

Abstract
A protein’s structure and function often depend not only on its primary sequence, but also the presence or absence of any number of non-coded posttranslational modifications. Complicating their study is the fact that the physiological consequences of these modifications are context-, protein-, and site-dependent, and there exist no purely biological techniques to unambiguously study their effects. To this end, protein semisynthesis has become an invaluable chemical biology tool to specifically install non-coded or non-native moieties onto proteins in vitro using synthetic and/or recombinant polypeptides. Here, we describe two facets of protein semisynthesis (solid-phase peptide synthesis and expressed protein ligation) and their use in generating site-specifically glycosylated small heat shock proteins for functional studies. The procedures herein require limited specialized equipment, employ mild reaction conditions, and can be extended to myriad other proteins, modifications, and contexts.
1. Introduction
O-GlcNAcylation is a required posttranslational modification (PTM) in mammals (Shafi et al., 2000; Yang et al., 2012), whereby intracellular protein substrates are decorated with monomers of β-linked N-acetylglucosamine at side-chain serine and threonine hydroxyls (Fig. 1A). This glycosylation event is unique in that it is not further elaborated with other carbohydrates, as seen in other forms of glycosylation, and the PTM has only one writer (O-GlcNAc transferase (OGT) (Haltiwanger, Holt, & Hart, 1990)) and one eraser (O-GlcNAcase (OGA) (Hanover et al., 2003)). There are many known substrates of this PTM, and they are involved in wide-ranging cellular functions such as transcriptional/translation regulation (Parker, Peterson, & Slawson, 2021; Xuexia et al., 2019), signaling (Ong, Han, & Yang, 2018), and stress response (Liu et al., 2021). Additionally, the UDP-GlcNAc donor sugar is generated via the hexosamine biosynthetic pathway, which requires an influx of biomolecules of many other metabolic pathways, thus linking the global levels of modification with the cell’s metabolic (Walgren, Vincent, Schey, & Buse, 2003), homeostatic, and disease states (Bolanle, Riches-Suman, Williamson, & Palmer, 2021; Ducheix, Magré, Cariou, & Prieur, 2018; Lee et al., 2020; Ma, Vocadlo, & Vosseller, 2013; Parker et al., 2021; Pinho, Correia, Perry, Ambrósio, & Moreira, 1865).
Fig. 1.
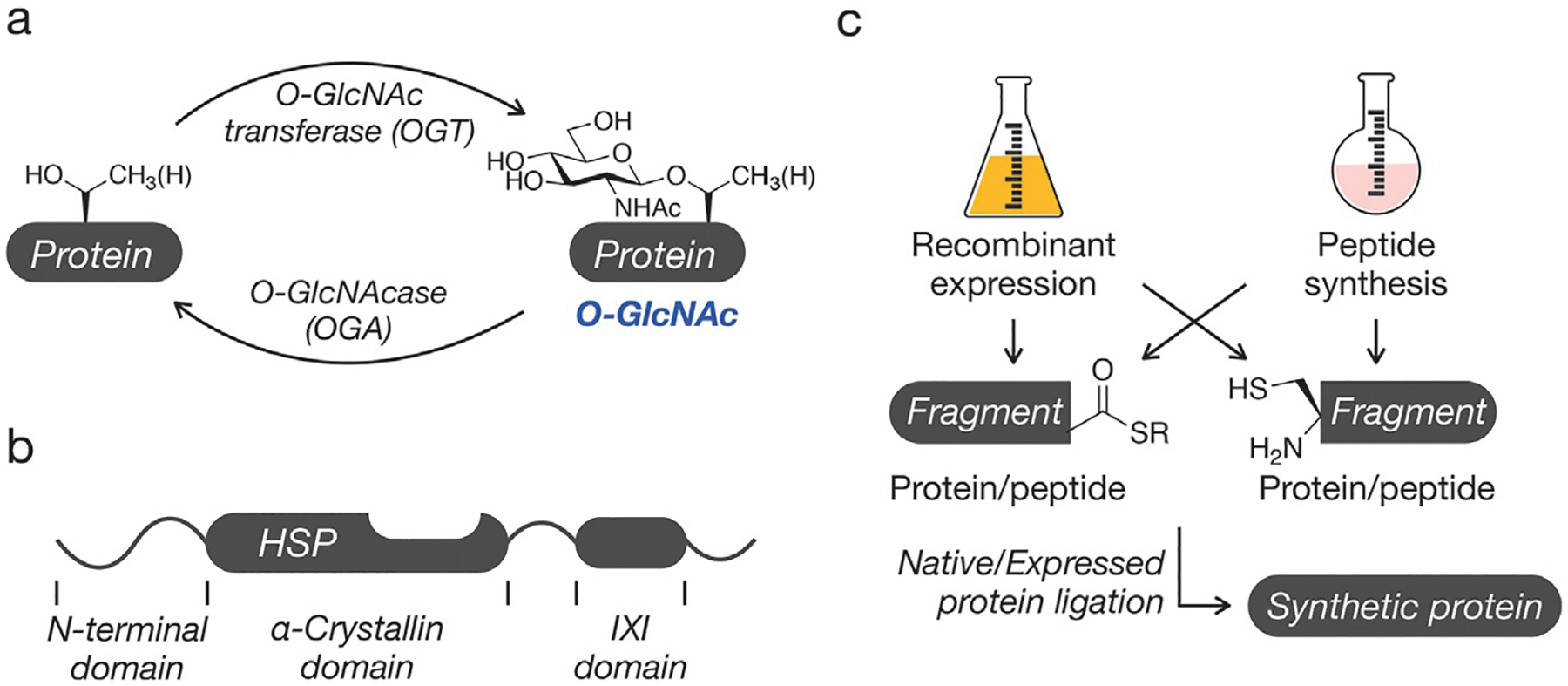
Synthesis of O-GlcNAc modified proteins. (A) O-GlcNAc is the dynamic addition of N-acetylglucosamine to serine/threonine residues of intracellular proteins. (B) The domain structure of O-GlcNAc modified small heat shock proteins (sHSPs). (C) The general approach to protein synthesis involves the use of native or expressed protein ligation reactions between protein fragments with C-terminal thioesters and other fragments with N-terminal cysteine residues.
