

Abstract
Cardiovascular disease (CVD) is the major cause of disability-adjusted life years (DALY) and death globally. The most common internal modification of mRNA is N6-adenosylate methylation (m6A). Recently, a growing number of studies have been devoted to researching cardiac remodeling mechanisms, especially m6A RNA methylation, revealing a connection between m6A and cardiovascular diseases. This review summarized the current understanding regarding m6A and elucidated the dynamic modifications of writers, erasers, and readers. Furthermore, we highlighted m6A RNA methylation related to cardiac remodeling and summarized its potential mechanisms. Finally, we discussed the potential of m6A RNA methylation in the treatment of cardiac remodeling.
Key words: RNA modification, m6A RNA methylation, cardiac remodeling, cardiac hypertrophy, heart failure
Introduction
Cardiovascular diseases result from complicated interactions between multiple genetic variations and environmental factors.[1] Common fatal cardiovascular diseases include ischemic heart disease (IHD),[2,3] hypertensive heart disease,[4] cardiomyopathies,[5] and heart failure (HF),[6,7] among others. One of the global health policy goals launched by World Health Organization is to reduce early mortality from noncommunicable diseases by 25% by 2025.[8] Therefore, it is of great significance to study the mechanisms of cardiovascular disease.
Cardiac hypertrophy is an important factor in the pathogenesis of cardiovascular diseases. Physiological cardiac hypertrophy is typically caused by exercise or pregnancy.[9] It is characterized by a slight increase in cardiac mass (10%–20%) and an increase in the length and width of individual cardiomyocytes.[10] However, the heart shape is normal, and this process is advantageous to the cardiac function. Pathological cardiac hypertrophy includes altered cardiac gene expression, cell death, fibrosis, imbalance in Ca2+ transport regulatory proteins, mitochondrial dysfunction, changes in sarcomere structure, and inadequate angiogenesis.[11] The signaling mechanisms that induce these responses contribute to maladaptive heart remodeling and dysfunction, ultimately leading to heart failure. Inhibiting concurrent signaling pathways may also have important therapeutic significance.[9]
RNAs can be modified after transcription, and more than 170 types of RNA posttranscriptional modifications have been discovered to date.[12] An increasing number of inner modifications of eukaryotic epigenetics have been explored in recent studies, including well-known markers named histone tails.[13,14] RNA modifications involve adenosine N6-methyladenosine (m6A), N1- methyladenosine (m1A), 5-methylcytosine (m5C), pseudothiopyrimidine (Ψ), N6, 2’-O-dimethyladenosine (m6Am),[15] the methylation of cytosine to 5-methylcytosine and its oxidation product 5-hydroxymethylcytosine (hm5C),[13] N7-methylguanosine (m7G),[16] N4-acetylcytidine (ac4C),[17] and ribose methylations (Nm).[18] The most extensive modification of mammalian mRNA, N6-methyladenosine (m6A), has aroused widespread interest and scrutiny in the field.[19] Scientists have isolated RNA from mammals and discovered that approximately 1‰–4‰ of adenosine was modified as m6A, which made up about half of the total ribonucleotide methylation.[20] m6A is also found in precursor RNAs (pre-RNAs) and long noncoding RNAs (lncRNAs).[21] Generally, m6A is embedded in the conserved sequence 5’-RRACU-3’,[22] and it mainly occurs in the beginning segment of the 3’-UTR, which is near the translation end codon.[23] Currently, extensive studies are being conducted to investigate the connection between m6A and various diseases.[24, 25, 26] One of the research hotspots is tumorigenesis, but research reports on the relationship between m6A modification and cardiovascular diseases are still limited.[27] This review summarizes m6A RNA methylation and the regulation of RNA stability in cardiac remodeling. It also focuses on how research advances in the relationship between m6A modification and cardiac remodeling provide new ideas for the prevention, early detection, and treatment of cardiac hypertrophy and heart failure.
m6A RNA methylation
RNA modified as m6A refers to the methylation of N6 in the nitrogenous base adenine.[28] There are three key enzymes mediating this process: methyltransferases (writers), demethylases (erasers), and methylation-reading proteins (readers).[29] We summarized their participation in biological dynamic modification and function, as shown in Figure 1.
Figure 1.
