

Summary
Despite their cytotoxic capacity, neutrophils are often co-opted by cancers to promote immunosuppression, tumor growth, and metastasis. Consequently, these cells have received little attention as potential cancer immunotherapeutic agents. Here, we demonstrate in mouse models that neutrophils can be harnessed to induce eradication of tumors and reduce metastatic seeding through the combined actions of tumor necrosis factor, CD40 agonist, and tumor-binding antibody. The same combination activates human neutrophils in vitro, enabling their lysis of human tumor cells. Mechanistically, this therapy induces rapid mobilization and tumor infiltration of neutrophils along with complement activation in tumors. Complement component C5a activates neutrophils to produce leukotriene B4, which stimulates reactive oxygen species production via xanthine oxidase, resulting in oxidative damage and T cell-independent clearance of multiple tumor types. These data establish neutrophils as potent anti-tumor immune mediators and define an inflammatory pathway that can be harnessed to drive neutrophil-mediated eradication of cancer.
eTOC Blurb
Linde et al. describe a cancer therapy that activates neutrophils to infiltrate and eradicate tumors and reduce metastatic seeding. The authors elucidate the responsible mechanism, which involves complement component C5a, leukotriene B4, and reactive oxygen species, and demonstrate the potential of harnessing neutrophils through inflammatory activation to drive tumor clearance.
Graphical Abstract
Introduction
While initially appreciated for their role in defense against microbial pathogens, neutrophils are now recognized to promote the growth and spread of many cancers1–3. Cancers are frequently accompanied by neutrophil recruitment to the tumor and expansion in the blood, which is associated with poor prognosis in most cases1,2,4,5. Studies of neutrophils from cancer patients and mouse models have established that neutrophils promote tumor growth6, angiogenesis7,8, and metastasis9–11 and inhibit anti-cancer T cell responses9,12,13. Furthermore, myeloid-derived suppressor cells (MDSCs), a heterogenous group of cells that overlap phenotypically with neutrophils1,2,14, are well-appreciated to induce T cell suppression and promote tumor growth and metastasis2,14–16.
Nonetheless, neutrophils have the potential to exert anti-tumor activity. Early studies demonstrated the ability of neutrophils to kill tumor cells in vitro17, and while neutrophils exert pro-tumor activity in most settings1,2, a growing number of reports support the potential for neutrophils to perform anti-tumor functions in certain contexts. Neutrophils naturally can inhibit some tumors during the early stages of tumor development18–21 or early metastasis22,23, and they are capable of promoting anti-tumor responses by other immune cells, including natural killer (NK) cells and multiple subsets of T cells19–21,24–26. The apparently contradictory roles of neutrophils in cancer are likely the result of differences in the tumor milieu impacting neutrophil maturation, activation, and functional states2,24,27.
Despite the natural capacity of neutrophils to inhibit cancer in certain contexts, little effort has been made to harness neutrophils as anti-tumor effector cells. Neutrophil-targeted treatments have generally focused on depleting MDSCs or blocking the recruitment of neutrophils and MDSCs to the tumor2,14,28,29. Recently, targeted inhibition of receptors on neutrophils has been shown to slow tumor growth by inhibiting pro-tumor functions of neutrophils such as the promotion of angiogenesis30 and T cell suppression25. Additionally, inhibition of certain suppressive signals can enhance killing of tumor cells by neutrophils ex vivo24, induce modest reductions in tumor growth31, and promote activation of CD8+ T cells24 or NK cells26 to inhibit tumor growth. Neutrophils can also kill antibody-bound tumor cells32 and mediate the effects of monoclonal antibody therapy initiated concurrently with tumor engraftment33. Despite these promising results, it is still not clear whether neutrophils can be harnessed therapeutically to drive regression of established tumors.
We set out to determine whether neutrophils in a tumor-bearing host could be mobilized and activated to attack tumors. Employing intratumoral injection of neutrophil stimuli and tumor-binding antibodies, we show that manipulation of the tumor milieu can result in the infiltration and activation of tumor-killing neutrophils that drive T cell-independent tumor clearance. We further reveal the mechanistic steps underlying this response, demonstrating the ability of neutrophil-mediated inflammation to generate a tumor-eradicating immune response and identifying multiple promising targets for therapeutic intervention.
Results
Neutrophil-activating therapy recruits activated neutrophils to the tumor
To determine if neutrophils might be harnessed therapeutically, we investigated how the tumor microenvironment could be modulated to optimally recruit neutrophils and activate their cytotoxic function. When we assessed the impact of various cytokines injected intratumorally in B16 melanoma, tumor necrosis factor (TNF) notably induced robust recruitment of neutrophils (Figure S1A–B) and upregulated neutrophil surface molecules consistent with activation34–36 (Figure S1C–D) in a dose-dependent manner (Figure S1E–G). However, TNF monotherapy failed to clear tumors in most mice (Figure S1H).
Given the promising neutrophil infiltration and activation induced by TNF but the failure to clear tumors, we sought complementary agents capable of enabling neutrophil-mediated tumor clearance in combination with TNF. Since cluster of differentiation 40 (CD40) agonists can activate neutrophils and promote neutrophil cytotoxicity37, we evaluated the effect of an agonistic anti-CD40 monoclonal antibody (mAb). As neutrophils are also potent mediators of antibody-dependent cellular cytotoxicity (ADCC) through ligation of their Fc receptors2,32, we also tested a mAb targeting the melanoma-associated antigen gp75. Strikingly, intratumoral treatment of tumors with two doses of 1 μg TNF + 100 μg anti-CD40 + 100 μg anti-gp75 two days apart induced durable clearance of B16 melanoma tumors (Figure 1A). In contrast, treatment with only one or two of these components failed to achieve the same frequency of tumor clearance (Figures S1I–J). Mice treated with TNF + anti-CD40 + anti-gp75 transiently lost a small amount of weight but rapidly recovered within two days of the second treatment (Figure S1K). Additionally, blood chemistry analysis one week after treatment completion and 60 days post-treatment revealed essentially normal liver and renal function (Figure S1L).
Figure 1: Neutrophil-activating therapy recruits activated neutrophils to the tumor.

(A) (Left) Tumor growth in B16-bearing mice following treatment with the indicated components, indicating mice with undetectable tumors at the conclusion of the study in parentheses. (Right) Survival of the mice shown in the left panel. Mice were euthanized when tumors exceeded 100mm2. (B-C) Neutrophil frequency (B) and numbers (C) in B16 tumors following treatment with TNF + anti-CD40 + anti-gp75. (n=5) (D) Neutrophil frequency in peripheral blood following treatment with this neutrophil-activating therapy. (n=5) (E) Immunofluorescence of neutrophil infiltration in B16 tumors following treatment with neutrophil-activating therapy. Scale bars = 500 μm. (F-H) Neutrophil frequency in the tumor (F) and blood (G-H) 4 hours (G) or 24 hours (F, H) after treatment with the indicated components. (n=4) (I) Frequencies of HSCs and progenitors in the bone marrow 24 hours after treatment with neutrophil-activating therapy. (anti-gp75 n=4, other groups n=5) (J) Representative histograms (top) and median fluorescent intensity (MFI) (bottom) of surface markers on neutrophils infiltrating B16 tumors 4 hours after treatment with the indicated components. (n=4) (K) Surface marker expression on B16 tumor-infiltrating neutrophils following treatment with the full neutrophil-activating therapy. (n=4). Statistics: Log-rank test with Bonferroni correction (A), One-way ANOVA with Tukey’s multiple comparisons test (B-D, F-K). For all dot plots, the line indicates the mean. Data are representative of 2 (B-E) or 3 (F-H) independent experiments or pooled from 2 experiments (A). See also Figures S1, S2.
Mice treated with the full three-component therapy consisting of TNF, anti-CD40, and tumor-binding antibody, hereafter referred to as neutrophil-activating therapy, exhibited rapid recruitment of neutrophils to tumors (Figure 1B–C), and neutrophils expanded in the blood with similar kinetics (Figure 1D). Treated neutrophils infiltrated throughout B16 tumors and were not merely confined to the periphery (Figure 1E). Evaluation of the impact of individual treatment components revealed that neutrophil recruitment to tumors was primarily induced by TNF (Figure 1F). While TNF induced a transient increase in neutrophil frequency in the blood (Figure 1G), anti-CD40 induced a large and sustained expansion of neutrophils in the blood (Figures 1H).
Flow cytometry analysis to identify hematopoietic stem cells (HSCs) and progenitors (Figure S1M–N) 38–40 in the bone marrow 24 hours after neutrophil-activating therapy revealed an increase in the frequencies of HSCs and multipotent progenitors (MPPs) (Figure 1I). While the full neutrophil-activating therapy had a minimal effect on the frequency of common lymphoid progenitors (CLPs), it induced a drastic reduction in common myeloid progenitors (CMPs), granulocyte-monocyte progenitors (GMPs), megakaryocyte-erythrocyte progenitors (MEPs), and common monocyte progenitors (cMoPs), while committed neutrophil progenitors (proNeu1s and proNeu2s) were not significantly altered (Figure 1I and S1O). These data indicate the induction of granulopoiesis. In addition, while the frequency in the bone marrow of late neutrophil precursors (preNeus) decreased with therapy, Ly6Glo immature neutrophils increased, and Ly6Ghi mature neutrophils decreased (Figure 1I), consistent with an increased differentiation of neutrophils in the bone marrow and a mobilization of mature neutrophils into the blood. Furthermore, the spleen displayed an expansion of neutrophils and similar alterations in the frequencies of HSCs and progenitors (Figure S1P), indicating extramedullary granulopoiesis.
In mice lacking TNF receptors (TNFR knockout (KO) mice), treatment with neutrophil-activating therapy failed to recruit or activate neutrophils (Figure S2A–C). While treatment induced cell death throughout tumors in wild-type (WT) mice, TNFR KO mice had reduced levels of cell death (Figure S2D–E) and failed to clear their tumors following treatment (Figure S2F). Tumor clearance was mediated through TNF receptor 1 (TNFR1) (Figure S2G) and was independent of TNFR1 expression on tumor cells (Figure S2H–I). These data demonstrate that TNF signaling in non-tumor cells is essential for neutrophil recruitment, tumor cell killing, and tumor clearance.
Neutrophil-activating therapy induced multiple alterations in the surface phenotype of tumor-infiltrating neutrophils, primarily in response to TNF (Figure 1J). These neutrophils upregulated CD11b and intercellular adhesion molecule (ICAM)-1, indicating activation or priming34–36, and increased expression of CD177, which has been associated with anti-tumor neutrophils in colon cancer41. Therapeutically activated neutrophils also had lower levels of signal regulatory protein-α (SIRPα), a myeloid checkpoint that inhibits neutrophil ADCC2,32, suggesting the capacity for enhanced tumor cell killing. Neutrophil-activating therapy did not alter the expression of Siglec F, a marker associated with pro-tumor neutrophils42, which remained low in all treatment conditions (Figure S2J). In contrast, treated neutrophils downregulated, but did not lose, expression of CD101 (Figure S2K). CD101-negative neutrophils are more immature and have been reported to correlate with tumor burden38. Neutrophils in tumors treated with neutrophil-activating therapy also upregulated CD14 and programmed cell death-ligand 1 (PD-L1), which have been reported to identify neutrophils with increased suppressive and reduced tumoricidal activity2,43,44. Neutrophils in the blood exhibited a very similar, but less extreme, pattern of changes in expression of these markers following neutrophil-activating therapy (Figure S2L). These alterations in neutrophil phenotype were transient, as neutrophils in both the tumor and blood increasingly reverted toward the phenotype of neutrophils in mock treated mice over the first week post-treatment (Figures 1K and S2M). Neutrophil-activating therapy of other tumors, including Sparkl.4640, a colon carcinoma cell line isolated from a genetically engineered mouse model (Figure S2N–O), and 4T1 mammary carcinoma (Figure S2P–Q) induced similar neutrophil expansion, recruitment, and activation.
Furthermore, treated tumor-infiltrating neutrophils possessed a mature morphology with hypersegmented nuclei (Figure S2R). Consistent with their activated status, they exhibited enhanced production of reactive oxygen species (ROS) (Figure S2S–U). While activated neutrophils can extrude neutrophil extracellular traps (NETs), neutrophils in the tumor and blood of treated mice exhibited no detectable increase in NETotic neutrophils45 (Figure S2V–W). Altogether, these data indicate that neutrophil-activating therapy induces an acute activation of tumor-infiltrating and circulating neutrophils, which exhibit a unique surface phenotype including markers associated with both anti-tumor and pro-tumor function.
Therapeutically activated neutrophils eradicate multiple tumor types and reduce metastatic seeding
To directly evaluate the anti-tumor activity of neutrophils following neutrophil-activating therapy, neutrophils were isolated from treated B16 tumors and co-cultured with B16 tumor cells ex vivo. Following stimulation ex vivo with neutrophil-activating therapy, these neutrophils mediated potent tumor cell killing (Figure 2A). In contrast, neutrophils isolated from the bone marrow of naïve mice failed to mediate significant tumor cell killing, even after ex vivo stimulation with neutrophil-activating therapy (Figure 2B). Notably, stimulation with all three components of the therapy was required for maximal cytotoxic function (Figure 2A). Tumor-infiltrating neutrophils required both anti-gp75 and Fc gamma receptors (FcγRs) to kill B16 tumor cells ex vivo (Figure 2C), and tumor-infiltrating neutrophils stimulated with neutrophil-activating therapy demonstrated enhanced uptake of anti-gp75 mAb (Figure 2D) and B16 cell membrane (Figure 2E–F), suggesting antibody-mediated trogocytosis32 may mediate ADCC by these activated neutrophils. Mice lacking functional activating FcγRs (Fcer1g−/−) exhibited reduced tumor clearance in vivo (Figure 2G and S3A), while neutrophil recruitment and CD11b upregulation were still maintained (Figure S3B–D). B16 tumors that recurred following therapy exhibited reduced levels of the antibody target antigen gp75, suggesting that ADCC may exert a selection pressure against the antibody target antigen (Figure S3E). Treatment of mice bearing B16 tumors with heterogenous expression of cell-surface enhanced green fluorescent protein (EGFP) with TNF + anti-CD40 + anti-EGFP induced tumor clearance at a comparable rate to tumors with homogenous EGFP expression (Figure S3F). Altogether, these data suggest that tumor-binding antibody-FcγR interactions enhance in vivo neutrophil killing of tumors through ADCC, but this is not absolutely required for tumor clearance.