One native O-GlcNAcylation substrate is HSP27, a small heat shock protein (sHSP) involved in many protective cellular pathways (Fig. 1B) (Hartl, Bracher, & Hayer-Hartl, 2011). The protein and its family members function as ATP-independent chaperones by interacting with partially or misfolded proteins to prevent their amyloidogenic or amorphous aggregation (Haslbeck, Weinkauf, & Buchner, 2019). sHSPs contain a conserved central α-crystallin domain (ACD) (Hochberg et al., 2014) responsible for substrate binding, as well as an IXI tripeptide motif at their C-termini (Delbecq, Jehle, & Klevit, 2012). The hydrophobic IXI domain has been shown to interact with the ACDs of other sHSPs to form large, dynamic oligomers, the size and heterogeneity of which are indicative of the proteins’ chaperone activities. Interestingly, the IXI domains of several sHSPs also have neighboring, conserved O-GlcNAc modification sites (T184 in HSP27) (Deracinois et al., 2018; Li et al., 2019; Wang et al., 2017). We hypothesized that the native role of this specific PTM event could be tuning the chaperone activity of these sHSPs by mediating the interaction between the ACD and IXI domains.
Because PTMs frequently impact their substrates in protein- and site-specific ways, the study of O-GlcNAcylation and other non-coded protein modifications has been hindered by a lack of biological techniques to access specifically modified polypeptides. In vivo/in cellulo treatment of OGT/OGA inhibitors alter global levels of the PTM, and the observed effects are difficult to trace to a single protein’s (de)glycosylation. Similarly, co-expression or co-incubation of a recombinant substrate and OGT imparts no control of the modification’s site or stoichiometry. Thus, our lab and others rely on chemical protein synthesis to generate site-specifically modified proteins for use in further in vitro experiments. Crucial to these ends are solid phase peptide synthesis (Palomo, 2014) (SPPS), expressed protein ligation (EPL) (Muir, Sondhi, & Cole, 1998), and native chemical ligation (NCL) (Agouridas et al., 2019; Dawson, Muir, Clark-Lewis, & Kent, 1994). SPPS allows the researcher to chemically control the identity and placement of non-coded elements within a synthetic peptide, while EPL is used to install specific chemical functionality onto recombinant polypeptides. Through NCL, the researcher can ligate synthetic and/or recombinant fragments to yield full-length proteins that are perfectly (or nearly perfectly) analogous to the modified protein found in nature (Fig. 1C).
Here, we describe the methodologies used to generate full-length, site-specifically O-GlcNAcylated (gT184) HSP27 in vitro to characterize the PTMs consequences through subsequent functional analyses (Fig. 2). We used SPPS to construct the protein’s C-terminus 1 (residues 173–205), importantly bearing an A-to-C mutation at position 173 and an Ac3O-GlcNAc-modified threonine at position 184. Using EPL, we generated the remainder of the protein’s sequence (1–172) as a protein thioester 2. The protein’s native cysteine (C137) was protected at this stage by Michael addition to N-methylmaleimide (NMM) (Vamisetti et al., 2020), yielding fragment 3. NCL was then used to join these fragments together to form the full-length product 4 via a native amide bond at the ligation junction. Subsequent desulfurization removed the C173A mutation, and deprotection of both the cysteine NMM and sugar acetate groups provided the semisynthetic protein 5.
Fig. 2.
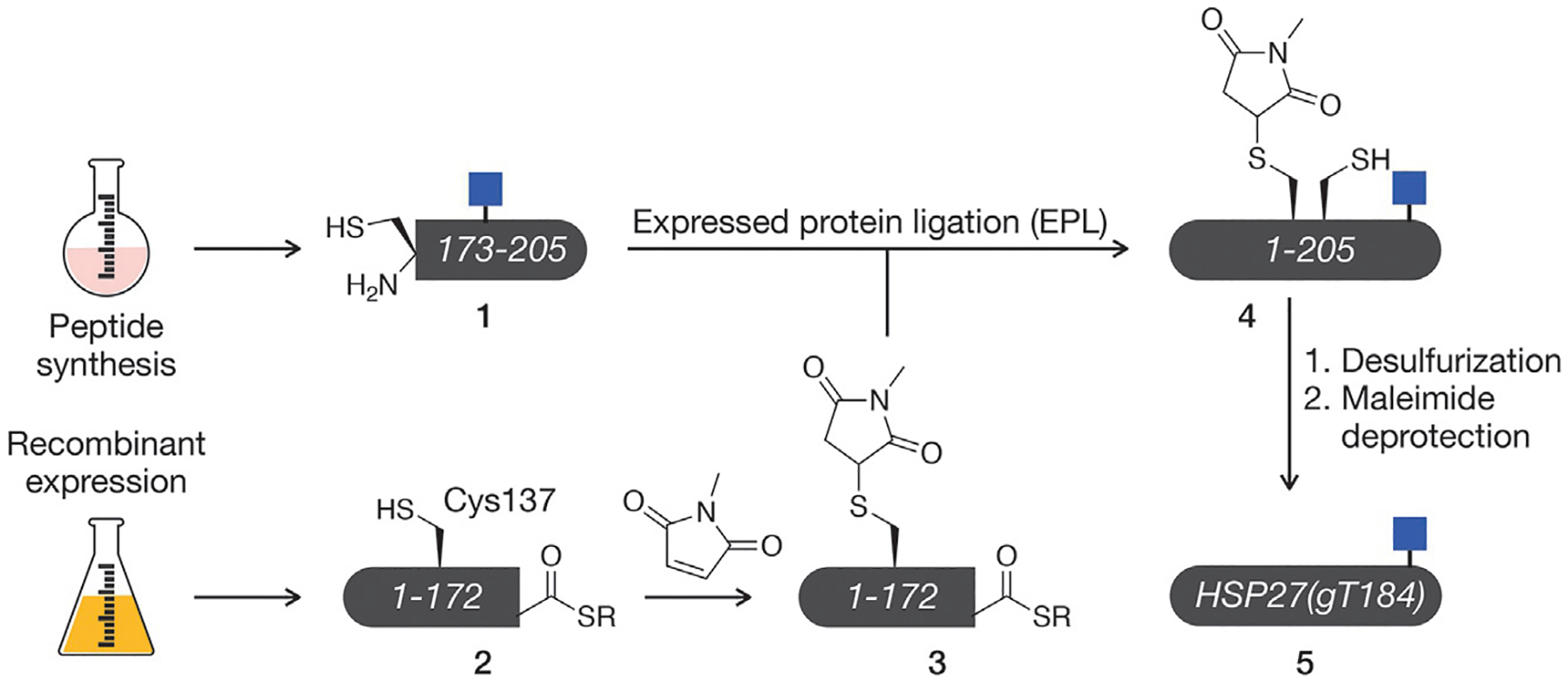
Semi-synthesis of O-GlcNAc modified HSP27. Our semisynthetic route to HSP27 with O-GlcNAc site specifically located at threonine 184, termed HSP27(gT184).
