

Summary
We assessed the concordance between immunohistochemistry (IHC) and gene expression profiling (GEP) for determining diffuse large B-cell lymphoma (DLBCL) cell of origin (COO) in the phase III PHOENIX trial of rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) with or without ibrutinib. Among 910 of 1114 screened patients with non-germinal centre B cell-like (non-GCB) DLBCL by IHC, the concordance with GEP for non-GCB calls was 827%, with 691 (759%) identified as activated B cell-like (ABC), and 62 (68%) as unclassified. Among 746 of 837 enrolled patients with verified non-GCB DLBCL by IHC, the concordance with GEP was 828%, with 567 (760%) identified as ABC and 51 (68%) unclassified; survival outcomes were similar regardless of COO or treatment, whereas among patients with ABC DLBCL aged <60 years, the overall and event-free survival were substantially better with ibrutinib versus placebo plus R-CHOP [hazard ratio (HR) 0365, 95% confidence interval (CI) 0147–0909, P = 00305; HR 0561, 95% CI 0326–0967, P = 00348, respectively]. IHC and GEP showed high concordance and consistent survival outcomes among tested patients, indicating centralised IHC may be used to enrich populations for response to ibrutinib plus R-CHOP.
Keywords: concordance, diffuse large B-cell lymphoma, gene expression profiling, IHC, subtyping
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of lymphoma, representing ~40% of lymphoma cases worldwide.1 It can be classified into distinct molecular subtypes based on the cell of origin (COO).2 Gene expression profiling (GEP) methods classify DLBCL into germinal centre B cell-like (GCB), activated B cell-like (ABC) and unclassified groups.2,3 Immunohistochemistry (IHC)-based methods are widely used and provide an approximation of GEP by dichotomising DLBCL into GCB and non-GCB subtypes. In IHC analysis by the ‘Hans’ algorithm, a non-GCB classification requires negative stains for cluster of differentiation 10 [CD10, also known as membrane metallo-endopeptidase [MME]) and B-cell lymphoma 6 (BCL-6), or a positive stain for multiple myeloma oncogene 1 (MUM1, also known as interferon regulatory factor 4 [IRF4]) if the BCL-6 stain is positive.4 More recently, four prominent genetic subtypes in DLBCL designated MCD, BN2, N1 and EZB have been identified.5 Additional genetic analyses identified five DLBCL subsets, which generally overlapped with the aforementioned subtypes and included ABC DLBCL subsets of extra-follicular/marginal zone origin; two subsets of GCB DLBCL; and an ABC/GCB-independent group.6 Nevertheless, all these recently identified subtypes are generally segregated into one of three GEP defined groups, highlighting the persistent clinical relevance of COO classification.
Many common genetic aberrations in GCB DLBCL were not reported in ABC DLBCL.7 In clinical studies, patients with ABC DLBCL showed poorer outcomes than those with GCB DLBCL with the standard front-line DLBCL treatment rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP).8 In more recent phase I and II trials, novel agents like ibrutinib (a Bruton tyrosine kinase inhibitor) or lenalidomide (an immunomodulatory drug) demonstrated preferential activity in patients with relapsed/refractory ABC DLBCL or in previously untreated patients with non-GCB DLBCL, respectively, versus those with GCB DLBCL.9,10 These data support the predictive significance of DLBCL subtyping, necessitating the development of rapid and accurate COO identification methods.
Microarray-based GEP was developed to subtype DLBCL COO.11 GEP is not routinely used in the clinical setting due to cost and technical issues, including a slow turnaround time of 2–3 weeks.2,7,12 Therefore, IHC remains widely adopted in clinical practice, although inter-laboratory variance limits comparison across sites,13,14 warranting the use of more reliable centralised assays. Despite the development of multiple IHC assays using different combinations of protein markers,13 the Hans method is most widely used in diagnostic clinical laboratories because of its simplicity and validated prognostic value.4,12 Several recently emerged simplified GEP-based methods can reproduce the original comprehensive microarray-based classification, which include the Lymphoma Subtyping Test (LST) assay (NanoString, Seattle, WA, USA) and the next-generation sequencing-based EdgeSeq DLBCL Cell-of-Origin Assay (HTG Molecular Diagnostics, Tucson, AZ, USA).12 Given these options, the optimal assay for selecting patients with poor prognosis DLBCL for clinical trials remains undetermined.
PHOENIX (NCT01855750) was a randomised, placebo-controlled phase III trial comparing the efficacy and safety of ibrutinib plus R-CHOP and placebo plus R-CHOP in previously untreated patients with IHC-selected non-GCB DLBCL.15 The trial did not meet its primary endpoint: the addition of ibrutinib to R-CHOP did not improve event-free survival (EFS) in the intent-to-treat (ITT) or ABC (by GEP) population.15 In an exploratory analysis, an improvement in EFS, progression-free survival (PFS) and overall survival (OS) with ibrutinib plus R-CHOP was observed in patients aged <60 years, but not in those aged ≥60 years, likely because of increased toxicity in the older population limiting completion of treatment.15 The current pre-specified analysis aims to assess the concordance between IHC and GEP by HTG EdgeSeq DLBCL Cell-of-Origin Assay for COO subtyping, both performed by central laboratory, and to investigate patient outcomes with COO GEP, which was tested following non-GCB selection by IHC, in the PHOE-NIX trial.
