

Abstract
Compromises in compensatory neurobiologic mechanisms due to aging and/or genetic factors (i.e. APOE gene) may influence BDNF val66met polymorphism effects on temporal lobe morphometry and memory performance. We studied two cohorts from ADNI: 175 healthy subjects and 222 with prodromal and established AD. Yearly structural MRI and cognitive performance assessments were carried out over 3 years of follow-up. Both cohorts had similar BDNF Val/Val and Met allele carriers’ (including both Val/Met and Met/Met individuals) distribution. In healthy subjects, a significant trend for thinner posterior cingulate and precuneus cortices were detected in Met carriers compared to Val homozygotes in APOE E4 carriers, with large and medium effect sizes respectively. The MCI/AD cohort showed a longitudinal decline in entorhinal thickness in BDNF Met carriers compared to Val/Val in APOE E4 carriers, with effect sizes ranging from medium to large. Also an effect of BDNF genotype was found in APOE E4 positive subjects for episodic memory (logical memory and ADAS-Cog) and semantic fluency measures, with Met carriers performing worse in all cases. These findings suggest a lack of compensatory mechanisms in BDNF Met carriers in APOE E4 carriers in healthy and pathological aging.
Keywords: BDNF, APOE, Healthy subjects, MCI, AD, brain morphometry, cognition
1. Introduction
Brain derived neuroptrophic factor (BDNF) is a neurotrophin that facilitates episodic memory function and storage through the promotion of both synaptic plasticity i.e. long-term potentiation (LTP) (Egan, et al., 2003), as well as neuronal survival and differentiation (Zuccato and Cattaneo, 2009). Specifically, BDNF expression is particularly high in the hippocampus (Binder and Scharfman, 2004), and is required for some forms of hippocampus-mediated plasticity (Tanaka, et al., 2008). A common missense polymorphism in the human BDNF gene produces an amino acid substitution (valine to methionine) at codon 66 (val66met). This single nucleotide polymorphism (SNP) impacts intracellular trafficking of BDNF, such that val proBDNF is more likely to be localized in neurites, whereas met BDNF aggregates in the cell body (Egan, et al., 2003). This polymorphism also impacts medial temporal lobe structural and functional integrity, as well as cognition (Goldberg, et al., 2008,Hariri, et al., 2003,Sambataro, et al., 2010).
Nonetheless, some reports have highlighted the fact that effects of the BDNF Val66Met variant on brain structure and function are complex, and may have a different impact when considering processes related to normal aging and pathological conditions. Sambataro et al illustrated this modulatory effect of BDNF on the trajectory of age-related changes in hippocampal function; Older met carriers showed impaired activation of the hippocampus during memory encoding and memory retrieval tasks compared to val/val individuals (Sambataro, et al., 2010). BDNF Met carriers have also been found to have reductions of hippocampal volumes associated with age. However, Voineskos et al found the opposite, in that Val/Val individuals were more susceptible to age related decline in late life, showing decreased thickness in the temporal lobe structures and episodic memory performance, whereas met carriers were more susceptible in early adult life (Voineskos, et al., 2011). Not surprisingly then, the effect of BDNF Met and hippocampal volume has been sometimes attributed to a “winners curse” effect (Molendijk, et al., 2012). More recently, Lim et al have shown prospectively that BDNF val66met met carriage affects brain volumes in older adults only in the presence of abnormally high levels of amyloid (Lim, et al., 2014b). Thus, brain amyloidosis may be mediating BDNF effects on brain structure.
In the context of preclinical AD, it has been reported more recently that healthy subjects carriers of the BDNF Met allele coupled with high PIB-PET AB uptake, showed accelerated cognitive decline and atrophy of the hippocampus (Lim, et al., 2014a). Furthermore, the same group found an epistatic interaction of BDNF and APOE genotypes on memory decline in the context of brain amyloidosis (Lim, et al., 2014b). They also showed similar findings in individuals with MCI who were amyloid positive (Lim, et al., 2014a). It is important to note that these studies used prospective designs.
However, in contrast to the majority of studies showing deleterious effects of met carriage, it must be acknowledged that a different hypothesis regarding the neurobiological effects of BDNF genotype has arisen from the complexities associated with bdnf molecular processing. Mature bdnf has generally been associated with LTP through its interaction with the TrkB receptor. In contrast, its precursor, pro-bdnf (a form of bdnf that includes a pro-domain containing val66met and the region of the mature protein) may be associated with apoptosis through interactions with p75 (Lu, 2003). Additionally the isolated pro-domain may, when it contains the met allele, be associated with various negative synaptic parameters (Anastasia, et al., 2013). Goldman and colleagues highlighted an advantageous role of Met allele in promoting recovery of executive function (Krueger, et al., 2011) and preservation of general cognitive functioning (Barbey, et al., 2014) after penetrating TBI. They attributed their finding to trafficking impairments associated with met allele which ultimately reduced apoptic effects. Therefore, Met allele may be protective in certain diseases (Zivadinov, et al., 2007).
We have proposed that BDNF val66met genotypic effects may be more clearly observed in older cohorts followed longitudinally both because age effects on genotypic differences (see below) and because declines may be characterized more accurately in within-subject designs (Goldberg and Mattay, 2009,Li, et al., 2010,Papenberg, et al., 2015,Sambataro, et al., 2010). Such a view is also consistent with increasing heritability for cognitive domains with age (Deary, et al., 2012). Here we comprehensively examine this proposal by: 1) testing the effects of BDNF genotype on age-related decline in cognition and brain morphometry measures of older healthy subjects (over 3 years of follow-up); and 2) by considering MCI/AD as a pathophysiological neurodegenerative state in which to examine BDNF effects. We critically stratified our results by APOE genetic variation, considering that E4 allele is associated with an increased OR for AD in comparison to E3 homozygotes, and APOE might act as a marker for neurodegeneration and risk for AD by influencing Abeta misprocessing. Our study extends prior work by Ward and colleagues (Ward, et al., 2014) that found APOE × BDNF effect on episodic memory in cross-sectional data.
2. Methods
2.1. Subjects
To test our hypotheses, we examined two cohorts from ADNI: 1) Healthy subjects (HS) (n= 175); and 2) AD patients and MCI individuals who progressed to AD in follow-up, thus showing evidence of prodromal AD at baseline (n= 222). Details of inclusion, exclusion, and sample selection criteria can be found elsewhere (Gomar, et al., 2011,Gomar, et al., 2014,Sousa, et al., 2015). Briefly, healthy participants were between 55–90 (inclusive) years old, had a Clinical Dementia Rating (CDR) (Morris, 1993) score of 0, a Mini Mental State Examination (MMSE) (Folstein, et al., 1975) score between 24 and 30 (inclusive), normal memory function according to Logical Memory II subscale (delayed Paragraph Recall) from the Wechsler Memory Scaled – Revised (Wechsler, 1987), no memory complaints, absence of significant impairment in other cognitive domains, and preserved activities of daily living. MCI patients had Mini Mental State Examination (MMSE) scores between 24 and 30 (inclusive), a memory complaint, objective memory loss as indicated by 1.5 standard deviations below the education adjusted cutoff on the Logical Memory II subscale, a Clinical Dementia Rating (CDR) score of 0.5, absence of significant impairment in other cognitive domains, and preserved activities of daily living. All MCI patients converted to AD at follow-up (mean time until conversion 20.44 months) (Gomar, et al., 2014). AD patients had MMSE scores between 20 and 26 (inclusive), memory complaint, objective memory loss, a CDR score of 0.5 or 1, and the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders (NINCDS/ADRDA) criteria for probable AD (McKhann, et al., 1984). All participants signed written informed consent for participation in ADNI, as approved by the institutional board at each participating center.
2.2. Genotyping
Genetic assessment for the functional single nucleotide polymorphism (SNP) of the BDNF gene at nucleotide 196 (rs6265) was performed using the Illumina Human610-Quad BeadChip (Illumina, Inc., San Diego, CA) and intensity data processed with GenomeStudio v2009.1. The two SNPs of the APOE gene (rs429358, rs7412) were genotyped from a 3 mL aliquot of blood taken in ethylenediaminetetraacetic acid (EDTA)-containing vacutainer tubes, and genomic DNA was extracted by Cogenics (now Beckman Coulter Genomics) using QIAamp DNA Blood Maxi Kit (Qiagen, Inc, Valencia, CA) following manufacturer’s protocol. For a more detailed description of the genotyping protocol see Saykin et al (Saykin, et al., 2010).
