Type ATP Synthase: Multiple Enzyme Functions Revealed by the Membrane-Embedded FO Structure. Crit. Rev. Biochem. Mol. Biol. 2020;55:309–321. doi: 10.1080/10409238.2020.1784084. [DOI] [PubMed] [Google Scholar]
- 49.Bonora M., Giorgi C., Pinton P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2021;23:266–285. doi: 10.1038/s41580-021-00433-y. [DOI] [PubMed] [Google Scholar]
- 50.Bernardi P. Looking Back to the Future of Mitochondrial Research. Front. Physiol. 2021;12:682467–682475. doi: 10.3389/fphys.2021.682467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izzo V., Bravo-San Pedro J.M., Sica V., Kroemer G., Galluzzi L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016;26:655–667. doi: 10.1016/j.tcb.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 52.Bernardi P., Di Lisa F., Fogolari F., Lippe G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ. Res. 2015;116:1850–1862. doi: 10.1161/CIRCRESAHA.115.306557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nesci S. The Mitochondrial Permeability Transition Pore in Cell Death: A Promising Drug Binding Bioarchitecture. Med. Res. Rev. 2020;40:811–817. doi: 10.1002/med.21635. [DOI] [PubMed] [Google Scholar]
- 54.Nesci S., Trombetti F., Algieri C., Pagliarani A. A Therapeutic Role for the F1FO-ATP Synthase. SLAS Discov. 2019;24:893–903. doi: 10.1177/2472555219860448. [DOI] [PubMed] [Google Scholar]
- 55.Pagliarani A., Nesci S., Ventrella V. Novel Drugs Targeting the C-Ring of the F1FO-ATP Synthase. Mini Rev. Med. Chem. 2016;16:815–824. doi: 10.2174/1389557516666160211120955. [DOI] [PubMed] [Google Scholar]
- 56.Bernardi P., Rasola A., Forte M., Lippe G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallagher N., Zangh H., Junghoefer T., Giangrisostomi E., Ovsyannikov R., Pink M., Rajca S., Casu M.B., Rajca A. Termally and magnetically robust triplet ground state diradical. J. Am. Chem. Soc. 2019;141:4764–4774. doi: 10.1021/jacs.9b00558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang C.-Y., Liu X.-H., Wang B.-L., Wang S.-H., Li Z.-M. Synthesis and antifungal activities of new pyrazole derivatives via 1,3-dipolar cycloaddition reaction. Chem. Biol. Drug. Des. 2010;75:489–493. doi: 10.1111/j.1747-0285.2010.00948.x. [DOI] [PubMed] [Google Scholar]
- 59.Dalpozzo R., De Nino A., Nardi M., Russo B., Procopio A. Erbium(III) Triflate: A Valuable Catalyst for the Synthesis of Aldimines, Ketimines, and Enaminones. Synthesis. 2006;7:1127–1132. doi: 10.1055/s-2006-926378. [DOI] [Google Scholar]
- 60.Xie C., Feng L., Li W., Ma X., Ma X., Liu Y., Ma C. Efficient synthesis of pyrrolo[1,2-a]quinoxalines catalyzed by a bronsted acid through cleavage of C-C bonds. Org. Biomol. Chem. 2016;14:8529–8535. doi: 10.1039/C6OB01401A. [DOI] [PubMed] [Google Scholar]
- 61.Liu K., Shang X., Cheng Y., Chang X., Li P., Li W. Regioselective [3+2]-annulation of hydrazonyl chlorides with 1,3-dicarbonyl compounds for assembling of polysubstituted pyrazoles. Org. Biomol. Chem. 2018;16:7811–7814. doi: 10.1039/C8OB02352B. [DOI] [PubMed] [Google Scholar]
- 62.Nesci S., Ventrella V., Trombetti F., Pirini M., Pagliarani A. The mitochondrial F1FO-ATPase desensitization to oligomycin by tributyltin is due to thiol oxidation. Biochimie. 2014;97:128–137. doi: 10.1016/j.biochi.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 63.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 64.Ventrella V., Nesci S., Trombetti F., Bandiera P., Pirini M., Borgatti A.R., Pagliarani A. Tributyltin inhibits the oligomycin-sensitive Mg-ATPase activity in Mytilus galloprovincialis digestive gland mitochondria. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2011;153:75–81. doi: 10.1016/j.cbpc.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 65.Algieri C., Trombetti F., Pagliarani A., Ventrella V., Bernardini C., Fabbri M., Forni M., Nesci S. Mitochondrial Ca2+ -activated F1FO -ATPase hydrolyzes ATP and promotes the permeability transition pore. Ann. N. Y. Acad. Sci. 2019;1457:142–157. doi: 10.1111/nyas.14218. [DOI] [PubMed] [Google Scholar]
- 66.Fiske C.H., Subbarow Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925;66:375–400. doi: 10.1016/S0021-9258(18)84756-1. [DOI] [Google Scholar]
