

Abstract
Pyrrolizidine alkaloid (PA)-containing plants are among the most common poisonous plants affecting humans, livestock, and wildlife worldwide. A large number of PAs are known to induce genetic damage after metabolic activation. In the present study, using a battery of fourteen newly developed TK6 cell lines, each expressing a single human cytochrome P450 (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C18, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7), we identified specific CYPs responsible for bioactivating three PAs – lasiocarpine, riddelliine, and senkirkine. Among the fourteen cell lines, cells expressing CYP3A4 showed significant increases in PA-induced cytotoxicity, evidenced by decreased ATP production and cell viability, and increased caspase 3/7 activities. LC-MS/MS analysis revealed the formation of 1-hydroxymethyl-7-hydroxy-6,7-dihydropyrrolizine (DHP), the main reactive metabolite of PAs, in CYP3A4-expressing TK6 cells. DHP was also detected in CYP3A5- and 3A7-expressing cells after PA exposure, but to a much lesser extent. Subsequently, using a high-throughput micronucleus assay, we demonstrated that PAs induced concentration-dependent increases in micronuclei and G2/M phase cell cycle arrest in three CYP3A variant-expressing TK6 cell lines. Using Western blotting, we observed that PA-induced apoptosis, cell cycle changes, and DNA damage were primarily mediated by CYP3A4. Benchmark dose (BMD) modeling demonstrated that lasiocarpine, of the three PAs, was the most potent inducer of micronuclei, with a BMD100 of 0.036 μM. These results indicate that our TK6 cell system holds promise for genotoxicity screening of compounds requiring metabolic activation, identifying specific CYPs involved in bioactivation, and discriminating the genotoxic compounds that have different chemical structures.
Keywords: TK6-derived cell lines, cytochrome P450, bioactivation, DNA damage, micronuclei, high-throughput assay
1. Introduction
Pyrrolizidine alkaloids (PAs) and their corresponding PA N-oxides are a class of phytochemicals found in over 6,000 plant species, representing about 2–3% of the world’s flowering plants (Fu, 2017; Schrenk et al., 2020). Plants usually contain mixtures of PAs, for example, comfrey contains as many as 14 different PAs (Mei et al., 2010). PAs were first reported more than five decades ago to induce liver tumors in rodents (Mattocks, 1968). Since then, other organs and tissues of rodents, such as lung, kidney, brain, bladder, skin, and pancreas, were also found to be targets of PA-induced toxicity (Chen et al., 2010). Particularly, the retronecine-, heliotridine-, and otonecine-types of PAs, which contain a double-bond at the C1-C2 positions of their necine bases (i.e., the 1,2-unsaturated PAs; Figure 1A), are observed to be toxic and genotoxic in animal models (Fu, 2017; Fu et al., 2004). Humans may be exposed through the consumption of PA-contaminated foods (e.g., honey, milk, and wheat flour), or plants and herbal products containing PAs (e.g., herbal teas and plant dietary supplements) (Dusemund et al., 2018; Ebmeyer et al., 2019). Due to the potential for PA-induced toxicity – hepatotoxicity in particular, as well as genotoxicity – the International Agency for Research on Cancer (IARC) has classified three PAs, lasiocarpine (heliotridine-type), monocrotaline (retronecine-type), and riddelliine (retronecine-type), as Group 2B possible human carcinogens (IARC, 2018).
Figure 1.
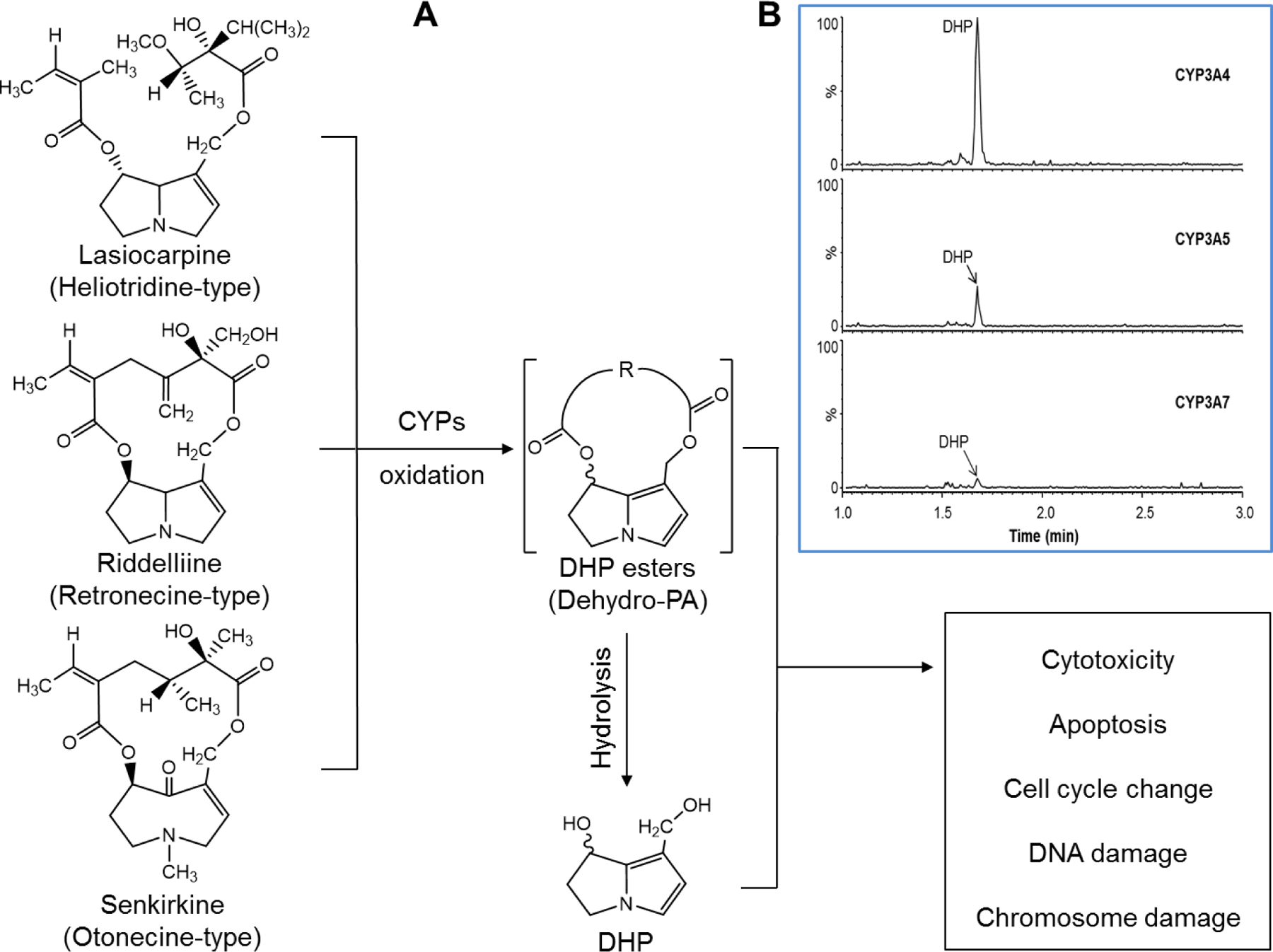
(A) Proposed metabolic activation pathway of three pyrrolizidine alkaloids. Lasiocarpine, riddelliine, and senkirkine undergo metabolic activation to their reactive metabolites including DHP esters and DHP that react with cellular macromolecules, leading to cytotoxicity and genotoxicity. (B) Representative LC-MS/MS chromatograms showing DHP formation in CYP-expressing TK6 cells exposed to 1 μM of lasiocarpine for 24 h.
PAs require metabolic activation to exert their cytotoxicity, genotoxicity, and carcinogenicity. Metabolism of retronecine- and heliotridine (the 7S-isomer of retronecine)-type PAs catalyzed by cytochrome P450 (CYP) enzymes forms the primary pyrrolic metabolites, dehydro-PAs (1-hydroxymethyl-7-hydroxy-6,7-dihydropyrrolizine esters, DHP esters), which then are hydrolyzed to form DHP (Figure 1A). Otonecine-type PAs require an initial oxidative N-demethylation step, potentially catalyzed by CYPs, to produce DHP esters. These reactive intermediates (DHP and DHP esters) can interact with cellular macromolecules, including proteins and DNA, to form pyrrole-protein adducts, pyrrole-DNA adducts, and protein-protein and protein-DNA cross-links, leading to hepatotoxic and genotoxic effects (Fu, 2017; Ma et al., 2018; Schrenk et al., 2020). Efforts have been made to identify the specific CYPs that are responsible for the bioactivation of PAs, and available data suggest that the CYP3A and 2B subfamilies mediate PA metabolic activation in rodents, but only CYP3A4 has the ability to activate PAs in humans (Li et al., 2011). For example, early in vivo studies using rats and guinea pigs suggested that the CYP3A subfamily of enzymes is the major enzyme group that accounts for the metabolic activation of PAs, such as clivorine, lasiocarpine, and riddelliine (Fu et al., 2004). Some evidence indicated that the CYP2B subfamily was involved in the metabolic activation of jacobine and senecionine (Chung and Buhler, 1994, 1995), but not clivorine (Lin et al., 2002). More recently, transfection with human CYP3A4 was shown to be required for lasiocarpine-induced genotoxicity in Chinese hamster lung V79 cells (Ebmeyer et al., 2019). In a cell-free system using human liver microsomes and recombinant CYP supersomes, ten human CYPs showed the ability to activate retronecine-type PAs, while only CYP3A4 and 3A5 activated otonecine-type PAs (Ruan et al. 2014). In addition, this study indicated that the capability of the major human CYPs to activate these PAs varied significantly. These findings require confirmation in in vitro and in vivo cell systems.
Genotoxicity assessment is a critical component in chemical hazard identification and risk assessment. Since many in vivo genotoxic carcinogens require metabolic activation by CYPs to exert their effects, in vitro genotoxicity assays must be conducted both in the absence and presence of a metabolizing rat S9 fraction (ICH, 2011; VICH, 2014). In addition to the absence of normal metabolism, the rodent cell lines most frequently used for in vitro genotoxicity testing are p53 and DNA-repair deficient, which may be a factor in the observed high levels of false positives seen in in vitro assays (Kirkland et al., 2007). Discussions on this topic at the 5th International Workshop on Genotoxicity Testing (IWGT) resulted in the recommendation that p53-competent (preferably human) cells be used for the in vitro micronucleus or chromosomal aberration assays (Pfuhler et al., 2011). Recently, participants at the 7th IWGT evaluated the state-of-the-science with respect to (1) technologies and innovations to improve existing assays using human lymphoblastoid TK6 cells and (2) novel and emerging technologies and approaches for in vitro mammalian cell mutagenicity test systems (Gollapudi et al., 2019). They recommended that any novel test system be metabolically competent, eliminating the need to use exogenous metabolic activation (Evans et al., 2019). These recommendations point to the growing need to develop new in vitro test systems that are metabolically competent and DNA repair proficient.