2.3. MRI acquisition and extraction of brain morphometry measures
Scans were obtained from 1.5 Tesla scanners at different sites involved in ADNI with minor variations in the MRI protocol based on the specific configuration of each scanner. Volumetric measures of the total brain, gray, white matter, and hippocampus, as well as cortical thickness measures of temporal lobe regions (middle temporal, inferior lateral temporal, parahippocampal, and entorhinal) were extracted. These measures were derived by Freesurfer (http://surfer.nmr.mgh.harvard.edu/) (Dale, et al., 1999,Fischl, et al., 2002). We also examined several other cortical thickness measures along the parietal and frontal lobes: posterior cingulate, precuneus, isthmus cingulate, anterior cingulate, middle frontal, lateral and medial orbitofrontal, in order to extend our analyses to key brain regions associated to neurodegeneration. Detailed descriptions of MRI protocol and methods are available at ADNI webpage and upon request of the authors. Individuals with either partial or total failure in the Freesurfer reconstruction stream outcome were excluded from further analysis.
2.4. Cognitive assessments
Several key measures of cognition were selected. First, we focused our analyses on both immediate and delayed episodic memory measures: immediate and delayed logical memory of the Wechsler Memory Scale (WMS) (Wechsler, 1987), and a composite score for ADAS-Cog memory items: word recall test, delayed word recall, and word recognition. Second, we also selected measures of working memory, language and semantic fluency, speed of processing, visuo-spatial abilities, and executive function (Goodglass and Kaplan, 1983,Wechsler, 1981,Wechsler, 1987). We also selected MMSE score as a measure of general cognition (Folstein, et al., 1975).
2.5. Statistical Analysis
Comparisons on demographic variables between BDNF Val/Val and Met carrier’s (including both Val/Met and Met/Met individuals) subgroups within each group (HS and MCI/AD) at baseline were performed with X square and t tests for dichotomous and quantitative variables respectively.
The analytic approach was as follows. Linear mixed models (SAS 9.3. PROC MIXED) were performed to examine the effect of BDNF genotype on temporal lobe integrity. Models included three factors: BDNF genotype group (Val/Val homozygotes and Met Carriers), Time (years since baseline), and a term for the interaction between BDNF × Time. Covariates for gender, education and year at each time point were included in all models. In these mixed models, the covariance pattern was set as heterogeneous autoregressive structure. Time was included as repeated factor and BDNF group (BDNF Val/Val vs BDNF Met carriers) as a between subject factor; subject was the random factor. All the analyses were performed first in the whole sample and second stratifying the sample by APOE genotype into two subgroups, APOE E3/E3 homozygotes and APOE E4 carriers; APOE E2 carriers were excluded because the association of this allele with neuroprotection (Conejero-Goldberg, et al., 2014). We also note that we used APOE4 to stratify the sample because it is a risk factor for neurodegeneration through multiple molecular mechanisms. Analyses were performed in HS and MCI/AD participants independently according to the hypothesis to be tested. Results were corrected for multiple comparisons through false discovery rate (FDR) method (p= 0.10). The method implemented by SAS PROC MULTTEST use FDR adjustment of multiple comparisons following Benjamini and Hochberg procedure (Benjamini, et al., 2001,Benjamini and Hochberg, 1995). Statistical significance was set at p <0.05 level. We believe that this approach in which we conducted separate analyses based on APOE E4 positivity, offered transparency and clarity in interpretation, as well as statistical rigor, due to FDR correction. Effect sizes were computed using Hedges and Olkin correction approach (Hedges and Olkin, 1985). We repeated the analyses including an APOE × BDNF interaction effect obtaining confirmatory results.
3. Results
3.1. Participant’s characteristics and BDNF genotype distribution
Within both HS and MCI/AD samples, distribution of age, gender and education were similar between BDNF Val/Val homozygotes and Met carriers (Table 1). Distribution of BDNF Val66Met polymorphism was similar between HS and MCI/AD patients, 31% of HS and 31% of MCI/AD were Met carriers (X2= 0.01, p= 0.94). These distributions did not change when dividing MCI and AD patients, 33% of MCI and 27% of AD were Met carriers. These characteristics remained unchanged when stratifying by APOE genotype and excluding APOE E2 carriers (Supplementary Table 1). Among HS, 106 subjects were APOE E3 homozygotes and 45 subjects were APOE E4 carriers (1 subject APOE E2 homozygote, 21 subjects APOE E2/E3, and 2 subjects APOE E2/E4). Among MCI/AD, 70 subjects were APOE E3 homozygotes and 138 subjects were APOE E4 carriers (6 subjects APOE E2/E3 and 8 subjects APOE E2/E4). These results were similar when stratifying for APOE genotype (Supplementary Table 2).
Table 1.
Sociodemographic and clinical status characteristics
| HC (N= 175) | MCI/AD (N= 222) | |||||
|---|---|---|---|---|---|---|
|
BDNF Val/Val
N= 120 |
BDNF Met Carriers
N= 55 |
Statistical Test |
BDNF Val/Val
N= 153 |
BDNF Met Carriers
N= 69 |
Statistical Test | |
| Age, Mean (SD) | 76 (5) Range: 60–88 |
76 (5) Range: 63–90 |
t173= −0.12 p= 0.91 |
75 (7) Range: 55–91 |
75 (7) Range: 55–88 |
t220= −0.24 p= 0.80 |
| Gender M/F | 62/58 | 31/24 | X2= 0.33 p= 0.56 |
59/94 | 34/35 | X2= 2.24 p= 0.13 |
| Education, Mean (SD) | 16 (2) Range: 10–20 |
16 (3) Range: 6–20 |
t173= 0.43 p= 0.67 |
15 (3) Range: 6–20 |
15 (3) Range: 8–20 |
t220= −0.60 p= 0.55 |
| CDR-SB, Mean (SD) | 0 (0) Range: 0–1.5 |
0 (0) Range: 0–1.5 |
t167= −0.31 p= 0.75 |
2.5 (1.6) Range: 0.5–9 |
2.7 (1.6) Range: 0.5–8 |
t220= −0.39 p= 0.70 |
3.2. Impact of BDNF genotype on brain integrity and cognitive markers in healthy subjects
In the HS sample, we found a marginally significant main effect of BDNF genotype in posterior cingulate thickness (F1, 41= 7.99, p= 0.07), in the APOE E4 carrier’s subsample. BDNF Met carriers APOE E4 carriers showed decreased thickness of the posterior cingulate area compared to APOE E3/E3 and BDNF Val/Val homozygotes (Figure 1 B). The effect size for this difference was medium (ES= 0.42, 95% CI −0.08–0.92). A similar pattern was also found in the precuneus area, although it was non-significant, with BDNF Met carriers and APOE E4 carriers showing decreased thickness compared to Val/Val homozygotes (F1,41= 5.47, p= 0.11) (Figure 1 D), but not in APOE E3 homozygotes (Figure 1C). Nevertheless, effect size for this difference was medium (ES= 0.46, 95% CI −0.07–0.97). Regarding cognitive performance, all measures were above the statistical threshold set for significance. Results of the linear mixed models for brain morphometry and cognitive measures are shown in Tables 2 and 3. No differences between BDNF Val/Val homozygotes and Met Carriers were evident at baseline for both HS and MCI/AD individuals.
Figure 1. Posterior cingulate and precuneus thickness by APOE and BDNF genotype in HS.
