

Abstract
This report involves a young woman with isolated cardiac paraganglioma that was diagnosed using 68Gallium-labeled [1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid]-1-NaI3-octreotide positron emission tomographic scintigraphy. For the preoperative evaluation, multimodality imaging accurately described the anatomic location of the tumor and its relationship with the surrounding tissues. The patient underwent successful surgical resection of the tumor along with right coronary artery bypass grafting. The 2-month follow-up scintigraphy was normal. Next-generation sequencing evaluation revealed a novel germline mutation for the succinate dehydrogenase subunit B gene.
Keywords: Pheochromocytoma; paraganglioma; SDHB protein, human; neuroendocrine tumor
Introduction
Isolated primary cardiac paragangliomas are rare,1,2 yet precise tumor locations are rather diverse in many cases.3–5 Diagnosis and management of these tumors are challenging to both medical and surgical teams. Therefore, it is important that a multidisciplinary investigation is conducted before any operation, and surgical tactics should be individualized accordingly. Paragangliomas can sporadically occur as part of an inherited predisposition syndrome. Only very few cases with succinyl dehydrogenase (SDH) gene mutation have been reported.6,7 This article presents a case of isolated cardiac paraganglioma with SDH subunit B (SDHB) gene mutation that was treated successfully using multimodality imaging.
Case Report
This case is a 30-year-old woman who had episodes of severe headache with chest heaviness, palpitations, anxiety, and diaphoresis over 1 year. She was determined to have hypertension, with a maximum blood pressure (BP) reaching up to 200/104 mm Hg. Her routine laboratory test results were within normal limits. Abdominal sonography and renal artery Doppler results were normal.
Upon further workup, echocardiography results revealed an unusual turbulence in the right ventricle (RV) cavity and RV outflow tract (RVOT). In view of severe hypertension with this echocardiographic finding, a multidetector computed tomography angiography was done, which revealed a lobulated heterogeneously enhancing mass (44 × 43 × 26 mm in size) in the atrioventricular (AV) groove extending superiorly into the aortopulmonary groove with complete encasement of the right coronary artery (RCA) with mild luminal narrowing (Fig. 1).
Fig. 1.
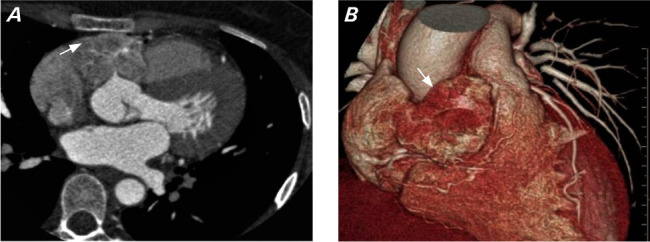
A) Multidetector computed tomography angiography image in axial view shows a lobulated heterogeneously enhancing mass (arrow) in the atrioventricular groove encasing ostia of the right coronary artery. B) Multidetector computed tomography angiography volume rendered image shows a mass in the right atrioventricular groove (arrow) extending superiorly between the aorta and pulmonary artery.
For further tumor delineation, the patient underwent cardiac magnetic resonance imaging (MRI), which showed a well-defined T2 iso- to hyperintense lesion with postcontrast enhancement in the region of the AV groove between the main pulmonary trunk and aortic root that was compressing the RVOT and abutting the RV wall posteriorly, with loss of interface between the lesion and RV wall without any pericardial involvement (Fig. 2).
Fig. 2.
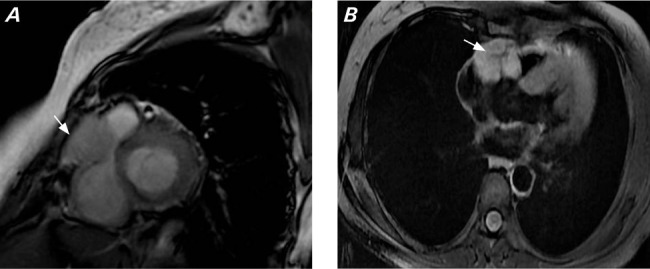
A) Cardiac magnetic resonance short-axis image (Fast Imaging Employing Steady-state Acquisition) shows a mass lesion (arrow) with intermediate to hyperintense signal in the right atrioventricular groove, compressing the right ventricular outflow tract and abutting the right ventricle posteriorly. B) Axial magnetic resonance image (postcontrast) shows homogenous enhancement of the mass (arrow) in the right atrioventricular groove.
Because of severe intermittent hypertension with a cardiac mass, the patient underwent a 24-hour urinary catecholamine measurement and was found to have elevated urinary normetanephrine levels with normal metanephrine levels.