Figure 2: Therapeutically activated neutrophils eradicate multiple tumor types and reduce metastatic seeding.

(A) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors and stimulated in vitro with the indicated components. (n=4) (B) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors or tumor-naïve bone marrow, stimulated in vitro with TNF + anti-CD40 + anti-gp75. (n=4) (C) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors in WT or Fcer1g−/− mice, stimulated in vitro with TNF + anti-CD40 + anti-gp75 or isotype control, with or without anti-CD16/CD32 to block FcγRs. (n=4) (D) Signal from anti-gp75-AlexaFluor 647 in neutrophils isolated from treated tumors or naïve bone marrow and cultured in vitro with B16 cells together with TNF + anti-CD40 + anti-gp75-AlexaFluor 647 or no stimulation. (BM n=3, Tumor n=4) (E-F) Percent DiD+ neutrophils (E) and DiD MFI in DiD+ neutrophils (F) following co-culture of treated tumor neutrophils with DiD-labeled B16 and stimulation in vitro with the indicated components. (Unstained/triple n=4, Unstimulated/double n=3) (G) Survival of B16-bearing WT or Fcer1g−/− mice following treatment with neutrophil-activating therapy. (n=10) (H) Regimen for neutrophil depletion and therapy. Treatment was performed 4 hours after administration of anti-Ly6G or isotype control on days 0 and 2. (I-J) Representative TUNEL immunofluorescence (I) and quantification (J) in B16 tumors 24 hours after treatment with neutrophil-activating therapy, following neutrophil depletion with anti-Ly6G or isotype control. Scale bars = 500 μm. (Isotype n=3, others n=4) (K) Survival of B16-bearing mice administered anti-Ly6G or isotype control prior to neutrophil-activating therapy. (n=10) (L-N) Survival of mice bearing LL/2 (L) (mock n=8, others n=10), 4T1 (M) (n=10), and Sparkl.4640 (N) (mock n=8, isotype n=10, anti-Ly6G n=9) tumors administered anti-Ly6G or isotype control prior to neutrophil-activating therapy. (O) Percent of MMTV-PyMT mice with treated tumors below the threshold of 100 mm2 following treatment of one tumor per mouse in the context of anti-Ly6G or isotype control (mock n=8, others n=6). (P) Representative images of B16-tdTomato fluorescence in the lung (left) and quantification of the number and average area of tdTomato+ lung metastases (right) in mice bearing s.c. B16 tumors that were injected intravenously through the tail vein with B16-tdTomato one week after tumor implantation. Ten hours after tail vein injection, s.c. tumors were treated with mock or neutrophil-activating therapy, and the lungs were harvested and imaged 9 days after the tail vein injection. Lung borders are outlined in white. Scale bars = 1 mm. (mock n=8, treated n=9) (Q) Representative image of India ink-stained lungs (left) and number of lung metastases (right) 30 days after orthotopic implantation of 4T1, in mice receiving neutrophil-activating therapy or mock treatment (mock n=8, treated n=9). Statistics: Two-way ANOVA with Tukey’s multiple comparisons test (A-D), One-way ANOVA with Tukey’s multiple comparisons test (E-F, J), Log-rank test (F, K), Log-rank test with Bonferroni correction (L-O), unpaired two-tailed t test (P-Q). For all dot plots, the line indicates the mean. Data are representative of 2 (A-J, P) or 3 (K) independent experiments or pooled from 2 (Q), 3 (L-N), or 6 (O) experiments. See also Figure S3.
To determine the role of therapeutically activated neutrophils in tumor clearance in vivo, neutrophils were depleted using anti-Ly6G antibodies (Figure 2H). While neutrophil depletion with anti-Ly6G did not completely remove neutrophils, this protocol blocked treatment-induced neutrophil tumor infiltration and reduced neutrophil expansion in the blood (Figure S3G–L). Similar results were obtained across multiple tumor models (Figure S3M–R).
Depletion of neutrophils with anti-Ly6G mAb prior to treatment markedly reduced cell death within tumors (Figure 2I–J). While neutrophil depletion did not alter tumor growth in untreated mice (Figure S3S), it completely prevented tumor clearance in response to neutrophil-activating therapy (Figure 2K and S3T), demonstrating a crucial role for neutrophils in tumor eradication. Treatment with neutrophil-activating therapy enabled neutrophils to clear multiple additional tumor types, including LL/2 lung carcinoma, 4T1 mammary carcinoma, and Sparkl.4640 colon carcinoma (Figure 2L–N and S3U–W). We next tested neutrophil-activating therapy in the MMTV-PyMT model of mammary carcinoma, in which tumors spontaneously develop in multiple breasts nearly simultaneously. Treatment of one tumor per mouse resulted in neutrophil-dependent regression of the treated tumors (Figure 2O and S3X), while tumors in untreated breasts did not regress. Tumors later grew out in treated breasts, which could represent either recurrences of the treated tumor or development of additional tumors in the same breast.
To determine whether neutrophil-activating therapy could restrict the growth of distant tumors in the context of metastasis, we injected B16 expressing tdTomato into the tail vein one week after subcutaneous implantation of B16. Treatment of the subcutaneous tumor ten hours after this seeding of the lungs resulted in a substantial reduction in the number and size of lung metastases (Figure 2P). Furthermore, treatment of orthotopically-implanted 4T1 mammary carcinoma with neutrophil-activating therapy reduced the number of spontaneous lung metastases (Figure 2Q). These data indicate that neutrophil-activating therapy can reduce metastatic seeding, restrict metastatic growth, and/or eliminate metastases.
Therapy activates antigen presenting cells and primes T cell memory
Neutrophil infiltration following treatment was accompanied by a TNF-dependent decrease in multiple other immune cell populations within the tumor (Figure 3A and S4A–C). Additionally, conventional dendritic cell (cDC) populations were skewed further toward cDC2s (Figure 3B), and the relative frequency of CD8+ T cells decreased slightly after treatment (Figure 3C). Treatment of TNFR KO mice with neutrophil-activating therapy did not induce the large alterations to immune cell populations seen in treated WT mice (Figure S4D–E). In contrast, treated Fcer1g−/− mice had alterations in immune subsets closely resembling treated WT mice (Figure S4F–G). The anti-CD40-dependent expansion of neutrophils in the blood (Figure 1H) was counterbalanced by a decrease in blood B cell frequency, and anti-CD40 increased the proportion of circulating CD4+ Forkhead box P3− (Foxp3−) T cells at the expense of CD8+ T cells (Figure S4H–J). Treatment of 4T1 and Sparkl.4640 tumors with neutrophil-activating therapy elicited similar changes in immune cell populations (Figure S4K–Q).
Figure 3: Therapy activates antigen-presenting cells and primes T cell memory.

(A) Frequency of immune cell subsets in B16 tumors 24 hours after treatment (n=4). (B) Percent of cDC2s out of total DCs in B16 tumors 24 hours after treatment (n=4). (C) Percent of T cell subsets out of total T cells in B16 tumors 24 hours after treatment (n=4). (D) Representative histograms and MFIs for markers expressed on APC populations in B16 tumors 24 hours after treatment (n=4). (E-F) Frequencies (E) and numbers (F) of T cells in the blood 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (G) Percent of T cell subsets out of total T cells in the blood 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (H) Memory and effector phenotypes of T cell subsets in the blood 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (I) Representative histograms and MFIs for markers expressed on T cell subsets in the blood 7 days after treatment (anti-gp75 n=4, others n=5). (J-K) Frequencies (J) and numbers (K) of T cells in the dLN 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (L) Percent of T cell subsets out of total T cells in the dLN 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (M) Memory and effector phenotypes of T cell subsets in the dLN 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (N) Representative histograms and MFIs for markers expressed on T cell subsets in the dLN 7 days after treatment (anti-gp75 n=4, others n=5). (O-P) Frequencies (O) and numbers (P) of T cells in the tumor 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (Q) Percent of T cell subsets out of total T cells in the tumor 7 days after treatment of B16 (anti-gp75 n=4, others n=5). (R) Survival of B16-bearing WT or Rag2−/− mice treated with neutrophil-activating therapy (n=15). (S) Survival of WT or Rag2−/− mice following implantation of B16 in tumor-naïve or B16-cleared mice 50 days after initial treatment with neutrophil-activating therapy (WT cleared n=14, Rag2−/− cleared n=18, WT naïve n=10, Rag2−/− naïve n=15). Statistics: Two-way ANOVA with Tukey’s multiple comparisons test (A, E-I, L-N, Q), One-way ANOVA with Tukey’s multiple comparisons test (B-D, J-K, O-P), Log-rank test (R), Log-rank test with Bonferroni correction (S). For all dot plots, the line indicates the mean. Data are representative of 2 (A-Q, S) or 3 (R) independent experiments. See also Figures S4, S5.
TNF and CD40 agonists are capable of activating antigen-presenting cells (APCs); indeed, both agents induced upregulation of major histocompatibility complex class II (MHCII), CD80, and CD86 on cDC1s in B16 tumors (Figure 3D), indicating activation. In contrast, there were minimal changes in activation markers in the more abundant cDC2s and macrophages, other than CD80 upregulation in cDC2s (Figure 3D). Opposing this activation, all three APC subsets upregulated PD-L1 with treatment (Figure 3D). Macrophage polarization, assessed by the ratio of CD206hi to MHCIIhi macrophages, did not change significantly following therapy (Figure S5A). Sparkl.4640 and 4T1 tumors treated with neutrophil-activating therapy exhibited mostly similar patterns of APC activation (Figure S5B–E). Altogether, these data indicate that neutrophil-activating therapy induces activation of APCs in the tumor, although this activation is mainly confined to the relatively rare cDC1 subset.
To determine the effects of neutrophil-activating therapy on T cells, we examined blood, tumor-draining lymph nodes (dLNs), and tumors one week after treatment of B16 tumors. While T cells in the blood did not expand (Figure 3E–F), there was an anti-CD40-dependent increase in the proportion of CD8+ T cells (Figure 3G), as well as a large shift in both CD8+ T cells and CD4+Foxp3− T cells from a naïve CD62L+CD44− phenotype to a CD62L−CD44+ Killer cell lectin-like receptor G1− (KLRG1−) effector memory/memory precursor effector phenotype and a CD62L−CD44+KLRG1+ phenotype that could encompass both short-lived effectors and future memory cells46 (Figure 3H). In addition, treatment activated CD8+ and CD4+Foxp3− T cells, indicated by expression of CD69, programmed cell death protein-1 (PD-1), and the proliferation marker Ki67 (Figure 3I). In the dLN, treatment induced an expansion of T cells and a similar bias toward CD8+ T cells (Figure 3J–L), in addition to increased memory differentiation (Figure 3M) and T cell activation (Figure 3N). The full neutrophil-activating therapy did not increase the numbers of tumor T cells, although the proportion of CD8+ T cells increased (Figure 3O–Q). Examination of T cells in the 4T1 model revealed a more blunted response, although certain commonalities were preserved, including T cell proliferation in the blood and dLN, activation and memory differentiation in the dLN, and elevated proportions of CD8+ T cells in the tumor (Figure S5F–Q).
To investigate the role of adaptive immunity in tumor clearance, we treated Rag2−/− mice, which lack mature T cells and B cells. Neutrophil-activating therapy cleared B16 tumors in both WT and Rag2−/− mice (Figure 3R and S5R–T). Nonetheless, WT mice that had previously cleared B16 were protected from re-challenge with the same tumor, in contrast to Rag2−/− mice or naïve WT mice (Figure 3S and S5U), indicating that neutrophil-activating therapy is capable of priming anti-tumor adaptive immune memory, even though this is not required for initial tumor clearance. These data are consistent with the low number of T cells in the tumor and the large-scale T cell activation, proliferation, and memory differentiation in the blood and dLN observed post-treatment.
Complement activates tumor-infiltrating neutrophils through C5AR1
To identify the mechanism by which neutrophil-activating therapy stimulates neutrophils, we investigated the complement system, which generates products that are well-known to stimulate neutrophil activation and recruitment47. Within four hours of treatment, complement component C3 was deposited on the surface of tumor-infiltrating neutrophils (Figure 4A) and throughout the tumor (Figure 4B–C), indicating local complement activation. Complement deposition throughout the tumor was dependent on TNF signaling (Figure S6A) with potential contributions from anti-CD40 and anti-gp75 (Figure S6B–D). Neutrophil depletion did not alter complement deposition (Figure S6E–G), suggesting that complement activation occurs upstream of neutrophil recruitment and activation.
Figure 4: Complement activates tumor-infiltrating neutrophils through C5AR1.

(A) Deposition of C3 on B16 tumor-infiltrating neutrophils following treatment with neutrophil-activating therapy (n=5). (B-C) Representative immunofluorescence (B) and quantification (C) of C3 staining in B16 tumors after treatment. Scale bars = 500 μm. (0h n=3, others n=4) (D-E) Representative immunofluorescence (D) and quantification (E) of TUNEL staining in B16 tumors 24 hours after treatment of mice that had received CVF or vehicle prior to treatment. Scale bars = 500 μm. (mock n=4, vehicle n=5, CVF n=6) (F) Survival of B16-bearing mice administered CVF prior to treatment with neutrophil-activating therapy. (n=5) (G-I) Survival of B16-bearing mice administered anti-Factor B (G) (n=7), anti-C5 (H) (isotype n=12, anti-C5 n=5), and anti-C5AR1 (I) (n=5) blocking antibodies prior to treatment. (J) Expression of CD11b on B16 tumor-infiltrating neutrophils 4 hours after treatment following CVF or vehicle administration (vehicle n=4, others n=5). (K) Expression of CD11b on naïve neutrophils following stimulation in vitro with the indicated factors (n=8). (L) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors and stimulated in vitro with the indicated factors (n=4). Statistics: Two-way ANOVA with Tukey’s multiple comparisons test (A, L), One-way ANOVA with Tukey’s multiple comparisons test (C, E, J-K), Log-rank test (F-I). For all dot plots, the line indicates the mean. Data are representative of 2 (A-C, F-G, I-L) or 3 (D-E) independent experiments or pooled from 2 experiments (H). See also Figures S6, S7.
Administration of cobra venom factor (CVF) to deplete complement prior to treatment prevented complement activation throughout tumors (Figure S6H–I), which mirrored the results seen in C3−/− mice (Figure S6J). CVF administration or C3 deficiency reduced tumor cell death following treatment (Figure 4D–E and S7A–B) and prevented tumor eradication (Figure 4F and S7C–D). Depletion of Factor B, which is required for complement activation through the alternative pathway (AP), as well as depletion or deficiency of the AP positive regulator properdin, blocked tumor clearance (Figure 4G and S7E–G), implicating AP complement activation as a crucial mediator of the treatment response.
