

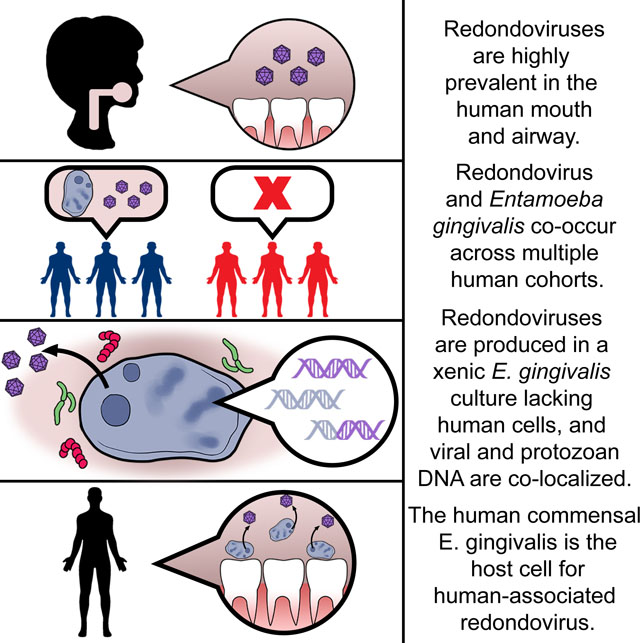
SUMMARY
Redondoviruses are circular Rep-encoding single-stranded DNA (CRESS) viruses of high prevalence in healthy humans. Redondovirus abundance is increased in oro-respiratory samples from individuals with periodontitis, acute illness, and severe COVID-19. We investigated potential host cells supporting redondovirus replication in oro-respiratory samples and uncovered the oral amoeba Entamoeba gingivalis as a likely host. Redondoviruses are closely related to viruses of Entamoeba and contain reduced GC nucleotide content, consistent with Entamoeba hosts. Redondovirus and E. gingivalis co-occur in metagenomic data from oral disease and healthy human cohorts. When grown in xenic cultures with feeder bacteria, E. gingivalis was robustly positive for redondovirus RNA and DNA. A DNA proximity-ligation assay (Hi-C) on xenic culture cells showed enriched cross-linking of redondovirus and Entamoeba DNA, supporting E. gingivalis as the redondovirus host. While bacteria are established hosts for bacteriophages within the human virome, this work shows that eukaryotic commensals also contribute an abundant human-associated virus.
Keywords: Redondovirus, Entamoeba gingivalis, CRESS virus, amoeba, virome, metagenomics, xenic culture
eTOC
Redondoviruses are widely present in human samples and are positively associated with several disease states. Keeler et al. report that redondoviruses are highly associated with Entamoeba gingivalis and appear to replicate within this commensal amoeba. Thus, a commensal eukaryote can serve as a host for members of the human-associated virome.
Graphical Abstract
INTRODUCTION
Redondoviruses were first identified as metagenomic “dark matter” in studies of human respiratory virome samples1,2. In samples from lung transplant donors and recipients1, a few virome sequence reads were initially found to align to porcine stool-associated circular virus 5, an uncharacterized viral genome identified in pig stool. Sequence assembly of the lung transplant samples yielded circular genomes of ~3 kb, with open reading frames (ORFs) distantly similar to capsid (Cap) and replication-associated (Rep) proteins of known CRESS viruses. The genomes recovered were ultimately classified into a newly-established family of viruses3,4 termed Redondoviridae, (from “redondo”; Spanish for circle), with two species, Vientovirus and Brisavirus (from “viento” and “brisa”, Spanish for wind and breeze). Redondoviruses were further confirmed to be CRESS viruses by demonstrating that purified Rep protein displayed the expected enzymatic activities in reconstructed reactions in vitro5.
Use of redondovirus genome sequences as alignment targets to query published metagenomic data revealed that redondoviruses are present almost exclusively in human samples from the oro-respiratory tract. Redondoviruses were found in healthy people from North America, Africa, Europe, and Asia, with prevalence ranging from 2% to >80%, depending on cohort and sample type1,2,5–7. Redondoviruses were detected at elevated levels in subjects with multiple conditions, including lung transplant donors and recipients, febrile patients, and individuals with periodontitis, rheumatoid arthritis, inflammatory bowel disease, HIV infection, respiratory and oropharyngeal disease, critical illness, and COVID-191,6–9.
Here we investigated the cell type responsible for hosting redondovirus replication. Redondoviruses do not appear to replicate in prokaryotic cells because i) redondovirus genomes do not contain the Shine-Dalgarno translation initiation sequence that is characteristic of bacteriophages10 and ii) redondovirus sequences are have not been found in arrays of prokaryotic CRISPR spacers1,11,12. Additionally, redondoviruses are phylogenetically distant from ssDNA bacteriophages (Figure S1A; Table S1). Given that redondoviruses have only been consistently detected in human samples, a simple model would be that redondoviruses replicate in human cells. However, evidence presented below identifies the human-associated amoeba Entamoeba gingivalis as the likely organism hosting redondovirus replication.
RESULTS
Redondovirus phylogeny and GC content are consistent with an Entamoeba host
A detailed phylogeny was recently proposed for CRESS viruses3,13, including Redondoviridae. To investigate Redondoviridae further, we updated this phylogeny to include three CRESS virus families recently described in 13 and generated a maximum-likelihood tree of 441 Rep protein sequences (Table S1) from members of the Cressdnaviricota phylum, annotated with probable host organisms (Figure 1A). A close neighbor to the Redondoviridae family is Naryaviridae, whose members have recently been proposed to replicate in Entamoeba parasites13, suggesting that an Entamoeba species could be a candidate host of redondovirus replication as well.
Figure 1.

Investigating the host cell supporting redondovirus replication. (A) Phylogenetic maximum-likelihood tree of Rep amino acid sequences from 441 members of the Cressdnaviricota phylum (Table S1). Rep sequences were aligned with MUSCLE53, followed by tree construction using RaxML54 and visualization using iTOL55. Clade coloring denotes the proposed host of the respective CRESS virus family. (B) Comparative analysis of GC content (% GC) of representative CRESS viruses and their hosts. Virus GC content positively correlates with host GC content (R2 = 0.63, Pearson’s Correlation Test p-value = 5.442e-10, n = 41). The Entamoeba arrows denote (from left to right): E. dispar (24.1%), E. histolytica (24.95%), E. nuttalli (25.1%), E. moshkovskii (26.5%), and E. invadens (30.3%). The dotted horizontal line represents the median GC content of Redondoviridae (34.25%). Figures adapted from Kinsella and coworkers 13.
Redondovirus DNA sequences are highly represented in sequencing data from samples of the gingival crevice of periodontitis patients1,9 and the amoeba E. gingivalis contributes the second most abundant ribosomal RNA (rRNA) detected in such samples, after human14. Thus E. gingivalis is a candidate for the cell hosting redondovirus replication.
The GC content (% GC) of DNA viruses, including CRESS viruses, often mimics that of their hosts13,15. We compared the GC content of CRESS virus lineages against those of known or proposed hosts using linear regression (Figure 1B; Table S2). This analysis confirmed the relationship between virus and host GC content (R2 = 0.63, Pearson’s Correlation Test p-value = 5.44e-10). The median GC contents of the two Redondoviridae species, Vientovirus and Brisavirus, are 34.6% and 33.9%, respectively. The genome of E. gingivalis has not been sequenced, thus redondovirus GC content was compared to that of the five Entamoeba species with complete publicly available genome sequences (E. histolytica, E. dispar, E. nuttalli, E. invadens, E. moshkovskii). Similar to redondovirus genomes, Entamoeba sequences are of low GC content, ranging from 24.1 to 30.3% depending on the species. In contrast, the GC content of the human genome is 40.9%16. This our analysis of GC content points to Entamoeba as a more likely host cell for redondovirus replication than human cells. Based on these findings, we investigated E. gingivalis as a candidate host for redondovirus replication.
Redondovirus and E. gingivalis co-occur in metagenomic data from subjects with oral disease and healthy controls
We quantified whether redondoviruses and E. gingivalis co-occurred over a variety of human-derived sample types, as would be expected if E. gingivalis is the host cell for redondovirus replication. We queried 58 metagenomic datasets encompassing 7,869 samples from diverse human body sites and disease states for the presence of redondovirus sequences, and then assessed the presence of E. gingivalis DNA (Table S3). For this, 81 complete redondovirus genomes and 28 E. gingivalis small subunit 18S rRNA genes were used as alignment targets (Table S4; because the genome of E. gingivalis has not been fully sequenced, we used the 18S rRNA gene in our queries). Four datasets (NCBI BioProject IDs: PRJEB4270117, PRJNA55229418, PRJNA50838518, PRJNA54771719) were positive for redondovirus DNA. The studies all described metagenomic analysis of samples from the oral cavity of healthy individuals and patients with peri-implantitis, mucositis, or periodontitis.
In metagenomic sequence studies of submucosal and subgingival plaque of healthy controls and patients with peri-implantitis or mucositis19, a distribution was seen of samples that were positive and negative for both E. gingivalis and redondovirus sequences, allowing statistical analysis of co-occurrence. In each case, redondoviruses occurrence was found to be highly correlated with that of E. gingivalis (Table 1, top three rows).