Recently, we developed eight TK6 cell lines each stably transduced with a specific human CYP (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C9, 2C19, and 3A4), and tested their performance using two prototypical genotoxicants, benzo[a]pyrene (B[a]P) and 7,12-dimethylbenz[a]anthracene (DMBA) (Li et al., 2020a). Without an added rat liver S9 fraction, the newly developed CYP1A1- and 1B1-expressing TK6 cell lines detected the genotoxicity of B[a]P and DMBA using a high-throughput (HT) micronucleus assay and quantification of γH2A.X protein expression. In the present study, we reported the development of another six human CYP-expressing TK6 cell lines, individually expressing CYP2C8, 2C18, 2D6, 2E1, 3A5, or 3A7. We then used this novel TK6-based cell system, along with our original eight CYP-expressing cell lines, to identify the specific CYP isoforms that account for the metabolic activation of PAs. Three PAs – lasiocarpine, riddelliine, and senkirkine – representing the heliotridine-, retronecine-, and otonecine-types of PAs, respectively, were selected as the test compounds. This study also extends the validation of our cell system for use in detecting pro-genotoxicants that require metabolic activation for their genotoxic activity.
2. Materials and Methods
2.1. Test chemicals
Lasiocarpine (CAS# 303–34-4), riddelliine (CAS# 23246–96-0), and senkirkine (CAS# 2318–18-5) were purchased from PhytoLab GmbH & Co. KG (Vestenbergsgreuth, Germany). DHP (6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine) standard was kindly provided by Dr. Peter Fu at the National Center for Toxicological Research (Jefferson, AR). All chemicals were stored at −80°C and protected from light, as recommended by the vendor.
2.2. Cell culture and treatment
Human lymphoblast TK6 cells, purchased from ATCC (Manassas, VA), were cultured at 37°C in a humidified atmosphere with 5% CO2 in RPMI 1640 medium supplemented with L-glutamine (Gibco, Gaithersburg, MD), 100 U/ml penicillin (Gibco), and 100 µg/ml streptomycin (Gibco), and 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA). Cells were routinely maintained at a density of 2 × 105 to 1.5 × 106 cells/ml with up to 10 passages and were treated at the density of 2 × 105 cells/ml (unless stated otherwise) with different concentrations of PAs dissolved in dimethyl sulfoxide (DMSO) for 24 h. The final concentration of DMSO did not exceed 0.5%.
2.3. Construction of CYP-expressing TK6 cell lines
Previously we created eight TK6 cell lines individually expressing CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C9, 2C19, and 3A4 with the detailed methods described (Li et al., 2020a). In the present study, we expanded our cell system and constructed an additional six cell lines stably transduced with CYP2C8, 2C18, 2D6, 2E1, 3A5, and 3A7 individually using the same methods described previously. Briefly, the six human CYP cDNAs were individually subcloned into the lentiviral expression vector pLV-EF1α-MCS-IRES-Bsd (Biosettia Inc., San Diego, CA) along with an in-frame c-Myc extension added to the carboxyl terminus of each recombinant CYP protein. The production of lentivirus was conducted following the manufacturer’s instructions. For lentiviral transduction, TK6 cells were seeded at a density of 2.5 × 105 cells per well in 6-well tissue culture plates and then were infected with lentivirus at a multiplicity of infection (MOI) of 10 with 8 µg/ml polybrene (Sigma, St. Louis, MO) by centrifuging at 1,000 × g for 60 min at room temperature. The transduction medium was replaced with fresh complete medium after centrifugation. Forty-eight hours after transduction, transduced cells were selected in the presence of 8 µg/ml blasticidin to generate stable TK6 cell lines expressing individual CYPs.
2.4. RNA extraction and quantitative real-time PCR
Quantitative real-time PCR (qPCR) was used to examine the expression of individual CYP enzymes in the newly constructed cell lines. Two million cells from each cell line were used for RNA extraction using the RNeasy Mini kit (Qiagen, Valencia, CA). The quantity and purity of RNA were measured with a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, DE). cDNA was synthesized from 2 µg RNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. The qPCR reactions were performed in a 20-µl volume according to the manufacturer’s protocol of FastStart Universal Probe Master (Rox) (Roche Applied Science, Indianapolis, IN) using a Bio-Rad CFX Real-Time PCR system (Bio-Rad Laboratories, Hercules, CA) under the following conditions: 10 min at 95°C followed by 40 cycles of 15 s at 95°C, and 1 min at 60°C. The following TaqMan probes (Applied Biosystems) were used: CYP2C8 (Hs00946140_g1), CYP2C18 (Hs00426403_m1), CYP2D6 (Hs00164385_m1), CYP2E1 (Hs00559368_m1), CYP3A5 (Hs00241417_m1), CYP3A7 (Hs00426361_m1), and human GAPDH (Hs02758991_g1). Quantitation cycle (Cq) was used to determine the relative expression levels of target genes, and Cq values were normalized to that of the endogenous control GAPDH. In order to calculate a relative amount, a Cq value of 35 was assigned to samples with non-detectable signals.
2.5. Western blot analysis
A total of 5 × 106 TK6 cells were lysed in RIPA buffer with Halt™ Protease Inhibitor Cocktail (ThermoFisher Scientific, Waltham, MA) for protein extraction. The protein concentrations of cell lysates were determined using the BCA assay (ThermoFisher Scientific). Equivalent amounts (20 μg) of total protein were used, and standard Western blot procedures were performed. Primary antibodies against CYP2C8, 2C18, 2D6, 2E1, 3A5, 3A7, phosphorylation of histone 2AX (γH2A.X), cleaved caspase 3, phospho-checkpoint kinase 1 (p-CHK1), phospho-checkpoint kinase 2 (p-CHK2), and GAPDH (loading control) were diluted in blocking reagent at a concentration of 1:1,000. Secondary antibodies conjugated with horseradish peroxidase (HRP) (Santa Cruz Biotechnology, Dallas, TX) were incubated at a dilution of 1:10,000. The protein signals were determined by FluorChem E System and quantified by AlphaView SA (ProteinSimple, San Jose, CA).
2.6. Measurement of cell viability, cellular ATP level, and caspase 3/7 activity
Cells were seeded in 24-well plates at a density of 2 × 105 cells/ml. After treatments with different PAs for 24 h, cell viability and cellular ATP levels were determined as described previously (Guo et al., 2015) using the CellTiter-Blue® fluorescent cell viability assay kit (Promega, Madison, WI) and the CellTiter-Glo® luminescent cell viability assay kit (Promega), respectively. The relative cell viability and cellular ATP content were calculated by comparing the signal intensity of the treated cells to that of the controls. Caspase 3/7 enzymatic activity was measured by the Caspase-Glo® 3/7 Assay System (Promega) following the manufacturer’s instructions and results were expressed as fold-change relative to the vehicle control group.
2.7. Measurement of DHP by LC-MS/MS
Cells were seeded at a density of 5 × 105 cells/ml in 1 ml phenol-free RPMI-1640 medium. For the measurement of DHP, cells were treated with 1 μM lasiocarpine, 10 μM riddelliine, or 50 μM senkirkine for 24 h at 37°C in a humidified atmosphere with 5% CO2. After incubation, the medium was collected by centrifuge, protein was removed with acetonitrile, then the final resultant was adjusted to a solution containing 10% acetonitrile and ready for LC-MS/MS analysis. A Waters ACQUITY UPLC coupled to a Waters Quattro Premier XE™ tandem mass spectrometer (Waters, Milford, MA) was used for quantitative analysis of DHP. An ACQUITY UPLC HSS T3 C18 column (2.1 × 50 mm, 1.8 μm) (Waters) was used and the column oven temperature was 40°C. The mobile phase was composed of solvent A (H2O containing 0.1% formic acid) and B (acetonitrile). The gradient was as follows: 0–0.5 min, 1% B; 0.5–3 min, 1–90% B; 3–3.5 min, 90% B; 3.5–3.6 min, 90–1% B; 3.6–5.5 min, 1% B for column re-equilibration. The mass spectrometer equipped with a heated electrospray ionization source was operated in positive ion mode. The ion source settings were optimized as follows: capillary, cone, and extractor voltages were set at 3000, 30, and 4 V, respectively; source temperature was 120°C; desolvation temperature was 400°C; desolvation gas was 1000 l/h. Multiple reaction monitoring (MRM) was employed for data acquisition. The collision gas flow was 0.3 ml/min, and the collision energy was 22 eV.
2.8. High-throughput micronucleus assay and cell cycle analysis
Cells were seeded in 96-well plates at a density of 2 × 105 cells/ml and were treated with PAs for 24 h, which allowed TK6 cells to divide for 1–2 cell cycles. After the treatments, cells were washed twice with sterile PBS, stained, and lysed following the protocol described in the In Vitro MicroFlow™ Kit (Litron Laboratories, Rochester, NY). The high-throughput (HT) micronucleus analysis was performed using a FACSCanto II flow cytometer equipped with a High Throughput Sampler (BD Biosciences, San Jose, CA). The stopping gate was set to record 10,000 intact nuclei and threshold parameters were set as recommended in the instruction manual. The percentage of micronucleated cells (%MN) was calculated as the ratio of micronucleus events to the total number of nucleated events. Cell cycle information was generated in the same run by retrieving histograms of SYTOX® Green nucleic acid stain fluorescence. The cell cycle analysis was conducted using FlowJo® (v10.1).
2.9. Bayesian benchmark dose analysis
In order to further compare the relative genotoxic potency of each PA, the micronucleus data in CYP3A4-expressing TK6 cells exposed to PAs were analyzed using the Bayesian Benchmark Dose (BBMD) modeling system (https://benchmarkdose.org) (Shao and Shapiro, 2018). Briefly, Markov chain Monte Carlo (MCMC) sampling was conducted in Pystan (version 2.19.0.0) with 30,000 iterations per chain, 50% warmup percent, and 0−99,999 random seed. The dose-response data were fitted by the eight standard models (Exponential 2, Exponential 3, Exponential 4, Exponential 5, Hill, Linear, Michaelis Menten, and Power) provided by the BBMD system for continuous data. The goodness-of-fit indicators were evaluated and models with posterior predictive P-values < 0.05 or > 0.95 were excluded. The choice of critical effective size (CES, also called benchmark response) has a large impact on the benchmark dose (BMD) metric, and this parameter is crucial for risk assessment (Zeller et al., 2017). While a single value of CES does not reflect an equivalent effect size for all toxicological endpoints (Slob, 2017), the CES has been suggested to be 0.7–0.8 for the micronucleus assay (Zeller et al., 2016). The BBMD modeling system gives the model-averaged BMD values that represent an ensemble of all eight models (Shao and Shapiro, 2018). BMD50 and BMD100 were calculated based on CESs of 0.5 and 1.0, which represent 50% or 100% (2-fold) increases in chromosomal damage levels over the background (vehicle controls), respectively.
2.10. Statistical analysis
All data are presented as mean ± 1 standard deviation (SD) from at least three independent experiments. Statistical analyses were performed using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA). One-way ANOVA followed by Dunnett’s post hoc test or two-way ANOVA followed by Bonferroni post hoc test were used to compare the differences between groups, with P < 0.05 as statistically significant.
