

Abstract
Objectives
Ruxolitinib, a Janus kinase (JAK) 1/2 inhibitor, demonstrates efficacy for treating steroid‐resistant acute graft‐versus‐host disease (SR‐aGVHD) following allogeneic stem cell transplantation (allo‐HSCT). Myeloid‐derived suppressor cells (MDSCs) have a protective effect on aGVHD via suppressing T cell function. However, the precise features and mechanism of JAK inhibitor‐mediated immune modulation on MDSCs subsets remain poorly understood.
Methods
A total of 74 SR‐aGVHD patients treated with allo‐HSCT and ruxolitinib were enrolled in the present study. The alterations of MDSC and regulatory T cell (Treg) populations were monitored during ruxolitinib treatment in responders and nonresponders. A mouse model of aGVHD was used to evaluate the immunosuppressive activity of MDSCs and related signalling pathways in response to ruxolitinib administration in vivo and in vitro.
Results
Patients with SR‐aGVHD who received ruxolitinib treatment achieved satisfactory outcomes. Elevation proportions of MDSCs before treatment, especially polymorphonuclear‐MDSCs (PMN‐MDSCs) were better to reflect the response to ruxolitinib than those in Tregs. In the mouse model of aGVHD, the administration of ruxolitinib resulted in the expansion and functional enhancement of PMN‐MDSCs and the effects could be partially reversed by an anti‐Gr‐1 antibody in vivo. Ruxolitinib treatment significantly elevated the suppressive function of PMN‐MDSCs through reactive oxygen species (ROS) production by Nox2 upregulation as well as bypassing the activated MAPK/NF‐κB signalling pathway. Additionally, ex vivo experiments demonstrated that ruxolitinib prevented the differentiation of mature myeloid cells and promoted the accumulation of MDSCs by inhibiting STAT5.
Conclusions
Ruxolitinib enhances PMN‐MDSCs functions through JAK/STAT and ROS‐MAPK/NF‐κB signalling pathways. Monitoring frequencies and functions of MDSCs can help evaluate treatment responses to ruxolitinib.
Keywords: acute graft‐versus‐host disease, JAK/STAT pathway, myeloid‐derived suppressor cells, ROS
Our results provide rationale for molecular modification of myeloid‐derived suppressor cells to augment the efficacy of ruxolitinib in patients with acute graft‐versus‐host disease, which may be applicable to other pathologies including autoimmunity, chronic inflammation and cancer.
Introduction
Allogeneic haematopoietic stem cell transplantation (allo‐HSCT) is a well‐established and effective therapeutic intervention for haematopoietic malignant and nonmalignant diseases. However, allo‐HSCT is associated with severe complications, including acute graft‐versus‐host disease (aGVHD). 1 , 2 aGVHD involves aberrant activation of donor‐derived naïve T cells, which recognise host antigens and subsequently attack target organs, including the skin, liver and gastrointestinal tract (GI). 3 Although substantial progress has been made in the development of prophylactic and immunosuppressive therapies for aGVHD in recent decades, 30–50% of patients undergoing allo‐HSCT develop aGVHD, which is associated with poor prognosis. 4 Further, approximately half of the patients treated with glucocorticoid as first‐line therapy for aGVHD develop steroid‐refractory acute GVHD (SR‐aGVHD), which accounts for significant mortality. 5 Ruxolitinib, a Janus kinase (JAK) 1/2 inhibitor, was recently approved for treating SR‐aGVHD as it can impair the differentiation of CD4+ T cells into IFN‐γ‐ and IL‐17A‐producing cells and increase the proportion of Foxp3+ regulatory T cells (Tregs). 6 , 7 However, the timing of ruxolitinib therapy standard indicator for evaluating treatment response to ruxolitinib is still largely unknown.
Myeloid‐derived suppressor cells (MDSCs) are a heterogeneous population of immunosuppressive myeloid cells that play a beneficial role in transplantation by suppressing alloreactive T cell responses, thereby exerting protective effects against the development of typical aGVHD and reducing transplantation‐related deaths. 8 , 9 MDSCs are defined as CD11b+Gr‐1+ and Lin−CD11b+HLA‐DR− cells in mice and humans, respectively. MDSCs comprise two subsets characterised by distinct morphological features: polymorphonuclear (PMN) and monocytic (M)‐MDSCs. 10 PMN‐MDSCs express high levels of reactive oxygen species (ROS) owing to the activity of nicotinamide adenine dinucleotide phosphate (NADPH), whereas M‐MDSCs contain nitric oxide (NO) and arginase‐1 (Arg‐1) because of the upregulation of signal transducer and activator of transcription 1 (STAT1) and inducible NO synthase (NOS). Both subsets exert potent suppressive effects on T cells and induce immunological tolerance following transplantation. 11 , 12
Myeloid‐derived suppressor cells are induced by various inflammatory factors, including granulocyte/macrophage colony‐stimulating factor (GM‐CSF), granulocyte CSF, vascular endothelial growth factor, interleukin‐6 (IL‐6), interferon‐γ (IFN‐γ), tumor necrosis factor‐α (TNF‐α), IL‐1β and toll‐like receptor ligands. 13 Most of these factors trigger signalling pathways in MDSCs that converge on JAK protein family members and STAT3/5, which are involved in the expansion and differentiation of MDSCs in cancer. 14 Ablation of STAT3 expression using conditional knockout mice or selective STAT3 inhibitors markedly reduces the expansion of MDSCs and increases T cell responses in tumor‐bearing mice. 15 Furthermore, persistent activation of STAT3 blocks the differentiation of myeloid progenitor cells by upregulating S100A8/9 and increases their survival by inducing the expression of B‐cell lymphoma xl (Bcl‐xl) and cyclin D1 in the tumor microenvironment (TME). 16 , 17 , 18 Meanwhile, STAT5 also contributes to the proliferation and survival of immature myeloid cells and prevents their differentiation into mature cells. 19 However, the role of JAK/STAT signalling of MDSCs in the pathogenesis of aGVHD has not been documented.
In this study, we evaluated the biological functions of MDSCs on therapeutic process of JAK1/2 inhibition interfering with aGVHD after allo‐HSCT. The administration of ruxolitinib as second‐line treatment of aGVHD may enhance the immunosuppressive function of MDSCs and block their differentiation into mature cells. PMN‐MDSCs were found to increase the levels of ROS in a MAPK/NF‐κB‐dependent manner. Considering the lack of standard indicators for evaluating treatment responses in patients with aGVHD, 20 we demonstrated that MDSC status can predict treatment response to ruxolitinib therapy in patients with SR‐aGVHD. These results provide further evidence that MDSCs are the therapeutic targets of ruxolitinib and the evaluation of MDSC population and function may have utility in patient selection for developing individualised treatment strategies.
Results
Outcomes following ruxolitinib treatment in patients with SR‐aGVHD
We enrolled 74 patients who received ruxolitinib as second‐line therapy for SR‐aGVHD. Patient characteristics are summarised in Supplementary table 1. The median age of patients was 35 years (range, 8–63 years), and 48.6% of them were men. Of these, 7 patients (9.5%) had grade II aGVHD, 33 (44.6%) had grade III aGVHD, and 34 (45.9%) had grade IV aGVHD. The overall response rate (ORR) at day 28 was 68.9% (51/74), with 29.7% of patients achieving CR after a median of 8 days (range, 1–40 days). The proportion of patients with durable ORR at day 56 was 63.5% (47/74), with 47.3% of patients achieving CR (35/74; Figure 1a). Over a median follow‐up period of 298 (7–1495) days, there was a significantly increased 6‐month OS in aGVHD patients with responders (68.6% ± 6.5%) compared with nonresponders (21.7% ± 8.6%; P < 0.001, Figure 1b).
Figure 1.
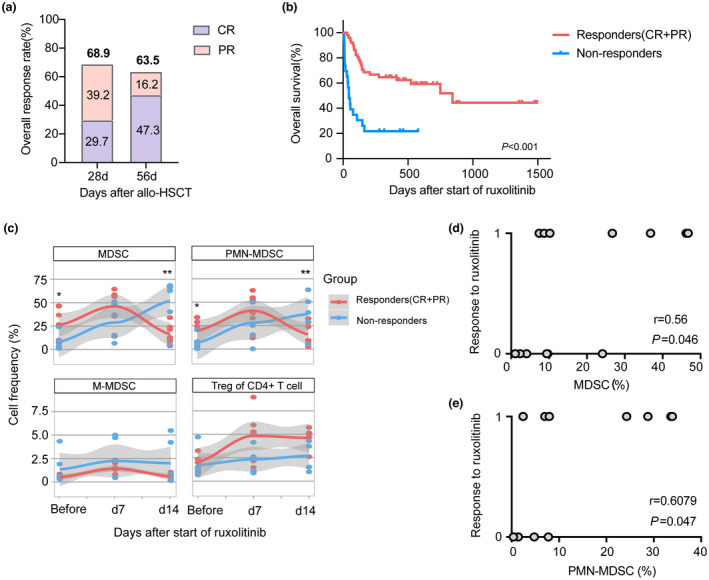
Ruxolitinib treatment improved the overall response of patients with SR‐aGVHD after allo‐HSCT along with alterations of myeloid‐derived suppressor cells (MDSCs). (a) The overall response rate (ORR) at day 28 and day 56 in patients with SR‐aGVHD who received ruxolitinib treatment after allo‐HSCT (n = 74). (b) The overall survival of patients with ruxolitinib for aGVHD according to the response of ruxolitinib (Responders who obtained PR or above, n = 51; Nonresponders who failed treatment, n = 23). (c) The percentages of MDSCs (CD11b+HLA‐DR−), PMN‐MDSCs (CD11b+HLA‐DR−CD14−CD15+), M‐MDSCs (CD11b+HLA‐DR−CD14+CD15−), Tregs (CD4+CD25+Foxp3+) from PB samples in responders (n = 7) and nonreponders (n = 6) before and within 7 days, 14 days after starting ruxolitinib. (d, e) Correlation analysis of the MDSCs and PMN‐MDSCs proportions before ruxolitinib administration compared with response to ruxolitinib treatment. Each dot represents an independent core. The response to ruxolitinib included response (CR + PR) and nonresponse (0 = nonresponse, 1 = response, respectively). r and P‐values were calculated using Spearman's correlation test. *P < 0.05, **P < 0.01.