Table 1.
Co-occurrence of redondoviruses (RV) and E. gingivalis (EG) in human clinical specimens.
| Subject status | Number of subjects | Sample type† | Redondovirus status | E. gingivalis status | p-value | Reference | Detection method | |
|---|---|---|---|---|---|---|---|---|
| EG+ | EG- | |||||||
| Peri-implantitis patients | 41 | Subgingival plaque |
RV+
RV- |
10 1 |
11 19 |
0.003626 | Ghensi et al., 2020 | Metagenomic sequencing |
| Mucositis patients | 37 | Subgingival plaque |
RV+
RV- |
7 1 |
4 25 |
0.0002265 | Ghensi et al., 2020 | Metagenomic sequencing |
| Healthy volunteers | 35 | Subgingival plaque |
RV+
RV- |
7 1 |
2 25 |
4.02E-5 | Ghensi et al., 2020 | Metagenomic sequencing |
| ICU patients | 38 | OP, NP, ETA |
RV+
RV- |
3 0 |
0 35 |
0.00012 | Merenstein et al., 2021; this study | qPCR |
| COVID ICU patients | 88 | OP, NP, ETA |
RV+
RV- |
9 5 |
1 73 |
3.30E-8 | Merenstein et al., 2021; this study | qPCR |
| Healthy volunteers | 50 | Saliva |
RV+
RV- |
15 7 |
1 27 |
9.85E-7 | Taylor et al., 2021; this study | qPCR |
OP, oropharyngeal; NP, nasopharyngeal; ETA, endotracheal aspirate.
For samples from periodontitis patients, two datasets were positive for redondovirus DNA and analyzed further17,18. E. gingivalis was detected in all samples in both datasets, precluding analysis of co-occurrence. We instead compared the maximum coverage of a redondovirus genome to the maximum coverage of an E. gingivalis 18S rRNA gene (Figure 2), revealing a strong positive association (R2 = 0.493, Pearson’s Correlation Test p-value = 2.501e-9, n = 130).
Figure 2.

Redondovirus and E. gingivalis abundance are positively associated in metagenomic DNA sequence data. Redondovirus relative abundance (measured using the maximum redondovirus genome coverage per sample) positively correlates with E. gingivalis relative abundance (measured using the maximum E. gingivalis 18S rRNA gene coverage per sample) in metagenomic data (NCBI BioProject IDs: PRJEB4270117, PRJNA55229418, PRJNA50838518) (R2 = 0.49, Pearson’s Correlation Test p-value = 2.501e-9, n = 130).
Redondovirus and E. gingivalis co-occur in critically ill patients, COVID-19 patients, and healthy controls when analyzed by quantitative PCR
We next investigated co-occurrence of redondovirus and E. gingivalis DNA in disease states previously found to be associated with elevated redondovirus prevalence using quantitative PCR (qPCR) (Table 1, bottom three rows). The PCR amplicons targeted the E. gingivalis 18S rRNA gene and a conserved region of the redondovirus Cap ORF.
Previously we reported that redondovirus levels were increased in upper respiratory samples from hospitalized patients with acute illness1. Here we surveyed oropharyngeal, nasopharyngeal, and endotracheal aspirate samples from 38 medical intensive care unit patients (Table S5). Where multiple samples were available per subject, if any one sample was positive for redondoviruses or E. gingivalis, we scored the patient as positive. Of the 38 patients, three were positive for redondoviruses, all of which were also positive for E. gingivalis (Table 1, row 4; Fisher’s Exact Test p-value = 0.00012).
We also revisited a cohort of 88 subjects hospitalized with COVID-19, where we previously reported an association between redondoviruses and disease severity (Table S5)8. Quantification of E. gingivalis prevalence showed a positive association between redondoviruses and E. gingivalis (Table 1, row 5; Fisher’s Exact Test p-value = 3.3e-8).
Lastly, we compared redondovirus and E. gingivalis prevalence in human saliva samples from 50 healthy volunteers in Philadelphia (Table S6). These samples were previously characterized for redondovirus prevalence5. Here we tested the samples for the presence of E. gingivalis DNA. Of the 16 samples that were positive for redondovirus DNA, 15 were also positive for E. gingivalis DNA, again showing co-occurrence (Table 1, row 6; Fisher’s Exact Test p-value = 9.85e-7).
Redondovirus and E. gingivalis co-occur in metatranscriptomic data derived from periodontitis patients
We reasoned that the cells hosting redondovirus replication should contain redondovirus RNA, and thus queried 18 publicly available RNA-seq datasets encompassing 1,879 samples from diverse human body sites (Table S7). We first screened for redondovirus sequences and then asked whether E. gingivalis sequences were detectable in the redondovirus-positive datasets. The alignment targets were those described above (Table S4). Two datasets 20,21, both of which contained samples from the gingival crevice of periodontitis patients, were positive for redondovirus RNA and were analyzed further.
The first study sequenced the metatranscriptome of matched healthy and diseased gingiva from three individuals with aggressive periodontitis20. All six samples (from both diseased and healthy sites) were positive for E. gingivalis RNA. Only the three samples from diseased sites were also positive for redondovirus RNA (Figure 3A–C). Substantially more RNA reads mapped to the E. gingivalis alignment targets in the diseased, redondovirus-positive samples (3,486.58 reads per kilobase per million (RPKM)) compared to the healthy, redondovirus-negative samples (33.25 RPKM).
Figure 3.

Detection of redondovirus and E. gingivalis RNA in metatranscriptomic datasets. Alignments show RNA reads from two datasets derived from the gingival crevice of periodontitis patients (NCBI BioProject IDs: PRJNA22162020, PRJNA31979021). Panels (A)-(D) represent individual samples from the datasets. The left side of each panel shows reads aligned to a redondovirus genome, with ORFs indicated at the bottom; the right side of each panel shows reads aligned to the E. gingivalis 18S rRNA gene. Panels (A)-(C) show samples from diseased gingival crevices of three patients sequenced in PRJNA22162020, and panel (D) shows pooled gingival tissues from periodontitis patients sequenced in PRJNA31979021. Plots were generated using the hisss pipeline (https://github.com/louiejtaylor/hisss). Query targets for alignment were (A) redondovirus NCBI accession MK059756.1 and E. gingivalis accession KX027290.1; (B) redondovirus NCBI accession MT482429.1 and E. gingivalis accession KX027290.1; (C) redondovirus accession MK059758.1 and E. gingivalis KX027293.1; (D) redondovirus accession MT482430.1 and E. gingivalis accession MG601094.1. Other samples from these datasets did not contain detectable redondovirus sequences and thus are not shown.
The second study analyzed the metatranscriptome of pooled gingival tissues from periodontitis patients and healthy controls21. Both the healthy and diseased pools were positive for E. gingivalis RNA, while redondovirus RNA was only detected in the diseased pool (Figure 3D). As before, more RNA reads mapped to the E. gingivalis 18S rRNA gene in the diseased, redondovirus-positive sample than in the healthy, redondovirus-negative sample (19.46 RPKM and 11.14 RPKM, respectively).
We then assessed the correlation between the maximum coverage to a redondovirus genome and an E. gingivalis 18S rRNA gene across both sequencing studies, which revealed a trend toward positive association (R2 = 0.57, Pearson’s Correlation Test p-value = 0.137; not shown).
Detection of redondovirus nucleic acids in a xenic E. gingivalis culture
E. gingivalis has not yet been grown in pure culture but can be grown in the presence of feeder bacteria. Such a xenic culture was obtained from ATCC and analyzed (E. gingivalis ATCC-30956). The culture contained cells with the morphology expected for Entamoeba trophozoites (Figure 4A) and the presence of E. gingivalis RNA and DNA was confirmed by qPCR (Figure 4B). The culture was also robustly positive for redondovirus nucleic acids, with 2.06e8 DNA copies/mL and 530.17 RNA copies/mL of culture fluid (Figure 4B). Quantitative PCR analysis targeting human GAPDH yielded no detectable signal, confirming the absence of human cells in the xenic culture. This finding implies that redondoviruses replicate in one of the unicellular organisms present in the xenic culture and not in human cells.
Figure 4.

Detection of redondovirus DNA and RNA in a xenic E. gingivalis culture. (A) Image of the xenic E. gingivalis culture (ATCC-30956) at 100X magnification stained using Kwik-Diff. Scale bar measures a cell (~20 um) that is morphologically consistent a E. gingivalis trophozoite (arrows). No cells with morphology expected for human cells were observed. (B) Detection of redondovirus and E. gingivalis DNA and RNA in the E. gingivalis culture by qPCR and RT-qPCR. (C) Agarose gel electrophoresis showing redondovirus genomic DNA amplification product of the expected size (~3 kb), generated by PCR of DNA from the xenic culture with “back-to-back” PCR primers targeting the circular redondovirus genome. (D) Genome map of the Vientovirus sequenced from the xenic E. gingivalis culture (RV-30956). The largest ORF (violet) encodes the putative capsid (Cap) protein, the second largest (salmon) encodes the Replication-associated (Rep) protein, and the third (grey) encodes a protein of unknown function (ORF3). (E) The RV-30956 sequence placed in a Redondoviridae phylogeny. Rep amino acid sequences from 37 redondoviruses were aligned with MUSCLE53. The tree was built using PhyML with branch support determined by approximate likelihood ratio test56 and visualized using iTOL55. (F) Metagenomic sequence analysis of the E. gingivalis xenic culture. Taxa were identified using Kraken257 and BLASTn58. Only results with reads that could be assigned taxonomically are shown.