3. Results
3.1. Confirmation of CYP expression in newly developed TK6-derived cell lines
The mRNA levels of the CYPs in the six newly constructed TK6 cell lines, individually transduced with human CYP2C8, 2C18, 2D6, 2E1, 3A5, and 3A7, were quantified using qPCR. No differences were found in CYP gene expression between parental TK6 cells and empty vector (EV) control transduced-TK6 cells; neither parental TK6 cells nor TK6 cells transduced with empty vector had detectable mRNA levels of CYP2C8, 2C18, 3A5, and 3A7 (Cq > 35), and both showed very low levels of CYP2B6 and 2E1 (Cq > 28). The −log2 fold-changes of all tested CYPs were above 9.5 (CYP2E1 being the lowest) in CYP-expressing cell lines when compared to parental TK6 cells or EV controls, indicating the mRNA level of the CYPs in these six cell lines increased more than 29.5-fold (Figure 2A). The protein expressions of the CYPs were also characterized by Western blot. While both parental TK6 cells and EV controls had negligible protein expression of the six CYPs studied, the specific CYP protein along with Myc (as transfection tag) was strongly expressed in the corresponding CYP-expressing cell lines (Figure 2B).
Figure 2.
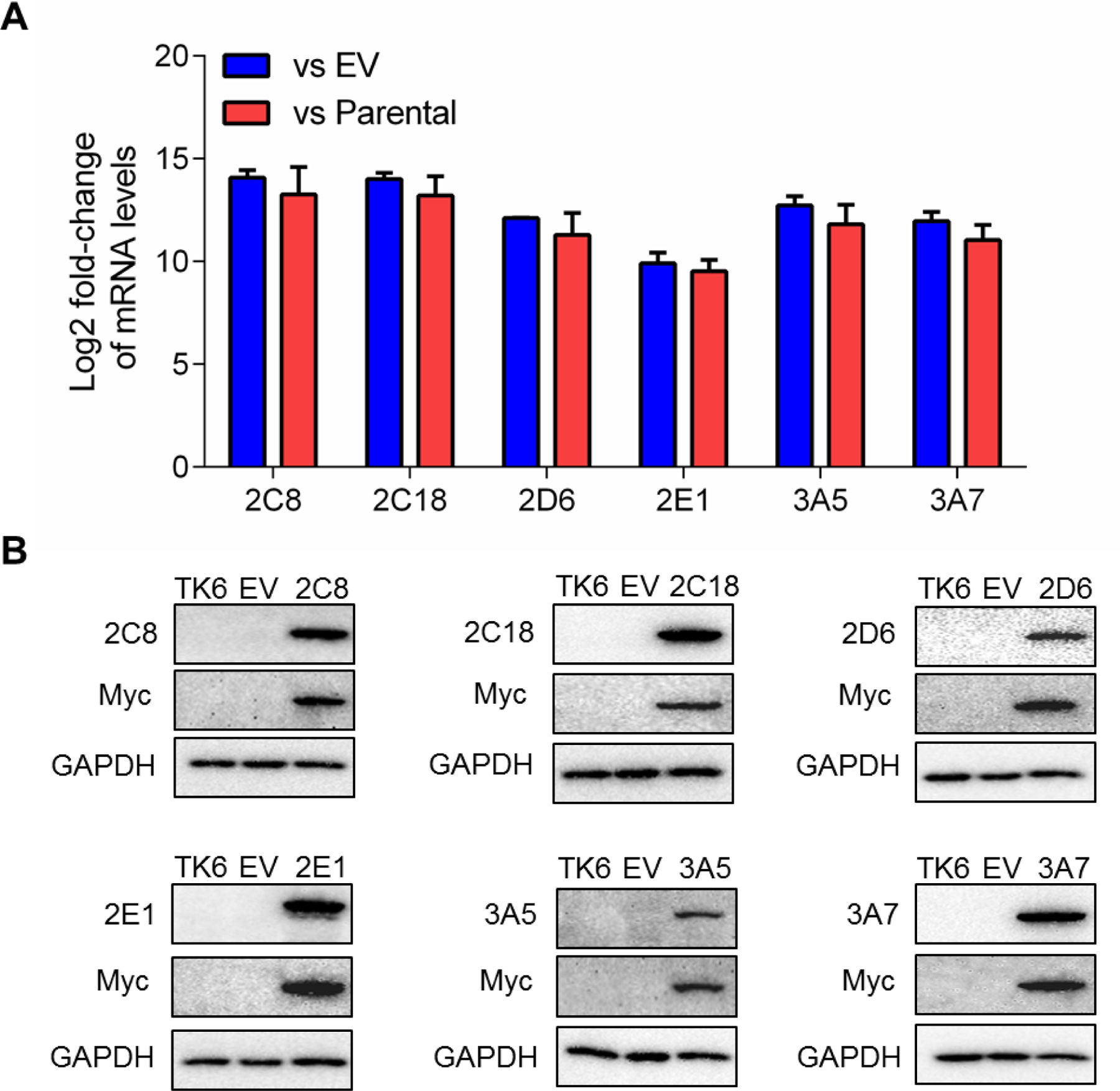
(A) Comparison of CYP mRNA levels between stably transduced TK6 cells with empty vector (EV) or parental TK6 controls. The Log2 fold change was calculated by ΔΔCt method using GAPDH as the housekeeping gene. (B) Characterization of CYP protein expression in stably transduced TK6 cells. The protein levels of CYP isoforms ranging in molecular weight between 45 and 60 kDa were detected by Western blotting. Myc was used as a tag for transfection and GAPDH (37 kDa) was used as a loading control. Similar results were obtained from three independent experiments.
3.2. The cytotoxicity of PAs in fourteen CYPs-expressing TK6 cells
Using these six newly developed cell lines (Figure 2) and eight previously generated TK6 cell lines (Li et al., 2020a), we screened the cytotoxicity of three PAs by measuring cell viability and cellular ATP production. Lasiocarpine appears to be the most potent after metabolic activation by CYP3A4, a concentration as low as 0.5 μM resulted in decreased cell viability in CYP3A4-expressing TK6 cells when compared to EV controls (Figure 3A). At the highest concentration tested (5 μM), over half of the CYP3A4-expressing cells were dead after the 24-h treatment. Riddelliine (up to 100 μM) had no overt cytotoxicity in EV controls. However, its cytotoxicity was significantly increased in CYP3A4-expressing TK6 cells at concentrations above 10 μM (Figure 3B). At 100 μM riddelliine, the cell viability and ATP content in CYP3A4-expressing cells decreased to 42% and 47% of the EV controls, respectively. The other thirteen CYPs, including CYP3A5 and 3A7, had no effect on the cytotoxicity of lasiocarpine and riddelliine at the concentrations tested (Figure 3 and Supplementary Figures 1 and 2).
Figure 3.
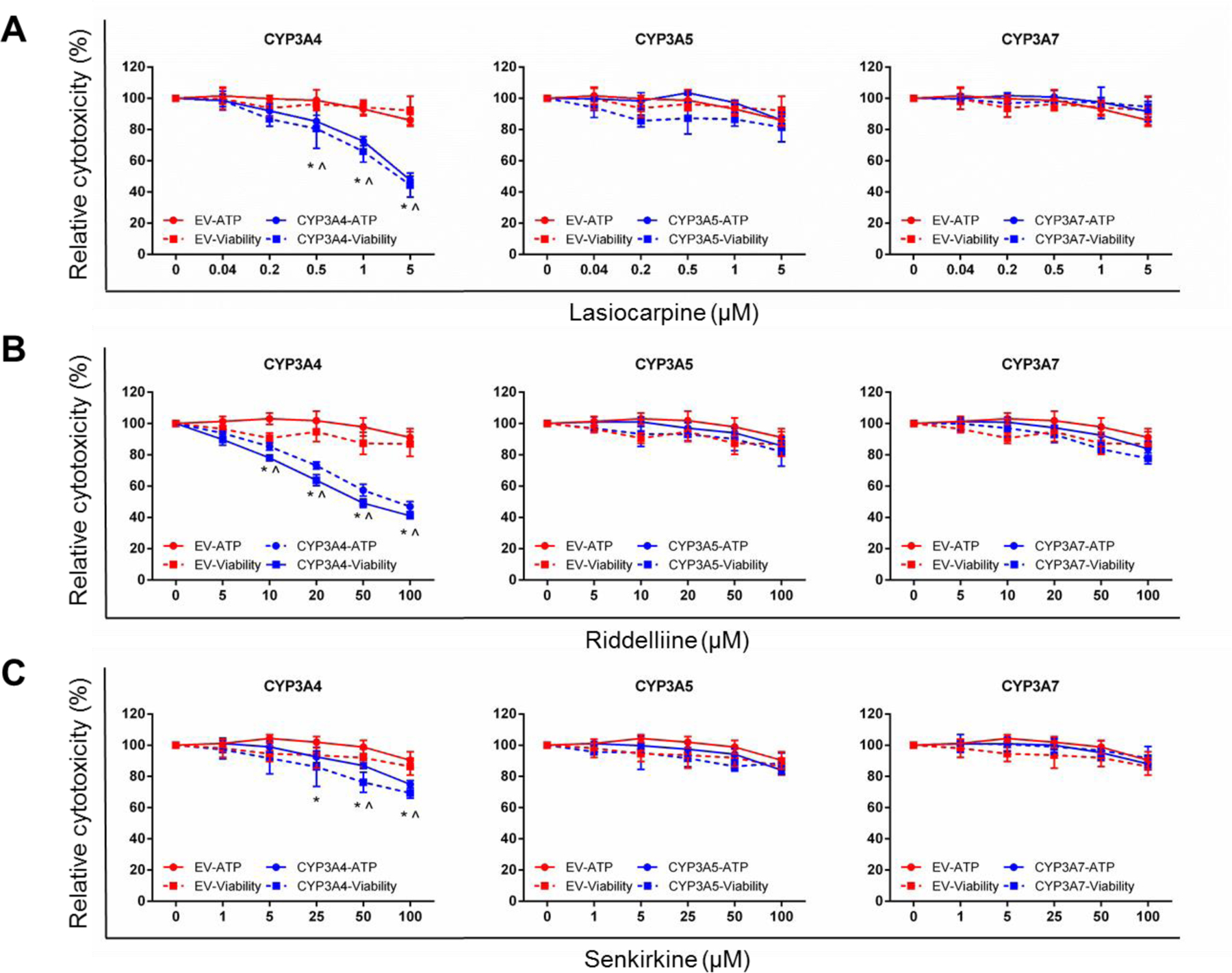
Cytotoxicity of lasiocarpine (A), riddelliine (B), and senkirkine (C) in TK6 cells transduced with empty vector (EV), CYP3A4, 3A5, and 3A7. Cells were treated at the indicated concentrations for 24 h. The cellular ATP level (circle, solid line) and cell viability (square, dashed line) were determined by the CellTiter-Glo assay and CellTiter-Blue assay, respectively. The data points represent the means ± SD from at least three independent experiments. * (ATP level) and ^ (cell viability) indicate P < 0.05 between a treatment group and the corresponding EV cells at the same concentration.
Senkirkine (up to 100 μM) had no significant cytotoxicity in EV controls (Figure 3C). Like lasiocarpine and riddelliine, senkirkine treatment also resulted in increased cytotoxicity only in CYP3A4-expressing TK6 cells, although the extent of toxicity was less than riddelliine. Compared to EV controls, ATP levels were significantly lower in CYP3A4-expressing TK6 cells after 25 μM senkirkine treatment, and significant decreases in cell viability were observed at concentrations above 50 μM. The cytotoxicity of senkirkine to TK6 cells was not affected by any of the other CYPs (Figure 3C and Supplementary Figure 3).