Cytopenias are common complications of ruxolitinib treatment. The incidence of grade III or IV anaemia, neutropenia and thrombocytopenia was 17.6% (13/74), 17.6% (13/74) and 14.9% (11/74), respectively. Bacterial, viral and fungal infections were common in patients with SR‐aGVHD receiving ruxolitinib treatment and included cytomegalovirus (CMV) viremia (58.1%), Epstein–Barr viremia (4.05%), fungal infections (5%) and bacterial/viral infections (10.8%). These findings highlight the importance of infectious surveillance in patients receiving ruxolitinib treatment (Supplementary table 2). Relapse of the underlying malignancy occurred in 18.9% (14/74) of patients during the follow‐up period.
The alterations of MDSCs, especially PMN‐MDSCs, were favorable indicators to evaluate the treatment response to ruxolitinib in aGVHD patients
Ruxolitinib is a promising treatment option for SR‐aGVHD and can achieve favorable ORR and OS without serious side effects. However, there are no consensual indicators to reflect treatment responses. To evaluate the role of circulating immunoregulatory cells obtained from PB samples, alterations in the levels of MDSCs and Tregs were monitored in patients with aGVHD before and after ruxolitinib treatment, including 7 responders and 6 nonresponders. As shown in Figure 1c, the number of MDSCs (CD11b+HLA‐DR− cells) significantly increased at day 7 and declined at day 14 in responders; however, the numbers were constantly increasing in nonresponders after ruxolitinib treatment. More importantly, we assessed two major MDSC subpopulations: CD11b+HLA‐DR− CD15+CD14− PMN‐MDSCs and CD11b+HLA‐DR− CD15−CD14+ M‐MDSCs. There was a similar tendency in PMN‐MDSCs. However, M‐MDSCs were not related to the treatment response of ruxolitinib. Patients who displayed a significant response to treatment had statistically higher frequencies of MDSCs and PMN‐MDSCs before the administration of ruxolitinib than patients who showed treatment failure (P < 0.05). Correlation analysis showed a significant positive correlation between the proportions of MDSCs and PMN‐MDSCs before ruxolitinib treatment and treatment response (P = 0.046 and 0.047, respectively, Figure 1d and e). A recent study suggested that Tregs play a critical role in allo‐HSCT, which was closely associated with the severity of aGVHD. 21 We further analysed the percentage of Tregs during ruxolitinib treatment. There was a higher frequency of Tregs in responders than in nonresponders within day 7 and day 14 after starting ruxolitinib treatment; however, this difference was not significant (Figure 1c). These data indicate that alterations in circulating MDSCs, especially PMN‐MDSCs, were more sensitive than those in Tregs and may predict and reflect the response of patients with SR‐aGVHD to ruxolitinib. Nevertheless, we need large‐scale cohort studies to verify this conclusion.
Ruxolitinib treatment alleviated aGVHD symptoms and induced the expansion of MDSCs, particularly PMN‐MDSCs
To determine the effects of ruxolitinib on established parameters of aGVHD, we employed a major histocompatibility complex (MHC)‐mismatched (C57BL/6 to BALB/c) murine model of aGVHD. Ruxolitinib or the vehicle was administered by oral gavage from day 0 to day 30 (Figure 2a). Ruxolitinib treatment resulted in significant improvements in survival as well as increased the body weight and clinical scores of mice after transplantation compared with the vehicle (Figure 2b–d). Mice that received BM only developed no signs of aGVHD and had a survival rate of 100% until the endpoint. Histological analysis of liver, intestine and colon tissues from day 7 revealed decreased pathological damage and inflammatory cell infiltration in mice treated with ruxolitinib (Figure 2e). No apparent cytopenias were observed upon blood routine test in ruxolitinib‐treated mice (Supplementary figure 1a–d).
Figure 2.
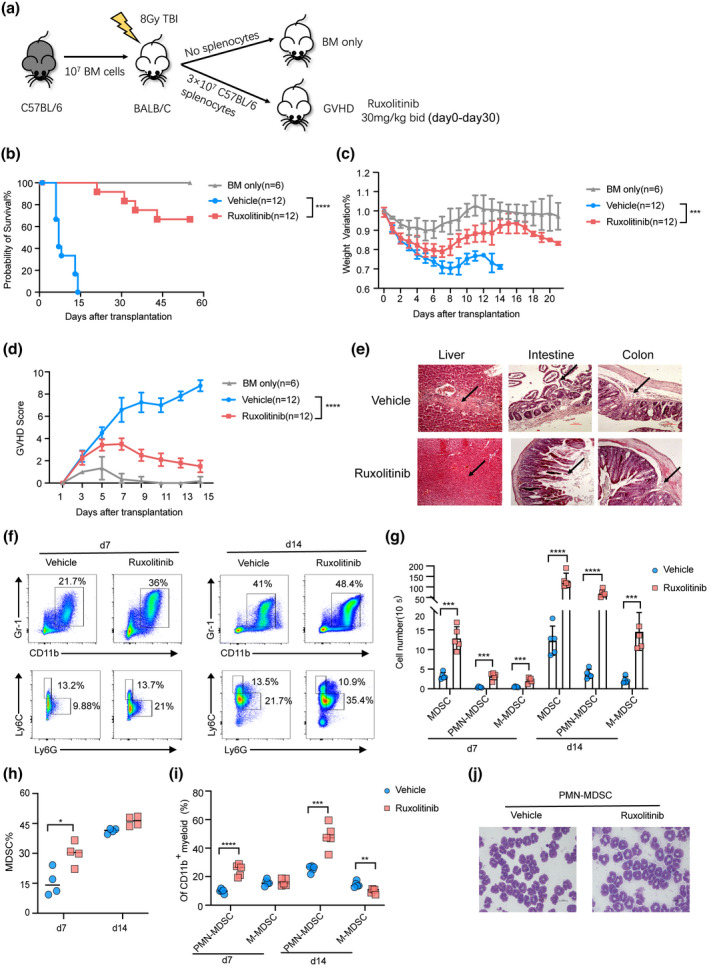
Ruxolitinib treatment reduced aGVHD severity and promoted myeloid‐derived suppressor cells (MDSCs) expansion. Lethally irradiated BALB/c mice were transplanted with 1 × 107 BM cells with or without 3 × 107 splenic cells from C57BL/6 mice. Recipient mice received vehicle or ruxolitinib (30 mg/kg) by oral gavage twice a day after transplantation. (a) Schematic representation of the experimental procedure. The overall survival (b), weight variation (c) and clinical GVHD score (d) were shown in each group (BM‐only group, n = 6; Vehicle group, n = 12; Ruxolitinib group, n = 12). (e) Representative haematoxylin & eosin‐stained sections of the liver, intestine and colon from vehicle and ruxolitinib‐treated mice at day 7 after transplantation (scale bar, 100 μm). The arrows were used to indicate the representative areas. (f–i) The percentages and absolute numbers of donor‐derived splenic MDSCs (CD11b+Gr‐1+), PMN‐MDSCs (CD11b+Ly6G+Ly6Clo) and M‐MDSC (CD11b+Ly6G−Ly6Chi) were measured with flow cytometry in vehicle treatment and ruxolitinib treatment groups at day 7 after transplantation (n = 4 or 5 per group). (j) The morphology of splenic‐derived PMN‐MDSCs isolated from vehicle‐ and ruxolitinib‐treated mice at day 7 after transplantation, which were visualised by Wright–Giemsa staining under light microscope (scale bar, 10 μm). *P < 0.05, **P <0.01, ***P < 0.001, ****P < 0.0001.
To determine the effect of ruxolitinib on T cell fate in mice with aGVHD, we performed donor‐derived T cell profiling. The proportions of activated CD4+CD69+ T and IFN‐γ‐producing CD4+ Th1 cells were significantly lower and the frequencies and absolute numbers of CD4+Foxp3+ Tregs in the spleen on day 7 were significantly higher in mice treated with ruxolitinib than in those treated with the vehicle, indicating attenuation of the inflammatory response in aGVHD (Supplementary figure 2a–d). In addition, ruxolitinib treatment considerably suppressed the production of proinflammatory cytokines, including IFN‐γ, TNF‐α, MCP‐1, IL‐12p70, IL‐6, IL‐17A and GM‐CSF and significantly increased the serum levels of anti‐inflammatory IL‐10 (Supplementary figure 2e). Consistent with the findings of a previous investigation, 6 these results demonstrate that the protective effect of ruxolitinib is mediated by reducing T cell pathogenicity and inflammatory cytokine production during the development of aGVHD.