A complete redondovirus genome was recovered from the xenic culture by PCR and DNA sequencing (Figure 4C–E). This genome, named RV-30956, is 3,162 bp in length and encodes the expected Cap, Rep, and ORF3 proteins. To compare redondovirus RV-30956 to previously determined redondovirus sequences, we integrated the RV-30956 Rep sequence into a maximum-likelihood tree of 36 reported redondovirus Rep sequences. This indicated that the redondovirus isolate from the xenic culture is a member of the Vientovirus species (Figure 4E). RV-30956 is close in sequence to redondoviruses that have been identified in other human specimens. For instance, RV-30956 Rep and its nearest relative, KY328746.1 Rep, which was isolated from a human respiratory specimen7, share 71.4% amino acid identity (Figure S1B).
Analysis of the redondovirus host using Hi-C
The detection of redondoviruses in a xenic E. gingivalis culture suggests that redondoviruses are capable of replication in the absence of human cells. However, given that the xenic culture was a mixture of cell types, it does not definitively establish E. gingivalis as the host cell. Metagenomic analysis showed that overall, the xenic culture was dominated by bacteria (Figure 4F). E. gingivalis ribosomal rRNA gene sequences were detectable (96.1% coverage), but not human rRNA gene sequences. Theoretically, one of the bacteria in the xenic culture might serve as the host for redondovirus replication, though as mentioned above, the lack of Shine-Dalgarno sequences in the redondovirus genome and absence of redondovirus sequences among bacterial CRISPR spacers argue against a bacterial host.
To investigate the identity the redondovirus host cell, we performed in situ DNA cross-linking using Hi-C22. Cells from the xenic E. gingivalis culture were partially permeabilized and then exposed to a DNA cross-linking agent to physically link DNA sequences in close proximity, such as redondovirus DNA and genomic DNA from the same cell. By this means, extrachromosomal DNA such as viral replication intermediates can be cross-linked to cellular genomic DNA. Following DNA cleavage and ligation, linked DNA fragments formed chimeric molecules that can be analyzed by paired-end sequencing, permitting the recovery of chimeric sequences containing extrachromosomal DNA (e.g., the redondovirus genome) linked to the host cell chromosome (Figure 5A). Although CRESS viruses are inferred to package single-stranded DNA in viral particles, replication inside host cells is thought to involve a double-stranded DNA intermediate23,24.
Figure 5.

Linking of redondovirus and E. gingivalis DNA in a xenic culture using DNA crosslinking and high-throughput sequencing (Hi-C). (A) Schematic diagram of the Hi-C chromatin conformation capture method (Phase Genomics). (B) Numbers of reads in cross-linked Hi-C pairs from the xenic culture where one read annotates as redondovirus, and the other annotates as redondovirus, Entamoeba, or bacteria. (C) Quantifying the degree of enrichment of Entamoeba reads among Hi-C reads linked to redondovirus sequences. The percentage of Hi-C reads linked to redondovirus mate pairs was compared to shotgun sequencing reads in total DNA from the xenic culture assigned to redondovirus, Entamoeba, and bacteria. Enrichment or depletion was determined by dividing the Hi-C percentage by the whole genome shotgun percentage and then applying a log10 transformation. Only 0.005% of total metagenomic reads in the culture could be identified as Entamoeba sequences using BLASTn; in contrast Parabacteroides represented 20% of all identifiable metagenomic reads in the culture.
A total of 114 Hi-C sequence reads matched redondoviruses and had mates that could be identified via BLASTn against the NCBI nucleotide database (Figure 5B). Of those, 101 were linked to mate pairs that also corresponded to redondovirus sequences. In 6 cases, redondovirus reads were paired with reads annotated as Entamoeba (Table S8), significantly more than would be expected by chance (in 10,000 random draws, only 10 contained one E. gingivalis read, none had more; Permutation test p-value <0.00001). Two reads were identified as linking to bacteria in the genus Parabacteroides. No other taxa were identified with more than one read linked to a redondovirus sequence and no reads were found linking redondovirus DNA to human DNA. The degree of enrichment of redondovirus-Entamoeba and redondovirus-redondovirus pairs is quantified in Figure 5C. Reads linking redondovirus sequences to bacteria may either arise due to artifactual ligation during sequence library preparation or possibly reflect the presence of bacterial DNA inside E. gingivalis cells associated with predation25. As a positive control for the Hi-C data, reads aligning to abundant bacterial species were analyzed and shown to be enriched in mate pairs aligning to the same bacterial species (Figure S2). These findings support E. gingivalis as a host for redondovirus replication.
DISCUSSION
Sequence analysis of viral particles isolated from human samples commonly yields a majority of reads that do not closely match any known viruses — the virome “dark matter.” Extensive efforts are under way to understand the origin and nature of the full human virome26–29. Here we present evidence that a newly annotated family of human-associated viruses, Redondoviridae, in fact appear to replicate in the human-associated amoeba Entamoeba gingivalis.
While considerable attention has focused on human virome constituents that infect the bacterial microbiome (i.e., bacteriophages), viruses of human eukaryotic commensals remain understudied. Viruses are known that infect a few eukaryotic parasites of humans, including Leishmania, Giardia, Trichomonas, Cryptosporidium, Plasmodium, and Entamoeba13,30–40. In rare cases these viruses have been detected in metagenomic samples from humans. For instance, the CRESS virus family Naryaviridae has been proposed to infect Entamoeba species and its members have been detected in human stool samples13. One of the viral endosymbionts of Leishmania, Leishmania RNA virus-1, is reported to enhance Leishmania virulence by promoting parasite persistence in the host41; it is unknown whether redondovirus infection influences E. gingivalis pathogenesis. Given the widespread distribution and high prevalence of redondoviruses in some surveys1,5–7, these findings underscore that, like viruses of the bacterial microbiome, viruses of human-associated eukaryotic commensals are potentially significant contributors to the human virome. This work further establishes that a virus of the human oro-respiratory tract can be traced to a eukaryotic commensal host.
Redondoviruses have been previously associated with several human disease states, such as periodontitis, critical illness, and COVID-191,8,9. These earlier observations suggested possible roles for redondoviruses in pathogenesis, but results reported here suggest that redondoviruses may instead be markers for E. gingivalis colonization or infection. It is increasingly appreciated that bacteriophages can have direct effects on human cells despite those cells not serving as hosts for infection and are even targets of immune recognition 42–44; it remains unclear whether redondoviruses have analogous direct effects on human cells.
Studies of periodontitis have previously reported an association of E. gingivalis with disease14,45–48 and results presented here based on tracking with redondovirus DNA raise the question of possible participation in additional disorders. E. gingivalis is a common inhabitant of the human oral cavity14,45–48 and has been detected, though infrequently, in the lungs49. Redondoviruses similarly have been commonly found in the oral cavity1,5,9 and respiratory tract 1,6,8. While it is possible that E. gingivalis may have a broader habitat in humans than has been recognized, it is also plausible that both E. gingivalis and redondoviruses translocate from the oral cavity to the lung during disease. Increased detection of redondoviruses and E. gingivalis in upper respiratory specimens in some conditions could also result from treatments that affect oral and oropharyngeal drainage, such as endotracheal intubation or alterations in oral hygiene in severe illness. Nevertheless, the importance of E. gingivalis colonization and infection in this setting is uninvestigated. Possibilities range from E. gingivalis being a benign transient or nonpathogenic colonizer of the respiratory tract to it being a contributor to pathogenesis and/or inflammation. E. gingivalis has been documented to ingest human cells48,50, and E. gingivalis can kill live epithelial cells by trogocytosis51, suggesting pathogenic potential. Our qPCR analysis supported an association of E. gingivalis and endotracheal intubation in COVID-19. Thus, it will be useful to investigate the importance of E. gingivalis in acute lung injury and respiratory failure more fully.
This study has several limitations. E. gingivalis has not been grown in pure culture, thus it was not possible to study redondovirus growth with experimental infections. The inability to grow E. gingivalis axenically has also hindered efforts to sequence the parasite genome, which considerably complicated our Hi-C analysis — despite this, we were able to find sufficient numbers of redondovirus sequences linked to recognizable Entamoeba sequences to make the association unlikely to be a result of chance. Within our study, difficulties working with the xenic E. gingivalis culture precluded further investigation such as fluorescence in situ hybridization to colocalize redondovirus and E. gingivalis sequences. So far, we have evidence for growth of redondoviruses in cells of E. gingivalis only, but our data do not rule out the possibility that redondoviruses may infect additional Entamoeba species or other hosts.
In summary, this study provides evidence that the widely prevalent, human-associated redondoviruses replicate in E. gingivalis cells. Most studies of the human virome report abundant viruses replicating in human-associated bacteria, but not viruses of eukaryotic commensals — thus our data focus attention on a little studied part of the human virome. Here we emphasize that human-associated eukaryotes can also contribute commonly encountered members of the human virome. These eukaryotic commensal viruses, in turn, can then be markers for the presence of their hosts, allowing investigation of virus/commensal/human interactions and microbe-disease associations, and may even allow for viral modulation of the eukaryotic microbiome in a manner analogous to phage therapy.