3.3. PAs-induced apoptosis in CYP3A4-expressing TK6 cells
To help characterize the nature of the cell death, the enzymatic activity of caspase 3/7 was measured. In CYP3A4-expressing TK6 cells, when compared to EV controls, all three PAs induced concentration-dependent increases in caspase 3/7 activity (Figure 4), beginning at 0.2 μM lasiocarpine, 5 μM riddelliine, and 25 μM senkirkine. These data clearly suggest that the bioactivation of PAs catalyzed by CYP3A4 resulted in apoptotic cell death in TK6 cells.
Figure 4.
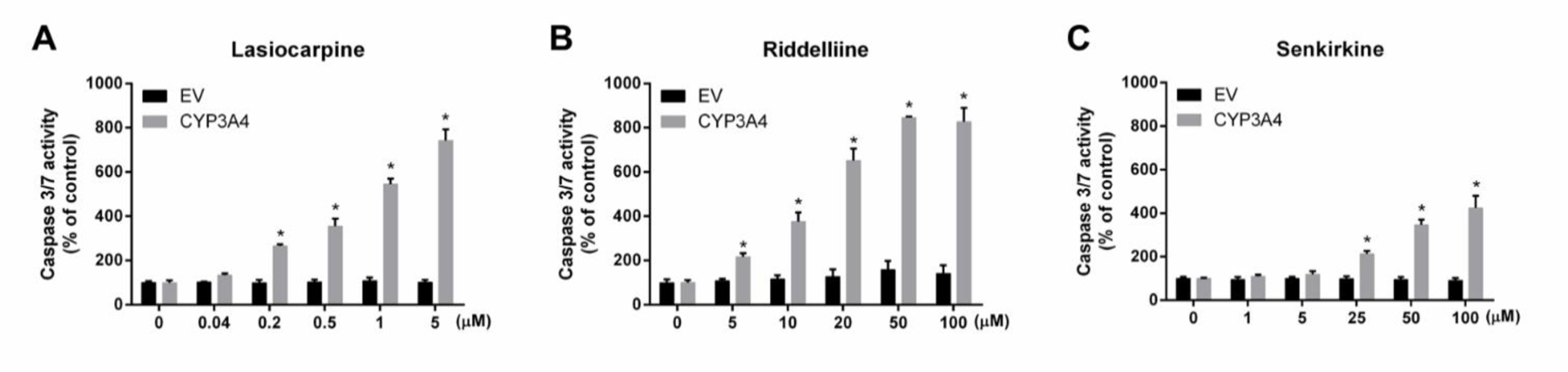
PAs increased caspase 3/7 enzymatic activity in CYP3A4-expressing TK6 cells. Cells were treated at the indicated concentrations for 24 h. Caspase 3/7 levels were determined by the Caspase-Glo 3/7 assay. The data points represent the means ± SD from three independent experiments. * indicates P < 0.05 comparing CYP3A4-expressing cells to the corresponding EV controls at the same concentration.
3.4. DHP formation in fourteen CYP-expressing TK6 cell lines exposed to PAs
The metabolic activation of PAs leading to their toxicity is catalyzed by CYPs. DHP esters (dehydro-PAs), the reactive primary pyrrolic metabolites of PAs, quickly undergo hydrolysis to a relatively more stable reactive product, DHP (Figure 1A). Herein, we employed LC-MS/MS to identify and quantify DHP levels in response to PA exposure. Representative LC-MS/MS chromatograms showing levels of DHP in CYP-expressing TK6 cells exposed to 1 μM of lasiocarpine for 24 h are presented in Figure 1B. Table 1 summarizes the quantitation of DHP formation detected in TK6 cells transduced with CYP3A4, 3A5, and 3A7; DHP was not detected in the other eleven CYP-transduced cell lines. As expected, CYP3A4 was the major enzyme involved in the metabolism of the 3 PAs we investigated. After exposure to lasiocarpine (1 μM), riddelliine (10 μM), and senkirkine (50 μM) for 24 h, the DHP levels in the media were found to be 5.76 ± 1.09, 4.72 ± 0.17, and 5.08 ± 0.59 nmol per 1 mg protein, respectively. To a lesser extent, CYP3A5 and 3A7 were also involved in PA metabolism. The relative efficiency of CYP3A families in metabolizing PAs can be ranked as follows: lasiocarpine, CYP3A4 >> 3A5 > 3A7; riddelliine, CYP3A4 >> 3A7 > 3A5; senkirkine, CYP3A4 >> 3A5 ≈ 3A7 (Table 1 and Figure 1B).
Table 1.
Levels of DHP formation in TK6 cells transduced with various CYPs after 24-h exposure to 1 µM lasiocarpine, 10 µM riddelliine, and 50 µM senkirkine*
| Transduced TK6 cells | DHP Formation (nmol/mg protein) |
|||
|---|---|---|---|---|
| Lasiocarpine | Riddelliine | Senkirkine | ||
| EV | < LOD | < LOD | < LOD | |
| CYP1A1 | < LOD | < LOD | < LOD | |
| CYP1A2 | < LOD | < LOD | < LOD | |
| CYP1B1 | < LOD | < LOD | < LOD | |
| CYP2A6 | < LOD | < LOD | < LOD | |
| CYP2B6 | < LOD | < LOD | < LOD | |
| CYP2C8 | < LOD | < LOD | < LOD | |
| CYP2C9 | < LOD | < LOD | < LOD | |
| CYP2C18 | < LOD | < LOD | < LOD | |
| CYP2C19 | < LOD | < LOD | < LOD | |
| CYP2D6 | < LOD | < LOD | < LOD | |
| CYP2E1 | < LOD | < LOD | < LOD | |
| CYP3A4 | 5.76 ± 1.09 | 4.72 ± 0.17 | 5.08 ± 0.59 | |
| CYP3A5 | 0.59 ± 0.14 | 0.22 ± 0.15 | 0.27 ± 0.04 | |
| CYP3A7 | 0.08 ± 0.04 | 0.67 ± 0.28 | 0.22 ± 0.06 | |
DHP released into the medium were quantified by LC–MS/MS and are expressed as nmol/mg protein.
EV, TK6 cells transduced with empty vector; LOD, lower limit of detection.
3.5. Micronuclei and G2/M cell cycle arrest induced by PAs in three CYP3A-expressing TK6 cell lines
After observing DHP formation induced by the 3 PAs in three different CYP3A variant-expressing TK6 cell lines, we used a HT micronucleus assay to evaluate whether these PAs induce chromosomal damages in these CYP3A-expressing cell lines. There were no changes in the percentage of micronucleated cells (%MN) following exposure to lasiocarpine in EV control cells (Figure 5A). In CYP3A4-expressing cells, lasiocarpine increased the %MN in a concentration-dependent manner, with the lowest-observed-adverse-effect level (LOAEL, defined as the lowest concentration that induces a statistically significant response from control) seen at 0.5 μM. Overt cytotoxicity was seen at the highest concentration tested (5 µM). In CYP3A5-expressing cells, lasiocarpine increased the %MN at concentrations of 1 and 5 µM, and in CYP3A7-expressing cells, at 5 µM only. The micronucleus assay results revealed a discernable dynamic range. In CYP3A4-expressing cells, 1 μM lasiocarpine increased the %MN by > 40-fold compared to untreated cells (Figure 5A). G2/M arrest in the cell cycle is a common response to many genotoxic agents that induce DNA damage and chromosome breakage (Brüsehafer et al., 2014; Chaudhary et al., 2013). Such a protective mechanism helps cells avoid entering mitosis with damaged DNA. As shown in Figures 6A & 6B, decreased cells in the S-phase and increased cells in G2/M were observed in three TK6 cell lines transduced with one of three CYP3A enzymes after lasiocarpine treatment. Increased cells in the sub-G1 phase, which indicates apoptotic cell death, were also seen in all three CYP3A-expressing cell lines after lasiocarpine exposure at various concentrations.
Figure 5.
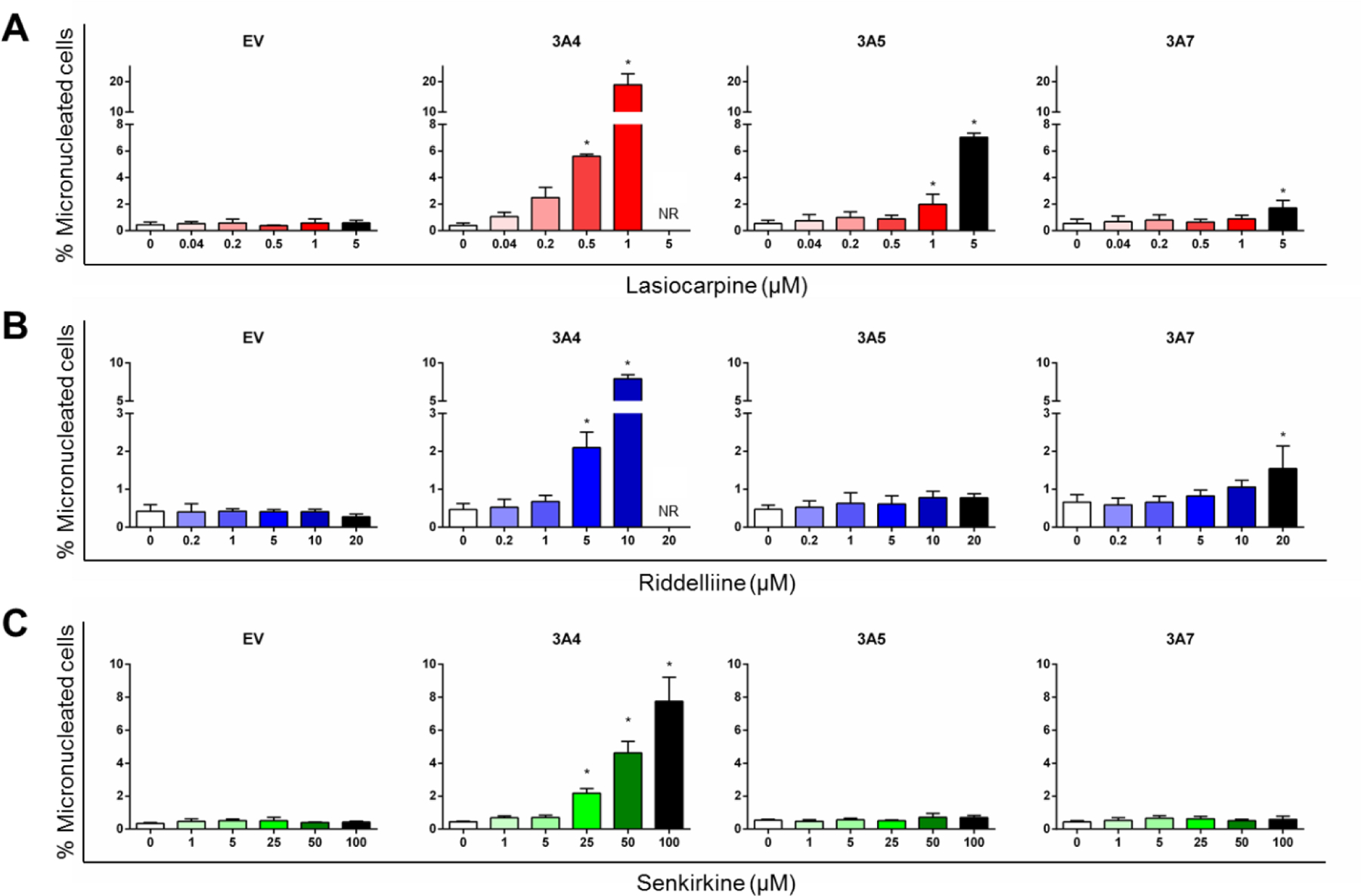
Induction of micronuclei by lasiocarpine (A), riddelliine (B), and senkirkine (C) in CYP-expressing TK6 cells using a high-throughput micronucleus assay. The stopping gate was set to record 10,000 intact nuclei. Data were expressed as means ± SD from at least three independent experiments. * indicates P < 0.05 comparing treated groups to DMSO control. NR: not reported due to > 55% cytotoxicity in at least one independent experiment.
