

Abstract
Transplantation is the only curative treatment for patients with kidney failure but it poses unique immunological challenges that must be overcome to prevent allograft rejection and ensure long-term graft survival. Alloreactive T cells are important contributors to graft rejection and a clearer understanding of the mechanisms by which these cells recognize donor antigens — through direct, indirect or semi-direct pathways — will facilitate their therapeutic targeting. Post-T cell priming rejection responses can also be modified by targeting pathways that regulate T cell trafficking, survival cytokines or innate immune activation. Moreover, the quantity and quality of donor-reactive memory T cells crucially shape alloimmune responses. Of note, many fundamental concepts in transplant immunology have been derived from models of infection. However, the programmed differentiation of allograft-specific T cell responses is probably distinct from that of pathogen-elicited responses, owing to the dearth of pathogen-derived innate immune activation in the transplantation setting. Understanding the fundamental (and potentially unique) immunological pathways that lead to allograft rejection is therefore a prerequisite for the rational development of therapeutics that promote transplantation tolerance.
Introduction
Alloreactive T cells are key mediators of allograft rejection1–3 and are readily detectable in naive animals and humans at surprisingly high frequencies — up to 10% of an individual’s TCR repertoire recognize alloantigen4,5. T cells have a pivotal role as both drivers and effectors of allograft rejection. For example, CD4+ helper T (TH) cells not only promote the differentiation of CD8+ T cells into cytotoxic effectors that mediate allograft rejection directly, but also provide help to B cells, enabling them to generate a humoral response and produce pro-inflammatory cytokines; these immune responses result in graft injury and rejection. Activation, differentiation and expansion of CD4+ and CD8+ T cells, require three signals: signal 1 refers to antigen recognition via T cell receptor (TCR)–MHC interactions; signal 2 involves costimulation, including binding of CD28 on T cells to CD80 or CD86 on antigen-presenting cells, or CD40–CD40L (also known as CD154) interactions among others; signal 3 comprises cytokine-mediated signals. Importantly, these processes can be strongly modulated by several factors, including the cell types that engage in alloantigen presentation, the location of the alloresponse and the allorecognition pathway involved. These factors influence the programming and responses of activated T cells by triggering different transcriptional pathways, some of which are specific to the setting of transplantation.
In this Review, we provide an update on the determinants that shape alloimmune T cell responses in transplantation, including the crosstalk between the innate and adaptive immune systems. Although models of infection have shaped the majority of our understanding of T cell differentiation and antigen-specific responses, here we examine the unique features of cytokine-mediated signals triggered after recognition of donor antigens compared with detection of microbial antigens. Lastly, we discuss the determinants that control T cell access to the graft, the latest advances in the identification of transplant-specific pathways that mediate T cell programming and novel therapeutic approaches.
Pathways of alloantigen presentation
Allograft rejection requires the recognition of donor antigens by the immune system of the recipient (FIG. 1). Therefore, elucidating the immunological mechanisms involved in allorecognition events has potential to guide the development of therapeutics to prevent rejection (FIG. 2). Below we discuss research advances that have led to a paradigm shift in understanding the mechanisms that underlie allorecognition, including insights into the interactions between professional antigen-presenting cells (APCs), such as dendritic cells (DCs), and alloreactive T cells.
Figure 1. Mechanisms of allorecognition at the priming stage.
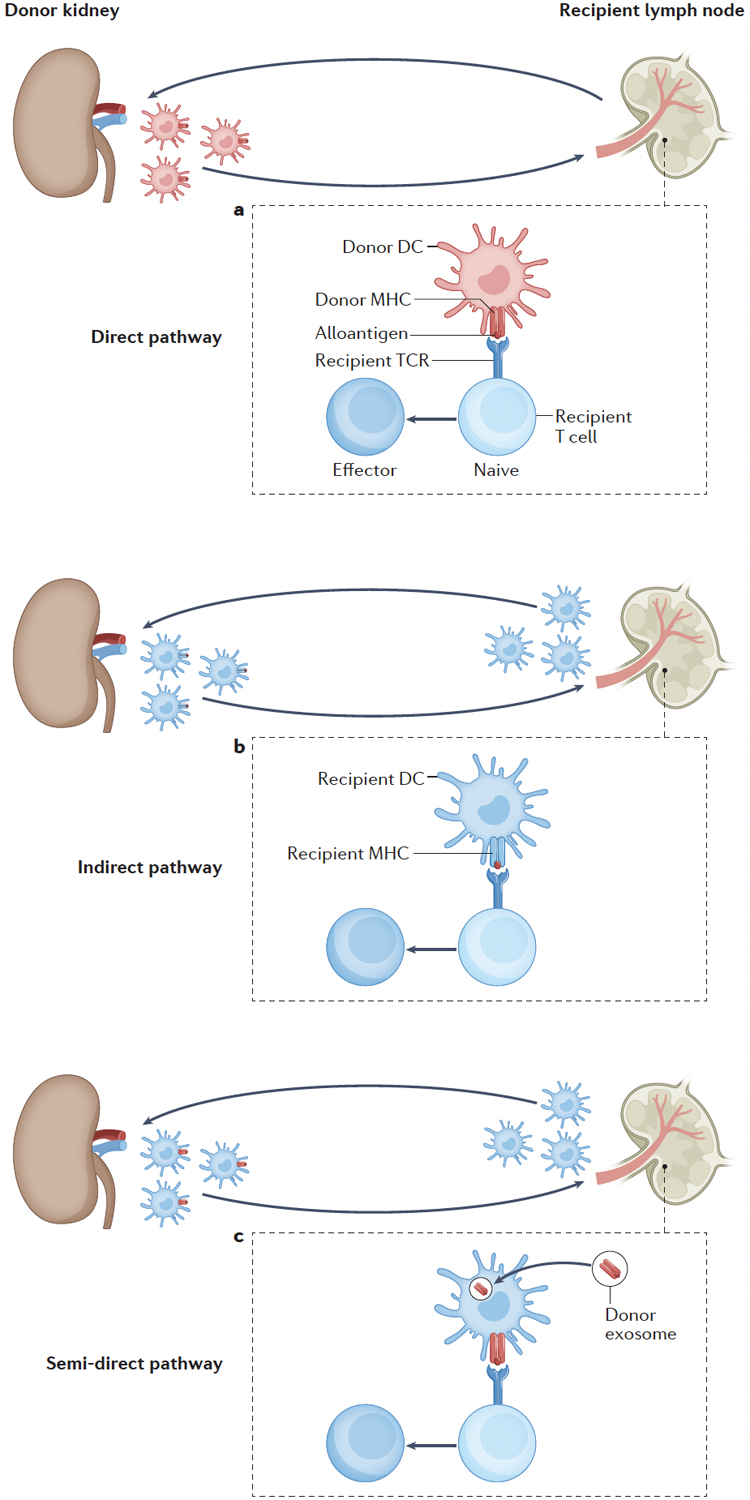
(A) Original hypothesis: More than thirty years ago, seminal publications hypothesized that the initial priming of alloreactive T cells was carried out by donor-derived dendritic cells (DCs) that migrated from the graft (termed passenger leukocytes) to the secondary lymphoid organs of the recipient, where they presented intact donor antigen–MHC complexes directly to recipient T cells (direct pathway of allorecognition) (B) Change of paradigm: Recipient-derived DCs have a key role in initial T cell priming by migrating to the graft, capturing donor antigen and presenting self-restricted alloantigen to recipient T cells in secondary lymphoid organs (indirect pathway of allorecognition) (C) Latest research suggests that ‘cross-decorated’ recipient-derived DCs (that is, recipient DCs that have acquired MHC–alloantigen complexes, notably via donor exosomes) present intact donor class I MHC-antigen complexes to recipient CD8+ T cells in secondary lymphoid organs (semi-direct pathway of allorecognition).
Figure 2. Pathways of allorecognition and therapeutic strategies.
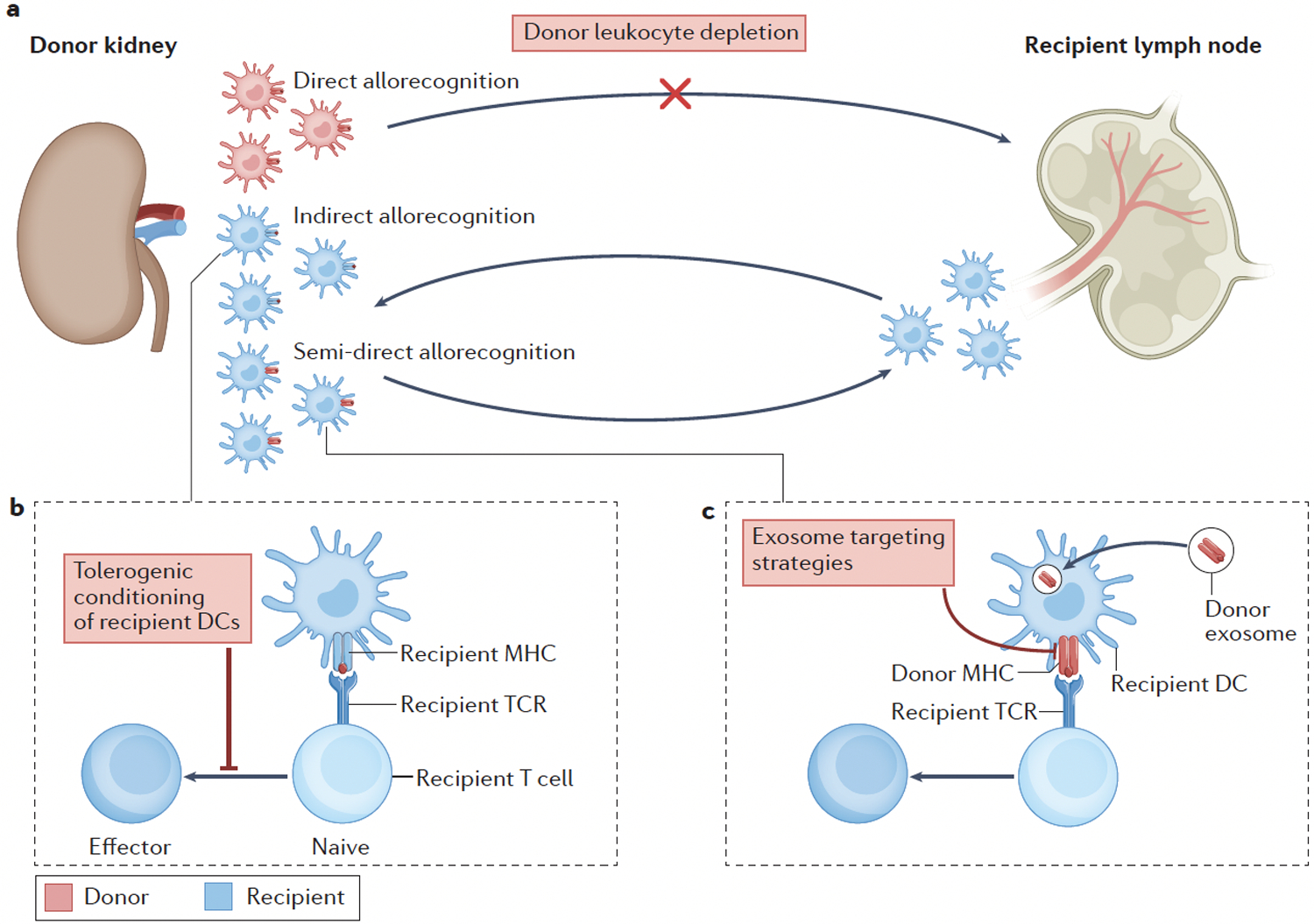
Different therapeutic approaches might target specific pathways of allorecognition. (A) Strategies that target the donor dendritic cells (DCs) that drive direct allorecognition include pre-transplant donor leukocyte depletion. (B) Strategies that target recipient DCs include pharmacological conditioning of recipient DCs in the presence of donor antigen to inhibit alloreactive T cell responses (that is, cell-based ‘negative vaccination’ strategies). (C) Strategies that block the release or capture of donor exosomes might prevent semi-direct allorecognition.