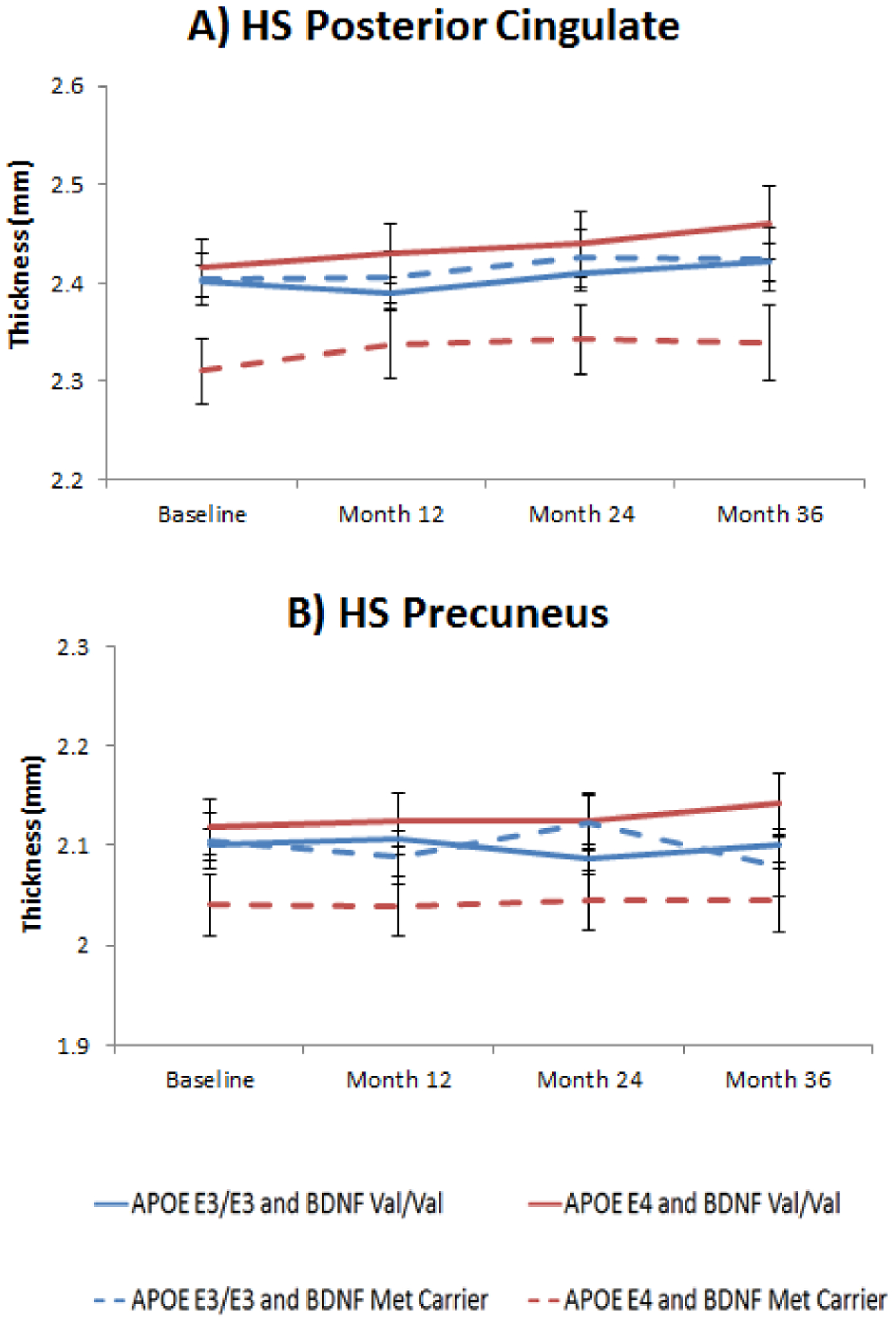
Figure 1A: Least square means from longitudinal mixed models of posterior cingulate thickness in HS according to APOE (E3/E3 and E4 carriers) and BDNF (Val/Val and Met carriers) interaction subgroups across 3 years of follow-up (BDNF main effect: F1, 41= 7.99, p= 0.07, ES= 0.42); Figure 1B: Least square means from longitudinal mixed models of longitudinal precuneus thickness in HS according to APOE (E3/E3 and E4 carriers) and BDNF (Val/Val and Met carriers) interaction subgroups across 3 years of follow-up (BDNF main effect: F1, 41= 5.47, p= 0.11, ES= 0.46). All p values have been FDR corrected. Error bars represent standard errors of the mean (SEM).
Table 2.
Linear mixed models results on brain morphometric measures for HS and MCI/AD groups stratified by APOE
| HS | MCI/AD | |||
|---|---|---|---|---|
| APOE E3/E3 | APOE E4 Carriers | APOE E3/E3 | APOE E4 Carriers | |
| Cerebral Cortex Volume | BDNF (F1,93 = 0.07/p= 0.79) TIME (F3,173= 4.01/p= 0.08) BDNF×TIME (F3,173= 1.10/p= 0.52) N= 97 |
BDNF (F1,38= 0.14/p= 0.94) TIME (F3,71= 0.16/p= 0.94) BDNF×TIME (F3,71= 0.13/p= 0.94) N= 42 |
BDNF (F1,53= 0.44/p= 0.72) TIME (F3,56= 3.82/p= 0.04) BDNF×TIME (F3,56= 0.52/p= 0.75) N= 57 |
BDNF (F1,111= 0.12/p= 0.78) TIME (F3,135= 12.42/p< 0.0001) BDNF×TIME (F3,135= 1.15/p= 0.55) N= 115 |
| Cerebral White Matter Volume | BDNF (F1,93= 3.52/p= 0.19) TIME( F3,173= 1.79/p= 0.34) BDNF×TIME (F3,173= 0.58/p= 0.77) N= 97 |
BDNF (F1,38= 0.79/p= 0.94) TIME (F3,71= 0.73/p= 0.94) BDNF×TIME (F3,71= 3.49/p= 0.18) N= 42 |
BDNF (F1,53= 0.06/p= 0.80) TIME (F3,56= 4.40/p= 0.034) BDNF×TIME (F3,56= 2.15/p= 0.24) N= 57 |
BDNF (F1,111= 0.83/p= 0.55) TIME (F3,135= 6.75/p< 0.0001) BDNF×TIME (F3,135= 0.36/p= 0.78) N= 115 |
| Hippocampus | BDNF (F1,93= 1.13/p= 0.52) TIME (F3,173= 3.18/p= 0.11) BDNF×TIME (F3,173= 0.49/p= 0.77) N= 97 |
BDNF (F1,38= 0.45/p= 0.94) TIME (F3,71= 0.50/p= 0.94) BDNF×TIME (F3,71= 0.19/p= 0.94) N= 42 |
BDNF (F1,53= 0.34/p= 0.72) TIME (F3,56= 5.61/p= 0.02) BDNF×TIME (F3,56= 0.71/p= 0.72) N= 57 |
BDNF (F1,111= 0.83/p= 0.55) TIME (F3,135= 35.01/p< 0.0001) BDNF×TIME (F3,135= 0.36/p= 0.78) N= 115 |
| Middle Temporal Thickness | BDNF (F1,99= 0.00/p= 0.96) TIME (F3,214= 0.65/p= 0.85) BDNF×TIME (F3,214= 0.15/p= 0.96) N= 103 |
BDNF (F1,39= 6.08/p= 0.27) TIME (F3,80= 0.25/p= 0.99) BDNF×TIME (F3,80= 0.03/p= 0.99) N= 42 |
BDNF (F1,57= 0.14/p= 0.82) TIME (F3,83= 7.18/p< 0.01) BDNF×TIME (F3,83= 0.83/p= 0.65) N= 61 |
BDNF (F1,121= 0.85/p= 0.60) TIME (F3,182= 30.20/p< 0.0001) BDNF×TIME (F3,182= 0.96/p= 0.62) N= 125 |
| Inferior Temporal Thickness | BDNF (F1,99= 0.24/p= 0.85) TIME (F3,214= 1.27/p= 0.85) BDNF×TIME (F3,214= 1.04/p= 0.85) N= 103 |
BDNF (F1,39= 2.23/p= 0.63) TIME (F3,80= 0.39/p= 0.99) BDNF×TIME (F3,80= 0.15/p= 0.99) N= 42 |
BDNF (F1,57= 1.38/p= 0.59) TIME (F3,83= 3.79/p= 0.04) BDNF×TIME (F3,83= 1.32/p= 0.59) N= 61 |
BDNF (F1,121= 0.31/p= 0.66) TIME (F3,182= 15.96/p<0.0001) BDNF×TIME (F3,182= 1.65/p= 0.38) N= 125 |
| Parahippocampal | BDNF (F1,99= 0.04/p= 0.96) TIME (F3,214= 2.21/p= 0.66) BDNF×TIME (F3,214= 0.27/p= 0.96) N= 103 |
BDNF (F1,39= 2.23/p= 0.63) TIME (F3,80= 0.39/p= 0.99) BDNF×TIME (F3,80= 0.15/p= 0.99) N= 42 |
BDNF (F1,57= 0.02/p= 0.89) TIME (F3,83= 5.78/p< 0.01) BDNF×TIME (F3,83= 0.88/p= 0.65) N= 61 |
BDNF (F1,121= 0.02/p= 0.88) TIME (F3,182= 26.09/p<0.0001) BDNF×TIME (F3,182= 1.50/p=0.41) N= 125 |
| Entorhinal | BDNF (F1,99= 1.77/p= 0.80) TIME (F3,214= 2.58/p= 0.66) BDNF×TIME (F3,214= 1.04/p= 0.81) N= 103 |
BDNF (F1,39= 1.62/p= 0.63) TIME (F3,80= 0.01/p= 0.99) BDNF×TIME (F3,80= 0.30/p= 0.99) N= 42 |
BDNF (F1,57= 0.05/p= 0.89) TIME (F3,83= 3.74/p= 0.04) BDNF×TIME (F3,83= 1.20/p= 0.59) N= 61 |
BDNF (F1,121= 0.02/p= 0.88) TIME (F3,182= 43.17/p<0.0001) BDNF×TIME (F3,182= 3.26/p= 0.06) N= 125 |
| Fusiform | BDNF (F1,99= 1.31/p= 0.80) TIME (F3,214= 0.88/p= 0.85) BDNF×TIME (F3,214= 0.74/p= 0.85) N= 103 |
BDNF (F1,39= 1.62/p= 0.63) TIME (F3,80= 0.10/p= 0.99) BDNF×TIME (F3,80= 1.17/p= 0.82) N= 42 |
