

Abstract
Associations between age-related neuropathological lesions and adult-onset lifetime major depressive disorder (a-MDD), late-life MDD (LLD), or depressive symptoms close to death (DS) were examined in a large community sample of non-demented older adults. Seven hundred forty-one individuals (age at death = 72.2 ± 11.7 years) from the Biobank for Aging Studies were analyzed. a-MDD was present in 54 (7.3%) participants, LLD in 80 (10.8%), and DS in 168 (22.7%). After adjustment for covariates and compared to controls, a-MDD, LDD and DS were associated with small vessel disease (p = 0.039, p = 0.003, and p = 0.003 respectively); LLD, and DS were associated with brain infarcts (p = 0.012, p = 0.018, respectively) and Lewy body disease (p = 0.043, p = 0.002, respectively). DS was associated with beta-amyloid plaque burden (p = 0.027) and cerebral amyloid angiopathy (p = 0.035) in cognitively normal individuals (Clinical Dementia Rating scale = 0). Vascular brain pathology was the strongest correlate of clinical depictions of depression in the absence of dementia, corroborating the vascular hypothesis of depression. Lewy body pathology underlay DS. An older adult with DS or LLD should be monitored for possible cognitive decline or neurodegenerative disorders.
Keywords: Depression, Alzheimer, Vascular, Lewy, Older adults, Post-mortem
1. Introduction
Depression predisposes to various medical illnesses, and medical illnesses increase the risk of late-life depression. The reciprocal relationships of depression with aging-related and disease-related processes (Alexopoulos, 2019; Chan et al., 2019; Korczyn and Halperin, 2009;) have strengthened the importance of further evaluating depression in older ages as it can be associated with underlying brain lesions. Moreover, chronic depression and stress are associated with excessive release of corticosteroids, which damage the brain (Korczyn and Halperin, 2009). Depression often precedes the disease-defining symptoms associated with neurodegenerative processes (Chan et al., 2019; Jellinger, 2013).
The literature proposes several non-mutually exclusive models for explaining depressive symptoms in people with cognitive decline: 1) emotional reaction to cognitive decline; 2) recurrence of early or midlife major depression precipitated by neurodegenerative lesions and, more specifically, possible early neurodegenerative pathology in mood-regulating brain areas; and 3) late-life cerebrovascular disease contributing to both dementia and depression (Alexopoulos, 2019; Jellinger, 2013; Korczyn and Halperin, 2009).
Regarding early neurodegenerative pathology in mood-regulating brain areas, relatively recent evidence of early (precortical) degeneration of neuromodulatory subcortical systems (NSS) (Braak et al., 2003) - especially serotonergic and noradrenergic neurons due to Alzheimer’s disease (AD) neurofibrillary tangles and Lewy body disease - reignited the hypothesis of depression as an early sign of a neurodegenerative condition. Such suspicion has been corroborated by work showing a clear association between depressive symptoms and pre-cortical AD tau deposits in the NSS (Ehrenberg et al., 2018). The neuro-biological basis of depression is likely multifactorial, and contributing causes may co-occur (Alexopoulos, 2019; Jellinger, 2013; Korczyn and Halperin, 2009; Suemoto et al., 2017). In line with these findings, other postmortem studies pinpoint a link between depression and AD lesions, Lewy body disease or cerebrovascular lesions (Meynen et al, 2010; Rapp et al., 2006), but not all studies have shown this association (Wilson et al., 2003). All things considered, studies investigating an association between depression and brain lesions show discrepant results (Jellinger, 2013; Kim et al., 2016; Taylor, 2017; van Dyck et al., 2021).
Several longitudinal studies suggest that depression is a risk factor for dementia (Chan et al., 2019), but few studies investigate whether depression is already a manifestation of the early stages of neurodegenerative conditions. Beta-amyloid neuritic plaque and neurofibrillary tangles burden were associated with higher depression scores in a cohort of 106 control individuals followed in 10 waves of assessment between 1991 and 2015 (only 30 were free of dementia at death) (Robinson et al., 2021). Association between plaque burden and depression scores was found in another longitudinal clinicopathological study of 161 older individuals free of dementia at death (Wennberg et al, 2019). Major depressive disorder (MDD) was also associated with a higher burden of ischemic brain lesions in a study with 20 older adults (Thomas et al., 2002). However, in the only clinicopathological study with a non-Caucasian cohort, comprising 393 Japanese males living in Hawaii, depressive symptoms and subsequent cognitive decline seem to be unrelated to AD neuropathologies (Royall and Palmer, 2013). Negative results were also found in a sample of 38 MDD cases; in this study, no association with cerebrovascular lesions was observed (Santos et al., 2010). In one of the few studies with a population-based sample (n = 153), MDD was associated with subcortical and hippocampal neuronal loss and subcortical Lewy-type pathology, but not with cerebrovascular or AD pathology (Tsopelas et al., 2011). Finally, in one of the largest neuropathological studies to date, including 72 MDD cases with no information on how many died free of dementia, beta-amyloid burden was associated with significant depressive symptoms, and no association was found with Lewy bodies, tau neurofibrillary tangle pathology, or infarcts (Wilson et al., 2016).
Therefore, more comprehensive investigations on the age-related neuropathological correlates of depression in cognitively normal individuals are still lacking. We aim to close this gap by investigating associations of neurodegenerative and cerebrovascular lesions with different forms of depression - adult-onset MDD (a-MDD), late-life MDD (LLD), and depressive symptoms (DS) - in a large community-based clinicopathological cohort of 741 non-demented older adults. As the literature suggests, the rationale behind investigating these groups separately was to test if there is more vascular pathology among individuals with depression at older ages (both the LLD and the DS groups). Moreover, we studied both LLD and DS because there are studies that analyze depressive symptoms (the DS group) but not the Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria for depression at an older age (the LLD group) and vice-versa, and we were able to analyze both constructs in the same sample.
2. Methods
2.1. Participants
Participants of this study were deceased individuals who underwent autopsy at the Sao Paulo Autopsy Service of the University of Sao Paulo (SVOC-USP) and had their brains donated to the Biobank for Aging Studies of the University of Sao Paulo (BAS-USP) from 2004 to 2019. SVOC-USP is a community-based autopsy service for individuals who died from natural (non-traumatic) causes. Furthermore, the SVOC-USP does not perform autopsies of forensic cases. In Brazil, an autopsy is mandatory when the non-traumatic cause of death is unclear due to lack of medical assistance or insufficient information before death, at no charge to the family. Autopsies start only when the next-of-kin family member arrives at SVOC. All autopsies are performed by medical pathologists assisted by nationally certified technicians. The SVOC-USP functions 24 h a day and performs around 13,000 autopsies per year within the city of Sao Paulo, Brazil, which has a population of 12 million residents and around 90,000 deaths per year (Grinberg et al, 2007).
Further information regarding methodological procedures of the BAS-USP can be reached elsewhere (Grinberg et al, 2007, 2009; Nunes et al., 2019; Suemoto et al., 2017). Cases were selected at random, during weekdays, between 8 and 18hs. Family members are asked if they consent to participate in the study while waiting for the autopsy to complete. The clinical and functional interview is conducted in a private room if consent is granted. Inclusion criteria for the BAS-USP were participants aged at death 50 years and older, and the next of kin being a knowledgeable informant who had had at least weekly contact with the deceased. Exclusion criteria for the BAS-USP were: (i) brain tissue unsuitable for neuropathological analyses (e.g., cerebrospinal fluid pH < 6.5, or significant acute brain lesions, such as hemorrhages or tumors); and (ii) inconsistent clinical data provided by the informant. All BAS-USP protocols, the informed consent form, and procedures follow international and Brazilian regulations for research involving humans and were approved by the local and federal research committees.