Direct pathway of allorecognition
Numerous studies support the concept that a large proportion of the alloreactive TCR repertoire is directed against donor-derived peptide–MHC complexes6, which comprise self-peptides from the donor complexed with allo-MHC molecules. Given the large number of peptide epitopes that can be presented by allo-MHC, the frequency of TCRs capable of recognizing allogeneic peptide–MHC is extremely high — approximately 1–10% of an individual’s entire TCR repertoire5. The mechanisms by which these donor-derived peptide–MHC complexes are presented to recipient T cells have been studied intensely, and have been at times controversial. More than thirty years ago, a series of seminal publications described the now largely discredited passenger leukocyte theory. This hypothesis postulated that allograft rejection was triggered by the migration of donor-derived DCs from the graft to the recipient’s lymph nodes, where these donor APCs presented donor-derived peptide–MHC complexes directly to recipient T cells (FIG. 1A). This theory was supported by the observations that leukocyte-depleted grafts were not rejected in murine models7–9 and that recipient lymph nodes contained donor-derived DCs10. For several decades, this process was considered to be the dominant pathway responsible for transplant rejection.
Given the initial observations that passenger leukocytes (that is, leukocytes contained within a transplanted organ) could migrate from the graft to recipient lymph nodes to prime alloreactive T cells directly — the direct pathway of allorecognition — depleting passenger leukocytes from the graft before transplantation emerged an approach to target donor-derived DCs and prevent graft rejection (FIG. 2a). In murine models, treating donors with sublethal total body irradiation (TBI) or anti-lymphocyte globulin prior to transplantation prolonged the survival of heart allografts significantly11. However, this approach had limited impact on kidney graft survival in porcine models12. This discrepancy is probably due to the fact that, unlike in mice and rats, endothelial cells in pigs (and in primates) not only express MHC class I, but also MHC class II and the costimulatory molecules required for triggering direct alloreactivity13,14. These characteristics therefore enable direct allorecognition to occur via the graft endothelial compartment even when donor leukocytes are depleted. These findings also highlight important differences between murine and human physiology that affect transplantation biology and must be considered when extrapolating findings from animal studies to humans (BOX 1).
Box 1: Optimizing experimental transplant models.
Current understanding of many aspects of transplantation biology is derived from murine experimental models. However, the limitations of these model systems must be noted because differences between murine and human physiology, as well as caveats of experimental design, have meant that mouse models do not always fully recapitulate human transplantation biology. For example, MHC class II and costimulatory molecules are expressed on human endothelial cells but less so in mice, which results in persistence of donor-derived class II MHC on functional antigen-presenting cells in human transplants13,14,150,167. Moreover, mice housed in specific pathogen-free (SPF) conditions have a low number of memory T cells, including tissue-resident memory T cells, compared with wild or pet store-derived mice, or humans168. These studies highlight the fact that most transplant studies involving SPF-housed mice fail to account for the contribution of environmentally-elicited immunologic memory. Finally, the presence of underlying disease (for example, type 2 diabetes, chronic kidney disease or autoimmunity) probably leads to additional differences in immune subset composition and differentiation status between experimental animals and human transplant recipients169. Addressing these issues in experimental models of transplantation might increase their relevance to human physiology, and hence the number of transplant therapies identified in mice that are successfully translated into clinical practice.
Indirect pathway of allorecognition
Subsequent studies reported that grafts are depleted of donor-derived DCs over time and that the graft APC compartment is repopulated by recipient DCs15–18. This observation suggested that alloantigens might also be recognized conventionally, that is, that donor self-peptides might be processed and presented by recipient APCs19,20. This process was termed the indirect pathway of allorecognition (FIG. 1B) and multiple studies underlined its essential role in graft rejection21–24. A change of paradigm occurred as several groups shifted their attention from donor DCs to the role of recipient DCs in the initial priming of T cells, which involved migration of recipient DCs to the graft, donor antigen capture and presentation of alloantigens to recipient T cells in secondary lymphoid organs18.
The relative contribution of distinct allorecognition pathways during transplant rejection was subsequently investigated. Experimental and human studies suggested that CD4+ T cell responses primed via the direct pathway were limited to the early post-transplant period (that is, within the first few weeks of transplantation25,26), which coincides with the limited lifespan of donor-derived DCs27,28. The new dogma postulated that short-lived donor-derived DCs were the primary drivers of acute allograft rejection via the direct pathway, whereas long-lasting recipient-derived DCs drove chronic rejection via the indirect pathway. However, these assumptions probably oversimplify the complex and dynamic processes that govern alloimmunity following transplantation.
The role of indirect allorecognition was highlighted by experiments involving mTOR inhibition, which could skew recipient DC differentiation and effector function (for example, inflammatory cytokine production) towards a tolerogenic profile and consequently reduce alloreactive T cell responses29. Recipient DCs conditioned with the mTOR inhibitor sirolimus and pulsed with cell-free lysate of donor splenocytes can induce hypo-responsiveness in antigen-specific CD4+ T cells30, inhibit the survival of alloantigen-specific CD8+ T cells31 and, when administered one week before transplant, prolong the survival of MHC-mismatched vascularized heart allografts in mice30. The ability of sirolimus-modified recipient DCs to inhibit T cell responses and promote tolerance in preclinical models of transplant suggests that a cell-based ‘negative vaccination’ strategy (that is, the use of DCs to reduce rather than augment immune responses) could be developed to prevent allograft rejection (FIG. 2b). However, this approach requires further investigation to assess its clinical applicability. Moreover, given the time required to tolerize alloreactive T cells prior to transplantation, this approach would probably only be useful in living donor organ transplantation.
Semi-direct pathway of allorecognition
Further investigations demonstrated that alloantigens could be transferred via extracellular vesicles32,33 from one cultured DC to another34, as well as between different cell types35–37. These in vitro experiments revealed that donor-derived antigen–MHC complexes could be presented by recipient-derived DCs via a semi-direct pathway (FIG. 1c), whereby intact donor-derived class I and class II peptide–MHC complexes ‘cross-decorate’ the surface of recipient APCs38. Extracellular vesicles include exosomes produced in the endosomal compartment, microvesicles or ectosomes derived from the plasma membrane, and apoptotic vesicles released during programmed cell death39. Exosomes and microvesicles contain proteins and RNA, whereas apoptotic vesicles also contain DNA. The surface of extracellular vesicles released by donor APCs can carry antigen–MHC complexes, costimulatory proteins and adhesion molecules. Several studies suggest that extracellular vesicles have a role in graft rejection. Among these, studies of heart transplantation in mice showed that recipient DCs acquired MHC molecules from donor parenchymal cells and presented them as intact molecules to alloreactive CD8+ T cells (that is, semi-direct presentation) and as peptides to CD4+ T cells (that is, indirect presentation), thereby promoting allograft rejection40. Experimental work in other vascularized allograft murine models also confirmed the acquisition of intact donor antigen–MHC complexes by recipient DCs40–44.
Subsequent observations32,33,39 suggested that priming of alloreactive T cells via intact donor antigen–MHC complexes is mostly driven by cross-decorated recipient-derived DCs (semi-direct presentation), rather than by donor-derived DCs (direct presentation), and might represent an essential element of alloimmune responses to transplanted vascularized solid organs. These findings therefore question the aforementioned classical distinction between the direct and indirect allorecognition pathways, and their contribution to acute versus chronic allograft rejection, given that donor-derived parenchymal cells can produce exosomes months or even years after the transplant14. Moreover, although MHC class II+ donor-derived APCs have relatively short lifespans and are lost over time post-transplantation, human graft endothelial cells provide a persistent source of donor-derived MHC class I and class II molecules capable of activating memory T cells directly, as well as a source of allo-MHC complexes for semi-direct recognition14. Alternatively, CD4+ T cells primed via the indirect pathway might interact with and activate recipient APCs that present, either simultaneously or sequentially, intact donor MHC class I molecules to alloreactive CD8+ T cells38.
Notably, extracellular vesicles have also been implicated in the induction of maternal tolerance to fetal alloantigens during pregnancy45–50 and in the spontaneous tolerance of liver allografts in mice51. Whether the ability to steer alloimmunity towards rejection or tolerance depends on the type and cellular origin of extracellular vesicles remains to be fully elucidated. Addressing these research questions will not only help to elucidate the mechanisms underlying allorecognition but might also lead to potential clinical applications. For example, several reports indicate that substantial differences in exosome content and surface markers exist between patients with and without rejection52–55, suggesting a potential role for extracellular vesicles as a biomarker of subclinical rejection. In a cross-sectional human study, the urinary exosomal proteome of patients with acute T cell-mediated rejection (TCMR) had higher levels of proteins such as tetraspanin-1 (encoded by TSPAN1) and hemopexin (encoded by HPX) than that of kidney recipients without rejection53. Another study showed that urinary T cell-derived CD3+ exosomes were also highly expressed in kidney recipients with TCMR compared with those without rejection56 and might reflect T cell infiltration of kidney tubules in the graft, which is associated with cellular rejection57. Another study reported that levels of plasma exosomal mRNA transcripts (IL6ST, CCL4, TNF, CAV1, ACKR1 and SH2D1B) could distinguish antibody-mediated rejection from TCMR in kidney transplant patients55. Of note, these blood samples were collected before the diagnosis of biopsy-proven rejection, suggesting that they might have predictive value and could provide an alternative to invasive kidney biopsies. Exosome-based monitoring post-transplantation might, in the future, help clinicians to diagnose transplant rejection at early stages non-invasively. However, further studies are needed to define the best source of extracellular vesicles (plasma versus urine), optimal molecular signatures (for example, RNA, protein, lipid or carbohydrate) and the most reliable and effective method of extracellular vesicle isolation (for example, ultracentrifugation, density gradient centrifugation, size exclusion chromatography or polymer-based precipitation) for clinical applications. Prospective clinical studies should be carried out to assess the diagnostic performance of these approaches.
In addition to their biomarker potential, exosomes might also have direct therapeutic applications. In a murine model, treatment of heart allograft recipients with exosomes derived from donor bone marrow DCs that were administered intravenously prolonged allograft survival58. Subsequent preclinical studies in rats have yielded similar results59 and emphasize the potential immunoregulatory role of exosomes. By contrast, blocking the release or capturing exosomes associated with rejection (FIG. 2c) might disrupt semi-direct T cell activation and promote tolerance.