2. General methods
Solvents and reagents were purchased from commercial sources and used without subsequent purification. Fmoc-Thr(Ac3GlcNAc)-OPfp was synthesized in-house via published procedures (De Leon, Lang, Saavedra, & Pratt, 2018), but is also available commercially as a protected free acid (Sigma). Aqueous solutions were made using deionized, filtered water. Protein concentrations were determined via Pierce™ BCA protein assay kit (Thermo).
Unless otherwise noted, purification and characterization of polypeptides was carried out via reverse-phase HPLC using an Agilent HPLC fitted with semipreparative or analytical C4 or C18 columns from Higgins Analytical (Buffer A: 0.1% trifluoroacetic acid in water, Buffer B: 0.1% trifluoroacetic acid, 90% acetonitrile in water). Mass spectrometry was performed using an Agilent HPLC/qTOF Spectrometer.
3. Solid-phase peptide synthesis (SPPS)
In typical protein semisynthesis schemes, the non-coded or non-native component of the desired protein product is installed via solid-phase peptide synthesis (SPPS) (Fig. 3A). In this technique, peptides can be generated in vitro by elongating a peptide chain grafted onto a solid support. In Fmoc SPPS, cycles of basic deprotection, activation, and coupling allow specifically protected amino acids to couple in precise orders from C-terminus to N-terminus, and the desired chemical moiety can be added using a pre-modified or non-native amino acid (synthesized separately in-house or commercially available) (Fig. 3B). Subsequent cleavage of the peptide from the resin using trifluoroacetic acid (TFA) affords the site-specifically modified peptide of interest. SPPS can be performed using an automated peptide synthesizer or manually with non-specialized equipment as described here.
Fig. 3.

Solid phase peptide synthesis of O-GlcNAc modified peptides. (A) Fmoc-based solid phase peptide synthesis. Peptides are built on solid-support (resin) using iterative cycles of Fmoc-deprotection and amino acid (Ax) coupling. At the completion of the synthesis the peptide can be cleaved from the resin and fully deprotected using trifluoroacetic acid (TFA). (B) O-GlcNAc serine/threonine can be directly incorporated into peptides using the corresponding, protected amino acids.
3.1. Equipment
Fume hood
Rocker or inverter
Polypropylene reaction vessels (Torviq Personal Chemistry Tools)
Centrifuge
RP-HPLC (see Section 2)
Lyophilizer
3.2. Reagents
Pre-loaded Fmoc-Lys(Boc)-Wang resin (P3 Biosystems)
Fmoc-amino acids (with compatible side chain protecting groups) (P3 Biosystems)
Fmoc-Thr(Ac3GlcNAc)-OPfp
HBTU (P3 Biosystems)
Diisopropylethylamine (DIPEA)
Dichloromethane (DCM)
N,N-dimethylformamide (DMF)
20% piperidine in DMF
Pyridine
Acetic anhydride
Trifluoroacetic acid (TFA)
Triisopropyl silane (TIPS)
H2O
Diethyl ether
3.3. Procedure
3.3.1. Resin preparation
Timing: 1 h
Transfer desired mass of resin to a clean reaction vessel. Resin loading (moles of reactive branching points/mass of resin) will vary by type, supplier, and lot. Typical SPPS scales range from 0.05 to 0.2 mmol
Wash resin 3 × with DCM, then incubate in DCM at room temperature for 30 min with agitation to swell
Wash resin 3 × with DMF, then incubate in DMF at room temperature for 30 min with agitation
3.3.2. Fmoc deprotection
Timing: 30 min
Incubate resin in 20% piperidine in DMF at room temperature for 15 min with agitation
Repeat 20% piperidine incubation to ensure full removal of N-terminal Fmoc protecting group
Wash the resin thoroughly with DMF four times, one minute each
3.3.3. Standard amino acid activation and coupling
Timing: 1–1.5 h
Weigh 5 eq of the subsequent amino acid (Fmoc-Ala-OH) into a separate reaction tube with 4.5 eq of HBTU. These dry mixtures can be prepared in advance and stored at 4 °C until needed
Add 2 eq of DIPEA and dissolve in 3–6 mL of DMF
Incubate at room temperature for 5 min with agitation. This step can be synchronized with the final wash step of Fmoc deprotection
Incubate the deprotected, washed resin in this solution at room temperature for 1 h with agitation. Couplings may be extended/repeated if needed
Wash the resin thoroughly with DMF four times, 1 min each
3.3.4. Peptide elongation
Timing: Length-dependent, 0–7 days
Repeat above deprotection and activation/coupling procedures cyclically as needed to synthesize the complete polypeptide
3.3.5. Fmoc-Thr(Ac3GlcNAc)-OPfp coupling
Timing: 12–16 h
Dissolve 2 eq of Fmoc-Thr(Ac3GlcNAc)-OPfp in 3 mL of DMF. HBTU and DIPEA are not needed as the Pfp ester is sufficiently activated
