

SUMMARY
TEAD1 and the mammalian Hippo pathway regulate cellular proliferation and function, though their regulatory function in β cells remains poorly characterized. In this study, we demonstrate that while β cell-specific TEAD1 deletion results in a cell-autonomous increase of β cell proliferation, β cell-specific deletion of its canonical coactivators, YAP and TAZ, does not affect proliferation, suggesting the involvement of other cofactors. Using an improved split-GFP system and yeast two-hybrid platform, we identify VGLL4 and MENIN as TEAD1 corepressors in β cells. We show that VGLL4 and MENIN bind to TEAD1 and repress the expression of target genes, including FZD7 and CCN2, which leads to an inhibition of β cell proliferation. In conclusion, we demonstrate that TEAD1 plays a critical role in β cell proliferation and identify VGLL4 and MENIN as TEAD1 corepressors in β cells. We propose that these could be targeted to augment proliferation in β cells for reversing diabetes.
In brief
Feng et al. report that YAP and TAZ do not have a physiological role in β cell proliferation and function. They identify VGLL4 and MENIN as TEAD1 corepressors in regulating β cell proliferation and propose that these may be targeted to augment proliferation in β cells for reversing diabetes.
Graphical abstract
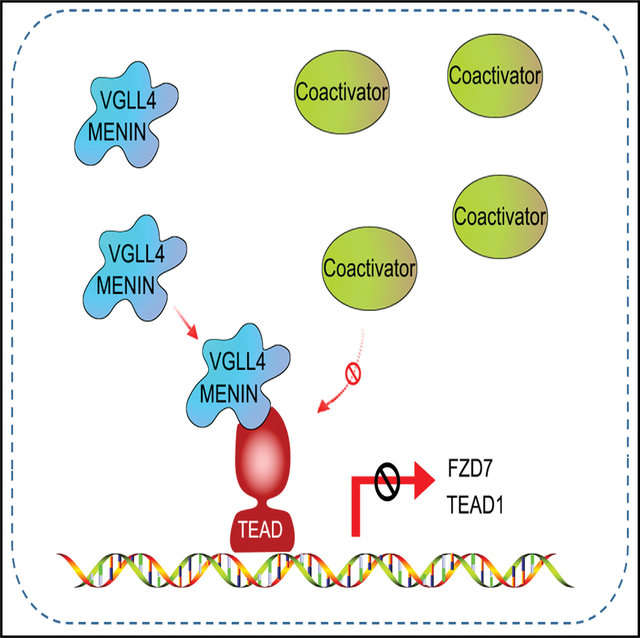
INTRODUCTION
Recent findings have challenged the dogma that all β cells are destroyed early in type 1 diabetes, with increasing evidence demonstrating the persistence of insulin-positive islets even decades after diagnosis.1 In type 2 diabetes, β cell mass significantly decreased with duration of clinical diabetes, being more than 54% lower in subjects with >15 years of overt diabetes.2 Increasing β cell mass by enhancing the proliferation of the remaining β cells would be ideal for reducing dependence on injectable insulin and improving the quality of life for many patients with diabetes. The mammalian Hippo pathway, with YAP and TAZ being the most studied downstream coactivators, regulates cell number by modulating cell proliferation, cell death, and cell differentiation.3 YAP and TAZ do not have their own DNA-binding domain and depend on binding transcription factors, including the TEAD family, to fulfill their gene regulatory functions.3 TEAD family of transcription factors thus play a critical role in connecting the upstream signals in Hippo pathway with downstream gene expression. In mammals, there are four genes encoding homologous members of the TEAD family, TEAD1–4, which recognize the MCAT gene sequence motif (GGAATG). TEADs do not have their own transcription activation or repression domain, so they need to bind to cofactors to regulate gene expression. Each TEAD has tissue-specific expression, which suggests tissue-specific roles for each TEAD.4 Our previous study showed that TEAD1 was preferentially expressed in adult mouse islets, whereas other members of the TEAD family are present at a 10-fold lower level (TEAD2) or not detected (TEAD3–4).5 By far, YAP and TAZ are the most studied coactivators of TEAD. Although YAP expression cannot be detected in islets,6,7 overexpression of YAP can increase β cell proliferation,8 suggesting TEAD1 pathway is preserved and ready to be reactivated. Our previous data showed that TAZ expression could be detected in mouse islets,5 but TAZ’s role in mature β cells remains unknown. Here, we demonstrate using in vivo β cell-specific deletion in mice that YAP and TAZ have no role in β cell function or proliferation. Using multiple strategies to identify functional TEAD1-binding partners, we identified VGLL4 and MENIN as corepressors that bind to TEAD1 in β cells. While MENIN’s loss of function leads to insulinomas, the role of VGLL4, a known TEAD corepressor shown to play an important role in cancer development,9 in β cells is unknown. We demonstrate that VGLL4 and MENIN bind to the C-terminal pocket region of TEAD1 and competitively inhibit co-activation of TEAD1, leading to a repression of β cell proliferation-related genes. Thus the balance between coactivators and these newly identified corepressors may play essential roles in modulating TEAD pathway activity and β cell proliferation and maturity. Therapeutic modulation of these regulatory pathways in diabetes could enhance β cell proliferation while preserving mature function.
RESULTS
YAP and TAZ are not required for β cell function in vivo
β cell proliferation was significantly reduced at 24 weeks compared with 4 weeks of age (Figure 1A), consistent with previous observations. Luciferase assay with HOPFLASH reporter (reporter with 8x MCAT) showed that TEAD pathway activities were significantly reduced in islets at 24 weeks compared with 4 weeks of age (Figure 1B). TAZ, TEAD1, and TEAD pathway classical target gene, CCN2 (CTGF), were significantly reduced in islets from 24 weeks compared with 4-week-old mice (Figure 1C), suggesting TEAD pathway may be less active with age. We generated a β cell-conditional YAP/TAZ double knockout (β-Y/T KO) mouse model to explore the role of YAP and TAZ in β cells proliferation and maturity (see STAR Methods). There were no changes in body weight between β-Y/T KO and control flox mice at 4 weeks (Figure S1A) or 6 months (Figure S1B). The fasting blood glucose level was not different between β-Y/T KO and control flox mice (Figure S1C). Intraperitoneal glucose tolerance test (IPGTT) showed blood glucose and serum insulin levels were not different at any time point between β-Y/T KO and controls on chow diet (Figure 1D) or after 8 weeks of high-fat diet feeding (Figure 1E). Immunostaining showed Ki67-positive β cell rate has no change between control and β-Y/T KO (Figure S1D). Immunostaining confirmed that TAZ was deleted in β cells successfully, while YAP was not expressed in islets. (Figure S1E). These results demonstrate that β cell YAP and TAZ were not required to maintain normal glucose homeostasis.
Figure 1. YAP/TAZ knockout does not affect β cell function and proliferation, while TEAD1 knockout results in β cell autonomous proliferation.
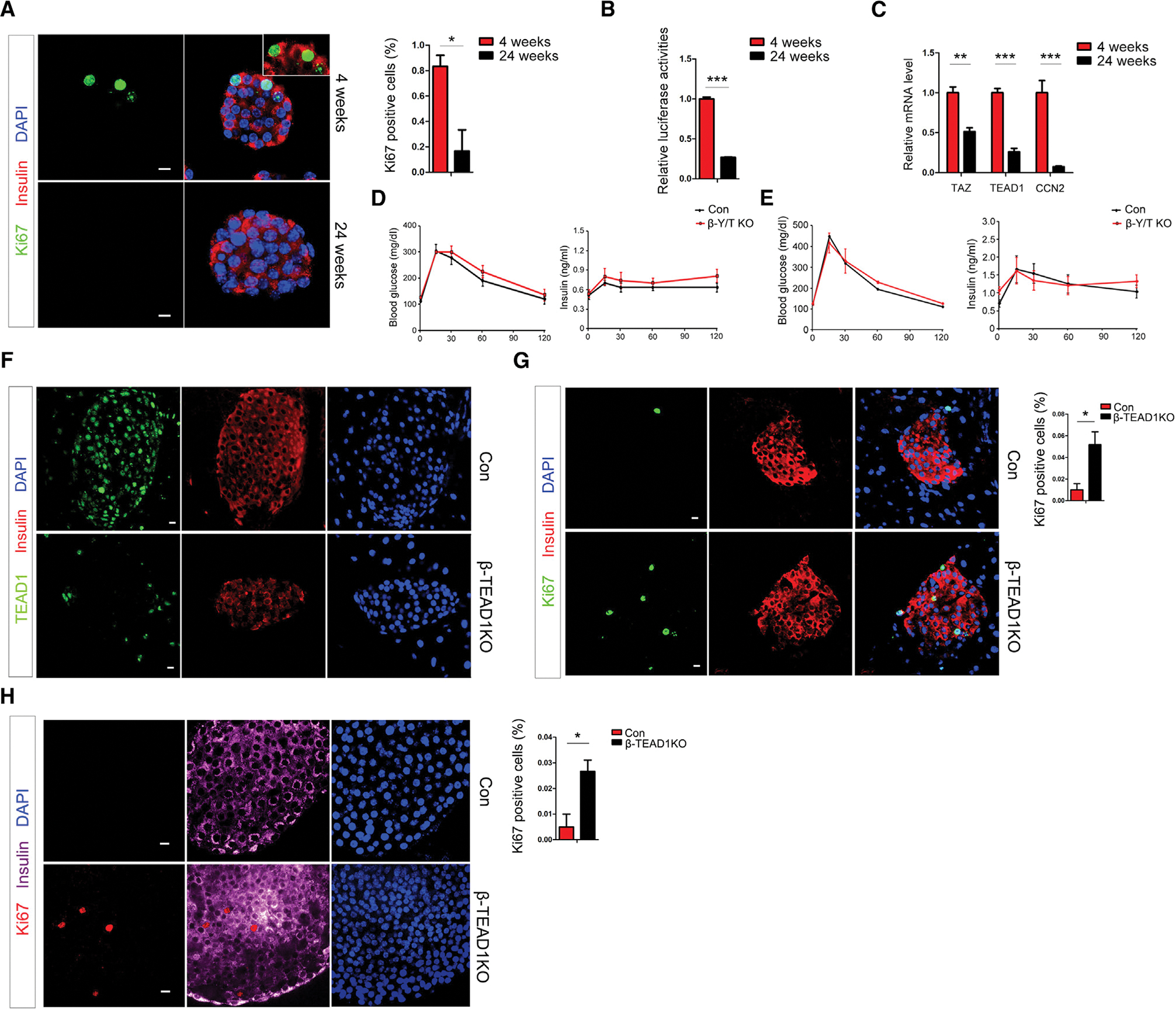
(A) Representative immunostaining image and quantitation of Ki67-positive β cells in wild-type mice at different ages (n = 3 mice/group).
(B) Luciferase assay showing TEAD pathway activities in wild-type mouse islets at different ages (n = 3 mice/group).
(C) mRNA levels of TAZ, TEAD1, and CCN2 in mouse β cells at different ages (n = 3 mice/group).
(D) IPGTT showing both blood glucose and plasma insulin levels in β-Y/T KO mice and control (n = 6 mice/group).
(E) IPGTT showing both blood glucose and plasma insulin levels at different time points in β-Y/T KO mice and control after 2-month high-fat diet feeding (n = 6 mice/group).