Note added in proof
While this work was under review Kinsella et al. reported based on bioinformatics analysis of published data that presence of redondoviruses correlated with presence of E. gingivalis 52.
STAR methods
RESOURCE AVAILABILITY
Lead contacts
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frederic Bushman (bushman@pennmedicine.upenn.edu).
Materials availability
This study did not generate any unique reagents.
Data and code availability
The metatranscriptomic and metagenomic screens used publicly available datasets (NCBI BioProject accessions are listed in Table S3 and Table S7, respectively). The redondovirus sequence obtained from the xenic E. gingivalis culture (RV-30956) has been deposited in GenBank under the accession ON986208. Metagenomic and Hi-C raw reads have been deposited in NCBI under the BioProject accession PRJNA858476.
This study does not report original code and all computational resources used are publicly available as of the date of publication and are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Saliva samples from healthy humans | Taylor et al., 2021 | N/A |
| Oropharyngeal, nasopharyngeal, and endotracheal aspirate samples subjects hospitalized with COVID-19 |
Merenstein et al., 2021 | N/A |
| Oropharyngeal, nasopharyngeal, and endotracheal aspirate samples from medical intensive care unit patients | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Phi29 polymerase | New England BioLabs | Cat#M0269 |
| Critical commercial assays | ||
| TaqMan Fast Universal PCR Master Mix | Thermo Fisher | Cat#4352042 |
| TaqMan Fast Virus 1-Step Master Mix | Thermo Fisher | Cat#5555532 |
| PowerUp SYBR Green Mix | Thermo Fisher | Cat#A25741 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| QIAamp DNA Mini Kit | Qiagen | Cat#51304 |
| SuperScript First-Strand Synthesis System Kit | Thermo Fisher | Cat#18080051 |
| Scientific Kwik-Diff™ Staining Kit | Thermo Fisher | Cat#9990700 |
| Phusion High-Fidelity PCR Kit | New England BioLabs | Cat#E0553S |
| Monarch DNA Gel Extraction Kit | New England BioLabs | Cat#T1020S |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC1311024 |
| Deposited data | ||
| Raw metagenomic sequencing reads | NCBI | Accession #: PRJNA858476 |
| Raw shotgun sequencing reads | NCBI | Accession #: PRJNA858476 |
| Redondovirus (RV-30956) genome | GenBank | Accession #: ON986208 |
| Experimental models: Organisms/strains | ||
| E. gingivalis | ATCC | ATCC-30956 |
| Oligonucleotides | ||
| qPCR, redondovirus Cap ORF (F: 5’-GGATGCCATGAAACTTTGATAC-3’; R: 5’TCTTCCTCCTTATTTGTATGGC-3’; probe: 5’-CCCATACTTACGCCGGTTACCTGC-3’) | Integrated DNA Technologies | N/A |
| qPCR, E. gingivalis 18S rRNA gene (F: 5’-TACCATACAAGGAATAGCTTTGTGAATAA-3’; R: 5’-ACAATTGTAAATTTGTTCTTTTTCT-3’) | Integrated DNA Technologies | N/A |
| Whole-genome PCR, redondovirus (Set A F: 5’CCTTTGGTCTCGAAATCTTCCTATACTGG-3’; Set A R: 5’-AGGCCTCTCTCCCTTCCATTTGG-3’; Set B F: 5’-GGTTATCGTTCATTTGATCATGCATTAGTACC-3’; Set B R: 5’-ACCAAGATGTTTAAGCCCTTTAGTTAATGTTTC-3’) | Integrated DNA Technologies | N/A |
| qPCR, human GAPDH (F: 5’-GGTGGTCTCCTCTGACTTCAACA-3’; R: 5’CCAGCCACATACCAGGAAATG-3’; probe: 5’CTGGCATTGCCCTCAACGACCAC-3’) | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pUC57 | BioBasic | Addgene 4509 |
| Software and algorithms | ||
| BLASTn v2.9.0 | Altschul et al., 1990 | https://www.ncbi.nlm.nih.gov/books/NBK279690/ |
| PhyML v3.0 | Guindon et al., 2010 | https://github.com/stephaneguindon/phyml |
| RaxML v8.2 | Stamatakis et al., 2005 | https://github.com/stamatak/standard-RAxML |
| MUSCLE v3.8.31 | Edgar, 2004 | https://github.com/rcedgar/muscle |
| iTOL v6 | Letunic and Bork, 2019 | https://itol.embl.de/ |
| VipTree v3.1 | Nishimura et al., 2017 | https://www.genome.jp/viptree/ |
| hisss v3.0 | Abbas et al., 2019 | https://github.com/louiejtaylor/hisss |
| IGV v2.9.0 | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| CAP3 v3.0 | Huang and Madan, 1999 | http://doua.prabi.fr/software/cap3 |
| MEGAHIT v1.2.9 | Li et al., 2015 | https://github.com/voutcn/megahit |
| FastQC v0.11.9 | Andrews, 2010 | https://github.com/s-andrews/FastQC |
| Trimmomatic v0.39 | Bolger et al., 2014 | https://github.com/usadellab/Trimmomatic |
| Sunbeam v2.1.0 | Clarke et al., 2019 | https://github.com/sunbeam-labs/sunbeam |
| Kraken2 v2.1.2 | Wood et al., 2019 | https://github.com/DerrickWood/kraken2 |
| R version v4.0.4 | Ihaka and Gentleman, 1996 | https://www.r-project.org/ |
| Snakemake v7.16.0 | Köster and Rahmann, 2012 | https://github.com/snakemake/snakemake |
| SAMtools v1.9 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| BEDtools v2.30.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human studies
Saliva samples were collected as previously described in 5 from healthy volunteers in Philadelphia following written informed consent under protocol #842613 approved by the institutional IRB. Endotracheal aspirate samples, oropharyngeal swab samples, and nasopharyngeal swabs were collected as previously described in 8 from patients in the medical intensive care unit of the Hospital of the University of Pennsylvania following written or verbal informed consent from patients or surrogates under protocol #823392 approved by the institutional IRB. Specimens were stored at −80°C until processing. Clinical data were extracted from the electronic medical record.
E. gingivalis xenic culture conditions
TYGM-9 medium (ATCC Medium 1171) was purchased from ATCC or prepared in-house according to the manufacturer’s instructions (ATCC). To prepare a rice starch solution (a media component), 0.5 g of rice starch (Sigma-Aldrich) was suspended in 9.5 mL of sterile phosphate buffered saline solution (pH 7.4). Undissolved rice starch was removed through centrifugation (100 × g for 5 min). Sterile TYGM-9 medium and starch solution were stored at 4°C.
Culture of E. gingivalis was carried out as instructed by staff at ATCC. A xenic E. gingivalis culture (ATCC-30956) was obtained from ATCC and upon arrival incubated at a 15° horizontal slant at 35°C for 3 hr. The tube was then gently inverted and centrifuged at 500 × g for 5 min to pellet E. gingivalis cells. To generate bacterized medium, 250 uL of the supernatant (containing bacteria) was removed from the culture and added to another tube containing 10 mL of fresh TYGM-9 medium. All but ~1 mL of the remaining supernatant was divided among eight 16 × 125 mm screw-capped tubes and fresh TYGM-9 medium was added to increase the volume of each tube to 8 mL. The remaining culture material (a cell pellet and ~1 mL of supernatant) was stored on ice for 5 min, inverted 20 times, and transferred as 0.1-mL aliquots to the eight tubes. The ratio of rice starch and bacterized medium was varied among subcultures in an effort to optimize growth. All subcultures, in addition to the tube of bacterized TYGM-9 medium, were incubated a 15° horizontal slant at 35°C. Every 24–48 hr, the procedure described above was repeated on all subcultures.
E. gingivalis culture viability was confirmed every 72 hr using trypan blue staining. After inverting each sub-culture 10 times, a 1-mL aliquot was centrifuged for 500 × g for 5 min, and the cell pellet was resuspended in 100 uL of PBS. 10 uL of 0.4% trypan blue (Sigma-Aldrich) was mixed with 10 uL of the cell suspension. After a 3 min incubation at room temperature, 10 uL of the mixture was deposited on a microscope slide and a coverslip was applied. An inverted binocular microscope was used to view the sample and permit the enumeration of viable (unstained) and non-viable (stained) E. gingivalis cells.
To image the xenic E. gingivalis culture, 500 uL of culture material was pelleted at 500 × g for 10 min and resuspended in 500 uL of sterile saline. Next 10 uL was deposited onto microscope slides using a Cytopro cytospin (ELITechGroup Inc) (1,000 × g for 5 min). Slides were air-dried, fixed in methanol, and stained using a Kwik Diff staining kit (Methylene blue and eosin) (Fisher Scientific) following the manufacturer’s instructions (Epredia) prior to being viewed under a microscope at 40X and 100X magnification (oil immersion lens).