2.2. Clinical postmortem evaluation
Briefly, after next-of-kin consenting, trained gerontologists applied the semi-structured clinical and functional assessments. The clinical evaluation assessed the deceased’s clinical and functional status in a time point 3 months before death and a life-time history of MDD. A validated semi-structured clinical interview (Ferretti et al., 2010) assessed demographics, neuropsychiatric symptoms, cognitive performance, and clinical medical history. We used the informant part of the Clinical Dementia Rating scale (CDR) (Morris, 1993) validated for postmortem use (Ferretti et al., 2010) to evaluate cognitive impairment. According to previous publications, CDR > 1 indicates dementia, and CDR = 0.5 is considered mild cognitive impairment but might as well include cases of mild dementia (Morris, 1993; Nunes et al., 2019).
The diagnosis of lifetime MDD was made using the Structured Clinical Interview for DSM-IV Disorders (SCID) for Axis I, informant part (Spitzer et al., 1992). The diagnosis of MDD was further confirmed according to DSM-5 criteria. The subgroup a-MDD was considered when the first MDD episode occurred in adulthood up until 60 years, and LLD was considered when the first MDD episode occurred after age 60.
Depressive and other neuropsychiatric symptoms around 3 months before death (to avoid the influence of peri-agonal events) were assessed using the Neuropsychiatric Inventory (NPI) (Cummings, 1997). The NPI measures 12 different neuropsychiatric symptoms, including depression. It is a structured interview applied to an informant, focusing on observable symptoms. The presence of significant depressive symptoms (DS) was considered positive when the NPI core for depressive symptoms was greater than zero, similarly to previous publications (Ehrenberg et al., 2018; Nunes et al., 2019; Peters et al., 2013).
2.3. Neuropathological assessment and definitions
The BAS-USP follows standardized protocols. Brain tissue was obtained within 24 hours of death. One hemisphere was fixed in paraformaldehyde. After fixation, samples from the following selected areas were embedded in paraffin: middle frontal gyrus, middle, and superior temporal gyri, angular gyrus, superior frontal, and anterior cingulate gyrus, visual cortex, hippocampal formation at the level of the lateral geniculate body, amygdala, basal ganglia at the level of the anterior commissure, thalamus, midbrain, pons, medulla oblongata, and cerebellum. Immunohistochemistry of selected sections was performed using antibodies against beta-amyloid, phosphorylated tau, and α-synuclein (MDV) (Ehrenberg et al., 2018). The pathologists (LTG and RDR) were blinded to demographics and clinical outcomes for all neuropathologic analyses.
Both hemispheres underwent gross assessment. Microvascular changes were analyzed semi-quantitatively using hematoxylin and eosin staining in all sampled areas. Small-vessel disease was diagnosed when there was widespread (more than 50%) and at least moderate small-vessel disease in white matter in at least 3 cortical regions out of those examined (superior frontal gyrus, medium frontal gyrus, inferior and medium temporal gyri, anterior cingulate gyrus, angular gyrus), and included small-vessel arteriosclerosis/atherosclerosis, arteriolosclerosis, and lipohyalinosis (Grinberg et al., 2009; Suemoto et al., 2017). Cases were classified as positive for cerebral amyloid angiopathy (CAA) when it was found diffusely in the parenchyma of at least 3 different cortical areas (Grinberg and Thal, 2010). Inclusion in the group “infarct” required 1 large chronic infarct (> 1 cm) or 3 lacunae in strategic areas - thalamus, frontocingular cortex, basal forebrain and caudate, medial temporal area, and angular gyrus (Grinberg and Thal, 2010; Jellinger and Attems, 2006).
AD-type neuropathology was evaluated using the Consortium to Establish a Registry for AD (CERAD) criteria for neuritic plaque burden (Mirra et al., 1991), and the Braak and Braak staging for neurofibrillary tangle pathology (Braak and Braak, 1991). The cohort was dichotomized into 2 groups: Braak stage 0 to II and III to VI
Lewy-type pathology was assessed using the Braak staging for Parkinson’s disease (Braak et al., 2003), and groups were divided as Lewy body disease present (Braak ≥ 0) or absent. According to this classification, cases were further analyzed as having limbic commitment (Braak ≥ 4) or not.
2.4. Statistical analysis
We used the Mann-Whitney test for quantitative variables and the chi-square test for categorical variables to compare participants in the groups. In addition, we used multivariable logistic regression models, adjusted for CDR, age at death, sex, and ethnicity to test the association of lifetime a-MDD, LLD or DS, and the neuropathological alterations. We also tested the association adjusting for CDR, age at death, sex, ethnicity, alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia. We further carried on sensitivity analyses excluding the group CDR = 0.5. Additional analyses were also performed considering possible differences among male and female groups. We used multivariable logistic regression models, adjusted for significant variables to test the association of sex and the neuropathological alterations. The level of significance was set at 0.05 in 2-tailed tests. The Statistical Package for the Social Sciences (SPSS) version 20.0 was used to perform the statistical analyses.
3. Results
We analyzed all participants with CDR < 1 (741 out of 1,068 individuals) included in the BAS from 2004 to 2019. CDR = 0 was present in 620 (83.7%) cases and CDR = 0.5 was present in 121 (16.3%) cases. In this sample, the mean age at death was 72.2 ± 11.7 years old, and 52.2% were male. a-MDD was present in 54 (7.3%) subjects and LLD in 80 (10.8%) cases, as seen in Fig. 1. DS were present in 168 (22.7%) subjects. Forty-two a-MDD cases were also present in the DS group, and 64 LLD cases were also present in the DS group.
Fig. 1.
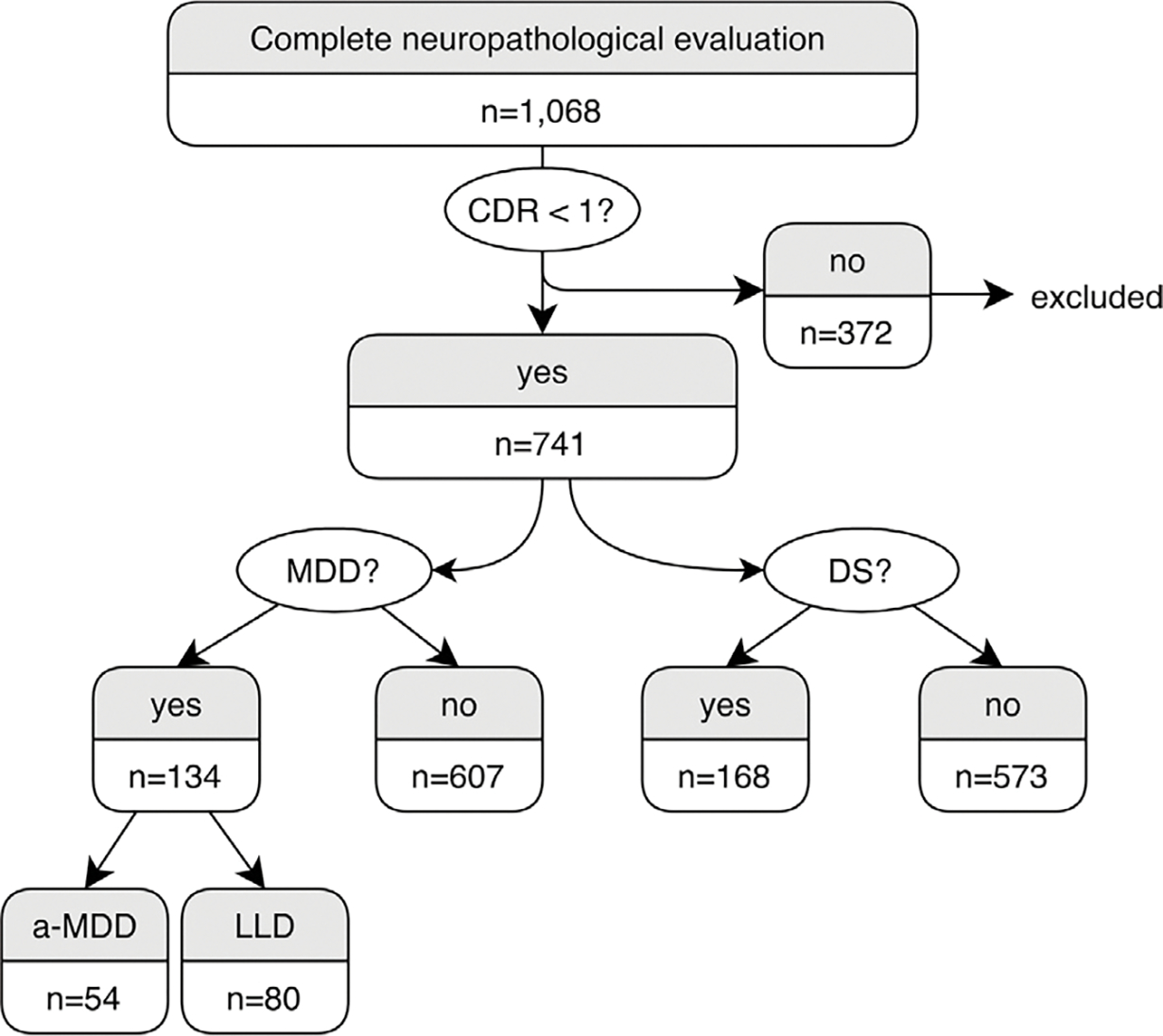
Flow chart of the participants of the study. Abbreviations: a-MDD, adult-onset major depressive disorder; DS, depressive symptoms 3 months prior to death; LLD, late life depression; MDD, major depressive disorder.