Post-priming alloantigen recognition
Following the priming of naive alloreactive T cells in recipient lymph nodes through the antigen presentation processes discussed above, CD4+ and CD8+ T cells differentiate into effector cells that have important roles in graft rejection. CD4+ cells differentiate into specific subsets (TH1 cells, TH2 cells, TH17 cells, follicular helper T cells, regulatory T (Treg) cells) depending on cytokine microenvironment and can carry out multiple functions including direct cytotoxicity, cytokine secretion, and the provision of help for cytotoxic CD8+ T cells and for B cells to produce alloantibodies. CD8+ T cells directly eliminate cells that present non-self epitopes by releasing cytotoxic molecules (for example, granzymes and perforin) or through cell surface interactions that induce apoptosis (mediated, for example, by the binding of Fas ligand (also known as CD95L) on T cells to Fas on target cells).
Activated cognate CD4+ and CD8+ T cells populations infiltrate the graft and orchestrate an inflammatory response within the kidney interstitium by engaging APCs (FIG. 3). These interactions result in the expression of inflammatory cytokines, particularly IFNγ, and IFNγ-induced genes, which induce macrophage activation, loss of parenchymal cell function and intimal arteritis60. The contribution of cytotoxic mechanisms to graft rejection have not been fully elucidated but the main transcriptomic signature associated with TCMR is an IFNγ signature; canonical cytotoxic T cell (CTL)-associated transcriptomic signatures are heterogeneous among patients with TCMR61. Of note, one study reported that expression of CTL-associated transcripts preceded the development of tubulitis in TCMR62. Importantly, post-priming antigen recognition within the graft can further modulate T cell effector function.
Figure 3-. Post-priming alloantigen recognition in the graft and potential therapeutic strategies.
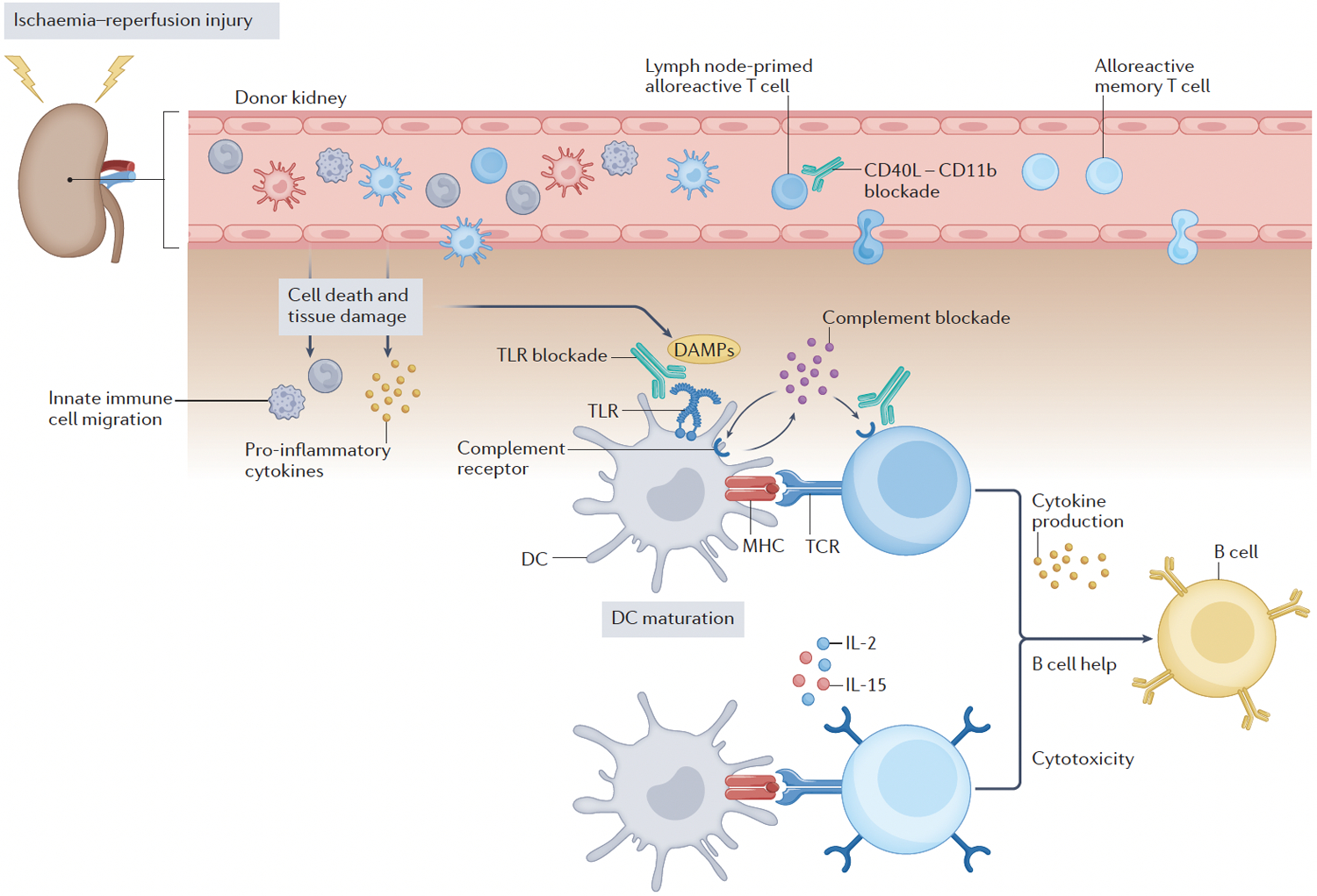
Beyond their contribution to the initial T cell activation in secondary lymphoid tissues, dendritic cells (DCs) have a crucial role in rejection by infiltrating the transplanted organ, forming cognate interactions with effector T cells (alloreactive cells primed in the lymph-nodes or memory T cells that migrate to the graft). Pharmacological blockade of CD40L–CD11b interactions might prevent leukocyte infiltration into the graft. Ischaemia–reperfusion injury in transplanted organs causes cells death and tissue damage, leading to the release of damage-associated molecular patterns (DAMPs). These DAMPs are recognized by pattern recognition receptors, such as Toll-like receptors (TLRs), which induces the activation of innate immune cells. TLR ligation induces autocrine and paracrine production of complement components (C3a and C5a) in DCs, which mediates their maturation and enables them to induce effector T cell activation, expansion and cytokine production, thereby promoting graft rejection. Activated CD8 T cells can exert direct cytotoxicity and CD4 T cells can also provide help to alloreactive B cells, enabling them to produce alloreactive antibodies. TLR and complement receptors therefore represent another potential therapeutic target in transplantation.
DCs and alloantigen presentation in the graft
Beyond their contribution to the initial activation of T cells in secondary lymphoid tissues, one study showed that recipient-derived DCs have a crucial role in rejection by infiltrating the transplanted organ, forming cognate interactions with effector T cells and sustaining T cell responses in situ63 (FIG. 3). Antigen presentation by recipient DCs in the graft involved both self-restricted alloantigen (that is, alloantigen presented on recipient MHC — indirect pathway, FIG. 1b) and intact donor MHC–antigen complexes (semi-direct pathway, FIG. 1c). The same study also provided novel insights into the nature, origin and function of graft-infiltrating recipient DCs. Most of these cells were non-conventional CD11b+ CD11c+ DCs that produced IL-12, originated from non-classical monocytes and were functionally distinct from other monocyte-derived cells in the graft63. These data are similar to those reported in studies showing that non-classical monocytes are present in sterile inflammation (for example, in arteriosclerosis64) and differ from findings in infection models, where the majority of DCs derive from classical monocytes and have an inflammatory tumour necrosis factor (TNF)-producing phenotype65–67.
Transplantation might represent a unique setting in which allogenic stimuli, combined with a sterile inflammation environment, induce a specific differentiation program in recipient monocytes. Recipient DCs might preferentially induce T cell differentiation towards a TH1 cell profile, which is the dominant T cell phenotype in rejection68. The additional signals received in the graft by the recipient DCs (leading to their differentiation) and by allogenic T cells upon re-engagement with recipient DCs (leading to further differentiation and establishment of effector function) remain to be elucidated. Targeting recipient DCs in the graft, or their monocyte precursors69 could theoretically interrupt or prevent rejection in patients that are already primed against donor alloantigen (that is, high-risk transplant recipients) and is therefore an area of active research70.
Allorecognition in memory T cells
Pre-existing alloreactive memory T cells represent a substantial challenge in transplantation. Recipient memory cells can be generated either by prior exposure to allogeneic MHC molecules (for example, owing to prior transplants, blood transfusions or pregnancy) or owing to crossreactivity of endogenous alloreactive memory T cells specific for microbial or environmental antigens that mimic alloantigen–MHC complexes (that is, heterologous immunity)71,72. Compared with their naive counterparts, memory T cells have a lower activation threshold and enhanced effector function73,74. Moreover, memory cells use the same mechanisms as effector T cells to access the graft75 and can therefore cause rejection without the need for priming in secondary lymphoid tissues76 (FIG. 3). These alloreactive memory cells are also resistant to several immunosuppressive treatments, such as costimulatory blockade (for example, inhibition of the CD28–CD80 or CD28–CD86 pathways with belatacept or blocking of the CD40–CD40L pathway)77–80. This resistance might be due to a reduced requirement for costimulation overall81 or the use of ‘non-classical’ costimulatory pathways such as those involving TNF ligand superfamily member 4 (also known as OX-40L) and TNF receptor superfamily member 9 (also known as 41BB)82. Moreover, high expression of the IL-2 and IL-15 receptors can enhance memory T cell survival and proliferation83. In infection models, DCs outside of secondary lymphoid organs are important initiators of memory T cell recall responses but whether these APCs have a similar role in transplantation following antigen recognition within the graft this remains to be demonstrated. A better understanding of the mechanisms underlying memory recall responses is an essential prerequisite for the development of clinical applications to improve transplant survival in sensitized patients84.
Several therapeutic approaches have been developed to target alloreactive memory T cell responses. Lympho-ablation during induction therapy at the time of transplant is commonly used in clinical practice to overcome pre-existing alloimmunity. Alefacept is a lymphocyte function-associated antigen 3 (LFA3)–IgG1 fusion protein that targets the LFA3 receptor CD2, which is upregulated on CD45RO+ effector or memory cells. This fusion protein is currently used to treat severe psoriasis. Preliminary work showed promise for alefacept in targeting and depleting alloreactive effector or memory cells in solid organ and bone marrow transplantation85–89 but its efficacy was not demonstrated in clinical trials. Limiting trafficking of alloreactive memory T cells is another therapeutic approach under investigation. Reagents blocking leukocyte function-associated antigen 1 (LFA1, also known as αLβ2 integrin) and very late antigen 4 (VLA4, also known as α4β1 integrin) prolonged allograft survival in experimental transplantation90–93 and efficacy was demonstrated in preliminary small cohort clinical studies94–96. However, identification of a risk of progressive multifocal leukoencephalopathy (PML) halted further development of these reagents for use in clinical transplantation97.