The effect of complement activation was mediated through complement component C5, as C5-depleted mice failed to clear tumors (Figure 4H and S7H). While cleavage of C5 both generates the anaphylatoxin C5a and catalyzes the formation of the membrane attack complex (MAC) through C5b47, C6−/− mice, which are incapable of forming the MAC, showed no deficit in tumor clearance (Figure S7I). In contrast, blocking complement C5a receptor 1 (C5AR1) prevented tumor eradication (Figure 4I and S7J), implicating C5a as the relevant complement effector. While complement deficiency did not induce a major deficit in neutrophil recruitment (Figure S7K–L), it reduced neutrophil activation in response to treatment (Figure 4J and S7M). In contrast, complement depletion did not reduce activation of other myeloid populations in the tumor (Figure S7N), suggesting that neutrophils are the main cell type activated by complement. Additionally, recombinant C5a activated neutrophils in vitro (Figure 4K), and neutrophils isolated from treated tumors required C5a or serum containing functional complement to lyse tumor cells (Figure 4L and S7O). Altogether, these data demonstrate that neutrophil-activating therapy induces complement activation through the AP, which in turn activates neutrophils through C5a-C5AR1 signaling to kill tumors.
Secretion of leukotriene B4 by C5a-activated neutrophils drives tumor clearance
To identify neutrophil-derived mediators that might contribute to tumor clearance, we considered the potent pro-inflammatory lipid mediator leukotriene B4 (LTB4) 48. Neutrophil-activating therapy induced a neutrophil-dependent increase in LTB4 in the tumor (Figure 5A and S8A), with neutrophils responsible for the majority of LTB4 production (Figure 5B). Furthermore, LTB4 production was dependent on TNF signaling and complement (Figure 5C and S8B–C), and stimulation of neutrophils with C5a in vitro induced LTB4 (Figure 5D). Inhibition of LTB4 production through the leukotriene A4 hydrolase (LTA4H) inhibitor SC57461A prevented treatment-induced tumor cell death (Figure 5E and S8D) and tumor clearance (Figure 5F and S8E). The effects of LTB4 were mediated through LTB4 receptor 1 (BLT1), as the BLT1 antagonist CP-105696 also prevented tumor clearance (Figure 5G and S8F), and SC57461A and CP-105696 both prevented ex vivo killing of tumor cells by neutrophils (Figure 5H). These data demonstrate that C5a-activated neutrophils mediate tumor eradication through LTB4 in response to neutrophil-activating therapy.
Figure 5: Secretion of leukotriene B4 by C5a-activated neutrophils drives tumor clearance.

(A) LTB4 levels in B16 tumors 24 hours after treatment with neutrophil-activating therapy following neutrophil depletion by anti-Ly6G (mock n=7, others n=6). (B) LTB4 produced ex vivo by cells harvested from B16 tumors 12 hours after treatment (n=11). (C) LTB4 levels in B16 tumors 24 hours after treatment following administration of CVF (n=6). (D) LTB4 production by naïve neutrophils following stimulation in vitro with the indicated factors (n=8). (E) Quantification of TUNEL staining in B16 tumors 24 hours after treatment with neutrophil-activating therapy following administration of SC57461A (vehicle n=3, others n=4). (F-G) Survival of B16-bearing mice after treatment following administration of SC57461A (F) (vehicle n=9, SC57461A n=8) or CP-105696 (G) (vehicle=9, CP-105696 n=10). (H) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors and stimulated in vitro with neutrophil-activating therapy together with the indicated inhibitors (n=4). Statistics: One-way ANOVA with Tukey’s multiple comparisons test (A, C-E), Repeated measures one-way ANOVA with Tukey’s multiple comparisons test (B), Log-rank test (F-G), Two-way ANOVA with Tukey’s multiple comparisons test (H). For all dot plots, the line indicates the mean. Data are representative of 2 (A, C-D, H) or 1 (E) independent experiments or pooled from 2 experiments (B, F-G). See also Figure S8.
LTB4-dependent induction of xanthine oxidase induces oxidative damage and tumor clearance
Since LTB448 and C5a49 can induce production of ROS by neutrophils, and since ROS are potent effectors of neutrophil-mediated cytotoxicity, we investigated the role of ROS in tumor clearance. Neutrophil-activating therapy induced neutrophil-dependent oxidative damage, as evidenced by oxidation of nucleic acids (Figure 6A–B) and endogenous glutathione (Figure 6C) within the tumor. This oxidation was dependent on TNF signaling, complement, and LTB4 (Figure 6D–E and S8G), suggesting that production of LTB4 by C5a-activated neutrophils drives the production of ROS and resulting oxidative damage in the tumor. Scavenging of ROS with reduced glutathione (GSH) or neutralization of hydrogen peroxide with catalase blocked ex vivo killing of tumor cells by tumor-infiltrating neutrophils (Figure 6F), demonstrating that ROS mediate neutrophil killing of tumor cells. Administration of GSH decreased tumor cell death in vivo (Figure S8H–I) and prevented tumor clearance (Figure S8J) following treatment. Furthermore, administration of catalase blocked tumor eradication in treated mice (Figure 6G and S8K). These data demonstrate a critical role for ROS, and specifically hydrogen peroxide, in neutrophil-dependent tumor clearance following neutrophil-activating therapy.
Figure 6: LTB4-dependent induction of xanthine oxidase induces oxidative damage and tumor clearance.

(A-B) Representative immunofluorescence (A) and quantification (B) of DNA/RNA oxidative damage in B16 tumors 24 hours post-treatment with neutrophil-activating therapy. Scale bars = 500 μm. (n=4) (C-E) Percent oxidized glutathione in B16 lysates 24 hours after treatment with neutrophil-activating therapy following administration of anti-Ly6G (C) (mock n=5, others n=6), CVF (D) (mock n=4, vehicle n=7, CVF n=6), or SC57461A (E) (mock n=5, vehicle n=8, SC57461A n=7). (F) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors and stimulated in vitro with neutrophil-activating therapy together with the indicated inhibitors (n=4). (G) Survival of B16-bearing mice treated with neutrophil-activating therapy following administration of catalase (n=9). (H-J) XO activity in the tumor 24 hours after treatment of B16 with neutrophil-activating therapy following administration of anti-Ly6G (H) (isotype n=7, others n=8), CVF (I) (n=7), or SC57461A (J) (n=7). (K) Percent oxidized glutathione in B16 lysates 24 hours after treatment following administration of topiroxostat (n=5). (L) Quantification of TUNEL staining in B16 tumors 24 hours after treatment with neutrophil-activating therapy following administration of topiroxostat (n=3). (M) Survival of B16-bearing mice treated with neutrophil-activating therapy following administration of topiroxostat (vehicle n=9, topiroxostat n=8). (N) Lysis of B16 cells co-cultured with neutrophils isolated from treated tumors and stimulated in vitro with neutrophil-activating therapy in the presence of topiroxostat (n=4). Statistics: One-way ANOVA with Tukey’s multiple comparisons test (B-E, H-L), Two-way ANOVA with Tukey’s multiple comparisons test (F, N), Log-rank test (G, M). For all dot plots, the line indicates the mean. Data are representative of 2 (F, I-K, M-N) or 1 (A-B, L) independent experiments or pooled from 2 experiments (C-E, G-H). See also Figure S8.
Despite the requirement for neutrophils and ROS, Ncf1−/− mice, which lack the p47 subunit of the phagocyte nicotinamide adenine dinucleotide phosphate oxidase complex (NOX) and fail to produce ROS through phagocyte NOX, were still able to clear tumors, albeit less efficiently than WT mice (Figure S8L). Given these results, we sought an additional source of ROS in the context of this treatment. Xanthine oxidoreductase is a bidirectional enzyme capable of both xanthine dehydrogenase (XDH) and xanthine oxidase (XO) activities, and XO can produce superoxide, serving as a source of ROS50. Neutrophil-activating therapy induced a neutrophil-dependent elevation in XO activity within the tumor (Figure 6H), and depletion of complement and inhibition of LTB4 prevented this increase in XO activity (Figure 6I–J). XO inhibition with topiroxostat did not prevent neutrophil infiltration or LTB4 production (Figure S8M–N), indicating that XO activation occurs downstream of neutrophil LTB4 production. Importantly, inhibition of XO by topiroxostat prevented oxidation in tumors (Figure 6K), demonstrating XO to be responsible for the ROS-mediated damage of the tumor induced by treatment. Inhibition of XO also reduced cell death (Figure 6L and S8O), prevented tumor clearance (Figure 6M and S8P), and inhibited ex vivo tumor cell killing (Figure 6N). Thus, following neutrophil-activating therapy, LTB4 production by complement-activated neutrophils induces ROS production through XO, leading to oxidative damage of tumor cells and subsequent tumor clearance.
Neutrophil-activating therapy activates human neutrophils to kill tumor cells
To determine whether neutrophils could clear human tumors, we treated Rag2−/− Il2rg −/− mice bearing A549 human lung carcinoma or orthotopic MDA-MB-231 human mammary carcinoma with neutrophil-activating therapy. While neutrophil-sufficient mice cleared the tumors, neutrophil-depleted mice were uniformly unable to do so (Figure 7A–B). We next examined whether human neutrophils could be activated in vitro using human versions of our neutrophil-activating therapy components. Stimulation of neutrophils isolated from the peripheral blood of healthy human donors with TNF + anti-CD40 + anti-EGFR mAb induced the upregulation of multiple activation markers, including CD11b and ICAM-1, which were also upregulated in mouse neutrophils (Figure 7C). The FcγR CD32 was upregulated, while CD16 was downregulated, consistent with reports that it can be cleaved from the surface upon neutrophil activation51. In addition, CD66b and CD63 were both upregulated on the cell surface, indicating degranulation, while increased DHR-123 staining indicated ROS production by the stimulated neutrophils. Consistent with our findings in mice, C5a induced upregulation of some activation markers and downregulation of C5AR1, while the combined effect of C5a together with neutrophil-activating therapy enhanced expression of CD63. To determine whether these activated neutrophils could kill human tumor cells, we co-cultured neutrophils from healthy donors with A549 human lung carcinoma cells. Stimulation with neutrophil-activating therapy induced high levels of tumor cell lysis, albeit at higher neutrophil-to-tumor ratios than were necessary with mouse neutrophils isolated from treated tumors (Figure 7D). In agreement with our findings with mouse neutrophils, all three components of neutrophil-activating therapy were required to achieve efficient lysis of tumor cells (Figure 7E). These data suggest that human neutrophils can be activated to kill tumor cells in the same manner we described for mouse neutrophils.
Figure 7: Neutrophil-activating therapy activates human neutrophils to kill tumors.

(A-B) Survival of Rag2−/− Il2rg−/− mice bearing subcutaneous A549 (A) (anti-Ly6G n=7, others n=6) or orthotopic MDA-MB-231 (B) (anti-Ly6G n=7, others n=6) treated with neutrophil-activating therapy following anti-Ly6G administration. (C) Cell surface markers on neutrophils from the peripheral blood of healthy human donors following stimulation with the indicated components for 30 minutes (ICAM-1 n=6, others n=8). (D-E) Lysis of A549 cells co-cultured with neutrophils isolated from healthy donor peripheral blood and stimulated in vitro with the indicated components (n=4). Statistics: Log-rank test with Bonferroni correction (A-B), One-way ANOVA with Tukey’s multiple comparisons test (C), Two-way ANOVA with Tukey’s multiple comparisons test (D-E). For all dot plots, the line indicates the mean. Data are representative of 2 independent experiments (D-E) or pooled from 2 (B-C) or 3 (A) experiments.
Discussion
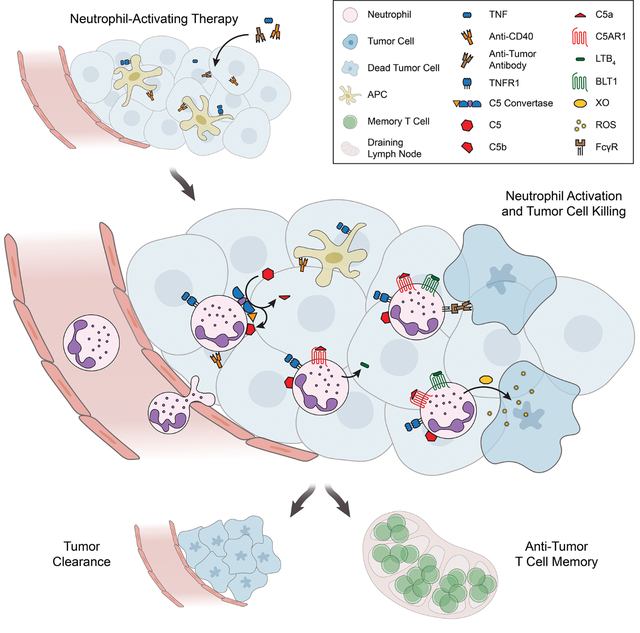
Based on the findings reported here, we propose a multistep mechanism by which neutrophils can be harnessed to eradicate multiple types of tumors (Figure 8). TNF signals through TNFR1 to induce neutrophil recruitment and activation in the tumor. Anti-CD40 promotes neutrophil cytotoxicity and granulopoiesis, and tumor-binding antibody enhances tumor clearance through FcγR-mediated ADCC, potentially by trogocytosis. These treatment components combine to activate complement through the AP, inducing production of C5a. C5a signaling through C5AR1 activates neutrophils to produce LTB4, which drives XO activity in the tumor microenvironment. ROS produced by XO induce oxidative damage and death of tumor cells, ultimately leading to tumor clearance. In contrast to most previous reports of neutrophil anti-tumor activity1,2, this mechanism relies on activation of neutrophils rather than inhibition of their suppressive effects. Moreover, it is capable of inducing eradication of multiple tumor types in immunocompetent mice, it is effective when therapy is initiated after tumors are already established, and it can reduce metastasis. Importantly, the same combination of therapeutic components that eradicates tumors in mice activates human neutrophils to kill tumors in vitro, suggesting that the therapy has the potential to prove effective in patients.
Figure 8: Proposed mechanism of neutrophil-dependent tumor eradication.