Our initial analysis demonstrated that the expansion of MDSCs, particularly PMN‐MDSCs, was closely correlated with treatment response to ruxolitinib during aGVHD progression. These results are consistent with those of a previous study, reporting that the JAK/STAT pathway plays an important role in the recruitment of MDSCs to inflammatory sites. 22 We therefore hypothesised that ruxolitinib affects MDSC accumulation in aGVHD. As shown in Figure 2f–i, the proportions and absolute numbers of MDSCs (CD11b+Gr‐1+) were significantly increased in the spleen of mice following treatment with ruxolitinib than those with vehicle treatment, particularly at day 7 after transplantation. Moreover, we sought to delineate the alterations in splenic PMN‐MDSCs (CD11b+Ly6G+Ly6Clo) and M‐MDSCs (CD11b+Ly6G‐Ly6Chi). Interestingly, the proportions and absolute numbers of PMN‐MDSCs in the spleen of ruxolitinib‐treated mice were significantly higher than those in the spleen of vehicle‐treated mice at day 7 and 14 after transplantation. Variation in the proportion of M‐MDSCs was less affected by ruxolitinib treatment than that in the proportion of PMN‐MDSCs.
Ruxolitinib enhanced the suppressive activity of MDSCs in aGVHD
We next evaluated the effect of ruxolitinib on the suppressive function of MDSCs and related subsets in aGVHD. MDSCs, PMN‐MDSCs and M‐MDSCs were isolated from splenocytes of vehicle and ruxolitinib group and cocultured with T cells purified from the spleen of wild‐type C57BL/6 at different ratios (MDSC: T = 0:1 to 1:2) for 3 days stimulated by anti‐CD3/CD28 beads. MDSCs isolated from vehicle‐treated mice showed reduced capacity to block T cell proliferation compared with those isolated from ruxolitinib‐treated mice. The immunosuppressive function of PMN‐MDSCs was significantly enhanced and that of M‐MDSCs was impaired in the ruxolitinib group compared with those in the vehicle group (Figure 3a–c). We further investigated the morphology of PMN‐MDSCs isolated from the two groups under light microscope. PMN‐MDSCs isolated from ruxolitinib‐treated mice had larger cell volumes and richer cytoplasm than those isolated from vehicle‐treated mice, which may contribute to the increased suppressive effects of T cells (Figure 2j).
Figure 3.
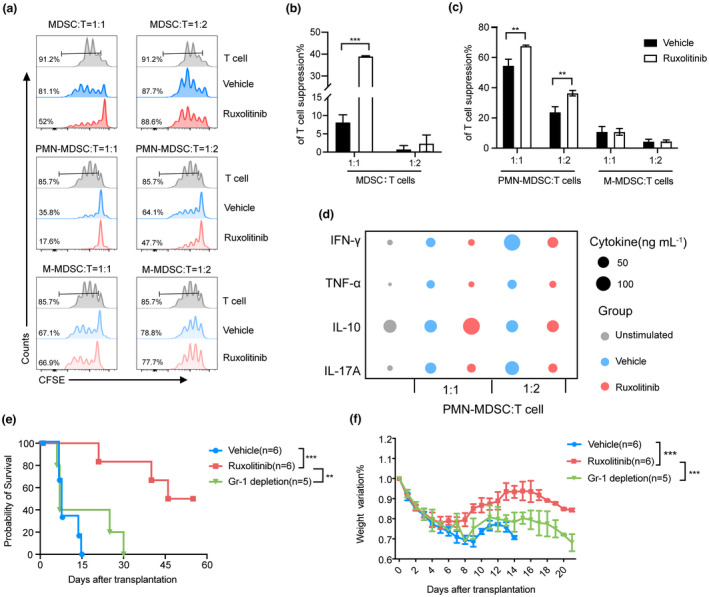
Ruxolitinib treatment enhanced immunosuppressive function of MDSCs, especially PMN‐MDSCs in aGVHD mice. Depletion of MDSCs exacerbated aGVHD lethality in ruxolitinib‐treated mice. (a–c) CFSE‐labelled CD3+ T cells (1 × 105 per well) from wild‐type C57BL/6 spleen were stimulated with CD3/28 beads and cocultured with different ratios of purified splenic MDSCs, PMN‐MDSCs and M‐MDSCs isolated from vehicle‐ and ruxolitinib‐treated mice at day 7 after transplantation for 72 h. Proliferation of CFSE‐labelled CD3+ T cells was measured with flow cytometry (Vehicle group, n = 3; Ruxolitinib group, n = 3). (d) The helper T cell‐related cytokines were detected in supernatants harvest from the above coculture system (n = 3). (e,f) aGVHD mouse models were built as described previously. Two hundred microgram anti‐Gr‐1 antibody was injected intraperitoneally into recipient mice with ruxolitinib treatment to deplete MDSCs as Gr‐1 depletion group from day 5 to day 29 every other day after transplantation. The overall survival and weight ratio were exhibited in each group (Vehicle group, n = 6; Ruxolitinib group, n = 6; Gr‐1 depletion group, n = 5). Data are expressed as mean ± standard error (SE) and from three independent experiments. **P < 0.01, ***P < 0.001.
Levels of helper T (Th) cell‐related cytokines in the supernatants derived from the PMN‐MDSCs suppression assay were examined using a multiplex kit. Compared with vehicle group, supernatants of PMN‐MDSCs from ruxolitinib group which cocultured with T cells exhibited apparent defects in the expression of proinflammatory cytokines such as Th1 cytokines‐IFN‐γ, TNF‐α and Th17 cytokines‐IL‐17. By contrast, the levels of anti‐inflammatory cytokines such as Treg cytokines (IL‐10) were increased in the supernatants of cultures containing PMN‐MDSCs isolated from the ruxolitinib group (Figure 3d). Overall, these results indicate that ruxolitinib alters the distribution of MDSCs towards PMN‐MDSCs, which modulate Th cell balance and exert immunosuppressive effects by inhibiting the proliferation of T cells.
MDSC depletion weakened the therapeutic efficacy of ruxolitinib in aGVHD
As we demonstrated the effect of ruxolitinib in maintaining MDSC function, we next evaluated the protective effect of MDSCs in GVHD following ruxolitinib treatment. CD11b+Gr‐1+ MDSCs in the BM, spleen and blood can reportedly be depleted using an anti‐Gr1 antibody. 23 Accordingly, we injected 200 μg of anti‐Gr‐1 antibodies into recipient mice treated with ruxolitinib every other day from day 5 to day 29 after transplantation, and flow cytometry revealed almost complete depletion of MDSCs (Supplementary figure 2f). As shown in Figure 3e and f, compared with mice receiving ruxolitinib alone, Gr‐1 depletion significantly decreased survival and accelerated weight loss caused by aGVHD in the ruxolitinib‐treated mice. This indicates that MDSCs are required at the stage of post‐transplantation for ruxolitinib treatment to suppress aGVHD.
Transcription signatures of PMN‐MDSCs in aGVHD mice with or without ruxolitinib treatment
To determine the mechanisms underlying the increased suppressive effects of PMN‐MDSCs in ruxolitinib‐treated mice with aGVHD, PMN‐MDSCs were isolated from mice with aGVHD treated with or without ruxolitinib at day 7 after transplantation. RNA‐seq was then used to evaluate the transcription signatures of PMN‐MDSCs. Pathway enrichment analysis was performed using gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes pathway database analysis and gene set enrichment analysis (GSEA). In PMN‐MDSCs isolated from ruxolitinib‐treated mice, GSEA analysis revealed that oxidative phosphorylation, Notch signalling and TGF‐β signalling were upregulated, whereas IFN‐γ and IL6‐JAK/STAT3 pathways were downregulated (Figure 4a). GO analysis indicated that differentially upregulated genes in the ruxolitinib group were particularly enriched in the negative regulation of apoptosis, ROS metabolic processes, NF‐κB signalling and MAPK activity. By contrast, PMN‐MDSCs from the vehicle group exerted impaired suppressive effects on T cells by promoting T cell activation, cellular responses to IFN‐γ and STAT phosphorylation (Figure 4b). Genes with elevated expression in PMN‐MDSCs from the ruxolitinib group were enriched for M2 anti‐inflammatory signatures compared with those from the vehicle group (Figure 4c). qRT–PCR demonstrated increased expression of M2 signatures, including genes related to ROS production (Nox2, Hif1α and Ho‐1) and immunosuppressive functions (Il10 and Tgfb1). No significant difference in mRNA expression of Arg1, Nos2 or Cox2 was observed between the two groups (Figure 4d).
Figure 4.