BDNF (F1,57= 0.55/p= 0.65) TIME (F3,83= 4.61/p= 0.02) BDNF×TIME (F3,83= 0.75/p= 0.66) N= 61 |
BDNF (F1,121= 0.37/p= 0.66) TIME (F3,182= 26.85/p<0.0001) BDNF×TIME (F3,182= 0.70/p= 0.66) N= 125 |
| Posterior Cingulate | BDNF (F1,100= 0.11/p= 0.83) TIME (F3,220= 1.98/p= 0.53) BDNF×TIME (F3,220= 1.22/p= 0.68) N= 104 |
BDNF (F1,41=
7.99/p= 0.07)
TIME (F3,99= 0.39/p= 0.87) BDNF×TIME (F3,99= 0.25/p= 0.87) N= 45 |
BDNF (F1,62= 0.01/p= 0.92) TIME (F3,104= 4.43/p= 0.02) BDNF×TIME (F3,104= 0.55/p= 0.92) N= 66 |
BDNF (F1,127= 0.08/p= 0.84) TIME (F3,215= 6.84/p< 0.0001) BDNF×TIME (F3,215= 0.28/p= 0.84) N= 131 |
| Precuneus | BDNF (F1,100= 0.00/p= 0.97) TIME (F3,220= 0.86/p= 0.76) BDNF×TIME (F3,220= 5.42/p=0.01) N= 104 |
BDNF (F1,41= 5.47/p= 0.11) TIME (F3,99= 0.28/p= 0.87) BDNF×TIME (F3,99= 0.24/p= 0.87) N= 45 |
BDNF (F1,62= 0.05/p= 0.92) TIME (F3,104= 2.47/p= 0.15) BDNF×TIME (F3,104= 1.41/p= 0.44) N= 66 |
BDNF (F1,127= 0.61/p= 0.65) TIME (F3,215= 16.00/ p<0.0001) BDNF×TIME (F3,215= 1.13/p= 0.61) N= 131 |
| Isthmus-Cingulate | BDNF (F1,100= 0.29/p= 0.76) TIME (F3,220= 0.71/p= 0.76) BDNF×TIME (F3,220= 1.58/p= 0.58) N= 104 |
BDNF (F1,41= 2.37/p= 0.39) TIME (F3,99= 0.32/p= 0.87) BDNF×TIME (F3,99= 0.99/p= 0.87) N= 45 |
BDNF (F1,62= 0.12/p= 0.92) TIME (F3,104= 5.64/p= 0.01) BDNF×TIME (F3,104= 2.45/p=0.15) N= 66 |
BDNF (F1,127= 1.79/p= 0.41) TIME (F3,215= 17.27/ p< 0.001) BDNF×TIME (F3,215= 0.33/p= 0.84) N= 131 |
| Rostral Anterior Cingulate | BDNF (F1,100= 5.28/p= 0.21) TIME (F3,220= 1.08/p= 0.82) BDNF×TIME (F3,220= 0.09/p= 0.99) N= 104 |
BDNF (F1,41= 0.15/p= 0.83) TIME (F3,99= 0.88/p= 0.82) BDNF×TIME (F3,99= 0.99/p= 0.81) N= 45 |
BDNF (F1,62= 4.14/p= 0.42) TIME (F3,105= 2.05/p= 0.45) BDNF×TIME (F3,105= 1.96/p= 0.45) N= 66 |
BDNF (F1,127= 0.16/p= 0.78) TIME (F3,215= 4.53/ p= 0.01) BDNF×TIME (F3,215= 0.53/p= 0.78) N= 131 |
| Caudal Anterior Cingulate | BDNF (F1,100= 1.31/p= 0.82) TIME (F3,220= 0.47/p= 0.99) BDNF×TIME (F3,220= 1.69/p= 0.77) N= 104 |
BDNF (F1,41= 5.86/p= 0.18) TIME (F3,99= 0.30/p= 0.83) BDNF×TIME (F3,99= 2.09/p= 0.48) N= 45 |
BDNF (F1,62= 0.31/p= 0.74) TIME (F3,105= 1.06/p= 0.60) BDNF×TIME (F3,105= 1.17/p= 0.58) N= 66 |
BDNF (F1,127= 4.38/ p= 0.11) TIME (F3,215= 1.56/ p= 0.45) BDNF×TIME (F3,215= 1.17/p= 0.58) N= 131 |
| Rostral Middle Frontal | BDNF (F1,100= 0.88/p= 0.82) TIME (F3,220= 0.19/p= 0.99) BDNF×TIME (F3,220= 0.44/p= 0.99) N= 104 |
BDNF (F1,41= 4.10/p= 0.30) TIME (F3,99= 1.15/p= 0.81) BDNF×TIME (F3,99= 0.39/p= 0.83) N= 45 |
BDNF (F1,62= 0.02/p= 0.89) TIME (F3,105= 2.95/p= 0.42) BDNF×TIME (F3,105= 1.52/p= 0.55) N= 66 |
BDNF (F1,127= 0.01/p= 0.95) TIME (F3,215= 12.56/ p<0.0001) BDNF×TIME (F3,215= 0.99/p= 0.65) N= 131 |
| Caudal Middle Frontal | BDNF (F1,100= 0.83/p= 0.82) TIME (F3,220= 1.68/p= 0.77) BDNF×TIME (F3,220= 3.66/p= 0.21) N= 104 |
BDNF (F1,41= 6.42/p= 0.18) TIME (F3,99= 0.38/p= 0.83) BDNF×TIME (F3,99= 0.99/p= 0.81) N= 45 |
BDNF (F1,62= 0.05/p= 0.88) TIME (F3,105= 2.42/p= 0.42) BDNF×TIME (F3,105= 1.68/p= 0.53) N= 66 |
BDNF (F1,127= 1.24/p= 0.53) TIME (F3,215= 11.69/ p<0.0001) BDNF×TIME (F3,215= 0.66/p= 0.78) N= 131 |
| Lateral Orbito-Frontal | BDNF (F1,100= 0.00/p= 1.00) TIME (F3,220= 0.85/p= 0.94) BDNF×TIME (F3,220= 0.48/p= 0.99) N= 104 |
BDNF (F1,41= 1.02/p= 0.81) TIME (F3,99= 0.67/p= 0.83) BDNF×TIME (F3,99= 0.40/p= 0.81) N= 45 |
BDNF (F1,62= 0.05/p= 0.88) TIME (F3,105= 1.39/p= 0.56) BDNF×TIME (F3,105= 0.53/p= 0.80) N= 66 |
BDNF (F1,127= 2.33/p= 0.33) TIME (F3,215= 7.87/ p<0.0001) BDNF×TIME (F3,215= 0.60/p= 0.78) N= 131 |
| Medial Orbito-Frontal | BDNF (F1,100= 0.08/p= 1.00) TIME (F3,220= 0.32/p= 0.99) BDNF×TIME (F3,220= 0.18/p= 1.00) N= 104 |
BDNF (F1,41= 0.05/p= 0.83) TIME (F3,99= 0.39/p= 0.83) BDNF×TIME (F3,99= 1.29/p= 0.81) N= 45 |
BDNF (F1,62= 0.34/p= 0.74) TIME (F3,105= 1.25/p= 0.58) BDNF×TIME (F3,105= 0.99/p= 0.60) N= 66 |
BDNF (F1,127= 0.00/p= 0.95) TIME (F3,215= 9.76/ p< 0.0001) BDNF×TIME (F3,215= 0.57/p= 0.78) N= 131 |
BDNF significant and trend effects are in bold (p<0.10); All p values have been FDR corrected
Table 3.