Patients and methods
Trial design and patients
A detailed methodology for this trial is published elsewhere.15 Briefly, patients aged ≥18 years who had previously untreated non-GCB DLBCL (n = 838) based on centralised IHC results at screening were randomly assigned at a 1:1 ratio to receive ibrutinib (560 mg daily) plus R-CHOP or placebo plus R-CHOP for six or eight cycles per local guidelines. The primary endpoint was investigator-assessed EFS in the ITT or ABC (by GEP) population. Secondary endpoints included PFS, complete response rate and OS in the ITT population.15
Cell of origin analysis and concordance evaluation
The COO analysis was performed as stated in the Supplementary Information. Concordance between IHC and GEP assays was estimated with reference to IHC data, as described in the Supplementary Information.
Validation of GEP prognostic value and prediction of treatment effect
The prognostic value of the GEP assay was assessed using probability scores derived from the expression levels of COO signature genes, housekeeping markers and positive controls16 using methods previously described, as stated in the Supplementary Information.
Survival outcomes analysis
Survival outcomes were estimated by Kaplan–Meier analysis with log-rank P value, as described in the Supplementary Information. The hazard ratios (HRs) and their Wald’s confidence intervals (CIs) were estimated using Cox proportional hazards regression modelling.
Results
Trial samples
Of 1490 patients screened, valid IHC results were obtained from 1338 (898%) tissue samples and showed 268 GCB and 1070 non-GCB cases. These samples were further tested by GEP with 1114 samples (833%) yielding valid GEP results. Figure S1 shows the percentage and concordance of screened and enrolled IHC-selected patients in each GEP subpopulation.
Concordance in screened patients
Of 1114 screened samples with evaluable results for both IHC and GEP, 910 were identified as non-GCB by IHC. Among these, 691 (759%) were identified as ABC, 62 (68%) unclassified and 157 (173%) GCB by GEP. These results led to a concordance of 827% (95% CI 802–851) for non-GCB calls between IHC and GEP (Table I). Of the 204 samples identified by IHC as GCB, 145, 49 and 10 were called GCB, ABC and unclassified by GEP, respectively, leading to a concordance of 711% (95% CI 645–769) for GCB calls between assays (Table I). The overall concordance between the assays was 806% (95% CI 782–828) (Table I).
Table I.
Concordance between IHC and GEP in screened patients (N = 1114).
| GEP assay, n (%) |
|||||
|---|---|---|---|---|---|
| ABC | Unclassified | GCB | Total | ||
|
| |||||
| IHC assay, n (%) | Non-GCB | 691 (75·9) | 62 (6·8) | 157 (17·3) | 910 (100·0) |
| GCB | 49 (24·0) | 10 (4· 9) | 145 (71·1) | 204 (100·0) | |
| Non-GCB concordance, % (95% CI) = 82·7 (80·2–85·1)* | |||||
| GCB concordance, % (95% CI) = 71·1 (64·5–76·9)* | |||||
| Overall concordance, % (95% CI) = 691 + 62 + 145/1114 = 80·6 (78·2–82·8)* | |||||
ABC, activated B cell-like; CI, confidence interval; GCB, germinal centre B cell-like; GEP, gene expression profiling; IHC, immunohistochemistry.
95% CI based on score method.
Concordance in enrolled patients
The PHOENIX trial enrolled 838 patients with non-GCB DLBCL, among whom one actually was GCB by IHC during further verification.15 Among 837 enrolled patients with non-GCB IHC-verified subtype, 746 (891%) samples were evaluable by GEP (Table II). All GEP-evaluable samples were CD10-negative because of the non-GCB pre-selection by IHC. Of these, 128 (172%), 567 (760%) and 51 (68%) were identified as CD10-negative GCB, ABC and unclassified by GEP, respectively (Fig 1). This led to a non-GCB concordance of 828%.
Table II.
Concordance between IHC and GEP in GEP-evaluable enrolled patients (N = 746).
| GEP assay, n (%) |
|||||
|---|---|---|---|---|---|
| ABC | Unclassified | GCB | Total | ||
|
| |||||
| IHC assay, n (%) | Non-GCB | 567 (76·0) | 51 (6·8) | 128 (17·2) | 746 (100·0) |
| Non-GCB concordance, % (95% CI) = 82·8 (80·0–85·4)* | |||||
ABC, activated B cell-like; CI, confidence interval; GCB, germinal centre B cell-like; GEP, gene expression profiling; IHC, immunohistochemistry.
95% CI based on score method.
Fig 1.