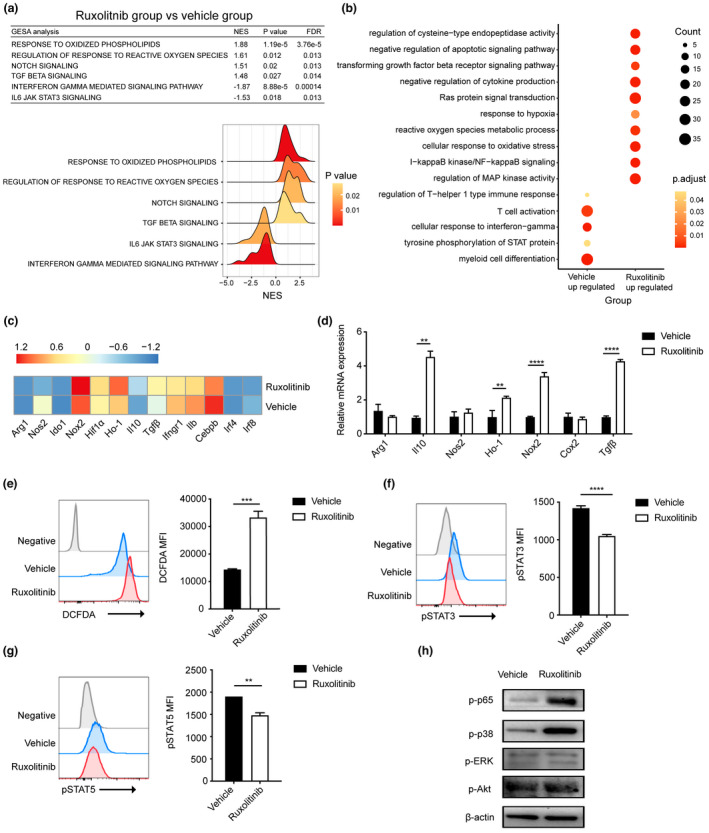
Transcriptional signatures and related protein levels of splenic PMN‐MDSCs in vehicle‐ and ruxolitinib‐treated mice at day 7 after transplantation. (a) Gene pathways that were differentially expressed in PMN‐MDSCs from two groups according to Gene Set Enrichment analysis. Gene sets were considered statistically significant at an FDR P‐value < 0.05 (n = 2 per group). (b) Dot graph shows the alterations of enriched GO pathways between two groups. The expression profile of PMN‐MDSCs function‐related genes between two groups according to (c) RNA sequencing and (d) real‐time PCR (n = 2 or 3 per group). (e) Flow cytometric detection of ROS in PMN‐MDSCs of vehicle‐ and ruxolitinib‐treated hosts on day 7 post‐transplantation. Representative DCFDA staining flow cytometry data gated on CD11b+Ly6G+Ly6Clo cells are shown. Geometric mean fluorescence intensity (MFI) values are plotted (n = 3). (f, g) The expression of pSTAT3 and pSTAT5 in PMN‐MDSCs of vehicle‐ and ruxolitinib‐treated hosts on day 7 post‐transplantation by phosflow techniques (n = 3). (h) The phosphorylation levels of p65, ERK, p38 and Akt were quantified by western blot assay in cell lysates of PMN‐MDSCs from vehicle‐ and ruxolitinib‐treated mice on day 7 post‐transplantation. β‐Actin was used as an internal control. Data are expressed as mean ± standard error (SE). **P < 0.01, ***P < 0.001, ****P < 0.0001. These results are representative of three independent experiments.
Enhanced ROS generation in PMN‐MDSCs isolated from ruxolitinib‐treated mice via the upregulation of the NF‐κB/MAPK‐p38 signalling pathway in vivo
Previous studies have reported that NADPH oxidase (NOX2) is a key mediator of PMN‐MDSC‐mediated T cell suppression by regulating ROS activity. 24 , 25 Therefore, we hypothesised that the immunosuppressive function of ruxolitinib is mediated by ROS generation. To test this hypothesis, PMN‐MDSCs were isolated from mice in the ruxolitinib and vehicle groups at day 7 after transplantation. ROS levels were determined using flow cytometric analysis of DCFDA staining, which revealed that the ROS levels in PMN‐MDSC subsets from the ruxolitinib group were significantly higher than those from the vehicle group (Figure 4e). Considering that ruxolitinib is a JAK1/2 inhibitor, we also measured the phosphorylation of STAT3, STAT5 and the downstream mediators of the JAK/STAT signalling pathway using phosflow technology. As expected, ruxolitinib treatment significantly reduced the phosphorylation levels of STAT3/5 in PMN‐MDSCs (Figure 4f and g). However, the JAK/STAT signalling pathway contributes to MDSC accumulation and facilitates ROS release in PMN‐MDSCs. 26 Accordingly, ruxolitinib treatment may exert a contradictory effect on PMN‐MDSCs by increasing ROS generation and inhibiting the JAK/STAT signalling pathway. We then focussed on the cytokine storm during aGVHD pathogenesis by STAT3 activation along with simulation of the NF‐κB pathway, 27 which is also involved in the expansion and accumulation of MDSCs. Interestingly, ruxolitinib treatment increased the phosphorylation levels of p65 and MAPK‐p38 but had no effect on p‐Erk and p‐Akt levels, as determined using western blot (Figure 4h). These results suggested that anti‐inflammatory effects of ruxolitinib on PMN‐MDSCs may enhance ROS generation through bypass activation of NF‐κB/MAPK‐p38 pathway during the development of aGVHD.
Ruxolitinib enhanced the immunosuppression of MDSCs by increasing ROS production in vitro
Myeloid‐derived suppressor cells were generated by treating BM cells isolated from C57BL/6 mice with GM‐CSF and IL‐6 for 4 days in vitro, according to previously described methods. Following treatment, 89.5% of cells were identified as CD11b+Gr‐1+ cells. CD11b+ cells can be subdivided into PMN‐ and M‐MDSCs according to cell surface marker expression and morphological features (Figure 5a). To determine whether ruxolitinib had a direct effect of maintaining the suppressive function of MDSCs on T cells, the proliferation of T cells stimulated with CD3/28 beads was analysed in the presence or absence of MDSCs pretreated with varying concentrations of ruxolitinib (0.1, 1 or 10 μm). As shown in Figure 5c, MDSCs pretreated with 10 μm ruxolitinib exerted stronger suppressive effects on T cells than those without ruxolitinib pretreatment. Next, we separated the two subsets of MDSCs from BM‐derived MDSCs pretreated with 10 μm ruxolitinib for 2 h and found that PMN‐MDSCs pretreated with ruxolitinib had substantially greater capacity to inhibit T cell proliferation than M‐MDSCs (Figure 5d and e).
Figure 5.
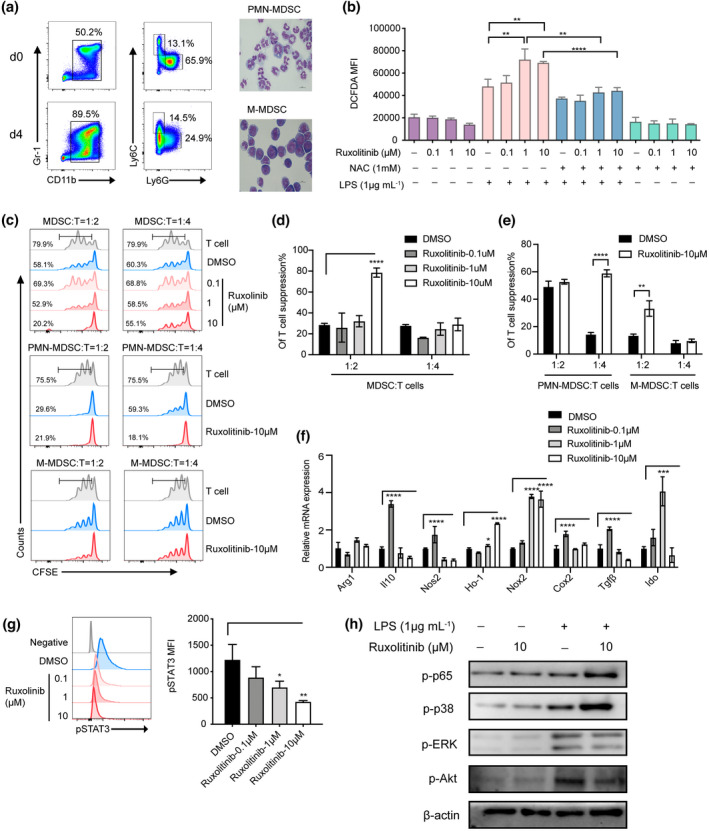
Ruxolitinib‐pretreated PMN‐MDSCs displayed remarkable immunosuppressive function by upregulation of Nox2 to regulate ROS generation via bypass activating NF‐κB/MAPK‐p38 pathways in vitro. MDSCs were generated in vitro from BM cells of C57BL/6 mice in the presence of 40 ng mL−1 GM‐CSF and IL‐6. After 4 days, cells were stained for CD11b and Gr‐1 expression or the distribution of MDSC subsets was gated on CD11b+ cells by the expression of Ly6C and Ly6G. (a) Data show one representative flow cytometric analysis and morphology of MDSCs subsets. (b) In vitro purified PMN‐MDSCs were pretreated with or without ruxolitinib (0.1 μm, 1 μm, 10 μm) for 2 h and then incubated with LPS (1 μg mL−1) in the presence or absence of ROS inhibitor NAC (1 mm) for another (h). The production of ROS was monitored by DCFDA flow cytometry in each group (n = 3). (c–e) CFSE‐labelled CD3+ T cells (1 × 105 per well) were stimulated by CD3/28 beads, then in vitro induced MDSCs, PMN‐MDSCs and M‐MDSCs were added at different ratios with or without ruxolitinib pretreated cocultured for 72 h. Proliferation of CFSE‐labelled CD3+ T cells was measured with flow cytometry. In vitro purified PMN‐MDSCs were pretreated with or without ruxolitinib (0.1 μm, 1 μm,10 μm) for 2 h and then stimulated with LPS (1 μg mL−1) (n = 3). (f) The immunosuppressive molecules of PMN‐MDSCs were detected by real‐time PCR (n = 3). The expression of pSTAT3, p‐p65, p‐p38, p‐ERK and p‐Akt were examined by phosflow analysis (g) and western blot assay (h). Data are expressed as mean ± standard error (SE). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. These results are representative of three independent experiments.