For further diagnostic and staging workup, whole-body 68Gallium-labeled [1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid]-1-NaI3-octreotide positron emission tomographic (Ga-68-DOTANOC PET) scintigraphy was done, which revealed a tracer-avid (standardized uptake maximum value, 4.66), ill-defined, soft-tissue density lesion in the groove between the aortic root and main pulmonary trunk, closely abutting the aorta, right pulmonary artery, and atrium. No other lesions were found (Fig. 3).
Fig. 3.
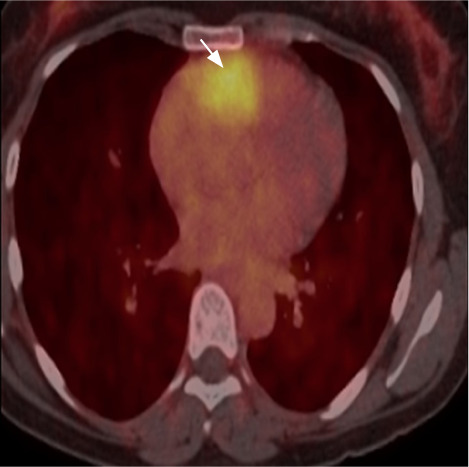
68Gallium-labeled [1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid]-1-NaI-3-octreotide positron emission tomographic scintigraphy scan shows tracer avidity (arrow) in the groove between the aortic root and pulmonary trunk.
After adequate preoperative preparation, the patient underwent surgery. After median sternotomy, the pericardium was separated and the tumor was localized in the aortopulmonary groove. It had well-defined margins and was adhered to the ascending aorta, RVOT, and pulmonary artery (Fig. 4A). A dissection plane was made between the aorta and tumor, which revealed feeders from the aorta. The patient immediately underwent cardiopulmonary bypass, during which the tumor was separated from the tumor bed and a saphenous vein graft was anastomosed between the aorta and posterior descending artery (Fig. 4B). The tumor was encasing the right coronary artery. The tumor was ligated and then excised (Fig. 5). The tumor bed was then repaired with a pericardial patch after glue fixation. During operation, marked fluctuations in BP were noted.
Fig. 4.
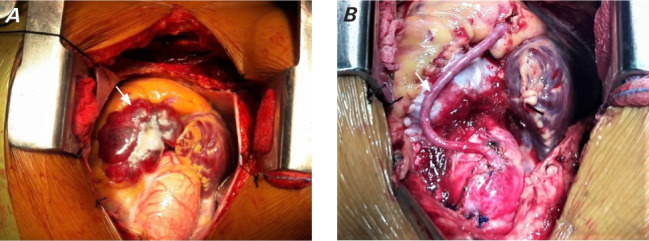
A) Intraoperative image of cardiac mass (arrow) in the right atrioventricular groove extending superiorly between the aorta and pulmonary artery. B) Intraoperative image after mass removal. Tumor was excised en bloc along with a saphenous vein graft to the right coronary artery (arrow).
Fig. 5.
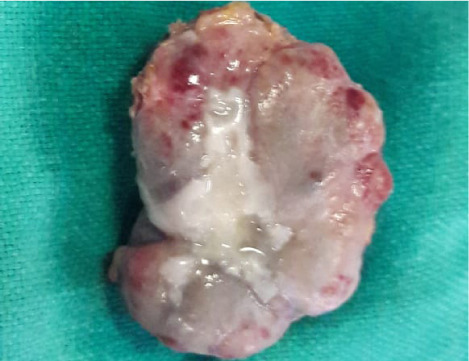
Image shows an excised cardiac paraganglioma.
Her postoperative course was uneventful. She was discharged after 7 days. After 1 month, the patient was asymptomatic and normotensive, and repeat urine catecholamine levels were in the normal range. Repeat Ga-68-DOTANOC PET scintigraphy was done after 2 months and was normal. Patient was asymptomatic at 1-year follow-up.
The resected tumor was confirmed to be paraganglioma by histopathologic examination (Fig. 6). On immunohistochemistry, the tumor cells were positive for vimentin, S-100, chromogranin, and synaptophysin.
Fig. 6.
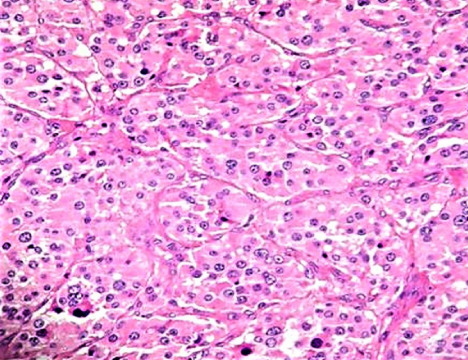
Histopathology of mass that shows tumor cells arranged in a zellballen pattern comprising nests separated by thin fibrovascular septa. The tumor cells are polygonal and display moderately pleomorphic nuclei with coarse chromatin, prominent nucleoli in certain places, and abundant eosinophilic cytoplasm. Hematoxylin-eosin stain, ×200 original magnification.