Incubate the deprotected, washed resin in this solution at room temperature overnight (12–16 h) with agitation
To cap unreacted, free N-termini, incubate the resin in an acetate capping solution (3:1:1 by volume DMF:pyridine:acetic anhydride) at room temperature for 15 min with agitation
Wash the resin thoroughly with DMF four times, one minute each
3.3.6. Peptide cleavage and purification
Timing: 16–24 h
Wash complete peptide with DCM 3 ×, then air-dry the resin for 10–20 min
Transfer the dried resin to a cleavage solution (4mL, 95% TFA, 2.5% TIPS, 2.5% water) in a glass reaction vessel. Incubate at room temperature for 4 h with agitation
Filter the cleavage reaction through a clean SPPS reaction vessel into pre-chilled diethyl ether at −80 °C. Store overnight at −80 °C
Remove supernatant by centrifugation at 7000 rcf, 0 °C, 20 min. If no precipitate formed, remove solvent by rotary evaporation
Air-dry to remove residual ether, dissolve pellet in HPLC buffers, and purify desired peptide via HPLC
Lyophilize and aliquot as needed
3.4. Notes
SPPS can be paused at any point following a thorough wash step, such as after a deprotection or a coupling. The peptide and resin are stable in clean DMF for ~1 week. If a longer pause is needed, the resin should be washed 3 × with DCM, air-dried, and stored at 4 °C
The acetate capping step can be performed after any sub-optimal coupling to remove unreacted chains from the reactive population and reduce deletion products
Extended coupling times (1.5 h) are used for β-branched amino acids (Fmoc-Thr-OH, Fmoc-Val-OH, and Fmoc-Ile-OH). Amino acids with large extents of steric hindrance (Fmoc-Pro-OH and Fmoc-Arg(Pbf)-OH) are typically double coupled 2 × 45 min with no Fmoc deprotection step in between
The authors recommend maintaining two working containers of DMF: one that is used for washing steps (and thus minimally contaminated by trace reagents on the reaction vessel’s inlet), and one that is used for extended exposure (such as storage and couplings)
The peptide can be characterized periodically during SPPS for correctness and purity via microcleavage. A very small portion can be scraped from the reaction vessel’s piston, air-dried, and added to 0.5 mL of cleavage solution. After 4 h, the solution can be diluted in HPLC buffers and characterized by HPLC and mass spectrometry
Our lab notices overall yields decline rapidly as peptides approach ~40 amino acids in length, although peptides of ~60–100 amino acids can be prepared in acceptable yield using an automated peptide synthesizer
4. Expression of protein thioesters
Recombinant expression of proteins and polypeptides has become a ubiquitous technique in biochemistry research. Therein, exogenous proteins of interest (POIs) are encoded into DNA vectors which are transformed into host cells resulting in the protein’s overexpression. Expressed protein ligation (EPL) circumvents the length restrictions of SPPS to generate unmodified protein acids and thioesters of any length. While many species of host cells can be used for recombinant expression, expression in bacteria is preferred in this application because of their cost, ease of use, and their inability to posttranslationally modify exogenous proteins. A common bacterial expression platform is the lac operon-T7 RNA polymerase system as found in BL21(DE3) E. coli.
Crucial to EPL is the fusion of the POI to a partially deactivated intein, in our case, AvaE (Fig. 4). Inteins are bacterial protein fragments capable of self-splicing from flanking exteins and are analogous to introns found in RNA. This mechanism proceeds through a branched thioester intermediate at the junction of the POI and the intein’s N-terminal cysteine. A key second catalytic cysteine of our intein is mutated to lock our fusion in this reactive intermediate state, and an excess of exogenous thiols such as sodium 2-mercaptoethanesulfonate (MESNa) are used thiolyze the intein and yield a thioester suitable for native chemical ligation (NCL). Because the intein is fully removed from the protein product, affinity tags such as the His6 tag can be appended to its C-terminus for facile, traceless purification of the fusion from genomic proteins in the bacterial lysate.
Fig. 4.
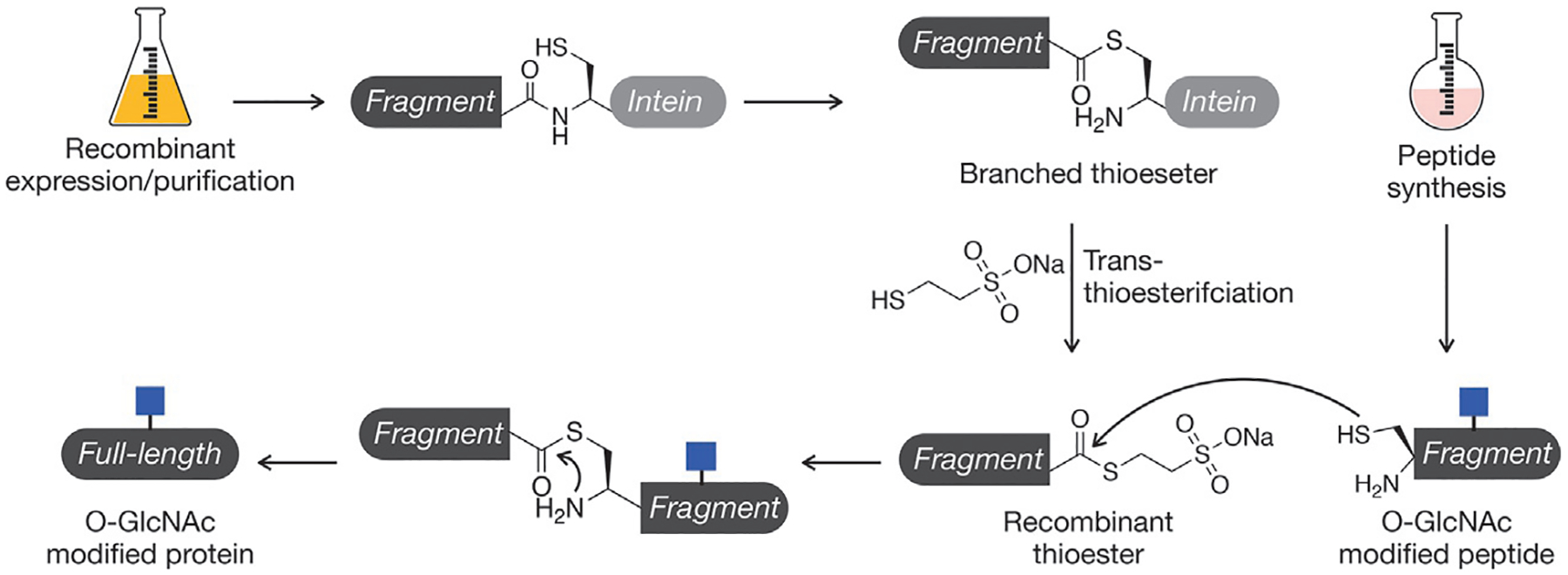
Expressed protein ligation (EPL). Recombinant protein thioesters can be generated through an in-frame fusion to an intein, which will generate a branched thioester and can be intercepted with exogenous thiols. This protein thioester can then be reacted with a synthetic peptide using the native chemical ligation reaction.