Table 1 shows demographics and clinical variable comparisons among a-MDD, LLD, and their comparison group, no MDD. a-MDD group was younger (p < 0.001), and had a prevalence of females of 59.3% as compared with 45.6% (p = 0.054) in the no MDD group. No difference between a-MDD and no MDD was found regarding education, ethnicity, alcohol misuse, smoking habits, hypertension, diabetes, or dyslipidemia. The LLD group was older (p = 0.001) and had less dyslipidemia (p = 0.042) than the group with no MDD. No difference between LLD and no MDD was found regarding education, gender, ethnicity, alcohol misuse, smoking habits, hypertension, or diabetes.
Table 1.
Comparison of a-MDD and LLD cases with cases with no MDD.
| a-MDD (n = 54) | LLD (n = 80) | no MDD (n = 607) | a-MDD × no MDD p | LLD × no MDD p | |
|---|---|---|---|---|---|
|
| |||||
| Age at death (years), mean (SD)a | 63.6 (10.8) | 76.4 (8.8) | 72.3 (11.7) | < 0.001 | 0.001 |
| Education (years), mean (SD)a | 4.4 (3.0) | 3.8 (3.3) | 4.6 (3.8) | 0.79 | 0.08 |
| Women, nb (%) | 32 (59.3%) | 45 (56.3%) | 277 (45.6%) | 0.054 | 0.07 |
| Ethnicity white, nb (%) | 37 (68.5%) | 55 (68.8%) | 420 (69.2%) | 0.92 | 0.67 |
| CDR = 0, nb (%) | 41 (75.9%) | 60 (75.0%) | 519 (85.5%) | 0.061 | 0.015 |
| Alcohol misuse, nb (%) | 12 (25.0%) | 16 (20.5%) | 100 (18.5%) | 0.27 | 0.64 |
| Smoking, nb (%) | 28 (52.8%) | 32 (40.5%) | 238 (41.5%) | 0.11 | 0.86 |
| Hypertension, nb (%) | 42 (77.8%) | 53 (66.3%) | 410 (68.4%) | 0.16 | 0.69 |
| Diabetes mellitus, nb (%) | 9 (16.7%) | 21 (26.3%) | 168 (28.0%) | 0.07 | 0.74 |
| Dyslipidemia, nb (%) | 5 (9.3%) | 3 (3.8%) | 68 (11.2%) | 0.64 | 0.042 |
Data are presented as means (SD) and for categorical values as the number of cases, and percentage (%) when indicated.
Key: a-MDD, adult-onset major depressive disorder; CDR, Clinical Dementia Rating scale; LLD, late life depression; MDD, major depressive disorder; SD, standard deviation.
Mann-Whitney test.
Qui-square test.
The demographics and clinical variables in the groups DS and no DS are shown in Table 2. The DS group had a higher prevalence of smoking habits (p = 0.035) than the group with no DS. No difference between the group with DS and the group with no DS was found regarding age at death, education, gender, ethnicity, alcohol misuse, hypertension, diabetes, or dyslipidemia.
Table 2.
Comparison of the group with DS with the group with no DS.
| DS (n = 168) | no DS (n = 573) | DS × no DS p | |
|---|---|---|---|
|
| |||
| Age at death (years), mean (SD)a | 71.7 (11.5) | 72.3 (11.7) | 0.57 |
| Education (years), mean (SD)a | 4.3 (3.2) | 4.6 (3.9) | 0.36 |
| Women, nb (%) | 88 (52.5%) | 265 (46.2%) | 0.17 |
| Ethnicity white, nb (%) | 119 (70.8%) | 392 (68.4%) | 0.47 |
| CDR = 0, nb (%) | 130 (77.4%) | 489 (85.5%) | 0.012 |
| Alcohol misuse, nb (%) | 31 (19.9%) | 97 (16.9%) | 0.82 |
| Smoking, nb (%) | 82 (49.9%) | 216 (37.9%) | 0.035 |
| Hypertension, nb (%) | 118 (70.2%) | 386 (67.3%) | 0.66 |
| Diabetes mellitus, nb (%) | 52 (31.0%) | 146 (25.4%) | 0.19 |
| Dyslipidemia, nb (%) | 12 (7.2%) | 64 (11.1%) | 0.12 |
Data are presented as means (SD) and for categorical values as the number of cases and percentage (%) when indicated.
Key: CDR, Clinical Dementia Rating scale; DS, depressive symptoms 3 months prior to death.
Mann-Whitney test.
Qui-square test.
Table 3 shows the association of neuropathologies with a-MDD, LLD, and DS. The logistic regression models adjusted for CDR, age at death, sex, and ethnicity reveal associations of neuropathologies in some of the groups. a-MDD, LLD, and DS were associated with small vessel disease, and the odds ratio (OR) for the association of small vessel disease with a-MDD, LLD, and DS were 2.5 (95% CI = 1.1–5.9; p = 0.037), 2.4 (95% CI = 1.3–4.5; p = 0.005), and 2.0 (95% CI = 1.2–3.3; p = 0.007), respectively. LLD and DS were associated with infarcts, and the OR for the association with LLD, and DS was 2.5 (95% CI = 1.3–5.1; p = 0.009), and 2.2 (95% CI = 1.3–4.2; p = 0.008), respectively. DS were associated with higher moderate to frequent neurofibrillary tangle burden (Braak’s stage III or above): OR = 1.7 (95% CI = 1.1–2.5; p = 0.018), and DS were also associated with Lewy body pathology: OR = 2.4 (95% CI = 1.4–4.1; p = 0.001). No differences were found for cerebral amyloid angiopathy or neuritic plaque burden (CERAD score).
Table 3.