Treatment with neutrophil-activating therapy recruits neutrophils to the tumor through TNFR1 signaling and activates the complement AP, generating C5a. C5a signals through C5AR1 in neutrophils and induces neutrophil activation and production of LTB4, which drives XO activity in the tumor environment. ROS produced by XO induce oxidative damage and death in tumor cells, driving tumor clearance. Tumor-binding antibody contributes to neutrophil killing of tumor cells by inducing ADCC, possibly involving trogocytosis. While not dependent on adaptive immunity, this process is capable of priming protective immune memory.
The identification of C5a and LTB4 as crucial mediators of tumor clearance in this mechanism is noteworthy, as previous studies have shown that these molecules generally promote cancer. Signaling through the C5a-C5AR1 axis recruits granulocytes and MDSCs to the tumor, stimulates secretion of immunosuppressive factors by these and other tumor-resident myeloid cells, and results in inhibition of T cell responses49,52–56. Similarly, LTB4 can promote tumor growth by recruiting suppressive neutrophils and MDSCs to the tumor57–59, and neutrophil-derived leukotrienes can support metastasis10.
However, in non-cancer contexts, C5a and LTB4 are potent inflammatory mediators that can induce neutrophil activation and ROS-mediated tissue damage. AP activation on the neutrophil surface can produce high local concentrations of C5a, inducing neutrophil activation and extravasation into inflamed tissue60,61. LTB4 promotes neutrophil “swarming” in the tissue and vasculature, amplifying neutrophil activation, inflammation, and tissue damage62–64. Elements of the mechanism we describe, such as AP-mediated complement activation, C5a-induced LTB4 production by neutrophils, and C5a-dependent production of ROS by neutrophils and XO, can contribute to tissue damage in pathologies as diverse as inflammatory arthritis60,65–67, fungal sepsis63, and acute lung injury68.
The immunotherapeutic strategy utilized in this study likely taps into the neutrophil’s capacity for potent cytotoxic activation that results in tissue damage in the context of these inflammatory pathologies. Although dysregulated inflammation in the tumor can induce pathological activation of neutrophils through chronic exposure to factors such as C5a and LTB4, our work demonstrates that these inflammatory mediators have the capacity to drive tumor-eradicating neutrophil responses if applied with the appropriate threshold and context. While inhibitors of C5AR1, LTB4 production, and BLT1 have been evaluated as cancer treatments49,59, our work here suggests an alternative approach by which these pathways can be exploited to direct neutrophil cytotoxic responses against the tumor.
Even though this mechanism of neutrophil-mediated tumor clearance is not dependent on adaptive immunity, it can still prime immune memory. Neutrophil-activating therapy induced activation of DC populations, and we have shown previously that treatment with tumor-binding antibody in an immunostimulatory context induces tumor antigen uptake by APCs and priming of T cells69,70. We observed activation, proliferation, and memory differentiation of T cells in the blood and dLN following neutrophil-activating therapy, and mice were protected from re-challenge with the same tumor. As such, combination with T cell-targeted treatments such as immune checkpoint blockade represents an attractive avenue for future study with the potential to further enhance therapeutic efficacy. Moreover, the ability of our neutrophil-activating approach to induce an inflammatory cascade resulting in large-scale neutrophil infiltration and cytotoxicity within the tumor may help overcome barriers to the efficacy of immune checkpoint blockade, such as “cold” tumors with poor T cell infiltration71. Additionally, while tumors could downregulate the antibody target antigen in response to therapy, since ADCC contributes to, but is not required for, tumor clearance, neutrophil-activating therapy is likely to be robust against acquired resistance that can limit the efficacy of other monoclonal antibody therapies.
This study lays the groundwork for a neutrophil-activating approach to cancer therapy with the potential to both reverse neutrophil-mediated immunosuppression and activate anti-cancer immunity. As neutrophils are numerous, plastic, and can amplify their own activation and recruitment, strategies to harness their potential to function as anti-cancer effector cells are an attractive option, and attempts to deplete or inhibit suppressive neutrophil populations may squander this powerful anti-cancer capacity. The present study defines therapeutic conditions and an in vivo mechanism by which neutrophils can be exploited to induce potent tumor eradication. Future work can build upon the data presented here to rationally design cancer therapies beyond the combination of agents used in this study.
Limitations of the study
This study raises some questions that should be addressed prior to potential clinical translation. First, the metastasis models were treated at time points shortly after the initiation of metastatic seeding72, so future work will be necessary to determine whether neutrophil-activating therapy can eliminate established metastases or whether inhibition of metastatic seeding is the primary anti-metastatic effect. Additionally, while blood chemistries and body weight were normal one week and 60 days following completion of treatment in mice, we did not perform a thorough analysis of the safety and tolerability of our therapy, which will be necessary prior to clinical translation. In this regard, systemically infused TNF has a history of toxicity in cancer patients73, although we used an intratumoral treatment approach that should reduce systemic exposure to the injected agents. On the other hand, while the intratumoral treatment approach was highly effective, it limits the use of this therapy to accessible tumors. Future work to develop systemic tumor-targeted delivery methods such as antibody conjugates and nanoparticles may broaden the applicability and utility of neutrophil-activating therapy.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Edgar Engleman (edengleman@stanford.edu).
Material Availability
This study did not generate new unique reagents.
Data Code and Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
Experimental Model and Subject Details
Animals
C57BL6/J (Jackson 000664), BALB/cJ (Jackson 000651), MMTV-PyMT (FVB/N-Tg(MMTV-PyVT)634Mul/J, Jackson 002374), Rag2−/− (B6(Cg)-Rag2tm1.1Cgn/J, Jackson 008449), TNFR KO (B6.129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx/J, Jackson 003243), C3−/− (B6.129S4-C3tm1Crr/J, Jackson 029661), and Ncf1−/− (B6N.129S2-Ncf1tm1Shl/J, Jackson 027331) mice were purchased from Jackson Laboratory. Fcer1g−/− mice (B6.129P2-Fcer1gtm1Rav N12, Taconic 583) were purchased from Taconic. Rag2−/− Il2rg−/− mice were generated by crossing B6(Cg)-Rag2tm1.1Cgn/J mice (Jackson 008449) with B6.129S4-Il2rgtm1Wjl/J mice (Jackson 003174). Cfp−/− mice67,74 and C6−/− mice75 were generated as previously described. 8–12 week old female mice were used, and mice of different experimental groups and genotypes were cohoused during all experiments, with the exception of immunocompromised Rag2−/− mice. Mice were randomly assigned to experimental groups. All animal studies were performed in accordance with the Stanford University Institutional Animal Care and Use Committee under protocol APLAC-17466. All mice were housed in an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility and maintained in specific pathogen-free conditions.
Cell Lines and Culture
The mouse melanoma cell line B16F10, mouse lung carcinoma line LL/2, mouse mammary carcinoma line 4T1, human lung carcinoma line A549, and human mammary carcinoma line MDA-MB-231 were purchased from ATCC. To generate the mouse colon carcinoma line Sparkl.4640 (Syngeneic P53 APC ROSA26-LSL-eYFP Kras Lgr5-CreERT2), crypts were harvested and expanded from the colon of an adult female Lgr5-EGFP-IRES-creERT2 Trp53fl/fl Apcfl/fl KrasLSL-G12D/+ ROSA26LSL-eYFP/LSL-eYFP mouse according to a published protocol76. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Cells were dissociated into a single cell suspension and then cultured in 4-hydroxytamoxifen (Sigma). Cells were washed and allowed to form colonies in Matrigel. After 1 week in culture, roughly 50 fluorescent colonies were collected and separated from non-fluorescent colonies under stereomicroscope with fluorescence attachment (Nikon). Colonies were dissociated and plated onto tissue culture treated plates in RPMI-1640 with 10% FBS. Notably, transformed cells were then able to be passaged in the absence of supplemental growth factors or Matrigel, both of which were required for the culture of crypts prior to treatment with 4-hydroxytamoxifen.
B16, LL/2, and A549 were cultured in DMEM (Gibco) supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco). Sparkl.4640, 4T1, and MDA-MB-231 were cultured in RPMI-1640 (Gibco) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were tested for endotoxins using LAL Chromogenic Endotoxin Quantitation Kit (Pierce) and for mycoplasma using PlasmoTest™ (InvivoGen), according to manufacturer’s instructions.
Human Blood
For studies involving human neutrophils, whole blood was obtained from de-identified healthy blood donors at Stanford Blood Center. Informed consent was obtained from all donors. Human neutrophils were isolated and used in assays immediately and were not maintained in culture.
Method Details
Tumor implantation, treatment, and survival
Cell lines were harvested with trypsin-EDTA (Gibco), washed once, and injected in 50 μl phenol red-free RPMI-1640 (Gibco). B16 (2.5×105 cells), Sparkl.4640 (5×105 cells), and LL/2 (1×105 cells) were injected subcutaneously (s.c.) into the flank of WT or KO mice on a syngeneic C57BL/6J background. 4T1 (1×105 cells) was injected s.c. into the flank of WT mice on a syngeneic BALB/cJ background. A549 (5×106 cells) was injected s.c. into the flank of Rag2−/− Il2rg−/− mice on a C57BL6/J background. MDA-MB-231 (5×106 cells) was injected orthotopically into the mammary fat pad of Rag2−/− Il2rg−/− mice on a C57BL6/J background. B16, Sparkl.4640, 4T1, and A549 tumors were allowed to grow for approximately 6 days prior to treatment, LL/2 was allowed to grow for approximately 8 days prior to treatment, and MDA-MB-231 was allowed to grow for approximately 12 days prior to treatment, at which point the tumors were approximately 10–30 mm2. MMTV-PyMT mice were monitored until palpable tumors developed in the breast, at approximately 8 weeks of age, and treatment was initiated when tumors reached approximately 4–10mm2.
Except where indicated otherwise, tumors were treated by intratumoral injection of tumor-binding antibody, 100 μg of agonistic anti-CD40 antibody (clone FGK4.5, Bio X Cell, Lebanon, NH), and 1 μg (ED50 0.5–3 pg/ml in L929 cytotoxicity assay) of recombinant mouse TNF (BioLegend) in 50 μl total volume in phosphate buffered saline (PBS), which is referred to in the text as neutrophil-activating therapy. As tumor-binding antibody, B16 received 100 μg anti-gp75 (clone TA99, Bio X Cell), B16-EGFP received 100 μg anti-GFP (clone F56–6A1.2.3, Bio X Cell), Sparkl.4640 and LL/2 received 10 μg anti-CD44 (DS-MB-00666, RayBiotech), 4T1 and MMTV-PyMT received 100 μg anti-MHC Class I (clone 34-1-2S, Bio X Cell), and A549 and MDA-MB-231 received 100 μg anti-human MHC Class I (clone W6/32, Bio X Cell). In all cases, tumor-binding antibodies were confirmed to bind the appropriate tumor cells by flow cytometry. Mock-treated mice received an intratumoral injection of 50 μl PBS (treatment vehicle). Treatment was administered twice, two days apart, designated as days 0 and 2 post-treatment. For MMTV-PyMT mice, treatment was repeated weekly for four cycles, so that mice were treated on days 0, 2, 7, 9, 14, 16, 21, and 23. Although tumors develop in multiple breasts in the MMTV-PyMT model, only one tumor was treated per mouse for the duration of the therapy, with the largest tumor at the time of treatment initiation chosen for treatment. For TNF dose response experiments, mice received doses of 10 ng, 50 ng, or 1 μg of TNF (BioLegend). Where indicated, tumors were injected with one or two of the three treatment components. In some experiments, tumors were injected with 1 μg recombinant mouse GM-CSF (BioLegend), 5 μg recombinant mouse IFNγ (BioLegend), 1 μg recombinant mouse IL-1β (BioLegend), or 1 μg recombinant mouse IL-17A (BioLegend). Tumor areas were measured three times per week, and mice were euthanized when treated tumors exceeded 100 mm2 or when tumors became ulcerated, with both indicated as a death event on the Kaplan Meier plots. Mice were censored from survival studies when they had to be euthanized for reasons unrelated to tumor progression, such as dermatitis, and were tumor-free. For re-challenge studies, C57BL/6J WT or Rag2−/− mice that had cleared B16 tumors were re-challenged with 5×104 B16 cells in the opposite flank 50 days after initial treatment.
Blood chemistry
Blood was collected from mice by retro-orbital bleed and allowed to clot for 30 minutes on ice. It was then centrifuged at 2000 × g for 20 minutes at 4°C. Serum was collected from the top of the clot and centrifuged again at 2000 × g for 10 minutes at 4°C to remove residual red blood cells. Chemistry analysis was performed on the Siemens Dimension EXL200/LOCI analyzer by the Stanford University Animal Diagnostic Lab.
Mouse in vitro cytotoxicity studies
B16 tumors were treated by intratumoral injection of TNF + anti-CD40 + anti-gp75 and harvested 12 hours post-treatment. Tumors were dissected away from any surrounding fat, minced, and digested in 5 mg/ml collagenase IV (Worthington) plus 0.1 mg/ml DNase I (Sigma) with continuous mixing by magnetic stir bars for 20 minutes at 37°C in RPMI-1640 (Gibco) with 2% FBS. Following digestion, tissue was mashed through a 70 μm cell strainer (Falcon) and washed. For some studies, bone marrow was harvested by grinding bones from tumor-naïve untreated mice in a mortar and pestle and mashing through a 70 μm strainer (Falcon). Neutrophils were isolated from tumor and bone marrow samples with the MojoSort Mouse Ly6G Selection Kit (BioLegend), according to the manufacturer’s instructions, and used in the cytotoxicity assay.