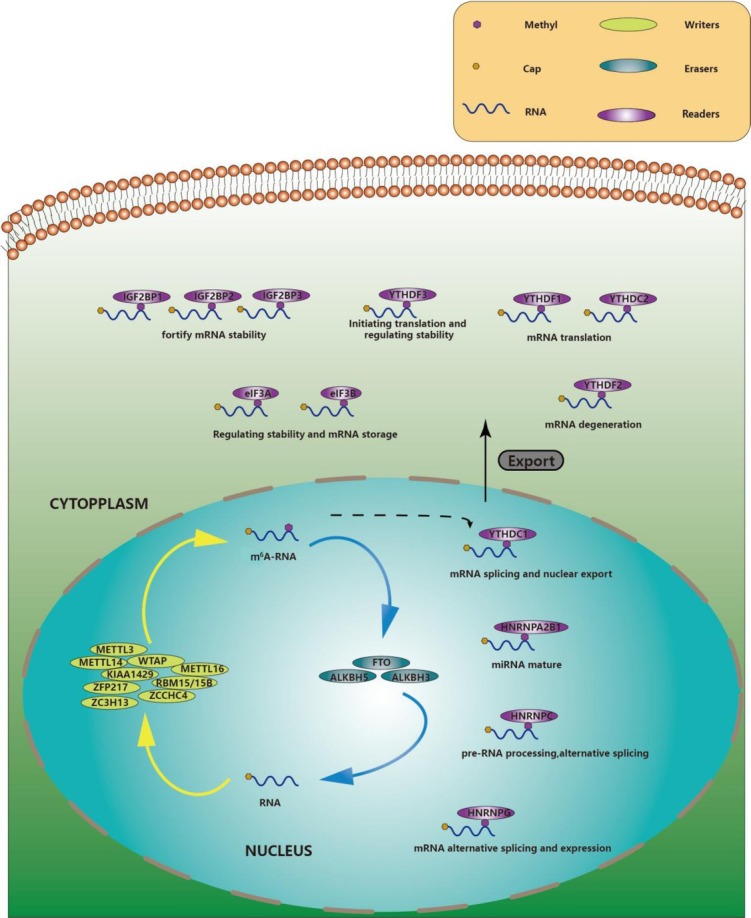
The dynamic modification of m6A. Writers (METTL3, METTL14, WTAP, METTL16, KIAA1429, RBM15/15B, ZFP127, ZC3H13, and ZCCHC4) can identify and methylate the N6 of RNA. Erasers (FTO, ALKBH5, ALKBH3) can catalyze m6A-RNA demethylation. m6A-RNA can be discerned by readers such as YTHDC1 for mRNA splicing. Other readers of m6A are located in the cytoplasm; for instance, YTHDF1, YTHDF2, YTHDF3, YTHDC2, HNRNPA2B1, HNRNPC/G, and IGF2BP1/2/3 are involved in the splicing, processing, translation, and degradation of m6A RNAs. METTL3: methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit; METTL14: methyltransferase 14, N6-adenosine-methyltransferase subunit; METTL16: methyltransferase 16, N6-methyladenosine; WTAP: WT1-associated protein; KIAA1429/VIRMA: vir-like m6A methyltransferase-associated; RBM15/15B: RNA-binding motif protein 15/15B; ZFP127/MKRN3: makorin ring finger protein 3; ZC3H13: zinc finger CCCH-type containing 13; ZCCHC4: zinc finger CCHC-type containing 4; FTO: FTO α-ketoglutarate dependent dioxygenase; ALKBH5: alkB homolog 5, RNA demethylase; ALKBH3: alkB homolog 3, RNA demethylase; YTHDC1/2: YTH domain-containing 1/2; YTHDF1/2/3: YTH N6-methyladenosine RNA binding protein 1/2/3; HNRNPA2B1: heterogeneous nuclear ribonucleoprotein A2/B1; HNRNPC/G: heterogeneous nuclear ribonucleoprotein C/G; IGF2BP1/2/3: insulin-like growth factor 2 mRNA-binding protein 1/2/3.
Writers
“Writers” refer to methyltransferases. Enzymes of this class mainly contain methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), methyltransferase-like 16 (METTL16), Wilms tumor 1-associated protein (WTAP),[30] vir-like m6A methyltransferase associated (KIAA1429/VIRMA),[31] zinc finger protein (ZFP217),[32] RNA-binding motif protein 15 (RBM15),[33] zinc finger CCCH-type containing 13 (ZC3H13),[34] zinc finger CCHC-type containing 4 (ZCCHC4),[35] and other components. They exist in the form of complexes and jointly catalyze the m6A modification of adenine on RNA. A steady formation can be achieved with METTL3 and METTL14,[36] which catalyze the epigenetic modification of m6A RNA in vitro and in vivo.[37] WTAP has no methyltransferase activity but can bind to METTL3 and METTL14.[38] These three proteins are colocalized in nuclear speckles and play important roles in regulating gene expression and alternative splicing.[39] METTL3, an m6A methyltransferase, also plays a key role in autophagy in non–small-cell lung cancer (NSCLC) cells.[40, 41, 42] This process reverses gefitinib resistance through β-elemene. Compared to paired normal tissues, METTL3 expression was increased in lung adenocarcinoma tissues and participated in gefitinib drug tolerance of NSCLC cells. The key genes in autophagy pathways, such as ATG7 and ATG5, are upregulated by METTL3.[43] The upregulation of inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin 1 beta (IL-1β), interleukin 6 (IL-6), and interleukin 18 (IL-18), and the inflammatory proteins TNF receptor associated factor 6 (TRAF6) and nuclear factor of kappa light polypeptide gene enhancer in B cells 1 (NF-κB) was observed in a microglial inflammation model mediated by lipopolysaccharide (LPS). Surprisingly, METTL3 expression levels were also upregulated alongside TRAF6 in this model. The TRAF6-NF-κB pathway is also activated when METTL3 is overexpressed. Therefore, METTL3 activates the TRAF6-NF-κB pathway and accelerates LPS-induced microglial inflammation.[44]
Erasers
Demethylases, also known as the “erasers,” remove the m6A modification of RNA. This process demonstrates the dynamic and reversible modification of m6A. It has been found that demethylases mainly include the genes Fat Mass and Obesity Associated (FTO)[45] and ALKBH5 (alkane hydroxylase homolog 5).[46] These two molecules are part of the α-ketoglutarate-dependent dioxygenase family.[47] m6A demethylation can be catalyzed in an Fe2+- and α-ketoglutarate-dependent manner.[48] A decrease in FTO and ALKBH5 expression was found to be coupled with an increase in m6A modification in mRNA.[45] FTO is associated with human obesity and is considered an obesity susceptibility gene.[49] It is related to body mass index through energy expenditure and intake.[50] Several studies have revealed that FTO is involved in m6A modifications. m6A demethylation catalyzed by FTO can regulate the stability of mRNA, regulate the efficiency of degradation and translation, and control the expression of protein levels. Research has shown that FTO is necessary for the normal development of the central nervous system[51] and the cardiovascular system.