To determine the pharmacological effects of ruxolitinib on ROS generation, BM‐derived MDSCs were cocultured with or without ruxolitinib for 2 h followed by incubation with 1 μg mL−1 LPS for 30 min. Cellular ROS were detected and labelled in the CD11b+Ly6G+Ly6Clo PMN‐MDSCs by flow cytometry. As shown in Figure 5b, ROS levels were significantly higher in PMN‐MDSCs pretreated with ruxolitinib. We next pretreated PMN‐MDSCs with NAC, a ROS inhibitor, which can specifically block ROS production. Immunosuppressive molecules, such as Nos2, Il‐10, Ho‐1, Nox2, Cox2, Tgf‐β and Ido, were strongly upregulated in ruxolitinib‐pretreated PMN‐MDSCs (Figure 5f). We also examined variations in related signalling pathways. Consistent with the results of our in vivo experiments, ruxolitinib‐pretreated PMN‐MDSCs exerted inhibition of STAT3 along with activation of NF‐κB/MAPK‐p38 by western blot detection (Figure 5g and h).
Ruxolitinib inhibited the differentiation of MDSCs into mature myeloid cells in vitro by inhibiting STAT5
After demonstrating the accumulation of MDSCs in the spleens of ruxolitinib‐treated aGVHD mice, we next determined the effect of ruxolitinib on the differentiation of MDSCs into mature myeloid cells, such as dendritic cells and macrophages. BM‐derived MDSCs were incubated with 10 ng mL−1 GM‐CSF for 5 days with or without ruxolitinib. Ruxolitinib‐treated MDSCs showed impaired differentiation capacity into CD11c+ and F4/80+ cells compared with DMSO‐treated MDSCs (Figure 6a and b). The expression of CD80+, a marker of the mature phenotype, was markedly decreased in ruxolitinib‐treated MDSCs compared with that in MDSCs treated with DMSO as a control (Figure 6c). Previous studies have suggested that GM‐CSF activates the JAK2/STAT5 pathway, thereby decreasing IRF8 transcription and altering the differentiation and function of MDSCs. 28 Our findings indicated that ruxolitinib suppressed the phosphorylation of STAT5 and increased the expression of Irf‐8 and Bcl‐xl, thereby inhibiting the differentiation and promoting survival of MDSCs under GM‐CSF stimulation (Figure 6d and e).
Figure 6.
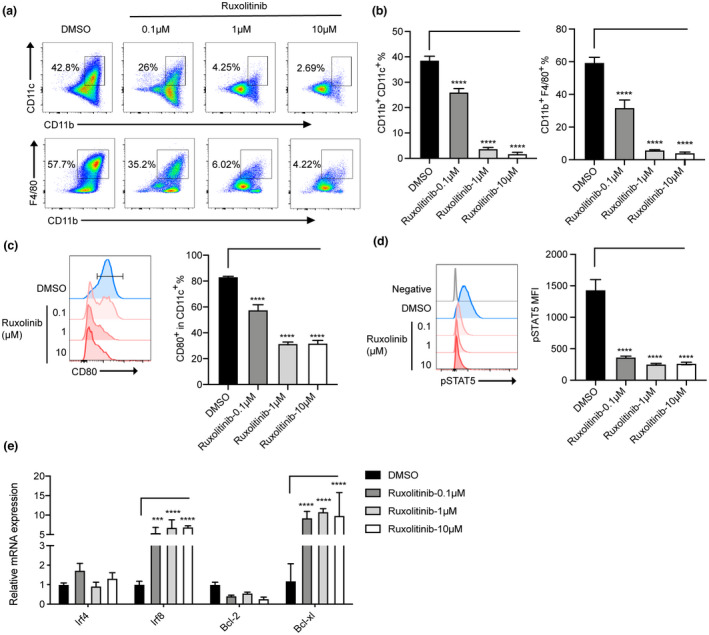
Ruxolitinib decreased the differentiation of MDSCs to mature cells via STAT5 inhibition in vitro. BM‐derived MDSCs in vitro were cultured with GM‐CSF with or without different concentrations of ruxolitinib (0.1 μm, 1 μm, 10 μm) for 5 days. (a) Representative plots of macrophages (CD11b+F4/80+) and dendritic cells (CD11b+CD11c+) were shown. The percentages of differentiated mature cells (b) and the expression of costimulatory molecules CD80+ gated on CD11b+ cells (c) were indicated (n = 3). (d) The phosphorylation of STAT5 was examined by phosflow techniques (n = 3). (e) The levels of transcriptional factors related to MDSCs differentiation were monitored by real‐time PCR (n = 3). Data are expressed as mean ± standard error (SE). ***P < 0.001, ****P < 0.0001. These results are representative of three independent experiments.
Discussion
Myeloid‐derived suppressor cells are predominantly defined by their capacity to inhibit T cell responses and promote Treg expansion, thereby exerting protective effects against the development of aGVHD. 29 Myeloid cells secrete molecules that stimulate a range of signalling pathways, including the JAK/STAT pathway which contributes to the expansion and function of myeloid cells. The results of the present study demonstrate that ruxolitinib, a JAK1/2 inhibitor, efficiently ameliorates the symptoms of aGVHD symptoms, thereby indicating the potential utility of ruxolitinib in the treatment of patients after allo‐HSCT. Alterations in the number and proportions of MDSCs may predict treatment responses to ruxolitinib in patients with aGVHD when assessed in combination with clinical symptoms. We further evaluated the effect ruxolitinib on the two major subsets of MDSCs in a mouse model of aGVHD and found that ruxolitinib increases the immunosuppressive effects of PMN‐MDSCs by increasing ROS generation via activation of the NF‐κB/MAPK‐p38 pathway in vivo and in vitro. In addition, inhibition of STAT5 hampered the differentiation of MDSCs. Further, MDSC depletion reduced the efficacy of ruxolitinib in treating aGVHD, which indicates that MDSC may play a pivotal role in mediating the therapeutic effects of ruxolitinib in aGVHD.
Ruxolitinib has been shown to be effective in treating SR‐aGVHD. 7 In our cohort, patients with SR‐aGVHD who received ruxolitinib treatment had favorable ORR and higher OS. Recent studies have reported that ruxolitinib alleviates aGVHD by inhibiting the JAK/STAT signalling pathway through multiple mechanisms, including inhibiting the production of proinflammatory cytokines; impairing the differentiation and maturation of dendritic cells; suppressing alloreactive T cell activation and promoting Treg expansion; and inhibiting IFN‐γ‐induced STAT1 phosphorylation in MSCs to maintain immune tolerance. 6 , 30 , 31 However, patients receiving ruxolitinib had a modestly higher incidence of infections and CMV reactivation. 7 Further, there are no standardised methods for monitoring treatment responses to ruxolitinib. Hence, the activation of JAK/STAT signalling in other immune cell types may account for the anti‐inflammatory effects of ruxolitinib in aGVHD or other autoimmune diseases 32 and therefore warrants further investigation.
Myeloid‐derived suppressor cells have been described as a biomarker of inflammation with remarkable immunosuppressive capacity. The proportions of MDSCs in the graft and PB after allo‐HSCT may be associated with the severity of aGVHD and disease‐free survival. 33 To determine the importance of measuring the proportion of MDSCs in monitoring treatment responses to ruxolitinib, we measured the frequencies of primary immunoregulatory PBMCs before and 7 and 14 days after ruxolitinib treatment. Interestingly, the levels of MDSCs, particularly PMN‐MDSCs, continued to increase until day 14 in nonresponders but returned to pretreatment levels by day 14 in responders. In particular, the frequencies of MDSCs and PMN‐MDSCs were substantially lower at day 14 in responders compared with those in nonresponders. However, we did not observe significant alterations in Treg and M‐MDSC populations, indicating that MDSCs were more sensitive with the extension of treatment time during aGVHD. We further observed that the proportions of MDSCs, particularly PMN‐MDSCs, before treatment were closely associated with treatment responses to ruxolitinib, indicating that alterations in MDSC levels may help improve the monitoring of treatment responses to ruxolitinib. Monitoring the proportion of MDSCs before and during the administration of ruxolitinib may aid in determining the optimal treatment regimens for specific patients with aGVHD. Despite these important findings, our study also has limitations, including the relatively small sample size of patients treated with ruxolitinib. Further, the retrospective nature of the current study and data obtained from a single centre may have introduced patient selection bias into the study analysis. Therefore, large cohort prospective investigations are warranted to identify specific subsets of MDSCs as biomarkers with prognostic impact. It is required further study for a comprehensive evaluation of MDSCs analysis by several methods, including FACS, immunohistochemistry and genomic/transcriptomic/methylation profiling and to confirm the pivotal status of MDSCs on treatment decision‐making for aGVHD.