A genetic evaluation was performed using peripheral blood leukocytes. Next-generation sequencing evaluation revealed a novel germline mutation: as c.386C>T on heterozygosis of exon 4 of the gene for SDHB.
Discussion
Cardiac paraganglioma is a primary neuroendocrine tumor. Paraganglion cells are nonneuronal cells that originate from the neural crest. These are widely distributed throughout the body and are either chromaffin or nonchromaffin types. Tumors that originate from chromaffin cells are catecholamine-secreting tumors and are known as chromaffin paragangliomas or secretary (active) paragangliomas or pheochromocytoma. Less than 2% of pheochromocytomas are found in the chest, with the heart being the least common site.1 Cardiac paragangliomas are the rarest form of primary cardiac tumors and constitute less than 1% of all cardiac tumors.2 They may arise from visceral autonomic paraganglia (atrium or interatrial septum) or branchiomeric paraganglia (branchial arch, coronary, pulmonary, or aortopulmonary). Tumors that originate from nonchromaffin cells are known as chemodectomas.
The most common site of this type of tumor's development inside the heart is the left atrium or at the base of the aortic root and pulmonary artery; occasionally, it is found in the right atrium, but rarely in the left ventricle and coronary sinus.3–5 In this case, the tumor originated from the root of the aorta and extended into the AV groove superiorly between the aorta and pulmonary artery, completely encasing the RCA. A tumor at this site is likely to have arisen from chromaffin cells of the paraganglia in the ventral connective tissues between the descending aorta and the pulmonary trunk. This paraganglion is known as a coronary paraganglion and receives its blood supply from the left coronary artery in adults. Boyd8 has pointed out that in the fetus the coronary paraganglion receives additional blood supply from the RCA. The fact that the tumor in this patient was mainly supplied from the RCA suggests that this artery may supply coronary paraganglia in adults, as well.
Imaging methods currently available for the evaluation of cardiac paraganglioma include multidetector computed tomography, MRI, and nuclear imaging. In this case, multimodality imaging was used for preoperative evaluation, yielding sufficient information to perform the procedure safely. Iodine metaiodobenzylguanidine, fluorodeoxyglucose F 18, and technetium TC 99m octreotide scans have been used as the imaging tools of choice for the initial localization of the tumor, or after localization to rule out metastasis and other coexistent neural crest-cell neoplasms.9 Newer somatostatin derivatives like Ga-68-DOTANOC PET have shown promising results in the imaging of somatostatin receptor–positive tumors as compared to the non-PET Indium In-111-pentetreotide scintigraphy.10 In this case, Ga-68-DOTANOC PET scintigraphy was helpful in not only localizing the site of the paraganglioma but also ruling out tumors in other organs and any metastasis. Multidetector computed tomography in combination with MRI excellently visualized the anatomic extent of the tumor and also defined the tissue planes for surgery, as has been reported previously.11
Because most cardiac paragangliomas are benign, recovery and prognosis are quite good after the complete tumor resection. Cardiac paragangliomas should be excised using cardiopulmonary bypass, as was done in the case discussed here. Such an operation allows complete excision of the tumor along with adequate cardiac repair, especially in cases in which the tumor has invaded the cardiac structures and the margins are not clear.12,13 Another benefit of cardiopulmonary bypass is that it dampens the BP fluctuations resulting from tumor manipulation. In addition, it allows for the safe revascularization of RCA.
Paragangliomas can sporadically occur as part of an inherited predisposition syndrome. More recently, genes that encode proteins in mitochondrial complex II, such as the SDH components subunit D (SDHD), SDHB, and subunit C, have been identified as a trigger for these syndromes.14 Only a few published cases of cardiac paraganglioma have been described in patients with the SDHD and SDHB mutations.6,7 The case presented here was associated with a mutation of the SDHB gene, which is exceedingly rare. In addition, the phenotypic consequences of SDHB gene mutation have not yet been completely defined.15 Malignant behavior in pheochromocytomas as well as the development of multifocal and extra-adrenal tumors is more common in patients with germline mutations in SDH genes. Monitoring for this patient includes family screening, regular BP monitoring, and annual monitoring of 24-hour urine catecholamines.