[52] This confirms that mutations in the alkb-related dioxygenase family of genes could cause severe polymalformation syndrome.[53] The Alkb family, which is enriched with iron- and 2-oxoglutarate-dependent nucleic acid oxygenase (NAOX), contains a member named ALKBH5. ALKBH5 catalyzes m6A demethylation.[54] According to a report, the double-stranded β-helix domain of ALKBH5 has a mutual effect on the ATP domain of the DEAD (Asp-Glu-Ala-Asp) box polypeptide 3 (DDX3). This domain participates in critical biological processes, such as the cell cycle, metabolism, and apoptosis.[55] Furthermore, it was revealed that both FTO and ALKBH5 are closely associated with single-nucleotide polymorphisms (SNPs).[56] In addition, it was reported that ALKBH3 could demethylate 1-meA and 3-meC; thus, the damage and incomplete methylation of DNA/RNA could be repaired.[57]
Readers
The major function of m6A-reading proteins is to recognize the bases that have been modified by m6A and to regulate the processing, transportation, translation, and stability of the modified RNA.[58] To date, the m6A reading proteins that have been identified include the YT521-B homology (YTH) family (YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2),[59] HNRNP family (HNRNPA2B1, HNRNPC, and HNRNPG),[60, 61, 62] IGF2BP (IGF2BP1, IGF2BP2, and IGF2BP3),[63] and eIF3A/B.[64] The YTHDFs, YTHDC2, IGF2BP, and eIF3A/B proteins are located in the cytoplasm,[65] whereas the YTHDC1 and HNRNP families can be found in the nucleus.[66] YTH N6-methyladenosine RNA-binding protein 2 (YTHDF2) was the first m6A reader to be discovered.[67] YTHDF2 accelerates the degradation of transcripts modified by m6A by directly enlisting the glucose-repressible alcohol dehydrogenase transcriptional effector (CCR4-NOT) deadenylase complex. In contrast, YTH N6-methyladenosine RNA binding protein 1 (YTHDF1) was initially shown to combine with the m6A site near the stop codon and then bind to the translation origination mechanism to enhance the translation efficiency of specific RNA in mammals.[68] YTH N6-methyladenosine RNA-binding protein 3 (YTHDF3) plays a crucial role in the original stages of translation and stability.[69] The YTH domain-containing 1 (YTHDC1) mediates m6A-regulated mRNA splicing,[70] nuclear transport, and gene translation silencing[71] as a nuclear RNA-binding protein.[72] YTH domain-containing 2 (YTHDC2) increases mRNA translation efficiency.[73] HNRNPA2B1 promotes miRNA maturation.[74] Heterogeneous nuclear ribonucleoprotein C (HNRNPC) participates in pre-mRNA processing[75] and alternative splicing.[76] Heterogeneous nuclear ribonucleoprotein G (HNRNPG) regulates alternative splicing and the abundance of target mRNAs.[77] Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), located in the cytoplasm as m6A readers, preferentially recognize m6A-modified mRNAs. They can reinforce mRNA stability and promote translational efficiency.[78]
m6A RNA methylation and pathological cardiac remodeling
Cardiac remodeling includes changes in genomic expression, molecules, cells, and the mesenchyme that clinically manifest as changes in cardiac size, shape, and function after injury.[79] Cardiac remodeling can be categorized into physiological remodeling and pathological remodeling. Physiological cardiac remodeling is often caused by exercise or pregnancy.[80] It manifests as a slight (15%) increase in heart weight and an increase in the length and width of individual cardiomyocytes. The shape of the heart is normal, which is beneficial to its function.[81] However, pathological cardiac hypertrophy can manifest as changes in cardiac gene expression, cell death, fibrosis, Ca2+ transport regulatory protein disorders,[9] mitochondrial dysfunction, metabolic maladjustment, restoration of antenatal gene expression, damaged protein quality assurance mechanisms, changes in sarcomere structure, and lack of angiogenesis.[82] The signaling mechanism inducing these reactions promotes maladaptive cardiac remodeling and dysfunction, eventually leading to heart failure (HF).[83] It has been reported that heart failure (HF) is a chronic disease that inflicts more than 20 million patients worldwide.[84,85] In the past several years, a growing number of studies have revealed the relationship between m6A modifications and cardiovascular diseases, including cardiac hypertrophy,[86] heart failure,[19] atherosclerosis, coronary heart disease,[87] ischemic cardiomyopathy, hypertension, and vascular disease.[88] Therefore, inhibiting concurrent signaling pathways will have important therapeutic significance for interventions in these cardiac diseases.