To determine the effects of ruxolitinib on MDSCs, we evaluated survival, body weight and clinical scores in an MHC‐mismatched mouse model of aGVHD. Ruxolitinib administration in vivo increased the survival of aGVHD mice and had a substantial inhibitory effect on CD4+ T cells by decreasing the levels of proinflammatory cytokines and promoting the expansion of Tregs, consistent with previous research. 34 We next evaluated the effect of ruxolitinib on the proportion of MDSCs in vivo. We observed that ruxolitinib increased the absolute numbers of MDSCs, particularly PMN‐MDSCs, and enhanced the suppressive effects of PMN‐MDSCs on T cells.
We then focussed on PMN‐MDSCs, an important cellular target through which ruxolitinib exerts protective effects against aGVHD. However, there is a lack of studies on the signalling pathways involved in PMN‐MDSC function. Using transcriptional profiling of PMN‐MDSCs isolated from ruxolitinib‐treated mice with aGVHD, GO and GSEA revealed enrichment of genes associated with MAPK signalling, NF‐κB signalling and ROS activity in PMN‐MDSCs isolated from ruxolitinib‐treated mice. We also observed significant upregulation of Nox2, which is known to be a critical regulator of ROS generation. These data demonstrate that PMN‐MDSCs generate high levels of cytosolic ROS under the activity of Nox2 to maintain the undifferentiated state with immunosuppressive properties of MDSCs in the TME as described previously. 35 , 36 However, the mechanisms underlying the regulation of ROS in PMN‐MDSCs are yet to be fully elucidated. Recent studies have indicated that the activation of the JAK/STAT pathway leads to the accumulation of MDSCs and prevents their differentiation into mature cell types. The activation of STAT3 is directly responsible for upregulating the transcription of Nox2 and increasing ROS production by MDSCs in tumor‐bearing mice. 24 Patients with oral squamous cell carcinoma have higher proportions of PMN‐MDSCs, with higher levels of ROS and pSTAT3 signalling abrogating T cell proliferation. 37
In the present study, ruxolitinib effectively inhibited the activation of JAK/STAT pathway and promoted the production of ROS in PMN‐MDSCs, which appears to contradict the findings of previous studies. As aGVHD is known to trigger the cytokine storm, aGVHD may involve the activation of several other inflammatory pathways in addition to the JAK/STAT pathway. Accordingly, other pathways may be involved in mediating the effect of ruxolitinib on MDSCs. Previous studies have revealed that ROS production in MDSCs is mediated by activation of the NF‐κB and MAPK‐p38 signalling pathways in lupus nephritis. 38 NF‐κB/MAPK‐p38 signalling may also play a critical role in the accumulation of MDSCs and mediating their activation in response to TNF‐α, IL‐1β and other inflammatory cytokines. 39 , 40 In the context of allo‐HSCT, the myeloid differentiation primary response gene 88 (MyD88)/NF‐κB pathway may also contribute to the expansion of donor MDSCs and initiation of aGVHD. 41 Myd88−/− MDSCs lose the ability to suppress T cell activity and release anti‐inflammatory cytokines such as IL‐10 and Arg‐1. 42 In the present study, we confirmed that upregulation of the NF‐κB/MAPK‐p38 pathway is involved in maintaining MDSC function in ruxolitinib‐treated PMN‐MDSCs in vivo and in vitro, which is consistent with the results of our transcriptome analysis.
The JAK/STAT signalling pathway may play a critical role in maintaining MDSC numbers and blocking their differentiation into mature cells. GM‐CSF triggers Tyr phosphorylation of the GM‐CSF receptor resulting in JAK/STAT5 activation and downregulation of Irf8, thereby promoting MDSC differentiation and survival. 43 Our results indicate that the inhibition of pSTAT5 by ruxolitinib may block the differentiation of immature MDSCs into macrophages or dendritic cells via upregulation of Irf8 and prevent apoptosis in MDSCs by upregulating Bcl‐2.
In conclusion, we evaluated the therapeutic effect of JAK1/2 inhibitor ruxolitinib in patients with SR‐aGVHD after allo‐HSCT and demonstrated that the monitoring of MDSCs is useful in assessing treatment responses to ruxolitinib and may aid in selecting patients suitable for alterations in treatment regimen. Using a mouse model of aGVHD and in vitro experiments, we observed that two signal transduction pathways mediate the expansion of MDSCs in response to ruxolitinib therapy. On the one hand, induced by proinflammatory cytokines, ruxolitinib could promote the differentiation of MDSCs into PMN‐MDSCs, which displayed strong immunosuppressive function by upregulating Nox2, thereby regulating ROS generation through bypass activation of NF‐κB/MAPK‐p38 pathways. On the other hand, MDSCs also required second signals to maintain their undifferentiated state via STAT5 influenced by ruxolitinib. The results of the present study demonstrate the direct immunoregulatory effect of ruxolitinib on MDSCs may also be relevant to pathological conditions other than aGVHD and facilitate the development of novel strategies for modifying MDSCs in the treatment of aGVHD. Patients exposed to long‐term treatment with ruxolitinib should be closely monitored for infectious complications or relapse of the underlying disease. However, considering their potential as therapeutic targets, the JAK/STAT and other signalling pathways involved in MDSC functional polarisation and phenotype warrant further investigation. Further studies are required to define the interactions between effector immune cells and MDSCs, which may improve therapeutic responses and disease outcomes.
Methods
Patient enrolment and data collection
We retrospectively analysed data from 74 patients with SR‐aGVHD who received ruxolitinib as second‐line therapy following allo‐HSCT at the Institute of Haematology, Chinese Academy of Medical Sciences, China, between May 2017 and December 2020. Eligibility criteria were aGVHD progression after 3 days or incomplete response after 7 days (SR‐aGVHD) of treatment with methylprednisolone at a dose of 2 mg/kg per day. 44 aGVHD grading was evaluated according to MAGIC criteria. 45 The clinical characteristics of patients are shown in Supplementary table 1. All patients were followed up through telephone and outpatient appointments until 31 January 2022.
The initial dose of ruxolitinib was 5 mg twice daily, and in cases with good tolerance and stable haematological parameters, the dose was increased to 10 mg twice daily at the physician's discretion. Based on a previous study, 7 the safety and efficacy of ruxolitinib was evaluated at 28 days after treatment initiation with ruxolitinib. Treatment responses were classified as complete response (CR), partial response (PR) or treatment failure. 46 The skin, GI and liver were the main target organs involved in aGVHD. CR was defined as the resolution of all clinical symptoms of GVHD. PR was defined as an improvement in at least one organ or site without progression at any other organ or site. Treatment failure was defined as the absence of improvement in aGVHD, disease progression in any organ or the need to start a new treatment for disease control. Overall survival (OS) was measured from the initiation of ruxolitinib treatment until death from any cause. Adverse events in patients who received ruxolitinib are summarised in Supplementary table 2. The severity of cytopenia was defined according to previously published criteria. 47
Peripheral blood (PB) samples were collected from patients with SR‐aGVHD who received ruxolitinib treatment between June and October 2020. Seven patients achieved PR or CR (responders), and six showed treatment failure (nonresponders). MDSCs and Tregs were isolated from PB mononuclear cells (PBMCs) using Ficoll–Hypaque density gradient centrifugation on the day before treatment initiation with ruxolitinib and at 7 and 14 days after treatment initiation. This study was approved by our centre's Medical Ethics Committee (Ethical approval No. IIT2020016‐EC‐3) and conducted in accordance with the Helsinki declaration. Written informed consent for collection data was obtained from each patient or their legal representative prior to enrolment.
Mice
C57BL/6 and BALB/c mice aged 8–9 weeks were purchased from Huafukang Company (Beijing, China) and maintained in specific pathogen‐free barrier facilities. All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Institute of Hematology, Chinese Academy of Medical Sciences.
Mouse models of bone marrow transplantation and aGVHD
Recipient BALB/c mice received total body (60Co source) irradiation at a dose of 8 Gy, which was split into two doses and applied at an interval of 4 h on day −1. Irradiated recipient mice were then intravenously injected with 1 × 107 bone marrow (BM) nucleated cells with or without 3 × 107 spleen cells obtained from age‐ and gender‐matched C57BL/6 mice within 24 h on day 0. BM and splenocyte suspensions were obtained from iliac, femoral, tibial and splenic samples. aGVHD scores were calculated according to the weight, posture, activity, fur texture and skin appearance of mice, as described previously. 48 Liver, intestine and colon specimens were collected on day 7 after transplantation. Specimens were fixed with 4% neutral formaldehyde, embedded in paraffin, sectioned, stained with haematoxylin and eosin and observed under a light microscope. In some experiments, mice were intraperitoneally injected with 200 μg of anti‐Gr‐1 depleting antibodies (BioXcell, New Hampshire, USA) every other day between days 5 and 29.