(F) Immunostaining to validate TEAD1 was successfully knocked out in β-TEAD1KO (TEAD1-conditional knockout) β cells.
(G) Representative immunostaining image and quantitation of Ki67-positive β cells in β-TEAD1KO and control (n = 3 mice/group).
(H) Representative immunostaining image and quantitation of Ki67- positive β cells that were cultured in vitro in β-TEAD1KO and control (n = 3 mice/group).
*p < 0.05; **p < 0.01; ***p < 0.001. p values are from Student’s t test. Error bars represent standard error of the mean (SEM). Scale bar, 10 μm.
TEAD1 knockout leads to β cell autonomous proliferation
A β cell-conditional TEAD1 knockout mouse model (β-TEAD1KO) was used to study the roles of TEAD1 in β cells proliferation and maturity.5 Immunostaining confirmed that TEAD1 was deleted in β cells successfully (Figure 1F). Immunostaining showed Ki67-positive β cells were significantly increased in β-TEAD1KO (Figure 1G). When isolated islets were cultured in the same condition, ex vivo, immunostaining showed Ki67-positive β cells were significantly increased in β-TEAD1KO (Figure 1H), which suggests that the proliferation caused by TEAD1 knockout was cell autonomous. The data from β-Y/T KO and β-TEAD1KO models suggested two possibilities: (1) there are unrevealed TEAD1 coactivators other than YAP and TAZ that play a functional role in β cells; (2) TEAD1 corepressors play dominant roles, compared with coactivators, in β cells.
Screen for TEAD1 cofactors in β cells
We utilized a yeast two-hybrid system (see STAR Methods) to screen new TEAD1 cofactors. Human islet cDNA library was used as “prey” and TEAD1 CDS sequence as the “bait” to perform an unbiased screen. Nine proteins, including VGLL4, TAZ, VGLL3, HIPK3, JUN, PIAS2, RNF111, RUNX1, and SUMO1, could bind TEAD1 directly, but the first three had higher binding scores (Figure 2A). VGLL3 and VGLL4 belong to the vestigial-like family members. In cancer cells, VGLL4 has opposing functions to VGLL3.9 Human islet cell sequencing showed that in whole islet, VGLL4 expression is much higher than the other VGLL family member (Figure S2A). Single-cell sequencing of human islets also showed VGLL4 expression to be higher in β cells than in α cells (Figure S2B). Since full-length human islet cDNA have 5′ and 3′ UTRs, which could result in frameshift, there could be high false-negative rates in a yeast two-hybrid system. Hence, we also used a split luciferase system10 to screen potential TEAD cofactors (Figure 2B). In this system, nanoluciferase was separated into two parts and was attached to TEAD1 and TAZ respectively. When TEAD1 binds TAZ, luciferase fragments are close to each other and form a complemented luciferase producing a luminescent signal in the presence of its substrate, luciferin. If another protein can also interact with TEAD1, this interaction will compete to interrupt TEAD1-TAZ binding, reducing the luminescent signal. Since it is known that SMAD411 and MENIN12 can repress β cell proliferation, we hypothesized that this was consistent with the possibility that they could also function as potential corepressors of TEAD. We tested if SMAD4 and MENIN, as well as VGLL4, can interrupt TEAD1-TAZ (TT) binding using this split luciferase system. As a positive control, YAP5SA, the constitutively active form of YAP, could reduce the TEAD1-TAZ binding signal in this split luciferase system. MENIN and VGLL4 also reduced TEAD1-TAZ binding signal, while SMAD4 could not (Figure 2C). qPCR showed VGLL4 level is significantly increased in islet at the age of 24 weeks compared with 4 weeks, while SMAD4 and MENIN displayed no change (Figure 2D). Similarly, single-cell sequencing of human islets demonstrated that VGLL4 expression is significantly increased in β cells at the age of 40–49 years compared with 20–29 years (Figure 2E). We then used a split-GFP system, an antibody-independent assay, to study real-time protein-protein interactions in living cells to check if SMAD4, VGLL4, and MENIN can bind TEAD1 directly. In the classical split-GFP system, the GFP protein with 11 β-stands is divided into three parts, GFP1–9, GFP10, and GFP11, with GFP10 and GFP11 being only 20 amino acids long. In the presence of GFP1–9, when two proteins of interest fused to GFP10 and GFP11 respectively (bait and prey) come in close proximity due to protein-protein interaction, the reassembly of the three parts of GFP leads to green fluorescence.13 However, the directionality requirement of GFP10 and GFP11 fusion sites on the interacting partners limits its efficient and easy applicability. Here we developed an improved split-GFP (iSG) system that has improved sensitivity and overcomes inefficiencies by having GFP10 and GFP11 on both terminals of the bait and prey proteins, respectively as an “all-in-one” plasmid including mCherry as a transfection tracking reporter to evaluate protein-protein interactions (Figures 2F and 2G). GFP11 and GFP10 were fused to TEAD1 and target protein with flexible linkers with a 15-mer linker (Gly-Gly-Gly-Gly-Ser) demonstrating superior sensitivity to a 30-mer linker (data not shown). We first validated the iSG system by testing the interaction of TEAD1 C terminus, the region that is known to interact with TAZ, (cTEAD1 aa200–426), and its known cofactor TAZ and obtained a robust GFP signal only in transfected cells, indicated by the expression of mCherry (red fluorescence) (Figure 2H). GFP10-TAZ12–56-GFP10 fragment was sufficient when expressed with GFP11-cTEAD1-GFP11 in the iSG system to display a very strong GFP signal detected in mCherry-positive cells (Figure 2I), providing further validation since TAZ uses its N terminus (aa13–57) to bind TEAD. As a negative control, no signal could be detected with co-expression of TAZ60–401 (which does not have the TEAD-interacting domain) and cTEAD1 (Figure 2J).
Figure 2. TEAD1 cofactor screen in β cells.
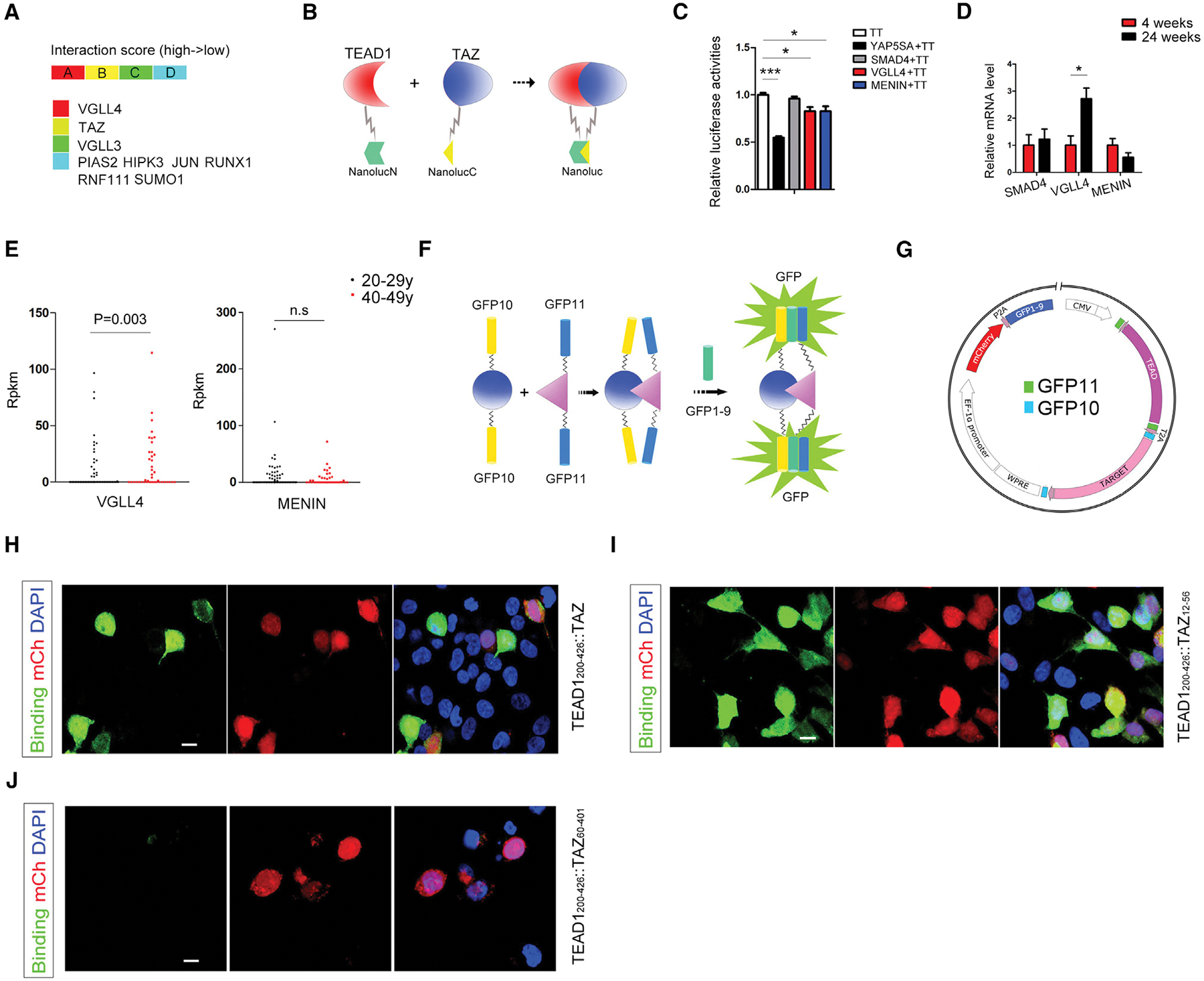
(A) Nine potential TEAD1 cofactors were found by yeast two-hybrid system using human islet cDNA library.
(B) Schematic representation for split luciferase system.
(C) Split luciferase system showed VGLL4 and MENIN can interrupt TAZ-TEAD1 binding.
(D) mRNA levels of VGLL4, SMAD4, and MENIN in wild-type β cells at different ages (n = 3 mice/group).
(E) Single β cell sequencing showing expression levels of VGLL4 at the age of 40–49 years (n = 46) compared with 20–29 years (n = 118)
(F and G) Schematic representation of the improved split-GFP system and the map of the “all-in-one” plasmid.
(H) Validation of iSG system using full-length TAZ as a positive control, where GFP signal was observed with C terminus of TEAD1 (aa200–429) and full-length TAZ in iSG. Red fluorescence was from mCherry. Nuclei were counterstained with DAPI (blue).
(I) GFP signal indicating binding between TEAD1 (aa200–429) and TAZ (aa12–56) in iSG.
(J) Negative control, where no GFP signal was observed with TEAD1 (aa200–429) and TAZ (aa60–401) in iSG. *p < 0.05; **p < 0.01; ***p < 0.001. (C and D) Graphs show mean (±SEM) from at least three biological repeat and triplicate replicate assays and show p value from Student’s t test. Scale bar, 10 μm.