Cardiac hypertrophy
In the presence of hemodynamic stress, cardiomyocytes adapt by becoming hypertrophic. This reaction plays a reparative role in improving cardiac function, decreasing the strain on the ventricular wall and oxygen expenditure.[89] Cardiac hypertrophy can be divided into two types: physiological and pathological. Physiological cardiac hypertrophy, which can maintain normal morphology and play a beneficial role in the heart, mostly results from exercise training or pregnancy.[90] In contrast, pathological cardiac hypertrophy causes many cardiovascular pathophysiological changes, such as ventricular remodeling, fibrosis, and cardiac gene expression alteration.[91]
Hinger et al.[92] found an increase in m6A content in human heart failure samples but showed a preserved distribution. The protein level of METTL3 was increased, and that of FTO was decreased, while there was no change in ALKBH5 levels. Afterward, human and hypertrophic neonatal rat ventricular myocytes obtained from heart failure samples were used to investigate whether there was conserved specificity in m6A events in cardiomyocytes across species. Their results showed stress-responsive m6A-transcripts between rats and humans were conservative. In both human hearts and rat cardiomyocytes, Western blotting showed that coronin 6 (CORO6) levels were reduced, whereas the expression of RE1 silencing transcription factor (REST) was increased. However, the mRNA levels of these two genes remained unaffected. Furthermore, they detected m6A content in both human heart failure samples and hypertrophic cardiomyocytes. They found that REST expression was increased, while CORO6 had greater m6A content in nonfailing heart and normal cardiomyocytes. Upon upregulation of METTL3, the translation levels of REST and CORO6 increased. Hence, posttranscriptional modifications may play a direct role in gene expression in cardiomyocytes.
Gao et al.[93] revealed a piRNA (PIWI-interacting RNA) named CHAPIR (cardiac-hypertrophy-associated piRNA), which regulates cardiac hypertrophy. Overexpression of CHAPIR using a mimic aggravated pathological hypertrophic response in a TAC mouse model, while the downregulation of CHAPIR notably attenuated cardiac hypertrophy and recovered cardiac function. In terms of mechanism, METTL3 combined with CHAPIR–PIWIL4 complexes suppressed Parp10 mRNA m6A methylation. The mRNA and protein expression levels of poly(ADP-ribose) polymerase family member 10 (PARP10) increased, which promoted mono-ADP-ribosylation of GSK3β and suppressed its kinase activity.[94] This process increased nuclear NFATC4 levels and led to the progression of pathological hypertrophy. Therefore, targeting the CHAPIRMETTL3-PARP10-NFATC4 signaling axis could be a therapeutic mechanism for improving cardiac hypertrophy.
Dorn et al.[95] discovered that the extent of m6A methylation increases in response to hypertrophic stimulation. The growth of hypertrophic cardiomyocytes was fully abolished upon stimulation, and they did not undergo hypertrophy when METTL3 was suppressed in vitro. However, the overexpression of METTL3 can cause spontaneous and compensatory hypertrophy. In vivo, cardiac-specific METTL3-knockout mice showed cardiac remodeling and heart failure followed by cardiac homeostasis disorders, whereas increased METTL3 levels caused cardiac hypertrophy.
Kmietczyk et al.[96] showed that the mechanism of m6A RNA methylation is dynamic and effective in cardiomyocytes undergoing pressure[97] and regulates gene expression and cellular proliferation in the heart. They found that METTL3 and FTO could participate in m6A RNA methylation by influencing transcript stability and regulating translational efficiency. In an in vitro model of neonatal rat cardiomyocytes (NRCM), the knockdown of METTL3 reduced m6A levels[98] and increased the cell size and the expression of the hypertrophic markers ANP and BNP. However, FTO-KO mice exhibited enhanced m6A levels and weakened NRCM hypertrophy. In an in vivo model of AAV9-mediated METTL3 overexpression in C57Bl6/N mice and TAC mice, METTL3 overexpression shrank the cross-sectional area of the myocytes and suppressed pathological hypertrophic cellular growth. Nevertheless, how METTL3 and FTO regulate gene expression and cellular growth and which specific target genes play an essential role in cardiomyocyte hypertrophy are still under study.
Heart failure
Berulava et al.[99] discovered that the level of m6A RNA methylation decreases during heart failure. The mRNA level of calmodulin 1 (calm1) remained unchanged, while the protein expression level of calm1 was reduced. In other words, m6A RNA methylation levels influenced protein levels rather than mRNA levels. m6A RNA methylation is directly proportional to ribosomal occupancy, indicating increased protein levels of hypermethylated transcripts and decreased protein levels of hypomethylated transcripts. A worsened cardiac phenotype in the FTO-knockout mice model after TAC was also observed, as the ejection fraction was reduced and the degree of dilatation was increased.
Mathiyalagan et al.[100] discovered that the demethylase FTO was associated with cardiac function during cardiac remodeling and repair. They detected reduced FTO expression levels in failing mammalian hearts and hypoxic cardiomyocytes; therefore, m6A RNA methylation increased. Sarco/endoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) is a contractile protein that exhibits less stability and lower efficiency to regulate translation when hypermethylated, eventually resulting in cardiomyocyte contractile function. However, FTO overexpression in human myocytes led to SERCA2a demethylation. Furthermore, cardiac contractile function improved with an increase in SERCA2a expression. They also found that FTO overexpression reduced fibrosis and promoted angiogenesis in mouse models of myocardial infarction. Hence, this mechanism provides novel insights into cardiac remodeling and repair.