T cell migration into the graft
Another key element of alloimmune responses is effector and memory T cell migration into the graft. Antagonism of the signal 2 CD40–CD40L pathway, which can induce costimulation blockade and induces long-term graft survival in preclinical models, might also block T cell graft infiltration98,99. In an allogenic murine skin graft model100, we found that CD40L blockade was more effective in prolonging graft survival than CD40 blockade, suggesting the involvement of an alternative CD40L receptor. Notably, CD40L had been reported to bind CD11b to mediate leukocyte recruitment in atherosclerosis101,102. We found that, in our murine skin graft model100, CD40L–CD11b antagonism reduced the frequency of graft-infiltrating CD8+ T cells and innate immune cells (FIG. 3 and 4a) and increased the efficacy of anti-CD40 in prolonging allograft survival significantly compared with anti-CD40 alone. Further investigation is required to determine what type of cells express CD11b and the underlying mechanisms by which the CD40L–CD11b interaction modulates T cell trafficking in the graft. Nonetheless, these observations suggest that targeting the CD40L–CD11b interaction could synergize with anti-CD40 treatment to prevent allograft rejection.
Figure 4. T cell migration to the graft.
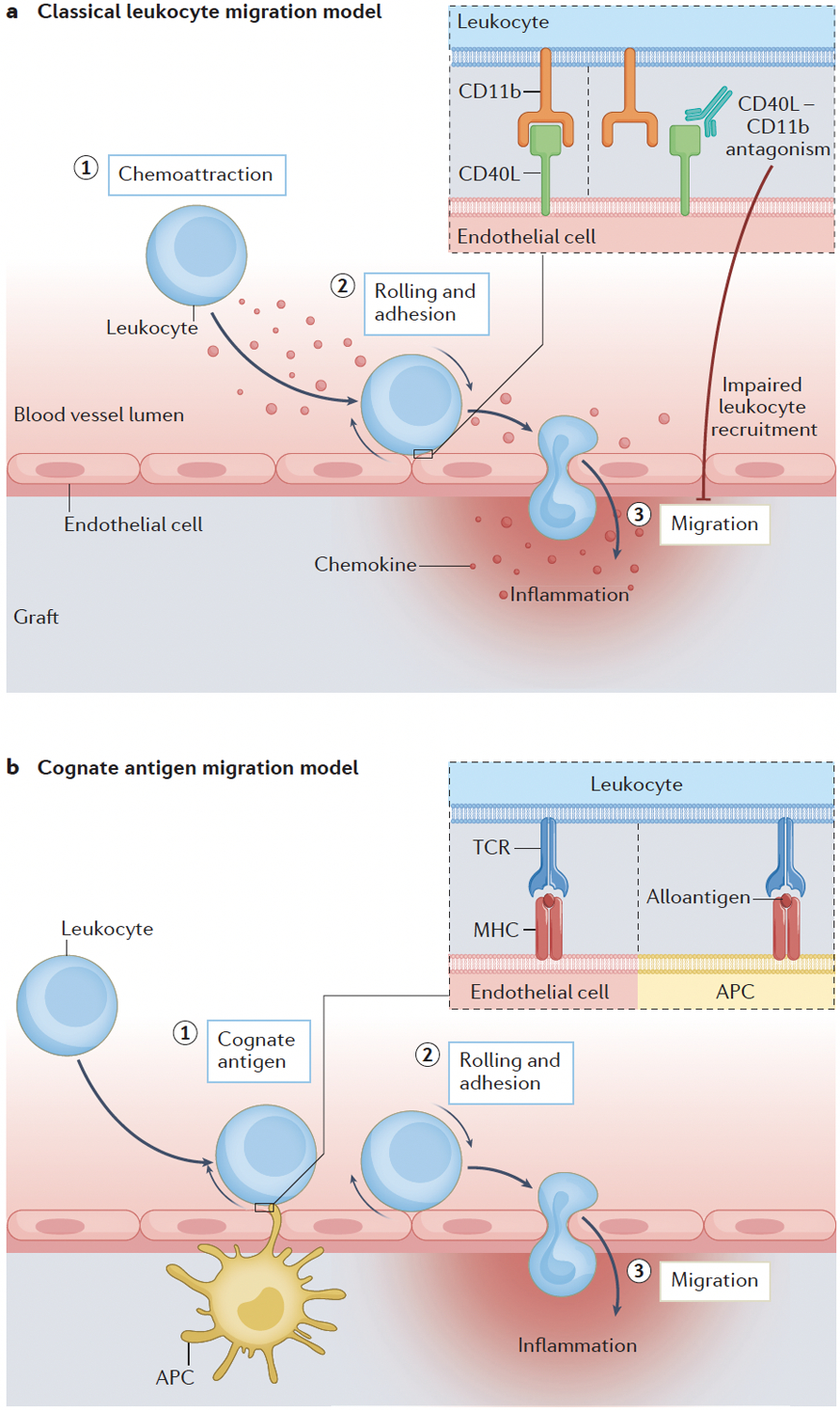
(A) Chemokines displayed on the inflamed endothelium attract T cells and trigger their migration to the graft. The interaction between CD11b and CD40L mediates rolling and adhesion, which facilitates leukocyte migration to the graft100. Pharmacological antagonism of CD40L–CD11b interactions reduces the frequency of graft-infiltrating CD8+ T cells and innate immune cells in murine models. (B) Antigens presented either by endothelial cells or bone marrow-derived antigen-presenting cells have a key role in T cell adhesion and transmigration to the graft75; this process is independent of chemokine receptor signalling.
The paradigm of classical leukocyte migration postulates that chemokines displayed on the inflamed endothelium engage Gαi-coupled chemokine receptors on rolling T cells and trigger their adhesion and transmigration via integrin-dependent mechanisms (FIG. 4a). However, targeting chemokine receptors had a negligible effect in reducing T cell graft infiltration103–105, which challenged this paradigm. An in vivo imaging study of murine models of heart and kidney transplantation75 showed that, in contrast to the prevailing view, the key step in allo-specific CD8+ effector T cell migration into the graft was the presentation of cognate antigen, either by graft endothelial cells or bone marrow-derived APCs (Figure 4b). This work demonstrated that interaction with cognate antigen was necessary for donor-specific T cell adhesion and transmigration to the graft, and that these steps were independent from Gαi signalling. Furthermore, this study showed that non-antigen-specific (that is, bystander) effector T cells remained dependent on Gαi and required the presence of antigen-specific effector T cells to infiltrate the graft. Targeting T cell allorecognition might be more effective than targeting chemokine receptor signalling to prevent T cell trafficking into allografts.
Innate–adaptive immunity crosstalk
The context and quality of antigen presentation is paramount to the activation and differentiation of alloreactive T cells but additional signals from innate immune pathways can fine-tune the programmed differentiation of alloreactive responses. As discussed below, crosstalk between innate and adaptive immunity can either enhance or suppress donor-reactive alloimmunity.
TLR-induced inflammation
Ischaemia–reperfusion injury (IRI) occurs at the time of transplantation when the graft blood supply is abruptly re-established after a period of interrupted perfusion; IRI induces sterile inflammation (FIG. 3). The cell death and tissue damage that occur after blood flow is restored to previously ischaemic tissues cause the release of endogenous damage-associated molecular patterns (DAMPs), which are usually hidden from the immune system inside cells and function as ‘danger signals’. DAMPs are recognized by the innate immune system via pattern recognitions receptors (PRRs) such as the Toll-like receptors (TLRs).
The binding of DAMPs to TLRs triggers a cascade of intracellular signalling events that results in the production of pro-inflammatory cytokines and migration of innate immune cells to the graft (FIG. 3). This sterile inflammation precedes T cell recruitment and helps to drive graft rejection106,107. In animal models and human studies, TLRs are upregulated in the graft following IRI108. The importance of TLRs in the transplant setting was demonstrated in a murine model of skin transplantation, where minor-antigen mismatched allograft rejection could not occur in the absence of MYD88; this adaptor protein is required for signalling downstream of all TLRs (except for TLR3)109,110. Interestingly, the same study showed that MYD88 deficiency did not prevent rejection in the setting of major MHC mismatch, suggesting that MYD88-dependent pathways might augment priming of T cells via the indirect pathway, for which the frequency of cognate T cells is typically low.
Subsequent studies have focused on the role of TLR2 and TLR4, given that several DAMPs associated with solid organ injury can be recognized by both of these TLRs111,112. In a model of murine lung transplantation, knockdown of TLR2 and TLR4 in the recipient inhibited DAMP-mediated acute rejection and reduced the generation of alloreactive effector T cells113. TLR expression patterns were also associated with worse clinical outcomes after transplantation. Kidney114,115 and liver116 transplant recipients with allograft rejection had higher levels of TLR2 and TLR4 than those with stable grafts.
Several research groups have focused on elucidating the mechanisms by which inflammation is induced after organ transplantation117,118 to guide the development of novel therapeutics. Several antibodies or small molecular antagonists targeting TLR2 and TLR4 signalling or their ligands are currently in development or being tested in clinical trials119,120 (FIG. 5a). Importantly, broad blockade of the innate immune response could compromise host defence profoundly and therefore increase the risk of infection and cancer.
Figure 5. Targeting the crosstalk between the innate and adaptive immune system.

(A) Toll-like receptors (TLRs) contribute to the activation of innate immune cells following graft-related ischaemia–reperfusion injury. MYD88-dependent TLR signalling leads to the activation of MAP kinases and nuclear factor κ-B (NF-κB) pathways, and the production of pro-inflammatory cytokines, therefore representing a potential therapeutic target. (B) TLR ligation induces dendritic cell (DC) autocrine and paracrine production of complement components (C3a and C5a), which mediates DC maturation, and effector T cell activation and expansion, thereby promoting graft rejection. Complement receptors represent another potential therapeutic target in transplantation.
T cell complement receptors
The complement cascade and its network of tightly regulated proteins has an important role in inflammation and host defence, but can also contribute to allograft rejection. Complement activation has been reported in both the donor (especially in deceased donors, in whom activation occurs rapidly after cardiac arrest or brain death) and the recipient (via donor-specific alloantibodies or as a result of ischaemia–reperfusion lesions)121. Binding of the anaphylatoxins C3a and C5a to their receptors, which are expressed by a large number of cells, including neutrophils, monocytes, macrophages, dendritic cells and endothelial cells122, can enhance inflammation, cytokine and chemokine production, and leukocyte recruitment. In addition to these traditional innate immune functions, the complement system also contributes to the adaptive immune response. Complement proteins such as C3a and C5a are produced locally by APCs and T cells during cognate interactions and participate fully in the T cell priming process via cytokine production and activation of both cell types, which results in the transduction of costimulatory and survival signals in naive T cells123,124.
DC maturation is commonly initiated by TLR signalling and enables the induction of pro-inflammatory T cell immunity. TLR-induced DC maturation has also been linked to complement activation. In a murine heart transplant model, ligation of TLR3, TLR4 or TLR9 induced DC-intrinsic production of complement components and TLR-mediated DC maturation required autocrine C3a and C5a receptor signaling125. Autocrine and/or paracrine C3a and C5a receptor signalling were also involved in activation and expansion of CD4 T cells124,125 (promoted TH1 cell differentiation126) and of CD8 T cells127, and therefore contributed to T cell-dependent transplant rejection (FIG. 5b). Moreover, complement components produced locally (for example, C3a and C5a) inhibit the generation and stability of Treg cells in mice and humans125,128–130, which further enhances the alloreactive effector T cell response and promotes rejection. Preclinical work showed that pharmacological blockade of C3 receptor 1 (C3R1) in combination with tacrolimus prolonged cardiac allograft survival in mice127. Importantly, the many physiological roles of complement might complicate its targeting for the control of T cell-mediated rejection.