Figure 6.
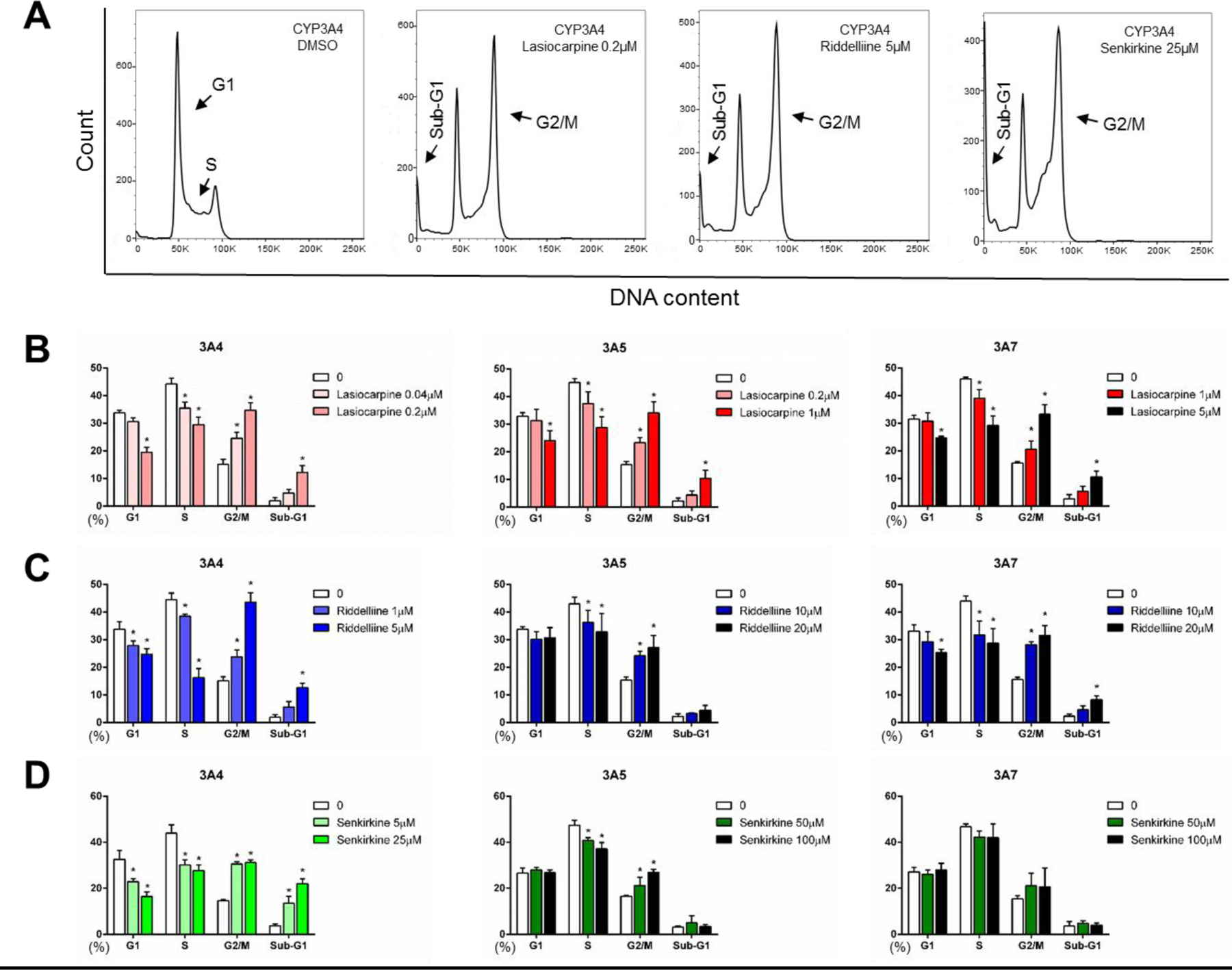
Cell cycle changes after treatments with 3 PAs in CYP-expressing TK6 cells. Representative images (A) show cell cycle distributions of different samples analyzed by flow cytometry. Histograms are DNA content analyses in the cells treated with lasiocarpine (B), riddelliine (C), and senkirkine (D). The stopping gate was set to record 10,000 intact nuclei. Data were expressed as means ± SD from at least three independent experiments. * indicates P < 0.05 comparing treated groups to DMSO control.
Riddelliine induced micronuclei in CYP3A4-expressing cells in a concentration-dependent manner (Figure 5B), although the response was less potent than the response seen with lasiocarpine. The LOAEL for %MN in CYP3A4-expressing cells was 5 μM. In CYP3A7-expressing cells, the LOAEL for %MN induced by riddelliine was 20 μM, while no induction of MN was observed in TK6 cells transduced with CYP3A5. Similar to lasiocarpine, we observed a trend of decreased cells in S-phase and G2/M arrest in all three CYP3A-expressing cell lines exposed to riddelliine (Figure 6C). Increased cells in the sub-G1 phase were seen in CYP3A4 and 3A7-expressing cell lines after 5 µM riddelliine treatment, suggestive of apoptotic cell death.
Senkirkine induced a concentration-dependent increase in %MN only in CYP3A4-expressing TK6 cells (Figure 5C). The LOAEL for %MN in CYP3A4-expressing cells was 25 μM. At 100 μM (the highest concentration tested), senkirkine induced an increase in MN of about 17-fold over the control cells. Cell cycle arrest at the G2/M checkpoint was also observed after bioactivation both by CYP3A4 and 3A5 (Figure 6D). No change was seen in the sub-G1 phase in cells with either CYP3A5 or 3A7, suggesting no induction of apoptosis.
3.6. Western blot analysis to confirm PA-induced cell cycle disruption, apoptosis, and DNA damage
To further confirm the effects of PAs on cell cycle changes, apoptosis, and DNA damage after bioactivation by the CYP3A subfamily of enzymes, we conducted Western blot analysis and measured the protein levels of p-CHK1, p-CHK2, cleaved caspase 3, and γH2A.X. As shown in Figure 7, in CYP3A4-expressing cells after 24-h treatments with the three PAs, expression of two DNA damage-responsive cell cycle checkpoint-related proteins (p-CHK1 and p-CHK2) significantly increased; the level of cleaved caspase 3, which reflects the activation of the key apoptosis-related protein caspase 3, was also significantly increased. These observations are in accord with the observed elevated protein level of γ-H2A.X, a hallmark of DNA damage response. These results confirmed that CYP3A4 is the major enzyme involved in the biotransformation of the three PAs to active genotoxicants in TK6 cells.
Figure 7.
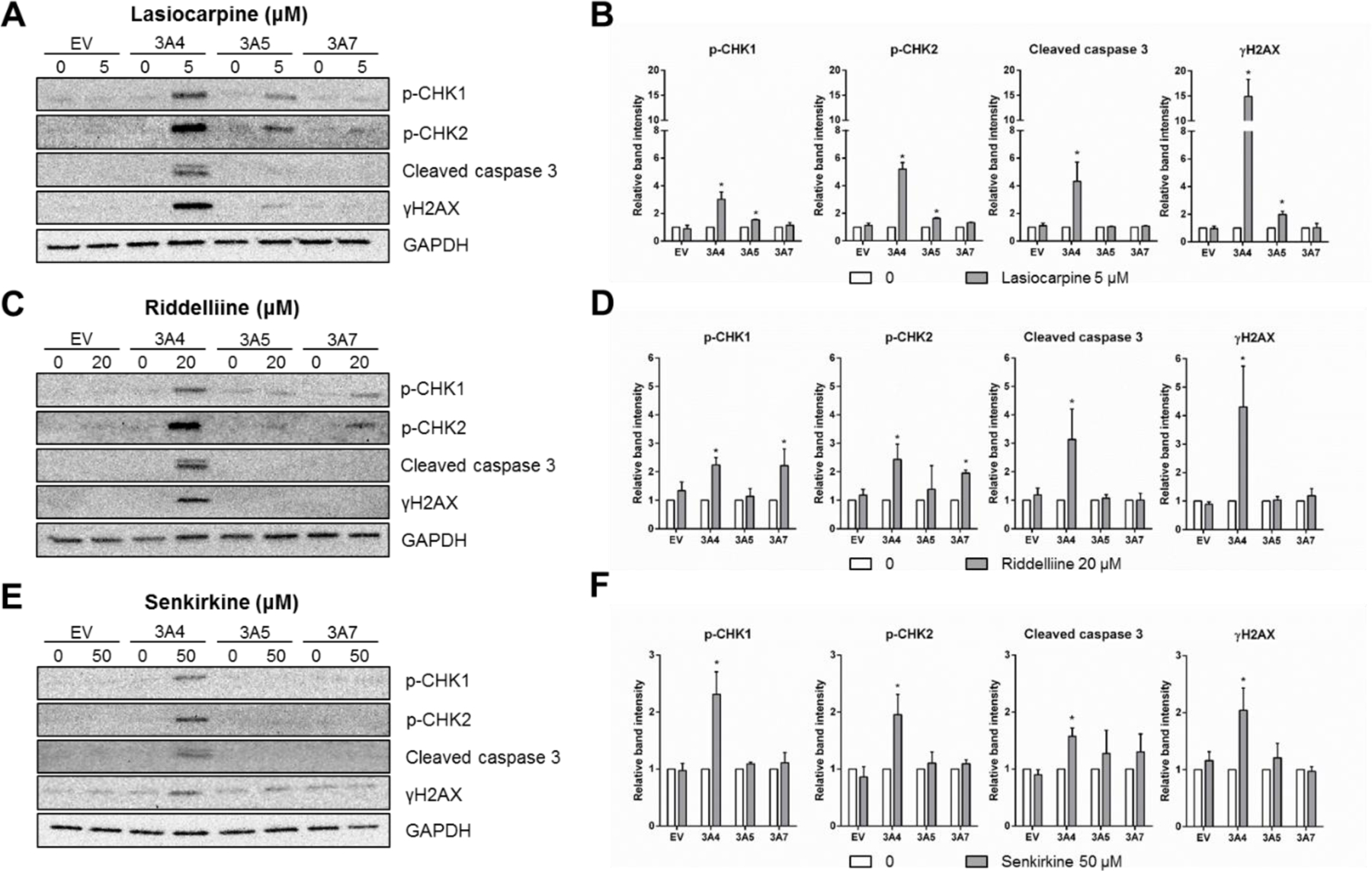
Western blotting confirmed DNA damage, cell cycle changes, and apoptosis induced by PAs. Cells were exposed to 5 µM lasiocarpine (A and B), 20 µM riddelliine (C and D), or 50 µM senkirkine (E and F) for 24 h. Total cellular protein (20 µg) was loaded for each lane and GAPDH was used as a loading control. Protein expression of p-CHK1, p-CHK2, cleaved caspase 3, and γH2A.X levels after PA treatments were quantified and expressed as the means ± SD from three independent experiments. Intensities of bands were normalized to the amount of GAPDH. * indicates P < 0.05 between the treatment group and corresponding vehicle control.