Association between MDD, LLD or DS and neuropathological alterations (n = 741) in CDR = 0 and CDR = 0.5 cases.
| Small vessel disease |
Crude OR (95% CI) |
p | Multivariatea OR (95% CI) |
p | Multivariateb OR (95% CI) |
p | ||
|---|---|---|---|---|---|---|---|---|
| No | Yes | |||||||
|
| ||||||||
| a-MDD × no MDD |
46 (85.2%) 549 (90.9%) |
8 (14.8%) 55 (9.1%) |
1.7 (0.8–3.9) | 0.18 | 2.5 (1.1–5.9) | 0.037 | 2.7 (1.1–6.8) | 0.039 |
| LLD × no MDD |
57 (80.3%) 549 (90.9%) |
14 (19.7%) 55 (9.1%) |
2.6 (1.5–4.9) | 0.001 | 2.4 (1.3–4.5) | 0.005 | 2.7 (1.4–5.2) | 0.003 |
| DS × no DS |
141 (83.9%) 516 (90.7%) |
27 (16.1%) 53 (9.3%) |
1.8 (1.1–3.1) | 0.013 | 2.0 (1.2–3.3) | 0.007 | 2.3 (1.3–4.0) | 0.003 |
|
Cerebral amyloid angiopathy
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
53 (98.1%) 582 (96.5%) |
1 (1.9%) 21 (3.5%) |
0.5 (0.1–4.0) | 0.53 | 0.4 (0.1–2.9) | 0.35 | 0.4 (0.1–3.4) | 0.39 |
| LLD × no MDD |
67 (94.4%) 582 (96.5%) |
4 (5.6%) 21 (3.5%) |
1.6 (0.6–5.0) | 0.32 | 1.3 (0.5–4.2) | 0.57 | 2.0 (0.6–6.4) | 0.24 |
| DS × no DS |
160 (95.2%) 550 (96.8%) |
8 (4.8%) 18 (3.2%) |
1.5 (0.7–3.6) | 0.33 | 1.5 (0.6–3.4) | 0.40 | 2.1 (0.8–5.2) | 0.13 |
|
Infarcts
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
51 (94.4%) 566 (94.0%) |
3 (5.6%) 36 (6.0%) |
0.9 (0.3–3.1) | 0.90 | 1.2 (0.3–4.2) | 0.82 | 0.6 (0.1–4.4) | 0.58 |
| LLD × no MDD |
59 (83.1%) 566 (94.0%) |
12 (16.9%) 36 (6.0%) |
3.2 (1.6–6.5) | 0.001 | 2.5 (1.3–5.1) | 0.009 | 2.7 (1.2–5.8) | 0.012 |
| DS × no DS |
146 (88.0%) 537 (94.4%) |
20 (12.0%) 32 (5.6%) |
2.3 (1.3–4.2) | 0.005 | 2.2 (1.3–4.2) | 0.008 | 2.3 (1.2–4.5) | 0.018 |
|
Neuritic plaques (beta-amyloid): moderate-frequent
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
51 (94.4%) 500 (82.4%) |
3 (5.6%) 107 (17.6%) |
0.3 (0.1–0.9) | 0.032 | 0.4 (0.1–1.5) | 0.19 | 0.5 (0.1–1.7) | 0.25 |
| LLD × no MDD |
59 (83.1%) 500 (82.4%) |
12 (16.9%) 107 (17.6%) |
1.0 (0.5–1.8) | 0.88 | 0.8 (0.4–1.5) | 0.41 | 0.8 (0.4–1.6) | 0.53 |
| DS × no DS |
135 (80.4%) 482 (84.3%) |
33 (19.6%) 90 (15.7%) |
1.3 (0.8–2.0) | 0.23 | 1.4 (0.9–2.2) | 0.18 | 1.4 (0.9–2.4) | 0.17 |
|
Neurofibrillary pathology (tau) III-VI
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
44 (81.5%) 433 (72.3%) |
10 (18.5%) 166 (27.7%) |
0.6 (0.3–1.2) | 0.15 | 1.2 (0.5–2.7) | 0.66 | 1.2 (0.5–2.9) | 0.70 |
| LLD × no MDD |
48 (60.0%) 433 (72.3%) |
32 (40.0%) 166 (27.7%) |
1.7 (1.1–2.8) | 0.023 | 1.2 (0.8–2.2) | 0.34 | 1.2 (0.7–2.2) | 0.45 |
| DS × no DS |
109 (65.3%) 416 (73.6%) |
58 (34.7%) 149 (26.4%) |
1.4 (1.0–2.1) | 0.035 | 1.7 (1.1–2.5) | 0.018 | 1.5 (1.0–2.4) | 0.06 |
|
Lewy body disease
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
48 (90.6%) 531 (91.4%) |
5 (9.4%) 50 (8.6%) |
1.1 (0.4–2.9) | 0.84 | 1.8 (0.6–4.9) | 0.28 | 2.1 (0.7–6.1) | 0.16 |
| LLD × no MDD |
57 (81.4%) 531 (91.4%) |
13 (18.6%) 50 (8.6%) |
2.4 (1.2–4.7) | 0.008 | 1.9 (0.9–3.6) | 0.07 | 2.0 (1.0–4.1) | 0.043 |
| DS × no DS |
138 (84.1%) 506 (92.3%) |
26 (15.9%) 42 (7.7%) |
2.3 (1.3–3.8) | 0.002 | 2.4 (1.4–4.1) | 0.001 | 2.5 (1.4–4.4) | 0.002 |
Key: a-MDD, adult-onset major depressive disorder; 95% CI, 95% Confidence Interval; DS, depressive symptoms 3 months prior to death; LLD, late life MDD; MDD, major depressive disorder.
Logistic regression models adjusted for CDR, age at death, sex, and ethnicity.
Logistic regression models adjusted for CDR, age at death, sex, ethnicity, alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia.
Results remained unaltered when alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia were also included in the regression models. In cases of individuals with Lewy body disease, no differences were found among the MDD, LLD, and DS groups regarding limbic involvement (Braak PD ≥ 4).
We further carried out sensitivity analyses to assess the specificity of the results excluding CDR = 0.5 cases as seen in Table 4. The logistic regression models adjusted for age at death, sex, and ethnicity revealed several similar associations of neuropathologies as seen in the analyses of cases with CDR = 0 and CDR = 0.5. a-MDD, LLD, and DS in CDR = 0 cases were associated with small vessel disease, and the odds ratio (OR) for the association of small vessel disease with a-MDD, LLD, and DS were 2.7 (95% CI = 1.1–6.7; p = 0.031), 2.7 (95% CI = 1.3–5.4; p = 0.004), and 2.2 (95% CI = 1.3–3.9; p = 0.005), respectively. LLD and DS were associated with infarcts, and the OR for the association with LLD, and DS were 2.5 (95% CI = 1.1–5.9; p = 0.035), and 2.1 (95% CI = 1.0–4.5; p = 0.042), respectively. DS were associated with higher moderate to frequent neurofibrillary tangle burden (Braak’s stage III or above): OR = 2.1 (95% CI = 1.3–3.3; p = 0.002), and DS were also associated with Lewy body pathology: OR = 2.6 (95% CI = 1.4–4.8; p = 0.002). No differences were found for cerebral amyloid angiopathy. Results remained unaltered when alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia were also included in the regression models, except from the fact that DS were associated with greater cerebral amyloid angiopathy OR = 2.7 (95% CI = 1.1–7.3; p = 0.035) and a-MDD were associated with Lewy body disease OR = 3.0 (95% CI = 1.0–8.8; p = 0.046).
Table 4.