4.1. Equipment
Autoclave
Bacterial incubator
Bunsen burner
Bacteria spreader
Visible spectrometer
Centrifuge
Vortexer
Econo-Pac® chromatography columns (Bio-Rad)
Rocker or inverter
Amicon™ Ultra-15 centrifugal filter units (10 kDa MWCO)
RP-HPLC (see Section 2)
Lyophilizer
4.2. Reagents
BL21(DE3) competent E. coli (EMD Millipore)
pTXB1 HSP27(1–172)-AvaE-His6 plasmid
H2O
Luria broth (LB) (RPI)
Ampicillin LB agar plate
Terrific broth (TB) (IBI Scientific)
Glycerol (for TB)
Ampicillin
Isopropyl β-d-1-thiogalactopyranoside (IPTG)
Dibasic sodium phosphate
Sodium chloride
Imidazole
Phenylmethylsulfonyl fluoride (PMSF)
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl)
Guanidine hydrochloride (Gu-HCl)
Cobalt agarose beads (GoldBio)
Sodium 2-mercaptoethanesulfonate (MESNa)
4.3. Procedure
4.3.1. Bacterial transformation
Timing: 14–18 h
Thaw aliquot of BL21(DE3) (~20–50 μL) E. coli on ice
Add ~10–50 ng of pre-chilled desired plasmid and gently mix
Incubate on ice for 30 min
Heat shock bacteria at 42 °C for 30 s, then return to ice for 2 min
Gently, aseptically add 500 μL LB and incubate at 37 °C for 1 h with agitation
Aseptically plate 100–150 μL of the bacteria to a pre-warmed ampicillin LB agar plate. Incubate the plate at 37 °C for 12–16 h
Store plate at 4 °C for up to 1 month or create a glycerol stock
4.3.2. Media and starter culture preparation
Timing: 14–18 h
Prepare and autoclave TB starter cultures (2 × 50 mL in 250 mL flasks) and TB growth cultures (6 × 500 mL in 3 L baffled flasks)
When cooled, aseptically add ampicillin to starter cultures at 100 μg/mL from a 100 mg/mL stock
Aseptically inoculate starter cultures with a single colony of transformed bacteria and incubate at 37 °C for 12–16 h with agitation
4.3.3. Bacterial growth, induction, and harvesting
Timing: 9–10 h
Combine confluent starter cultures in a single flask
Aseptically add ampicillin to growth cultures at 100 μg/mL from a 100 mg/mL stock. Reserve 1–2 mL of media from a marked flask to serve as an optical blank
Inoculate each 500 mL growth culture with 15 mL of confluent starter culture and incubate at 37 °C with agitation until an OD600 of 0.6–0.8 is observed (~2h).
Induce expression of the protein fusion with 1 mM IPTG from a 1 M stock. Incubate the cultures at 37 °C 6 h with agitation to allow POI expression
Harvest the bacteria via centrifugation at 7000 rcf for 15min, 4 °C. Store pellets at −20 °C until lysis
4.3.4. Lysis and His6 purification
Timing: 24–36 h
Vortex to resuspend bacterial pellets in 25 mL lysis buffer (50 mM phosphate, 300 mM NaCl, 5 mM imidazole, 2 mM PMSF, 1 mM TCEP, pH7.5)
Add 28 g of Gu-HCl (6 M) and vortex to dissolve. The volume of the resuspension will increase to ~40–50 mL
Lyse bacteria by tip sonication and clear lysate via centrifugation at 7000 rcf for 45 min at 4 °C. Remove lysate from the pellet and adjust its pH to 7–7.4
Incubate lysate with 2 mL of washed and equilibrated cobalt agarose resin in two chromatography columns at 4 °C for 1 h with agitation
Elute flowthrough and wash each column with 25 mL of wash buffer (4 M urea, 50 mM phosphate, 300 mM NaCl, 20 mM imidazole, 1 mM TCEP, pH 7.2) in 5 mL portions
Elute POI from each column with 15 mL of elution buffer (4 M urea, 50 mM phosphate, 300 mM NaCl, 250 mM imidazole, 1 mM TCEP, pH 7.2)
Concentrate eluted fractions and thoroughly buffer exchange into transfer buffer (4 M urea, 50 mM phosphate, 1 mM TCEP, pH 7.2) using centrifugal filters
Dilute with additional transfer buffer to 10 mL, add 200 mM MESNa, adjust pH to 7, and incubate at room temperature for 12–16 h with agitation to thiolyze
Purify desired protein thioester via HPLC. Lyophilize and aliquot as needed
4.4. Notes
Time of bacterial growth varies by construct but requires ~1–2 h at the growth culture inoculation described here. Inoculant volume can be changed to account for the specific construct and to fit researcher’s timeline
Expression yield is sequence-dependent, and the induction step should be optimized for individual protein constructs. Induction can also be performed overnight at room temperature with half the concentration of IPTG, or for 2–8 h at elevated temperature. Additionally, not all expression products are sequestered in inclusion bodies; those that are not do not require guanidine/urea during lysis/purification
Prior to tip sonication, resuspended bacteria can be gently centrifuged (200 rcf, 1 min) in order to remove air bubbles from vortexing and increase lysis efficiency
E. coli endogenously express a methionine aminopeptidase which cleaves initiator methionine residues provided the residue at the second position is small and polar (Ser, Thr, Cys). During mass spectrometry characterization, expect the mass of the protein with or without this methionine residue
Intein fusions and MES thioesters are susceptible to hydrolysis. To minimize this, the pH of the protein’s environment must be carefully controlled (<7.5), and the protein should be kept on ice as much as possible throughout this protocol
Bacterial expression via intein fusion can also be used to generate full-length proteins and C-terminal protein fragments for NCL. If a C-terminal acid is desired, the intein can be hydrolyzed post-purification in 150 mM DTT, pH 8 with incubation at 37 °C for 36–40 h with agitation. If an N-terminal Cys is desired, the initiator methionine of the translation product will be cleaved by methionine aminopeptidase (see above). The resulting N-terminal Cys is reactive toward endogenous aldehydes, but the cysteine’s thiol and amine can be freed by 12–16-h, room temperature incubation with 150 mM methoxyamine at pH 4