Linear mixed models results on cognitive measures for HS and MCI/AD groups stratified by APOE
| HC | MCI/AD | |||
|---|---|---|---|---|
| APOE E3/E3 N= 106 |
APOE E4 Carriers N= 45 |
APOE E3/E3 N= 70 |
APOE E4 Carriers N= 138 |
|
| MMSE | BDNF (F1,102= 0.00/p= 1.00) TIME (F3,291= 0.18/p= 0.99) BDNF×TIME(F3,291=1.37/p=0.99) |
BDNF (F1,41= 0.24/p= 0.86) TIME (F3,119= 0.46/p= 0.87) BDNF×TIME(F3,119=0.05/p=0.98) |
BDNF (F1,66= 0.20/p=0.77) TIME (F3,155= 19.74/p<0.001) BDNF×TIME(F3,155=0.42/p=0.77) |
BDNF (F1,134= 1.92/p= 0.30) TIME (F3,315= 45.68/ p<0.0001) BDNF×TIME(F3,315=0.94/p=0.55) |
| Logical Memory Immediate | BDNF (F1,102= 2.61/p= 0.65) TIME (F3,291= 5.45/ p=0.04) BDNF×TIME(F3,291= 0.43/p=0.99) |
BDNF (F1,41= 1.38/p=0.63) TIME (F3,119= 2.28/p=0.50) BDNF×TIME(F3,119=1.15/p=0.63) |
BDNF (F1,66= 3.81/p=0.15) TIME (F3,154=6.59/p=0.001) BDNF×TIME(F3,154=0.65/p=0.77) |
BDNF (F1,134= 5.63/p=0.05)
TIME (F3,313= 19.16/ p<0.0001) BDNF×TIME(F3,313=0.10/p=0.96) |
| ADAS Memory | BDNF (F1,102= 0.47/p= 0.99) TIME (F3,289= 0.57/p= 0.99) BDNF×TIME(F3,289= 1.28/p=0.99) |
BDNF (F1,41= 0.03/p= 0.90) TIME (F3,119= 2.64/p= 0.40) BDNF×TIME(F3,119=1.82/p=0.63) |
BDNF (F1,66= 0.37/p= 0.77) TIME (F3,152= 16.78/ p<0.001) BDNF×TIME(F3,152=0.37/p=0.77) |
BDNF (F1,134= 5.72/p= 0.05)
TIME (F3,313= 28.45/ p<0.0001) BDNF×TIME(F3,313=0.67/p=0.71) |
| Logical Memory Delayed | BDNF (F1,102= 3.50/p= 0.48) TIME (F3,290= 0.81/p= 0.99) BDNF×TIME(F3,290= 0.24/p=0.99) |
BDNF (F1,41= 2.32/p= 0.63) TIME (F3,119= 0.77/p= 0.77) BDNF×TIME(F3,119=1.53/p=0.63) |
BDNF (F1,66= 2.35/p= 0.28) TIME (F3,154= 5.95/ p<0.01) BDNF×TIME(F3,154=1.87/p=0.28) |
BDNF (F1,134= 3.27/p= 0.15) TIME (F3,312= 7.74/ p<0.0001) BDNF×TIME(F3,312=1.64/p=0.30) |
| ADAS No Memory | BDNF (F1,102= 0.09/p= 0.99) TIME (F3,286= 0.98/p= 0.99) BDNF×TIME(F3,286= 1.06/p=0.99) |
BDNF (F1,41= 0.95/p= 0.63) TIME (F3,117= 2.72/p= 0.40) BDNF×TIME(F3,117=0.45/p=0.87) |
BDNF (F1,66= 0.53/p= 0.77) TIME (F3,149= 21.20/ p<0.001) BDNF×TIME(F3,149=1.04/p=0.67) |
BDNF (F1,134= 0.99/p= 0.46) TIME (F3,306= 30.63/ p<0.0001) BDNF×TIME(F3,306=0.11/p=0.96) |
| Digit Span | BDNF (F1,102= 0.02/p= 0.99) TIME (F3,291= 0.50/p= 0.99) BDNF×TIME(F3,291=0.40/p= 0.99) |
BDNF (F1,41= 0.13/p= 0.87) TIME (F3,119= 1.13/p= 0.63) BDNF×TIME(F3,119=0.91/p=0.74) |
BDNF (F1,66= 0.13/p= 0.77) TIME (F3,152= 12.37/p<0.001) BDNF×TIME(F3,152=2.25/p=0.21) |
BDNF (F1,134= 4.95/p= 0.06)
TIME (F3,310= 15.43/ p<0.0001) BDNF×TIME(F3,310=0.17/p=0.96) |
| Digit-Symbol | BDNF (F1,102= 0.11/p= 0.99) TIME (F3,288= 2.99/p= 0.32) BDNF×TIME(F3,288= 0.05/p=0.99) |
BDNF (F1,41= 0.60/p= 0.74) TIME (F3,119= 1.26/p= 0.63) BDNF×TIME(F3,119=0.38/p=0.88) |
BDNF (F1,66= 0.15/p= 0.77) TIME (F3,148= 14.63/p<0.001) BDNF×TIME(F3,148=0.73/p=0.77) |
BDNF (F1,134= 1.02/p= 0.46) TIME (F3,308= 20.45/ p<0.0001) BDNF×TIME(F3,308=0.34/p=0.89) |
| Semantic Fluency | BDNF (F1,102= 0.54/p= 0.99) TIME (F3,291= 0.71/p= 0.99) BDNF×TIME(F3,291=1.16/p= 0.99) |
BDNF (F1,41= 1.19/p= 0.63) TIME (F3,119= 0.84/p= 0.75) BDNF×TIME(F3,119=0.24/p=0.90) |
BDNF (F1,66= 0.14/p= 0.77) TIME (F3,154= 9.06/ p<0.001) BDNF×TIME(F3,154=0.38/p=0.77) |
BDNF (F1,134= 5.53/p= 0.05)
TIME (F3,311= 38.99/ p<0.0001) BDNF×TIME(F3,311=2.00/p=0.21) |
| Trail A | BDNF (F1,102= 0.00/p= 0.99) TIME (F3,290= 3.72/p= 0.18) BDNF×TIME(F3,290= 0.22/p=0.99) |
BDNF (F1,41= 0.17/p= 0.87) TIME (F3,119= 3.76/p= 0.19) BDNF×TIME(F3,119=0.26/p=0.90) |
BDNF (F1,66= 1.06/p= 0.58) TIME (F3,148= 7.60/ p<0.001) BDNF×TIME(F3,148=1.70/p=0.34) |
BDNF (F1,134= 0.65/p= 0.55) TIME (F3,308= 15.55/ p<0.0001) BDNF×TIME(F3,308=0.34/p=0.89) |
| Trail B | BDNF (F1,102= 0.19/p= 0.99) TIME (F3,290= 1.59/p= 0.95) BDNF×TIME(F3,290=0.51/p= 0.99) |
BDNF (F1,41= 1.31/p= 0.63) TIME (F3,117= 6.33/p= 0.01) BDNF×TIME(F3,117=1.36/p=0.63) |
BDNF (F1,66= 0.13/p= 0.77) TIME (F3,132= 7.72/ p<0.001) BDNF×TIME(F3,132=0.46/p=0.77) |
BDNF (F1,134= 1.28/p= 0.41) TIME (F3,274= 16.12/ p<0.0001) BDNF×TIME(F3,274=0.52/p=0.80) |
BDNF significant and trend effects are in bold (p<0.10); All p values have been FDR corrected
3.3. Impact of BDNF on brain integrity and cognitive markers in MCI/AD patients
In the MCI/AD sample a BDNF × Time interaction effect was evident for entorhinal thickness (F3, 298= 5.16, p= 0.004), that resulted in a marginally significant effect in the APOE E4 carriers subgroup (F3, 182= 3.26, p= 0.06), i.e. BDNF Met carriers and APOE E4 carriers showed greater atrophy compared to Val/Val homozygotes and APOE E3 homozygotes over 3 years in the entorhinal cortex (Figure 2 A). Effect sizes went from 0.48 to 1.03 (medium to large) between baseline and third year of follow-up in BDNF Met and APOE E4 carriers, as compared to 0.31 to 0.77 in Val/Val homozygotes. As expected in the context of MCI/AD neuropathology, several brain areas showed an effect of time, i.e. decreasing volume or thickness through 3 years: cortical volume, hippocampus, and other key regions of the temporal and parietal cortex. This atrophy pattern was evident independent of APOE genotype (i.e. both APOE subgroups showed significant atrophy over time). However, this was not the case for frontal cortex thickness measures, where only APOE E4 carriers showed significant atrophy, specifically in regions such as the rostral part of the anterior cingulate cortex, middle frontal area, lateral and medial orbitofrontal cortex (see Supplement)
Figure 2. APOE and BDNF interaction effects in entorhinal thickness and cognitive measures in MCI/AD patients.