FcγR2b inhibitory pathway
Expression of the inhibitory Fc receptor FcγR2b was initially only observed on B cells131 and innate immune cells132, but was later also detected on memory T cells at the mRNA and protein level133–135. Furthermore, we showed that, after transplantation, a subset of effector CD8+ T cells expressed FcγR2b following activation and multiple rounds of division, both in mice and humans136. Unexpectedly, FcγR2b did not bind to IgG but rather interacted with the immunosuppressive cytokine fibrinogen-like 2 (FGL2)137, which induced CD8+ T cell apoptosis via activation of caspases 3 and 7. Deletion of Fcgr2b specifically on CD8+ T cells in murine models led to the accumulation of CD8+ effector T cells and accelerated graft rejection. By contrast, increased expression of FcγR2b on CD8+ T cells correlated with lack of rejection following immunosuppressant withdrawal in a kidney transplantation clinical trial138. FGL2 can be produced by both effector and Treg cells139,140 but the conditions under which FcγR2b and FGL2 are upregulated to promote tolerance versus rejection are still being elucidated. Nonetheless, these findings suggest that strategies to promote FcγR2b–FGL2 interactions might enable therapeutic manipulation of CD8+ T cell responses in transplantation.
Role of survival cytokines:
The cytokine microenvironment is another key determinant of alloimmune responses and survival cytokines seem to have an important role in supporting memory responses following transplantation. In the setting of infection, maintenance of immunological memory is crucial to ensure host protection in the event of re-infection. However, in transplantation, persistence of such memory responses owing to an allo-sensitizing event or heterologous immunity71,141 represents a threat to graft tolerance and survival142.
IL-7
In a murine model143, transplantation preferentially induced a killer cell lectin-like receptor subfamily G member 1 (KLRG1)lo CD127hi (also known as the IL-7 receptor α-chain) differentiation program in antigen-specific CD8+ T cells. Specifically, CD8+ T cells primed by a skin graft expressed higher levels of CD127 than cells specific for a murine homolog of Epstein-Barr virus (EBV), which is known to elicit allo-crossreactive T cells141,144,145. These phenotypic differences between graft- and microbe-elicited T cells were present at the same acute phase timepoint, when antigen was still present. Thus, the data suggest that allograft-elicited CD8+ T cell responses might be more reliant on IL-7-mediated signals than microbially-activated CD8+ T cells. The cellular and molecular pathways by which sensitization history and priming conditions affect memory differentiation might provide novel biomarkers and therapeutic targets in transplantation. For example, expression of CD127 on memory cells could be used to predict the strength of subsequent recall responses and stratify the immunological risk following transplantation. Furthermore, this study also showed that donor-reactive memory T cells induced by prior alloimmunization might be more susceptible to combined CD127 and costimulation blockade than infection-induced donor-reactive memory T cells143. Given its role in memory cell maintenance in preclinical transplant models, the IL-7 signalling pathway might represent a target for therapeutic intervention (FIG. 6a).
Figure 6. Targeting survival cytokines in preclinical model.

(A) CD127 (IL-7 receptor ɑ-chain) blockade. One study143 showed that ovalbumin (OVA)-specific CD8+ memory cells expressed different levels of CD127 depending on whether the animals had been infected with an OVA-expressing virus or received an OVA-expressing skin graft. After re-challenged with an OVA expressing skin graft, treatment with anti-CD127 combined with costimulation blockade (CTLA4-Ig, belatacept and anti-CD40L) improved graft survival in mice that had been pre-challenged with the OVA-expressing skin graft but exacerbated rejection in the virus-infected animals. (B) CD122 (common β-chain for IL-2R and IL-15R) blockade. A combination of belatacept and anti CD122 prolonged survival compared with belatacept alone in non-human primates that underwent bi-nephrectomy and subsequent kidney transplantation with a fully MHC mismatched allograft83.
IL-15
Belatacept, which is a high-affinity version of CTLA4–Ig and blocks CD80 or CD86 ligation to CD28, has been approved for use in kidney transplant recipients146. Despite the long-term benefits associated with its use in kidney transplant recipients, including improved estimated glomerular filtration rate and lower cardiovascular risk, a subset of patients treated with belatacept have higher rates of acute T cell-mediated rejection, suggesting a role for costimulation-independent memory T cell alloreactivity147. Signal 3 cytokines IL-2 and IL-15 can support costimulation-independent rejection83. Interestingly, studies in mouse models showed that costimulation blockade-resistant rejection was independent of the high-affinity IL-2 receptor α chain (IL-2Rα; also known as CD25), which highlighted IL-15 as a potential key mediator of co-stimulation-independent responses148. Of note, this work raises questions about the utility of basiliximab (non-depleting treatment targeting CD25) induction therapy because it suggests that it might not be effective in controlling alloreactive memory T cells. This finding is in line with clinical data collected in patients showing that basiliximab induction is associated with higher rates of acute rejection than anti-thymoglobulin (polyclonal T cell-depleting antibodies) induction149. However, pharmacological blockade of both IL-2 and IL-15 signalling with a single humanized monoclonal antibody against CD122 (the shared β-chain of the IL-2 and IL-15 receptors) in combination with belatacept prolonged kidney allograft survival in non-human primates (NHPs) compared with belatacept monotherapy83. Moreover, in vitro assays demonstrated that human CD28neg memory T cells, which are independent of the CD28 checkpoint, required IL-15 for proliferation148. Of note, in contrast to other cytokines that are secreted into the extracellular environment, IL-15 is displayed in trans on the surface of the APC or target cell, in a complex with the IL-15Rα chain150. This process is termed trans-presentation and IL-15 might therefore be more functionally similar to a surface-bound costimulatory ligand than a soluble cytokine. These preclinical findings suggest that targeting CD122 might be useful as adjunct immunosuppression to optimize costimulation blockade therapy after transplantation (FIG. 6b).
Transplant-specific T cell programming
As discussed above, transcriptional networks regulate distinct programs of gene expression and eventually control the differentiation and function of effector or memory T cells. Importantly, several reports in the past few years have suggested that context-dependent programs exist and thus information from models of infection might not be globally applicable to alloimmune responses. Understanding the transplant-specific role of these differentiation programs and transcriptional pathways in donor-specific T cells is essential to develop future therapeutic strategies that can suppress alloimmunity while preserving protective anti-microbial immune responses.
mTOR inhibition
Evidence of the differential T cell programs elicited by grafts versus pathogens is very clearly observed in the mammalian target of rapamycin (mTOR) pathway. The mTOR inhibitor rapamycin is a well-known effective inhibitor of alloreactive T cell proliferation and allograft rejection151. Seminal studies in NHP models investigated the effect of rapamycin immunosuppression on vaccine responses and unexpectedly reported that mTOR inhibition enhanced vaccinia-specific CD8+ T cell responses in rhesus macaques152. Mechanistic studies in mouse models also showed that, in the context of infection, mTOR inhibition improved memory CD8+ T cell responses and protective anti-viral immunity by promoting the differentiation of CD127hi KLRG-1lo memory precursor effector cells (MPECs)153. By contrast, mTOR inhibition did not enhance MPEC differentiation when CD8+ T cells were stimulated in the context of transplantation154. However, the molecular signals that underlie these differential CD8+ T cell responses to mTOR inhibition are unclear.
Coronin 1–PDE4–cAMP axis
Coronin 1, which is a tryptophan-aspartic acid (WD) repeat family member, regulates T cell homeostasis, especially in naive T cells155–157. One study158 identified a coronin 1–cAMP-specific 3’,5’-cyclic phosphodiesterase 4 (PDE4)–cAMP axis that, when disrupted, induced transplant tolerance while maintaining anti-pathogen immunity. Mechanistically, coronin 1-deficiency suppressed allo-specific responses by raising cAMP levels; cAMP is a key regulator of T cell immunity159–161. However, ligation of CD28 by CD80 upregulated on microbe-infected APCs enabled T cells to overcome cAMP-mediated immunosuppression and maintain anti-pathogen immunity. These observations suggest that targeting coronin 1 or the coronin 1-dependent cAMP pathway could promote transplantation tolerance without impairing host anti-microbial immunity162. Importantly, coronin 1 is expressed in a variety of cell types163 and any therapeutic intervention would therefore have to consider potential off-target toxicities carefully.
Transplant-specific role of IRF4
In 2017, two studies reported a role for the TCR-responsive transcription factor interferon regulatory factor 4 (IRF4) in T cell exhaustion. Specifically, IRF4 promoted exhaustion in chronic viral infection164 but dampened it in response to tissue allografts165. The allograft study165 showed that IRF4 was a key promoter of CD4+ T cell responses during transplantation and that Irf4 deletion in mice resulted in long-term allograft survival. Specifically, mice with fully MHC-mismatched heart allografts survived indefinitely when Irf4 was deleted in CD4+ T cells and without the use of other immunosuppressive therapies. By contrast, in the model of chronic viral infection, IRF4 deficiency prevented CD8+ T cell exhaustion and enabled functional CD8+ T cell responses. Mechanistically, whereas IRF4 seems to repress programmed cell death protein 1 (PD-1) expression on CD4+ T cells in the context of transplantation, it promotes PD-1 expression on CD8+ T cells in the context of chronic viral infection. These data suggest either a differential role for IRF4 on CD4+ and CD8+ T cells, or that targeting IRF4 might inhibit alloimmunity and achieve transplant tolerance while enabling protective immunity. In support of the latter hypothesis, a preclinical study166 showed that the proteasome inhibitor bortezomib suppressed acute rejection in mice by downregulating IRF4 expression, which decreased the proliferation and differentiation of CD4+ follicular helper T cells.
Conclusions
Understanding of how T cells recognize donor-derived alloantigens and of how these allorecognition events contribute to allograft rejection has advanced substantially over the last 20 years. Many studies have clarified where and when direct, indirect and semi-direct alloantigen presentation takes place and how these distinct pathways modulate the quantity and quality of alloreactive T cell priming. T cell allorecognition in solid organ transplantation is clearly complex and is modified by distinct but overlapping antigen processing and presentation pathways. Elucidating the allorecognition mechanisms and the transplant-specific pathways that are involved in alloimmune responses will yield novel strategies to overcome the barriers to allograft tolerance. Transplant rejection can involve directly-primed CD4+ and CD8+ T cells, indirectly-primed CD4+ T cells and semi-directly primed CD8+ T cells, which are probably primed by donor peptide–MHC complexes that originate from many graft cell types, including hematopoietic, parenchymal, and stromal cells. These observations revealed the diversity and complexity of the alloreactive TCR repertoire. Notably, this diversity in the mechanisms underlying antigen selection, processing and presentation complicates the identification of immunodominant alloreactive T cell epitopes to facilitate the tracking of donor-reactive T cells, similar to what has been achieved in tracking virus-specific T cells in viral immunology.