VGLL4 and MENIN are TEAD1 functional cofactors in β cells
GFP signal can be observed between VGLL4-TEAD1 and MENIN-TEAD1 in contrast to SMAD4-TEAD1, indicated by the iSG system (Figure 3A). To further validate the interactions detected by the iSG system, we confirmed binding by coIP of TEAD1 and VGLL4/MENIN protein (Figure 3B). Proximity ligation assay (PLA) also showed the binding signal between TEAD1 and VGLL4/MENIN in situ (Figures 3C and S3A). Since it is possible that the MENIN-TEAD1 interaction could be occurring only when bound/tethered to the DNA by either protein (DNA dependent) or occurring independent of DNA binding, using the inherent advantage of our split-GFP system, we tested if MENIN-TEAD1 and VGLL4-TEAD1 binding is DNA dependent. After the DNase treatment, the known DNA-dependent SOX2-OCT4 binding signal is significantly reduced (positive control). In contrast, MENIN-TEAD1 and VGLL4-TEAD1, as well as TAZ-TEAD1 binding signal, had no significant change, which suggests that MENIN-TEAD1 and VGLL4-TEAD1 binding, similar to TAZ-TEAD1, is DNA independent (Figure 3D). We then used pyDockWEB14 to determine the binding interface between MENIN and TEAD1 (Figure 3E). To validate this prediction, we deleted aa359–371 of TEAD1 (ΔTEAD1), which led to a loss of the binding signal between ΔTEAD1 and MENIN in the split-GFP system (Figure S3B), suggesting that α3 helix of TEAD1 is important for MENIN-TEAD1 binding. Since α3 helix of TEAD1 is also important for YAP/TAZ-TEAD1 binding,15 we hypothesized that MENIN-TEAD1 binding will compete with YAP/TAZ-TEAD1 binding. Luciferase assay showed that MENIN, as well as VGLL4, can decrease the luciferase activities activated by TAZ-TEAD1, indicated by HOPFLASH reporter (Figure 3F) or CCN2 promoter reporter (Figure 3G), while SMAD4 could not (Figure S3C). JUND is an important cofactor for MENIN16; however our data showed that JUND cannot affect the repression effect of MENIN (Figure S3D). The binding signal between TEAD1 and TAZ was significantly reduced when MENIN-BFP was overexpressed in the split-GFP system (Figure 3H). Relative GFP (GFP intensity divided by mCherry intensity) was significantly reduced when MENIN was overexpressed in the split-GFP system, indicated by flow cytometry sorting experiments (Figure 3I). Western blotting showed that overexpression of MENIN cannot affect TEAD1 and TAZ expression in the iSG system (Figure S3E). No binding signals can be observed between MENIN and TAZ in the iSG system (Figure S3F). These experiments indicated that MENIN could block TEAD1-TAZ binding directly, not at the translation level or by MENIN-TAZ binding.
Figure 3. VGLL4 and MENIN bind TEAD1 directly.
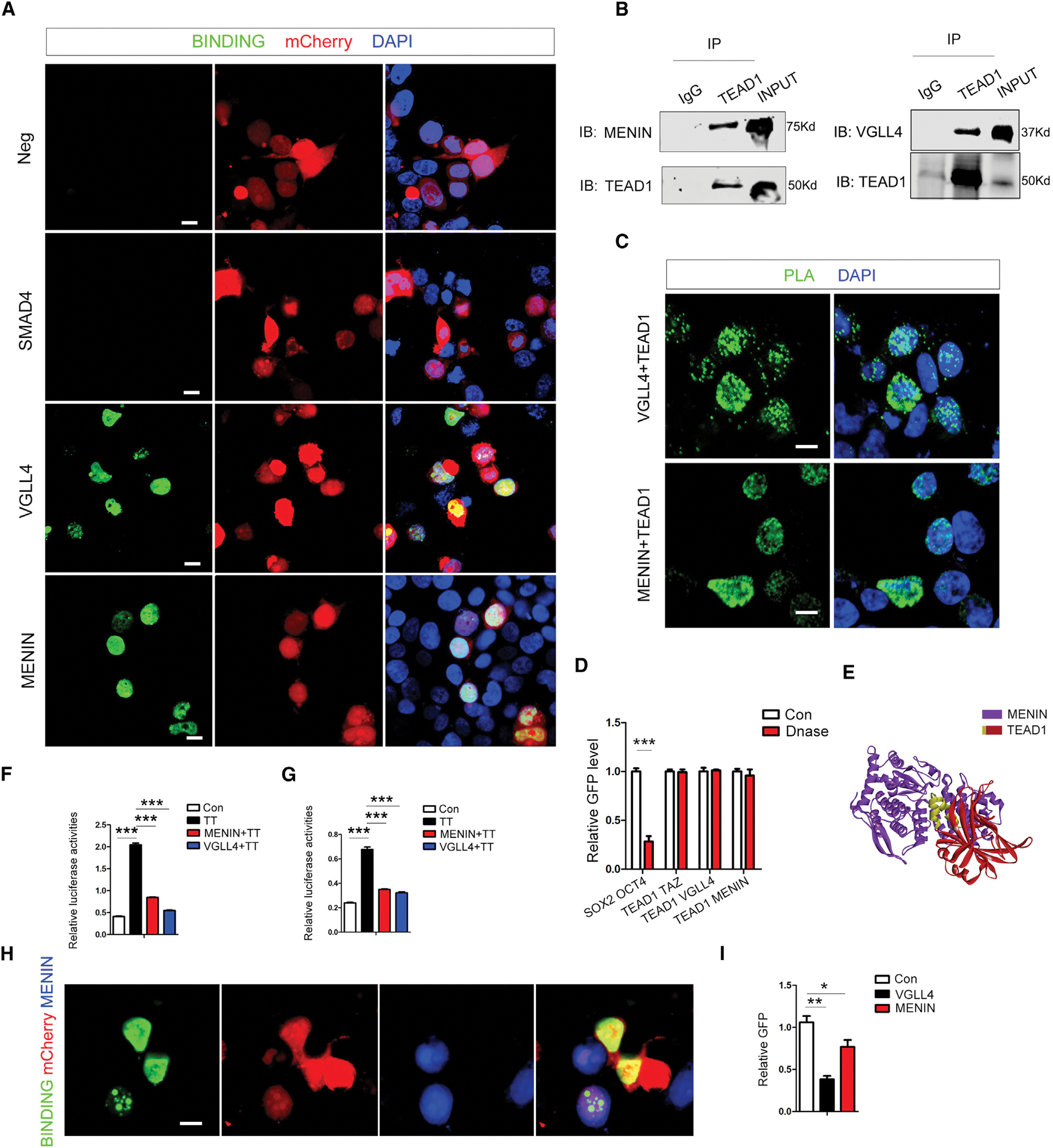
(A) GFP indicating binding between VGLL4-TEAD1 and MENIN-TEAD1 in split-GFP system.
(B) CoIP experiments for MENIN-TEAD1 and VGLL4-TEAD1 binding.
(C) Proximity ligation assay (PLA) for MENIN-TEAD1 and VGLL4-TEAD1 binding (green dots); see also Figure S3A.
(D) SOX2-OCT4 relative binding signal (GFP divided by mCherry value) after DNase treatment. No significant change was observed for TEAD1-TAZ and MENIN-TEAD1 binding.
(E) pyDockWEB prediction model of TEAD1 and MENIN.
(F) Luciferase assay using HOPFLASH reporter for assessing VGLL4 and MENIN inhibition of TAZ-TEAD1 (TT) pathway activity.
(G) Luciferase assay using CCN2 promoter reporter for assessing VGLL4 and MENIN inhibition of TT pathway activity.
(H) TT binding signal in split-GFP system with MENIN overexpression.
(I) Relative GFP value from TT (GFP intensity divided by mCherry intensity) after MENIN overexpression analyzed by flow cytometry sorting. *p < 0.05; **p < 0.01;***p < 0.001. (D, F, G, I) Graphs show mean (±SEM) from at least three biological repeats and triplicate replicate assays and show p value from Student’s t test. cale bar, 10 μm.
VGLL4 can regulate β cell proliferation
It is known that MENIN can regulate β cell proliferation; however the function of VGLL4 in β cells is as yet undetermined. To determine the function of VGLL4 in β cells, we generated a lentiviral plasmid in which VGLL4 was connected with GFP by porcine teschovirus-1 2A (P2A) (Figure S4A) that also allowed evaluation of infection efficiency. Compared with only GFP-expressing control cells, VGLL4 overexpression, confirmed by western blotting (Figure S4B) in INS1 cells, led to a significant reduction in cell number on day 6 after infection (Figure 4A, with quantitation by fluorescence-activated cell sorting [FACS] shown on the right). As opposed to GFP control cells that had an expected increase in cell numbers, the percentage of VGLL4-expressing GFPhigh cells was not changed between day 3 and day 4 by FACS analysis (Figure 4B), suggesting that overexpression of VGLL4 led to a decrease in proliferation of INS1 cells. qPCR demonstrated CCN2, the classical TEAD pathway target gene, was decreased, as would be expected with overexpression of VGLL4, a corepressor of the TEAD pathway. Interestingly, many of the genes associated with mature function such as MAFA, GLUT2 (Slc2a2), NEUROD1, NKX6.1, and PDX1 were not changed with VGLL4 overexpression on day 3 (Figure 4C), suggesting short-term VGLL4 overexpression does not affect β cell maturation status. Immunostaining also showed that Ki67+ VGLL4 + cells were significantly reduced in INS1 cells, suggesting that VGLL4 can repress proliferation in INS1 cells (Figure 4D). Similarly, Ki67- and BRDU-positive cells were significantly reduced after VGLL4 overexpression in INS2 cells (Figures 4E and 4F). Apoptosis was unchanged after VGLL4 overexpression, compared with controls, in INS1 cells, indicated by flow cytometry experiments (Figure S4C). To test the requirement of VGLL4 in β cells, we generated a lentiviral plasmid for mouse VGLL4 knockdown with the shRNA targeting VGLL4 driven by U6 promoter with a CMV promoter-driven GFP as a tracking reporter and a scrambled shRNA lentivirus serving as a control. This shRNA was effective in knocking down VGLL4 in INS2 cells by 70% by qPCR (Figure S4D). Though shRNA lentivirus transduction rate was ~35% in mouse islets, VGLL4 knockdown in adult mouse islets significantly increased β cell proliferation by 4-fold (Figure 4G). Similar results were obtained in human islets (Figure 4H). This suggested that VGLL4 was required for maintaining the low proliferation in adult β cells, and a loss of VGLL4-TEAD function could be mediating the de-repression of proliferative quiescence in adult β cells.
Figure 4. VGLL4 can regulate β cell proliferation.