Cytotoxicity assays were conducted using the EuTDA assay from the DELFIA TRF cytotoxicity kit (PerkinElmer) according to the manufacturer’s instructions. Briefly, B16 cells were labeled for 10 minutes with BATDA, and 5×103 labeled cells were added per well to a 96 well V-bottom plate in RPMI-1640. Neutrophils were added at a ratio of 10:1 unless otherwise stated. All co-cultures were conducted in the presence of TNF (10 ng/ml), anti-CD40 (1 μg/ml), anti-gp75 (1 μg/ml), and 10% active mouse C57BL/6 complement serum (Innovative Research), except where indicated otherwise. Mouse C57BL/6 complement serum was heat-inactivated for 40 minutes at 57°C where inactivation is specified. In some experiments, mouse IgG2a isotype control (1 μg/ml, isotype control for anti-gp75, clone C1.18.4, Bio X Cell), anti-mouse CD16/CD32 (10 μg/ml, clone 2.4G2, Bio X Cell), recombinant mouse C5a (50 nM, R&D Systems), anti-C5a (25 μg/ml, clone 295108, R&D Systems), rat IgG2a isotype control (25 μg/ml, isotype control for anti-C5a, clone 2A3, Bio X Cell), anti-C5AR1 (5 μg/ml, clone 20/70, BioLegend), rat IgG2b isotype control (5 μg/ml, isotype control for anti-C5AR1, clone LTF-2, Bio X Cell), SC57461A (10 μM, Cayman Chemical), CP-105696 (1 μM, Sigma), topiroxostat (10 μM, MedChem Express), or DMSO (vehicle for SC57461A, CP-105696, and topiroxostat) were added to the wells with the B16 and neutrophils. After 4 hours of co-culture at 37°C, supernatant was taken from the wells, Europium solution was added, and TDA released from lysed B16 cells was detected by TRF on a Victor X4 fluorescence microplate reader (PerkinElmer). Percent maximal lysis was determined by calculating the specific release of TDA using the formula: (experimental release – spontaneous release) / (maximum release – spontaneous release), where spontaneous release was determined by wells containing no neutrophils and maximum release was determined by wells with lysis buffer added.
For trogocytosis studies investigating transfer of tumor-binding antibody, anti-gp75 labeled with AF647 (clone TA99, Novus Biologicals) was used in place of unlabeled anti-gp75. For studies investigating transfer of B16 cell membrane, DiD’ Solid (ThermoFisher Scientific) was reconstituted in DMSO at 10 mg/ml and diluted to a working solution of 5 μg/ml in serum-free DMEM. B16 cells were labeled in this solution for 20 minutes at 37°C at 1×106 cells/ml. Labeled B16 was washed 3 times in warm media before being added to the co-culture wells with the neutrophils. Following co-culture, flow cytometry was used to identify DiD signal in neutrophils.
Depletion and inhibition studies
In neutrophil depletion experiments, 500 μg anti-Ly6G (clone 1A8, Bio X Cell) or isotype control (clone 2A3, Bio X Cell) was administered intraperitoneally (i.p.) on days −2, 0, and 2 relative to treatment, with administration 4 hours prior to treatment on days 0 and 2. For MMTV-PyMT mice, this administration pattern was continued for four weeks for each of the treatment cycles. For neutrophil depletion studies in untreated mice, anti-Ly6G or isotype control administration began 4 days after tumor inoculation (equivalent to day –2 in treated mice), and administration continued every 2 days until the mice were euthanized. Graphing of survival for these mice began at day 6 post tumor inoculation (equivalent to day 0 in treated mice).
For TNFR blocking experiments, 100 μg of anti-TNFR1 (clone 55R-170, BioLegend), anti-TNFR2 (clone TR75-54.7, BioLegend), or isotype control (Armenian Hamster IgG, Bio X Cell) was administered i.p. once per day on days −1 through 4 relative to treatment, with administration 1 hour prior to treatment on days 0 and 2. Anti-factor B (clone 1379) was produced from a hybridoma (PTA-6230, ATCC) with serum-free CD Hybridoma Medium (Gibco) in a 1L CELLine bioreactor flask, purified with HiTrap Protein G HP columns, and buffer-exchanged to PBS in an Amicon Ultra 100 kDa centrifugal filter (Millipore). Anti-factor B or isotype control (clone MOPC-21, Bio X Cell) was administered i.p. at 1 mg once per day on days −1 through 2 relative to treatment, with administration 1 hour prior to treatment on days 0 and 2. Anti-properdin (clone 14E1) and anti-C5 (clone BB5.1) were produced as previously described74. For properdin blocking experiments, 1 mg of anti-properdin or isotype control (clone MOPC-21, Bio X Cell) was administered i.p. on days −1 and 1 relative to treatment. For C5 blocking experiments, 800 μg of anti-C5 or isotype control (clone MOPC-21, Bio X Cell) was administered i.p. once daily on days −1 through 2 relative to treatment, with administration 1 hour prior to treatment on days 0 and 2. For C5AR1 blocking experiments, anti-C5AR1 (clone 20/70, BioLegend) and isotype control (clone LTF-2, Bio X Cell) were deglycosylated with the deGlycIT kit (Genovis) prior to administration in order to abrogate Fc receptor binding and prevent depletion of anti-C5AR1-bound neutrophils77, and 100 μg of anti-C5AR1 or isotype control were administered daily on days −1 through 3 relative to treatment, with administration 1 hour prior to treatment on days 0 and 2.
Reduced L-glutathione (Sigma) was dissolved in PBS and administered i.p. at 500 mg/kg at hours −1, 0, 2, 4, and 8 relative to treatment on days 0 and 2, as well as twice per day on days 1 and 3. Catalase (C40, Sigma) was dissolved in PBS and administered i.p. at 500 mg/kg twice per day on days 0, 1, 2, and 3 relative to treatment, with administrations on days 0 and 2 coming immediately prior and 4 hours after treatment. CVF (from Naja naja kaouthia, Millipore), which depletes complement components C3 and C5 from blood through fluid-phase activation78, was diluted in PBS and administered at 50 μg i.p. daily on days –2 through 2 relative to treatment, with administration 30 minutes prior to treatment on days 0 and 2. SC57461A (Cayman Chemical, Ann Arbor, MI) was dissolved in DMSO at 150 mg/ml, diluted in PBS, and administered i.p. at 75 mg/kg twice per day on days −1 through 3 relative to treatment, with administration 1 hour prior and 4 hours after treatment on days 0 and 2. CP-105696 (Sigma) was dissolved in DMSO at 400 mg/ml, diluted in 25% 2-hydroxypropyl-β-cyclodextrin (Cayman Chemical), and administered i.p. at 100 mg/kg twice per day on days 0 through 3 relative to treatment, with administration 1 hour prior and 4 hours after treatment on days 0 and 2. Topiroxostat (MedChemExpress) was dissolved in 0.2N NaOH in PBS, the pH was adjusted with HCl, and it was administered i.p. at 150 mg/kg on days 0 and 2, 3 hours prior to treatment.
Metastasis studies
For B16 experimental metastasis studies, mice were implanted with 2.5×105 B16 cells expressing tdTomato s.c. in the flank. Seven days after implantation, 2×105 B16 cells expressing tdTomato were injected i.v. by the tail vein. Ten hours after tail vein injection, the s.c. tumors were treated with PBS or TNF + anti-CD40 + anti-gp75, and the primary tumors were treated again two days later according to the standard treatment protocol. Nine days after tail vein injection, the mice were euthanized, the lungs harvested, and fluorescence images were acquired under a stereomicroscope with fluorescence attachment (Nikon). Discrete fluorescent metastases visible on the exterior of the lungs were counted to obtain metastasis counts. The average nodule area for metastases was determined using ImageJ by thresholding the image to remove background, using the analyze particles function to obtain the total area of the metastases, and dividing this area by the number of metastases.
For 4T1 metastasis studies, 1×105 4T1 tumor cells were implanted orthotopically in the mammary fat pad. One week post-implantation, when the primary tumor had reached a size of 16–30mm2, the primary tumor was treated by intratumoral injection of neutrophil-activating therapy or PBS mock treatment, with two injections two days apart, according to the standard protocol. The mice were euthanized 30 days post-implantation and pulmonary metastases were enumerated as previously described79 by intra-tracheal injection of India ink (15% India Ink, 85% PBS, 0.1% NaOH). India ink injected lungs were washed in 3mL Fekete’s solution (50% ethanol, 6% formaldehyde, and 3% glacial acetic acid) and then placed 5 mL fresh Fekete’s solution overnight. White tumor nodules against a black lung background were counted manually.
Knockout of TNFR1 in B16 using CRISPR-Cas9
pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138) 80. Two sgRNA target sequences for mouse Tnfrsf1a were chosen from the Brie library81: AGACCTAGCAAGATAACCAG and GATGGGGATACATCCATCAG, referred to in the text as Tnfrsf1a sgRNA1 and Tnfrsf1a sgRNA2, respectively. An sgRNA targeting the irrelevant E. coli β-galactosidase gene (LacZ) was also included as a control. Oligos for these sgRNA target sequences were synthesized by the Stanford Protein and Nucleic Acid facility, with end overhangs to enable cloning into the BbsI site of the PX458 backbone. Oligos were phosphorylated with T4 PNK (NEB) and annealed in a thermocycler at 37°C for 30 minutes, followed by 95°C for 5 minutes, and ramping down to 25°C at 5°C/minute. PX458 was digested with BbsI (NEB) and gel purified using the QIAquick Gel Extraction Kit (Qiagen). Phosphorylated and annealed oligos were ligated into BbsI-cut PX458 with T4 DNA ligase (NEB) and transformed into Stellar Electrocompetent Cells (Clontech) by electroporation with the GenePulser Xcell (BioRad). Plasmids were prepared with the Plasmid Plus Maxi Kit (Qiagen) and transfected into B16 cells using Lipofectamine 2000 (Thermo Fisher Scientific). Successfully transfected cells positive for expression of GFP were sorted on the FACSAria II (BD), followed by three successive rounds of sorting for cells negative for both GFP and TNFR1 staining using APC anti-TNFR1 (clone 55R-286, BioLegend), to achieve a population of cells lacking expression of TNFR1 with the transient expression of GFP and CRISPR machinery removed.
B16-EGFP
To generate B16 cells expressing EGFP on their surface, DNA encoding a fusion of mouse Igκ signal peptide, EGFP, and the transmembrane domain of mouse PDGFR was synthesized using GeneArt Gene Synthesis (Invitrogen). The synthesized gene was digested with BamHI and EcoRI (NEB) and gel purified using the QIAquick Gel Extraction Kit (Qiagen). This was ligated into pLVX-EF1α-IRES-Puro (Clontech) with T4 DNA ligase (NEB) and transformed into Stellar Electrocompetent Cells (Clontech) by electroporation with the GenePulser Xcell (BioRad). Plasmids were prepared with the Plasmid Plus Maxi Kit (Qiagen) and transfected into 293T cells together with the psPax2 and pCMV-VSV-G plasmids using Lipofectamine 2000 (Thermo Fisher Scientific). Virus was collected and used to transduce B16 cells, and successful transductants were selected using 1 μg/ml puromycin (ThermoFisher Scientific).
Flow cytometry
Tumors were digested with collagenase IV and Dnase I as described above. Following digestion, tissue was mashed through a 70 μm cell strainer (Falcon), washed, red blood cells were lysed with ACK buffer for 1 minute, and cells were washed again prior to antibody staining. Blood was harvested into PBS plus 20 mM EDTA by cardiac puncture or retro-orbital bleed, lysed in ACK buffer for 5 minutes, and washed prior to antibody staining. Bone marrow was harvested by grinding the femur and tibia in a mortar and pestle and mashing through a 70 μm strainer (Falcon), or by flushing femurs with a syringe and needle, then lysed in ACK buffer for 5 minutes and washed prior to antibody staining. Spleens were harvested by mashing through a 70 μm cell strainer (Falcon), then lysed in ACK buffer for 5 minutes and washed prior to antibody staining.
Live cells were stained with antibodies on ice for 20 minutes in FACS buffer (HBSS 1% BSA 5mM EDTA) with Brilliant Stain Buffer Plus (BD). Following staining, cells were washed twice in FACS buffer and resuspended in 1 μg/ml DAPI plus AccuCount Fluorescent Particles (Spherotech) for absolute count determination. In some experiments, live cells were stained with Live/Dead Fixable Blue Stain (Invitrogen) in HBSS on ice for 20 minutes prior to antibody staining, and DAPI was not used. Cells for the HSC/progenitor experiment were viability stained with 800CW NHS Ester (Li-Cor #929-70020) at a 1:4000 dilution of a 1 mg/ml DMSO stock, for 20 minutes in PBS on ice prior to antibody staining. For assessment of neutrophil depletion efficiency and experiments analyzing Foxp3 and Ki67, cells were stained with Live/Dead Fixable Blue Stain (Invitrogen) in HBSS on ice for 20 minutes, cells were fixed and permeabilized prior to antibody staining using the Foxp3/Transcription Factor Staining Kit (eBioscience) or the True-Nuclear Transcription Factor Set (BioLegend) according to the manufacturer’s instructions, and DAPI was not used. This staining following fixation/permeabilization allows identification of neutrophils with depleting antibody-bound extracellular Ly6G by staining intracellular Ly6G. Samples were acquired on an LSRFortessa (BD), except for the HSC/progenitor data, which was acquired on a Cytek Aurora using SpectroFlo V2.2.0.3. The following antibodies were used: BUV395 anti-CD45 (clone 30-F11, BD), BV421 anti-ICAM-1 (clone YN1/1.7.4, BioLegend), BV480 anti-MHC II (clone M5/114.15.2, BD), BV650 anti-CD11b (clone M1/70, BioLegend), PE-Cy7 or BUV737 anti-Ly6G (clone 1A8, BD/BioLegend), PE or AF647 anti-C3 (clone 11H9, Novus Biologicals), APC-R700 anti-CD11c (clone N418, BD), APC-Cy7 or BV785 anti-Ly6C (clone HK1.4, BioLegend), CD177 AF647 (clone Y127, BD Biosciences), SIRPα FITC (clone P84, BioLegend), Siglec F BV480 (clone E50-2440, BD Biosciences), CD101 PE-Cy7 (clone Moushi101, ThermoFisher Scientific), CD14 APC-Cy7 (clone Sa14-2, BioLegend), PD-L1 BV421 or BV711 or PE-Cy7 (clone 10F.9G2, BioLegend), XCR1 APC (clone ZET, BioLegend), F4/80 APC-Cy7 (clone BM8, BioLegend), CD206 FITC (clone C068C2, BioLegend), CD80 PerCP-eF710 (clone 16-10A1, ThermoFisher Scientific), CD86 BUV737 (clone GL1), B220 BV711 (clone RA3-6B2, BioLegend), TCRβ BV421 (clone H57-597, BioLegend), NK1.1 PE-Cy7 (clone PK136, BioLegend), CD8α BV510 (clone 53-6.7, BioLegend), CD4 BV711 (clone RM4-5, BioLegend), Foxp3 AF488 (clone MF23, BD Biosciences), CD25 BV650 (clone PC61, BioLegend), CD62L PerCP-Cy5.5 (clone MEL-14, BioLegend), CD44 APC-eF780 (clone IM7, ThermoFisher Scientific), KLRG1 PE (clone 2F1/KLRG1, BioLegend), PD-1 BV785 (clone 29F.1A12, BioLegend), CD69 APC (clone H1.2F3, BD Biosciences), Ki67 AF700 (clone SolA15, ThermoFisher Scientific), CD34 eF450 or AF700 (clone RAM34, ThermoFisher Scientific), IL-7Rα AF700 (clone A7R34, ThermoFisher Scientific), Sca-1 BV711 (clone D7, BioLegend), Sca-1 PE-Cy7 (clone E13-161/7, BioLegend), cKit FITC (clone 2B8, ThermoFisher Scientific), CD16/32 PE (clone S17011E, BioLegend), CD3ε PE-Cy7 (clone 17A2, ThermoFisher Scientific), CD4 PE-Cy7 (clone (GK1.5, ThermoFisher Scientific), CD8α PE-Cy7 (clone 53–6.7, ThermoFisher Scientific), CD11b PE-Cy7 (clone M1/70, ThermoFisher Scientific), B220 PE-Cy7 (clone RA3-6B2, ThermoFisher Scientific), NK1.1 PE-Cy7 (clone PK136, ThermoFisher Scientific), Flt3 APC (clone A2F10, BioLegend), CD106 Pacific Blue (clone 429 (MVCAM.A), BioLegend), CD115 BV605 (clone AFS98, BioLegend), Ly6G PE-Cy5 (clone 1A8, ThermoFisher Scientific), CD81 PerCP-Cy5.5 (clone Eat-2, BioLegend), H-2Kb PE (clone AF6-88.5, BD Biosciences), H-2Db APC (clone KH95, BioLegend), fluorescein anti-gp75 (clone TA99, Bio X Cell, labeled using NHS-Fluorescein (Thermo Scientific)), MPO FITC (clone 2D4, Abcam), citrullinated Histone H3 (Abcam ab5103), donkey anti-rabbit IgG PE (BioLegend Poly4064), and unconjugated anti-CD16/CD32 (clone 2.4G2, Bio X Cell) to block Fc receptors. For dihydrorhodamine-123 (DHR-123) staining of tumor samples, 5mM DHR-123 in DMSO (Invitrogen) was added to the collagenase mixture at a dilution of 1:4000 and allowed to stain for the duration of the 20-minute collagenase digestion. Flow cytometry data was analyzed using FlowJo software (BD). Staining levels were quantified using the median fluorescence intensity (MFI).