Treatment with ruxolitinib
The mouse models of aGVHD were administered with ruxolitinib (Novartis, Basel, Switzerland) dissolved in PEG300 and 5% dextrose (at a ratio of 1:3) by oral gavage at a dose of 30 mg kg– 1 twice daily from day 0 until day 30, as described previously. 6 The vehicle group was only administered with PEG300:5% dextrose (1:3). For the in vitro study, MDSCs were pretreated with various concentrations of ruxolitinib (0.1, 1 and 10 μm), which was diluted with dimethyl sulfoxide (DMSO) for 2 h.
Isolation and generation of MDSCs
Total MDSCs (CD11b+Gr‐1+), PMN‐MDSCs (CD11b+Ly6G+Ly6Clo) and M‐MDSCs (CD11b+Ly6G‐Ly6Chi) were isolated from single‐cell suspensions of splenic samples using a FACS Arial III cell sorter. The purity was typically over 90%. For in vitro experiments, MDSCs were generated from BM cells obtained from C57BL/6 wild‐type mice and maintained in culture medium supplemented with 40 ng mL−1 murine IL‐6 and 40 ng mL−1 GM‐CSF (Peprotech, Rochy Hill, USA) for 4 days. 49 The purity ranged between 80% and 90%, as assessed via flow cytometry. PMN‐ and M‐MDSC subpopulations were purified using MDSC Isolation Kit (Miltenyi Biotec, Kӧln, Germany) or FACS Arial III cell sorter.
Myeloid‐derived suppressor cell suppression assay
Splenic CD3+ T cells isolated from C57BL/6 mice using CD3 microbead kit (Miltenyi Biotec) were incubated with 5 μm CFSE (Biolegend, San Diego, USA) for 8 min in phosphate‐buffered saline (PBS) at room temperature and then washed with RPMI 1640 containing 10% foetal bovine serum (FBS). Next, 1 x 105 labelled T cells were seeded into 96‐well round‐bottom plates with anti‐CD3/CD28 beads (Invitrogen, New York, USA) and cocultured with or without different ratios of purified MDSCs, PMN‐MDSCs or M‐MDSCs in RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin and 50 ng mL−1 recombinant IL‐2 for 72 h. Cell proliferation was measured using flow cytometry. Cell suppression (%) was calculated using the following formula:
Myeloid‐derived suppressor cell differentiation assay
BM‐derived MDSCs were cultured in the presence of 10 ng mL−1 recombinant GM‐CSF (Peprotech) for 5 days. In some experiments, cell cultures were treated with varying concentrations of ruxolitinib (0.1, 1, or 10 μm) or DMSO. Subsequently, cell phenotypes were assessed using flow cytometry.
Flow cytometry
For the analysis of cell surface molecules, human blood cells were isolated and fluorescently stained for 30 min at 4°C in the dark with the following antibodies: APC‐Cy7 anti‐human CD4, PE‐Cy7 anti‐human CD8, APC anti‐human CD25, PE anti‐human CD11b, FITC anti‐human HLA‐DR, PE/Cy7 anti‐human CD15 and APC/Cy7 anti‐human CD14 (Biolegend). In mice, single‐cell suspensions were prepared from BM, spleen and cell cultures in vitro. The following monoclonal antibodies were added to the cell suspensions: Percp/Cy 5.5 anti‐mouse H2‐Kb, APC anti‐mouse CD11b, PE anti‐mouse Gr‐1, APC‐Cy7 anti‐mouse ly6C, PE‐Cy7 anti‐mouse Ly‐6G, PE anti‐mouse CD69, APC anti‐mouse CD25, APC‐Cy7 anti‐mouse CD4, PE‐Cy7 anti‐mouse CD8, Percp/Cy5.5 anti‐mouse CD11b, APC anti‐mouse CD80, PE‐Cy7 anti‐mouse CD11c and PE anti‐mouse F4/80 (Biolegend). For Foxp3 staining, cells were resuspended in fixation/permeabilisation buffer (eBioscience, New York, USA) according to the manufacturer's protocol. Cells were incubated with Foxp3 antibodies at a concentration of 1:100 at room temperature for 1 h under protection from light. For intracellular cytokine staining, cells were stimulated with cell stimulation cocktail (500x, Invitrogen) for 4–5 h and blocked with Brefeldin A solution (1000×, Invitrogen). For phosphorylate staining, cells were fixed with IC fixation buffer (Invitrogen) and then exposed to precooled methanol before incubation with pSTAT3 and pSTAT5 antibodies. Viability was assessed using Fixable Viability Dye EF506 (Invitrogen). Finally, stained cells were washed with PBS containing 2% FBS or permeabilisation buffer (eBioscience) and analysed using FACS Canto II. Data were analysed using FlowJo software.
Reactive oxygen species measurements
Myeloid‐derived suppressor cells were incubated in serum‐free medium at 37°C in the presence or absence of 1 μg mL−1 LPS (Sigma, St. Louis, USA) for 30 min. Intracellular ROS levels were measured via flow cytometry using 2.5 mm DCFDA, an oxidation‐sensitive dye (Beyotime Biotechnology, Shanghai, China). For determining the inhibition of ROS, cells were incubated with 1 mm N‐acetyl‐L‐cysteine (NAC; Beyotime Biotechnology) for 2 h.
Reverse transcription real‐time polymerase chain reaction (RT–PCR) and bulk RNA sequencing (RNA‐seq)
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using One‐Step RT–PCR SuperMix (Transgen. Inc, Beijing, China) to synthesise cDNA. Quantitative RT–PCR was performed using GeneAmp 7500 Sequence Detection System (Applied Biosystems). The relative abundance of each gene was calculated using 2−ΔΔCT method upon normalisation to β‐actin expression. RNA‐seq was performed using the BGIseq500 platform (BGI, Wuhan, China). The PCR primers used in this experiment are listed in Supplementary table 3.
Western blot
Cells were lysed with RIPA lysis buffer (Beyotime Biotechnology) containing protease and phosphatase inhibitors according to standard techniques. 50 Antibodies against phospho‐p38 MAPK, phospho‐Akt (Ser 473), phospho‐p44/42 MAPK (Erk1/2 and Thr202/Tyr204), phospho‐NF‐κb p65 (Ser 536) and HRP‐linked anti‐rabbit IgG antibodies were purchased from Cell Signaling Technology (Danvers, USA). Protein bands were visualised using ECL plus chemiluminescent substrate (Invitrogen). β‐actin was used as the internal control.
Cytokine profiling
Serum was collected from at least three mice per group. Supernatants were collected after centrifugation of the culture media from MDSCs from control and ruxolitinib‐treated mice cocultured with T cells. According to the manufacturer's instructions, cytokine profiling was performed using the LEGENDplex™ Mouse Inflammation Panel (Biolegend). Measured levels of mouse cytokines are reported in ng mL−1.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9.0 and R 4.0.0. Results are reported as the mean ± standard error of the mean. The Student's t‐test was used to compare differences between two groups. One‐ or two‐way ANOVA was used to compare multiple groups. The correlation between categorical variables was calculated using Spearman's correlation. Survival curves were estimated using the Kaplan–Meier method and compared using the log‐rank test. A P‐value of < 0.05 was considered statistically significant: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
AUTHOR CONTRIBUTIONS
Yigeng Cao: Conceptualization; data curation; formal analysis; funding acquisition; writing – review and editing. Jiali Wang: Data curation; formal analysis; methodology; writing – original draft. Shan Jiang: Data curation; formal analysis. Mengnan Lyu: Formal analysis; methodology. Fei Zhao: Data curation; formal analysis. Jia Liu: Data curation; formal analysis. Mingyang Wang: Investigation; methodology. Xiaolei Pei: Methodology; resources; software. Weihua Zhai: Data curation; funding acquisition. Xiaoming Feng: Resources; validation. Sizhou Feng: Data curation; investigation. Mingzhe Han: Methodology; supervision; visualization. Yuanfu Xu: Funding acquisition; methodology; supervision; writing – review and editing. Erlie Jiang: Conceptualization; funding acquisition; investigation; project administration; writing – review and editing.
Conflict of interest
The authors declare no competing interests.
Supporting information
Supporting information
Acknowledgments
This work was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021‐I2M‐1‐017, 2020‐I2M‐C&T‐B‐088), Fundamental Research Funds for the Central Universities (3332020052), the National Natural Science Foundation of China (81970107, 82070192).
Contributor Information
Mingzhe Han, Email: mzhan@ihcams.ac.cn.
Yuanfu Xu, Email: xuyf@ihcams.ac.cn.