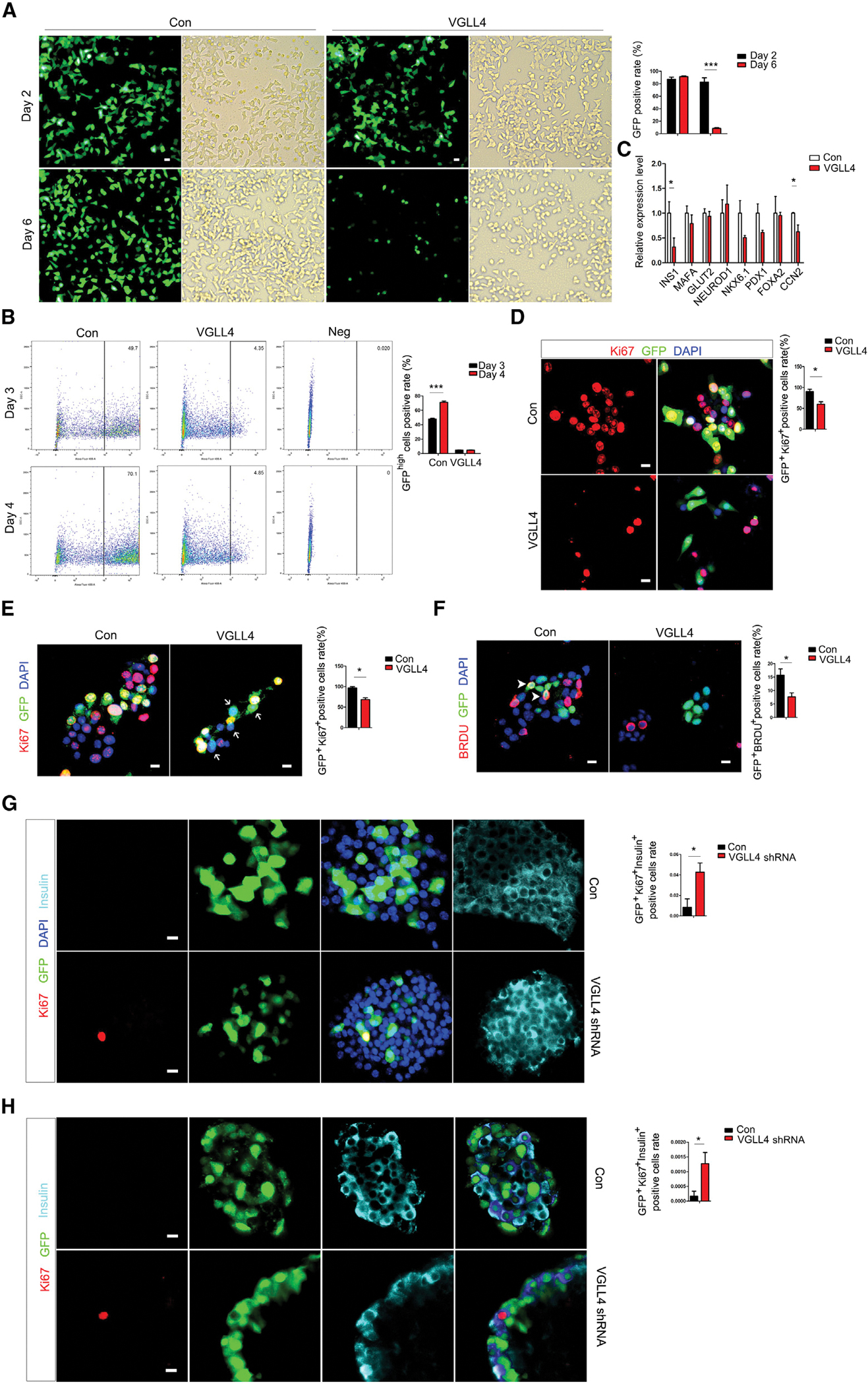
(A) Quantitation of INS1 cell number after VGLL4 overexpression on day 6.
(B) Quantitation of VGLL4-overexpression INS1 cells by flow cytometry sorting analysis.
(C) mRNA levels of INS1, CCN2, MAFA, GLUT2, NEUROD1, NKX6.1, PDX1, and FOXA2 after VGLL4 overexpression in INS1 cells.
(D) Representative immunostaining image and quantitation of Ki67+VGLL4+ cells in INS1 cells.
(E) Representative immunostaining image and quantitation of Ki67+VGLL4+ cells in INS2 cells (arrow).
(F) Representative immunostaining image and quantitation of BRDU+VGLL4+ cells in INS2 cells (arrow head).
(G) Representative immunostaining image and quantitation of Ki67-positive β cells after VGLL4 knockdown in mouse islet.
(H) Representative immunostaining image and quantitation of Ki67-positive β cells after VGLL4 knockdown in human islet. *p < 0.05; **p < 0.01; ***p < 0.001. (A–G) n = at least three per condition with three technical replicates. Error bars represent standard error of the mean (SEM). p values are from Student’s t test. Scale bar, 10 μm.
FZD7 is the target of VGLL4/MENIN-TEAD1 pathway in β cells
We next sought to determine the targets of VGLL4/MENIN-TEAD1 in β cells. Since these genes would be subjected to repression by VGLL4/MENIN-TEAD1, we would expect an upregulation of expression of these target genes with loss of VGLL4/MENIN-TEAD1 function. While β-TEAD1KO islets RNA-seq showed many β cell maturity-associated genes to be significantly reduced, such as MAFA, FOXA1, SLC2a2, NKX2.2, NKX6.1, PDX1, NEUROD1, and FOXA2, many proliferation-associated genes including Mki67, Myc, Mycn, POU3F1, and BMP4 were significantly increased (Figure 5A), suggesting that there was a loss of β cell functional maturity with loss of TEAD1 function. Since Myc, Mycn, and BMP4 are known WNT pathway target genes17–19 and the inactivation of WNT pathway plays a critical role in β cell maturation,20,21 we hypothesized that in β cells, corepressors, such as VGLL4 and MENIN, bind TEAD1 to repress WNT-associated genes’ expression to block proliferation and promote acquisition of mature function. Interestingly, of the “frizzled” (FZD) gene family members, which are receptors for WNT pathway, in β-TEAD1KO islets, qPCR demonstrated expressions of FZD4, FZD5, and FZD7 were significantly increased, while that of FZD10 was reduced (Figure 5B). Analysis of ChIP-seq data from public databases revealed that there were TEAD1 binding peaks in the promoter regions of FZD7 in different cell lines (Figure S5A), including pancreatic progenitor cells (Figure S5B). One classical TEAD binding motif (GGAATG) and one non-classical motif (GGAATT) were found in the sequences extracted from the reported binding peaks (Figure S5C), which were close to the transcription start site (TSS). qPCR showed that similar to CCN2, a known TEAD1 target, FZD7 expression was significantly reduced in islets at the age of 24 weeks compared with 4 weeks (Figure 5C). We performed TEAD1-ChIP-seq in adult mouse islets and found a binding peak in the FZD7 promoter area (Figure 5D). To determine if VGLL4/MENIN-TEAD1 binding to FZD7 promoter was resulting in functional repression, we generated a luciferase reporter using the native FZD7 promoter sequence. Overexpression of VGLL4 or MENIN can significantly reduce the luciferase activities (Figure 5E), indicating that TEAD1 with VGLL4 or MENIN was sufficient to repress FZD7 expression. Consistent with this, a re-analysis of RNA-seq in MENIN-conditional knockout islets22 showed that FZD7 was significantly increased (Figures 5F and S5D). ChIP experiments showed that TEAD1, VGLL4, and MENIN can pull down FZD7 promoter sequences in INS2 cells (Figure 5G). Overexpression VGLL4 in INS1 cells repressed FZD7 expression (Figure 5H). To confirm this regulation of FZD7, we generated a TEAD1 inhibitor to determine if MENIN repression of FZD7 expression is TEAD1 dependent. The N-terminal domain (aa13–57) of TAZ binds helix α3–4 of TEAD1,15 while helix α3 is important for MENIN-TEAD1 binding. We generated a TEAD1-TAZ inhibitor (TTi) peptide incorporating TAZ12–66 connected with a FLAG tag, 2XNLS (nuclear localization sequence) and T2A mCherry (TTi) to block MENIN-TEAD1 binding interface (Figure 5I). TEAD1 reporter luciferase assay showed that TTi had a stronger inhibition of the HOPFLASH reporter than sTDU, another TEAD1 inhibitor that was reported before23 (Figure S5E). qPCR showed FZD7 expression was significantly reduced after MENIN overexpression, which was rescued when co-expressed with TTi (Figure 5J), suggesting that MENIN repression of FZD7 was TEAD1 dependent. Moreover, when we performed public ChIP-seq analysis, we found strong TEAD1 binding peaks in the TEAD1 promoter area in many cell types (Figure S5F), suggesting auto-regulation. This binding of TEAD1 to TEAD1 promoter was especially strong, with a high binding peak observed in pancreatic progenitor cells (Figure S5G). Since TEAD1 expression and activity were lower in islets from mice at 24 weeks compared with 4 weeks of age, we investigated the potential regulators of TEAD1 expression. Interestingly, our ChIP-seq data also showed a high TEAD1 binding peak in TEAD1 promoter area, and this peak covered the TSS (Figure 5K). Two tandem MCATs were found in the peak sequences, and the second one encompassed the TSS. This tandem motif is conserved in human TEAD1 and TEAD4 and mouse TEAD1, while in mouse TEAD4, it is a little far away from TSS (Figure S5H). To test the functional regulation of this binding, we generated a construct wherein the TEAD1 promoter region, from −650 to TSS, was connected to 5′ UTR and CDS0–18 of TEAD1, and then it was fused with firefly luciferase to generate a luciferase reporter (T1R) (Figure 5L). As a control, we also generated a mutation reporter (T1mR), in which we mutated the second MCAT (Figure 5L). Luciferase assay of T1R showed that overexpression of TEAD1 significantly reduced luciferase activities, and co-expression of VGLL4 and TEAD1 could enhance this repression. However, in the T1mR experiment, due to a reduction in basal luciferase activity, VGLL4 or TEAD1 overexpression could not further repress the T1mR luciferase activity (Figure 5M), suggesting the tandem TEAD1 binding motifs play a critical role in the auto-regulation of TEAD1 expression and that this regulation for TEAD1 expression is MCAT dependent. After overexpression of VGLL4 and human TEAD1 in INS2 cells, qPCR showed the endogenous mTEAD1 was significantly reduced (Figure 5N). Western blot showed that VGLL4 and MENIN were successfully overexpressed in the luciferase assay (Figure S5I). Taken together, these data demonstrate that corepressors (VGLL4 and MENIN) bind TEAD1 to inhibit downstream target genes, including FZD7, CCN2, and TEAD1, to reduce TEAD pathway activities that relate to maintaining proliferative quiescence in adult β cells by inhibiting pro-proliferative genes (Figure 5O).
Figure 5. VGLL4/MENIN-TEAD1 pathway target genes screen.