Consistent with the results from other endpoints (Table 1, Figures 5 & 6), CYP3A5 and 3A7 also manifested some activity in metabolizing PAs. For example, the expression of proteins, p-CHK1 and p-CHK2, was significantly increased in CYP3A7-expressing cells when exposed to riddelliine, although the fold-changes were less than those in CYP3A4-expressing cells (Figures 7C and 7D). On the other hand, CYP3A5 appeared to be more efficient than CYP3A7 in metabolizing lasiocarpine (Figures 7A and 7B). Cell cycle perturbation and induction of DNA damage were evidenced by increased levels of p-CHK1, p-CHK2, and γ-H2A.X in CYP3A5-expressing cells after lasiocarpine exposure. Notably, none of the three PAs changed the protein-expression level of cleaved caspase 3 in TK6 cells transduced with CYP3A5 and 3A7. This was consistent with the results from our cytotoxicity screening (Figure 3), further suggesting that CYP3A4 is likely the dominant player in PA-induced apoptosis.
3.7. Bayesian benchmark dose (BBMD) modeling comparing the relative potency of PAs
To investigate the relative potency of PAs, the micronucleus data in CYP3A4-expressing TK6 cells exposed to lasiocarpine, riddelliine, or senkirkine were used for the BBMD modeling. All eight standard models for continuous data were included for the dose-response model fitting with prior weight set as 0.125 for each model. The best fitting curve of the model (i.e., the curve with the highest posterior weight) for each PA is shown in Figure 8A. The linear model provided the best fit for lasiocarpine and senkirkine while the exponential model appeared to be the best fit for riddelliine. Model averaging was used to estimate BMD values to address model uncertainty (Shao and Gift, 2014). As expected, lasiocarpine had the lowest BMD estimates at CESs of 50% and 100%, with BMD50 = 0.019 μM and BMD100 = 0.036 μM (Figure 8B). In other words, a 24-h treatment with 0.036 μM lasiocarpine may produce a 2-fold increase in %MN in CYP3A4-expressing cells. Riddelliine is less potent with a BMD100 of 2.56 μM and senkirkine is the weakest with a BMD100 = 7.73 μM (Figure 8B).
Figure 8.
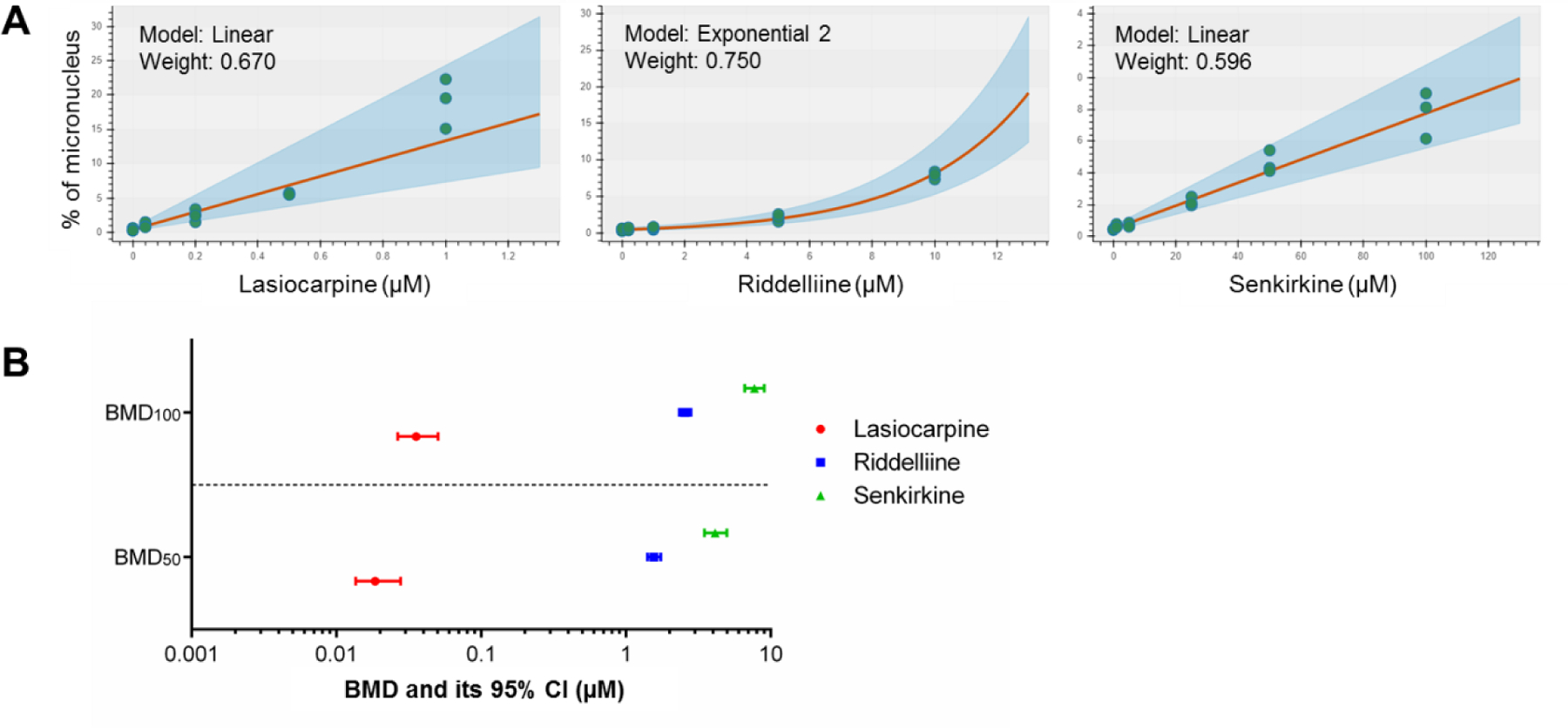
Bayesian benchmark dose (BBMD) modeling evaluating the potency of PAs. The micronucleus (MN) data in CYP3A4-expressing TK6 cells exposed to lasiocarpine, riddelliine, or senkirkine were used for the BMD modeling. (A) The model with the highest posterior weight (i.e., the best fitting curve) for each PA is presented. (B) The BMD (BMD50 and BMD100) estimates producing a 50% or 100% increase above the controls. The bars represent the calculated lower and upper 95% confidence interval (CI) for each BMD value (i.e., BMDL and BMDU).
4. Discussion
Traditional approaches to chemical risk assessment worldwide are undergoing a transformation to alternative methods designed to reduce animal testing and provide more mechanistic data (EPA, 2019; NRC, 2007). In vitro genotoxicity testing has long been a critical component of chemical toxicity evaluation and relies heavily on the use of rat liver S9 as an enzyme source for metabolic transformation (Guo et al., 2020; Kirkland et al., 2007). Recently, the U.S. Tox21 Program used a battery of quantitative HT in vitro assays for screening more than ten thousand chemicals for DNA damage potential (Hsieh et al., 2019); however, no source of exogenous metabolic activation was used in that screening, due to the inability to incorporate rat liver S9 mix at the usual concentrations to the assay formats used. In light of these challenges, we previously developed a battery of human TK6 cell lines, each stably transduced with a human CYP, with the goal to use this novel system as a potential alternative for the use of rat liver S9 in genotoxicity screening (Li et al., 2020a). To expand this TK6 cell system, in the present study we generated six more TK6 cell lines and in each established cell line, the increased expression and metabolic function of each individual CYP were confirmed using real-time PCR and Western blotting (Figure 2). Currently, our TK6 cell system contains fourteen TK6-derived cell lines, each individually expressing one human CYP (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7).
To verify the functions of these CYP-expressing TK6 cell lines, we previously used two prototypical polyaromatic hydrocarbon mutagens, B[a]P and DMBA (Li et al., 2020a). Our goal moving forward is to test more chemicals from different categories that require metabolic activation to elicit their genotoxicity. We previously investigated the mutagenicity of riddelliine using transgenic Big Blue rats (Mei et al., 2004a; Mei et al., 2004b) and riddelliine-induced gene expression profile changes (Mei et al., 2007). Therefore, in the present study we selected riddelliine along with two other PAs, lasiocarpine and senkirkine, to validate these newly generated cell lines and to identify the specific CYP enzymes that account for the genotoxicity of three different types of PAs. Among the fourteen CYPs studied, CYP3A4 was the major CYP enzyme accounting for the metabolic activation of the 3 PAs, based on the results of cytotoxicity tests (Figure 3 and Supplementary Figures 1-3); the main reactive metabolite of these 3 PAs was only detected in three CYP3A variant-expressing TK6 cell lines (Table 1). Therefore, we used these three CYP3A variant-expressing cell lines to further investigate the toxicity of the 3 PAs (Table 2).
Table 2.
Summary of the toxicity of of lasiocarpine, riddelliine, and senkirkine in three CYP3A variant-expressing TK6 cells
| Pyrrolizidine alkaloid |
Endpoint | CYP3A-expressing TK6 cells | ||
|---|---|---|---|---|
| CYP3A4 | CYP3A5 | CYP3A7 | ||
|
Lasiocarpine (0–5 μM) |
Cytotoxicity 1 | ++ | − | − |
| Caspase 3/7 2 | ++ | − | − | |
| Cell cycle changes 3 | + | + | + | |
| p-CHK1/2 4 | ++ | + | − | |
| Micronuclei 5 | +++ | + | + | |
| γH2A.X 4 | +++ | + | − | |
|
Riddelliine (0–100 μM) |
Cytotoxicity | ++ | − | − |
| Caspase 3/7 | ++ | − | − | |
| Cell cycle changes | + | + | + | |
| p-CHK1/2 | + | − | + | |
| Micronuclei | ++ | − | + | |
| γH2A.X | ++ | − | − | |
|
Senkirkine (0–100 μM) |
Cytotoxicity | + | − | − |
| Caspase 3/7 | + | − | − | |
| Cell cycle changes | + | + | − | |
| p-CHK1/2 | + | − | − | |
| Micronuclei | + | − | − | |
| γH2A.X | + | − | − | |
−, No significant difference when compared to its EV control.
+, Significant changes (increase or decrese) after treatments.
++ or +++, Relative effects compared with the responses observed with serkirkine (based on the concentrations and effects).
See Figure 3 (cell viability and cellular ATP levels).
See Figure 6 (cell cycle analyzed by flow cytometry).
See Figure 7 (Western blot analysis).
See Figure 5 (high-throughput micronucleus assay).