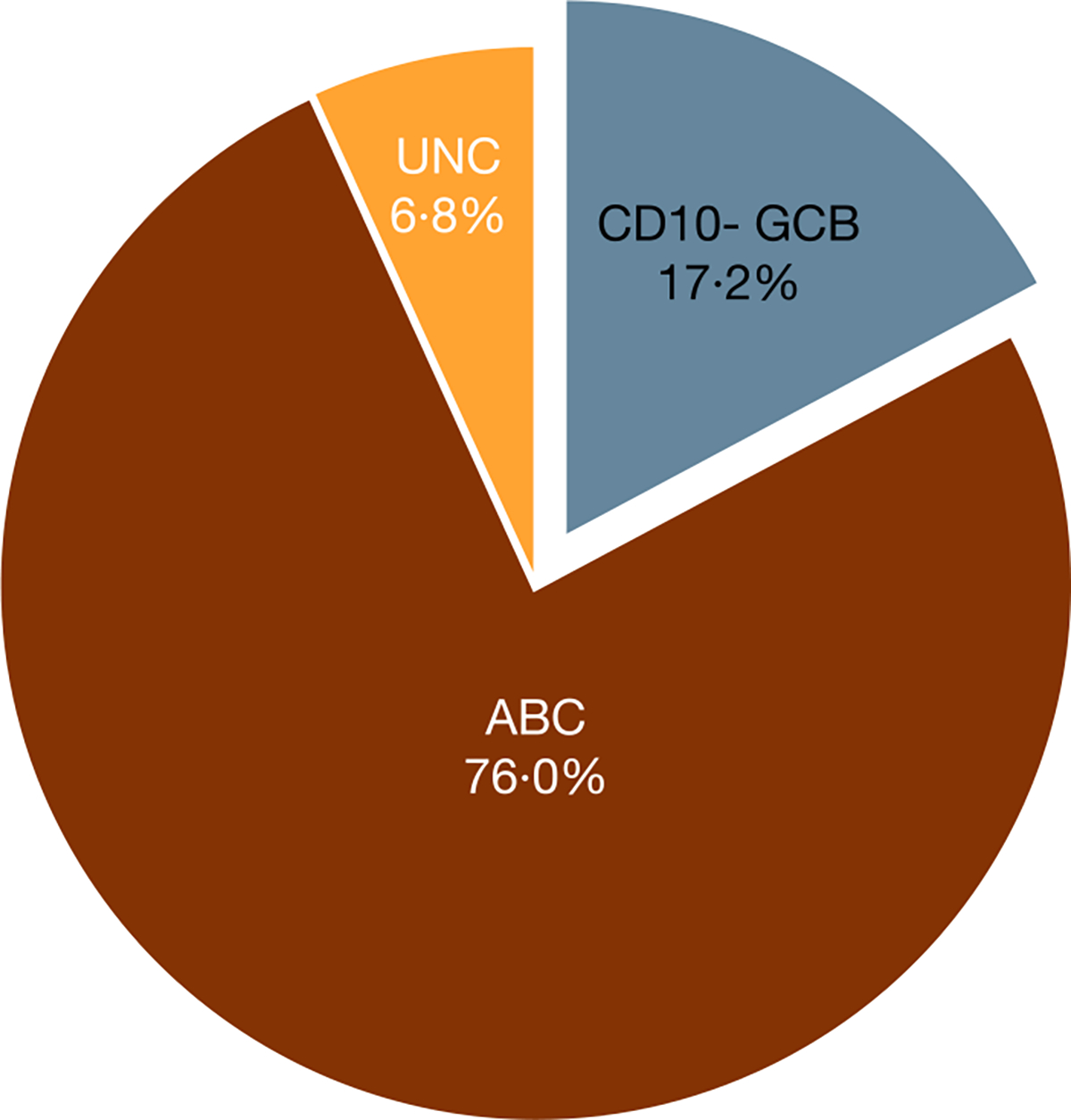
IHC-selected patients in each GEP COO category in enrolled patients. The proportion of patients in the CD10-negative GCB, ABC and UNC subpopulations assessed by GEP are described. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; COO, cell of origin; GCB, germinal centre B cell-like; GEP, gene expression profiling; IHC, immunohistochemistry; UNC, unclassified.
Of non-GCB IHC-selected samples evaluable by GEP, 301 were from patients aged <60 years and 445 were from those aged ≥60 years. Among patients aged <60 years, the number of CD10-negative GCB, ABC and unclassified by GEP samples was 70, 205 and 26, respectively (Figure S2A), leading to a non-GCB concordance of 767%. In patients aged ≥60 years, the number of CD10-negative GCB, ABC and unclassified by GEP samples was 58, 362 and 25, respectively (Figure S2B), resulting in a non-GCB concordance of 868%.
OS comparison
In the ITT population, OS was similar between treatment arms regardless of ABC (HR 1035, 95% CI 0706–1518; P = 08584) or CD10-negative GCB (HR 1027, 95% CI 0382–2759; P = 09585) subpopulations (Fig 2A). There was a non-significant trend towards better OS for CD10-negative GCB versus ABC subpopulation in both the ibrutinib plus R-CHOP (P = 02532) and placebo plus R-CHOP (P = 02183) arms (Fig 2A). In patients aged <60 years, OS was significantly better in the ibrutinib plus R-CHOP versus placebo plus R-CHOP arm in the ABC subpopulation (HR 0365, 95% CI 0147–0909; P = 00305) but was similar between arms in the CD10-negative GCB subpopulation (HR 0266, 95% CI 0031–2281; P = 02272) (Fig 2B). However, within each treatment arm, as expected, patients in the CD10-negative GCB subpopulation showed a non-significant trend for better OS than those in the ABC subpopulation [ibrutinib plus R-CHOP (P = 04906); placebo plus R-CHOP P = 04163)], but the between-subpopulation difference appeared to be more pronounced with placebo plus R-CHOP.
