

Abstract
Kyphoscoliotic Ehlers–Danlos syndrome (kEDS) is a rare genetic disorder combining congenital hypotonia, congenital/early onset and progressive kyphoscoliosis, and generalized joint hypermobility. Vascular fragility is another characteristic of the disease rarely described. We report a severe case of kEDS‐PLOD1 with several vascular complications leading to difficulties in disease management.
Keywords: case report, kyphoscoliotic Ehlers–Danlos syndrome, PLOD1 gene, vascular complications
This case report corroborates the increased risk of vascular complications in patients with PLOD1‐related kyphoscoliotic Ehlers–Danlos syndrome complicating disease management.

1. INTRODUCTION
Kyphoscoliotic Ehlers–Danlos syndrome (kEDS) is a rare autosomal recessive connective tissue disorder characterized by congenital muscle hypotonia, congenital or early onset and progressive kyphoscoliosis, generalized joint hypermobility, skin and scleral fragility, facial dysmorphia, and vascular fragility. 1 , 2 Kyphoscoliotic Ehlers–Danlos syndrome can be divided into two subtypes based on genotyping. kEDS‐PLOD1 (OMIM 225400) results from lysyl‐hydroxylase‐1 (LH1) deficiency due to pathogenic variants of PLOD1 (procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase 1) gene. kEDS‐FKBP14 (OMIM 614557) is due to pathogenic variants of FKBP14 gene. LH1 is a posttranslational modification enzyme that plays an important role in the formation of intra‐ and intermolecular collagen cross‐links. 3 Lack of LH1 leads to an under hydroxylation of collagen lysyl residues and ultimately to impaired formation of collagen cross‐links, leading to unstable affected tissues. 4
Severe and life‐threatening complications such as spontaneous aneurysms or spontaneous ruptures of medium‐ to large‐sized arteries have been reported in patients affected by kEDS‐PLOD1. 5 , 6
We hereby report the case of a 29‐year‐old female diagnosed with kEDS‐PLOD1, due to two novel PLOD1 mutations, who presented several vascular manifestations. This case illustrates the severity of kEDS and the complexity of disease management associated with this condition.
2. CLINICAL CASE
A 29‐year‐old female was referred to our department with a suspicion of kEDS. She was born with muscle hypotonia, joint hypermobility, and congenital vertical talus. She walked without assistance at 24 months. Myopia and kyphoscoliosis were diagnosed during childhood. Despite early disease management, kyphoscoliosis progressed rapidly and required four successive interventions for arthrodesis over a period of 6 years. She developed restrictive lung disease with a forced vital capacity of 40% leading to treatment by noninvasive ventilation. Falls were frequent and she had difficulty climbing stairs. The fourth spinal surgery at the age of 14 improved mobility, but she has never been able to run. Joint instability was moderate. She also had lower incisor root resorption.
In her 20s, myopia worsened (−10.75 and −13.75 diopters) with the development of open‐angle glaucoma. She required anticoagulant treatment for an idiopathic pulmonary embolism (thrombophilia screening was negative). She developed progressive weakness in the lower limbs with a positive Gower's sign. Walking, balance, and autonomy became increasingly difficult. A muscle biopsy was performed and revealed an asymmetric axonal sensorimotor neuropathy and a demyelinating neuropathy in the legs, which was probably responsible for the development of bilateral perforating plantar ulcers. She developed urinary and bowel incontinence with a renal angiomyolipoma.
At clinical examination at the age of 29, arm span ratio was 1.09. She presented minor pectus carinatum, pes planus, talipes varus, arachnodactyly with positives thumb and wrist signs, and atrophy of thenar and hypothenar eminences. She had blue sclera and mandibular retrognathia (Figure 1). Her skin was velvety and hyperelastic with multiple abnormal scars (atrophic/papyraceous or dystrophic). She had varicose veins and hyperpigmentation of bruises on the legs (Figure 1). In addition, she had generalized joint hypermobility with a Beighton score of 6/9. She complained of muscle weakness which was more marked in her legs.