May-Grünwald-Giemsa staining
Tumors were harvested, processed, and stained for flow cytometry as described above, 24 hours after treatment or mock treatment. Cells were stained with FITC anti-CD45 (clone 30-F11, BioLegend), APC-Cy7 anti-CD11b (clone M1/70, BioLegend), and PE anti-Ly6G (clone 1A8, BioLegend), and CD45+CD11b+Ly6G+ neutrophils were sorted on a FACSAria II (BD). Sorted neutrophils were resuspended at 5×105 cells/ml in FACS buffer, and 200 μl was spun onto a slide using the StatSpin CytoFuge 2 at 850 rpm for 10 minutes. Slides were dried and then stained for 4 minutes in May-Grünwald stain solution (Electron Microscopy), transferred directly into 4% Giemsa stain solution (Electron Microscopy) for 4 minutes, and washed twice with water for 30 seconds each. Slides were dried and coverslips were mounted using Cytoseal 60 (Richard Allen Scientific). Stained cells were imaged on a Keyence BZ-X810 microscope (Keyence) with the 20X objective and a resolution of 1920 × 1440 pixels.
Immunofluorescence
Tumors were dissected away from surrounding fat, fixed in 2% paraformaldehyde for 2 hours at 4°C, equilibrated in a 30% sucrose solution at 4°C, and embedded and frozen in O.C.T. Compound (Tissue-Tek). Slides were cut to 6 μm, blocked with 1% BSA and 10% serum matched to the secondary antibody species. The following antibodies were used for immunofluorescence: PE anti-Ly6G (clone 1A8, BioLegend), DyLight 650 anti-gp75 (clone TA99, Bio X Cell, labeled using DyLight 650 NHS Ester (Thermo Scientific)), fluorescein anti-gp75 (clone TA99, Bio X Cell, labeled using NHS-Fluorescein (Thermo Scientific)), FITC anti-DNA/RNA damage (clone 15A3, recognizing 8-hydroxy-2’-deoxyguanosine/8-oxo-7,8-dihydroguanine/8-oxo-7,8-dihydroguanosine, Abcam), and AF647 anti-C3 (clone 11H9, Novus Biologicals). DAPI (Invitrogen) was stained at 1 μg/ml. FITC/fluorescein signal was amplified using AF488 anti-FITC (ThermoFisher Scientific), and PE was amplified using biotin anti-PE (clone PE001, BioLegend) followed by DyLight 594 streptavidin (BioLegend) or DyLight 649 streptavidin (BioLegend). Prior to use of biotinylated antibodies, endogenous biotin was blocked using the Avidin/Biotin Blocking Kit (Vector Laboratories). Prior to DNA/RNA damage staining, sections were permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate. TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) staining was performed using the In Situ Cell Death Detection Kit, TMR Red (Roche) according to the manufacturer’s instructions. Briefly, tissue sections were fixed with 4% paraformaldehyde for 20 minutes on ice prior to treatment with 0.1% Triton X-100 in 0.1% sodium citrate for permeabilization. Sections were washed in PBS before incubation for 60 minutes at 37°C with antibodies and TdT enzyme, followed by washing. Images were acquired by tile scanning using a Zeiss LSM 700 confocal laser scanning microscope (Carl Zeiss Microscopy) or a Keyence BZ-X810 microscope (Keyence) using the 20X objective and a resolution of 960 × 720 pixels per tile. Tumor immunostainings were repeated independently at least 2 times in at least biological triplicate and whole tissue section images were acquired. Stitching of images acquired with the LSM 700 was performed using ZEN software, and stitching of images acquired with the BZ-X810 was performed using BZ-X800 Analyzer software. Multiple tumors from the same experiment were embedded together in the same block, stained together in the same section, and acquired together in a tile scan across the entire section. Individual tumors were then cropped from the full tile scan image for display in figures. Images were overlaid and color channel levels were adjusted in Photoshop (Adobe), with individual color channels receiving individual level adjustments based on the staining intensity. All parameters that were quantified were acquired with identical microscope settings and adjusted identically in Photoshop, and all adjustments were applied equally across the entire tumor. Quantification was performed in ImageJ, using the wand tool on the overlaid multichannel image to draw a border around the tumor and then measuring the percent area within that border with signal for the channel of the marker quantified.
LTB4 ELISA
Tumors were dissected away from surrounding fat 24 hours post-treatment and lysed in PBS using 3 mm zirconium beads (Benchmark Scientific) in the BeadBug Microtube Homogenizer (Benchmark Scientific) for 2 cycles of 45 seconds at 3000 rpm. Crude lysate was centrifuged for 15 minutes at 16000 × g at 4°C to obtain clarified lysate. Clarified lysate was deproteinized by ethanol precipitation by adding 4 volumes of 100% ethanol, incubating on ice for 5 minutes, and centrifuging for 10 minutes at 3000 × g at 4°C. Deproteinized supernatant was transferred to a new tube, ethanol was removed by evaporation at room temperature, and samples were brought to the appropriate volume in ELISA assay buffer. LTB4 content was determined with the Leukotriene B4 ELISA kit (Cayman Chemical 520111) according to the manufacturer’s instructions and read with a Victor X4 fluorescence microplate reader (PerkinElmer).
Ex vivo neutrophil LTB4 assay
B16 tumors were harvested from mice 12 hours after treatment with TNF + anti-CD40 + anti-gp75 and digested with collagenase IV and DNase I as described above. Tumor samples were then split into two halves, with one half undergoing selection with the MojoSort Mouse Ly6G Selection Kit (BioLegend) to generate Ly6G+ and Ly6G-depleted tumor samples and the other half undergoing depletion with an isotype control antibody and streptavidin nanobeads (BioLegend) to generate the “all cells” condition. These selected samples were then plated in 200μl of Opti-MEM (Gibco) and incubated for 30 minutes at 37°C. Following the incubation, the supernatant was collected, centrifuged to remove cells and debris, and analyzed for LTB4 using the Leukotriene B4 ELISA kit (Cayman Chemical 520111) as described above.
Mouse in vitro neutrophil stimulations
Bone marrow was harvested by grinding bones in a mortar and pestle and mashing through a 70 μm strainer (Falcon). Neutrophils were isolated by negative selection with the MojoSort Mouse Neutrophil Isolation Kit (BioLegend). Following isolation, neutrophils were plated in Opti-MEM (Gibco) at 1×105 cells in 100 μl and stimulated with 10 ng/ml TNF, 1 μg/ml anti-CD40, 1 μg/ml anti-gp75, and/or 50 nM recombinant mouse C5a (R&D Systems) for 30 minutes at 37°C. After 30 minutes, the supernatant was collected, centrifuged to remove cells and debris, and analyzed for LTB4 using the Leukotriene B4 ELISA kit (Cayman Chemical 520111) as described above. Additionally, stimulated neutrophils were stained for activation markers and analyzed by flow cytometry as described above.
Determination of oxidized glutathione content
Tumors were dissected away from surrounding fat 24 hours post-treatment and lysed in mammalian lysis buffer (Abcam ab179835) using 3 mm zirconium beads (Benchmark Scientific) in the BeadBug Microtube Homogenizer (Benchmark Scientific) for 2 cycles of 45 seconds at 3000 rpm. Crude lysate was centrifuged for 15min at 16000 × g at 4°C, and clarified lysate was deproteinized using the Deproteinizing Sample Preparation Kit – TCA (Abcam ab204708) according to the manufacturer’s instructions. Glutathione was detected using the GSH/GSSG Ratio Detection Assay Kit II (Abcam ab205811) according to the manufacturer’s instructions, reading the resulting signal with a Victor X4 fluorescence microplate reader. The percentage of oxidized glutathione was calculated from the reduced glutathione and total glutathione values determined by the kit.
XO assay
Tumors were dissected away from surrounding fat 24 hours post-treatment and lysed in XO assay buffer (Abcam) using 3 mm zirconium beads (Benchmark Scientific) in the BeadBug Microtube Homogenizer (Benchmark Scientific) for 2 cycles of 45 seconds at 3000 rpm. Crude lysate was centrifuged for 10 minutes at 16000 × g at 4°C to obtain clarified lysate. XO activity of the lysate was determined using the Xanthine Oxidase Activity Assay kit (Abcam), setting up the fluorometric assay and performing calculations according to the manufacturer’s instructions, and reading fluorescence with a Victor X4 fluorescence microplate reader.
ROS assays
For the luminol assay, B16-bearing mice were anesthetized 4 hours post-treatment, and 50 μL luminol sodium salt (Sigma-Aldrich) at 20 mg/mL in PBS was administered intratumorally. Mice were immediately imaged using the IVIS Lumina system (Xenogen). Signal intensity was quantified as photons/second (p/s) over a one-minute exposure in equally sized regions of interest placed over the tumor, using Living Image software (Caliper Life Sciences).
For the OxyBurst assay, neutrophils were isolated from naïve bone marrow or treated tumors, and 1×105 neutrophils were plated in a 96-well plate together with 5×103 B16 tumor cells. The cells were stimulated as in the cytotoxicity studies and cultured for 4 hours at 37°C in the presence of 10 μg/ml OxyBurst Green H2HFF BSA. After co-culture, the plate was read on a Victor X4 fluorescence microplate reader (PerkinElmer) with 485 nm excitation and 535 nm emission.
Human neutrophil studies
Human neutrophils were isolated from whole blood obtained from de-identified blood donors using the EasySep Direct Human Neutrophil Isolation Kit (StemCell Technologies) according to the manufacturer’s instructions. For activation marker studies, neutrophils were plated at 1×106 cells/ml in Opti-MEM (Gibco) and stimulated for 30 minutes at 37°C with human TNF (50 ng/ml, BioLegend), anti-human CD40 (1 μg/ml, clone G28.5, Bio X Cell), anti-human EGFR (1 μg/ml, Cetuximab biosimilar, Bio X Cell), or human C5a (50 nM, R&D Systems), as indicated in the figures. In some experiments, 5mM DHR-123 in DMSO (Invitrogen) was added to the stimulation well at a dilution of 1:4000. Following stimulation, the cells were stained with antibodies on ice for 20 minutes in FACS buffer (HBSS 1% BSA 5mM EDTA) with Brilliant Stain Buffer Plus (BD). Following staining, cells were washed twice in FACS buffer and resuspended in 1 μg/ml DAPI. Samples were acquired on an LSRFortessa (BD). The following antibodies were used: APC-Fire 750 anti-CD11b (clone M1/70, BioLegend), ICAM-1 FITC (clone HA58, BioLegend), CD16 BV711 (BioLegend, clone 3G8), CD32 (clone FLI8.26, BD Biosciences), CD66b AF647 (clone G10F5, BioLegend), CD63 BV510 (clone H5C6, BD Biosciences), and anti-C5AR1 PE (clone S5/1, BioLegend).
Cytotoxicity assays were performed with the EuTDA assay from the DELFIA TRF cytotoxicity kit (PerkinElmer) according to the manufacturer’s instructions. A549 cells were labeled for 30 minutes with BATDA, and 1×104 labeled cells were added per well to a 96 well V-bottom plate in RPMI-1640. Neutrophils were added at a ratio of 50:1 unless specified otherwise. All co-cultures were conducted in the presence of TNF (10 ng/ml), anti-CD40 (1 μg/ml), anti-gp75 (1 μg/ml), and 10% pooled human complement serum (Innovative Research) for 1 hour, except where indicated otherwise.