Fig 2.
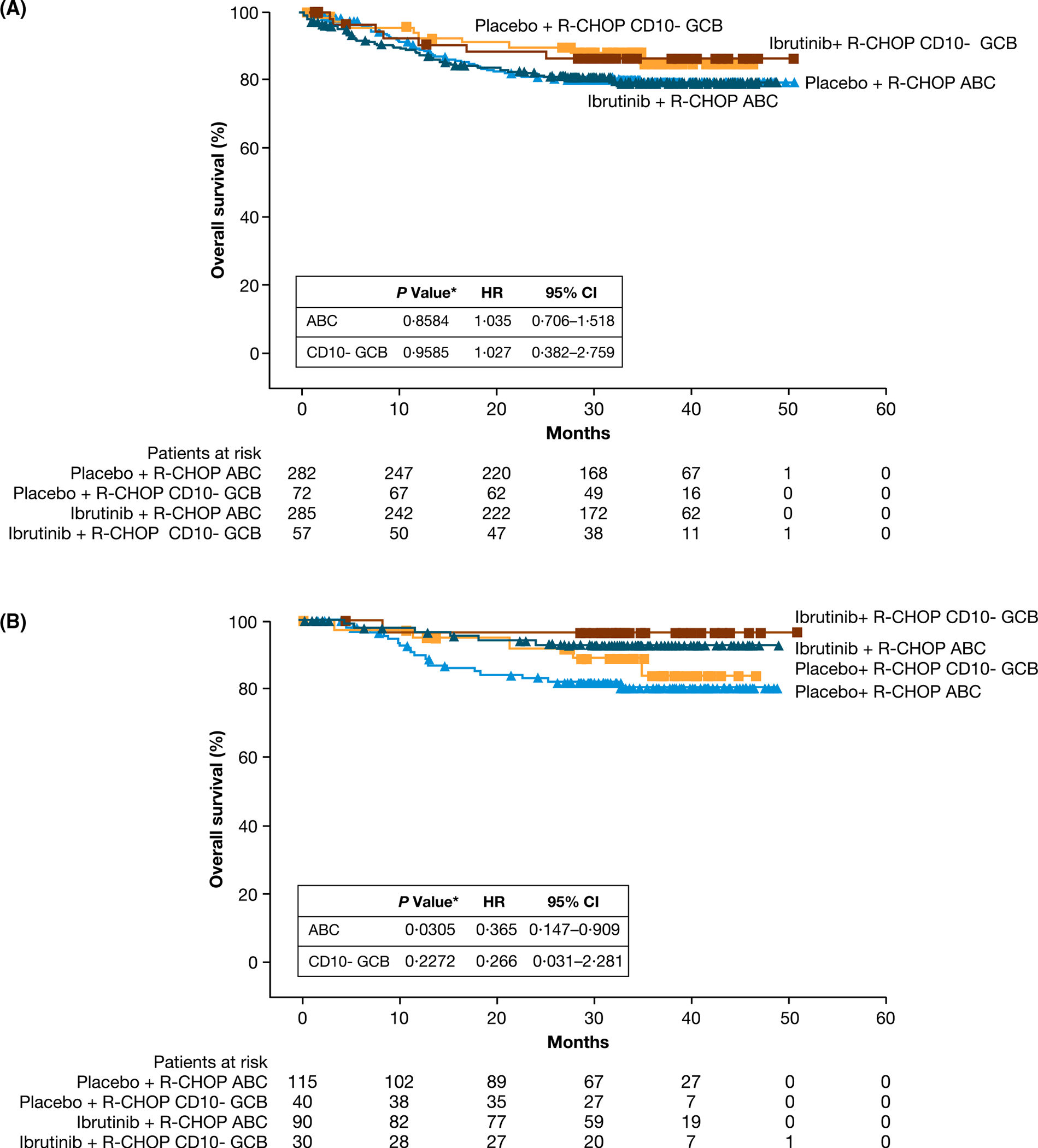
OS by GEP subpopulations in (A) ITT population and (B) patients aged <60 years. The OS is described in both CD10-negative GCB and ABC subpopulations for patients treated with either placebo plus R-CHOP or ibrutinib plus R-CHOP. *P values are from an exploratory analysis. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; ITT, intent to treat; OS, overall survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Within-arm EFS comparison by GEP subpopulations
In an exploratory analysis, EFS for CD10-negative GCB, ABC and unclassified subpopulations by GEP was compared within each treatment arm in the ITT population and by the 60-year age cut-off (Fig 3). In the placebo plus R-CHOP arm, although statistically non-significant (P = 05319) among the GEP CD10-negative GCB, ABC and unclassified subpopulations, there was a trend towards better EFS in patients with CD10-negative GCB versus ABC DLBCL in the ITT population (Fig 3A); the difference was more pronounced in patients aged <60 years (P = 04115) (Fig 3B). While also statistically non-significant in the GEP subpopulations, in the ibrutinib plus R-CHOP arm, the difference in EFS between the CD10-negative GCB and ABC subpopulations were less pronounced versus the placebo plus R-CHOP arm for both the ITT population (P = 06338) and patients aged <60 years (P = 06902) (Fig 3C–D). Specifically, beyond 24 months in the ABC and CD10-negative GCB subpopulations, there was little progression in the ibrutinib plus R-CHOP arm, while progression was seen in the placebo plus R-CHOP arm in the ITT population (Fig 3A–3C) and in patients aged <60 years (Fig 3B–3D).