In CYP3A4-expressing TK6 cells, lasiocarpine (0.5–5 µM)-induced apoptosis was detected after a 24-h treatment (Figure 4), DNA damage was confirmed by γH2A.X protein expression (Figure 7A), and chromosome damage was observed in a high-throughput MN assay (Figure 5A). To a lesser extent, CYP3A5-expressing cells treated with lasiocarpine demonstrated cell cycle changes and increases in γH2A.X and MN induction, while CYP3A7-expressing cells treated with lasiocarpine only showed an increase in the %MN (Figures 5A, 6B, & 7B). These results are consistent with those from a recent study showing that lasiocarpine (10–500 µM), in a cell-free system, was activated by exogenous human supersomes containing CYP3A4 or 3A5 enzymes (CYP3A7 was not tested in this system), and in Chinese hamster lung V79-hCYP3A4 cells, lasiocarpine induced γH2A.X formation and increases in micronuclei (Ebmeyer et al., 2019). In another cell-free system using human recombinant CYP supersomes, the formation of pyrrole–protein adducts after incubation with 200 µM lasiocarpine was shown to be mediated predominantly by CYP3A4 and 3A5 (Ruan et al., 2014).
Human and rat liver microsomes were shown to be similarly effective in metabolizing riddelliine, as evidenced by a CYP3A4 inhibitor that significantly reduced the formation of DHP with both sets of microsomes (Xia et al., 2003). In the present study we provide strong evidence that the metabolic activation of riddelliine (5–100 µM) is catalyzed by human CYP3A4 (induction of cytotoxicity, apoptosis, DNA damage, and chromosome damage; Figures 3, 4, 5B, 7C, & 7D) followed by CYP3A7 (induction of cell cycle changes and chromosome damage; Figures 5B & 6C) and 3A5 (cell cycle changes only; Figure 6C). These results confirmed previous observations in a cell-free system (Xia et al., 2003) and suggest that our TK6 cell system is valid and effective in detecting the specific enzymes that are responsible for the bioactivation of pro-genotoxicants.
Senkirkine, an otonecine-type of PA, is a rodent liver carcinogen and was shown to induce chromosomal aberrations in V79 cells and mutations in S. typhimurium TA100 with S9 (Chen et al., 2010). Senkirkine is generally less toxic compared to the heliotridine-type and retronecine-type PAs (Fu, 2017), and there is little information on the specific enzymes that catalyze its biotransformation. In this study we demonstrated that senkirkine exhibits cytotoxicity and genotoxicity in CYP3A4-expressing TK6 cells. In CYP3A5-expressing TK6 cells, senkirkine only induced cell cycle changes (Figure 6D). Although low levels of DHP were detected after senkirkine exposure in CYP3A5- and 3A7-expressing TK6 cells (Table 1), no increases in the %MN or γ-H2A.X formation were observed at the concentrations tested (Figures 5C, 7E, & 7F), suggesting that any DNA damage that might have occurred, was either rapidly repaired or below the level of detection. Previously, using human recombinant CYP supersomes in a cell-free system, senkirkine and another otonecine-type PA (clivorine), tested at 200 µM, were shown to induce pyrrole–protein adducts mediated by CYP3A4 and 3A5 (Ruan et al., 2014). Taken together, these studies suggest that CYP3A4 is the primary enzyme responsible for the bioactivation of the otonecine-type of PAs.
TK6 cells have wild-type p53, the key regulator in cells’ DNA-repair signaling pathways. In response to genotoxic stress, cells with DNA repair capability activate checkpoint genes to help maintain the integrity of the genome (Branzei and Foiani, 2008). Specifically, phosphorylation of CHK1 and CHK2 in response to p53 signaling triggers cell-cycle arrest at G2/M, allowing the cell time to repair genotoxicant-induced DNA lesions (Kondo et al., 2010). We previously showed that PAs, after metabolic activation, formed DHP-dG and DHP-dA adducts in vivo (F344 rats) and in vitro (human lung carcinoma cell line A549) (He et al., 2019; Xia et al., 2013). In p53-competent cells, the removal of DNA adducts is mainly mediated by nucleotide-excision repair (NER), which can activate key checkpoint pathways (Marini et al., 2006). In the present study we observed that expression of p-CHK1, p-CHK2, and γ-H2A.X was increased in CYP3A4-expressing TK6 cells after treatments with all 3 PAs, supporting our hypothesis that DNA and chromosome damage observed in TK6 cells was induced by the reactive metabolites formed by CYP3A4 (Figure 1).
In our TK6 cell system, since the CYP cDNAs were transduced into the TK6 cell genome with lentiviral expression vectors, CYP expression is propagated to daughter cells. Therefore, minimal handling procedures (i.e., no additional transfection) are required before initiation of treatments, enhancing the compatibility of our cell system for quantitative HT in vitro screening. The dynamic range of the HT micronucleus assay can be as high as 40-fold at the maximum concentration tested (Figure 5A). Therefore, our novel TK6 cell system, in combination with HT genotoxicity assays, is a promising tool to evaluate compound libraries for genotoxic potential, as well as to identify the specific CYPs that may be involved in biotransformation.
The field of genetic toxicology is at a crossroads, transitioning from assays that provide qualitative hazard identification to those that include data suitable for quantitative risk assessment (Guo and Mei, 2018; Heflich et al., 2020). Comparing the relative genotoxic potentials among chemicals is an important component in risk assessment. BMD modeling is a promising method for quantitative genotoxicity evaluation (Li et al., 2020b; White et al., 2020). In the present study, in CYP3A4-expressing TK6 cells, lasiocarpine, riddelliine, and senkirkine significantly decreased cell viability at concentrations of 0.5 μM, 10 μM, and 25 μM, respectively (Figure 3) and showed a LOAEL for MN induction of 0.5 μM, 5 μM, and 25 μM, respectively (Figure 5). When BMD modeling was applied to these data (Figure 8A), lasiocarpine was shown to be the most potent PA with a BMD100 of 0.04 µM, a value that was about 64- and 193-fold lower than the BMD100 values for riddelliine and senkirkine, respectively (Figure 8B). The BMD approach explicitly takes the shape of the concentration-response curve into account; in the present study, the best-fitting model for the riddelliine MN data showed a non-linear concentration-response (i.e., exponential shape), reflecting the fact that riddelliine did not induce an increase in %MN at lower concentrations and then induced a sharp increase in %MN over a concentration range from 5 µM to 10 µM. For risk assessment, it was assumed that all PAs had similar genotoxic potentials (Geburek et al., 2020). However, when comparing the genotoxic potency of 15 PAs in human liver HepaRG cells which express metabolizing enzymes, lasiocarpine was found to be the most potent PA, with a BMD100 (measured as a 2-fold increase in MN induction) that was distinctly lower than the other 14 PAs tested (Allemang et al., 2018). Using the γH2AX In Cell Western (ICW) assay in HepaRG cells, the genotoxic potencies of 37 PAs were also shown to be remarkably different (Louisse et al., 2019). PAs display a strong relationship between structure and toxicity, and their relative potencies may span several orders of magnitude (Lester et al., 2019; Louisse et al., 2019). Results of our study in TK6 cells provide additional evidence of the differing genotoxic potencies of PAs.
In summary, we demonstrated that the genotoxicity of lasiocarpine, riddelliine, and senkirkine is primarily catalyzed by CYP3A4, one out of the fourteen CYPs in our newly developed cell system. CYP3A5 and 3A7 are also involved in the biotransformation of these PAs at a much lower magnitude. We showed that lasiocarpine (the heliotridine-type of PA) had the highest cytotoxic and genotoxic potency, followed by riddelliine (the retronecine-type of PA) and then senkirkine (the otonecine-type of PA). These results indicate that our newly developed TK6 cell system individually expressing fourteen different CYPs is useful in screening for cytotoxicity and genotoxicity of chemicals that require metabolic activation. Furthermore, this TK6 cell system is also useful in identifying the specific CYP isozymes that are involved in the biotransformation and discriminating genotoxicity potency of PAs that have different structural PA types.
Supplementary Material
Acknowledgments
This work was supported by the U.S. FDA, National Center for Toxicological Research (NCTR, project number E0765701), and in part by the U.S. NIH, National Institute of Environmental Health Sciences (NIEHS). XL was supported by an appointment to the Postgraduate Research Program at the NCTR administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. FDA. We thank Drs. Sreeni Ramaiahgari from NIEHS, Alexander Alund, Peter P. Fu, and Qingsu Xia from NCTR for their critical review of this manuscript.
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest. The information in this manuscript is not a formal dissemination of information by the U.S. Food and Drug Administration (FDA) or the U.S. National Institutes of Health (NIH) and does not constitute an advisory opinion, does not necessarily represent the formal position of FDA and NIH, and does not bind or otherwise obligate or commit the agency to the views expressed.