5. Cysteine protection and protein semisynthesis
Native chemical ligation (NCL) has become a workhorse in the field of protein chemistry for its ability to regioselectively form peptide backbone amide bonds in the absence of side chain protecting groups under mild, aqueous conditions. Our lab and others routinely leverage this reaction to build native proteins from recombinant or synthetic polypeptides bearing compatible NCL functional groups: a C-terminal thioester and a free, N-terminal cysteine (Fig. 4). In the reaction, the thiol of the thioester is displaced by the cysteine side chain in a reversible transthioesterification. Subsequently, an S,N-acyl shift is mediated by a five-membered ring tetrahedral intermediate to form the desired amide bond. Our lab uses MES-thioesters as starting materials due to their enhanced stability toward hydrolysis but catalyzes the reaction in situ with 4-mercaptophenylacetic acid (MPAA), as the MPAA-thioester is more reactive toward transthioesterification due to the MPAA’s much lower pKa.
Crucial to the NCL mechanism is the cysteine residue C-terminal to the ligation junction. While cysteine’s relatively low abundance in the proteome significantly limited the generalizability of this technique in terms of accessible proteins and ligation sites, the field has developed a number of routes to circumvent the requirement of native cysteines at particular primary sequence positions. Primary among these is radical desulfurization, wherein an initiator generates a cysteine radical which transfers its sulfur to a molecule of TCEP. The resulting methylene radical is quenched by exogenous thiols (such as glutathione) to generate an alanine side chain. In this way, NCL can be performed at sites that are natively alanines, and the use of many other commercially available β-thio amino acid derivatives has allowed for ligation at other junctions. A disadvantage to the global desulfurization protocol is that it cannot be used in the presence of any native cysteines within the protein. Recently, the Brik lab demonstrated the use of N-methylmaleimide as protecting groups capable of shielding native cysteines from desulfurization conditions (Vamisetti et al., 2020). The moiety is easily added via a chemoselective Michael addition to free thiols, and the group can be removed mildly via palladium chemistry following deprotection of non-native cysteines. In this way, our lab has synthesized HSP27 with a site-specific glycosylation event while maintaining the integrity of its primary sequence and amide backbone.
5.1. Equipment
Rocker or inverter
RP-HPLC (see Section 2)
Lyophilizer
Amicon™ Ultra 0.5 mL centrifugal filter units (10 kDa MWCO)
FPLC system (Amersham Pharmacia Biotech)
HiTrap Q HP (1 mL) FPLC column (Cytiva)
5.2. Reagents
HSP27 2–172-MES (see Section 4)
Guanidine hydrochloride (Gu-HCl)
Dibasic sodium phosphate
TCEP-HCl
N-methylmaleimide (NMM) (Sigma)
4-mercaptophenylacetic acid (MPAA)
HSP27 173–205 A173C, gT184 (see Section 3)
Glutathione
2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) (Sigma)
Magnesium chloride
Palladium (II) chloride
Dithiothreitol (DTT)
Hydrazine monohydrate
Glacial acetic acid
Urea
Bis-Tris
5.3. Procedure
5.3.1. Cysteine protection
Timing: 1 h
Resuspend the N-terminal protein thioester to 1–4 mM in degassed 6 M Gu-HCl, 200 mM sodium phosphate, 1 eq TCEP-HCl, pH 7
Add 3 equivalents of NMM, readjust pH to 7, and incubate at room temperature for 30 min with agitation
Purify NMM-protected N-terminal protein thioester by HPLC and lyophilize
5.3.2. Ligation
Timing: 6–18 h
Resuspend the NMM-protected N-terminal protein thioester to 1–4 mM in degassed 6 M Gu-HCl, 200 mM sodium phosphate, 30 mM TCEP-HCl, 30 mM MPAA, pH 7
Adjust the pH of the solution to 7–7.2, then use this solution to resuspend the C-terminal peptide fragment (1–2 eq)
Incubate fragments at 37 °C with agitation until ligation is complete as monitored by analytical HPLC (typically 4–16 h)
Purify the coeluting N-terminal fragment and full-length product by HPLC and lyophilize
5.3.3. Desulfurization
Timing: 1–2 h
Resuspend the mixture of unreacted N-terminal fragment and ligation product to 1 mM with degassed 6 M Gu-HCl, 200 mM sodium phosphate, 175 mM TCEP-HCl, 40 mM glutathione, pH 7
Add 40 mM VA-044 and adjust pH to 7
Incubate at 37 °C with agitation until desulfurization is complete as monitored by analytical HPLC (typically ~1 h)
Purify desulfurized mixture of ligation products via HPLC and lyophilize
5.3.4. Cysteine and acetate deprotection
Timing: 3–4 h
Resuspend the mixture of unreacted N-terminal fragment and desulfurized ligation product to 1–4 mM with degassed 6 M Gu-HCl, 200 mM sodium phosphate, pH 7, then add 200 mM MgCl2 and 40 mM PdCl2
Incubate at 37 °C with agitation for 1.5 h
Quench reaction with 200 mM DTT. Remove resulting solids by centrifugation
Remove metal chlorides, freed cysteine protecting groups, and DTT by buffer exchange into 6 M Gu-HCl, 200 mM sodium phosphate, pH 7 using centrifugal filters