In addition to the pathways of antigen presentation, the priming environment present during alloantigen recognition, which is distinct to that of infection, uniquely modulates the differentiation programs of alloreactive T cells and might offer promising therapeutic targets. Perhaps the differential expression and function of the mTOR, IRF4 and coronin 1 pathways observed in alloreactive versus pathogen-elicited T cells are due to differences in the abundance or quality of PRR signalling during T cell priming. Alternatively, these differences might result from unique features of the transplant-specific antigen-presentation pathways discussed above. Acknowledging these discrepancies between infection and transplantation is important, especially as many inferences about transplant immunology have been extrapolated from observations made in infectious models. These findings highlight the paramount importance of testing fundamental aspects of T cell priming, differentiation, trafficking and survival in relevant transplant models.
Key points.
Direct, indirect and semi-direct alloantigen presentation all have important and potentially distinct roles in priming effective alloimmune responses. Semi-direct presentation occurs when recipient dendritic cells acquire donor peptide–MHC complexes in graft-draining secondary lymphoid organs by capturing clusters of donor-derived extracellular vesicles
Allospecific T cell responses encounter antigen and undergo programmed differentiation in secondary lymphoid organs but their effector response is fine-tuned by further antigen presentation within the graft
Pre-existing alloreactive memory T cells represent a substantial challenge in transplantation given their low activation threshold and resistance to costimulatory blockade. Pre-clinical data show that pharmacological blockade of the IL-2 and IL-15 receptors might be useful as adjunct immunosuppression to optimize costimulation blockade therapy after transplantation.
Innate–adaptive immunity crosstalk has an important role in transplant rejection and these pathways might be a source of potential therapeutic targets.
Key differences in priming conditions can induce distinct differentiation programs in graft-elicited versus microbe-elicited T cell responses, including the differential expression and function of pathways involving mammalian target of rapamycin, interferon regulatory factor 4 and coronin 1.
Footnotes
Competing interests
M.L.F. has received speaking honoraria from Veloxis Pharmaceuticals and Sanofi Inc. The other authors declare no competing interests.
REFERENCES
- 1.Singer A, Munitz TI, Golding H, Rosenberg AS & Mizuochi T Recognition requirements for the activation, differentiation and function of T-helper cells specific for class I MHC alloantigens. Immunol Rev 98, 143–170 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Bolton EM, Armstrong HE, Briggs JD & Bradley JA Cellular requirements for first-set renal allograft rejection. Transplant Proc 19, 321–323 (1987). [PubMed] [Google Scholar]
- 3.Bolton EM, Gracie JA, Briggs JD, Kampinga J & Bradley JA Cellular requirements for renal allograft rejection in the athymic nude rat. J Exp Med 169, 1931–1946, doi: 10.1084/jem.169.6.1931 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashwell JD, Chen C & Schwartz RH High frequency and nonrandom distribution of alloreactivity in T cell clones selected for recognition of foreign antigen in association with self class II molecules. J Immunol 136, 389–395 (1986). [PubMed] [Google Scholar]
- 5.Suchin EJ et al. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol 166, 973–981 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Sherman LA & Chattopadhyay S The molecular basis of allorecognition. Annual Review of Immunology 11, 385–402 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Talmage DW, Dart G, Radovich J & Lafferty KJ Activation of transplant immunity: effect of donor leukocytes on thyroid allograft rejection. Science 191, 385–388, doi: 10.1126/science.1082167 (1976). [DOI] [PubMed] [Google Scholar]
- 8.Lechler RI & Batchelor JR Restoration of immunogenicity to passenger cell-depleted kidney allografts by the addition of donor strain dendritic cells. J Exp Med 155, 31–41, doi: 10.1084/jem.155.1.31 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietra BA, Wiseman A, Bolwerk A, Rizeq M & Gill RG CD4 T cell-mediated cardiac allograft rejection requires donor but not host MHC class II. J Clin Invest 106, 1003–1010, doi: 10.1172/JCI10467 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen CP, Morris PJ & Austyn JM Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med 171, 307–314, doi: 10.1084/jem.171.1.307 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oluwole S et al. Donor pretreatment: rat heart allograft survival and measurement of passenger leukocyte depletion with indium-111. Transplantation 30, 31–33 (1980). [PubMed] [Google Scholar]
- 12.Tai HC et al. Attempted depletion of passenger leukocytes by irradiation in pigs. J Transplant 2011, 928759, doi: 10.1155/2011/928759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrahimi P et al. Blocking MHC class II on human endothelium mitigates acute rejection. JCI insight 1, doi: 10.1172/jci.insight.85293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage CO, Hughes CC, McIntyre BW, Picard JK & Pober JS Human CD4+ T cells proliferate to HLA-DR+ allogeneic vascular endothelium. Identification of accessory interactions. Transplantation 56, 128–134, doi: 10.1097/00007890-199307000-00024 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Grau V, Herbst B & Steiniger B Dynamics of monocytes/macrophages and T lymphocytes in acutely rejecting rat renal allografts. Cell Tissue Res 291, 117–126, doi: 10.1007/s004410050985 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Penfield JG et al. Transplant surgery injury recruits recipient MHC class II-positive leukocytes into the kidney. Kidney Int 56, 1759–1769, doi: 10.1046/j.1523-1755.1999.00741.x (1999). [DOI] [PubMed] [Google Scholar]
- 17.Saiki T, Ezaki T, Ogawa M & Matsuno K Trafficking of host- and donor-derived dendritic cells in rat cardiac transplantation: allosensitization in the spleen and hepatic nodes. Transplantation 71, 1806–1815, doi: 10.1097/00007890-200106270-00017 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Celli S, Albert ML & Bousso P Visualizing the innate and adaptive immune responses underlying allograft rejection by two-photon microscopy. Nat Med 17, 744–749, doi: 10.1038/nm.2376 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Benichou G, Takizawa PA, Olson CA, McMillan M & Sercarz EE Donor major histocompatibility complex (MHC) peptides are presented by recipient MHC molecules during graft rejection. J Exp Med 175, 305–308, doi: 10.1084/jem.175.1.305 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benichou G et al. Limited T cell response to donor MHC peptides during allograft rejection. Implications for selective immune therapy in transplantation. J Immunol 153, 938–945 (1994). [PubMed] [Google Scholar]
- 21.Gallon L et al. The indirect pathway of allorecognition. The occurrence of self-restricted T cell recognition of allo-MHC peptides early in acute renal allograft rejection and its inhibition by conventional immunosuppression. Transplantation 59, 612–616 (1995). [PubMed] [Google Scholar]
- 22.Harris PE, Cortesini R & Suciu-Foca N Indirect allorecognition in solid organ transplantation. Rev Immunogenet 1, 297–308 (1999). [PubMed] [Google Scholar]
- 23.Gould DS & Auchincloss H Direct and indirect recognition: the role of MHC antigens in graft rejection. Immunol Today 20, 77–82, doi: 10.1016/s0167-5699(98)01394-2 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Ali JM, Bolton EM, Bradley JA & Pettigrew GJ Allorecognition pathways in transplant rejection and tolerance. Transplantation 96, 681–688, doi: 10.1097/TP.0b013e31829853ce (2013). [DOI] [PubMed] [Google Scholar]
- 25.Baker RJ et al. Loss of direct and maintenance of indirect alloresponses in renal allograft recipients: implications for the pathogenesis of chronic allograft nephropathy. J Immunol 167, 7199–7206, doi: 10.4049/jimmunol.167.12.7199 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Haynes LD et al. Donor-specific indirect pathway analysis reveals a B-cell-independent signature which reflects outcomes in kidney transplant recipients. Am J Transplant 12, 640–648, doi: 10.1111/j.1600-6143.2011.03869.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali JM et al. Diversity of the CD4 T Cell Alloresponse: The Short and the Long of It. Cell Rep 14, 1232–1245, doi: 10.1016/j.celrep.2015.12.099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreisel D et al. Vascular endothelium does not activate CD4+ direct allorecognition in graft rejection. J Immunol 173, 3027–3034, doi: 10.4049/jimmunol.173.5.3027 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Hackstein H et al. Rapamycin inhibits IL-4--induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 101, 4457–4463 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Taner T, Hackstein H, Wang Z, Morelli AE & Thomson AW Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am J Transplant 5, 228–236, doi: 10.1046/j.1600-6143.2004.00673.x (2005). [DOI] [PubMed] [Google Scholar]
- 31.Fischer RT, Turnquist HR, Wang Z, Beer-Stolz D & Thomson AW Rapamycin-conditioned, alloantigen-pulsed myeloid dendritic cells present donor MHC class I/peptide via the semi-direct pathway and inhibit survival of antigen-specific CD8(+) T cells in vitro and in vivo. Transpl Immunol 25, 20–26, doi: 10.1016/j.trim.2011.05.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J Clin Invest 126, 2805–2820, doi: 10.1172/JCI84577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marino J et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Science immunology 1, doi: 10.1126/sciimmunol.aaf8759 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russo V et al. Acquisition of intact allogeneic human leukocyte antigen molecules by human dendritic cells. Blood 95, 3473–3477 (2000). [PubMed] [Google Scholar]
- 35.Joly E & Hudrisier D What is trogocytosis and what is its purpose? Nat Immunol 4, 815, doi: 10.1038/ni0903-815 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Knight SC, Iqball S, Roberts MS, Macatonia S & Bedford PA Transfer of antigen between dendritic cells in the stimulation of primary T cell proliferation. Eur J Immunol 28, 1636–1644, doi: (1998). [DOI] [PubMed] [Google Scholar]
- 37.Wykes M, Pombo A, Jenkins C & MacPherson GG Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J Immunol 161, 1313–1319 (1998). [PubMed] [Google Scholar]
- 38.Herrera OB et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol 173, 4828–4837, doi: 10.4049/jimmunol.173.8.4828 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Benichou G, Wang M, Ahrens K & Madsen JC Extracellular vesicles in allograft rejection and tolerance. Cell Immunol 349, 104063, doi: 10.1016/j.cellimm.2020.104063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harper SJ et al. CD8 T-cell recognition of acquired alloantigen promotes acute allograft rejection. Proc Natl Acad Sci U S A 112, 12788–12793, doi: 10.1073/pnas.1513533112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown K, Sacks SH & Wong W Extensive and bidirectional transfer of major histocompatibility complex class II molecules between donor and recipient cells in vivo following solid organ transplantation. FASEB J 22, 3776–3784, doi: 10.1096/fj.08-107441 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Brown K, Sacks SH & Wong W Coexpression of donor peptide/recipient MHC complex and intact donor MHC: evidence for a link between the direct and indirect pathways. Am J Transplant 11, 826–831, doi: 10.1111/j.1600-6143.2011.03437.x (2011). [DOI] [PubMed] [Google Scholar]
- 43.Sivaganesh S et al. Copresentation of intact and processed MHC alloantigen by recipient dendritic cells enables delivery of linked help to alloreactive CD8 T cells by indirect-pathway CD4 T cells. J Immunol 190, 5829–5838, doi: 10.4049/jimmunol.1300458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smyth LA, Lechler RI & Lombardi G Continuous Acquisition of MHC:Peptide Complexes by Recipient Cells Contributes to the Generation of Anti-Graft CD8. Am J Transplant 17, 60–68, doi: 10.1111/ajt.13996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frängsmyr L et al. Cytoplasmic microvesicular form of Fas ligand in human early placenta: switching the tissue immune privilege hypothesis from cellular to vesicular level. Mol Hum Reprod 11, 35–41, doi: 10.1093/molehr/gah129 (2005). [DOI] [PubMed] [Google Scholar]
- 46.LeMaoult J et al. Immune regulation by pretenders: cell-to-cell transfers of HLA-G make effector T cells act as regulatory cells. Blood 109, 2040–2048, doi: 10.1182/blood-2006-05-024547 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Brown R et al. CD86+ or HLA-G+ can be transferred via trogocytosis from myeloma cells to T cells and are associated with poor prognosis. Blood 120, 2055–2063, doi: 10.1182/blood-2012-03-416792 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Caumartin J et al. Trogocytosis-based generation of suppressive NK cells. EMBO J 26, 1423–1433, doi: 10.1038/sj.emboj.7601570 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.HoWangYin KY et al. Proper regrafting of Ig-like transcript 2 after trogocytosis allows a functional cell-cell transfer of sensitivity. J Immunol 186, 2210–2218, doi: 10.4049/jimmunol.1000547 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Tilburgs T, Evans JH, Crespo  & Strominger JL The HLA-G cycle provides for both NK tolerance and immunity at the maternal-fetal interface. Proc Natl Acad Sci U S A 112, 13312–13317, doi: 10.1073/pnas.1517724112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ono Y et al. Graft-infiltrating PD-L1. Hepatology 67, 1499–1515, doi: 10.1002/hep.29529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sigdel TK et al. Perturbations in the urinary exosome in transplant rejection. Front Med (Lausanne) 1, 57, doi: 10.3389/fmed.2014.00057 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim JH et al. Novel urinary exosomal biomarkers of acute T cell-mediated rejection in kidney transplant recipients: A cross-sectional study. PLoS One 13, e0204204, doi: 10.1371/journal.pone.0204204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tower CM et al. Plasma C4d+ Endothelial Microvesicles Increase in Acute Antibody-Mediated Rejection. Transplantation 101, 2235–2243, doi: 10.1097/TP.0000000000001572 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Zhang H et al. Plasma Exosomes From HLA-Sensitized Kidney Transplant Recipients Contain mRNA Transcripts Which Predict Development of Antibody-Mediated Rejection. Transplantation 101, 2419–2428, doi: 10.1097/TP.0000000000001834 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Park J et al. Integrated Kidney Exosome Analysis for the Detection of Kidney Transplant Rejection. ACS Nano 11, 11041–11046, doi: 10.1021/acsnano.7b05083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Racusen LC et al. The Banff 97 working classification of renal allograft pathology. Kidney Int 55, 713–723, doi: 10.1046/j.1523-1755.1999.00299.x (1999). [DOI] [PubMed] [Google Scholar]