Erlie Jiang, Email: jiangerlie@ihcams.ac.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
References
- 1. Xu L, Feng J, Xu X et al. IL‐17‐producing γδT cells ameliorate intestinal acute graft‐versus‐host disease by recruitment of gr‐1+CD11b+ myeloid‐derived suppressor cells. Bone Marrow Transplant 2021; 56: 2389–2399. [DOI] [PubMed] [Google Scholar]
- 2. Guo WW, Su XH, Wang MY, Han MZ, Feng XM, Jiang EL. Regulatory T cells in GVHD therapy. Front Immunol 2021; 12: 697854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zeiser R. Advances in understanding the pathogenesis of graft‐versus‐host disease. Br J Haematol 2019; 187: 563–572. [DOI] [PubMed] [Google Scholar]
- 4. Toubai T, Magenau J. Immunopathology and biology‐based treatment of steroid‐refractory graft‐versus‐host disease. Blood 2020; 136: 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cordes S, Mokhtari Z, Bartosova M et al. Endothelial damage and dysfunction in acute graft‐versus‐host disease. Haematologica 2021; 106: 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spoerl S, Mathew NR, Bscheider M et al. Activity of therapeutic JAK 1/2 blockade in graft‐versus‐host disease. Blood 2014; 123: 3832–3842. [DOI] [PubMed] [Google Scholar]
- 7. Zeiser R, von Bubnoff N, Butler J et al. Ruxolitinib for glucocorticoid‐refractory acute graft‐versus‐host disease. N Engl J Med 2020; 382: 1800–1810. [DOI] [PubMed] [Google Scholar]
- 8. Koehn BH, Blazar BR. Role of myeloid‐derived suppressor cells in allogeneic hematopoietic cell transplantation. J Leukoc Biol 2017; 102: 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blazar BR, MacDonald KPA, Hill GR. Immune regulatory cell infusion for graft‐versus‐host disease prevention and therapy. Blood 2018; 131: 2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bronte V, Brandau S, Chen SH et al. Recommendations for myeloid‐derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid‐derived suppressor cells. Onco Targets Ther 2016; 5: e1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang C, Wang S, Li J et al. The mTOR signal regulates myeloid‐derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death Dis 2017; 8: e2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trikha P, Carson WE 3rd. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta 2014; 1846: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kortylewski M, Kujawski M, Wang T et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11: 1314–1321. [DOI] [PubMed] [Google Scholar]
- 16. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand‐Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid‐derived suppressor cells. J Immunol 2008; 181: 4666–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joshi S, Sharabi A. Targeting myeloid‐derived suppressor cells to enhance natural killer cell‐based immunotherapy. Pharmacol Ther 2022; 235: 108114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nefedova Y, Nagaraj S, Rosenbauer A, Muro‐Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic‐selective inhibition of the janus‐activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 2005; 65: 9525–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid‐derived suppressor cell differentiation and function. Trends Immunol 2011; 32: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Penack O, Marchetti M, Ruutu T et al. Prophylaxis and management of graft versus host disease after stem‐cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol 2020; 7: e157–e167. [DOI] [PubMed] [Google Scholar]
- 21. Su X, Wang Q, Guo W et al. Loss of Lkb1 impairs Treg function and stability to aggravate graft‐versus‐host disease after bone marrow transplantation. Cell Mol Immunol 2020; 17: 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sido JM, Yang X, Nagarkatti PS, Nagarkatti M. Delta9‐tetrahydrocannabinol‐mediated epigenetic modifications elicit myeloid‐derived suppressor cell activation via STAT3/S100A8. J Leukoc Biol 2015; 97: 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li L, Zhang T, Diao W et al. Role of myeloid‐derived suppressor cells in glucocorticoid‐mediated amelioration of FSGS. J Am Soc Nephrol 2015; 26: 2183–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Corzo CA, Cotter MJ, Cheng P et al. Mechanism regulating reactive oxygen species in tumor‐induced myeloid‐derived suppressor cells. J Immunol 2009; 182: 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all‐trans retinoic acid effect on tumor‐associated myeloid‐derived suppressor cells. Cancer Res 2007; 67: 11021–11028. [DOI] [PubMed] [Google Scholar]
- 26. Cheng P, Corzo CA, Luetteke N et al. Inhibition of dendritic cell differentiation and accumulation of myeloid‐derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 2008; 205: 2235–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanchez‐Valdepenas C, Casanova L, Colmenero I et al. Nuclear factor‐kappaB inducing kinase is required for graft‐versus‐host disease. Haematologica 2010; 95: 2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waight JD, Netherby C, Hensen ML et al. Myeloid‐derived suppressor cell development is regulated by a STAT/IRF‐8 axis. J Clin Invest 2013; 123: 4464–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vendramin A, Gimondi S, Bermema A et al. Graft monocytic myeloid‐derived suppressor cell content predicts the risk of acute graft‐versus‐host disease after allogeneic transplantation of granulocyte colony‐stimulating factor‐mobilized peripheral blood stem cells. Biol Blood Marrow Transplant 2014; 20: 2049–2055. [DOI] [PubMed] [Google Scholar]
- 30. Stickel N, Hanke K, Marschner D et al. MicroRNA‐146a reduces MHC‐II expression via targeting JAK/STAT signaling in dendritic cells after stem cell transplantation. Leukemia 2017; 31: 2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryan MM, Patel M, Hogan K et al. Ruxolitinib inhibits IFNγ licensing of human bone marrow derived mesenchymal stromal cells. Transplant Cell Ther 2021; 27: 389.e1–389.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fleischmann R, Kremer J, Cush J et al. Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012; 367: 495–507. [DOI] [PubMed] [Google Scholar]
- 33. Yin J, Wang C, Huang M, Mao X, Zhou J, Zhang Y. Circulating CD14+ HLA‐DR−/low myeloid‐derived suppressor cells in leukemia patients with allogeneic hematopoietic stem cell transplantation: novel clinical potential strategies for the prevention and cellular therapy of graft‐versus‐host disease. Cancer Med 2016; 5: 1654–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carniti C, Gimondi S, Vendramin A et al. Pharmacologic inhibition of JAK1/JAK2 signaling reduces experimental murine acute GVHD while preserving GVT effects. Clin Cancer Res 2015; 21: 3740–3749. [DOI] [PubMed] [Google Scholar]
- 35. Brandau S, Trellakis S, Bruderek K et al. Myeloid‐derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol 2011; 89: 311–317. [DOI] [PubMed] [Google Scholar]
- 36. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid‐derived suppressor cells in tumor‐bearing mice. J Leukoc Biol 2012; 91: 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong LM, Liu ZG, Zhou X et al. Expansion of PMN‐myeloid derived suppressor cells and their clinical relevance in patients with oral squamous cell carcinoma. Oral Oncol 2019; 95: 157–163. [DOI] [PubMed] [Google Scholar]
- 38. Zhang D, Xu J, Ren J et al. Myeloid‐derived suppressor cells induce podocyte injury through increasing reactive oxygen species in lupus nephritis. Front Immunol 2018; 9: 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu X, Li B, Li X et al. Transmembrane TNF‐alpha promotes suppressive activities of myeloid‐derived suppressor cells via TNFR2. J Immunol 2014; 192: 1320–1331. [DOI] [PubMed] [Google Scholar]
- 40. Tu S, Bhagat G, Cui G et al. Overexpression of interleukin‐1beta induces gastric inflammation and cancer and mobilizes myeloid‐derived suppressor cells in mice. Cancer Cell 2008; 14: 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim JY, Lee YK, Lee SE et al. MyD88 in donor bone marrow cells is critical for protection from acute intestinal graft‐vs.‐host disease. Mucosal Immunol 2016; 9: 730–743. [DOI] [PubMed] [Google Scholar]
- 42. Arora M, Poe SL, Oriss TB et al. TLR4/MyD88‐induced CD11b+gr‐1intF4/80+ non‐migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol 2010; 3: 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhan Y, Lew AM, Chopin M. The pleiotropic effects of the GM‐CSF rheostat on myeloid cell differentiation and function: more than a numbers game. Front Immunol 2019; 10: 2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohty M, Holler E, Jagasia M et al. Refractory acute graft‐versus‐host disease: a new working definition beyond corticosteroid refractoriness. Blood 2020; 136: 1903–1906. [DOI] [PubMed] [Google Scholar]
- 45. Schoemans HM, Lee SJ, Ferrara JL et al. EBMT‐NIH‐CIBMTR task force position statement on standardized terminology & guidance for graft‐versus‐host disease assessment. Bone Marrow Transplant 2018; 53: 1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Escamilla Gomez V, Garcia‐Gutierrez V, Lopez Corral L et al. Ruxolitinib in refractory acute and chronic graft‐versus‐host disease: a multicenter survey study. Bone Marrow Transplant 2020; 55: 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zeiser R, Burchert A, Lengerke C et al. Ruxolitinib in corticosteroid‐refractory graft‐versus‐host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia 2015; 29: 2062–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reichenbach DK, Schwarze V, Matta BM et al. The IL‐33/ST2 axis augments effector T‐cell responses during acute GVHD. Blood 2015; 125: 3183–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marigo I, Bosio E, Solito S et al. Tumor‐induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity 2010; 32: 790–802. [DOI] [PubMed] [Google Scholar]
- 50. Egelston C, Kurko J, Besenyei T et al. Suppression of dendritic cell maturation and T cell proliferation by synovial fluid myeloid cells from mice with autoimmune arthritis. Arthritis Rheum 2012; 64: 3179–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.