Research progress on new techniques in detecting m6A RNA methylation
Researchers are actively exploring the role of m6A modification-related molecules in cardiovascular disease; however, many problems and challenges still need to be resolved. For example, transcriptome-wide mapping used in m6A can help us better understand catalog m6A targets and reveal the underlying epigenetic modification mechanisms. In 2012, Nature and Cell published a method for the whole transcriptome sequencing of m6A modification via m6A-specific antibody enrichment (MeRIP-seq or m6A-seq);[19,101] however, MeRIP-seq has an insufficient resolution (about 100 nt). However, insurmountable weaknesses in principle, such as low repeatability, large sample demand, and cumbersome operation, have caused significant problems in m6A research in recent years.
In 2015, Nature Methods proposed a new method for the high-resolution detection of the localization of N6-methyladenosine in eukaryotic RNA called m6A single-nucleotide resolution cross-linking and immunoprecipitation (miCLIP).[102] Mutations would occur when the cross-linking of the RNA-m6A antibody-binding sites is reverse-transcribed. The mutated sequences had unique features (e.g., C-T transition or truncation) that could pinpoint m6A. miCLIP can perform high-resolution detection of individual m6A residues and m6A cluster analysis of the total RNA. In particular, miCLIP is suitable for small nucleolar RNA (snoRNA).
In a recent study, Zhang et al. published a research paper titled “Single-base mapping of m6A by an antibody-independent method,”[103] which described a new principle of m6A detection technology named m6A-REF-seq (m6A-sensitive RNA-endoribonuclease-facilitated sequencing). This technology used the sensitivity of the newly discovered RNA endonuclease to m6A, which eliminated the dependence of traditional methods on antibodies and achieved accurate detection of m6A across the transcriptome.[104] New methods must be implemented in the m6A field with the development of better scientific methods and technological advances. However, whether other types of m6A modification have some links to cardiac remodeling is still to be discovered. Finally, drugs targeting m6A are promising for the clinical treatment of cardiovascular diseases.
We hope that consistent studies in this field can further deepen our understanding of the processes surrounding heart failure and approach the reality of discovering new treatments, thereby improving the quality of life of patients with heart failure.
Conclusions and future perspectives
The most abundant RNA modification in RNA epigenetics is m6A methylation.[105] m6A methylation studies have currently gained significant popularity in scientific research.[106] In this review, we focused on cardiac remodeling, summarized the classification of m6A RNA methylases, and discussed their dynamic modification (Figure 1) in detail. Furthermore, we surveyed m6A RNA modifications in cardiac remodeling, including cardiac hypertrophy and heart failure (Table 1). The mechanisms regarding the development of cardiac hypertrophy are intricate; however, what we currently know is just the tip of the iceberg, and further research is needed to elucidate the epigenetic mechanisms underlying heart failure.[107] In the past few years, we have opened new areas for advancing the known mechanisms and identifying the unknown pathways involved in cardiac remodeling. Heart failure is still difficult to cure in the clinical setting and its prevalence rate increases with age.[108] m6A has potential applications in the diagnosis and treatment of heart failure. Research focus should be placed on the abnormal expression of some m6A enzymes, such as METTL3 and FTO, because they are related to cardiac hypertrophy or heart failure since the early detection of these abnormalities will help in the early diagnosis of heart failure. It is also possible that we interfere with the expression of methylases, such as METTL3 and FTO, to prevent heart failure.
Table 1.
m6A and cardiac remodeling
| Types of cardiac remodeling | Effector | Type of effector | Expression | Target genes | Mechanism | Reference |
|---|---|---|---|---|---|---|
| Cardiac | METTL3 | Writer | Upregulation | REST | Protein expression was higher in condition of greater | [92] |
| hypertrophy | FTO | Eraser | Downregulation | CORO6 | m6A content, and overexpression of METTL3 was sufficient to positively affect the translation of REST and CORO6 | |
| METTL3 | Writer | Reduce activity of METTL3 | PARP10 | CHAPIR-PIWIL4 → METTL3 → m6A-PARP10 → PARP10 (mRNA and protein) → mono-ADP-ribosylation of GSK3β → GSK3β kinase activity → NFATC4 → pathological hypertrophy | [93] | |
| METTL3 | Writer | Upregulation | MAP3K6/ | In vitro: METTL3 → prevent pathological hypertrophy | [95] | |
| MAP4K5/ MAPK14/ Nppa/Nppb | METTL3 → spontaneous and compensate hypertrophy In vivo: METTL3-KO → remodeling and heart failure → cardiac homeostasis disorder METTL3 → cardiac hypertrophy | |||||
| METTL3 | Writer | Downregulation | Unknown | In vitro: METTL3-KO → m6A level → cell size and level of Nppa/Nppb; FTO-KO → m6A level → hypertrophy of NRCM In vivo: METTL3-overexpression → myocytes cross-sectional area → pathological hypertrophic cellular growth | [96] | |
| Heart failure | FTO | Eraser | Downregulation | Calm1 | Calm1 protein expression regulation in heart failure occurs partially only on translational level and without changes in DNA to RNA transcription | [99] |
| FTO | Eraser | Downregulation | SERCA2a | In failing mammalian hearts and hypoxic cardiomyocyte, FTO SERCA2a mRNA is hypermethylated cardiomyocytes contractile function | [100] |
METTL3: methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit; REST: RE1 silencing transcription factor; CORO6: coronin 6; PARP10: poly (ADP-ribose) polymerase family member 10; MAP3K6/5/14: mitogen-activated protein kinase kinase kinase 6/5/14; Nppa: natriuretic peptide A; Nppb: natriuretic peptide B; FTO: FTO α-ketoglutarate-dependent dioxygenase; Calm1: calmodulin 1; SERCA2a: sarco/endoplasmic reticulum Ca2+-ATPase.