To remove the sugar’s hydroxyl acetate protecting groups, add 5% v/v hydrazine monohydrate and incubate at room temperature with agitation for 1 h. Quench with 5% v/v acetic acid
5.3.5. Purification
Timing: 2–3 days
Remove all salts and small molecules from the mixture via HPLC. Lyophilize
Resuspend in anion exchange Buffer 1 (4 M urea, 20 mM Bis-Tris, pH 7)
Remove unreacted N-terminal fragment via anion exchange FPLC (Buffer 2: 4 M urea, 500 mM NaCl, 20 mM Bis-Tris, pH 7)
5.4. Notes
Multi-fragment syntheses can be directed via both N- and C-terminal protecting and masking groups. Thiazolidine (Thz), an N-terminal protecting group can be installed during SPPS and deprotected with methoxyamine (150 mM, pH 4, 12–16 h). Further, a hydrazide, a common C-terminal masking group, can be installed during EPL (by supplementing the intein thiolysis with 10% v/v hydrazine monohydrate) or during SPPS (by pre-functionalizing 2-chlorotrityl resin with hydrazine prior to loading) and converted to an active thioester with sodium nitrite (Zheng, Tang, Guo, Chang, & Liu, 2012), or, more mildly, acetylacetone (Flood et al., 2018), and an exogenous, acidic thiol
Selenocysteine ligation and deselenization (Mitchell, Kulkarni, Malins, Wang, & Payne, 2017) can be employed to generate ligation site alanines or serines in the presence of native cysteines. While the amino acid is commercially available (Carbosynth) and synthesizable in-house (Shimodaira & Iwaoka, 2016), the fragment, ligation, and deselenization are quite sensitive to the redox environment
HPLC purification can, in some cases, be replaced with buffer exchange steps. Additionally, the anion exchange purification can be performed following the ligation step (provided all ionic species are removed from the solution via HPLC first). Replacing chromatography purification steps with buffer exchange significantly reduces the amount of product lost throughout the synthetic scheme, but is less thorough than
If reactant fragments do not coelute with the ligation product during HPLC purification, the anion exchange step is not required
6. Refolding
Protein semisynthesis is performed in the presence of denaturing agents (guanidine or urea) in very high concentrations to ensure that the polypeptide fragments remain soluble in solutions of varying ionic strength and pH. Beyond this, proteins are also denatured by organic solvents during HPLC purification. Proteins with no inherent structure or proteins that are extremely well folded need only be resuspended in the desired assay buffer in order to be used for subsequent analyses, but many proteins require a distinct refolding step to uptake their native structures. In our refolding protocol, purified proteins are denatured in an aqueous solution of 6 M guanidine and dialyzed against a refolding buffer lacking denaturing agents. Through this process, the denaturing agent decreases in concentration over time to allow the protein to slowly uptake its structure through a much more ordered pathway, thus giving our semisynthetic protein its native structure and function.
6.1. Equipment
Tube-O-DIALYZER™ dialysis devices (Medi, 8 kDa MWCO)
Amicon™ Ultra 0.5 mL centrifugal filter units (10 kDa MWCO)
6.2. Reagents
HSP27 2–205 gT184 (see Section 5)
Guanidine hydrochloride (Gu-HCl)
Dithiothreitol (DTT)
Dibasic sodium phosphate
6.3. Procedure
6.3.1. Refolding
Timing: 12–16 h
Concentrate FPLC-purified HSP27 2–205 gT184 to 0.5 mg/mL and buffer exchange into 6 M Gu-HCl, 2 mM DTT, pH 7
Transfer to Tube-O-Dialysis tube
Invert sealed tube in 1.5 L of refolding buffer (10 mM sodium phosphate, 2 mM DTT, pH 7.4)
Incubate dialysis setup at 4 °C for 12–16 h with stirring
Concentrate as needed using centrifugal filters
6.4. Notes
The components of the refolding buffer can be changed as desired to suit subsequent experimental conditions
7. Summary and conclusions
Semi-synthetic strategies like those described above are extremely powerful tools to prepare site-specifically modified versions of proteins like sHSPs and interrogate their function. For example, our lab has successfully synthesized full-length Hsp27 that is site-specifically O-GlcNAc-modified at four serine and threonine residues near its autoregulatory IXI motif. We used these semisynthetic proteins to show that each of the modification events yielded a chaperone with increased activity toward a panel of different clients, particularly those that form amyloid aggregates that characterize neurodegenerative diseases. We continued to show that this increase in chaperone activity was due to the sugar’s interruption of the ACD-IXI interaction. Similarly, the Becker lab used SPPS/EPL to determine the consequences of another, nonenzymatic PTM, argpyrimidine (Matveenko, Cichero, Fossa, & Becker, 2016).
The protocols described here are generally accessible given that they rely on reagents and equipment common to chemistry and biochemistry labs and employ mild, mostly aqueous conditions. They can also be used to construct theoretically any protein of interest bearing many other non-coded elements (such as PTMs and various tags). As the PTMome is still a relatively under-studied facet of molecular biology, particularly in a site-specific fashion, we hope that these techniques and improvements thereof will continue to elucidate the implications of many more physiologically and disease-relevant protein PTMs.