Fig 3.
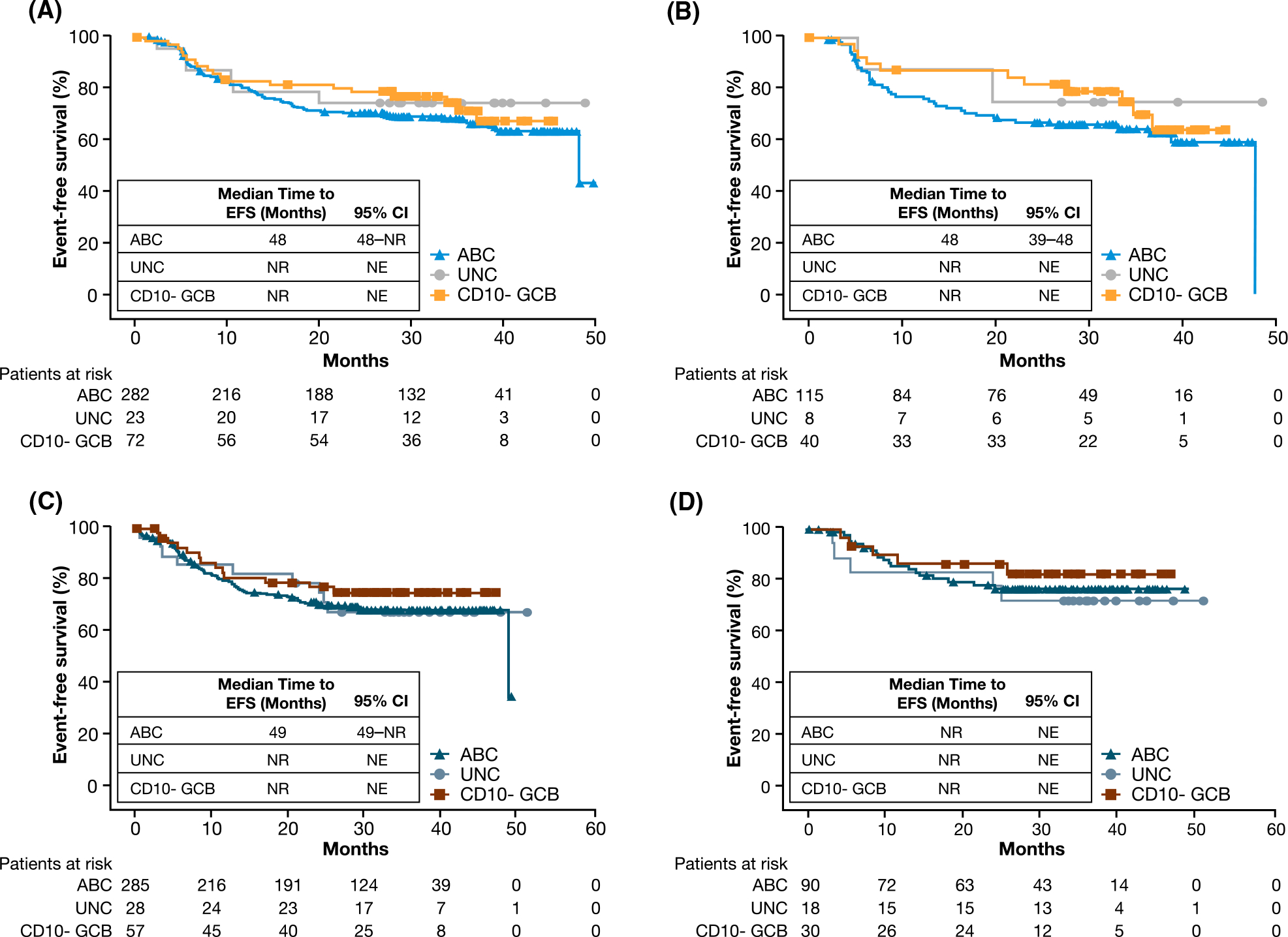
EFS by GEP subpopulations within each treatment arm. (A) Shows EFS in the ITT population by CD10-negative GCB, ABC and UNC subpopulations in the placebo plus R-CHOP arm. (B) Shows EFS in patients aged <60 years by CD10-negative GCB, ABC and UNC subpopulations in the placebo plus R-CHOP arm. (C) Shows EFS in the ITT population by CD10-negative GCB, ABC and UNC subpopulations in the ibrutinib plus R-CHOP arm. (D) Shows EFS in patients aged <60 years by CD10-negative GCB, ABC and UNC subpopulations in the ibrutinib plus R-CHOP arm. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; ITT, intent to treat; NE, not evaluable; NR, not reached; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone; UNC, unclassified.