Quantification and Statistical Analysis
Statistics
Statistical tests were performed in Prism (GraphPad Software, Inc.). Statistical tests are used are listed in the figure legends. In cases where conditions were compared across multiple time points or cell types, statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test, comparing only the conditions within each time point or cell type. For in vitro cytotoxicity studies with multiple conditions and groups, statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test, comparing all conditions and groups with all other conditions and groups. Plots display individual biological replicates obtained from distinct mice, with a line at the mean. For all figures, * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001, **** denotes p < 0.0001, and n.s. indicates not significant, with the exception of Kaplan Meier plots with multiple comparisons, in which case the asterisks are assigned based on Bonferroni-corrected p values. For all experiments, n represents the number of mice or the number of samples. Exact n values and the number of independent experiments are provided in the figure legends.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BUV395 anti-CD45 | BD Biosciences | Cat# 564279 |
| BV421 anti-ICAM-1 | BioLegend | Cat# 116141 |
| BV480 anti-MHC II | BD Biosciences | Cat# 566086 |
| BV650 anti-CD11b | BioLegend | Cat# 101259 |
| PE-Cy7 anti-Ly6G | BioLegend | Cat# 127618 |
| PE anti-C3 | Novus Biologicals | Cat# NB200-540PE |
| AF647 anti-C3 | Novus Biologicals | Cat# NB200-540AF647 |
| APC-R700 anti-CD11c | BD Biosciences | Cat# 565872 |
| APC-Cy7 anti-Ly6C | BioLegend | Cat# 128026 |
| Purified anti-CD16/CD32 | Bio X Cell | Cat# BE0307 |
| FITC anti-CD45 | BioLegend | Cat# 103108 |
| PE anti-Ly6G | BioLegend | Cat# 127608 |
| APC-Cy7 anti-CD11b | BioLegend | Cat# 101216 |
| APC anti-TNFR1 | BioLegend | Cat# 113006 |
| Purified anti-gp75 | Bio X Cell | Cat# BE0151 |
| FITC anti-DNA/RNA damage | Abcam | Cat# ab183393 |
| AF488 anti-FITC | ThermoFisher Scientific | Cat# A11096 |
| APC-Fire 750 anti-CD11b | BioLegend | Cat# 101262 |
| FITC anti-ICAM-1 | BioLegend | Cat# 353108 |
| BV711 anti-CD16 | BioLegend | Cat# 302044 |
| BV786 anti-CD32 | BD Biosciences | Cat# 564840 |
| AF647 anti-CD66b | BioLegend | Cat# 305110 |
| BV510 anti-CD63 | BD Biosciences | Cat# 740182 |
| PE anti-C5AR1 | BioLegend | Cat# 344304 |
| AF647 anti-CD177 | BD Biosciences | Cat# 566599 |
| FITC anti-SIRPa | BioLegend | Cat# 144006 |
| BV480 anti-Siglec F | BD Biosciences | Cat# 746668 |
| PE-Cy7 anti-CD101 | ThermoFisher Scientific | Cat# 25-1011-82 |
| APC-Cy7 anti-CD14 | BioLegend | Cat# 123316 |
| BV711 anti-PD-L1 | BioLegend | Cat# 124319 |
| APC anti-XCR1 | BioLegend | Cat# 148206 |
| APC-Cy7 anti-F4/80 | BioLegend | Cat# 123118 |
| FITC anti-CD206 | BioLegend | Cat# 141704 |
| PerCP-eF710 anti-CD80 | ThermoFisher Scientific | Cat# 46-0801-82 |
| BUV737 anti-CD86 | BD Biosciences | Cat# 741737 |
| BV421 anti-PD-L1 | BioLegend | Cat# 124315 |
| BUV737 anti-Ly6G | BD Biosciences | Cat# 741813 |
| BV785 anti-Ly6C | BioLegend | Cat# 128041 |
| BV421 anti-TCRb | BioLegend | Cat# 109230 |
| PE-Cy7 anti-NK1.1 | BioLegend | Cat# 108714 |
| PE anti-KLRG1 | BioLegend | Cat# 138408 |
| BV510 anti-CD8a | BioLegend | Cat# 100752 |
| BV711 anti-CD4 | BioLegend | Cat# 100557 |
| B220 anti-BV711 | BioLegend | Cat# 103255 |
| AF488 anti-Foxp3 | BD Biosciences | Cat# 560403 |
| BV650 anti-CD25 | BioLegend | Cat# 102038 |
| PerCP-Cy5.5 anti-CD62L | BioLegend | Cat# 104432 |
| APC-eF780 anti-CD44 | ThermoFisher Scientific | Cat# 47-0441-82 |
| BV785 anti-PD-1 | BioLegend | Cat# 135225 |
| APC anti-CD69 | BD Biosciences | Cat# 560689 |
| AF700 anti-Ki67 | ThermoFisher Scientific | Cat# 56-5698-82 |
| eF450 anti-CD34 | ThermoFisher Scientific | Cat# 48-0341-82 |
| AF700 anti-IL-7Ra | ThermoFisher Scientific | Cat# 56-1271-82 |
| BV711 anti-Sca-1 | BioLegend | Cat# 108131 |
| FITC anti-cKit | BD Biosciences | Cat# 553354 |
| PE anti-CD16/32 | BioLegend | Cat# 156606 |
| PE-Cy7 anti-CD3e | ThermoFisher Scientific | Cat# 25-0032-82 |
| PE-Cy7 anti-CD4 | ThermoFisher Scientific | Cat# 25-0041-82 |
| PE-Cy7 anti-CD8a | ThermoFisher Scientific | Cat# 25-0081-82 |
| PE-Cy7 anti-CD11b | ThermoFisher Scientific | Cat# 25-0112-82 |
| PE-Cy7 anti-B220 | ThermoFisher Scientific | Cat# 25-0452-82 |
| APC anti-Flt3 | BioLegend | Cat# 135310 |
| Pacific Blue anti-CD106 | BioLegend | Cat# 105722 |
| PE-Cy5 anti-Ly6G | ThermoFisher Scientific | Cat# 15-9668-82 |
| PerCP-Cy5.5 anti-CD81 | BioLegend | Cat# 104911 |
| PE-Cy7 anti-NK1.1 | ThermoFisher Scientific | Cat# 25-5941-82 |
| PE-Cy7 anti-Sca-1 | BioLegend | Cat# 122514 |
| AF700 anti-CD34 | ThermoFisher Scientific | Cat# 56-0341-82 |
| AF647 anti-gp75 | Novus Biologicals | Cat# NBP2-34720AF647 |
| PE-Cy7 anti-PD-L1 | BioLegend | Cat# 124314 |
| PE anti-H-2Kb | BioLegend | Cat# 116508 |
| APC anti-H-2Db | BioLegend | Cat# 111514 |
| FITC anti-MPO | Abcam | Cat# ab90812 |
| Purified anti-citrullinated H3 | Abcam | Cat# ab5103 |
| PE Donkey anti-rabbit IgG | BioLegend | Cat# 406421 |
| Biotin anti-PE | BioLegend | Cat# 408104 |
| Purified anti-CD40 | Bio X Cell | Cat# BE0016-2 |
| Purified anti-CD44 | RayBiotech | Cat# DS-MB-00666 |
| Purified anti-MHC Class I | Bio X Cell | Cat# BE0180 |
| Purified anti-human MHC Class I | Bio X Cell | Cat# BE0079 |
| Purified mouse IgG2a isotype control | Bio X Cell | Cat# BE0085 |
| Purified anti-Ly6G | Bio X Cell | Cat# BE0075-1 |
| Purified rat IgG2a isotype control | Bio X Cell | Cat# BE0089 |
| Purified anti-TNFR1 | BioLegend | Cat# 112906 |
| Purified anti-TNFR2 | BioLegend | Cat# 113307 |
| Purified Armenian Hamster IgG | Bio X Cell | Cat# BE0091 |
| Purified anti-Factor B, produced from ATCC hybridoma | ATCC | Cat# PTA-6230 |
| Purified mouse IgG1 isotype control | Bio X Cell | Cat# BE0083 |
| Purified anti-properdin | Produced as in Miwa et al., 2013 | N/A |
| Purified anti-C5 | Produced as in Miwa et al., 2013 | N/A |
| Purified anti-C5AR1 | BioLegend | Cat# 135816 |
| Purified rat IgG2b isotype control | Bio X Cell | Cat# BE0090 |
| Purified anti-CD40 | Bio X Cell | Cat# BE0189 |
| Purified anti-EGFR | Bio X Cell | Cat# SIM0002 |
| Purified anti-GFP | Bio X Cell | Cat# RT0265 |
| Biological samples | ||
| De-identified human whole blood | Stanford Blood Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant mouse TNF | BioLegend | Cat# 575208 |
| Recombinant mouse GM-CSF | BioLegend | Cat# 576302 |
| Recombinant mouse IFNγ | BioLegend | Cat# 575304 |
| Recombinant mouse IL-1β | BioLegend | Cat# 575102 |
| Recombinant mouse IL-17A | BioLegend | Cat# 576002 |
| Recombinant mouse C5a | R&D Systems | Cat# 2150-C5-025/CF |
| Recombinant human TNF | BioLegend | Cat# 570104 |
| Recombinant human C5a | R&D Systems | Cat# 2037-C5-025/CF |
| Cobra venom factor | Millipore Sigma | Cat# 233552-1MG |
| SC57461A | Cayman Chemical | Cat# 10108 |
| CP-105696 | Millipore Sigma | Cat# PZ0363-25MG |
| Reduced L-glutathione | Millipore Sigma | Cat# G4251-25G |
| Catalase | Millipore Sigma | Cat# C40-500MG |
| Topiroxostat | MedChem Express | Cat# HY-14874 |
| Luminol sodium salt | Sigma Aldrich | Cat# A4685 |
| DiD’ solid | ThermoFisher Scientific | Cat# D7757 |
| Critical commercial assays | ||
| In Situ Cell Death Detection Kit, TMR Red | Millipore Sigma | Cat# 12156792910 |
| OxyBURST Green H2HFF BSA | ThermoFisher Scientific | Cat# O13291 |
| Leukotriene B4 ELISA Kit | Cayman Chemical | Cat# 520111 |
| GSH/GSSG Ratio Detection Kit II | Abcam | Cat# ab205811 |
| Xanthine oxidase activity kit | Abcam | Cat# ab102522 |
| DELFIA EuTDA cytotoxicity reagents | PerkinElmer | Cat# AD0116 |
| Experimental models: Cell lines | ||
| Mouse: B16F10 | ATCC | Cat# CRL-6475 |
| Mouse: LL/2 | ATCC | Cat# CRL-1642 |
| Mouse: 4T1 | ATCC | Cat# CRL-2539 |
| Human: A549 | ATCC | Cat# CRM-CCL-185 |
| Human: MDA-MD-231 | ATCC | Cat# CRM-HTB-26 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6/J | Jackson Laboratory | Cat# 000664 |
| Mouse: BALB/cJ | Jackson Laboratory | Cat# 000651 |
| Mouse: MMTV-PyMT: FVB/N-Tg(MMTV-PyVT)634Mul/J | Jackson Laboratory | Cat# 002374 |
| Mouse: Rag2−/−: B6(Cg)-Rag2tm1.1Cgn/J | Jackson Laboratory | Cat# 008449 |
| Mouse: TNFR KO: B6.129S-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J | Jackson Laboratory | Cat# 003243 |
| Mouse: C3−/−: B6.129S4-C3tm1Crr/J | Jackson Laboratory | Cat# 029661 |
| Mouse: Ncf1−/−: B6N.129S2-Ncf1tm1Shl/J | Jackson Laboratory | Cat# 027331 |
| Mouse: Rag2−/− Il2rg−/−: B6(Cg)-Rag2tm1.1Cgn/J crossed with B6.129S4-Il2rgtm1Wjl/J | Jackson Laboratory | Cat# 008449 × Cat# 003174 |
| Mouse: Cfp−/− | Kimura et al., 2010 | N/A |
| Mouse: C6−/− | Ueda et al., 2019 | N/A |
| Mouse: Fcer1g−/−: B6.129P2-Fcer1gtm1Rav N12 | Taconic | Cat# 583 |
| Oligonucleotides | ||
| See Table S1 for oligonucleotide information | ||
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene # 48138; RRID:Addgene_48138 |
| pLVX-EF1a-IRES-Puro | Clontech | Cat# 631988 |
| Software and algorithms | ||
| BD FACSDiva (v8) | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software |
| Zen black edition | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Zen blue edition | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| BZ-X800 Viewer | Keyence | https://www.keyence.com/ |
| BZ-X800 Analyzer | Keyence | https://www.keyence.com/ |
| WorkOut 2.5 | PerkinElmer | https://www.perkinelmer.com/ |
| FlowJo (v10) | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| SpectroFlo V2.2.0.3 | Cytek Biosciences | https://cytekbio.com/blogs/resources/spectroflo-v2-2-0-3-release-notes |
| Living Image | Caliper Life Sciences | https://www.perkinelmer.com/Product/li-software-for-lumina-1-seat-add-on-128110 |
| ImageJ | Fiji | https://imagej.net/software/fiji/downloads |
| Photoshop CS6 | Adobe | https://www.adobe.com/products/photoshop.html |
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| GraphPad Prism (v9) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Therapeutically activated neutrophils infiltrate and eradicate multiple tumor types
Intratumoral TNF + anti-CD40 + anti-tumor antibodies induce an inflammatory cascade
Neutrophil C5AR1 signaling stimulates LTB4 release, driving ROS production via XO
Neutrophil-mediated oxidative damage drives T cell-independent tumor clearance
Acknowledgements
The authors thank Hannah Wastyk, Raghav Kumar, and Tyler Zhang for experimental assistance; Okmi Choi, and Nancy Wu for expert technical support; Sameera Kongara for critical evaluation of the manuscript; and all members of the Engleman lab for helpful discussions. This work was supported by NIH grants U54 CA209971 (EGE), R01 CA222969 (EGE), R01 CA233958 (EGE), P01 CA244114 (EGE), R01 AI085596 (W-CS), R01 AI117410 (W-CS), R01 AI146162 (W-CS), F31 CA196029 (ILL), F30 CA196145 (TRP), T32 HL120824 (GP), and F32 CA189408 (NERF).