Association between MDD, LLD or DS and neuropathological alterations (n = 620) in CDR = 0 cases.
| Small vessel disease |
Crude OR (95% CI) |
p | Multivariatea OR (95% CI) |
p | Multivariateb OR (95% CI) |
p | ||
|---|---|---|---|---|---|---|---|---|
| No | Yes | |||||||
|
| ||||||||
| a-MDD × no MDD |
34 (82.9%) 471 (91.1%) |
7 (17.1%) 46 (8.9%) |
2.1 (0.9–5.0) | 0.09 | 2.7 (1.1–6.7) | 0.031 | 3.3 (1.3–8.6) | 0.012 |
| LLD × no MDD |
46 (76.7%) 471 (91.1%) |
14 (23.3%) 46 (8.9%) |
3.1 (1.5–6.1) | 0.001 | 2.7 (1.3–5.4) | 0.004 | 3.0 (1.5–6.1) | 0.002 |
| DS × no DS |
107 (82.3%) 443 (91.0%) |
23 (17.7%) 44 (9.0%) |
2.2 (1.2–3.7) | 0.006 | 2.2 (1.3–3.9) | 0.005 | 2.6 (1.5–4.7) | 0.001 |
|
Cerebral amyloid angiopathy
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
40 (97.6%) 500 (96.9%) |
1 (2.4%) 16 (3.1%) |
0.8 (0.1–6.0) | 0.81 | 0.6 (0.1–5.1) | 0.68 | 0.6 (0.1–5.1) | 0.64 |
| LLD × no MDD |
56 (93.3%) 500 (96.9%) |
4 (6.7%) 16 (3.1%) |
2.2 (0.7–6.9) | 0.16 | 2.3 (0.7–7.4) | 0.16 | 2.6 (0.7–8.4) | 0.13 |
| DS × no DS |
122 (93.8%) 473 (97.3%) |
8 (6.2%) 13 (2.7%) |
2.4 (1.0–5.8) | 0.06 | 2.3 (1.0–5.9) | 0.06 | 2.7 (1.1–7.3) | 0.035 |
|
Infarcts
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
40 (97.6%) 490 (95.1%) |
1 (2.4%) 25 (4.9%) |
0.5 (0.1–3.7) | 0.49 | 0.8 (0.1–5.2) | 0.70 | 0.8 (0.1–6.6) | 0.88 |
| LLD × no MDD |
52 (86.7%) 490 (95.1%) |
8 (13.3%) 25 (4.9%) |
3.0 (1.3–7.0) | 0.011 | 2.5 (1.1–5.9) | 0.035 | 2.3 (0.9–5.7) | 0.08 |
| DS × no DS |
116 (90.6%) 465 (95.5%) |
12 (9.4%) 22 (4.5%) |
2.2 (1.1–4.5) | 0.036 | 2.1 (1.0–4.5) | 0.042 | 2.1 (1.0–4.6) | 0.06 |
|
Neuritic plaques (beta-amyloid): moderate-frequent
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
38 (92.7%) 433 (83.4%) |
3 (7.3%) 86 (16.6%) |
0.4 (0.1–1.3) | 0.13 | 0.6 (0.2–2.0) | 0.39 | 0.6 (0.2–2.3) | 0.47 |
| LLD × no MDD |
47 (78.3%) 433 (83.4%) |
13 (21.7%) 86 (16.6%) |
1.4 (0.7–2.6) | 0.32 | 1.0 (0.5–2.0) | 0.96 | 1.1 (0.5–2.2) | 0.89 |
| DS × no DS |
99 (76.2%) 419 (85.7%) |
31 (23.8%) 70 (14.3%) |
1.8 (1.2–3.0) | 0.010 | 1.9 (1.1–3.1) | 0.013 | 1.8 (1.1–3.2) | 0.027 |
|
Neurofibrillary pathology (tau) III-VI
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
33 (80.5%) 381 (74.6%) |
8 (19.5%) 130 (25.4%) |
0.7 (0.3–1.6) | 0.40 | 1.4 (0.5–3.4) | 0.52 | 0.7 (0.2–1.7) | 0.38 |
| LLD × no MDD |
35 (58.3%) 381 (74.6%) |
25 (41.7%) 130 (25.4%) |
2.1 (1.2–3.6) | 0.009 | 1.5 (0.8–2.6) | 0.22 | 1.5 (0.8–2.7) | 0.23 |
| DS × no DS |
81 (62.8%) 368 (76.3%) |
48 (37.2%) 114 (23.7%) |
1.9 (1.3–2.9) | 0.002 | 2.1 (1.3–3.3) | 0.002 | 1.9 (1.2–3.2) | 0.009 |
|
Lewy body disease
|
||||||||
| No | Yes | |||||||
| a-MDD × no MDD |
35 (87.5%) 453 (91.9%) |
5 (12.5%) 40 (8.1%) |
1.6 (0.6–4.4) | 0.34 | 2.6 (0.9–7.3) | 0.08 | 3.0 (1.0–8.8) | 0.046 |
| LLD × no MDD |
49 (83.1%) 453 (91.9%) |
10 (16.9%) 40 (8.1%) |
2.3 (1.1–4.9) | 0.029 | 1.9 (0.9–4.1) | 0.10 | 2.0 (0.9–4.3) | 0.09 |
| DS × no DS |
105 (83.3%) 431 (92.7%) |
21 (16.7%) 34 (7.3%) |
2.5 (1.4–4.6) | 0.002 | 2.6 (1.4–4.8) | 0.002 | 2.6 (1.4–4.9) | 0.002 |
Key: a-MDD, adult-onset major depressive disorder; 95% CI, 95% Confidence Interval; DS, depressive symptoms 3 months prior to death; LLD, late life MDD; MDD, major depressive disorder.
Logistic regression models adjusted for age at death, sex, and ethnicity.
Logistic regression models adjusted for age at death, sex, ethnicity, alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia.
The main difference in the results from this sensitivity analysis, excluding CDR = 0.5 cases, was that DS were associated with neuritic plaque burden (beta-amyloid): OR = 1.9 (95% CI = 1.1–3.1, p = 0.013) for the logistic regression models adjusted for age at death, sex, and ethnicity and OR = 1.8 (95% CI = 1.1–3.2; p = 0.027) for the logistic regression models adjusted for age at death, sex, ethnicity, alcohol misuse, smoking, hypertension, diabetes mellitus, and dyslipidemia.
Additional analyses were also performed considering possible differences among male and female groups. First we analyzed demographics and clinical variables between genders as shown in Table 5. The female group was older (p = 0.002), less educated (p < 0.001), had a smaller prevalende of alcohol misuse (p < 0.001), smoking habits (p < 0.001), but a higher prevalence of hypertension (p < 0.001).
Table 5.
Comparisons between female and male groups.
| Male (n = 387) | Female (n = 354) | p | |
|---|---|---|---|
|
| |||
| Age at death (years), mean (SD)a | 70.9 (11.4) | 73.5 (11.8) | 0.002 |
| Education (years), mean (SD)a | 5.1 (3.9) | 4.0 (3.5) | <0.001 |
| Ethnicity white, nb (%) | 278 (71.8%) | 334 (66.1%) | 0.09 |
| Alcohol misuse, nb (%) | 109 (32.1%) | 19 (5.8%) | <0.001 |
| Smoking, nb (%) | 205 (54.5%) | 93 (28.3%) | <0.001 |
| Hypertension, nb (%) | 226 (59.5%) | 279 (79.0%) | <0.001 |
| Diabetes mellitus, nb (%) | 96 (25.3%) | 102 (28.8%) | 0.28 |
| Dyslipidemia, nb (%) | 37 (9.8%) | 39 (11.0%) | 0.58 |
| a-MDD, nb (%) | 22 (6.3%) | 32 (10.4%) | 0.054 |
| LLD, nb (%) | 35 (9.6%) | 45 (14.0%) | 0.07 |
| DS, nb (%) | 80 (20.7%) | 88 (24.9%) | 0.17 |
| CDR = 0.5, nb (%) | 59 (15.2%) | 62 (17.5%) | 0.40 |
Data are presented as means (SD) and for categorical values as the number of cases and percentage (%) when indicated.
Key: a-MDD, adult-onset major depressive disorder; CDR, Clinical Dementia Rating scale; DS, depressive symptoms 3 months prior to death; LLD, late life MDD; MDD, major depressive disorder.
Mann-Whitney test.
Qui-square test.
When we used multivariable logistic regression models, adjusted for significant variables (age at death, schooling, alcohol and smoke use, hypertension), no associations between gender and neuropathological alterations were found, as seen in Table 6.
Table 6.