- 58.Pêche H, Heslan M, Usal C, Amigorena S & Cuturi MC Presentation of donor major histocompatibility complex antigens by bone marrow dendritic cell-derived exosomes modulates allograft rejection. Transplantation 76, 1503–1510, doi: 10.1097/01.TP.0000092494.75313.38 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Ma B et al. Combining Exosomes Derived from Immature DCs with Donor Antigen-Specific Treg Cells Induces Tolerance in a Rat Liver Allograft Model. Sci Rep 6, 32971, doi: 10.1038/srep32971 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halloran PF, Einecke G, Sikosana MLN & Madill-Thomsen K The Biology and Molecular Basis of Organ Transplant Rejection. Handb Exp Pharmacol, doi: 10.1007/164_2021_557 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Hidalgo LG et al. The transcriptome of human cytotoxic T cells: measuring the burden of CTL-associated transcripts in human kidney transplants. Am J Transplant 8, 637–646, doi: 10.1111/j.1600-6143.2007.02129.x (2008). [DOI] [PubMed] [Google Scholar]
- 62.Einecke G et al. Expression of CTL associated transcripts precedes the development of tubulitis in T-cell mediated kidney graft rejection. Am J Transplant 5, 1827–1836, doi: 10.1111/j.1600-6143.2005.00974.x (2005). [DOI] [PubMed] [Google Scholar]
- 63.Zhuang Q et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nature communications 7, 12623, doi: 10.1038/ncomms12623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tacke F et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117, 185–194, doi: 10.1172/JCI28549 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA & Pamer EG TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19, 59–70, doi: 10.1016/s1074-7613(03)00171-7 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Aldridge JR et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A 106, 5306–5311, doi: 10.1073/pnas.0900655106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Trez C et al. iNOS-producing inflammatory dendritic cells constitute the major infected cell type during the chronic Leishmania major infection phase of C57BL/6 resistant mice. PLoS Pathog 5, e1000494, doi: 10.1371/journal.ppat.1000494 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D’Elios MM et al. Predominant Th1 cell infiltration in acute rejection episodes of human kidney grafts. Kidney Int 51, 1876–1884, doi: 10.1038/ki.1997.256 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Li J et al. The Evolving Roles of Macrophages in Organ Transplantation. J Immunol Res 2019, 5763430, doi: 10.1155/2019/5763430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van den Bosch TP, Kannegieter NM, Hesselink DA, Baan CC & Rowshani AT Targeting the Monocyte-Macrophage Lineage in Solid Organ Transplantation. Front Immunol 8, 153, doi: 10.3389/fimmu.2017.00153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adams AB et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest 111, 1887–1895, doi: 10.1172/JCI17477 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams AB, Pearson TC & Larsen CP Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev 196, 147–160, doi:082 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 73.Jameson SC & Masopust D Diversity in T cell memory: an embarrassment of riches. Immunity 31, 859–871, doi: 10.1016/j.immuni.2009.11.007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Croft M, Bradley LM & Swain SL Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J Immunol 152, 2675–2685 (1994). [PubMed] [Google Scholar]
- 75.Walch JM et al. Cognate antigen directs CD8+ T cell migration to vascularized transplants. J Clin Invest 123, 2663–2671, doi: 10.1172/JCI66722 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Benichou G, Gonzalez B, Marino J, Ayasoufi K & Valujskikh A Role of Memory T Cells in Allograft Rejection and Tolerance. Front Immunol 8, doi: 10.3389/fimmu.2017.00170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Y, Heeger PS & Valujskikh A In vivo helper functions of alloreactive memory CD4+ T cells remain intact despite donor-specific transfusion and anti-CD40 ligand therapy. J Immunol 172, 5456–5466 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Valujskikh A, Pantenburg B & Heeger PS Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant 2, 501–509, doi: 10.1034/j.1600-6143.2002.20603.x (2002). [DOI] [PubMed] [Google Scholar]
- 79.Welsh RM et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol 74, 2210–2218, doi: 10.1128/jvi.74.5.2210-2218.2000 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhai Y, Meng L, Gao F, Busuttil RW & Kupiec-Weglinski JW Allograft rejection by primed/memory CD8+ T cells is CD154 blockade resistant: therapeutic implications for sensitized transplant recipients. J Immunol 169, 4667–4673 (2002). [DOI] [PubMed] [Google Scholar]
- 81.Krummey SM & Ford ML Heterogeneity within T Cell Memory: Implications for Transplant Tolerance. Front Immunol 3, 36, doi: 10.3389/fimmu.2012.00036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krummey SM & Ford ML New insights into T-cell cosignaling in allograft rejection and survival. Curr Opin Organ Transplant 20, 43–48, doi: 10.1097/MOT.0000000000000151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mathews DV et al. CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. J Clin Invest 128, 4557–4572, doi: 10.1172/JCI95914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hartigan CR, Sun H & Ford ML Memory T-cell exhaustion and tolerance in transplantation. Immunol Rev 292, 225–242, doi: 10.1111/imr.12824 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Shapira MY et al. Rapid response to alefacept given to patients with steroid resistant or steroid dependent acute graft-versus-host disease: a preliminary report. Bone Marrow Transplant 36, 1097–1101, doi: 10.1038/sj.bmt.1705185 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Shapira MY et al. A new induction protocol for the control of steroid refractory/dependent acute graft versus host disease with alefacept and tacrolimus. Cytotherapy 11, 61–67, doi: 10.1080/14653240802644669 (2009). [DOI] [PubMed] [Google Scholar]
- 87.Shapira MY et al. Alefacept treatment for refractory chronic extensive GVHD. Bone Marrow Transplant 43, 339–343, doi: 10.1038/bmt.2008.324 (2009). [DOI] [PubMed] [Google Scholar]
- 88.Lo DJ et al. Selective targeting of human alloresponsive CD8+ effector memory T cells based on CD2 expression. Am J Transplant 11, 22–33, doi: 10.1111/j.1600-6143.2010.03317.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weaver TA et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med 15, 746–749, doi:nm.1993 [pii] 10.1038/nm.1993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kitchens WH, Larsen CP & Ford ML Integrin antagonists for transplant immunosuppression: panacea or peril? Immunotherapy 3, 305–307, doi: 10.2217/imt.10.113 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kitchens WH et al. Integrin antagonists prevent costimulatory blockade-resistant transplant rejection by CD8(+) memory T cells. Am J Transplant 12, 69–80, doi: 10.1111/j.1600-6143.2011.03762.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Setoguchi K et al. LFA-1 Antagonism Inhibits Early Infiltration of Endogenous Memory CD8 T Cells into Cardiac Allografts and Donor-Reactive T Cell Priming. Am J Transplant 11, 923–935, doi: 10.1111/j.1600-6143.2011.03492.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iida S et al. Peritransplant VLA-4 blockade inhibits endogenous memory CD8 T cell infiltration into high-risk cardiac allografts and CTLA-4Ig resistant rejection. Am J Transplant 19, 998–1010, doi: 10.1111/ajt.15147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turgeon NA et al. Experience with a Novel Efalizumab-Based Immunosuppressive Regimen to Facilitate Single Donor Islet Cell Transplantation. Am J Transplant, doi:AJT3212 [pii] 10.1111/j.1600-6143.2010.03212.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Posselt AM et al. Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am J Transplant 10, 1870–1880, doi:AJT3073 [pii] 10.1111/j.1600-6143.2010.03073.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vincenti F et al. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation. Am J Transplant 7, 1770–1777, doi:AJT1845 [pii] 10.1111/j.1600-6143.2007.01845.x (2007). [DOI] [PubMed] [Google Scholar]
- 97.Carson KR et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 10, 816–824, doi:S1470–2045(09)70161–5 [pii] 10.1016/S1470-2045(09)70161-5 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Kirk AD et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med 5, 686–693, doi: 10.1038/9536 (1999). [DOI] [PubMed] [Google Scholar]
- 99.Larsen CP et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 381, 434–438, doi: 10.1038/381434a0 (1996). [DOI] [PubMed] [Google Scholar]
- 100.Liu D & Ford ML CD11b is a novel alternate receptor for CD154 during alloimmunity. Am J Transplant, doi: 10.1111/ajt.15835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolf D et al. Binding of CD40L to Mac-1’s I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis--but does not affect immunity and thrombosis in mice. Circ Res 109, 1269–1279, doi: 10.1161/circresaha.111.247684 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michel NA, Zirlik A & Wolf D CD40L and Its Receptors in Atherothrombosis-An Update. Frontiers in cardiovascular medicine 4, 40, doi: 10.3389/fcvm.2017.00040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hancock WW, Gao W, Faia KL & Csizmadia V Chemokines and their receptors in allograft rejection. Curr Opin Immunol 12, 511–516, doi: 10.1016/s0952-7915(00)00130-8 (2000). [DOI] [PubMed] [Google Scholar]
- 104.Halloran PF & Fairchild RL in Am J Transplant Vol. 8 1578–1579 (2008). [DOI] [PubMed] [Google Scholar]
- 105.Oberbarnscheidt MH et al. Memory T cells migrate to and reject vascularized cardiac allografts independent of the chemokine receptor CXCR3. Transplantation 91, 827–832, doi: 10.1097/TP.0b013e31820f0856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hoffmann SC et al. Molecular and immunohistochemical characterization of the onset and resolution of human renal allograft ischemia-reperfusion injury. Transplantation 74, 916–923 (2002). [DOI] [PubMed] [Google Scholar]
- 107.Mori DN, Kreisel D, Fullerton JN, Gilroy DW & Goldstein DR Inflammatory triggers of acute rejection of organ allografts. Immunol Rev 258, 132–144, doi: 10.1111/imr.12146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ponticelli C Ischaemia-reperfusion injury: a major protagonist in kidney transplantation. Nephrol Dial Transplant 29, 1134–1140, doi: 10.1093/ndt/gft488 (2014). [DOI] [PubMed] [Google Scholar]
- 109.Pandey S, Kawai T & Akira S Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol 7, a016246, doi: 10.1101/cshperspect.a016246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goldstein DR, Tesar BM, Akira S & Lakkis FG Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest 111, 1571–1578, doi: 10.1172/jci17573 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Messmer D et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol 173, 307–313, doi: 10.4049/jimmunol.173.1.307 (2004). [DOI] [PubMed] [Google Scholar]
- 112.McNulty S et al. Heat-shock proteins as dendritic cell-targeting vaccines--getting warmer. Immunology 139, 407–415, doi: 10.1111/imm.12104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tesar BM et al. The role of hyaluronan degradation products as innate alloimmune agonists. Am J Transplant 6, 2622–2635, doi: 10.1111/j.1600-6143.2006.01537.x (2006). [DOI] [PubMed] [Google Scholar]
- 114.Braudeau C et al. Contrasted blood and intragraft toll-like receptor 4 mRNA profiles in operational tolerance versus chronic rejection in kidney transplant recipients. Transplantation 86, 130–136, doi: 10.1097/TP.0b013e31817b8dc5 (2008). [DOI] [PubMed] [Google Scholar]
- 115.Sharbafi MH et al. TLR-2, TLR-4 and MyD88 genes expression in renal transplant acute and chronic rejections. Int J Immunogenet 46, 427–436, doi: 10.1111/iji.12446 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Deng JF et al. The role of toll-like receptors 2 and 4 in acute allograft rejection after liver transplantation. Transplant Proc 39, 3222–3224, doi: 10.1016/j.transproceed.2007.02.102 (2007). [DOI] [PubMed] [Google Scholar]
- 117.Braza F, Brouard S, Chadban S & Goldstein DR Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat Rev Nephrol 12, 281–290, doi: 10.1038/nrneph.2016.41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kulkarni HS, Scozzi D & Gelman AE Recent advances into the role of pattern recognition receptors in transplantation. Cell Immunol 351, 104088, doi: 10.1016/j.cellimm.2020.104088 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.(https://ClinicalTrials.gov/show/NCT01794663).