Conclusion
Isolated cardiac paraganglioma is an extremely rare tumor and poses diagnostic and treatment dilemmas. Multimodality imaging is helpful in diagnosis and surgical planning. Complete surgical excision on cardiopulmonary bypass is the treatment of choice and is curative. All patients diagnosed with this condition should be screened for SDH gene mutation.
Abbreviations and Acronyms
- AV
atrioventricular
- BP
blood pressure
- Ga-68-
Gallium-labeled [1,4,7,10-tetraazacy
- DOTANOC
clododecane-1,4,7,10-tetraacetic
- PET
acid]-1-NaI3-octreotide positron emission tomography
- MRI
magnetic resonance imaging
- RCA
right coronary artery
- RV
right ventricle
- RVOT
RV outflow tract
- SDH
succinyl dehydrogenase
- SDHB
SDH subunit B
Funding Statement
Funding/Support: None
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Vander Salm TJ. Unusual primary tumors of the heart. Semin Thorac Cardiovasc Surg . 2000;12(2):89–100. doi: 10.1053/ct.2000.5080. [DOI] [PubMed] [Google Scholar]
- 2.Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med . 1993;117(10):1027–1031. [PubMed] [Google Scholar]
- 3.Kennelly R, Aziz R, Toner M, Young V. Right atrial paraganglioma: an unusual primary cardiac tumour. Eur J Cardiothorac Surg . 2008;33(6):1150–1152. doi: 10.1016/j.ejcts.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 4.David TE, Lenkei SC, Marquez-Julio A, Goldberg JA, Meldrum DA. Pheochromocytoma of the heart. Ann Thorac Surg . 1986;41(1):98–100. doi: 10.1016/S0003-4975(10)64507-9. [DOI] [PubMed] [Google Scholar]
- 5.Palubinskas AJ, Roizen MF, Conte FA. Localization of functioning pheochromocytomas by venous sampling and radioenzymatic analysis. Radiology . 1980;136(2):495–496. doi: 10.1148/radiology.136.2.7403529. [DOI] [PubMed] [Google Scholar]
- 6.Del Forno B, Zingaro C, Di Palma E, Capestro F, Rescigno G, Torracca L. Cardiac paraganglioma arising from the right atrioventricular groove in a paragangliomapheochromocytoma family syndrome with evidence of SDHB gene mutation: an unusual presentation. Ann Thorac Surg . 2016;102(3):e215–e216. doi: 10.1016/j.athoracsur.2016.01.072. [DOI] [PubMed] [Google Scholar]
- 7.Petramala L, Cotesta D, Filetti S, Letizia C. Pigmented ‘black' cardiac paraganglioma in a patient with a novel germ-line SDHD mutation. Eur J Cardiothorac Surg . 2009;35(1):189. doi: 10.1016/j.ejcts.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Boyd JD. The inferior aortico-pulmonary glomus. Br Med Bull . 1961;17(2):127–131. doi: 10.1093/oxfordjournals.bmb.a069886. [DOI] [Google Scholar]
- 9.Quint LE, Glazer GM, Francis IR, Shapiro B, Chenevert TL. Pheochromocytoma and paraganglioma: comparison of MR imaging with CT and I-131 MIBG scintigraphy. Radiology . 1987;165(1):89–93. doi: 10.1148/radiology.165.1.3628794. [DOI] [PubMed] [Google Scholar]
- 10.Buchmann I, Henze M, Engelbrecht S et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging . 2007;34(10):1617–1626. doi: 10.1007/s00259-007-0450-1. [DOI] [PubMed] [Google Scholar]
- 11.Araoz PA, Mulvagh SL, Tazelaar HD, Julsrud PR, Breen JF. CT and MR imaging of benign primary cardiac neoplasms with echocardiographic correlation. Radiographics . 2000;20(5):1303–1319. doi: 10.1148/radiographics.20.5.g00se121303. [DOI] [PubMed] [Google Scholar]
- 12.Yang T, Li Y, Zhang HM, Wang HY, Song YH. Resection of cardiac pheochromocytoma with cardiopulmonary bypass. Ann Thorac Surg . 2021;111(3):e153–e155. doi: 10.1016/j.athoracsur.2020.06.035. [DOI] [PubMed] [Google Scholar]
- 13.Zhou J, Chen HT, Xiang J, Qu XH, Zhou YQ, Zang WF. Surgical treatment of cardiac pheochromocytoma: a case report. Ann Thorac Surg . 2009;88(1):278–281. doi: 10.1016/j.athoracsur.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 14.Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med . 2009;266(1):19–42. doi: 10.1111/j.1365-2796.2009.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astuti D, Latif F, Dallol A et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet . 2001;69(1):49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]