- 67.Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974;137:143–144. doi: 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nesci S., Algieri C., Trombetti F., Ventrella V., Fabbri M., Pagliarani A. Sulfide affects the mitochondrial respiration, the Ca2+-activated F1FO-ATPase activity and the permeability transition pore but does not change the Mg2+-activated F1FO-ATPase activity in swine heart mitochondria. Pharmacol. Res. 2021;166:105495–105504. doi: 10.1016/j.phrs.2021.105495. [DOI] [PubMed] [Google Scholar]
- 69.Liu H., Jia H., Wang B., Xiao Y., Guo H. Synthesis of spirobidihydropyrazole through double 1,3-dipolar cycloaddition of nitrilimines with allenoates. Org. Lett. 2017;19:4714–4717. doi: 10.1021/acs.orglett.7b01961. [DOI] [PubMed] [Google Scholar]
- 70.Jamieson C., Livingstone K. The Nitrile Imine 1,3-Dipole–Properties, Reactivity and Applications. Springer; Berlin, Germany: 2020. pp. 1–152. [Google Scholar]
- 71.Bakavoli M., Moeinpour F., Davoodnia A., Morsali A. 1,3-dipolar cycloaddition of N-[4-nitrophenyl]-C-[2-furyl] nitrilimine with some dipolarophiles: A combined experimental and theoretical study. J. Mol. Struct. 2010;969:139–144. doi: 10.1016/j.molstruc.2010.01.059. [DOI] [Google Scholar]
- 72.Gaspar F.V., Azevedo M.F.M.F., Carneiro L.S.A., Ribeiro S.B., Esteves P.M., Buarque C.D. 1,3-Dipolar cycloaddition reactions of enaminones and azides: Synthesis of 4-acyl-1,2,3-triazoles and mechanistic studies. Tetrahedron. 2022;120:132856–132866. doi: 10.1016/j.tet.2022.132856. [DOI] [Google Scholar]
- 73.Goossens K., Lava K., Bielawski C.W., Binnemans K. Ionic liquid crystals: Versatile materials. Chem. Rev. 2016;116:4643–4807. doi: 10.1021/cr400334b. [DOI] [PubMed] [Google Scholar]
- 74.Bini R., Chiappe C., Mestre V.L., Pomelli C.S., Welton T. A theoretical study of the solvent effect on Diels-Alder reaction in room temperature ionic liquids using a supramolecular approach. Theor. Chem. Acc. 2009;123:347–352. doi: 10.1007/s00214-009-0525-0. [DOI] [Google Scholar]
- 75.Algieri V., Algieri C., Maiuolo L., De Nino A., Pagliarani A., Tallarida M.A., Trombetti F., Nesci S. 1,5-Disubstituted-1,2,3-triazoles as inhibitors of the mitochondrial Ca2+-activated F1Fo-ATP(hydrol)ase and the permeability transition pore. Ann. N. Y. Acad. Sci. 2021;1485:43–85. doi: 10.1111/nyas.14474. [DOI] [PubMed] [Google Scholar]
- 76.Algieri C., Bernardini C., Marchi S., Forte M., Tallarida M.A., Bianchi F., La Mantia D., Algieri V., Stanzione R., Cotugno M., et al. 1,5-disubstituted-1,2,3-triazoles counteract mitochondrial dysfunction acting on F1FO-ATPase in models of cardiovascular diseases. Pharmacol. Res. 2023;187:106561. doi: 10.1016/j.phrs.2022.106561. [DOI] [PubMed] [Google Scholar]
- 77.Neginskaya M.A., Solesio M.E., Berezhnaya E.V., Amodeo G.F., Mnatsakanyan N., Jonas E.A., Pavlov E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019;26:11–17. doi: 10.1016/j.celrep.2018.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Algieri C., Trombetti F., Pagliarani A., Ventrella V., Nesci S. Phenylglyoxal Inhibition of the Mitochondrial F1FO-ATPase Activated by Mg2+ or by Ca2+ Provides Clues on the Mitochondrial Permeability Transition Pore. Arch. Biochem. Bioph. 2020;681:108258–108264. doi: 10.1016/j.abb.2020.108258. [DOI] [PubMed] [Google Scholar]
- 79.Nesci S. Protein Folding and Unfolding: Proline Cis-Trans Isomerization at the c Subunits of F1 FO -ATPase Might Open a High Conductance Ion Channel. Proteins. 2022;90:2001–2005. doi: 10.1002/prot.26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nesci S. What Happens When the Mitochondrial H+-Translocating F1FO-ATP(Hydrol)Ase Becomes a Molecular Target of Calcium? The Pore Opens. Biochimie. 2022;198:92–95. doi: 10.1016/j.biochi.2022.03.012. [DOI] [PubMed] [Google Scholar]
- 81.Mnatsakanyan N., Park H.-A., Wu J., He X., Llaguno M.C., Latta M., Miranda P., Murtishi B., Graham M., Weber J., et al. Mitochondrial ATP Synthase C-Subunit Leak Channel Triggers Cell Death upon Loss of Its F1 Subcomplex. Cell Death Differ. 2022;29:1874–1887. doi: 10.1038/s41418-022-00972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bernardi P., Carraro M., Lippe G. The Mitochondrial Permeability Transition: Recent Progress and Open Questions. FEBS J. 2021;289:7051–7074. doi: 10.1111/febs.16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Šileikytė J., Devereaux J., de Jong J., Schiavone M., Jones K., Nilsen A., Bernardi P., Forte M., Cohen M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. Chem. Med. Chem. 2019;14:1771–1782. doi: 10.1002/cmdc.201900376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.