METHOD DETAILS
DNA isolation
DNA was extracted from human specimens (e.g., saliva, oropharyngeal swabs, nasopharyngeal swabs, endotracheal aspirates) using the DNeasy Blood and Tissue Kit (Qiagen). When saliva was used as the starting material, the following modifications to the manufacturer’s protocol were made: 250 uL of saliva were used as the starting input in Step 1, 250 uL of ethanol were used in Step 5, and a repeat elution step was performed using the initial eluate to maximize DNA yield. When an aliquot of E. gingivalis xenic culture material was used as the starting material, the following modifications to the manufacturer’s protocol were made: 200 uL of liquid culture were used as the starting input in Step 1 and a repeat elution step was performed using the initial eluate to maximize DNA yield. When human peripheral blood mononuclear cells or human 293T cells were used as the starting material, the protocol was followed with no modification. DNA purity was determined using a Nanodrop 2000/200C spectrophotometer (Thermo Fisher) and DNA yield was measured using PicoGreen (Affymetrix) quantification. Isolated DNA was stored at −20°C.
RNA isolation
RNA was extracted using the RNeasy Mini Kit (Qiagen). When an aliquot of E. gingivalis culture was used as the starting material, the following modification to the manufacturer’s protocol was made: 300 uL of liquid media were used as the starting material and 300 uL of Buffer RLT were used for Step 1. The optional on-column DNase digestion referenced in Step 5 was performed using DNase I Reaction Buffer (10X, 1X final concentration) (New England Biolabs, NEB) and RNase-free H2O. RNA purity and yield were determined using an Eppendorf BioPhotometer D30 (Eppendorf). Isolated RNA was stored at −80°C.
Multiple displacement amplification and quantitative PCR
For redondovirus detection (presence/absence), extracted DNA was subjected to multiple displacement amplification (MDA), which is sequence-unbiased but preferentially amplifies circular DNA, followed by qPCR; for quantification, extracted DNA was subject to qPCR without prior MDA. MDA and qPCR were performed as previously described1,59,60.
In brief, MDA was carried out using Phi29 Buffer (10X, 1X final concentration) (NEB), BSA (20 mg/mL, 0.1 mg/mL final concentration) (NEB), Phi29 DNA Polymerase (10 units/uL, 10 units final concentration) (NEB), random hexamers (50 uM, 2 uM final concentration) (Invitrogen), dNTPs (10 mM, 1mM final concentration) (NEB), and molecular grade H2O. MDA used the following conditions on a Veriti 96-well thermocycler (Thermo Fisher): 35°C for 5 min, 34°C for 10 min, 33°C for 15 min, 32°C for 20 min, 31°C for 30 min, 30°C for 16 h, and a final extension at 65°C for 15 min.
For the detection and quantification of redondovirus DNA, MDA-amplified and unamplified DNA samples were run in duplicate in a real-time qPCR assay using TaqMan Fast Universal Master Mix (2X, 1X final concentration) (Thermo Fisher), primer-probe mix (Integrated DNA Technologies (IDT)) based on conserved segments of the redondovirus genome (F: 5’-GGATGCCATGAAACTTTGATAC-3’; R: 5’-TCTTCCTCCTTATTTGTATGGC-3’; probe: 5’-CCCATACTTACGCCGGTTACCTGC-3’), and molecular grade H2O. The following conditions were used for the PCR reaction: 50°C for 2 min, 95°C for 10 min; 40 cycles of 95°C for 15 sec and 60°C for 1 min; and a final extension at 4°C.
For the detection and quantification of redondovirus RNA, RNA samples were run in duplicate in a real-time RT-qPCR assay using TaqMan Fast Virus 1-Step Master Mix (4X, 1X final concentration) (Thermo Fisher), primer-probe mix (IDT) targeting the Cap ORF (F: 5’-GGATGCCATGAAACTTTGATAC-3’; R: 5’-TCTTCCTCCTTATTTGTATGGC-3’; probe: 5’-CCCATACTTACGCCGGTTACCTGC-3’), and molecular grade H2O. Reverse transcription was carried out at 50°C for 5 min. The following conditions were used for the PCR reaction: 95°C for 20 sec; 40 cycles of 95°C for 3 sec and 60°C for 30 sec; and a final extension at 4°C.
For redondovirus qPCR and RT-qPCR, a standard curve was generated from serial dilutions of a plasmid (pUC57) containing the cloned genome of brisavirus AA. qPCRs were run on a QuantStudio5 (Thermo Fisher) machine using the “Fast” mode. Negative controls consistently showed a cycle threshold (CT) of >40 cycles, so positive samples were defined as samples with any CT value <40 cycles. Non-template controls and extraction controls were included in qPCR assays; no negative controls showed amplification. In cases where multiple samples were queried per patient, if one sample was found to be positive for redondoviruses, the patient was scored as positive.
E. gingivalis qPCR was performed as previously described in 47. For E. gingivalis DNA detection, samples were run in duplicate in a real-time qPCR assay using PowerUp SYBR Green Mix (2X, 1X final concentration) (Thermo Fisher), MgCl2 (25 mM, 3 mM final concentration), primer mix (IDT) targeting the 18S rRNA gene (F: 5’-TACCATACAAGGAATAGCTTTGTGAATAA-3’; R: 5’-ACAATTGTAAATTTGTTCTTTTTCT-3’), and molecular grade H2O. The following conditions were used for the PCR reaction: 50°C for 2 min, 95°C for 2 min; 40 cycles of 95°C for 15 sec, 60°C for 15 sec, 72°C for 1 min; 95°C for 15 sec, 66°C for 1 min, 40°C for 30 sec; and a final extension at 4°C. Isolated RNA was reverse transcribed into cDNA using a SuperScript III First-Strand Synthesis System Kit (Invitrogen). For E. gingivalis RNA detection and quantification, the qPCR protocol described above was used with cDNA as the template.
For E. gingivalis qPCR, a standard curve made from serial dilutions of a synthetic gene block (IDT) encoding an E. gingivalis 18S rRNA gene (NCBI accession: KX027295.1) was included in each run. qPCRs were run on a QuantStudio5 (Thermo Fisher) machine. Negative controls consistently showed a cycle threshold (CT) value of ≥40 cycles, so positive samples were defined as samples with any CT value <40 cycles. Non-template controls and extraction controls were included in qPCR assays. In cases where multiple samples were queried per patient, if one sample was found to be positive for E. gingivalis, the patient was scored as positive.
For the detection of human DNA, DNA samples were run in triplicate in a real-time qPCR assay using TaqMan Fast Universal Master Mix (2X, 1X final concentration) (Applied Biosystems), primer-probe mix (IDT) targeting the GAPDH gene (F: 5’-GGTGGTCTCCTCTGACTTCAACA-3’; R: 5’-CCAGCCACATACCAGGAAATG-3’; probe: 5’-CTGGCATTGCCCTCAACGACCAC-3’ 61), and molecular grade H2O. The following conditions were used for the PCR reaction: 50°C for 2 min, 95°C for 10 min; 40 cycles of 95°C for 15 sec and 60°C for 1 min; and a final extension at 4°C. Two standard curves were generated from serial dilutions of DNA isolated from human peripheral blood mononuclear cells and human 293T cells. Negative controls consistently showed a CT value of ≥40 cycles, thus positive samples were defined as samples with any CT value <40 cycles.
Redondovirus whole-genome sequencing from the xenic culture
To recover the complete redondovirus genome from the xenic culture, two whole-genome PCRs were performed with nonoverlapping primer sets (Set A F: 5’-CCTTTGGTCTCGAAATCTTCCTATACTGG-3’; Set A R: 5’-AGGCCTCTCTCCCTTCCATTTGG-3’; Set B F: 5’GGTTATCGTTCATTTGATCATGCATTAGTACC-3’; Set B R: 5’-ACCAAGATGTTTAAGCCCTTTAGTTAATGTTTC-3’) using the Phusion PCR Kit (NEB) and the following PCR settings: 98°C for 30 sec, then 35 cycles of 98°C for 10 sec, 55°C for 15 sec, and 72°C for 1 min 30 sec, followed by a final extension of 10 min at 72°C. The ~3-kb PCR products were visualized on a 1% agarose gel, excised, and purified from gels using a Monarch DNA Gel Extraction Kit (NEB). The manufacturer’s protocol was used with the modification of a 15-uL H2O final elution. After gel extraction, libraries were prepared from PCR products using the Nextera XT DNA Library Preparation Kit (Illumina). Libraries were sequenced using the Illumina MiSeq platform (Illumina).
Read processing and genome assembly
Sunbeam (v2.1)62, a Snakemake-based pipeline63, was used for quality control, host decontamination, and contig assembly as previously described1,62. Trimmomatic (v0.39)64 and FastQC (v0.11.9) were used for adapter trimming and read quality control, respectively. Contigs were assembled from quality-controlled reads using MEGAHIT (v1.2.9)65 and annotated using BLAST against a database of published redondovirus genome sequences1,58. A Sunbeam extension (sbx_select_contigs, https://github.com/ArwaAbbas/sbx_select_contigs ) was used to extract contigs with homology to redondoviruses (as annotated by BLAST). Extracted contigs were overlap-assembled using CAP3 (v3.0)66, circularized based on the overlaps identified by sbx_select_contigs, and polished by aligning quality-controlled reads to the draft genomes. Visualization of alignments in Integrated Genomics Viewer (IGV) (v2.9.0)67 permitted the manual correction of assembly errors, which were rare.