References
- Allemang A, Mahony C, Lester C, Pfuhler S, 2018. Relative potency of fifteen pyrrolizidine alkaloids to induce DNA damage as measured by micronucleus induction in HepaRG human liver cells. Food Chem Toxicol 121, 72–81. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M, 2008. Regulation of DNA repair throughout the cell cycle. Nature reviews Molecular cell biology 9, 297–308. [DOI] [PubMed] [Google Scholar]
- Brüsehafer K, Rees BJ, Manshian BB, Doherty AT, O’Donovan MR, Doak SH, Jenkins GJS, 2014. Chromosome breakage induced by the genotoxic agents mitomycin C and cytosine arabinoside is concentration and p53 dependent. Toxicological sciences 140, 94–102. [DOI] [PubMed] [Google Scholar]
- Chaudhary P, Sharma R, Sahu M, Vishwanatha JK, Awasthi S, Awasthi YC, 2013. 4-Hydroxynonenal induces G2/M phase cell cycle arrest by activation of the ataxia telangiectasia mutated and Rad3-related protein (ATR)/checkpoint kinase 1 (Chk1) signaling pathway. Journal of Biological Chemistry 288, 20532–20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Mei N, Fu PP, 2010. Genotoxicity of pyrrolizidine alkaloids. J Appl Toxicol 30, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WG, Buhler DR, 1994. The effect of spironolactone treatment on the cytochrome P450-mediated metabolism of the pyrrolizidine alkaloid senecionine by hepatic microsomes from rats and guinea pigs. Toxicol Appl Pharmacol 127, 314–319. [DOI] [PubMed] [Google Scholar]
- Chung WG, Buhler DR, 1995. Major factors for the susceptibility of guinea pig to the pyrrolizidine alkaloid jacobine. Drug Metab Dispos 23, 1263–1267. [PubMed] [Google Scholar]
- Dusemund B, Nowak N, Sommerfeld C, Lindtner O, Schafer B, Lampen A, 2018. Risk assessment of pyrrolizidine alkaloids in food of plant and animal origin. Food Chem Toxicol 115, 63–72. [DOI] [PubMed] [Google Scholar]
- Ebmeyer J, Braeuning A, Glatt H, These A, Hessel-Pras S, Lampen A, 2019. Human CYP3A4-mediated toxification of the pyrrolizidine alkaloid lasiocarpine. Food Chem Toxicol 130, 79–88. [DOI] [PubMed] [Google Scholar]
- EPA, 2019. Efforts to reduce animal testing at EPA. U.S. Environmental Protection Agency, https://www.epa.gov/research/efforts-reduce-animal-testing-epa (accessed on 9 July 2020). [Google Scholar]
- Evans SJ, Gollapudi B, Moore MM, Doak SH, 2019. Horizon scanning for novel and emerging in vitro mammalian cell mutagenicity test systems. Mutat Res 847, 403024. [DOI] [PubMed] [Google Scholar]
- Fu PP, 2017. Pyrrolizidine Alkaloids: Metabolic Activation Pathways Leading to Liver Tumor Initiation. Chem Res Toxicol 30, 81–93. [DOI] [PubMed] [Google Scholar]
- Fu PP, Xia Q, Lin G, Chou MW, 2004. Pyrrolizidine alkaloids--genotoxicity, metabolism enzymes, metabolic activation, and mechanisms. Drug Metab Rev 36, 1–55. [DOI] [PubMed] [Google Scholar]
- Geburek I, Preiss-Weigert A, Lahrssen-Wiederholt M, Schrenk D, These A, 2020. In vitro metabolism of pyrrolizidine alkaloids - Metabolic degradation and GSH conjugate formation of different structure types. Food Chem Toxicol 135, 110868. [DOI] [PubMed] [Google Scholar]
- Gollapudi BB, White PA, Honma M, 2019. The IWGT in vitro Mammalian Cell Gene Mutation (MCGM) assays working group-Introductory remarks & consensus statements. Mutat Res 848, 403061. [DOI] [PubMed] [Google Scholar]
- Guo X, Chen S, Zhang Z, Dobrovolsky VN, Dial SL, Guo L, Mei N, 2015. Reactive oxygen species and c-Jun N-terminal kinases contribute to TEMPO-induced apoptosis in L5178Y cells. Chem Biol Interact 235, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Mei N, 2018. Benchmark Dose Modeling of In Vitro Genotoxicity Data: a Reanalysis. Toxicol Res 34, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Seo JE, Li X, Mei N, 2020. Genetic toxicity assessment using liver cell models: past, present, and future. J Toxicol Environ Health B Crit Rev 23, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Xia Q, Wu Q, Tolleson WH, Lin G, Fu PP, 2019. Primary and secondary pyrrolic metabolites of pyrrolizidine alkaloids form DNA adducts in human A549 cells. Toxicology in Vitro 54, 286–294. [DOI] [PubMed] [Google Scholar]
- Heflich RH, Johnson GE, Zeller A, Marchetti F, Douglas GR, Witt KL, Gollapudi BB, White PA, 2020. Mutation as a Toxicological Endpoint for Regulatory Decision-Making. Environ Mol Mutagen 61, 34–41. [DOI] [PubMed] [Google Scholar]
- Hsieh JH, Smith-Roe SL, Huang R, Sedykh A, Shockley KR, Auerbach SS, Merrick BA, Xia M, Tice RR, Witt KL, 2019. Identifying Compounds with Genotoxicity Potential Using Tox21 High-Throughput Screening Assays. Chem Res Toxicol 32, 1384–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC, 2018. Agents classified by the IARC monographs, Volumes 1–123. The International Agency for Research on Cancer https://monographs.iarc.fr/wp-content/uploads/2018/2009/List_of_Classifications.pdf (accessed on 9 July 2020). [Google Scholar]
- ICH, 2011. ICH guidance S2(R1) on genotoxicity testing and data interpretation for pharmaceuticals intened for human use https://www.ema.europa.eu/en/ich-s2-r1-genotoxicity-testing-data-interpretation-pharmaceuticals-intended-human-use (accessed 9 July 2020). [Google Scholar]
- Kirkland D, Pfuhler S, Tweats D, Aardema M, Corvi R, Darroudi F, Elhajouji A, Glatt H, Hastwell P, Hayashi M, Kasper P, Kirchner S, Lynch A, Marzin D, Maurici D, Meunier JR, Muller L, Nohynek G, Parry J, Parry E, Thybaud V, Tice R, van Benthem J, Vanparys P, White P, 2007. How to reduce false positive results when undertaking in vitro genotoxicity testing and thus avoid unnecessary follow-up animal tests: Report of an ECVAM Workshop. Mutation research 628, 31–55. [DOI] [PubMed] [Google Scholar]
- Kondo N, Takahashi A, Ono K, Ohnishi T, 2010. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids 2010, 543531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester C, Troutman J, Obringer C, Wehmeyer K, Stoffolano P, Karb M, Xu Y, Roe A, Carr G, Blackburn K, Mahony C, 2019. Intrinsic relative potency of a series of pyrrolizidine alkaloids characterized by rate and extent of metabolism. Food Chem Toxicol 131, 110523. [DOI] [PubMed] [Google Scholar]
- Li N, Xia Q, Ruan J, Fu PP, Lin G, 2011. Hepatotoxicity and tumorigenicity induced by metabolic activation of pyrrolizidine alkaloids in herbs. Current drug metabolism 12, 823–834. [DOI] [PubMed] [Google Scholar]
- Li X, Chen S, Guo X, Wu Q, Seo JE, Guo L, Manjanatha MG, Zhou T, Witt KL, Mei N, 2020a. Development and Application of TK6-derived Cells Expressing Human Cytochrome P450s for Genotoxicity Testing. Toxicol Sci 175, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Guo XQ, Wang HR, Chen T, Mei N, 2020b. Aristolochic Acid-Induced Genotoxicity and Toxicogenomic Changes in Rodents. World J Tradit Chin Med 6, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Cui YY, Liu XQ, Wang ZT, 2002. Species differences in the in vitro metabolic activation of the hepatotoxic pyrrolizidine alkaloid clivorine. Chem Res Toxicol 15, 1421–1428. [DOI] [PubMed] [Google Scholar]
- Louisse J, Rijkers D, Stoopen G, Holleboom WJ, Delagrange M, Molthof E, Mulder PPJ, Hoogenboom R, Audebert M, Peijnenburg A, 2019. Determination of genotoxic potencies of pyrrolizidine alkaloids in HepaRG cells using the gammaH2AX assay. Food Chem Toxicol 131, 110532. [DOI] [PubMed] [Google Scholar]
- Ma J, Xia Q, Fu PP, Lin G, 2018. Pyrrole-protein adducts - A biomarker of pyrrolizidine alkaloid-induced hepatotoxicity. Journal of food and drug analysis 26, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, Muzi Falconi M, 2006. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc Natl Acad Sci USA 103, 17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattocks AR, 1968. Toxicity of pyrrolizidine alkaloids. Nature 217, 723–728. [DOI] [PubMed] [Google Scholar]
- Mei N, Chou MW, Fu PP, Heflich RH, Chen T, 2004a. Differential mutagenicity of riddelliine in liver endothelial and parenchymal cells of transgenic Big Blue rats. Cancer Lett 215, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei N, Guo L, Fu PP, Fuscoe JC, Luan Y, Chen T, 2010. Metabolism, genotoxicity, and carcinogenicity of comfrey. J Toxicol Environ Health B Crit Rev 13, 509–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei N, Guo L, Liu R, Fuscoe JC, Chen T, 2007. Gene expression changes induced by the tumorigenic pyrrolizidine alkaloid riddelliine in liver of Big Blue rats. BMC Bioinformatics 8 Suppl 7, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei N, Heflich RH, Chou MW, Chen T, 2004b. Mutations induced by the carcinogenic pyrrolizidine alkaloid riddelliine in the liver cII gene of transgenic Big Blue rats. Chem Res Toxicol 17, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC, 2007. Toxicity testing in the 21st century: A vision and a strategy National Research Council, National Academy Press, Washington, DC., https://www.nap.edu/read/11970/chapter/11971 (accessed on 9 July 12020). [Google Scholar]
- Pfuhler S, Fellows M, van Benthem J, Corvi R, Curren R, Dearfield K, Fowler P, Frotschl R, Elhajouji A, Le Hegarat L, Kasamatsu T, Kojima H, Ouedraogo G, Scott A, Speit G, 2011. In vitro genotoxicity test approaches with better predictivity: summary of an IWGT workshop. Mutat Res 723, 101–107. [DOI] [PubMed] [Google Scholar]
- Ruan J, Yang M, Fu P, Ye Y, Lin G, 2014. Metabolic activation of pyrrolizidine alkaloids: insights into the structural and enzymatic basis. Chemical research in toxicology 27, 1030–1039. [DOI] [PubMed] [Google Scholar]
- Schrenk D, Gao L, Lin G, Mahony C, Mulder PPJ, Peijnenburg A, Pfuhler S, Rietjens I, Rutz L, Steinhoff B, These A, 2020. Pyrrolizidine alkaloids in food and phytomedicine: Occurrence, exposure, toxicity, mechanisms, and risk assessment - A review. Food Chem Toxicol 136, 111107. [DOI] [PubMed] [Google Scholar]
- Shao K, Gift JS, 2014. Model Uncertainty and Bayesian Model Averaged Benchmark Dose Estimation for Continuous Data. Risk Anal 34, 101–120. [DOI] [PubMed] [Google Scholar]
- Shao K, Shapiro AJ, 2018. A web-based system for Bayesian benchmark dose estimation. Environmental health perspectives 126, 017002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slob W, 2017. A general theory of effect size, and its consequences for defining the benchmark response (BMR) for continuous endpoints. Critical reviews in toxicology 47, 342–351. [DOI] [PubMed] [Google Scholar]
- VICH, 2014. Studies to evaluate the safety of residues of veterinary drugs in human food: genotoxicity testing, VICH GL23(R). International Cooperation on Harmonization of Technical Requirements for Registration of Veterinary Medicinal Products (VICH) https://vichsec.org/en/guidelines/pharmaceuticals/pharma-safety/toxicology.html (accessed on 9 July 2020). [Google Scholar]
- White PA, Long AS, Johnson GE, 2020. Quantitative Interpretation of Genetic Toxicity Dose-Response Data for Risk Assessment and Regulatory Decision-Making: Current Status and Emerging Priorities. Environ Mol Mutagen 61, 66–83. [DOI] [PubMed] [Google Scholar]
- Xia Q, Chou MW, Kadlubar FF, Chan PC, Fu PP, 2003. Human liver microsomal metabolism and DNA adduct formation of the tumorigenic pyrrolizidine alkaloid, riddelliine. Chem Res Toxicol 16, 66–73. [DOI] [PubMed] [Google Scholar]
- Xia Q, Zhao Y, Von Tungeln LS, Doerge DR, Lin G, Cai L, Fu PP, 2013. Pyrrolizidine alkaloid-derived DNA adducts as a common biological biomarker of pyrrolizidine alkaloid-induced tumorigenicity. Chem Res Toxicol 26, 1384–1396. [DOI] [PubMed] [Google Scholar]
- Zeller A, Duran-Pacheco G, Guerard M, 2017. An appraisal of critical effect sizes for the benchmark dose approach to assess dose-response relationships in genetic toxicology. Arch Toxicol 91, 3799–3807. [DOI] [PubMed] [Google Scholar]
- Zeller A, Tang L, Dertinger SD, Funk J, Duran-Pacheco G, Guerard M, 2016. A proposal for a novel rationale for critical effect size in dose-response analysis based on a multi-endpoint in vivo study with methyl methanesulfonate. Mutagenesis 31, 239–253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.