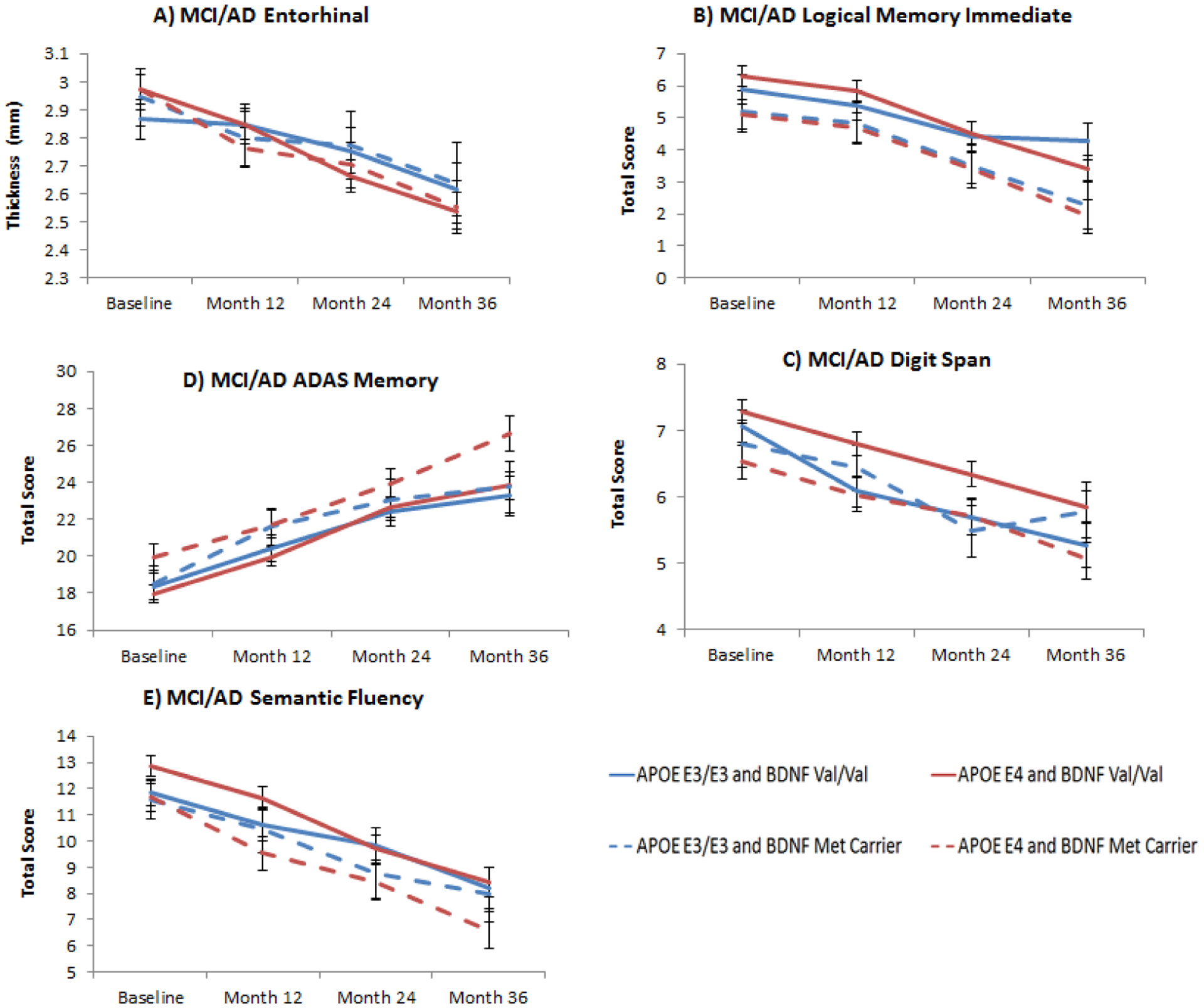
Figure 2A: Least square means from longitudinal mixed models of entorhinal atrophy over 3 years in MCI/AD according to APOE (E3/E3 and E4 carriers) and BDNF (Val/Val and Met carriers) interaction subgroups (BDNF×TIME effect: F3,182= 3.26, p= 0.06); Figure 2B: Least square means from longitudinal mixed models of logical memory immediate scores in MCI/AD according to APOE (E3/E3 and E4 carriers) and BDNF (Val/Val and Met carriers) interaction subgroups (BDNF main effect: F1,134= 5.63, p= 0.05); Figure 2C: Least square means from longitudinal mixed models of working memory (digit span) showed that APOE E4 and BDNF Met carriers were also more impaired compared to APOE E3/E3 and BDNF Val/Val (BDNF main effect: F1, 134= 4.95, p= 0.06) Figure 2D: Least square means from longitudinal mixed models of ADAS Memory score showed that BDNF Met carriers had higher scores (higher scores correspond to worse performance) compared to Val homozygotes in the APOE E4 subgroup compared to APOE E3/E3 (BDNF main effect: F1,134= 5.72, p= 0.05); Figure 2E: Similar result for least square means from longitudinal mixed models of semantic fluency, where BDNF Met carriers showed lower score compared to Val/Val in the APOE E4 subgroup compared to APOE E3/E3 (BDNF main effect: F1,134= 5.53, p= 0.05). All p values have been FDR corrected. Error bars represent standard errors of the mean (SEM).
Regarding cognition, we found several measures that showed a main effect of BDNF genotype in the APOE E4 subgroup: Logical memory immediate (F1, 134= 5.63, p= 0.05) (Figure 2B), ADAS Memory (F1, 134= 5.72, p= 0.05) (Figure 2C), semantic fluency (F1, 134= 5.53, p= 0.05) (Figure 2D), and digit span (F1, 134= 4.95, p= 0.06). In all cases BDNF Met carriers showed poorer performance compared to Val/Val homozygotes. Differences in effect sizes were medium for all three measures: 0.38 (95% CI 0.02–0.74) for logical memory immediate, 0.26 (95% CI −0.10–0.62) for ADAS-Memory, 0.38 (95% CI 0.02–0.74) for semantic fluency and 0.36 (95% CI −0.00–0.72) for digit span. Practically all cognitive measures showed an effect of Time irrespective of APOE status, suggesting, as expected, a lack of the ability to benefit from practice of repeated cognitive testing in established AD as well as in MCI progressors, and/or frank decline. Results of the linear mixed models for brain morphometry and cognitive measures are shown in Tables 2 and 3.
4. Discussion
Taken together our findings suggest lack of compensatory mechanisms in carriers of the BDNF Met allele and carriers of the APOE E4 allele. In the context of healthy aging, posterior cingulate and precuneus thickness showed a trend for significantly decreases in BDNF Met carriers compared to Val/Val homozygotes. In MCI/AD carriers of the BDNF Met allele and APOE E4 allele, a significantly steeper rate of atrophy of the entorhinal cortex over 3 years was evident compared to Val/Val homozygotes. This was accompanied by a significant impairment in several cognitive functions (specifically memory and semantic fluency).