Phylogenetic analysis
For the ssDNA virus phylogenetic tree (Figure S1), we used ViPTree (v3.1) to generate a “proteomic tree” of 2,164 viral genome sequences (10 redondovirus genomes and 2,154 genomes of eukaryotic and prokaryotic viruses in the ViPTree ssDNA database; Table S1) based on genome-wide sequence similarities computed by tBLASTx68. iTOL (v6) was used for tree visualization and clade collapsing (based on average BRL (>0.4))55.
For the Cressdnaviricota tree (Figure 1A), alignments of CRESS virus Rep sequences were performed using MUSCLE (v3.8.31)53. From the alignments, trees were constructed using RAxML (v8.2)54 and visualized using iTOL (v6)55.
For the Redondoviridae tree (Figure 4E), redondovirus Rep amino acid sequences were aligned using MUSCLE (v3.8.31)53. Phylogenetic trees were constructed from sequence alignments using PhyML (v3.0)56 and visualized using iTOL (v6)55.
Hi-C and shotgun library generation and sequencing
A chromatin interaction (Hi-C) library was generated using the Proximo Hi-C (Microbe) Kit (Phase Genomics). After starting with DNA from 200 uL of E. gingivalis culture material, the ProxiMeta Hi-C protocol (v4.0) was followed without modification using proprietary materials supplied with the kit. Cells were partially permeabilized and exposed to a DNA crosslinker. Cells were lysed to release the crosslinked DNA into the supernatant and enable DNA recovery by centrifugation. Endonucleases were then used to fragment the crosslinked DNA. Fragmented DNAs were biotinylated and ligated to create chimeric junctions between adjacent sequences (i.e., sequences that originated from the same cell). Crosslinks were then reversed and the DNA was purified using a streptavidin bead pull down. Streptavidin-bound DNA was quantified using Qubit dsDNA HS, which was used to determine the subsequent adapter dilution and number of PCR cycles. Next a Hi-C library was prepared and on-bead amplification was performed using the Nextera XT DNA Library Preparation Kit (Illumina). In addition, a standard shotgun library of total DNA isolated from the E. gingivalis culture was prepared using the Nextera XT DNA Library Preparation Kit (Illumina) and sequenced to compare to the Hi-C library. Both were sequenced using the Illumina NextSeq 500 platform (Illumina), generating 150 bp paired-end reads. Proximity-ligated reads were mapped against shotgun sequencing data to inform in-cell DNA interactions. 29,415,700 and 109,616,621 reads were obtained from sequencing the shotgun and Hi-C libraries, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Querying public sequence data for redondovirus and E. gingivalis DNA and RNA
To investigate the presence of redondovirus DNA, we queried 58 metagenomic datasets (Table S3). 81 redondovirus virus genomes and 28 E. gingivalis 18S rRNA genes downloaded from GenBank (NCBI) were used as local alignment targets (Table S4)69,70. Alignments were performed using the hisss pipeline (https://github.com/louiejtaylor/hisss) 1, which uses Bowtie 2 (option, –very-sensitive-local)71 to align reads to target genomes, SAMtools72 and BEDtools73 to calculate the coverage of the reads to the target genomes, and ggplot2 in R (v3.3.6) to visualize the alignments. Positive identification was defined as ≥5% coverage to any redondovirus genome or 18S rRNA gene sequence.
To investigate the presence of redondovirus RNA, we queried 18 metatranscriptomic datasets (Table S7) for redondovirus and E. gingivalis RNA (Table S4). The hisss pipeline was used as described above to perform alignments, calculate read coverage to target sequences, and visualize alignments. Positive identification was prospectively defined as ≥0.05 fractional coverage (≥5% coverage) to a redondovirus genome or an E. gingivalis 18S rRNA gene.
Analysis of xenic culture metagenome
Taxonomic assignment of the quality-controlled reads was performed with Kraken2 (v2.1.2)57 using the Standard database (archaea, bacteria, viral, plasmid, human, UniVec_Core) and the PlusPF database (standard database plus protozoa and fungi) (https://benlangmead.github.io/aws-indexes/k2). To supplement the Kraken2 results and capture low-abundance taxa of interest (Redondoviridae, Entamoeba), we performed BLASTn on 10 million reads before estimating relative abundance58.
We used 18S rRNA genes belonging to E. gingivalis and Homo sapiens to query of the metagenomic data obtained from the E. gingivalis culture. The hisss pipeline was used as described above to align the sequencing reads to the alignment targets and calculate coverage of the target sequences.
Hi-C analysis
To assess whether redondovirus genomes resided in the same cells as the E. gingivalis genome, we assessed Hi-C crosslinking between redondovirus and Entamoeba sequences. We created a custom BLASTn database of all available redondovirus genomes from the NCBI nt database58. All Hi-C reads were aligned to this database, identifying 246 forward or reverse reads classified as Redondoviridae. The read mate pairs for each Redondoviridae read were then aligned via BLASTn to the complete nt database (downloaded January 2022). Because there is no whole genome sequence available for Entamoeba gingivalis, we accepted alignments to any member of the Entamoeba genus. We identified 6 chimeric reads indicative of Entamoeba-redondovirus cross-linking (Figure 5; Table S8). To determine the probability of 6 reads aligning to Entamoeba by chance in our data, we took 100,000 random draws of 246 reads and counted Entamoeba reads using the same BLASTn search. As a positive control, reads were identified matching the most abundant bacterial genera in the xenic culture, and mate pairs verified to be highly enriched in sequences aligning to the same bacterial genus (Figure S2). Alignments were carried out using BLAST querying the NCBI database.
Statistical tests
Co-occurrence of redondoviruses and E. gingivalis was assessed using Fisher’s Exact Test (Table 1). Association of virus and host GC content was assessed using Pearson’s Correlation Test (Figure 1). Association of redondovirus and E. gingivalis relative abundance was assessed using Pearson’s Correlation Test (Figure 2). The probability of identifying Hi-C mate pairs by chance was calculated by a permutation test based on 100,000 random draws of 246 reads and counting Entamoeba reads using a BLASTn search.
Supplementary Material
Table S8. Hi-C results for chimeric reads with one pair matching redondovirus and the other matching Entamoeba; related to Figure 5
Table S1. Viral genomes used to generate the ssDNA phylogeny; related to Figure S1A and Figure 1A.
Highlights.
Redondoviruses are highly prevalent in human respiratory samples.
Redondoviruses are associated with periodontitis, critical illness, and COVID-19.
In human samples, redondoviruses co-occur with the amoeba Entamoeba gingivalis.
Redondoviruses are found in E. gingivalis cultures, specifying amoebas as hosts.
ACKNOWLEDGEMENTS
We are grateful to the individuals who provided samples for analysis. We thank members of the Bushman and Collman labs for helpful discussions and suggestions, Laurie Zimmerman and Arwa Abbas for help with illustrations, and staff at ATCC for guidance on management of the E. gingivalis xenic culture. This work was supported by NIH grant R33-HL137063 (R.G.C., F.D.B.); the PennCHOP Microbiome Program; and NIH grant R35 GM134957-01; National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00015 (CEIRR); and American Diabetes Association Pathway to Stop Diabetes grant #1-19-VSN-02 (ST). We acknowledge assistance from multiple cores of the Penn Center for AIDS Research (P30-AI45008) and the Penn Center for Research on Coronaviruses and Other Emerging Pathogens.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Abbas AA, Taylor LJ, Dothard MI, Leiby JS, Fitzgerald AS, Khatib LA, Collman RG, and Bushman FD (2019). Redondoviridae, a Family of Small, Circular DNA Viruses of the Human Oro-Respiratory Tract Associated with Periodontitis and Critical Illness. Cell Host Microbe 26, 297. 10.1016/j.chom.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui L, Wu B, Zhu X, Guo X, Ge Y, Zhao K, Qi X, Shi Z, Zhu F, Sun L, and Zhou M.(2017). Identification and genetic characterization of a novel circular single-stranded DNA virus in a human upper respiratory tract sample. Arch Virol 162, 3305–3312. 10.1007/s00705-017-3481-3. [DOI] [PubMed] [Google Scholar]