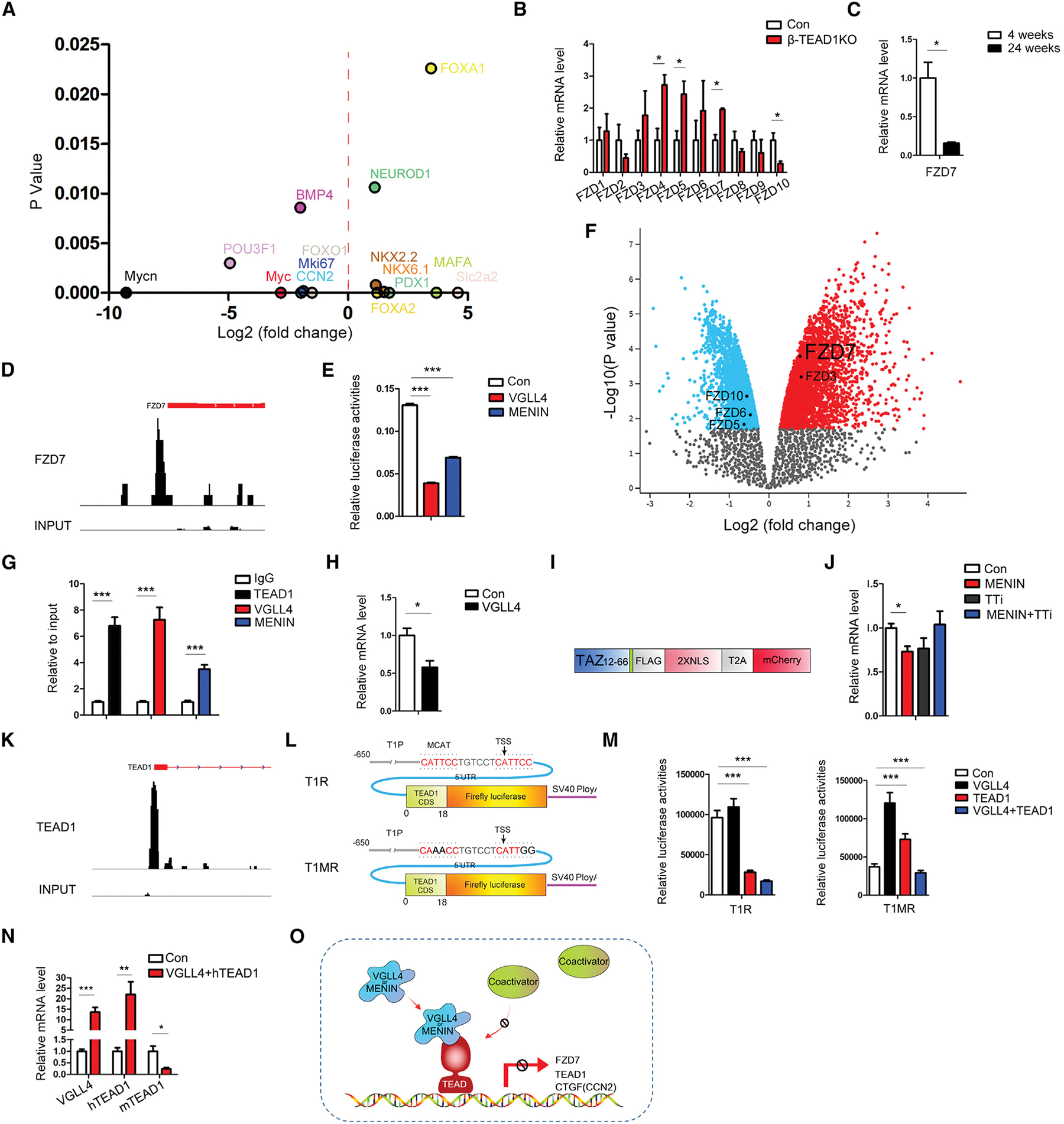
(A) RNA-seq (TEAD1flox/flox vs. TEAD1−/−) showing the changes in FOXA1, NEUROD1, NKX2.2, MAFA, NKX6.1, PDX1, Slc2a2, Mki67, FOXO1, POU3F1, CCN2, Myc, Mycn, and BMP4 in β-TEAD1KO islet.
(B) mRNA levels of FZD family members in β-TEAD1KO and control islet (n = 3 mice/group).
(C) mRNA level of FZD7 in mouse islets at the different ages (n = 3 mice/group).
(D) ChIP-seq in mouse islet showing a TEAD1 binding peak in the promoter area of FZD7.
(E) Luciferase assay using a FZD7 promoter reporter for assessing VGLL4 and MENIN repression.
(F) Re-analysis of RNA-seq (MEN1−/− vs. MEN1flox/flox) showing FZD7 expression in MENIN knockout islets. See also Figure S5D.
(G) Validation of TEAD1, VGLL4, and MENIN binding to the FZD7 promoter region by ChIP experiments.
(H) mRNA level of FZD7 after VGLL4 overexpression in INS1 cells.
(I) Schematic representation for TEAD inhibitor (TTi) structure. N terminus of TAZ (aa12–66) was connected with FLAG tag, NLS, T2A, and mCherry.
(J) mRNA level of FZD7 after MENIN overexpression or co-overexpression of MENIN and TTi in INS1 cells.
(K) ChIP-seq in mouse islet showing a high TEAD1 binding peak in the promoter area of TEAD1.
(L) Schematic representation for TEAD1 promoter reporter (T1R) and TEAD1 promoter reporter with MCAT mutation (T1MR).
(M) The changes of luciferase activities after TEAD1 overexpression or TEAD1-VGLL4 co-expression indicated by T1R and T1mR in luciferase assay.
(N) mRNA level of endogenous TEAD1 (mTEAD1) after the co-expression of human TEAD1 (hTEAD1) and VGLL4 in INS2 cells.
(O) The schematic model for VGLL4/MENIN and TEAD1 interaction. *p < 0.05; **p < 0.01; ***p < 0.001. (B–E, G, H, J, M) n = at least three per condition with three technical replicates. Error bars represent standard error of the mean (SEM). p values are from Student’s t test and ANOVA.
DISCUSSION
β cells proliferate significantly less as they acquire mature function. Deciphering the regulation of this delicate balance is critical in any β cell replacement therapy for diabetes. The mammalian Hippo-TEAD1 pathway has been implicated in oncogenesis in many cell types, with TEAD1 and its canonical coactivators YAP and TAZ playing a critical role in proliferation and differentiation. TEAD1 does not have a transactivation domain and depends on cofactors, such as YAP and TAZ, to activate gene transcription. However, while TEAD1 has a critical role in β cell function and regulation of proliferation, we demonstrate here that YAP and TAZ deletion has no effect under basal or under metabolic stress induced by a high-fat diet. This raised the possibility that other TEAD1 cofactors have a functional role in maintaining mature function in β cells. Data from the current study demonstrate that compared with coactivators, TEAD1 corepressors play a dominant role in the regulation of β cell proliferation. In our quest to identify cofactors of TEAD1 in β cells, we have identified VGLL4 and MENIN as important TEAD1-binding corepressors using multiple complementary approaches. While VGLL4-TEAD interaction is known to play important roles in cancer24 and cardiac development,25 its role in β cells has been previously unknown. The current study demonstrates the role of VGLL4TEAD1 interaction in the maintenance of proliferative quiescence in mature β cells.
In addition, we also identified that MENIN is a corepressor of TEAD1 in β cells, suggesting an additional role for MENIN in mature β cells. Though VGLL4’s in vivo role in β cells has largely been unexplored, MENIN deletion has been shown to increase β cell proliferation, leading to insulinomas.26 Interestingly and concordant with our study, acute Men1 excision that led to amelioration/reversal in three models of diabetes (STZ, db/db, and high-fat diet) mouse models can reverse preexisting glucose intolerance in diabetic mice.27 MENIN can bind TEAD1 and repress the expression of the WNT receptor, FZD7, in the basal state. This is also consistent with the WNT pathway activation seen in insulinomas associated with loss of MENIN function.26 We thus link MENIN as an additional link between the mammalian Hippo-TEAD1 pathway and the WNT pathway, two critical developmental and oncogenesis pathways.28
VGLL4 and MENIN can bind the pocket region of TEAD1 in its C terminus. Their repressive activities appear to depend on competing with YAP and TAZ to occupy this pocket region. Interestingly, VGLL4 and MENIN cannot repress TEAD1 pathway activity independently, suggesting that VGLL4 and MENIN play more important roles in modulating the co-activation induced by other factors such as YAP and TAZ. Since the binding affinity between TEAD and VGLL4 (Kd = 6.8 nM) is much lower than between TEAD and YAP (Kd = 2.1 nM),23 the relative level of nuclear VGLL4 would be important for its repressive effects on TEAD1 signaling. The levels of nuclear VGLL4 are regulated not only by transcriptional regulation but have been shown to be subject to nucleo-cytoplasmic shuttling in cardiac myocytes.29 Whether similar regulation exists in β cells would be of significant interest and needs further study.
Since VGLL4 and MENIN’s repressive effects depend on competitively binding to the pocket area, this raises the intriguing question as to what other TEAD1-binding coactivators would be signaling in β cells since YAP and TAZ are redundant to normal β cell function. Our yeast two two-hybrid screen showed that JUN could be a cofactor for TEAD1 in human islets. JUND, a member of the JUN family, can bind TEAD4 and then regulate cancer cell migration and invasion.24 Unpublished data from our lab indicate that JUND can bind TEAD1 directly, and a high level of JUND can activate the TEAD1 pathway (data not shown). Whether JUND is a functional co-activator of TEAD1 and plays a role in proliferation and mature function in β cells deserves more study. SMAD4, a tumor-suppressor and an intracellular signaling mediator of the TGF-β pathway, has also been reported to bind TEAD, indicated by coIP experiments, to form a regulatory complex in stem cells.30 However, our split-GFP system data showed that SMAD4 could not bind TEAD1 directly nor possess the ability to directly repress TAZ-TEAD1 pathway activity, suggesting that SMAD4-TEAD1 interaction is dependent on the presence of other mediators.
β cell proliferation decreases with age. Here we demonstrate that this is concordant with the decreased TEAD1 activity as a result of increased expression of VGLL4 and a decrease in expression of TEAD1. While regulating proliferation-related genes, TEAD1 also regulates genes that maintain mature function. Indeed, different gene networks may be differentially regulated by TEAD1. For instance, many of the transcription factors and genes regulating β cell maturity and identity, such as PDX1, MAFA, and NKX6.1, have TEAD1 binding sites in their promoter and are activated by TEAD1 and its coactivators,5 while many others are activated, suggesting repression by TEAD1 in the basal state. It is also possible that there may be other cofactors that bind to TEAD1 in a pocket-independent manner, and hence not regulated by VGLL4/MENIN, to regulate gene expression and would be the subjects of further study.
Auto-regulation is an integral mechanism of many regulatory pathways. Feedback inhibition allows a steady activation level, while feedforward activation allows the maintenance of a high activation level. This has been demonstrated in the HippoTEAD1 pathway, wherein the expression of LATS2, an inhibitor of YAP, is directly activated by YAP-TEAD1.31 Here, we demonstrate an additional feedback loop wherein TEAD1 binds to its own promoter to repress its activation in the basal state. This suggests that this is a potential regulatory node to modulate the activity of the TEAD1 pathway.
In conclusion, this study identified VGLL4 and MENIN as corepressors of TEAD1 in β cells that repress pro-proliferative genes to maintain proliferative quiescence in mature β cells in a TEAD1-dependent manner without significantly repressing mature function-related genes. This raises the possibility that modulating this regulatory pathway could increase β cell proliferation while preserving mature function in patients with diabetes.