Posterior cingulate and precuneus are considered to be important brain regions associated specifically with preclinical AD, and additionally are key regions forming part of the default mode network that has been found to be disrupted in pathological aging (Greicius, et al., 2004,Rami, et al., 2012,Sperling, et al., 2014). The BDNF effects in APOE E4 carriers that we have found in our sample, i.e. Met carriers having thinner posterior cingulate and precuneus cortex, suggest compromises in this neurotrophic factor in one of the brain regions undergoing primary manifestations of pathological aging associated to AD. Indeed, APOE E4 increases amyloid and tau misprocessing plus brain atrophy in healthy older adults and in MCI. This is broadly consistent with the Lim et al findings in AIBL that BDNF effects were conditioned by the presence/absence of amyloid (Lim, et al., 2014b).
Entorhinal cortex is a key region in both memory formation (Squire, et al., 2004) and development of AD pathophysiology (Desikan, et al., 2009,Small, et al., 2011). It seems to be also a crucial area for BDNF expression, as Nagahara et al demonstrated that increasing BDNF expression in the entorhinal cortex ameliorates neurodegeneration in animal models of AD and aging, (Nagahara, et al., 2009). Furthermore, BDNF protein has been found to be reduced in entorhinal cortex of AD patients (at post-mortem) (Connor, et al., 1997). Our findings suggest that improper trafficking and/or reduction of secretion of BDNF in Met carriers may contribute to the progressive neuronal atrophy of the entorhinal cortex in prodromal and established AD.
These findings might reflect a mechanism by which BDNF and APOE contribute to AD related localized brain atrophy, or alternatively cannot compensate for, or ameliorate AD related neurodegeneration. It has been suggested that BDNF Met allele may accelerate the progression of AD; BDNF Met effect on longitudinal cognition and hippocampal volume is only present if coupled with high AB amyloid levels (Lim, et al., 2013). Furthermore, epistatic interaction between BDNF and APOE E4 on disease progression in preclinical AD has also been reported (Adamczuk, et al., 2013,Hashimoto, et al., 2009). As stated in the introduction, this is in contrast to the findings that Met allele may have advantageous effects in the context of traumatic brain injury, even though it remains possible that these two neuropathological states, one consisting in a one-time trauma and the other consisting of progressive brain changes might result in BDNF Val/Met differential effects on brain morphometry and cognition.
In addition, our findings also highlight the important role of BDNF genotype in episodic memory performance (Goldberg, et al., 2008,Hariri, et al., 2003), especially in the context of neurodegeneration in APOE E4 individuals. In our MCI/AD sample, we found episodic memory (plus semantic fluency) to be impaired in Met carriers compared to Val/Val homozygotes, as others have also found (Lim, et al., 2014a), but interestingly in our case only in APOE E4 carriers. Consistent with prior research logical memory is probably one of the more sensitive measures for identifying BDNF genotypic modulatory effects on memory. However, we did not find BDNF/APOE effects on episodic memory in our older healthy sample, as others have recently reported using cross-sectional data; It should be noted that in this study APOE E2 carriers seem to drive the difference found in episodic memory performance (Ward, et al., 2014). Semantic fluency performance is thought to engage temporal lobe areas (Henry and Crawford, 2004,Troyer, et al., 1998) that are known to be at brain region in which bdnf expression is high.
One of the main conclusions that might be drawn from this set of data is that in the examination of BDNF genotype effects on temporal lobe morphometry and memory performance, APOE status should be considered along with BDNF genotype. Recently, taking an analytic approach in with HS, MCI and AD individuals were combined, Honea et al in ADNI context found a BDNF interaction with age in whole-brain volume (Older Val/Val had smaller volume), and greater rate of hippocampal and whole-brain atrophy over two years (Honea, et al., 2013), such that Val/Met declined more quickly. Their analytic strategy was to pool HS, MCI and AD individuals. Alternatively, we took a different approach, first by examining diagnostic groups separately, and second by stratifying by APOE. This strategy allowed us to observe APOE modulatory effects on BDNF that otherwise would have been masked by pooling the diagnostic groups. Our findings are also broadly consistent with a similar set of conclusions drawn from another recent study for the Australian Imaging Biomarkers & Lifestyle Study of Aging (AIBL) that found faster episodic memory decline in healthy subjects with both BDNF Met and APOE E4 alleles who were amyloid positive (Lim, et al., 2014b). As our APOE stratification analysis indicated, positive BDNF findings were uniquely evident in APOE E4 carriers. Therefore, this signal points towards a greater deleterious effect of Met BDNF only with an APOE E4 background, suggesting that both genetic factors interact to impact cortical morphology of important brain regions in pathological aging. In this respect, it is important to note that E4 produces robust decreases in CSF AB and increases in tau in both HS and MCI groups in ADNI (Conejero-Goldberg, et al., 2014,Schuff, et al., 2009,Shaw, et al., 2009).
Several caveats in our study must be acknowledged. First, longitudinal follow-up may also be too short to have sensitively detected more generalized BDNF patterns of brain atrophy and cognitive decline. Second, cell size at the two levels of stratification of genotype association (BDNF and APOE genotype) could have been too small for detecting subtle effects. We acknowledge the possibility of false positive errors and the moderate effect size d value. Finally, we were unable to directly study the effect of brain amyloid burden; however given APOE E4 decreases CSF AB, we believe APOE might act as marker for AB related neurodegeneration.
In conclusion, our set of findings suggests lack of neural compensatory mechanisms in BDNF Met carriers and APOE E4 carriers. This was less robust in the brain cortical integrity of healthy aging, but nevertheless in key characteristic areas for pathological aging (posterior-cingulate/precuneus). In MCI/AD, BDNF/APOE effects were stronger, especially in the form of both entorhinal atrophy and cognitive performance.
Supplementary Material
HIGHLIGHTS.
BDNF and APOE polymorphisms impact age-related brain morphometry and cognition.
We examined interactions between BDNF and APOE in healthy aging (HA) and MCI/AD.
In HA, BDNF Met/APOE4 carriers showed atrophy in posterior cingulate cortex.
In MCI/AD, this allele combination led to entorhinal atrophy and impaired memory.
Reduction of CNS compensatory mechanisms results in large BDNF effects.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s disease Cooperative Study at the University of California San Diego. ADNI data are disseminated by the Laboratory for Neuro-Imaging at the University of California, Los Angeles. This research was also supported by the Litwin-Zucker Research Center and by grant RO1 AG038734 (Goldberg TE, PI) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
Dr. Goldberg receives royalties for the use of the Brief Assessment of Cognition in Schizophrenia (BACS) in clinical trials.