- 120.(https://ClinicalTrials.gov/show/NCT01808469).
- 121.Biglarnia AR, Huber-Lang M, Mohlin C, Ekdahl KN & Nilsson B The multifaceted role of complement in kidney transplantation. Nat Rev Nephrol 14, 767–781, doi: 10.1038/s41581-018-0071-x (2018). [DOI] [PubMed] [Google Scholar]
- 122.Mathern DR & Heeger PS Molecules Great and Small: The Complement System. Clin J Am Soc Nephrol 10, 1636–1650, doi: 10.2215/CJN.06230614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lalli PN et al. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112, 1759–1766, doi: 10.1182/blood-2008-04-151068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Strainic MG et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28, 425–435, doi: 10.1016/j.immuni.2008.02.001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sheen JH et al. TLR-Induced Murine Dendritic Cell (DC) Activation Requires DC-Intrinsic Complement. J Immunol 199, 278–291, doi: 10.4049/jimmunol.1700339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arbore G et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 352, aad1210, doi: 10.1126/science.aad1210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mathern DR, K Horwitz J & Heeger PS Absence of recipient C3aR1 signaling limits expansion and differentiation of alloreactive CD8. Am J Transplant 19, 1628–1640, doi: 10.1111/ajt.15222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kwan WH, van der Touw W, Paz-Artal E, Li MO & Heeger PS Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med 210, 257–268, doi: 10.1084/jem.20121525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Strainic MG, Shevach EM, An F, Lin F & Medof ME Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol 14, 162–171, doi: 10.1038/ni.2499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Touw W et al. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol 190, 5921–5925, doi: 10.4049/jimmunol.1300847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ono M, Bolland S, Tempst P & Ravetch JV Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 383, 263–266, doi: 10.1038/383263a0 (1996). [DOI] [PubMed] [Google Scholar]
- 132.Nimmerjahn F & Ravetch JV Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8, 34–47, doi: 10.1038/nri2206 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Wirth TC et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 33, 128–140, doi: 10.1016/j.immuni.2010.06.014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Starbeck-Miller GR, Badovinac VP, Barber DL & Harty JT Cutting edge: Expression of FcγRIIB tempers memory CD8 T cell function in vivo. J Immunol 192, 35–39, doi: 10.4049/jimmunol.1302232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269, doi: 10.1038/s41586-019-1326-9 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Morris AB et al. Signaling through the Inhibitory Fc Receptor FcγRIIB Induces CD8. Immunity 52, 136–150.e136, doi: 10.1016/j.immuni.2019.12.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu H et al. The FGL2-FcgammaRIIB pathway: a novel mechanism leading to immunosuppression. Eur J Immunol 38, 3114–3126, doi: 10.1002/eji.200838338 (2008). [DOI] [PubMed] [Google Scholar]
- 138.Hricik DE et al. Adverse Outcomes of Tacrolimus Withdrawal in Immune-Quiescent Kidney Transplant Recipients. J Am Soc Nephrol 26, 3114–3122, doi: 10.1681/ASN.2014121234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hu J et al. The Duality of Fgl2 - Secreted Immune Checkpoint Regulator Versus Membrane-Associated Procoagulant: Therapeutic Potential and Implications. International reviews of immunology 35, 325–339, doi: 10.3109/08830185.2014.956360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Joller N et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581, doi: 10.1016/j.immuni.2014.02.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Amir AL et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood 115, 3146–3157, doi: 10.1182/blood-2009-07-234906 (2010). [DOI] [PubMed] [Google Scholar]
- 142.Nadazdin O et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med 3, 86ra51, doi:3/86/86ra51 [pii] 10.1126/scitranslmed.3002093 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bozeman AM, Laurie SJ, Haridas D, Wagener ME & Ford ML Transplantation preferentially induces a KLRG-1(lo) CD127(hi) differentiation program in antigen-specific CD8(+) T cells. Transpl Immunol 50, 34–42, doi: 10.1016/j.trim.2018.06.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Burrows SR et al. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J Exp Med 182, 1703–1715, doi: 10.1084/jem.182.6.1703 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Barton E, Mandal P & Speck SH Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29, 351–397, doi: 10.1146/annurev-immunol-072710-081639 (2011). [DOI] [PubMed] [Google Scholar]
- 146.Larsen CP et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant 5, 443–453 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Floyd TL et al. Limiting the amount and duration of antigen exposure during priming increases memory T cell requirement for costimulation during recall. J Immunol 186, 2033–2041, doi: 10.4049/jimmunol.1003015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Traitanon O et al. IL-15 induces alloreactive CD28(−) memory CD8 T cell proliferation and CTLA4-Ig resistant memory CD8 T cell activation. Am J Transplant 14, 1277–1289, doi: 10.1111/ajt.12719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Asderakis A et al. Thymoglobulin Versus Alemtuzumab Versus Basiliximab Kidney Transplantation From Donors After Circulatory Death. Kidney Int Rep 7, 732–740, doi: 10.1016/j.ekir.2022.01.1042 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xie CB et al. Complement-activated interferon-gamma-primed human endothelium transpresents interleukin-15 to CD8+ T cells. J Clin Invest 130, 3437–3452, doi: 10.1172/JCI135060 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ferrer IR, Araki K & Ford ML Paradoxical aspects of rapamycin immunobiology in transplantation. Am J Transplant 11, 654–659, doi: 10.1111/j.1600-6143.2011.03473.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Turner AP et al. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant 11, 613–618, doi: 10.1111/j.1600-6143.2010.03407.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Araki K et al. mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112, doi:nature08155 [pii] 10.1038/nature08155 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ferrer IR et al. Cutting Edge: Rapamycin Augments Pathogen-Specific but Not Graft-Reactive CD8+ T Cell Responses. J Immunol 185, 2004–2008, doi:jimmunol.1001176 [pii] 10.4049/jimmunol.1001176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Föger N, Rangell L, Danilenko DM & Chan AC Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science 313, 839–842, doi: 10.1126/science.1130563 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Mueller P et al. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat Immunol 9, 424–431, doi: 10.1038/ni1570 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Shiow LR et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immunol 9, 1307–1315, doi: 10.1038/ni.1662 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jayachandran R et al. Disruption of Coronin 1 Signaling in T Cells Promotes Allograft Tolerance while Maintaining Anti-Pathogen Immunity. Immunity 50, 152–165.e158, doi: 10.1016/j.immuni.2018.12.011 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Bourne HR et al. Modulation of inflammation and immunity by cyclic AMP. Science 184, 19–28, doi: 10.1126/science.184.4132.19 (1974). [DOI] [PubMed] [Google Scholar]
- 160.Bopp T et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med 204, 1303–1310, doi: 10.1084/jem.20062129 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mosenden R & Taskén K Cyclic AMP-mediated immune regulation--overview of mechanisms of action in T cells. Cell Signal 23, 1009–1016, doi: 10.1016/j.cellsig.2010.11.018 (2011). [DOI] [PubMed] [Google Scholar]
- 162.Ford ML Coronin-1, King of Alloimmunity. Immunity 50, 3–5, doi: 10.1016/j.immuni.2018.12.030 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Moshous D et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol 131, 1594–1603, doi: 10.1016/j.jaci.2013.01.042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Man K et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141 e1125, doi: 10.1016/j.immuni.2017.11.021 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Wu J et al. Ablation of Transcription Factor IRF4 Promotes Transplant Acceptance by Driving Allogenic CD4. Immunity 47, 1114–1128.e1116, doi: 10.1016/j.immuni.2017.11.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu L et al. Bortezomib ameliorates acute allograft rejection after renal transplant by inhibiting Tfh cell proliferation and differentiation via miR-15b/IRF4 axis. Int Immunopharmacol 75, 105758, doi: 10.1016/j.intimp.2019.105758 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Epperson DE & Pober JS Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol 153, 5402–5412 (1994). [PubMed] [Google Scholar]
- 168.Beura LK et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516, doi: 10.1038/nature17655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Winterberg PD & Ford ML The effect of chronic kidney disease on T cell alloimmunity. Curr Opin Organ Transplant 22, 22–28, doi: 10.1097/MOT.0000000000000375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]