Limitations of the study
We have identified VGLL4 and MENIN as important corepressors of the TEAD1 pathway using multiple lines of evidence, using β cell lines and mouse and human islets. While MENIN’s role in vivo in mouse and human islets has been extensively studied, in vivo evidence for VGLL4 in β cells is lacking and beyond the scope of this study. The in vivo role of VGLL4 in β cells using loss of function studies in basal and under metabolic stress would be of great interest and awaits future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yechoor Vijay K (yechoorv@pitt.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
RNAseq and ChIPseq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal to TEF-1 | BD Biosciences | Cat# 610922, RRID:AB_398237 |
| Mouse Anti-Myc-Tag Monoclonal Antibody | Cell Signaling Technology | Cat# 2276, RRID:AB_331783 |
| Rabbit polyclonal to VGLL4 | Novus | Cat# NBP1-81543, RRID:AB_11003050 |
| Rabbit monoclonal to Menin | Abcam | Cat# ab92443, RRID:AB_10564144 |
| Rabbit monoclonal to GAPDH | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| Rabbit monoclonal to Ki67 | Abcam | Cat# ab16667, RRID:AB_302459 |
| Mouse monoclonal to Insulin | Abcam | Cat# ab6995, RRID:AB_305690 |
| Rabbit monoclonal to TAZ | Cell Signaling Technology | Cat# 72804, RRID:AB_2904134 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq in C57/BI6 Wild and β-TEAD1KO islets | This paper | GSE139228, GSE139152 |
| TEAD1 ChIPseq in C57/BI6 mouse islets | This paper | GSE157513 |
| Single cell sequencing related to Figure S2 | Åsa Segerstolpe, et al.32 | E-MTAB-5061, E-MTAB-5060 |
| TEAD1 ChIPseq data in pancreatic progenitor cells | Inês Cebola, et al.33 | E-MTAB-1990 E-MTAB-3061 |
| Expression data from pancreatic islets from MEN1flf RIP-Cre mice and matched control | Wenchu Lin, et al.22 | GSE26978 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Subcloning EfficiencyTM DH5a Competent Cells | Thermo Fisher | 18265017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase from Clostridium histolyticum | Sigma | C7657 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Dual-Luciferase® Reporter Assay | Promega | E1910 |
| Duolink® In Situ Orange Starter Kit Mouse/Rabbit | Sigma | DUO92102 |
| EZ-Magna ChIP™ HiSens Chromatin Immunoprecipitation Kit | Sigma | 17-10461 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | CRL-3216 | ATCC |
| INS1 | SCC207 | Sigma |
| INS2 | Gift from Akio Koizumi’s lab | |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Wwtr1tm1Hmc Yap1tm1Hmc/WranJ | 030532 | THE JACKSON LABORATORY |
| INS1-Cre (B6(Cg)-Ins1tm1.1(cre)Thor/J) | 026801 | THE JACKSON LABORATORY |
| TEAD1flox/flox mice | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Mouse VGLL4 shRNA (5′-ACACATGGCTTCAGATCAAAG-3′) | Sigma | TRCN0000250410 |
| Human VGLL4 shRNA (5′-CAGGAGCCTGGGCAAGAATTA-3′) | Sigma | TRCN0000231109 |
| PCR and RT-qPCR primers | This paper | Table S1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| GFP10 (GGCATGGATTTACCAGACGACCATTACCTGTCAACACAAACTATCCTTTCGAAAGATCTCAAC) | This paper | N/A |
| GFP11 (GAAAAGCGTGACCACATGGTCCTTCTTGAGTATGTAACTGCTGCTGGGATTACAGATGCTAGC) | This paper | N/A |
| Backbone for Split GFP | Addgene | 80347 |
| Hopflash reporter | Addgene | 83467 |
| TEAD1 plasmid | Addgene | 33109 |
| YAP5SA plasmid | Addgene | 27371 |
| Menin plasmid | Addgene | 32079 |
| VGLL4 plasmid | GenScript | OHu15880 |
| Lenti-VGLL4-P2A-GFP plasmid | This paper | Addgene #194159 |
| Lenti-MENIN-P2A-GFP plasmid | This paper | N/A |
| SMAD4 plasmid | Addgene | 80888 |
| GFP1-9 | This paper | N/A |
| TAZ plasmid | Addgene | 128351 |
| EF1a-mCherry-P2A | Addgene | 135003 |
|
| ||
| Software and algorithms | ||
|
| ||
| CLC genomics workbench | QIAGEN | https://www.qiagen.com/ |
| Integrative genomics viewer | N/A | https://software.broadinstitute.org/software/igv/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
| BIOVIA Discovery Studio Visualizer | Dassault Systemes BIOVIA | https://discover.3ds.com/ |
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of.
University of Pittsburgh. TEAD1flox/flox mice5 were bred with mice expressing Cre recombinase driven by the mouse INS1 promoter (Mi1p-Thorens INS1-Cre) to generate the β-TEAD1KO mice. Mice were genotyped using TEAD1-flox-forward (GCCTTCTGAGTGCTGGCATTAAAGG) and reverse (AAGGCAGACTCCTTCATTGGAATGG) primers. TAZflox/flox/YAPflox/flox mice were purchased from Jackson Laboratory (#030532), and were bred with Rip-Cre to generate the β-Y/T KO mice. The following primer pair was used for genotyping, CTTCCAAGGTGCTTCAGAGA and GGAGAGGTA AAGCCCACCAG. The β-Y/T KO and control mice were fed a standard chow diet or an HFD (research diets) for 2 months. 8-week male mice were used to perform experiments, and they were maintained under standard 12-h light-dark cycles with ad lib access to food and water, unless specified otherwise.
Human islets
Human islets (distributed by IIDP) obtained from human pancreas, obtained from non-diabetic donor (brain death after head trauma), with informed consent for transplant or research use from relatives of the donor. All studies involving the use of human islets were approved by the Institutional Review Board of University of Pittsburgh. Received human islets were collected by centrifugation, selected by hand picking and cultured in CMRL1066 (Mediatech, Corning) supplemented with 10% human serum (Sigma), 1% P/S and 2 mM L-glutamine. For lentivirus infection, human islets were disassociated with 0.05% Trypsin for 3 min and subjected to infection at 20 MOI. Three individual non-diabetic donor islets (Male, 26yrs, BMI-29.2; Male 44 yrs, BMI-42; Female, 37 yrs, BMI-30.2) were used in this study.
Human pancreatic islets and/or other resources were provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) (RRID:SCR_014387) at City of Hope, NIH Grant # 2UC4DK098085.
Cell lines
INS1 and INS2 cell line were cultured and maintained as previously described.5 For HEK293T cells, transfection were performed using the Lipofectamine 3000 kit (Thermo Fisher) according to the manufacturer’s instructions. For INS1 and INS2 cells, transduction were performed with lentivirus.
METHOD DETAILS
Plasmids for Split-GFP system
The backbone vector used for Split-GFP system is Plenti from Addgene (#80347, a gift from Erik Rodriguez & Roger Tsien34). The sequences for GFP10 is GGCATGGATTTACCAGACGACCATTACCTGTCAACACAAACTATCCTTTCGAAAGATCTCAAC, and GFP11 is GAAAAGCGTGACCACATGGTCCTTCTTGAGTATGTAACTGCTGCTGGGATTACAGATGCTAGC. The sequences for “GFP11-linker-Myc tag-XbaI:SgsI-linker-GFP11-T2A-GFP10-linker-Pfl23II:SfaAI-Flag tag-linker-GFP10″ were synthesized form Integrated DNA Technologies (IDT) directly. This fragment was inserted into Plenti by BamHI:EcoRI. The CDS of TEAD1 were inserted by XbaI:SgsI using In-Fusion HD Cloning Kit (Takara Bio, #638946). The CDS of TAZ, SMAD4, VGLL4, and MENIN were inserted by Pfl23II:SfaAI. The fragment, EF1a-mCherry-P2A, which was subcloned from an Addgene plasmid (#135003, a gift from Prashant Mali35), was ligated with GFP1–9 by overlap PCR first and then inserted into Plenti by XhoI:Acc65I using In-Fusion HD Cloning Kit.
“All-in-one” plasmid to evaluate TEAD signaling activities
The backbone vector used for Split-GFP system is Plenti from Addgene (#80347). Renilla luciferase was ligated with CMV promoter by overlap PCR and then inserted into Plenti by XhoI:Acc65I. Firefly luciferase was inserted into Plenti by BamHI:EcoRI. The original HOP-FLASH plasmid was from Addgene (#83467), from which we subcloned 8X MCAT and inserted it into our “all-in-one” reporter backbone. This plasmid were packaged into lentivirus and transduced into islets to evaluate TEAD signaling activities.
IPGTT
Glucose tolerance tests (GTTs) were performed in 12 h overnight fasted mice administrated 1.5 g/kg D-glucose i.p.
Islet isolation and culture
Mouse islets were isolated as previously described.26 Briefly, 1 mg/mL type XI collagenase (Sigma) in Hank’s buffered saline solution was injected into the pancreas of the mouse models through the bile duct. The pancreas was removed and incubated for 18 min at 37°C and dissociated by mechanical pipetting. The islets were hand-picked under a dissecting microscope. Islet were cultured in a modified culture medium36: Minimum essential medium (MEM) with GlutaMAX (Gibco), 11 mM glucose, 5% FBS, 1 mM sodium pyruvate, 10 mM HEPES and 1x B-27 Supplement53 (Gibco). Before transduction with lentivirus, islet were dissociated with a trypsin EDTA solution (Invitrogen) for 3 min and cultured overnight.
Lentivirus packaging
HEK293T packaging cells were seeded at 8×106 cells per plate in DMEM and incubated at 37°C, 5% CO2 for 20 h. A mixture of the 3 transfection plasmids: psPAX2 (1.3 pmol), pMD2.G (0.72 pmol) and transfer plasmid (1.64 pmol) was prepared. The plasmids were transfected into 293T cells using Lipofectamine 3000 kit. After 12 h of transfection, media was replaced with fresh media without antibiotic. Lentivirus was harvested at 48 h post transfection and filtered through a 0.45μm PES filter. The lentiviral supernatant was concentrated using Lenti-X Concentrator (Takara Bio, # 631232) according to the manufacturer’s instructions.
Immunostaining
Immunostaining was performed as previously described.5 Primary antibodies used were TEAD1 (rabbit polyclonal, 1:200; Abcam), Ki67 (rabbit polyclonal, 1:50; Abcam), BRDU (rat polyclonal, 1:1,000; Abcam), TAZ (WWTR1) (mouse monoclonal, 1:100, Cell signaling). Tyramide SuperBoost Kits (Thermofisher) were used for TAZ (WWTR1) staining according to the manufacturer’s instructions in islet. The images were taken by the Nikon confocal microscope.
Western blotting
Western blotting was performed as described.5 Briefly, cell protein samples were prepared in ice-cold lysis buffer (100 mmol/L TrisHCl pH7.4, 10 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 10% glycerol, 0.5% deoxycholate, 1% Triton X-100, 0.1% SDS, 20 mmol/L Na4P2O7, 2 mmol/L Na3VO4, 1 mmol/L NaF, 1 mmol/L PMSF) supplemented with 1X protease inhibitor and phosphatase inhibitor (Roche) when necessary. Equal amount of protein samples were loaded and fractionated by SDS-PAGE, then transferred to 0.45 μm pore-size nitrocellulose membranes (Millipore). Membranes were blocked in 5% non-fat milk-TBS (w/v), then incubated with indicated primary antibodies. The following antibodies were used: TEAD1 (BD, #610922), Myc tag (Cell signaling, #2276), VGLL4 (Novus Biologicals, #NBP1–81543), MENIN (Abcam, # ab92443) and GAPDH. The blots were imaged using Licor Odyssey Clx and quantified by Image Studio ver 5.2 software.
Real-time PCR
Total RNA was extracted and cleaned up using RNeasy Mini Kit (QIAGEN) according to manufacturer’s protocol. cDNA were synthesized using q-Script cDNA Supermix kit (Quanta Biosci.). Realtime PCR was performed using Perfecta SYBR Green Supermix (Quanta Biosci.) in a Roche 480 Light Cycler machine. Relative mRNA expression levels were determined by normalization of target genes to 18S as internal control. All the primers were designed by Primer3 online tool (https://primer3.ut.ee/) using the CDS of the target genes.