References
- Adamczuk K, De Weer AS, Nelissen N, Chen K, Sleegers K, Bettens K, Van Broeckhoven C, Vandenbulcke M, Thiyyagura P, Dupont P, Van Laere K, Reiman EM, Vandenberghe R. 2013. Polymorphism of brain derived neurotrophic factor influences beta amyloid load in cognitively intact apolipoprotein E epsilon4 carriers. Neuroimage Clin 2, 512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasia A, Deinhardt K, Chao MV, Will NE, Irmady K, Lee FS, Hempstead BL, Bracken C. 2013. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat Commun 4, 2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbey AK, Colom R, Paul E, Forbes C, Krueger F, Goldman D, Grafman J. 2014. Preservation of general intelligence following traumatic brain injury: contributions of the Met66 brain-derived neurotrophic factor. PLoS One 9(2), e88733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. 2001. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125(1–2), 279–84. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57(1), 289–300. [Google Scholar]
- Binder DK, Scharfman HE. 2004. Brain-derived neurotrophic factor. Growth Factors 22(3), 123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, Chen S, Davies P, Goldberg TE. 2014. APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol Psychiatry 19(11), 1243–50. [DOI] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. 1997. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res Mol Brain Res 49(1–2), 71–81. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI. 1999. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage 9(2), 179–94. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Yang J, Davies G, Harris SE, Tenesa A, Liewald D, Luciano M, Lopez LM, Gow AJ, Corley J, Redmond P, Fox HC, Rowe SJ, Haggarty P, McNeill G, Goddard ME, Porteous DJ, Whalley LJ, Starr JM, Visscher PM. 2012. Genetic contributions to stability and change in intelligence from childhood to old age. Nature 482(7384), 212–5. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Cabral HJ, Hess CP, Dillon WP, Glastonbury CM, Weiner MW, Schmansky NJ, Greve DN, Salat DH, Buckner RL, Fischl B. 2009. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease. Brain 132(Pt 8), 2048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. 2003. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112(2), 257–69. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. 2002. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33(3), 341–55. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. 1975. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12(3), 189–98. [DOI] [PubMed] [Google Scholar]
- Goldberg TE, Iudicello J, Russo C, Elvevag B, Straub R, Egan MF, Weinberger DR. 2008. BDNF Val66Met polymorphism significantly affects d’ in verbal recognition memory at short and long delays. Biol Psychol 77(1), 20–4. [DOI] [PubMed] [Google Scholar]
- Goldberg TE, Mattay VS. 2009. The Genetics of Cognitive Neuroscience. in: Goldberg TE, Weinberger DR (Eds.). Masschussets Institute of Technocology, Cambridge, MA, pp 159–74. [Google Scholar]
- Gomar JJ, Bobes-Bascaran MT, Conejero-Goldberg C, Davies P, Goldberg TE. 2011. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer’s disease neuroimaging initiative. Arch Gen Psychiatry 68(9), 961–9. [DOI] [PubMed] [Google Scholar]
- Gomar JJ, Conejero-Goldberg C, Davies P, Goldberg TE. 2014. Extension and refinement of the predictive value of different classes of markers in ADNI: four-year follow-up data. Alzheimers Dement 10(6), 704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E. 1983. The assessment of aphasia and related disorders Lea & Febiger, Philadelphia. [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V. 2004. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A 101(13), 4637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR. 2003. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci 23(17), 6690–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Hirata Y, Asada T, Yamashita F, Nemoto K, Mori T, Moriguchi Y, Kunugi H, Arima K, Ohnishi T. 2009. Effect of the brain-derived neurotrophic factor and the apolipoprotein E polymorphisms on disease progression in preclinical Alzheimer’s disease. Genes Brain Behav 8(1), 43–52. [DOI] [PubMed] [Google Scholar]
- Hedges LV, Olkin L, editors. Statistical Methods for Meta-Analysis. London: Academic Press, INC; 1985. [Google Scholar]
- Henry JD, Crawford JR. 2004. A meta-analytic review of verbal fluency performance following focal cortical lesions. Neuropsychology 18(2), 284–95. [DOI] [PubMed] [Google Scholar]
- Honea RA, Cruchaga C, Perea RD, Saykin AJ, Burns JM, Weinberger DR, Goate AM. 2013. Characterizing the role of brain derived neurotrophic factor genetic variation in Alzheimer’s disease neurodegeneration. PLoS One 8(9), e76001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Pardini M, Huey ED, Raymont V, Solomon J, Lipsky RH, Hodgkinson CA, Goldman D, Grafman J. 2011. The role of the Met66 brain-derived neurotrophic factor allele in the recovery of executive functioning after combat-related traumatic brain injury. J Neurosci 31(2), 598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Chicherio C, Nyberg L, von Oertzen T, Nagel IE, Papenberg G, Sander T, Heekeren HR, Lindenberger U, Backman L. 2010. Ebbinghaus revisited: influences of the BDNF Val66Met polymorphism on backward serial recall are modulated by human aging. J Cogn Neurosci 22(10), 2164–73. [DOI] [PubMed] [Google Scholar]
- Lim YY, Villemagne VL, Laws SM, Ames D, Pietrzak RH, Ellis KA, Harrington K, Bourgeat P, Bush AI, Martins RN, Masters CL, Rowe CC, Maruff P. 2014a. Effect of BDNF Val66Met on memory decline and hippocampal atrophy in prodromal Alzheimer’s disease: a preliminary study. PLoS One 9(1), e86498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YY, Villemagne VL, Laws SM, Ames D, Pietrzak RH, Ellis KA, Harrington KD, Bourgeat P, Salvado O, Darby D, Snyder PJ, Bush AI, Martins RN, Masters CL, Rowe CC, Nathan PJ, Maruff P. 2013. BDNF Val66Met, Abeta amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol Aging 34(11), 2457–64. [DOI] [PubMed] [Google Scholar]
- Lim YY, Villemagne VL, Laws SM, Pietrzak RH, Snyder PJ, Ames D, Ellis KA, Harrington K, Rembach A, Martins RN, Rowe CC, Masters CL, Maruff P. 2014b. APOE and BDNF polymorphisms moderate amyloid beta-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B 2003. Pro-region of neurotrophins: role in synaptic modulation. Neuron 39(5), 735–8. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. 1984. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34(7), 939–44. [DOI] [PubMed] [Google Scholar]
- Molendijk ML, Bus BA, Spinhoven P, Kaimatzoglou A, Oude Voshaar RC, Penninx BW, van IMH, Elzinga BM. 2012. A systematic review and meta-analysis on the association between BDNF val(66)met and hippocampal volume--a genuine effect or a winners curse? Am J Med Genet B Neuropsychiatr Genet 159B(6), 731–40. [DOI] [PubMed] [Google Scholar]
- Morris JC. 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43(11), 2412–4. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. 2009. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15(3), 331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenberg G, Salami A, Persson J, Lindenberger U, Backman L. 2015. Genetics and Functional Imaging: Effects of APOE, BDNF, COMT, and KIBRA in Aging. Neuropsychol Rev 25(1), 47–62. [DOI] [PubMed] [Google Scholar]
- Rami L, Sala-Llonch R, Sole-Padulles C, Fortea J, Olives J, Llado A, Pena-Gomez C, Balasa M, Bosch B, Antonell A, Sanchez-Valle R, Bartres-Faz D, Molinuevo JL. 2012. Distinct functional activity of the precuneus and posterior cingulate cortex during encoding in the preclinical stage of Alzheimer’s disease. J Alzheimers Dis 31(3), 517–26. [DOI] [PubMed] [Google Scholar]
- Sambataro F, Murty VP, Lemaitre HS, Reed JD, Das S, Goldberg TE, Callicott JH, Weinberger DR, Mattay VS. 2010. BDNF modulates normal human hippocampal ageing [corrected]. Mol Psychiatry 15(2), 116–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, Risacher SL, Nho K, Huentelman MJ, Craig DW, Thompson PM, Stein JL, Moore JH, Farrer LA, Green RC, Bertram L, Jack CR Jr., Weiner MW. 2010. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement 6(3), 265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR Jr., Weiner MW. 2009. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain 132(Pt 4), 1067–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. 2009. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65(4), 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. 2011. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci 12(10), 585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa A, Gomar JJ, Goldberg TE. 2015. Neural and behavioral substrates of disorientation in Mild Cognitive Impairment and Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Mormino E, Johnson K. 2014. The Evolution of Preclinical Alzheimer’s Disease: Implications for Prevention Trials. Neuron 84(3), 608–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Stark CE, Clark RE. 2004. The medial temporal lobe. Annu Rev Neurosci 27, 279–306. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. 2008. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319(5870), 1683–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyer AK, Moscovitch M, Winocur G, Alexander MP, Stuss D. 1998. Clustering and switching on verbal fluency: the effects of focal frontal- and temporal-lobe lesions. Neuropsychologia 36(6), 499–504. [DOI] [PubMed] [Google Scholar]
- Voineskos AN, Lerch JP, Felsky D, Shaikh S, Rajji TK, Miranda D, Lobaugh NJ, Mulsant BH, Pollock BG, Kennedy JL. 2011. The brain-derived neurotrophic factor Val66Met polymorphism and prediction of neural risk for Alzheimer disease. Arch Gen Psychiatry 68(2), 198–206. [DOI] [PubMed] [Google Scholar]
- Ward DD, Summers MJ, Saunders NL, Janssen P, Stuart KE, Vickers JC. 2014. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav Brain Res 271, 309–15. [DOI] [PubMed] [Google Scholar]
- Wechsler D 1981. Wechsler Adult Intelligence Scale-Revised. The Psychological Corporation, Sant Antonio, TX. [Google Scholar]
- Wechsler D 1987. Wechsler Memory Scale-Revised. The Psychological Corporation, San Antonio, TX. [Google Scholar]
- Zivadinov R, Weinstock-Guttman B, Benedict R, Tamano-Blanco M, Hussein S, Abdelrahman N, Durfee J, Ramanathan M. 2007. Preservation of gray matter volume in multiple sclerosis patients with the Met allele of the rs6265 (Val66Met) SNP of brain-derived neurotrophic factor. Hum Mol Genet 16(22), 2659–68. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. 2009. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5(6), 311–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.