Association between gender and neuropathological alterations (n = 741).
| Small vessel disease |
Crude OR (95% CI) |
p | Multivariatea OR (95% CI) |
p | ||
|---|---|---|---|---|---|---|
| No | Yes | |||||
|
| ||||||
| Female × male |
311 (88.1%) 347 (90.1%) |
42 (11.9%) 38 (9.9%) |
0.8 (0.5–1.3) | 0.38 | 0.7 (0.4–1.2) | 0.18 |
|
Cerebral amyloid angiopathy
|
||||||
| No | Yes | |||||
| Female × male |
336 (95.5%) 375 (97.4%) |
16 (4.5%) 10 (2.6%) |
0.6 (0.3–1.3) | 0.16 | 0.5 (0.2–1.4) | 0.19 |
|
Infarcts
|
||||||
| No | Yes | |||||
| Female × male |
325 (92.6%) 359 (93.2%) |
26 (7.4%) 26 (6.8%) |
0.9 (0.5–1.6) | 0.73 | 1.1 (1.0–2.2) | 0.82 |
|
Neuritic plaques (beta-amyloid): moderate-frequent
|
||||||
| No | Yes | |||||
| Female × male |
284 (80.2%) 333 (86.0%) |
70 (19.8%) 54 (14.0%) |
0.7 (0.5–1.0) | 0.035 | 0.7 (0.5–1.2) | 0.19 |
|
Neurofibrillary pathology (tau) III-VI
|
||||||
| No | Yes | |||||
| Female × male |
234 (66.7%) 291 (76.2%) |
117 (33.3%) 91 (23.8%) |
0.6 (0.5–0.9) | 0.004 | 0.4 (0.1–1.1) | 0.07 |
|
Lewy body disease
|
||||||
| No | Yes | |||||
| Female × male |
307 (90.3%) 338 (90.6%) |
33 (9.7%) 35 (9.4%) |
1.0 (0.6–1.6) | 0.88 | 1.0 (0.6–1.9) | 0.89 |
Key: a-MDD, adult-onset major depressive disorder; 95% CI, 95% Confidence Interval; DS, depressive symptoms 3 months prior to death; LLD, late life MDD; MDD, major depressive disorder.
Logistic regression models adjusted for age at death, education, alcohol misuse, smoking, and hypertension.
4. Discussion
In analyses comparing current and previous depressive episodes in a large population-based clinicopathological cohort of 741 individuals without dementia (CDR < 1), we found that the presence of small vessel disease was twice more prevalent in individuals with any of the 3 depression subtypes than in individuals with no depression. In addition, LLD and DS groups had similar results for infarcts. LLD and DS were associated with twice more Lewy body disease. When cases with CDR = 0.5 were excluded, results remained alike for most analyses. Nonetheless, an association of DS with cerebral amyloid angiopathy, neuritic plaques (beta-amyloid), and neurofibrillary pathology (AD-tau burden) was found. It may be the case that the exclusion of CDR = 0.5 leads to the exclusion of controls with pathology and cases without pathology, increasing the contrast between groups.
This work brings several insights into the present understanding of the neurobiological bases of depression in older adults. It provided evidence of possible vascular origins of depression and gives insights into a hypothesis in which mood disorders may be pre-cognitive symptoms in AD and pre-parkinsonism symptoms in Lewy body disease (Ehrenberg et al., 2018; Kim et al., 2016; Mariani et al, 2021). In addition, beyond the social determinants of health leading to depression, the heterogeneity of neuropathologic lesions associated with depression highlights its multifactorial biological origins. This may help explain why it is still so challenging to find specific biomarkers for depression, as childhood trauma, sex differences, lifestyle, and demographic variables are also important factors (Mariani et al, 2021).
Smaller neuropathological and clinical-only studies have pointed to a possible correlation between cerebrovascular lesions and depression (Aizenstein et al., 2016; Diniz et al., 2013; Jamieson et al., 2019; Kim et al., 2016). The association of LLD with an increased risk for vascular disease in clinical-only samples is supported by both meta-analysis (Diniz et al., 2013) and other studies such as a 5-year follow-up of 1,949 community older adults free of dementia or baseline depressive symptoms (van Sloten et al., 2015). In this prospective analysis, the onset of depressive symptoms in 10% of the sample was associated with increased white matter hyperintensities and with new subcortical infarcts, detected through magnetic resonance imaging scans. Interestingly, in our sample, the younger age at death and a lower percentage of diabetes mellitus in the a-MDD might have contributed to the lack of association of a-MDD with brain infarcts, differently from what was found in the LDD group and the DS group. Still, the small a-MDD sample size could have contributed to these negative results. The association of white matter lesions with small vessel disease is well supported by the literature (Aizenstein et al., 2016; Grinberg and Thal, 2010; Jellinger, 2013; Korczyn and Halperin, 2009). In an interesting study with 20 older adults with a history of MDD and their matched controls, magnetic resonance imaging and neuropathological analysis were performed in the same 3 slices of brain tissue (Thomas et al., 2002). Deep white matter hyperintensities due to cerebral ischemia were more frequent in depressed subjects. This is in agreement with our results from a large cohort and infuses confidence in the veracity of this relationship. Moreover, we were able to use neuropathological analysis to evaluate small vessel disease, a type of lesion still underestimated in neuroimaging studies. Emerging evidence suggests that microvascular dysfunction may contribute to depression (Empana et al., 2021). However, as authors argue, more research is needed to fully characterize the association between microvascular dysfunction and specific depressive symptoms. This will help to further define microvascular depression as a specific subtype of depression. In addition, clinical studies are needed to evaluate whether stratification of patients according to the presence of microvascular dysfunction could identify subgroups more likely to respond to specific clinical therapies, including agents that improve microvascular function (Empana et al., 2021).
Previous studies investigating cognitively normal individuals failed to find a relationship between neuropathological changes and depression, but the sample size regarding depression cases and especially dementia-free cases was significantly smaller when compared to our study’s, ranging from 36 to 74 individuals (Santos et al., 2010; Tsopelas et al., 2011; Wilson et al., 2016) or even less (Aizenstein et al., 2016). Other studies only measured the intensity of depressive symptoms; moreover, median sample scores were below the cut-off for depression (Royall and Palmer, 2013; Wennberg et al., 2019). Given the significant variability on how depression was established and limited sample sizes, we hypothesize that previous negative results may be due to type II error.
Interestingly, depression is the most common prodromal psychiatric symptom in Lewy body disease patients, often preceding the onset of motor symptoms (Jellinger, 2013). Depression is also associated with cognitive impairment as well as with a faster rate of cognitive decline in Lewy body disease (Patterson et al., 2019). We found an association between LLD and DS and Lewy body disease. There are few studies of Lewy-type pathology in individuals with incidental Lewy body disease but no dementia. However, neuropathological studies show that mood-modulating nuclei such as the locus coeruleus and dorsal raphe nucleus develop Lewy bodies before or simultaneously with the substantia nigra, suggesting depression might be an early non-motor symptom in Lewy body disease. The association of LLD and DS with Lewy body disease can also be partially explained by a spread of Lewy-type pathology in mesolimbic and mesostriatal dopaminergic neurons, impairing dopamine metabolism and transmission, as seen in a postmortem study of Lewy body disease cases (Patterson et al., 2019). In line with our results, positive associations were also found in a sample of 153 older adults with no dementia, 36 of whom had depressive symptoms above the cut-off score (Jellinger and Attems, 2006). In a sample of 124 older adults with no dementia, higher densities of brainstem Lewy bodies were associated with a higher level of depressive symptoms (Wilson et al., 2016). That association was observed in 3 of the 4 brainstem nuclei (locus coeruleus, dorsal raphe nucleus, and substantia nigra). In another study from the same research group, however, no relationship with Lewy-type pathology was found in a sample of 72 individuals with MDD, although more than half of the entire sample had dementia (Wilson et al., 2016).
Finally, we found a relationship between neurofibrillary tangle burden and DS (p = 0.06 and p = 0.027 when cases with CDR = 0.5 were excluded). Other studies are in line with this finding (Ehrenberg et al., 2018; Rapp et al., 2006; Robinson et al., 2021; Wilson et al., 2013). AD-tau pathology and degeneration of locus coeruleus and dorsal raphe nucleus are the recognizable signs of AD pathology in the human brain; these nuclei are also the main producers of norepinephrine and serotonin in the central nervous system (Grinberg et al., 2009). Furthermore, cognitively normal individuals at I–II Braak stage for neurofibrillary pathology have more than twice the chance of showing depressive symptoms than individuals at Braak 0 (Ehrenberg et al., 2018).