Co-immunoprecipitation (CoIP)
Cells were lysed using Pierce IP Lysis Buffer (Thermo Fisher, #87787). Cells lysates (samples) were incubated with primary antibody and rotated for 6 h at 4°C. Use Normal mouse IgG as control and run alongside the primary antibody samples. Magnetic beads (BiaRad) were washed 3 times in 1.5mL tubes, and then incubated with samples with beads, and rotated for 20 min at room temperature. After washing the immunomagnetic beads 5 times, supernatant was removed and eluted with 15μL elution buffer and 5 μL 4× SDS-PAGE loading buffer and heated for 10 min at 100°C. Western Blotting were performed to detect target proteins in elution buffer. Antibodies from different species were used to avoid visualizing IgG heavy and light chains bands in Western blotting. The following antibodies were used in CoIP: TEAD1 (BD, mouse monoclonal, #610922), TEAD1 (Abcam, rabbit polyclonal, #133533), MENIN (Abcam, rabbit polyclonal, #ab92443), Mouse IgG isotype control (Novusbio, #NBP1–97019).
In situ proximity ligation assay (PLA)
HEK293T cells were seeded on fibronectin-coated glass chamber slides and transfected with TEAD1, MENIN or empty backbone as negative control. After 24 h, cells were fixed in 4% PFA for 15 min at RT. In situ PLA was performed with DuoLink In Situ Reagents (Sigma) according to manufacturer’s instructions. Primary antibodies used in the PLA are: TEAD1 (BD, # 610922), MENIN (Abcam, #ab92443). The images were taken by the Nikon confocal microscope.
RNAi
The original mouse and human VGLL4 shRNA plasmid were purchased from Sigma. The target gene for mouse VGLL4 is ACACATGGCTTCAGATCAAAG and for human is CAGGAGCCTGGGCAAGAATTA. We subcloned the sequence of U6-shRNA from the original plasmids into lentiviral backbones with CMV-GFP to facilitate the tracking of transfection. VGLL4 shRNA plasmids were packaged into lentivirus and then transduced in islets cells or β cell lines. After 36-h transduction, islets or β cell lines were used for further experiments. Knockdown efficiency was validated by qPCR.
Luciferase assay
The original HOPFLASH reporter plasmid was from Addgene (#83467, a gift from Barry Gumbiner37). We subcloned the 8X MCAT from HOPFLASH into a lentiviral backbone. The sequence from human CTGF between −600 to 0 and human FZD7 promoter area between −597 to −123 were amplified by PCR and cloned to generate CTGF and FZD7 luciferase reporter. Human TEAD1 promoter between −650 to +404 were amplified by PCR and then connected by overlap PCR with the left part of 5′UTR and CDS0–18 which were synthesized by IDT directly to generate T1R. PCR and overlap PCR were performed using primers with MCAT mutation and T1R as template to generate T1MR. Luciferase assay were performed with Dual-Luciferase KIT (Promega) according to the manufacturer’s instructions. Briefly, cells were transfected with luciferase reporters and the target genes for 24 h, and then luciferase (Firefly and Renilla) activity was measured from 20 μL cell lysate with the Dual-luciferase Reporter Assay System on a multilabel plate reader (Wallac Victor, PerkinElmer). The relative luciferase activity was calculated dividing the value obtained for Firefly luciferase activity for each well by the Renilla luciferase activity or total protein from the same well. Total protein level were measured by BCA method.
Chromatin immunoprecipitation (ChIP)
INS1 cells were transduced by the lentivirus which express TEAD1, Myc-VGLL4 and MENIN respectively for 36 h, and then cell protein from one 10-cm tissue-culture dish was fixed in 1.5% formaldehyde and collected using SDS-lysis buffer. The chromatin was fragmented to sizes ranging from 200 to 1000bp by sonication, immunoprecipitated by 1 μg normal mouse IgG or indicated antibody and protein A/G beads. Precipitated chromatin fragments were then purified. PCR was carried out for detection of the DNA enrichment. TEAD1 (BD Biosciences, #610922), Myc (Cell Signaling Technology, #2276) and MENIN (Abcam, #ab92443) antibody or control IgG for pulldown. Primers, 5′-CCCTCGGTCTCAGGTTACTTTCATA-3′ and 5′- GAGAAAGGGGTGCGACTAAGAAT-3′, were used for FZD7 promoter amplification in ChIP experiment.
RNAseq
Total RNA samples with RNA integrity number (RIN) ≥8 were used for transcriptome sequencing. Total RNA (10ng) from a pooled sample from 1 year old mice (n = 3) each for two independent samples for each group was used for amplified double-stranded cDNA that was sheared to 200–300bp and ligated to Illumina paired-end adaptors using the Illumina TruSeq DNA library preparation kit according to the manufacturer’s instructions (Illumina, San Diego, CA). PCR amplification was performed to obtain the final cDNA library using Illumina kits. Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) analysis was used to verify fragment size after amplification, library size and concentration before clustering. A total of 10pM of the library was then used for paired-end sequencing on the HiSeq 2500 at the Sequencing core at Brigham Young University. All further analysis were performed using the CLC genomics workbench ver 12. Raw RNA sequencing reads were mapped to the mouse reference genome build 10 (UCSCmm10/GRCm38). Mapped reads were counted using the feature counts and differential expression between the samples analyzed using multiple hypothesis testing. Significance was assessed by analyzing signal-tonoise ratio and gene permutations based on 1,000 permutations. Molecular signature database (MSigDB) 3.0 curated gene sets for hallmark and canonical pathways were used for the analysis. Significant gene sets with enrichment score and a q value cutoff of 0.05 are presented.
Yeast two hybrid screen
Yeast two-hybrid screening was performed by Hybrigenics Services, S.A.S., Evry, France (http://www.hybrigenics-services.com). The coding sequence for Homo sapiens TEAD1 (NCBI reference NM_021961.3) was PCR-amplified and cloned into pB27 as a C-terminal fusion to LexA (LexA-TEAD1). The construct was checked by sequencing the entire insert and used as a bait to screen a random-primed Human Islets Langerhans cDNA library constructed into pP6. 55 million clones (5-fold the complexity of the library) were screened using a mating approach with YHGX13 (Y187 ade2–101:loxP-kanMX-loxP, mat alpha) and L40deltaGal4 (mata) yeast strains. 128 His + colonies were selected on a medium lacking tryptophan, leucine and histidine, and supplemented with 0.5 mM 3-aminotriazole to handle bait autoactivation. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5′ AND-3′ junctions. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI) using a fully automated procedure. A confidence score (PBS, for Predicted Biological Score) was attributed to each interaction. The PBS relies on two different levels of analysis. Firstly, a local score takes into account the redundancy and independency of prey fragments, as well as the distribution of reading frames and stop codons in overlapping fragments. Secondly, a global score takes into account the interactions found in all the screens performed at Hybrigenics using the same library. This global score represents the probability of an interaction being nonspecific. For practical use, the scores were divided into four categories, from A (highest confidence) to D (lowest confidence). Finally, several of these highly connected domains have been confirmed as false-positives of the technique and are now tagged as F. The PBS scores have been shown to positively correlate with the biological significance of interactions.38
pyDockWEB prediction
The MENIN structure data used in pyDockWEB (https://life.bsc.es/pid/pydockweb/) prediction was from rcsb.org (3RE2).39 TEAD1 structure data were from rcsb.org (4RE1).40
Public ChIPseq, RNAseq and islet single cell sequencing data analysis
Processed BED and WIG files were download from ChIP-atlas (http://ChIP-atlas.org/).41 WIG files were opened in UCSC Genome Browser (https://genome.ucsc.edu/) or integrative genomics viewer (IGV) to obtain the “PEAK” graph. CLC Genomics Workbench 12 was used to extract the target gene information about “PEAK score”, annotation and nearby gene information. TEAD1 ChIPseq data in pancreatic progenitor cells were download from ArrayExpress (E-MTAB-1990 and E-MTAB-3061).33 Expression data from pancreatic islets from MEN1flf RIP-Cre mice and matched control22 were download from GEO database (GSE26978), and were analyzed by GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE26978). Raw data for human single-cell and whole-islet RNA-seq were download from ArrayExpress (E-MTAB-5061 and E-MTAB-5060).32
Chromatin Immunoprecipitation for ChIPseq
Frozen tissue was sent to Active Motif Services (Carlsbad, CA) to be processed for ChIPseq. In brief, tissue was submersed in PBS +1% formaldehyde, cut into small pieces and incubated at room temperature for 15 min. Fixation was stopped by the addition of 0.125 M glycine (final). The tissue pieces were then treated with a TissueTearer and finally spun down and washed 2x in PBS. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield. An aliquot of chromatin (10 ug for islets and 30 ug for hearts) was precleared with protein G agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 20 ul of antibody against Tead1. Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65 C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation. Quantitative PCR (qPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad). The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using Input DNA.
ChIP sequencing (Illumina)
Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. Steps were performed on an automated system (Apollo 342, Wafergen Biosystems/Takara). After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single end). Reads were aligned to the mouse genome (mm10) using the BWA algorithm (default settings). Duplicate reads were removed and only uniquely mapped reads (mapping quality R 25) were used for further analysis. Alignments were extended in silico at their 3′-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library, and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of p value = 1e-7. Peaks that were on the ENCODE blacklist of known false ChIPseq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations.
QUANTIFICATION AND STATISTICAL ANALYSIS
The data are presented as means ± SEM, the in vitro data are representative of at least three independent experiments and is indicated in the respective figure legends. Significance tests were determined by the two-tailed, unpaired Student’s t-test, or two-way ANOVA followed by Sidak’s multiple comparisons test. p < 0.05 was considered statistically significant and labeled with *. p < 0.01, **. p < 0.001, ***. GraphPad Prism 7.04 software was used for statistical analyses.
Supplementary Material
Highlights.
YAP and TAZ do not have a physiological role in β cell function and proliferation
VGLL4 and MENIN are important repressors of TEAD1 activity in β cells
TEAD1 inhibits its own expression in an auto-feedback loop
Improved split-GFP (iSG) system is a sensitive assay for unknown protein interactions
ACKNOWLEDGMENTS
This work was supported by grants from the VA-ORD - I01BX002678 (V.Y.), National 13 Institutes of Health R01 DK130499 (V.Y.), National Institutes of Health R01-HL14794614 (M.M.), and The Pittsburgh Foundation (V.Y.).
Footnotes
DECLARATION OF INTERESTS
No conflicts of interest, financial or otherwise, are declared by the authors.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111904.