FIGURE 1.
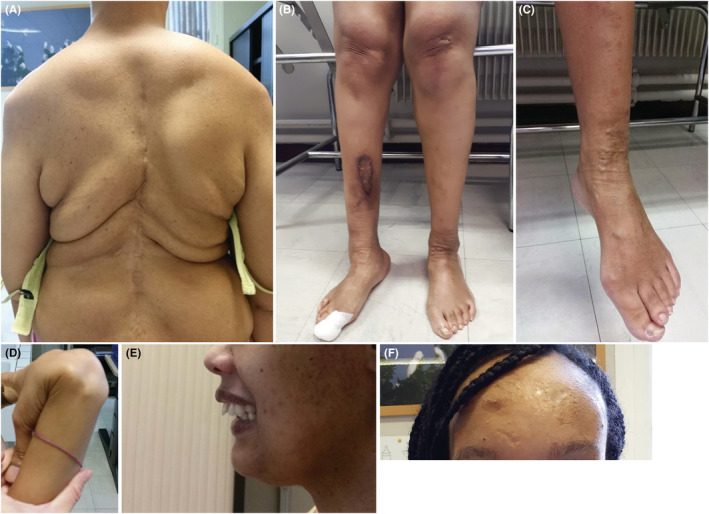
Clinical phenotype of the kEDS patient. (A) Patient at the age of 29 years after multiple surgical corrections of severe thoracolumbar scoliosis. (B) Pes planovarus and scar hyperpigmentation. (C) Varicose veins of the left lower limb; (D) Joint hyperlaxity: passive apposition of the thumb on the forearm. (E) Mandibular retrognathia. (F) Atrophic scars on the forehead
Based on the clinical history and examination, she was diagnosed with kyphoscoliotic EDS. Her parents did not met criteria for any type of EDS.
3. INVESTIGATIONS
Genetic testing by next generation sequencing‐targeted panel, identified pathogenic heterozygous compound variants in PLOD1 gene with c.1756–13 C > A paternally inherited in exon 17, probably disrupting the acceptor site of intron 16, and a 5‐bp deletion c.814_818delGAAGG in exon 8 maternally inherited, leading to a premature stop codon p.(Glu272Leufs*10). The pathogenicity of the identified PLOD1 variants (NM_000302.4) was assessed through a set of criteria according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Genomic Sanger sequencing in the parents confirmed the localization in trans of these variants. They have not been described to date (PubMed, LOVD, and HGMD Pro) and their allelic frequencies are unknown (GnomAD). The c.814_818delGAAGG variant leads to a truncated protein in exon 9 and c.1756–13 C > A probably results in splicing alteration in the C part of the protein disturbing the catalytic site of PLOD1. 7
4. FOLLOW‐UP
After diagnosis, symptoms progressively worsened. She developed bilateral scleromalacia, bilateral corneal thinning (432 μm in the right eye and 465 μm in the left eye), and peripheral retinal fragility. Osteodensitometry showed femoral osteopenia (T‐score: −1.9 and − 1.3). X‐ray revealed acetabular protrusions and a disc herniation at L5 (Figure 2c), which was responsible for pain and neuropathy of the lower limbs. Subsequently, spinal cord compression with spinal cord atrophy was identified. A systematic cardiac and vascular evaluation revealed splenic infarctions without identified trauma and a small ectasia of the splenic artery (Figure 2d). The Doppler ultrasonography of the lower extremity arteries revealed several complications. On the right leg, she had a false thrombosed occlusive aneurysm of the proximal anterior tibial artery and a dissection of the posterior tibial artery without stenosis (Figure 3). On the left leg, she had a dissecting aneurysm of the tibial‐peroneal trunk with a parietal hematoma without significant hemodynamic stenosis (Figure 4), an occlusion and a dysplasia of posterior tibial artery on a probable occlusive dissection. The left anterior tibial artery was of irregular size, with a stenosing dissection and a parietal hematoma, also moderately stenosing. Venous examination showed an incontinence of the long saphenous vein at the saphenofemoral junction. Echocardiography revealed a myxoid mitral valve without prolapse. Left ventricular ejection fraction was reduced to 43%.