Notably, our study found no relationship of depression subtypes with neuritic plaque burden (beta-amyloid), except when cases with CDR = 0.5 were excluded. When we carried out the analysis of the n = 620 cases with CDR = 0, DS were associated with higher neuritic plaque burden. Associations were found in a postmortem study of 161 individuals with no dementia, there was an association between a higher burden of neuritic plaques and depressive symptoms (Wennberg et al., 2019). Higher beta-amyloid plaque burden was also found to be associated with higher likelihood of MDD diagnosis, but not with the intensity of depressive symptoms (Wilson et al., 2016). However, negative associations between amyloid severity and depression have also been reported (Royall and Palmer, 2013; Tsopelas et al., 2011; Wilson et al., 2014), and indeed the literature brings conflicting findings (Jamieson et al., 2019). There are some possible explanations for our negative findings in some of our analysis. For example, in early AD stages, beta-amyloid pathology is confined to neocortical regions less likely to modulate depressive symptoms than the subcortical regions already carrying substantial neurofibrillary tangle burden, making possible correlations between beta-amyloid and depression very weak and the results spurious (Ehrenberg et al., 2018).
Our study results were less consistent regarding cerebral amyloid angiopathy, as positive associations were found only in the DS group in the analysis of cases with CDR = 0. These inconsistent results might be due to low statistical power as the sample size of positive cases was the smallest in our sample: 26 cases, as compared to 52 cases with infarcts, 68 with Lewy body disease, 80 cases with small vessel disease, 123 cases with neuritic plaques (beta-moderate-frequent and 208 cases with neurofibrillary pathology III–VI. Cerebral amyloid angiopathy studies in depression are also lacking in literature; we found only 1 study with 10 individuals with LLD that found a trend toward less severe amyloid angiopathy (Sweet et al., 2004). Future studies with large samples size are needed to fill this literature gap.
Despite our effort s to minimize weaknesses in study design, remaining shortcomings should be noted. Unfortunately, many MDD cases in our sample were not under antidepressant treatment as the inclusion criteria were lifetime MDD and not current MDD. No specific association of antidepressant treatment with neuropathological alterations was found. We also lacked the information on more severe depression cases as well as the cause of death. This is, however, an important question to be addressed, and further studies, including previous long-term antidepressant use, are warranted. The CDR used in this study evaluates cognitive function and functional performance, but it does not evaluate specific cognitive domains. The inherent cross-sectional design of the study with a retrospective collection of data may also be a limitation. To minimize this limitation, only informants that had at least 1 weekly contact with the deceased and that were able to convey reliable information were included. Moreover, the clinical interview with informants used in this study was validated in clinical settings, using simple and straight-forward questions (Ferretti et al., 2010) that were used in previous studies (Ehrenberg et al., 2018; Grinberg et al., 2009; Nunes et al., 2019; Suemoto et al., 2017). Clinical information from the clinical interview and the SCID was further checked by a geriatric psychiatrist (PVN) to confirm the diagnosis of depression and to exclude secondary causes of depression. Finally, some sort of selection of lower social class may have occurred in BAS as in Brazil, high-income individuals (approximately 3% of the population) (IBGE, 2017) usually have better access to medical assistance before death and some might not need an autopsy.
This study also has strengths. We studied a unique, large, population-based clinicopathologic sample free of biases typically found in convenience samples, such as the over-representation of more severe cases. Moreover, neuropathology remains the gold standard for diagnosing neurodegenerative disease; despite recent advances, methods for staging AD pathological markers in vivo fail to reach the same level of sensitivity and specificity of the pathological prediction, and there are no neuroimaging markers for Lewy body disease. This discrepancy is particularly prominent at early AD stages when, for instance, tau burden is primarily subcortical.
5. Conclusions
Findings from this large community sample suggest that depression in older adults with no dementia might be associated with some forms of organic pathology and neurodegeneration and, therefore, may be a prodrome of dementia. Neuropathological studies should be large if they are to detect the association between depression and brain lesions, given the often-multifactorial nature of depression. An older adult with depressive symptoms or a complete clinical picture of MDD should be monitored for possible cognitive decline, especially in the case of LLD. Moreover, possible disease-modifying treatments or preventive measures for cerebrovascular disease, like physical activity and optimized control of glycemia or dyslipidemia, will likely benefit older adults with depression, even before the clinical onset of dementia. The same might be true when disease-modifying treatments for tau and alpha-synucleinopathies become available.
Acknowledgements
This study was supported by Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) [grant numbers 2018/16626-0 and 2016/24326-0]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [grant number 466763/2014-0]; and Alzheimerś Association Research Fellowship [AARF 18-566005]; and by a generous private donation from Paulo Sérgio Galvão to the USP Bipolar Disorder Research Program (PROMAN). CN was supported by FAPESP [2017/07089-8]. LTG is supported by NIA K24AG053435.
Footnotes
Declaration of Competing Interest
None.
CRediT authorship contribution statement
Paula Villela Nunes: Conceptualization, Methodology, Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Visualization, Project administration, Funding acquisition. Claudia Kimie Suemoto: Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – review & editing, Project administration, Funding acquisition. Roberta Diehl Rodriguez: Methodology, Investigation, Writing – review & editing, Funding acquisition. Renata Elaine Paraizo Leite: Investigation, Data curation, Writing – review & editing. Camila Nascimento: Formal analysis, Writing – original draft, Writing – review & editing. Carlos Augusto Pasqualucci: Methodology, Resources, Writing – review & editing, Funding acquisition. Ricardo Nitrini: Methodology, Resources, Writing – review & editing, Funding acquisition. Wilson Jacob-Filho: Methodology, Resources, Writing – review & editing, Funding acquisition. Lea T. Grinberg: Methodology, Resources, Writing – review & editing, Project administration, Funding acquisition. Beny Lafer: Conceptualization, Resources, Writing – review & editing, Visualization, Supervision, Funding acquisition.
References
- Alexopoulos GS., 2019. Mechanisms and treatment of late-life depression. Transl Psychiatry 9 (1), 188. doi: 10.1038/s41398-019-0514-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenstein HJ, Baskys A, Boldrini M, Butters MA, Diniz BS, Jaiswal MK, Jellinger KA, Kruglov LS, Meshandin IA, Mijajlovic MD, Niklewski G, Pospos S, Raju K, Richter K, Steffens DC, Taylor WD, Tene O, 2016. Vascular depression consensus report - a critical update. BMC Med 14 (1), 161. doi: 10.1186/s12916-016-0720-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E, 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Chan JYC, Yiu KKL, Kwok TCY, Wong SYS, Tsoi KKF., 2019. Depression and antidepressants as potential risk factors in dementia: a systematic review and meta–analysis of 18 longitudinal studies. J Am Med Dir Assoc 20, 279–286. [DOI] [PubMed] [Google Scholar]
- Cummings JL., 1997. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology 48, S10–S16. [DOI] [PubMed] [Google Scholar]
- Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF 3rd, 2013. Late-life depression and risk of vascular dementia and Alzheimer’s disease: systematic review and meta-analysis of community-based cohort studies. Br J Psychiatry 202, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empana JP, Boutouyrie P, Lemogne C, Jouven X, van Sloten TT., 2021. Microvascular contribution to late-onset depression: mechanisms, current evidence, association with other brain diseases, and therapeutic perspectives. Biol Psychiatry 90, 214–225. [DOI] [PubMed] [Google Scholar]
- Ehrenberg AJ, Suemoto CK, França Resende EP, Petersen C, Leite REP, Rodriguez RD, Ferretti-Rebustini REL, You M, Oh J, Nitrini R, Pasqualucci CA, Jacob-Filho W, Kramer JH, Gatchel JR, Grinberg LT, 2018. Neuropathologic correlates of psychiatric symptoms in alzheimer’s disease. J Alzheimers Dis 66, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti REL, Damin AE, Brucki SMD, Morillo LS, Perroco TR, Campora F, Moreira EG, Balbino ÉS, Lima MDCA, Battela C, Ruiz L, Grinberg LT, Farfel JM, Leite REP, Suemoto CK, Pasqualucci CA, Rosemberg S, Saldiva PHN, Jacob–Filho W, Nitrini R, 2010. Post-Mortem diagnosis of dementia by informant interview. Dement Neuropsychol 4, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg LT, Ferretti RE, Farfel JM, Leite R, Pasqualucci CA, Rosemberg S, Nitrini R, Saldiva PH, Filho WJ Brazilian Aging Brain Study Group, 2007. Brain bank of the Brazilian aging brain study group - a milestone reached and more than 1,600 collected brains. Cell Tissue Bank 8, 151–162. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Rüb U, Ferretti RE, Nitrini R, Farfel JM, Polichiso L, Gierga K, Jacob–Filho W, Heinsen H Brazilian Brain Bank Study Group, 2009. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 35, 406–416. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Thal DR., 2010. Vascular pathology in the aged human brain. Acta Neuropathol 119, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBGE (Instituto Brasileiro de Geografia e Estatistica), 2017. Available at: https://www.ibge.gov.br. Accessed in May 17th, 2022.