Footnotes
Source of Funding
This study was funded by the National Natural Science Foundation of China (No. 8197021725) and the Shenyang Science and Technology Project (No. 19-112-4-003).
Conflict of Interest
Yingxian Sun is an Associate-Editor-in-Chief of the journal. The article was subject to the journal's standard procedures, and peer review was handled independently of this editor and his research groups.
References
- 1.De Backer G. Epidemiology and prevention of cardiovascular disease: Quo vadis? Eur J Prev Cardiol. 2017;24:768–72. doi: 10.1177/2047487317691875. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Chen J, Cowan DB, Wang DZ. Noncoding RNAs in cardiac regeneration: Mechanism of action and therapeutic potential. Semin Cell Dev Biol. 2021;118:150–62. doi: 10.1016/j.semcdb.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duan B. Concise Review: Harnessing iPSC-derived Cells for Ischemic Heart Disease Treatment. J Transl Intern Med. 2020;8:20–5. doi: 10.2478/jtim-2020-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saliba LJ, Maffett S. Hypertensive Heart Disease and Obesity: A Review. Heart Fail Clin. 2019;15:509–17. doi: 10.1016/j.hfc.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: An Overview. Int J Mol Sci. 2021;22:7722. doi: 10.3390/ijms22147722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gedela M, Khan M, Jonsson O. Heart Failure. SD Med. 2015;68:403–5. 407–9. [PubMed] [Google Scholar]
- 7.Li C, Wang DW, Zhao C. Cardiovascular Involvement in Patients with 2019 Novel Coronavirus Disease. J Transl Intern Med. 2021;9:152–60. doi: 10.2478/jtim-2021-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ Res. 2017;121:677–94. doi: 10.1161/CIRCRESAHA.117.308903. et al. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 10.Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. 2020;98:74–84. doi: 10.1139/cjpp-2019-0566. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Liang Y, Zhu Y, Zhang Y, Bei Y. Noncoding RNAs in Cardiac Hypertrophy. J Cardiovasc Transl Res. 2018;11:439–49. doi: 10.1007/s12265-018-9797-x. [DOI] [PubMed] [Google Scholar]
- 12.Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:303–7. doi: 10.1093/nar/gkx1030. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu M, Ding Q, Lin Z, Chen X, Chen S, Zhu Y. New insights of epigenetics in vascular and cellular senescence. J Transl Intern Med. 2021;9:239–48. doi: 10.2478/jtim-2021-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma CJ, Ding JH, Ye TT, Yuan BF, Feng YQ. AlkB Homologue 1 Demethylates N(3)-Methylcytidine in mRNA of Mammals. ACS Chem Biol. 2019;14:1418–25. doi: 10.1021/acschembio.8b01001. [DOI] [PubMed] [Google Scholar]
- 16.Cockman E, Anderson P, Ivanov P. TOP mRNPs: Molecular Mechanisms and Principles of Regulation. Biomolecules. 2020;10:969. doi: 10.3390/biom10070969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karthiya R, Wasil SM, Khandelia P. Emerging role of N4-acetylcytidine modification of RNA in gene regulation and cellular functions. Mol Biol Rep. 2020;47:9189–99. doi: 10.1007/s11033-020-05963-w. [DOI] [PubMed] [Google Scholar]
- 18.Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet. 2021;22:119–31. doi: 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- 19.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613. doi: 10.1016/j.biopha.2019.108613. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606. doi: 10.1016/j.ccell.2017.02.013. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Wu K, Quan W, Yu L, Chen S, Cheng C. The dynamics of FTO binding and demethylation from the m6A motifs. RNA Biol. 2019;16:1179–89. doi: 10.1080/15476286.2019.1621120. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6. doi: 10.1038/nature11112. et al. [DOI] [PubMed] [Google Scholar]
- 24.Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12:121. doi: 10.1186/s13045-019-0805-7. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176. doi: 10.1186/s12943-019-1109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong J, Flavell RA, Li HB. RNA m6A modification and its function in diseases. Front Med. 2018;12:481–9. doi: 10.1007/s11684-018-0654-8. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Wei X, Yi X, Jiang DS. RNA Modification by m6A Methylation in Cardiovascular Disease. Oxid Med Cell Longev. 2021;2021:8813909. doi: 10.1155/2021/8813909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li LJ, Fan YG, Leng RX, Pan HF, Ye DQ. Potential link between m6A modification and systemic lupus erythematosus. Mol Immunol. 2018;93:55–63. doi: 10.1016/j.molimm.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Zhang W, Qian Y, Jia G. The detection and functions of RNA modification m6A based on m6A writers and erasers. J Biol Chem. 2021;297:100973. doi: 10.1016/j.jbc.2021.100973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichel M, Köster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol. 2019;11:899–910. doi: 10.1093/jmcb/mjz085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu W, Wang JZ, Wei JF, Lu C. Role of m6A methyltransferase component VIRMA in multiple human cancers (Review) Cancer Cell Int. 2021;21:172. doi: 10.1186/s12935-021-01868-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, Zhao Y, Wu R, Jiang Q, Cai M, Bi Z. ZFP217 regulates adipogenesis by controlling mitotic clonal expansion in a METTL3-m6A dependent manner. RNA Biol. 2019;16:1785–93. doi: 10.1080/15476286.2019.1658508. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20. doi: 10.1038/nature12730. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415–29. doi: 10.1101/gad.309146.117. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R. N(6-) Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88–94. doi: 10.1038/s41589-018-0184-3. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5. doi: 10.1038/nchembio.1432. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. 2016;63:306–17. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selberg S, Žusinaite E, Herodes K, Seli N, Kankuri E, Merits A. HIV Replication Is Increased by RNA Methylation METTL3/METTL14/ WTAP Complex Activators. ACS Omega. 2021;6:15957–63. doi: 10.1021/acsomega.1c01626. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89. doi: 10.1038/cr.2014.3. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Liu S, Zhao T, Dang C. METTL3‑mediated m6A modification of Bcl-2 mRNA promotes non‑small cell lung cancer progression. Oncol Rep. 2021;46:163. doi: 10.3892/or.2021.8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li M, Wang Q, Zhang X, Yan N, Li X. CircPUM1 promotes cell growth and glycolysis in NSCLC via upregulating METTL3 expression through miR-590-5p. Cell Cycle. 2021;20:1279–94. doi: 10.1080/15384101.2021.1934625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin D, Guo J, Wu Y, Du J, Yang L, Wang X. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2021;14:32. doi: 10.1186/s13045-021-01048-8. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S, Li Q, Li G, Zhang Q, Zhuo L, Han X. The mechanism of m6A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020;11:969. doi: 10.1038/s41419-020-03148-8. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen L, Sun W, Xia D, Wang Y, Li J, Yang S. The m6A methyltransferase METTL3 promotes LPS-induced microglia inflammation through TRAF6/NF-κB pathway. Neuroreport. 2022;33:243–51. doi: 10.1097/WNR.0000000000001550. [DOI] [PubMed] [Google Scholar]
- 45.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7. doi: 10.1038/nchembio.687. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu G, Yan Y, Cai Y, Peng B, Li J, Huang J. ALKBH1-8 and FTO: Potential Therapeutic Targets and Prognostic Biomarkers in Lung Adenocarcinoma Pathogenesis. Front Cell Dev Biol. 2021;9:633927. doi: 10.3389/fcell.2021.633927. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huo FC, Zhu ZM, Pei DS. N(6) -methyladenosine (m(6) A) RNA modification in human cancer. Cell Prolif. 2020;53:e12921. doi: 10.1111/cpr.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Hambly BD, McLachlan CS. FTO associations with obesity and telomere length. J Biomed Sci. 2017;24:65. doi: 10.1186/s12929-017-0372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuzbashian E, Asghari G, Chan CB, Hedayati M, Safarian M, Zarkesh M. The association of dietary and plasma fatty acid composition with FTO gene expression in human visceral and subcutaneous adipose tissues. Eur J Nutr. 2021;60:2485–94. doi: 10.1007/s00394-020-02422-x. et al. [DOI] [PubMed] [Google Scholar]
- 51.Pan T, Wu F, Li L, Wu S, Zhou F, Zhang P. The role m6A RNA methylation is CNS development and glioma pathogenesis. Mol Brain. 2021;14:119. doi: 10.1186/s13041-021-00831-5. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu C, Mou S, Pan C. The FTO gene rs9939609 polymorphism predicts risk of cardiovascular disease: a systematic review and meta-analysis. PLoS One. 2013;8:e71901. doi: 10.1371/journal.pone.0071901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–11. doi: 10.1016/j.ajhg.2009.06.002. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou B, Han Z. Crystallization and preliminary X-ray diffraction of the RNA demethylase ALKBH5. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:1231–4. doi: 10.1107/S1744309113024858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah A, Rashid F, Awan HM, Hu S, Wang X, Chen L. The DEAD-Box RNA Helicase DDX3 Interacts with m6A RNA Demethylase ALKBH5. Stem Cells Int. 2017;2017:8596135. doi: 10.1155/2017/8596135. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piette ER, Moore JH. Identification of epistatic interactions between the human RNA demethylases FTO and ALKBH5 with gene set enrichment analysis informed by differential methylation. BMC Proc. 2018;12:59. doi: 10.1186/s12919-018-0122-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271. doi: 10.1038/srep42271. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patil DP, Pickering BF, Jaffrey SR. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018;28:113–27. doi: 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–9. doi: 10.1038/nchembio.1654. et al. [DOI] [PubMed] [Google Scholar]
- 60.Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299–308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013;152:453–66. doi: 10.1016/j.cell.2012.12.023. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Ż Pan JN. Regulation of Co-transcriptional Pre-mRNA Splicing by m6A through the Low-Complexity Protein hnRNPG. Mol Cell. 2019;76:70–81. doi: 10.1016/j.molcel.2019.07.005. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang X, Zhang H, Guo X, Zhu Z, Cai H, Kong X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol Oncol. 2018;11:88. doi: 10.1186/s13045-018-0628-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi H, Chai P, Jia R, Fan X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol Cancer. 2020;19:78. doi: 10.1186/s12943-020-01194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu N, Pan T. N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol. 2016;23:98–102. doi: 10.1038/nsmb.3162. [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–24. doi: 10.1038/s41422-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JY, Lu AQ. The biological function of m6A reader YTHDF2 and its role in human disease. Cancer Cell Int. 2021;21:109. doi: 10.1186/s12935-021-01807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–99. doi: 10.1016/j.cell.2015.05.014. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–7. doi: 10.1038/cr.2017.10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu S, Zhang S, Wu X, Zhou X. m6A RNA Methylation in Cardiovascular Diseases. Mol Ther. 2020;28:2111–9. doi: 10.1016/j.ymthe.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73. doi: 10.1038/nature19342. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roundtree IA, He C. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Trends Genet. 2016;32:320–1. doi: 10.1016/j.tig.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27. doi: 10.1038/cr.2017.99. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cieniková Z, Damberger FF, Hall J, Allain FH, Maris C. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J Am Chem Soc. 2014;136:14536–44. doi: 10.1021/ja507690d. [DOI] [PubMed] [Google Scholar]