FIGURE 2.
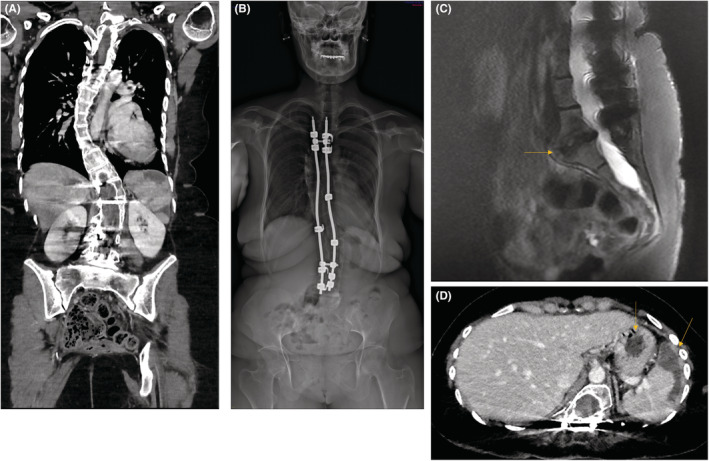
Radiological findings at the age of 29 and 30 years. (A and B) Left thoracolumbar kyphoscoliosis corrected by arthrodesis. (C) L5‐S1 disc herniation. (D) Splenic infarction (full arrow) and angiomyolipoma (dashed arrow)
FIGURE 3.
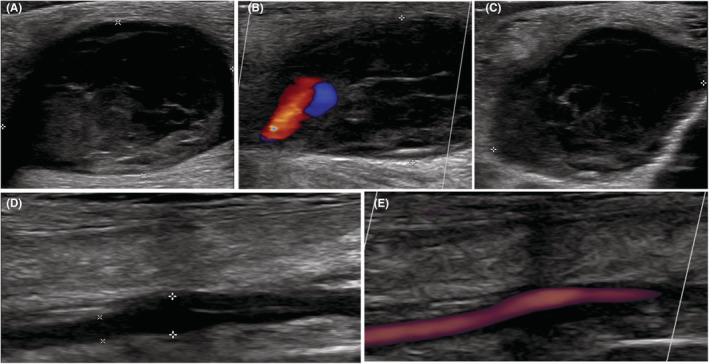
Arterial ultrasonography and Doppler of the false thrombosed occlusive aneurysm (proximal right anterior tibial artery). (A) and (B): Longitudinal view. (C): Transverse view. (D) Longitudinal view on ultrasonography and (E) Doppler ultrasonography of the dissection of the right posterior tibial artery
FIGURE 4.
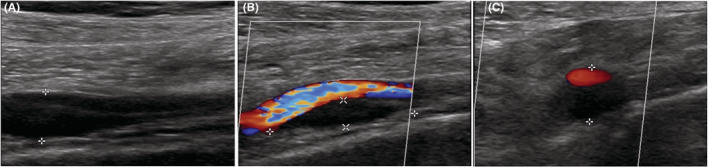
Arterial ultrasonography and Doppler of the dissecting aneurysm of the left tibial‐peroneal trunk. (A) and (B): Longitudinal view. (C): Transversal view
The patient was referred to a vascular medicine department for management and follow‐up of her vascular complications. However, these incidents considerably disrupted management of her lumbar pain, which became extremely challenging due to pain killer resistance. The indications for treatment via infiltrations or new spinal surgery were intensively discussed, the benefit–risk balance was considered unfavorable. Palliative therapeutic strategies have been preferred.