References
- Agouridas V, et al. (2019). Native chemical ligation and extended methods: Mechanisms, catalysis, scope, and limitations. Chemical Reviews, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]
- Bolanle IO, Riches-Suman K, Williamson R, & Palmer TM (2021). Emerging roles of protein O-GlcNAcylation in cardiovascular diseases: Insights and novel therapeutic targets. Pharmacological Research, 165, 105467. [DOI] [PubMed] [Google Scholar]
- Dawson PE, Muir TW, Clark-Lewis I, & Kent SB (1994). Synthesis of proteins by native chemical ligation. Science (80-.), 266. 776 LP–779. [DOI] [PubMed] [Google Scholar]
- De Leon CA, Lang G, Saavedra MI, & Pratt MR (2018). Simple and efficient preparation of O- and S-GlcNAcylated amino acids through InBr3-catalyzed synthesis of β-N-acetylglycosides from commercially available reagents. Organic Letters, 20, 5032–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq SP, Jehle S, & Klevit R (2012). Binding determinants of the small heat shock protein, αB-crystallin: Recognition of the ‘IxI’motif. The EMBO Journal, 31, 4587–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deracinois B, et al. (2018). O-GlcNAcylation site mapping by (azide-alkyne) click chemistry and mass spectrometry following intensive fractionation of skeletal muscle cells proteins. Journal of Proteomics, 186, 83–97. [DOI] [PubMed] [Google Scholar]
- Ducheix S, Magré J, Cariou B, & Prieur X (2018). Chronic O-GlcNAcylation and diabetic cardiomyopathy: The bitterness of glucose. Frontiers in Endocrinology, 9, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood DT, et al. (2018). Leveraging the Knorr pyrazole synthesis for the facile generation of thioester surrogates for use in native chemical ligation. Angewandte Chemie International Edition, 57, 11634–11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltiwanger RS, Holt GD, & Hart GW (1990). Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. The Journal of Biological Chemistry, 265, 2563–2568. [PubMed] [Google Scholar]
- Hanover JA, et al. (2003). Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Archives of Biochemistry and Biophysics, 409, 287–297. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, & Hayer-Hartl M (2011). Molecular chaperones in protein folding and proteostasis. Nature, 475, 324–332. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Weinkauf S, & Buchner J (2019). Small heat shock proteins: Simplicity meets complexity. The Journal of Biological Chemistry, 294, 2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg GKA, et al. (2014). The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proceedings of the National Academy of Sciences of the United States of America, 111, E1562–E1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BE, et al. (2020). O-GlcNAcylation regulates dopamine neuron function, survival and degeneration in Parkinson disease. Brain, 143, 3699–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. (2019). An isotope-coded photocleavable probe for quantitative profiling of protein O-GlcNAcylation. ACS Chemical Biology, 14, 4–10. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. (2021). O-GlcNAcylation: The “stress and nutrition receptor” in cell stress response. Cell Stress & Chaperones, 26, 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Vocadlo DJ, & Vosseller K (2013). Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. The Journal of Biological Chemistry, 288, 15121–15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveenko M, Cichero E, Fossa P, & Becker CFW (2016). Impaired chaperone activity of human heat shock protein Hsp27 site-specifically modified with argpyrimidine. Angewandte Chemie: International Edition, 55, 11397–11402. [DOI] [PubMed] [Google Scholar]
- Mitchell NJ, Kulkarni SS, Malins LR, Wang S, & Payne RJ (2017). One-pot ligation-oxidative deselenization at selenocysteine and selenocystine. Chemistry: A European Journal, 23, 946–952. [DOI] [PubMed] [Google Scholar]
- Muir TW, Sondhi D, & Cole PA (1998). Expressed protein ligation: A general method for protein engineering. Proceedings of the National Academy of Sciences of the United States of America, 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong Q, Han W, & Yang X (2018). O-GlcNAc as an integrator of signaling pathways. Frontiers in Endocrinology (Lausanne), 9, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo JM (2014). Solid-phase peptide synthesis: An overview focused on the preparation of biologically relevant peptides. RSC Advances, 4, 32658–32672. [Google Scholar]
- Parker MP, Peterson KR, & Slawson C (2021). O-GlcNAcylation and O-GlcNAc cycling regulate gene transcription: Emerging roles in cancer. Cancers, 13, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho TS, Correia SC, Perry G, Ambrósio AF & Moreira PI Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. Biochimica et Biophysica Acta—Molecular Basis of Disease 1865, 2048–2059 (2019). [DOI] [PubMed] [Google Scholar]
- Shafi R, et al. (2000). The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proceedings of the National Academy of Sciences of the United States of America, 97, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira S, & Iwaoka M (2016). Improved synthetic routes to the selenocysteine derivatives useful for Boc-based peptide synthesis with benzylic protection on the selenium atom. ARKIVOC, 2017, 260–271. [Google Scholar]
- Vamisetti GB, et al. (2020). On-demand detachment of succinimides on cysteine to facilitate (semi)synthesis of challenging proteins. Journal of the American Chemical Society, 142, 19558–19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walgren JLE, Vincent TS, Schey KL, & Buse MG (2003). High glucose and insulin promote O-GlcNAc modification of proteins, including α-tubulin. American Journal of Physiology-Endocrinology and Metabolism, 284, E424–E434. [DOI] [PubMed] [Google Scholar]
- Wang S, et al. (2017). Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. The Journal of Pathology, 243, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuexia L, et al. (2019). O-GlcNAcylation of core components of the translation initiation machinery regulates protein synthesis. Proceedings of the National Academy of Sciences of the United States of America, 116, 7857–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YR, et al. (2012). O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell, 11, 439–448. [DOI] [PubMed] [Google Scholar]
- Zheng J-S, Tang S, Guo Y, Chang H-N, & Liu L (2012). Synthesis of cyclic peptides and cyclic proteins via ligation of peptide hydrazides. Chembiochem: A European Journal of Chemical Biology, 13, 542–546. [DOI] [PubMed] [Google Scholar]