Within GEP subpopulation EFS comparison by treatment arms
In the ITT population, EFS was similar between arms among patients with ABC (HR 0937, 95% CI 0697–1259; P = 06637) or CD10-negative GCB DLBCL (HR 0916, 95% CI 0452–1856; P = 08081) by GEP (Figure S3).
Among patients aged <60 years, EFS was significantly better in the ibrutinib plus R-CHOP versus placebo plus R-CHOP arm among patients with ABC DLBCL (HR 0561, 95% CI 0326–0967; P = 00348) (Figure S4A). While this was not significant in the CD10-negative GCB subpopulation (HR 0644, 95% CI 0223–1858; P = 04119), the numbers were small; beyond 2 years, there was little progression with ibrutinib plus R-CHOP, unlike with placebo plus R-CHOP (Figure S4B).
Among patients aged ≥60 years, EFS was similar between treatment arms in both the ABC (HR 1188, 95% CI 0822–1715; P = 03583) and CD10-negative GCB (HR 1279, 95% CI 0480–3408; P = 06217) subpopulations (Figure S5), although patient numbers were small in the latter.
Validation of prognostic value and prediction of treatment effect
Assessment of the prognostic value of the GEP assay and correlation of the predetermined cut-off with treatment effect prediction included 559 patients (total samples: 419 ABC, 41 unclassified, 99 CD10-negative GCB). The mean GCB probability scores in all patients were 0103, 0494 and 0811 for the ABC, unclassified and CD10-negative GCB by GEP subpopulations, respectively; similar results were obtained for the separate treatment arms (Table SI). The GEP assay’s prognostic value was confirmed by showing the likelihood of patient survival increased proportionately with the CD10-negative GCB probability score. A higher score was associated with longer survival in the placebo plus R-CHOP arm; this non-significant trend was less clear for the ibrutinib plus R-CHOP arm (Figures S6–S8). Lowering the cut-off of the CD10-negative GCB score did not significantly alter the outcomes (Table SII; Figure S9).
Discussion
In all screened patients, the concordance between IHC and GEP for non-GCB calls was high (827%). The overall concordance (proportion of patients of non-GCB or GCB concordance in relation to patients with test results from both GEP and IHC assays) between the assays in screened patients was 806%. In enrolled patients with non-GCB DLBCL, concordance was also high (827%) in the ITT population, but greater for patients aged ≥60 years (868%) than those aged <60 years (767%). These results were generally consistent with the reported overall concordance between the Hans-based IHC method and the DNA microarray-based GEP, LST and EdgeSeq COO assays (796%, 800% and 781%, respectively).12
Validation of the GEP method showed a higher score was associated with longer survival for the placebo plus R-CHOP arm; this non-significant trend was less defined for the ibrutinib plus R-CHOP arm. Lowering the cut-off of the CD10-negative GCB score did not significantly alter the HR for the between-arm comparison (Supplementary Table SII; Figure S9).
Studies have shown patients with distinctive DLBCL subtypes respond differently to chemotherapy or chemoimmunotherapy regimens.8,9 R-CHOP was associated with poorer outcomes in non-GCB or ABC than GCB DLBCL.8,9 With this regimen, patients with GCB DLBCL showed a higher probability of PFS and OS versus those with ABC DLBCL.8 Similarly, patients with non-GCB DLBCL treated with R-CHOP reportedly had an inferior 2-year PFS and OS versus patients with GCB DLBCL.9
In interpreting the data, we should consider the small sample size. However, in our present analysis, patients aged <60 years in the ABC subpopulation showed significantly better OS and EFS with ibrutinib plus R-CHOP versus placebo plus R-CHOP. Supporting our present findings, ibrutinib demonstrated preferential activity in ABC versus GCB (by GEP) DLBCL in a phase I/II trial,10 with objective responses in 37% of patients with ABC DLBCL versus 5% with GCB DLBCL, despite the small sample size (N = 80). Recent DLBCL genetic classifications5,6,17 have suggested a molecular basis for the increased activity of B-cell receptor (BCR) inhibitors in ABC and unclassified (or non-GCB) DLBCL. The exact molecular definitions are not established, but it is apparent the activity of these compounds may span multiple genetic subtypes, suggesting a continued role for COO in DLBCL prognostication. Also, in the enrolled patients of the present study, the GCB subpopulation by GEP was CD10 negative because of the non-GCB preselection by IHC. Although the clinical features of CD10-negative versus CD10-positive GCB DLBCL have not been well studied, it is possible CD10-negative GCB DLBCL by GEP may identify or enrich a genetic GCB DLBCL subtype with higher sensitivity to BCR inhibitors than CD10-positive GCB disease, which constitutes the majority of GCB DLBCL tumours. Until these GCB subtypes are better characterised, the generalisability of the results should be interpreted with these considerations.