5. DISCUSSION
PLOD1‐related kyphoscoliotic EDS is a rare autosomal‐recessive connective tissue disorder needing appropriate follow‐up to manage complications such as progressive kyphoscoliosis with restrictive lung disease, skin fragility, scleral and ocular complications, and vascular fragility.
We report the natural history of a woman with a severe kEDS phenotype in whom the occurrence of vascular events complicated management and clinical care of her other complications of kEDS. The development in her 30s of arterial manifestations illustrated underlying vascular fragility that was not present during her childhood. Because of the extreme fragility of the condition, we strongly recommended regular vascular and cardiac monitoring in order to detect aneurysms and dissections.
Our case underlines the increased risk of vascular accidents that may occur in kEDS‐PLOD1. Although vascular events are more common in vascular EDS, ruptures, dissections, or dissecting aneurysms of large‐ or medium‐sized arteries have been reported in several studies focused on kEDS‐PLOD1, supporting the presence of these potential life‐threatening complications in the diagnostic criteria. 5 , 6 , 8 , 9 Several cases have been reported. One patient died from an arterial rupture at the age of 14. 2 Aortic or arterial aneurysms have been found in five patients, two of them died. 2 , 10 , 11 , 12 , 13 , 14 One patient had an acute cardiac failure due to a spontaneous dissection of the left anterior descending artery. 4 Other vascular events (mitral valve prolapse, aortic dilatation, aortic stenosis, deep venous thrombosis, antenatal/neonatal intracranial hemorrhage, etc.) underline the variability of vascular fragility in these patients. 5 , 6 Overall, these reports demonstrate that patients with kEDS‐PLOD1 are at risk of developing severe vascular complications at any age and should be offered cardiovascular management similar to vascular EDS patient. After diagnosis, we advise regular cardiovascular follow‐up to screen for complications that may occur at any age. 5 Blood pressure monitoring is recommended as well as early beta‐blocker therapy for the secondary prevention of arterial complications. Doctors and surgeons should be advised about the potential risks of some interventions. 5
Pain is a common symptom among EDS patients. 15 However, patients may not respond to standard pain medications. In our case, the management of severe pain due to the lumbar disc herniation was a problem as pain was pharmaco‐resistant. Invasive treatments of the herniated disc such as infiltrations or conventional discectomy were considered, but finally excluded due to the high risk in a patient with vascular fragility. Palliative therapeutic strategies were preferred, with annual hospitalization in a physical medicine and rehabilitation unit for follow‐up and disease management (including scoliosis, lower limb pain, mobility, and balance support).
Because of the possible occurrence of vascular events, involvement of the multidisciplinary team is essential not only to explore treatment decisions, avoid iatrogenesis, but also to address pain and osteoarticular complications. Care providers should be aware of the risks of complications of invasive procedures that could occur in EDS patients with vascular fragility, independently of the type.
In conclusion, kEDS is a rare disease impacting multiple organ systems requiring the involvement of a specialized multidisciplinary team to optimize management.
AUTHOR CONTRIBUTIONS
Malika Foy: Project administration; writing – original draft; writing – review and editing. Corinne Métay: Formal analysis; investigation; resources; writing – review and editing. Michael Frank: Investigation; resources; writing – review and editing. Nicolas Denarié: Investigation; resources; writing – review and editing. Salma Adham: Investigation; resources. Clarisse Billon: Investigation. Anne Legrand: Investigation. Xavier Jeunemaitre: Investigation. Fabrice Gillas: Investigation; resources; writing – review and editing. Karen Gaudon: Investigation. Philippe De Mazancourt: Investigation; resources. Ahmed Mekki: Investigation; resources. Robert Carlier: Investigation; resources. Karelle Benistan: Conceptualization; formal analysis; investigation; resources; supervision; writing – original draft; writing – review and editing.
FUNDING INFORMATION
Not applicable.
CONFLICT OF INTEREST
No conflict of interest to disclose.