- 76.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63. doi: 10.1093/nar/gkx141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:28595. doi: 10.1038/s41556-018-0045-z. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 80.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 81.Bernardo BC, McMullen JR. Molecular Aspects of Exercise-induced Cardiac Remodeling. Cardiol Clin. 2016;34:515–30. doi: 10.1016/j.ccl.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 82.Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond) 2017;131:2319–45. doi: 10.1042/CS20171167. et al. [DOI] [PubMed] [Google Scholar]
- 83.Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–38. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 84.Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J. 2014;35:2950–9. doi: 10.1093/eurheartj/ehu299. [DOI] [PubMed] [Google Scholar]
- 85.Kapiloff MS, Emter CA. The cardiac enigma: current conundrums in heart failure research. F1000Res. 2016;5:F1000. doi: 10.12688/f1000research.7278.1. Faculty Rev-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The AlkB Family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J Biol Chem. 2015;290:20734–42. doi: 10.1074/jbc.R115.656462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu Y, Zhan S, Xu Y, Gao X. RNA modifications in cardiovascular diseases, the potential therapeutic targets. Life Sci. 2021;278:119565. doi: 10.1016/j.lfs.2021.119565. [DOI] [PubMed] [Google Scholar]
- 88.Paramasivam A, Vijayashree Priyadharsini J, Raghunandhakumar S. N6-adenosine methylation (m6A): a promising new molecular target in hypertension and cardiovascular diseases. Hypertens Res. 2020;43:153–4. doi: 10.1038/s41440-019-0338-z. [DOI] [PubMed] [Google Scholar]
- 89.Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:h8–h16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- 90.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 91.Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–62. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 92.Hinger SA, Wei J, Dorn LE, Whitson BA, Janssen PML, He C. Remodeling of the m6A landscape in the heart reveals few conserved post-transcriptional events underlying cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2021;151:46–55. doi: 10.1016/j.yjmcc.2020.11.002. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao XQ, Zhang YH, Liu F, Ponnusamy M, Zhao XM, Zhou LY. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N(6)-methyladenosine methylation of Parp10 mRNA. Nat Cell Biol. 2020;22:1319–31. doi: 10.1038/s41556-020-0576-y. et al. [DOI] [PubMed] [Google Scholar]
- 94.Huang B, Ding C, Zou Q, Wang W, Li H. Cyclophosphamide Regulates N6-Methyladenosine and m6A RNA Enzyme Levels in Human Granulosa Cells and in Ovaries of a Premature Ovarian Aging Mouse Model. Front Endocrinol (Lausanne) 2019;10:415. doi: 10.3389/fendo.2019.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M. The N(6)-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation. 2019;139:533–45. doi: 10.1161/CIRCULATIONAHA.118.036146. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kmietczyk V, Riechert E, Kalinski L, Boileau E, Malovrh E, Malone B. m6A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci Alliance. 2019;2:e201800233. doi: 10.26508/lsa.201800233. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Y, Kong Y, Fan W, Tao T, Xiao Q, Li N. Principles of RNA methylation and their implications for biology and medicine. Biomed Pharmacother. 2020;131:110731. doi: 10.1016/j.biopha.2020.110731. et al. [DOI] [PubMed] [Google Scholar]
- 98.Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124. doi: 10.1038/s41419-017-0129-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail. 2020;22:54–66. doi: 10.1002/ejhf.1672. et al. [DOI] [PubMed] [Google Scholar]
- 100.Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N. FTO-Dependent N(6)-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation. 2019;139:518–532. doi: 10.1161/CIRCULATIONAHA.118.033794. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D. m6A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273–6. doi: 10.1038/nature23883. et al. [DOI] [PubMed] [Google Scholar]
- 102.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–72. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Z, Chen LQ, Zhao YL, Yang CG, Roundtree IA, Zhang Z. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5:eaax0250. doi: 10.1126/sciadv.aax0250. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao Y, Liu X, Wu B, Wang H, Xi F, Kohnen MV. Quantitative profiling of N(6)-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biol. 2021;22:22. doi: 10.1186/s13059-020-02241-7. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meyer KD, Jaffrey SR. Rethinking m6A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol. 2017;33:319–42. doi: 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu ZX, Li LM, Sun HL, Liu SM. Link Between m6A Modification and Cancers. Front Bio Biotechnol. 2018;6:89. doi: 10.3389/fbioe.2018.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morissens M, Besse-Hammer T, Azerad MA, Efira A, Rodriguez JC. Evaluation of Cardiac Function in Patients with Sickle Cell Disease with Left Ventricular Global Longitudinal Strain. J Transl Intern Med. 2020;8:41–7. doi: 10.2478/jtim-2020-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu S, Qiu Y, Tao J. The challenges and optimization of cell-based therapy for cardiovascular disease. J Transl Intern Med. 2021;9:234–8. doi: 10.2478/jtim-2021-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]