Nonetheless, in the overall population, survival outcomes were similar regardless of COO or treatment. In the ITT population, although there was a non-significant trend towards better survival in patients with CD10-negative GCB DLBCL versus ABC DLBCL in both treatment arms, the differences between subpopulations were smaller with ibrutinib plus R-CHOP than placebo plus R-CHOP. Consistently, there was a non-significant trend towards a higher EFS rate beyond 2 years of study in the ibrutinib plus R-CHOP versus the placebo plus R-CHOP arm among patients with CD10-negative GCB DLBCL, implying some clinical benefit, although numbers were small, especially in patients aged <60 years.
The overall similar survival outcomes in the present analysis are consistent with the high concordance between IHC and GEP, which continues to highlight the clinical relevance of COO classification and the use of centralised IHC to select patients with an inferior prognosis or enrich BCR-inhibitor sensitive populations in future clinical trials. Ongoing trials are investigating the impact of COO subtyping on treatment efficacy in previously untreated or relapsed/refractory DLBCL.18–21
Determining the utility of IHC as a surrogate for GEP profiling is important because the time and infrastructure required for GEP are challenging for the real-time management of patients with DLBCL. These results show that centralised IHC is sufficiently concordant with GEP in our present study, as both represent complex molecular entities. Therefore, centralised diagnostic assays such as IHC could serve as a reasonable surrogate for GEP methods to enrich populations for response to ibrutinib plus R-CHOP.
Supplementary Material
Table SI. Summary of predicted probability score. ABC, activated B-cell–like; GCB, germinal centre B-cell–like; GEP, gene expression profiling; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; SD, standard deviation.
Table SII. Hazard ratios in all patients treated with ibrutinib plus R-CHOP versus R-CHOP by probability score. CI, confidence interval; EFS, event-free survival; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Fig S1. Percentage and concordance of screened and enrolled IHC-selected patients in each GEP subpopulation. CI, confidence interval; COO, cell of origin; GCB, germinal centre B cell-like; GEP, gene expression profiling; IHC, immunohistochemistry; UNC, unclassified.
Fig S2. Percentage of enrolled immunohistochemistry-selected patients in each GEP subpopulation by those patients aged (A) aged <60 years and (B) aged ≥60 years. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; GCB, germinal centre B cell-like; GEP, gene expression profiling; UNC, unclassified.
Fig S3. EFS by GEP subpopulation in both treatment arms in the ITT population by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from an exploratory analysis. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S4. EFS by GEP subpopulations in both treatment arms in patients aged <60 years by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from exploratory analysis. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S5. EFS by GEP subpopulations in both treatment arms in patients aged ≥60 years by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from an exploratory analysis. ABC, activated B cell-like; CD10-GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S6. EFS probability score by quartile and treatment. EFS, event-free survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S7. OS probability score by quartile and treatment. OS, overall survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S8. PFS probability score by quartile and treatment. PFS, progression-free survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S9. HR by different HTG COO assay-derived GCB probability score cut-off. CI, confidence interval; COO, cell of origin; EFS, event-free survival; GCB, germinal centre B cell-like; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Acknowledgements
This trial was funded by Janssen Research and Development. The authors would like to thank Dr Anas Younes’ contributions towards the PHOENIX study and all patients included in this analysis. Writing assistance was provided by Ian Phillips, PhD, Min Yu, MD, and Liqing Xiao, PhD, of Parexel, and was funded by Janssen Research and Development.
Footnotes
Conflict of interest
Sriram Balasubramanian: Janssen employment and stock ownership from Johnson & Johnson, Gilead Sciences, Celgene, Vertex and AbbVie; Songbai Wang, Christopher Major, Michael Schaffer, Jodi Carey, S. Martin Shreeve, Steven Sun and Jessica Vermeulen: Janssen employment and stock ownership from Johnson & Johnson; Brendan Hodkinson and John Gerecitano: Janssen employment; Laurie H. Sehn: honoraria from Amgen, Apobiologix, Abb-Vie, Celgene, Gilead Sciences, Janssen-Ortho, Karyopharm Therapeutics, Kite Pharma, Lundbeck, Merck, Roche/Genentech, Seattle Genetics, Takeda Pharmaceuticals, TEVA Pharmaceuticals Industries and TG Therapeutics, research funding from Roche/Genentech, consulting or an advisory role with Karyopharm Therapeutics, Kite Pharma, Merck, Takeda Pharmaceuticals, TEVA Pharmaceuticals Industries and TG Therapeutics; Peter Johnson: honoraria from Bristol Myers Squibb, Takeda, Novartis, Celgene, Janssen, Epizyme, Boehringer Ingelheim, Kite, Genmab and Incyte, research funding from Epizyme and Janssen, and consulting or advisory role with Janssen; Pier Luigi Zinzani: advisory roles for Verastem, Janssen, Takeda, TG Therapeutics, Bristol Myers Squibb, Roche, Gilead, Novartis and Celltrion; Wyndham Wilson and Louis M. Staudt: have no competing interests.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Previous presentation: part of the study results were presented as an oral presentation at the 2019 International Conference on Malignant Lymphoma, Lugano, Switzerland.