CONSENT
Written informed consent for publication was obtained from the patient for publication of this case report and accompanying images.
ACKNOWLEDGMENTS
Authors thank the patient for her contribution. The authors also thank Sarah Hartley, MD, for English editing.
Foy M, Métay C, Frank M, et al. A severe case of PLOD1 ‐related kyphoscoliotic Ehlers–Danlos syndrome associated with several arterial and venous complications: A case report. Clin Case Rep. 2023;11:e06760. doi: 10.1002/ccr3.6760
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers‐Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175(1):8‐26. [DOI] [PubMed] [Google Scholar]
- 2. Yeowell HN, Walker LC, Farmer B, Heikkinen J, Myllyla R. Mutational analysis of the lysyl hydroxylase 1 gene (PLOD) in six unrelated patients with Ehlers‐Danlos syndrome type VI: prenatal exclusion of this disorder in one family. Hum Mutat. 2000;16(1):90. [DOI] [PubMed] [Google Scholar]
- 3. Hyland J, Ala‐Kokko L, Royce P, Steinmann B, Kivirikko KI, Myllylä R. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers–Danlos syndrome type VI. Nat Genet. 1992;2(3):228‐231. [DOI] [PubMed] [Google Scholar]
- 4. Rohrbach M, Vandersteen A, Yi U, et al. Phenotypic variability of the kyphoscoliotic type of Ehlers‐Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation. Orphanet J Rare Dis. 2011;6(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D'Hondt S, Van Damme T, Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers‐Danlos syndrome: a systematic review. Genet Med. 2018;20(6):562‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malfait F. Vascular aspects of the Ehlers‐Danlos syndromes. Matrix Biol. 2018;71–72:380‐395. [DOI] [PubMed] [Google Scholar]
- 7. Yeowell HN, Steinmann B. PLOD1‐related Kyphoscoliotic Ehlers‐Danlos syndrome. GeneReviews®; 2018. [Google Scholar]
- 8. Henneton P, Legrand A, Giunta C, Frank M. Arterial fragility in kyphoscoliotic Ehlers‐Danlos syndrome. BMJ Case Rep. 2018;2018:bcr2018224423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zieminski P, Risse J, Legrand A, et al. Vascular manifestations and kyphoscoliosis due to a novel mutation of PLOD1 gene. Acta Cardiol. 2021;76(5):557‐558. [DOI] [PubMed] [Google Scholar]
- 10. Beighton P. Serious ophthalmological complications in the Ehlers‐Danlos syndrome. Br J Ophthalmol. 1970;54(4):263‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Busch A, Suellner J, Anger F, et al. Critical care of kyphoscoliotic type ehlers‐danlos syndrome with recurrent vascular emergencies. Vasa ‐ Eur J Vasc Med. 2014;43(3):216‐221. [DOI] [PubMed] [Google Scholar]
- 12. Giunta C, Randolph A, Al‐Gazali LI, Brunner HG, Kraenzlin ME, Steinmann B. Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers‐Danlos syndrome (EDS VIA). Am J Med Genet Part A. 2005;133A(2):158‐164. [DOI] [PubMed] [Google Scholar]
- 13. Voermans NC, Van Alfen N, Pillen S, et al. Neuromuscular involvement in various types of Ehlers‐Danlos syndrome. Ann Neurol. 2009;65(6):687‐697. [DOI] [PubMed] [Google Scholar]
- 14. Gok E, Goksel OS, Alpagut U, Dayioglu E. Spontaneous brachial pseudo‐aneurysm in a 12‐year‐old with kyphoscoliosis‐type ehlers‐danlos syndrome. Eur J Vasc Endovasc Surg. 2012;44(5):482‐484. [DOI] [PubMed] [Google Scholar]
- 15. Bénistan K, Martinez V. Pain in hypermobile Ehlers‐Danlos syndrome: new insights using new criteria. Am J Med Genet Part A. 2019;179(7):1226‐1234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.