REFERENCES
- 1.Oram RA, Sims EK, and Evans-Molina C (2019). Beta cells in type 1 diabetes: mass and function; sleeping or dead? Diabetologia 62, 567–577. 10.1007/s00125-019-4822-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahier J, Guiot Y, Goebbels RM, Sempoux C, and Henquin JC (2008). Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 10, 32–42. 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 3.Misra JR, and Irvine KD (2018). The hippo signaling network and its biological functions. Annu. Rev. Genet. 52, 65–87. 10.1146/annurev-genet-120417-031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin KC, Park HW, and Guan KL (2017). Regulation of the hippo pathway transcription factor TEAD. Trends Biochem. Sci. 42, 862–872. 10.1016/j.tibs.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J, Liu R, Kim BS, Zhang Y, Li F, Jagannathan R, Yang P, Negi V, Melissa Perez-Garcia E, Saha PK, et al. (2022). Tead1 reciprocally regulates adult β-cell proliferation and function. Preprint at bioRxiv. 10.1101/2020.03.05.979450. [DOI] [Google Scholar]
- 6.George NM, Day CE, Boerner BP, Johnson RL, and Sarvetnick NE (2012). Hippo signaling regulates pancreas development through inactivation of Yap. Mol. Cell Biol. 32, 5116–5128. 10.1128/MCB.01034-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao T, Zhou D, Yang C, Singh T, Penzo-Méndez A, Maddipati R, Tzatsos A, Bardeesy N, Avruch J, and Stanger BZ (2013). Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology 144, 1543–1553.e1. 10.1053/j.gastro.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George NM, Boerner BP, Mir SUR, Guinn Z, and Sarvetnick NE (2015). Exploiting expression of hippo effector, yap, for expansion of functional islet mass. Mol. Endocrinol. 29, 1594–1607. 10.1210/me.2014-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi N (2020). Multiple roles of vestigial-like family members in tumor development. Front. Oncol. 10, 1266. 10.3389/fonc.2020.01266. eCollection 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azad T, Tashakor A, and Hosseinkhani S (2014). Split-luciferase complementary assay: applications, recent developments, and future perspectives. Anal. Bioanal. Chem. 406, 5541–5560. 10.1007/s00216-014-7980-8. [DOI] [PubMed] [Google Scholar]
- 11.Simeone DM, Zhang L, Treutelaar MK, Zhang L, Graziano K, Logsdon CD, and Burant CF (2006). Islet hypertrophy following pancreatic disruption of Smad4 signaling. Am. J. Physiol. Endocrinol. Metab. 291, E1305–E1316. 10.1152/ajpendo.00561.2005. [DOI] [PubMed] [Google Scholar]
- 12.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, and Kim SK (2005). Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 102, 14659–14664. 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabantous S, Nguyen HB, Pedelacq J-D, Koraïchi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, and Waldo GS (2013). A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 3, 2854. 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimé nez-Garcıá, B , Pons C, and Fernández-Recio J. (2013). PyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 29, 1698–1699. 10.1093/bioinformatics/btt262. [DOI] [PubMed] [Google Scholar]
- 15.Kaan HYK, Chan SW, Tan SKJ, Guo F, Lim CJ, Hong W, and Song H (2017). Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 7, 2035. 10.1038/s41598-017-02219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, et al. (1999). Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96, 143–152. 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 17.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, and Kinzler KW (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512. 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 18.ten Berge D, Brugmann SA, Helms JA, and Nusse R (2008). Wnt and FGF signals interact to coordinate growth with cell fate specification during limb development. Development 135, 3247–3257. 10.1242/dev.023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J-S, Crooks H, Dracheva T, Nishanian TG, Singh B, Jen J, and Waldman T (2002). Oncogenic beta-catenin is required for bone morphogenetic protein 4 expression in human cancer cells. Cancer Res. 62, 2744–2748. [PubMed] [Google Scholar]
- 20.Sharon N, Vanderhooft J, Straubhaar J, Mueller J, Chawla R, Zhou Q, Engquist EN, Trapnell C, Gifford DK, and Melton DA (2019). Wnt signaling separates the progenitor and endocrine compartments during pancreas development. Cell Rep. 27, 2281–2291.e5. 10.1016/j.celrep.2019.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henley KD, Gooding KA, Economides AN, and Gannon M (2012). Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am. J. Physiol. Endocrinol. Metab. 303, E752–E761. 10.1152/ajpendo.00531.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, et al. (2011). Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci. USA 108, 13379–13386. 10.1073/pnas.1110104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, et al. (2014). A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25, 166–180. 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park J-S, et al. (2016). Tead and AP1 coordinate transcription and motility. Cell Rep. 14, 1169–1180. 10.1016/j.celrep.2015.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu W, Ma X, Xu J, Heumüller AW, Fei Z, Feng X, Wang X, Liu K, Li J, Cui G, et al. (2019). VGLL4 plays a critical role in heart valve development and homeostasis. PLoS Genet. 15, e1007977. 10.1371/journal.pgen.1007977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Cao Y, Li F, Su Y, Li Y, Peng Y, Cheng Y, Zhang C, Wang W, and Ning G (2014). Targeting β-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat. Commun. 5, 5809. 10.1038/ncomms6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Gurung B, Wu T, Wang H, Stoffers DA, and Hua X (2010). Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene. Proc. Natl. Acad. Sci. USA 107, 20358–20363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M, and Jho EH (2014). Cross-talk between Wnt/β-catenin and Hippo signaling pathways: a brief review. BMB Rep. 47, 540–545. 10.5483/bmbrep.2014.47.10.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen HH, Mullett SJ, and Stewart AF (2004). Vgl-4, a novel member of the vestigial-like family of transcription co-factors, regulates alpha1-adrenergic activation of gene expression in cardiac myocytes. J. Biol. Chem. 279, 30800–30806. [DOI] [PubMed] [Google Scholar]
- 30.Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, and Wrana JL (2013). Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 5, 1611–1624. 10.1016/j.celrep.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 31.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, and Guan KL (2015). A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 29, 1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. (2016). Single-cell transcriptome profiling of human pancreatic islets in Health and type 2 diabetes. Cell Metab. 24, 593–607. 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cebola I, RodrÍguez-SeguÍ SA, Cho CH-H, Bessa J, Rovira M, Luengo M, Chhatriwala M, Berry A, Ponsa-Cobas J, Maestro MA, et al. (2015). TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol. 17, 615–626. 10.1038/ncb3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez EA, Tran GN, Gross LA, Crisp JL, Shu X, Lin JY, and Tsien RY (2016). A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein. Nat. Methods 13, 763–769. 10.1038/nmeth.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parekh U, Wu Y, Zhao D, Worlikar A, Shah N, Zhang K, and Mali P (2018). Mapping cellular reprogramming via pooled overexpression screens with paired fitness and single-cell RNA-sequencing readout. Cell Syst. 7, 548–555.e8. 10.1016/j.cels.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phelps EA, Cianciaruso C, Santo-Domingo J, Pasquier M, Galliverti G, Piemonti L, Berishvili E, Wiederkehr A, Hubbell JA, Baekkeskov S, and Burri O (2017). Advances in pancreatic islet monolayer culture on glass surfaces enable super-resolution microscopy and insights into beta cell ciliogenesis and proliferation. Sci. Rep. 7, 45961. 10.1038/srep45961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim NG, and Gumbiner BM (2015). Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 210, 503–515. 10.1083/jcb.201501025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wojcik J, Boneca IG, and Legrain P (2002). Prediction, assessment and validation of protein interaction maps in bacteria. J. Mol. Biol. 323, 763–770. 10.1016/s0022-2836(02)01009-4. [DOI] [PubMed] [Google Scholar]
- 39.Murai MJ, Chruszcz M, Reddy G, Grembecka J, and Cierpicki T (2011). Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 286, 31742–31748. 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Z, Hu T, Xu Z, Lin Z, Zhang Z, Feng T, Zhu L, Rong Y, Shen H, Luk JM, et al. (2015). Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J 29, 724–732. 10.1096/fj.14-262980. [DOI] [PubMed] [Google Scholar]
- 41.Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, Kawaji H, Nakaki R, Sese J, and Meno C (2018). ChIP-Atlas: a data-mining suite powered by full integration of public ChIPseq data. EMBO Rep. 19, e46255. 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq and ChIPseq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal to TEF-1 | BD Biosciences | Cat# 610922, RRID:AB_398237 |
| Mouse Anti-Myc-Tag Monoclonal Antibody | Cell Signaling Technology | Cat# 2276, RRID:AB_331783 |
| Rabbit polyclonal to VGLL4 | Novus | Cat# NBP1-81543, RRID:AB_11003050 |
| Rabbit monoclonal to Menin | Abcam | Cat# ab92443, RRID:AB_10564144 |
| Rabbit monoclonal to GAPDH | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| Rabbit monoclonal to Ki67 | Abcam | Cat# ab16667, RRID:AB_302459 |
| Mouse monoclonal to Insulin | Abcam | Cat# ab6995, RRID:AB_305690 |
| Rabbit monoclonal to TAZ | Cell Signaling Technology | Cat# 72804, RRID:AB_2904134 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq in C57/BI6 Wild and β-TEAD1KO islets | This paper | GSE139228, GSE139152 |
| TEAD1 ChIPseq in C57/BI6 mouse islets | This paper | GSE157513 |
| Single cell sequencing related to Figure S2 | Åsa Segerstolpe, et al.32 | E-MTAB-5061, E-MTAB-5060 |
| TEAD1 ChIPseq data in pancreatic progenitor cells | Inês Cebola, et al.33 | E-MTAB-1990 E-MTAB-3061 |
| Expression data from pancreatic islets from MEN1flf RIP-Cre mice and matched control | Wenchu Lin, et al.22 | GSE26978 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Subcloning EfficiencyTM DH5a Competent Cells | Thermo Fisher | 18265017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase from Clostridium histolyticum | Sigma | C7657 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Dual-Luciferase® Reporter Assay | Promega | E1910 |
| Duolink® In Situ Orange Starter Kit Mouse/Rabbit | Sigma | DUO92102 |
| EZ-Magna ChIP™ HiSens Chromatin Immunoprecipitation Kit | Sigma | 17-10461 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | CRL-3216 | ATCC |
| INS1 | SCC207 | Sigma |
| INS2 | Gift from Akio Koizumi’s lab | |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Wwtr1tm1Hmc Yap1tm1Hmc/WranJ | 030532 | THE JACKSON LABORATORY |
| INS1-Cre (B6(Cg)-Ins1tm1.1(cre)Thor/J) | 026801 | THE JACKSON LABORATORY |
| TEAD1flox/flox mice | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Mouse VGLL4 shRNA (5′-ACACATGGCTTCAGATCAAAG-3′) | Sigma | TRCN0000250410 |
| Human VGLL4 shRNA (5′-CAGGAGCCTGGGCAAGAATTA-3′) | Sigma | TRCN0000231109 |
| PCR and RT-qPCR primers | This paper | Table S1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| GFP10 (GGCATGGATTTACCAGACGACCATTACCTGTCAACACAAACTATCCTTTCGAAAGATCTCAAC) | This paper | N/A |
| GFP11 (GAAAAGCGTGACCACATGGTCCTTCTTGAGTATGTAACTGCTGCTGGGATTACAGATGCTAGC) | This paper | N/A |
| Backbone for Split GFP | Addgene | 80347 |
| Hopflash reporter | Addgene | 83467 |
| TEAD1 plasmid | Addgene | 33109 |
| YAP5SA plasmid | Addgene | 27371 |
| Menin plasmid | Addgene | 32079 |
| VGLL4 plasmid | GenScript | OHu15880 |
| Lenti-VGLL4-P2A-GFP plasmid | This paper | Addgene #194159 |
| Lenti-MENIN-P2A-GFP plasmid | This paper | N/A |
| SMAD4 plasmid | Addgene | 80888 |
| GFP1-9 | This paper | N/A |
| TAZ plasmid | Addgene | 128351 |
| EF1a-mCherry-P2A | Addgene | 135003 |
|
| ||
| Software and algorithms | ||
|
| ||
| CLC genomics workbench | QIAGEN | https://www.qiagen.com/ |
| Integrative genomics viewer | N/A | https://software.broadinstitute.org/software/igv/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
| BIOVIA Discovery Studio Visualizer | Dassault Systemes BIOVIA | https://discover.3ds.com/ |
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.