References
- 1.International Agency for Research on Cancer. World Cancer Report 2014. [Internet]. [cited 5 February 2020]. Available from: http://www.who.int/cancer/publications/WRC_2014/en/
- 2.Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123:1214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 4.Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–82. [DOI] [PubMed] [Google Scholar]
- 5.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32. [DOI] [PubMed] [Google Scholar]
- 8.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, Nelson GD, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-cell lymphoma: a phase II study. J Clin Oncol. 2015;33:251–7. [DOI] [PubMed] [Google Scholar]
- 10.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott DW. Cell-of-origin in diffuse large B-cell lymphoma: are the assays ready for the clinic? Am Soc Clin Oncol Educ Book. 2015;e458–66. [DOI] [PubMed] [Google Scholar]
- 12.Schaffer M, Chaturvedi S, Alvarez JD, Frans S, Aquino R, Hall B, et al. Comparison of immunohistochemistry assay results with gene expression profiling methods for diffuse large B-cell lymphoma subtype identification in matched patient samples. J Mol Biomark Diagn. 2018;9:2. [Google Scholar]
- 13.Coutinho R, Clear AJ, Owen A, Wilson A, Matthews J, Lee A, et al. Poor concordance among nine immunohistochemistry classifiers of cell-of-origin for diffuse large B-cell lymphoma: implications for therapeutic strategies. Clin Cancer Res. 2013;19:6686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reber R, Banz Y, Garamvolgyi E, Perren A, Novak U. Determination of the molecular subtypes of diffuse large B-cell lymphomas using immunohistochemistry: a case series from the Inselspital, Bern, and a critical appraisal of this determination in Switzerland. Swiss Med Wkly. 2013;143:w13748. [DOI] [PubMed] [Google Scholar]
- 15.Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol. 2019;37:1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rimsza LM, Wright G, Schwartz M, Chan WC, Jaffe ES, Gascoyne RD, et al. Accurate classification of diffuse large B-cell lymphoma into germinal center and activated B-cell subtypes using a nuclease protection assay on formalin-fixed, paraffin-embedded tissues. Clin Cancer Res. 2011;17:3727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–68 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ClinicalTrials.gov. A study of the Bruton’s tyrosine kinase inhibitor, PCI-32765 (ibrutinib), in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone in patients with newly diagnosed non-germinal center B-cell subtype of diffuse large B-cell lymphoma. [Internet]. [cited 5 February 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT01855750
- 19.ClinicalTrials.gov. S9704-S0014-S0313A studying genes in samples from patients with limited or advanced diffuse large B-cell lymphoma. [Internet]. [cited 5 February 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT01563861
- 20.ClinicalTrials.gov. Rituximab and combination chemotherapy with or without lenalidomide in treating patients with newly diagnosed stage II-IV diffuse large B cell lymphoma. [Internet]. [cited 5 February 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT01856192
- 21.ClinicalTrials.gov. Phase II copanlisib in relapsed/refractory diffuse large B-cell lymphoma (DLBCL). [Internet]. [cited 5 February 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT02391116
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Summary of predicted probability score. ABC, activated B-cell–like; GCB, germinal centre B-cell–like; GEP, gene expression profiling; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; SD, standard deviation.
Table SII. Hazard ratios in all patients treated with ibrutinib plus R-CHOP versus R-CHOP by probability score. CI, confidence interval; EFS, event-free survival; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Fig S1. Percentage and concordance of screened and enrolled IHC-selected patients in each GEP subpopulation. CI, confidence interval; COO, cell of origin; GCB, germinal centre B cell-like; GEP, gene expression profiling; IHC, immunohistochemistry; UNC, unclassified.
Fig S2. Percentage of enrolled immunohistochemistry-selected patients in each GEP subpopulation by those patients aged (A) aged <60 years and (B) aged ≥60 years. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; GCB, germinal centre B cell-like; GEP, gene expression profiling; UNC, unclassified.
Fig S3. EFS by GEP subpopulation in both treatment arms in the ITT population by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from an exploratory analysis. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S4. EFS by GEP subpopulations in both treatment arms in patients aged <60 years by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from exploratory analysis. ABC, activated B cell-like; CD10- GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S5. EFS by GEP subpopulations in both treatment arms in patients aged ≥60 years by (A) ABC and (B) CD10-negative GCB subpopulations. *P values are from an exploratory analysis. ABC, activated B cell-like; CD10-GCB, CD10-negative GCB; CI, confidence interval; EFS, event-free survival; GCB, germinal centre B cell-like; GEP, gene expression profiling; HR, hazard ratio; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S6. EFS probability score by quartile and treatment. EFS, event-free survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S7. OS probability score by quartile and treatment. OS, overall survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S8. PFS probability score by quartile and treatment. PFS, progression-free survival; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone.
Fig S9. HR by different HTG COO assay-derived GCB probability score cut-off. CI, confidence interval; COO, cell of origin; EFS, event-free survival; GCB, germinal centre B cell-like; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.