- 3.Krupovic M, Varsani A, Kazlauskas D, Breitbart M, Delwart E, Rosario K, Yutin N, Wolf YI, Harrach B, Zerbini FM, et al. (2020). Cressdnaviricota: a Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J Virol 94. 10.1128/JVI.00582-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbas A, Taylor LJ, Collman RG, Bushman FD, and Ictv Report C.(2021). ICTV Virus Taxonomy Profile: Redondoviridae. J Gen Virol 102. 10.1099/jgv.0.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor LJ., Dothard MI., Rubel MA., Allen AA., Hwang Y., Roche AM., Graham-Wooten J., Fitzgerald AS., Khatib LA., Ranciaro A., et al. (2021). Redondovirus Diversity and Evolution on Global, Individual, and Molecular Scales. J Virol 95, e0081721. 10.1128/JVI.00817-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spezia PG, Macera L, Mazzetti P, Curcio M, Biagini C, Sciandra I, Turriziani O, Lai M, Antonelli G, Pistello M, and Maggi F.(2020). Redondovirus DNA in human respiratory samples. J Clin Virol 131, 104586. 10.1016/j.jcv.2020.104586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazaro-Perona F, Dahdouh E, Roman-Soto S, Jimenez-Rodriguez S, Rodriguez-Antolin C, de la Calle F, Agrifoglio A, Membrillo FJ, Garcia-Rodriguez J, and Mingorance J.(2020). Metagenomic Detection of Two Vientoviruses in a Human Sputum Sample. Viruses 12. 10.3390/v12030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merenstein C, Liang G, Whiteside SA, Cobian-Guemes AG, Merlino MS, Taylor LJ, Glascock A, Bittinger K, Tanes C, Graham-Wooten J, et al. (2021). Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. mBio 12, e0177721. 10.1128/mBio.01777-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Wang C, Feng X, Chen X, and Zhang W.(2021). Redondoviridae and periodontitis: a case-control study and identification of five novel redondoviruses from periodontal tissues. Virus Evol 7, veab033. 10.1093/ve/veab033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnamurthy SR, and Wang D.(2018). Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 516, 108–114. 10.1016/j.virol.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Gophna U, and Brodt A.(2012). CRISPR/Cas systems in archaea: What array spacers can teach us about parasitism and gene exchange in the 3rd domain of life. Mob Genet Elements 2, 63–64. 10.4161/mge.19907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero DA., and Horvath P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 13.Kinsella CM, Bart A, Deijs M, Broekhuizen P, Kaczorowska J, Jebbink MF, van Gool T, Cotten M, and van der Hoek L.(2020). Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat Commun 11, 4620. 10.1038/s41467-020-18474-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng ZL, Szafranski SP, Jarek M, Bhuju S, and Wagner-Dobler I.(2017). Dysbiosis in chronic periodontitis: Key microbial players and interactions with the human host. Sci Rep 7, 3703. 10.1038/s41598-017-03804-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardinale DJ, and Duffy S.(2011). Single-stranded genomic architecture constrains optimal codon usage. Bacteriophage 1, 219–224. 10.4161/bact.1.4.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piovesan A, Pelleri MC, Antonaros F, Strippoli P, Caracausi M, and Vitale L.(2019). On the length, weight and GC content of the human genome. BMC Res Notes 12, 106. 10.1186/s13104-019-4137-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plachokova AS, Andreu-Sanchez S, Noz MP, Fu J, and Riksen NP (2021). Oral Microbiome in Relation to Periodontitis Severity and Systemic Inflammation. Int J Mol Sci 22. 10.3390/ijms22115876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altabtbaei K., Maney P., Ganesan SM., Dabdoub SM., Nagaraja HN., and Kumar PS. (2021). Anna Karenina and the subgingival microbiome associated with periodontitis. Microbiome 9, 97. 10.1186/s40168-021-01056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghensi P, Manghi P, Zolfo M, Armanini F, Pasolli E, Bolzan M, Bertelle A, Dell’Acqua F, Dellasega E, Waldner R, et al. (2020). Strong oral plaque microbiome signatures for dental implant diseases identified by strain-resolution metagenomics. NPJ Biofilms Microbiomes 6, 47. 10.1038/s41522-020-00155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, and Whiteley M.(2014). Metatranscriptomics of the human oral microbiome during health and disease. mBio 5, e01012–01014. 10.1128/mBio.01012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YG, Kim M, Kang JH, Kim HJ, Park JW, Lee JM, Suh JY, Kim JY, Lee JH, and Lee Y.(2016). Transcriptome sequencing of gingival biopsies from chronic periodontitis patients reveals novel gene expression and splicing patterns. Hum Genomics 10, 28. 10.1186/s40246016-0084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, and Lander ES (2010). Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp. 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor LJ, Keeler EL, Bushman FD, and Collman RG (2022). The enigmatic roles of Anelloviridae and Redondoviridae in humans. Curr Opin Virol 55, 101248. 10.1016/j.coviro.2022.101248. [DOI] [PubMed] [Google Scholar]
- 24.Zhao L, Rosario K, Breitbart M, and Duffy S.(2019). Eukaryotic Circular Rep-Encoding Single-Stranded DNA (CRESS DNA) Viruses: Ubiquitous Viruses With Small Genomes and a Diverse Host Range. Adv Virus Res 103, 71–133. 10.1016/bs.aivir.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Garcia LS (2001). Diagnostic Medical Parasitology, 4th ed. Edition (ASM Press; ). [Google Scholar]
- 26.Liang G, and Bushman FD (2021). The human virome: assembly, composition and host interactions. Nat Rev Microbiol 19, 514–527. 10.1038/s41579-021-00536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumata R, Ito J, Takahashi K, Suzuki T, and Sato K.(2020). A tissue level atlas of the healthy human virome. BMC Biol 18, 55. 10.1186/s12915-020-00785-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tisza MJ, and Buck CB (2021). A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2023202118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishnamurthy SR, and Wang D.(2017). Origins and challenges of viral dark matter. Virus Res 239, 136–142. 10.1016/j.virusres.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Widmer G, Comeau AM, Furlong DB, Wirth DF, and Patterson JL (1989). Characterization of a RNA virus from the parasite Leishmania. Proc Natl Acad Sci U S A 86, 5979–5982. 10.1073/pnas.86.15.5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantanhede LM., Fernandes FG., Ferreira GEM., Porrozzi R., Ferreira RGM., and Cupolillo E. (2018). New insights into the genetic diversity of Leishmania RNA Virus 1 and its species-specific relationship with Leishmania parasites. PLoS One 13, e0198727. 10.1371/journal.pone.0198727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheffter SM, Ro YT, Chung IK, and Patterson JL (1995). The complete sequence of Leishmania RNA virus LRV2–1, a virus of an Old World parasite strain. Virology 212, 84–90. 10.1006/viro.1995.1456. [DOI] [PubMed] [Google Scholar]
- 33.Nalcaci M, Karakus M, Yilmaz B, Demir S, Ozbilgin A, Ozbel Y, and Toz S.(2019). Detection of Leishmania RNA virus 2 in Leishmania species from Turkey. Trans R Soc Trop Med Hyg 113, 410–417. 10.1093/trstmh/trz023. [DOI] [PubMed] [Google Scholar]
- 34.Grybchuk D, Macedo DH, Kleschenko Y, Kraeva N, Lukashev AN, Bates PA, Kulich P, Lestinova T, Volf P, Kostygov AY, and Yurchenko V.(2020). The First Non-LRV RNA Virus in Leishmania. Viruses 12. 10.3390/v12020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang AL, and Wang CC (1986). Discovery of a specific double-stranded RNA virus in Giardia lamblia. Mol Biochem Parasitol 21, 269–276. 10.1016/0166-6851(86)90132-5. [DOI] [PubMed] [Google Scholar]
- 36.Goodman RP, Ghabrial SA, Fichorova RN, and Nibert ML (2011). Trichomonasvirus: a new genus of protozoan viruses in the family Totiviridae. Arch Virol 156, 171–179. 10.1007/s00705-010-0832-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodman RP., Freret TS., Kul T., Geller AM., Talkington MW., Tang-Fernandez V., Suciu O., Demidenko AA., Ghabrial SA., Beach DH., et al. (2011). Clinical isolates of Trichomonas vaginalis concurrently infected by strains of up to four Trichomonasvirus species (Family Totiviridae). J Virol 85, 4258–4270. 10.1128/JVI.00220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khramtsov NV, Woods KM, Nesterenko MV, Dykstra CC, and Upton SJ (1997). Virus-like, double-stranded RNAs in the parasitic protozoan Cryptosporidium parvum. Mol Microbiol 26, 289–300. 10.1046/j.1365-2958.1997.5721933.x. [DOI] [PubMed] [Google Scholar]
- 39.Nibert ML, Woods KM, Upton SJ, and Ghabrial SA (2009). Cryspovirus: a new genus of protozoan viruses in the family Partitiviridae. Arch Virol 154, 1959–1965. 10.1007/s00705-009-0513-7. [DOI] [PubMed] [Google Scholar]
- 40.Charon J, Grigg MJ, Eden JS, Piera KA, Rana H, William T, Rose K, Davenport MP, Anstey NM, and Holmes EC (2019). Novel RNA viruses associated with Plasmodium vivax in human malaria and Leucocytozoon parasites in avian disease. PLoS Pathog 15, e1008216. 10.1371/journal.ppat.1008216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ives A, Ronet C, Prevel F, Ruzzante G, Fuertes-Marraco S, Schutz F, Zangger H, Revaz-Breton M, Lye LF, Hickerson SM, et al. (2011). Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331, 775–778. 10.1126/science.1199326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweere JM, Van Belleghem JD, Ishak H, Bach MS, Popescu M, Sunkari V., Kaber G., Manasherob R., Suh GA., Cao X., et al. (2019). Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 363. 10.1126/science.aat9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, Ajami NJ, Wong MC, Ghazaryan A, Valentine JF, et al. (2019). Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 25, 285–299 e288. 10.1016/j.chom.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popescu M, Van Belleghem JD, Khosravi A, and Bollyky PL (2021). Bacteriophages and the Immune System. Annu Rev Virol 8, 415–435. 10.1146/annurev-virology-091919-074551. [DOI] [PubMed] [Google Scholar]