Footnotes
Declaration of Interests
The authors declare the following competing interests: ILL, TRP, and EGE are co-inventors on a patent application filed by Stanford University related to this work. W-CS owns equity, receives a consultant fee and research grant from, and is an inventor of patents licensed to, Kira Pharmaceuticals and Aevitas Therapeutics. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coffelt SB, Wellenstein MD, and de Visser KE (2016). Neutrophils in cancer: neutral no more. Nature reviews. Cancer 16, 431–446. 10.1038/nrc.2016.52. [DOI] [PubMed] [Google Scholar]
- 2.Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, and Mantovani A (2020). Neutrophil diversity and plasticity in tumour progression and therapy. Nature Reviews Cancer 20, 485–503. 10.1038/s41568-020-0281-y. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Cassatella MA, Costantini C, and Jaillon S (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nature Reviews Immunology 11, 519–531. 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 4.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature Medicine 21, 938–945. 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaul ME, and Fridlender ZG (2019). Tumour-associated neutrophils in patients with cancer. Nature reviews. Clinical oncology 16, 601–620. 10.1038/s41571-019-0222-4. [DOI] [PubMed] [Google Scholar]
- 6.Pekarek LA, Starr BA, Toledano AY, and Schreiber H (1995). Inhibition of tumor growth by elimination of granulocytes. The Journal of experimental medicine 181, 435–440. 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jablonska J, Leschner S, Westphal K, Lienenklaus S, and Weiss S (2010). Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. The Journal of Clinical Investigation 120, 1151–1164. 10.1172/JCI37223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nozawa H, Chiu C, and Hanahan D (2006). Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proceedings of the National Academy of Sciences 103, 12493–12498. 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, and de Visser KE (2015). IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348. 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wculek SK, and Malanchi I (2015). Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417. 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R, Singer J, et al. (2019). Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557. 10.1038/s41586-019-0915-y. [DOI] [PubMed] [Google Scholar]
- 12.Schmielau J, and Finn OJ (2001). Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res 61, 4756–4760. [PubMed] [Google Scholar]
- 13.Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, Singhal S, Albelda SM, Granot Z, and Fridlender ZG (2014). Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. International journal of cancer 135, 1178–1186. 10.1002/ijc.28770. [DOI] [PubMed] [Google Scholar]
- 14.Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nature Reviews Immunology 21, 485–498. 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusmartsev S, Nefedova Y, Yoder D, and Gabrilovich DI (2004). Antigen-Specific Inhibition of CD8<sup>+</sup> T Cell Response by Immature Myeloid Cells in Cancer Is Mediated by Reactive Oxygen Species. The Journal of Immunology 172, 989–999. 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 16.Youn J-I, Nagaraj S, Collazo M, and Gabrilovich DI (2008). Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. The Journal of Immunology 181, 5791–5802. 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark RA, and Klebanoff SJ (1975). Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. The Journal of experimental medicine 141, 1442–1447. 10.1084/jem.141.6.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, and Erlebacher A (2015). Neutrophils Oppose Uterine Epithelial Carcinogenesis via Debridement of Hypoxic Tumor Cells. Cancer cell 28, 785–799. 10.1016/j.ccell.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, Akimova T, Vachani A, Litzky L, Hancock WW, et al. (2014). Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest 124, 5466–5480. 10.1172/jci77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singhal S, Bhojnagarwala Pratik S., O’Brien S, Moon Edmund K., Garfall Alfred L., Rao Abhishek S., Quatromoni Jon G., Stephen Tom L., Litzky L, Deshpande C, et al. (2016). Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer cell 30, 120–135. 10.1016/j.ccell.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ponzetta A, Carriero R, Carnevale S, Barbagallo M, Molgora M, Perucchini C, Magrini E, Gianni F, Kunderfranco P, Polentarutti N, et al. (2019). Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell 178, 346–360.e324. 10.1016/j.cell.2019.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costanzo-Garvey DL, Keeley T, Case AJ, Watson GF, Alsamraae M, Yu Y, Su K, Heim CE, Kielian T, Morrissey C, et al. (2020). Neutrophils are mediators of metastatic prostate cancer progression in bone. Cancer immunology, immunotherapy : CII 69, 1113–1130. 10.1007/s00262-020-02527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Granot Z, Henke E, Comen EA, King TA, Norton L, and Benezra R (2011). Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer cell 20, 300–314. 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, and Albelda SM (2009). Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer cell 16, 183–194. 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. (2019). Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569, 73–78. 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Kumar A, Vilgelm AE, Chen S-C, Ayers GD, Novitskiy SV, Joyce S, and Richmond A (2018). Loss of CXCR4 in Myeloid Cells Enhances Antitumor Immunity and Reduces Melanoma Growth through NK Cell and FASL Mechanisms. Cancer immunology research 6, 1186–1198. 10.1158/2326-6066.Cir-18-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sagiv Jitka Y., Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov Ronit V., et al. (2015). Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Reports 10, 562–573. 10.1016/j.celrep.2014.12.039. [DOI] [PubMed] [Google Scholar]
- 28.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. (2016). CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer cell 29, 832–845. 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. (2012). A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178. 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pylaeva E, Harati MD, Spyra I, Bordbari S, Strachan S, Thakur BK, Höing B, Franklin C, Skokowa J, Welte K, et al. (2019). NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. International journal of cancer 144, 136–149. 10.1002/ijc.31808. [DOI] [PubMed] [Google Scholar]
- 31.Shrestha S, Noh JM, Kim S-Y, Ham H-Y, Kim Y-J, Yun Y-J, Kim M-J, Kwon M-S, Song D-K, and Hong C-W (2016). Angiotensin converting enzyme inhibitors and angiotensin II receptor antagonist attenuate tumor growth via polarization of neutrophils toward an antitumor phenotype. Oncoimmunology 5, e1067744. 10.1080/2162402X.2015.1067744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, Franke K, Schornagel K, Verkuijlen P, Janssen H, et al. (2018). Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Reports 23, 3946–3959.e3946. 10.1016/j.celrep.2018.05.082. [DOI] [PubMed] [Google Scholar]
- 33.Albanesi M, Mancardi DA, Jönsson F, Iannascoli B, Fiette L, Di Santo JP, Lowell CA, and Bruhns P (2013). Neutrophils mediate antibody-induced antitumor effects in mice. Blood 122, 3160–3164. 10.1182/blood-2013-04-497446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miralda I, Uriarte SM, and McLeish KR (2017). Multiple Phenotypic Changes Define Neutrophil Priming. Frontiers in Cellular and Infection Microbiology 7. 10.3389/fcimb.2017.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodfin A, Beyrau M, Voisin M-B, Ma B, Whiteford JR, Hordijk PL, Hogg N, and Nourshargh S (2016). ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 127, 898–907. 10.1182/blood-2015-08-664995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Condliffe AM, Chilvers ER, Haslett C, and Dransfield I (1996). Priming differentially regulates neutrophil adhesion molecule expression/function. Immunology 89, 105–111. 10.1046/j.1365-2567.1996.d01-711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan SY, Kelher MR, Heal JM, Blumberg N, Boshkov LK, Phipps R, Gettings KF, McLaughlin NJ, and Silliman CC (2006). Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood 108, 2455–2462. 10.1182/blood-2006-04-017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, Sieow JL, Penny HL, Ching GC, Devi S, et al. (2018). Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 48, 364–379.e368. 10.1016/j.immuni.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, Evrard M, Li JLY, Tran HTN, Tan Y, et al. (2020). Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity 53, 303–318.e305. 10.1016/j.immuni.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 40.Challen GA, Boles N, Lin K-YK, and Goodell MA (2009). Mouse hematopoietic stem cell identification and analysis. Cytometry Part A 75A, 14–24. 10.1002/cyto.a.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou G, Peng K, Song Y, Yang W, Shu W, Yu T, Yu L, Lin M, Wei Q, Chen C, et al. (2017). CD177+ neutrophils suppress epithelial cell tumourigenesis in colitis-associated cancer and predict good prognosis in colorectal cancer. Carcinogenesis 39, 272–282. 10.1093/carcin/bgx142. [DOI] [PubMed] [Google Scholar]
- 42.Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, Rickelt S, Severe N, Baryawno N, Faget J, et al. (2017). Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science (New York, N.Y.) 358, eaal5081. 10.1126/science.aal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E, Kossenkov A, Mulligan C, Nam B, Masters G, et al. (2021). Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. The Journal of experimental medicine 218. 10.1084/jem.20201803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yajuk O, Baron M, Toker S, Zelter T, Fainsod-Levi T, and Granot Z (2021). The PD-L1/PD-1 Axis Blocks Neutrophil Cytotoxicity in Cancer. Cells 10, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gavillet M, Martinod K, Renella R, Harris C, Shapiro NI, Wagner DD, and Williams DA (2015). Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. American journal of hematology 90, 1155–1158. 10.1002/ajh.24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, et al. (2018). KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48, 716–729.e718. 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunkelberger JR, and Song WC (2010). Complement and its role in innate and adaptive immune responses. Cell research 20, 34–50. 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 48.Brandt SL, and Serezani CH (2017). Too much of a good thing: How modulating LTB(4) actions restore host defense in homeostasis or disease. Seminars in immunology 33, 37–43. 10.1016/j.smim.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roumenina LT, Daugan MV, Petitprez F, Sautès-Fridman C, and Fridman WH (2019). Context-dependent roles of complement in cancer. Nature Reviews Cancer 19, 698–715. 10.1038/s41568-019-0210-0. [DOI] [PubMed] [Google Scholar]
- 50.Battelli MG, Polito L, Bortolotti M, and Bolognesi A (2016). Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med 5, 546–557. 10.1002/cam4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Wu J, Newton R, Bahaie NS, Long C, and Walcheck B (2013). ADAM17 cleaves CD16b (FcγRIIIb) in human neutrophils. Biochim Biophys Acta 1833, 680–685. 10.1016/j.bbamcr.2012.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, Bértolo C, Lasarte JJ, Vicent S, Hoehlig K, et al. (2017). A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discovery 7, 694–703. 10.1158/2159-8290.Cd-16-1184. [DOI] [PubMed] [Google Scholar]
- 53.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, and Pio R (2012). Anaphylatoxin C5a Creates a Favorable Microenvironment for Lung Cancer Progression. The Journal of Immunology 189, 4674–4683. 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, Manne S, Fairlie DP, Gorczyca W, Almanza O, et al. (2014). Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res 74, 3454–3465. 10.1158/0008-5472.Can-14-0157. [DOI] [PubMed] [Google Scholar]
- 55.Piao C, Cai L, Qiu S, Jia L, Song W, and Du J (2015). Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-mediated Inflammatory Cell Infiltration. The Journal of biological chemistry 290, 10667–10676. 10.1074/jbc.M114.612622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, and Lambris JD (2008). Modulation of the antitumor immune response by complement. Nature immunology 9, 1225–1235. 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satpathy SR, Jala VR, Bodduluri SR, Krishnan E, Hegde B, Hoyle GW, Fraig M, Luster AD, and Haribabu B (2015). Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nature communications 6, 7064. 10.1038/ncomms8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yokota Y, Inoue H, Matsumura Y, Nabeta H, Narusawa M, Watanabe A, Sakamoto C, Hijikata Y, Iga-Murahashi M, Takayama K, et al. (2012). Absence of LTB4/BLT1 axis facilitates generation of mouse GM-CSF-induced long-lasting antitumor immunologic memory by enhancing innate and adaptive immune systems. Blood 120, 3444–3454. 10.1182/blood-2011-10-383240. [DOI] [PubMed] [Google Scholar]
- 59.Tian W, Jiang X, Kim D, Guan T, Nicolls MR, and Rockson SG (2020). Leukotrienes in Tumor-Associated Inflammation. Front Pharmacol 11. 10.3389/fphar.2020.01289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akk A, Springer LE, Yang L, Hamilton-Burdess S, Lambris JD, Yan H, Hu Y, Wu X, Hourcade DE, Miller MJ, and Pham CTN (2019). Complement activation on neutrophils initiates endothelial adhesion and extravasation. Molecular immunology 114, 629–642. 10.1016/j.molimm.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, and Halbwachs-Mecarelli L (2011). Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood 117, 1340–1349. 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 62.Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, and Germain RN (2013). Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498, 371–375. 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee EKS, Gillrie MR, Li L, Arnason JW, Kim JH, Babes L, Lou Y, Sanati-Nezhad A, Kyei SK, Kelly MM, et al. (2018). Leukotriene B4-Mediated Neutrophil Recruitment Causes Pulmonary Capillaritis during Lethal Fungal Sepsis. Cell Host & Microbe 23, 121–133.e124. 10.1016/j.chom.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 64.Uderhardt S, Martins AJ, Tsang JS, Lämmermann T, and Germain RN (2019). Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 177, 541–555.e517. 10.1016/j.cell.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sadik CD, Kim ND, Iwakura Y, and Luster AD (2012). Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling. Proceedings of the National Academy of Sciences of the United States of America 109, E3177–3185. 10.1073/pnas.1213797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim ND, Chou RC, Seung E, Tager AM, and Luster AD (2006). A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. The Journal of experimental medicine 203, 829–835. 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kimura Y, Zhou L, Miwa T, and Song W-C (2010). Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. The Journal of Clinical Investigation 120, 3545–3554. 10.1172/JCI41782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubo H, Morgenstern D, Quinian WM, Ward PA, Dinauer MC, and Doerschuk CM (1996). Preservation of complement-induced lung injury in mice with deficiency of NADPH oxidase. J Clin Invest 97, 2680–2684. 10.1172/jci118718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, Davidson MG, Kenkel JA, Segal E, Pusapati GV, et al. (2015). Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 521, 99–104. 10.1038/nature14424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502.e415. 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, and Depil S (2019). Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol 10, 168. 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arroyo-Crespo JJ, Armiñán A, Charbonnier D, Deladriere C, Palomino-Schätzlein M, Lamas-Domingo R, Forteza J, Pineda-Lucena A, and Vicent MJ (2019). Characterization of triple-negative breast cancer preclinical models provides functional evidence of metastatic progression. International journal of cancer 145, 2267–2281. 10.1002/ijc.32270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roberts NJ, Zhou S, Diaz LA Jr., and Holdhoff M (2011). Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2, 739–751. 10.18632/oncotarget.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miwa T, Sato S, Gullipalli D, Nangaku M, and Song W-C (2013). Blocking Properdin, the Alternative Pathway, and Anaphylatoxin Receptors Ameliorates Renal Ischemia-Reperfusion Injury in Decay-Accelerating Factor and CD59 Double-Knockout Mice. The Journal of Immunology 190, 3552–3559. 10.4049/jimmunol.1202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ueda Y, Miwa T, Ito D, Kim H, Sato S, Gullipalli D, Zhou L, Golla M, Song D, Dunaief JL, et al. (2019). Differential contribution of C5aR and C5b-9 pathways to renal thrombic microangiopathy and macrovascular thrombosis in mice carrying an atypical hemolytic syndrome-related factor H mutation. Kidney international 96, 67–79. 10.1016/j.kint.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 77.Sjögren J, Lood R, and Nägeli A (2020). On enzymatic remodeling of IgG glycosylation; unique tools with broad applications. Glycobiology 30, 254–267. 10.1093/glycob/cwz085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van den Berg CW, Aerts PC, and Van Dijk H (1991). In vivo anti-complementary activities of the cobra venom factors from Naja naja and Naja haje. Journal of immunological methods 136, 287–294. 10.1016/0022-1759(91)90015-8. [DOI] [PubMed] [Google Scholar]
- 79.Paschall AV, and Liu K (2016). An Orthotopic Mouse Model of Spontaneous Breast Cancer Metastasis. J Vis Exp. 10.3791/54040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.