- Jamieson A, Goodwill AM, Termine M, Campbell S, Szoeke C, 2019. Depression related cerebral pathology and its relationship with cognitive functioning: a systematic review. J Affect Disord 250, 410–418. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J, 2006. Prevalence and impact of cerebrovascular pathology in Alzheimer’s disease and parkinsonism. Acta Neurol Scand 114, 38–46. [DOI] [PubMed] [Google Scholar]
- Jellinger KA., 2013. Organic bases of late-life depression: a critical update. J Neural Transm (Vienna) 120, 1109–1125. [DOI] [PubMed] [Google Scholar]
- Kim HK, Nunes PV, Oliveira KC, Young LT, Lafer B, 2016. Neuropathological relationship between major depression and dementia: a hypothetical model and review. Prog Neuropsychopharmacol Biol Psychiatry 67, 51–77. [DOI] [PubMed] [Google Scholar]
- Korczyn AD, Halperin I, 2009. Depression and dementia. J Neurol Sci 283, 139–142. [DOI] [PubMed] [Google Scholar]
- Mariani N, Cattane N, Pariante C, Cattaneo A, 2021. Gene expression studies in Depression development and treatment: an overview of the underlying molecular mechanisms and biological processes to identify biomarkers. Transl Psychiatry 11 (1), 354. doi: 10.1038/s41398-021-01469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynen G, Van Stralen H, Smit JH, Kamphorst W, Swaab DF, Hoogendijk WJ., 2010. Relation between neuritic plaques and depressive state in Alzheimer’s disease. Acta Neuropsychiatr 22, 14–20. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L, 1991. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Morris JC., 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- Nunes PV, Schwarzer MC, Leite REP, Ferretti-Rebustini REL, Pasqualucci CA, Nitrini R, Rodriguez RD, Nascimento CF, Oliveira KC, Grinberg LT, Jacob-Filho W, Lafer B, Suemoto CK., 2019. Neuropsychiatric inventory in community-dwelling older adults with mild cognitive impairment and dementia. J Alzheimers Dis 68, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson L, Rushton SP, Attems J, Thomas AJ, Morris CM., 2019. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol 29, 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters ME, Rosenberg PB, Steinberg M, Norton MC, Welsh-Bohmer KA, Hayden KM, Breitner J, Tschanz JT, Lyketsos CG Cache County Investigators, 2013. Neuropsychiatric symptoms as risk factors for progression from CIND to dementia: the Cache County Study. Am J Geriatr Psychiatry 21, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, Gorman JM, Haroutunian V, 2006. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry 63, 161–167. [DOI] [PubMed] [Google Scholar]
- Robinson AC, Roncaroli F, Davidson YS, Minshull J, Heal C, Montaldi D, Payton A, Horan MA, Pendleton N, Mann DMA., 2021. Mid to late-life scores of depression in the cognitively healthy are associated with cognitive status and Alzheimer’s disease pathology at death. Int J Geriatr Psychiatry 36, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Palmer RF., 2013. Alzheimer’s disease pathology does not mediate the association between depressive symptoms and subsequent cognitive decline. Alzheimers Dement 9, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos M, Gold G, Kövari E, Herrmann FR, Hof PR, Bouras C, Giannakopoulos P, 2010. Neuropathological analysis of lacunes and microvascular lesions in late-onset depression. Neuropathol Appl Neurobiol 36, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer RL, Williams JB, Gibbon M, First BM., 1992. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry 49, 624–629. [DOI] [PubMed] [Google Scholar]
- Suemoto CK, Ferretti-Rebustini RE, Rodriguez RD, Leite RE, Soterio L, Brucki SM, Spera RR, Cippiciani TM, Farfel JM, Chiavegatto Filho A, Naslavsky MS, Zatz M, Pasqualucci CA, Jacob-Filho W, Nitrini R, Grinberg LT, 2017. Neuropathological diagnoses and clinical correlates in older adults in Brazil: a cross-sectional study. PLoS Med 14, e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Hamilton RL, Butters MA, Mulsant BH, Pollock BG, Lewis DA, Lopez OL, DeKosky ST, Reynolds CF 3rd, 2004. Neuropathologic correlates of late-onset major depression. Neuropsychopharmacology 29, 2242–2250. [DOI] [PubMed] [Google Scholar]
- Taylor WD., 2017. Lack of a role for Alzheimer’s disease pathology in late-life depression, or just no relationship with amyloid? Am J Psychiatry 174, 197–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AJ, O’Brien JT, Davis S, Ballard C, Barber R, Kalaria RN, Perry RH., 2002. Ischemic basis for deep white matter hyperintensities in major depression: a neuropathological study. Arch Gen Psychiatry 59, 785–792. [DOI] [PubMed] [Google Scholar]
- Tsopelas C, Stewart R, Savva GM, Brayne C, Ince P, Thomas A, Matthews FE Medical Research Council Cognitive Function and Ageing Study, 2011. Neuropathological correlates of late-life depression in older people. Br J Psychiatry 198, 109–114. [DOI] [PubMed] [Google Scholar]
- van Dyck CH, O’Dell RS, Mecca AP, 2021. Amyloid-associated depression-or not? Biol Psychiatry 89, 737–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Sloten TT, Sigurdsson S, van Buchem MA, Phillips CL, Jonsson PV, Ding J, Schram MT, Harris TB, Gudnason V, Launer LJ., 2015. Cerebral small vessel disease and association with higher incidence of depressive symptoms in a general elderly population: the AGES-Reykjavik study. Am J Psychiatry 172, 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennberg AM, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Machulda MM, Petrucelli L, Mielke MM, Jack CR Jr, Knopman DS, Parisi JE, Petersen RC, Dickson DW, Josephs KA., 2019. The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging 77, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Schneider JA, Bienias JL, Arnold SE, Evans DA, Bennett DA., 2003. Depressive symptoms, clinical AD, and cortical plaques and tangles in older persons. Neurology 61, 1102–1107. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, Shah RC, Schneider JA, Arnold SE, Bennett DA., 2013. Brainstem aminergic nuclei and late-life depressive symptoms. JAMA Psychiatry 70, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Capuano AW, Boyle PA, Hoganson GM, Hizel LP, Shah RC, Nag S, Schneider JA, Arnold SE, Bennett DA., 2014. Clinical-pathologic study of depressive symptoms and cognitive decline in old age. Neurology 83, 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Boyle PA, Capuano AW, Shah RC, Hoganson GM, Nag S, Bennett DA., 2016. Late-life depression is not associated with dementia-related pathology. Neuropsychology 30, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]