- 45.Yaseen A, Mahafzah A, Dababseh D, Taim D, Hamdan AA, Al-Fraihat E, Hassona Y, Sahin GO, Santi-Rocca J, and Sallam M.(2021). Oral Colonization by Entamoeba gingivalis and Trichomonas tenax: A PCR-Based Study in Health, Gingivitis, and Periodontitis. Front Cell Infect Microbiol 11, 782805. 10.3389/fcimb.2021.782805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kikuta N, Yamamoto A, and Goto N.(1996). Detection and identification of Entamoeba gingivalis by specific amplification of rRNA gene. Can J Microbiol 42, 1248–1251. 10.1139/m96-161. [DOI] [PubMed] [Google Scholar]
- 47.Trim RD, Skinner MA, Farone MB, Dubois JD, and Newsome AL (2011). Use of PCR to detect Entamoeba gingivalis in diseased gingival pockets and demonstrate its absence in healthy gingival sites. Parasitol Res 109, 857–864. 10.1007/s00436-011-2312-9. [DOI] [PubMed] [Google Scholar]
- 48.Bonner M., Amard V., Bar-Pinatel C., Charpentier F., Chatard JM., Desmuyck Y., Ihler S., Rochet JP., Roux de La Tribouille V, Saladin L, et al. (2014). Detection of the amoeba Entamoeba gingivalis in periodontal pockets. Parasite 21, 30. 10.1051/parasite/2014029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jian B, Kolansky AS, Baloach ZW, and Gupta PK (2008). Entamoeba gingivalis pulmonary abscess - diagnosed by fine needle aspiration. Cytojournal 5, 12. 10.4103/1742-6413.43179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bao X, Wiehe R, Dommisch H, and Schaefer AS (2020). Entamoeba gingivalis Causes Oral Inflammation and Tissue Destruction. J Dent Res 99, 561–567. 10.1177/0022034520901738. [DOI] [PubMed] [Google Scholar]
- 51.Bao X, Weiner J 3rd, Meckes O, Dommisch H, and Schaefer AS (2021). Entamoeba gingivalis Exerts Severe Pathogenic Effects on the Oral Mucosa. J Dent Res 100, 771–776. 10.1177/00220345211004498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kinsella CM, Deijs M, Becker C, Broekhuizen P, van Gool T, Bart A, Schaefer AS, and van der Hoek L.(2022). Host prediction for disease-associated gastrointestinal cressdnaviruses. Virus Evol 8. 10.1093/ve/veac087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamatakis A, Ludwig T, and Meier H.(2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463. 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 55.Letunic I, and Bork P.(2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259. 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guindon S., Lethiec F., Duroux P., and Gascuel O. (2005). PHYML Online--a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33, W557–559. 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wood DE, Lu J, and Langmead B.(2019). Improved metagenomic analysis with Kraken 2. Genome Biol 20, 257. 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Leichty AR, and Brisson D.(2014). Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics 198, 473–481. 10.1534/genetics.114.165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, et al. (2002). Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A 99, 5261–5266. 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yi Y, Shaheen F, and Collman RG (2005). Preferential use of CXCR4 by R5X4 human immunodeficiency virus type 1 isolates for infection of primary lymphocytes. J Virol 79, 1480–1486. 10.1128/JVI.79.3.14801486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clarke EL., Taylor LJ., Zhao C., Connell A., Lee JJ., Fett B., Bushman FD., and Bittinger K. (2019). Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 7, 46. 10.1186/s40168-019-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koster J, and Rahmann S.(2012). Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 64.Bolger AM, Lohse M, and Usadel B.(2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H, and Lam TW (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 66.Huang X, and Madan A.(1999). CAP3: A DNA sequence assembly program. Genome Res 9, 868–877. 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, and Goto S.(2017). ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380. 10.1093/bioinformatics/btx157. [DOI] [PubMed] [Google Scholar]
- 69.Taylor LJ., Abbas A., and Bushman FD. (2020). grabseqs: simple downloading of reads and metadata from multiple next-generation sequencing data repositories. Bioinformatics 36, 3607–3609. 10.1093/bioinformatics/btaa167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coordinators NR (2013). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 41, D8–D20. 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S8. Hi-C results for chimeric reads with one pair matching redondovirus and the other matching Entamoeba; related to Figure 5
Table S1. Viral genomes used to generate the ssDNA phylogeny; related to Figure S1A and Figure 1A.
Data Availability Statement
The metatranscriptomic and metagenomic screens used publicly available datasets (NCBI BioProject accessions are listed in Table S3 and Table S7, respectively). The redondovirus sequence obtained from the xenic E. gingivalis culture (RV-30956) has been deposited in GenBank under the accession ON986208. Metagenomic and Hi-C raw reads have been deposited in NCBI under the BioProject accession PRJNA858476.
This study does not report original code and all computational resources used are publicly available as of the date of publication and are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Saliva samples from healthy humans | Taylor et al., 2021 | N/A |
| Oropharyngeal, nasopharyngeal, and endotracheal aspirate samples subjects hospitalized with COVID-19 |
Merenstein et al., 2021 | N/A |
| Oropharyngeal, nasopharyngeal, and endotracheal aspirate samples from medical intensive care unit patients | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Phi29 polymerase | New England BioLabs | Cat#M0269 |
| Critical commercial assays | ||
| TaqMan Fast Universal PCR Master Mix | Thermo Fisher | Cat#4352042 |
| TaqMan Fast Virus 1-Step Master Mix | Thermo Fisher | Cat#5555532 |
| PowerUp SYBR Green Mix | Thermo Fisher | Cat#A25741 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| QIAamp DNA Mini Kit | Qiagen | Cat#51304 |
| SuperScript First-Strand Synthesis System Kit | Thermo Fisher | Cat#18080051 |
| Scientific Kwik-Diff™ Staining Kit | Thermo Fisher | Cat#9990700 |
| Phusion High-Fidelity PCR Kit | New England BioLabs | Cat#E0553S |
| Monarch DNA Gel Extraction Kit | New England BioLabs | Cat#T1020S |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC1311024 |
| Deposited data | ||
| Raw metagenomic sequencing reads | NCBI | Accession #: PRJNA858476 |
| Raw shotgun sequencing reads | NCBI | Accession #: PRJNA858476 |
| Redondovirus (RV-30956) genome | GenBank | Accession #: ON986208 |
| Experimental models: Organisms/strains | ||
| E. gingivalis | ATCC | ATCC-30956 |
| Oligonucleotides | ||
| qPCR, redondovirus Cap ORF (F: 5’-GGATGCCATGAAACTTTGATAC-3’; R: 5’TCTTCCTCCTTATTTGTATGGC-3’; probe: 5’-CCCATACTTACGCCGGTTACCTGC-3’) | Integrated DNA Technologies | N/A |
| qPCR, E. gingivalis 18S rRNA gene (F: 5’-TACCATACAAGGAATAGCTTTGTGAATAA-3’; R: 5’-ACAATTGTAAATTTGTTCTTTTTCT-3’) | Integrated DNA Technologies | N/A |
| Whole-genome PCR, redondovirus (Set A F: 5’CCTTTGGTCTCGAAATCTTCCTATACTGG-3’; Set A R: 5’-AGGCCTCTCTCCCTTCCATTTGG-3’; Set B F: 5’-GGTTATCGTTCATTTGATCATGCATTAGTACC-3’; Set B R: 5’-ACCAAGATGTTTAAGCCCTTTAGTTAATGTTTC-3’) | Integrated DNA Technologies | N/A |
| qPCR, human GAPDH (F: 5’-GGTGGTCTCCTCTGACTTCAACA-3’; R: 5’CCAGCCACATACCAGGAAATG-3’; probe: 5’CTGGCATTGCCCTCAACGACCAC-3’) | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pUC57 | BioBasic | Addgene 4509 |
| Software and algorithms | ||
| BLASTn v2.9.0 | Altschul et al., 1990 | https://www.ncbi.nlm.nih.gov/books/NBK279690/ |
| PhyML v3.0 | Guindon et al., 2010 | https://github.com/stephaneguindon/phyml |
| RaxML v8.2 | Stamatakis et al., 2005 | https://github.com/stamatak/standard-RAxML |
| MUSCLE v3.8.31 | Edgar, 2004 | https://github.com/rcedgar/muscle |
| iTOL v6 | Letunic and Bork, 2019 | https://itol.embl.de/ |
| VipTree v3.1 | Nishimura et al., 2017 | https://www.genome.jp/viptree/ |
| hisss v3.0 | Abbas et al., 2019 | https://github.com/louiejtaylor/hisss |
| IGV v2.9.0 | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| CAP3 v3.0 | Huang and Madan, 1999 | http://doua.prabi.fr/software/cap3 |
| MEGAHIT v1.2.9 | Li et al., 2015 | https://github.com/voutcn/megahit |
| FastQC v0.11.9 | Andrews, 2010 | https://github.com/s-andrews/FastQC |
| Trimmomatic v0.39 | Bolger et al., 2014 | https://github.com/usadellab/Trimmomatic |
| Sunbeam v2.1.0 | Clarke et al., 2019 | https://github.com/sunbeam-labs/sunbeam |
| Kraken2 v2.1.2 | Wood et al., 2019 | https://github.com/DerrickWood/kraken2 |
| R version v4.0.4 | Ihaka and Gentleman, 1996 | https://www.r-project.org/ |
| Snakemake v7.16.0 | Köster and Rahmann, 2012 | https://github.com/snakemake/snakemake |
| SAMtools v1.9 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| BEDtools v2.30.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |