

Abstract
As the understanding of immune responses in Alzheimer’s disease (AD) is in its early phases, there remains an urgency to identify the cellular and molecular processes driving chronic inflammation. In AD, a subpopulation of astrocytes acquires a neurotoxic phenotype which prompts them to lose typical physiological features. While the underlying molecular mechanisms are still unknown, evidence suggests that myeloid differentiation primary response 88 (MyD88) adaptor protein may play a role in coordinating these cells’ immune responses in AD. Herein, we combined studies in human postmortem samples with a conditional genetic knockout mouse model to investigate the link between MyD88 and astrocytes in AD. In silico analyses of bulk and cell-specific transcriptomic data from human postmortem brains demonstrated an upregulation of MyD88 expression in astrocytes in AD versus non-AD individuals. Proteomic studies revealed an increase in glial fibrillary acidic protein in multiple brain regions of AD subjects. These studies also showed that although overall MyD88 steady-state levels were unaffected by AD, this protein was enriched in astrocytes near amyloid plaques and neurofibrillary tangles. Functional studies in mice indicated that the deletion of astrocytic MyD88 protected animals from the acute synaptic toxicity and cognitive impairment caused by the intracerebroventricular administration of β-amyloid (Aβ). Lastly, unbiased proteomic analysis revealed that loss of astrocytic MyD88 resulted in altered astrocyte reactivity, lower levels of immune-related proteins, and higher expression of synaptic-related proteins in response to Aβ. Our studies provide evidence of the pivotal role played by MyD88 in the regulation of astrocytes response to AD.
Keywords: astrocytes, MyD88, inflammation, β-amyloid, synaptic toxicity, cognitive impairment, Alzheimer’s disease
1. Introduction
A dysregulated inflammatory response has emerged as a key target for therapeutic intervention in Alzheimer’s disease (AD). Current research activities focus primarily on deciphering the role of microglia, central nervous system’s resident immune cells and macrophages, because of their immunological role within the brain and their altered immune capacity in genetic forms of AD (Lewcock et al., 2020). However, other cell types in the brain also harbor the biological machinery to participate in immune responses, highlighting that a more expansive approach is required to successfully reveal the complete picture of how inflammation contributes to AD (Newcombe et al., 2018). Astrocytes, which account for 20–40% of glial cells in the brain, are well-known for their involvement in modulating information processing, signal transmission, and neural and synaptic plasticity (Azevedo et al., 2009; Pelvig et al., 2008; Santello et al., 2019). Nevertheless, although some progress has been made regarding their participation in brain immunity, the overall role of astrocytes in immune regulation and neurodegeneration in AD remains relatively unexplored.
Astrocytes have the capability to alleviate and accelerate neurodegeneration. These cells provided neuroprotection in models of neurodegeneration through the activation of distinct signaling pathways, including nuclear factor erythroid 2-related factor 2 (NRF2), signal transducer and activator of transcription 3 (STAT3)/thrombospondin 1 (TSP1), interleukin-3 (IL-3), interleukin-33 (IL-33)/aquaporin 4 (AQP4), endocannabinoid, and lipoxin (Dunn et al., 2015; Hu et al., 2022; Jiwaji et al., 2022; McAlpine et al., 2021; Medeiros et al., 2013; Tyzack et al., 2014; Wu et al., 2021). Contrariwise, astrocyte reactivity can trigger detrimental neurological effects via α2-Na+/K+ adenosine triphosphatase (α2-NKA), calcineurin (CN)/nuclear factor of activated T cells 4 (NFAT4), apolipoprotein E4 (ApoE4), and nuclear factor-κB (NF-κB)/complement component 3 (C3), among other signaling pathways (Jackson et al., 2021; Lian et al., 2015a; Mann et al., 2022; Sompol et al., 2017; Wang et al., 2021). Therefore, decoding the underlying molecular mechanisms of astrocyte reactivity could unveil cell-specific targets with meaningful translational benefits in AD.
Myeloid differentiation primary response 88 (MyD88) is an adapter protein that participates in inflammatory responses driven by toll-like receptors (TLRs, except TLR3) and receptors of the interleukin-1 (IL-1) family of cytokines. While astrocytes express these receptors constitutively, their levels are upregulated during disease processes (Liddelow & Barres, 2017; Linnerbauer et al., 2020). Mechanistically, MyD88 acts as a bridge between these receptors and IL-1 receptor-associated kinases (IRAKs), ultimately amplifying inflammatory signals through the activation of transcriptional factors NF-κB and activator protein-1 (AP-1) (Deguine & Barton, 2014). Evidence of the relevance of MyD88 in AD has been presented both indirectly and directly. For example, TLRs and components of the IL-1 family of cytokines signaling have been linked to the pathogenesis of AD, and β-amyloid (Aβ) has been shown to directly activate some of these receptors (Ghosh et al., 2013; Halle et al., 2008; Kitazawa et al., 2011b; Liu et al., 2012; Reed-Geaghan et al., 2010; Reed-Geaghan et al., 2009; Shaftel et al., 2007; Vollmar et al., 2010). Pharmacological inhibition of TLR2 and MyD88 interaction attenuated inflammation and AD-like pathology in the 5xFAD mouse model (Rangasamy et al., 2018). At the cell-specific level, it has been shown that deletion of MyD88 in microglia reduces inflammation and β-amyloid (Aβ) accumulation in the APP/PS1 mouse model (Quan et al., 2021). Conversely, another study has indicated that total MyD88 genetic deletion in APP/PS1 mice does not alter AD-like pathology (Weitz et al., 2014). The reason for the divergence is unknown, but it could be related to the distinct approaches applied in the studies. Hence, additional research is needed to fully elucidate the impact of MyD88 in AD, and more specifically its role in modulating the function of astrocytes.
We investigated whether MyD88 plays a role in the response of astrocytes to AD. First, we performed studies using human postmortem samples that revealed that MyD88 is upregulated in astrocytes in AD and strongly detected in cells near amyloid plaques and neurofibrillary tangles (NFTs) – the key pathological hallmarks of AD. Furthermore, we selectively deleted MyD88 from astrocytes in a conditional knockout mouse model and found that these mice were protected against the synaptic toxicity and cognitive impairment induced by the intracerebroventricular injection of Aβ. Ultimately, our study suggested that astrocytes employ MyD88 to respond to Aβ and that this signaling is associated with their pathological phenotype in AD.
2. Materials and Methods
2.1. Human databases
Normalized transcriptomic data from human temporal cortex (TCX, non-AD = 78, AD = 82) and cerebellum (CBL, non-AD = 77, AD = 81) were obtained from the MayoPilot RNAseq study deposited in the AD Knowledge Portal (ID: syn5550404) (Allen et al., 2016). All AD cases had a definite diagnosis of AD according to the National Institute of Neurological and Communicative Diseases (NINCDS) and Alzheimer’s Disease and Related Disorders Association (ADRDA) criteria and had a Braak NFT stage of IV or greater. Non-AD subjects had a Braak NFT stage of III or less, neuritic and cortical plaque densities of 0 (none) or 1 (sparse) according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) and lacked any other major neurodegenerative disorder. Our study used normalized data of MyD88 and astrocyte markers glial fibrillary acidic protein (GFAP) and aldehyde dehydrogenase-1 family member L1 (ALDH1L1) to calculate the z-score of individual samples. Data were analyzed through an unpaired t-test, with a confidence level of 95%, using Prism (GraphPad Software, San Diego, CA, USA). The accepted level of significance for the tests was P < 0.05. The association between MyD88 and GFAP or ALDH1L1 was determined using linear regression and Pearson correlation using Prism.
Cell-specific single-cell ribonucleic acid-sequencing (RNA-seq) data, including the statistical significance, were obtained from Mathys and colleagues (Supplementary Table 1) (Mathys et al., 2019). Cells were obtained from individuals with no-pathology (N, individuals with no or very low Aβ burden or other pathologies, n = 24), early-AD (EAD, individuals with Aβ burden, but modest neurofibrillary tangles and modest cognitive impairment, n = 15), and late-AD (LAD, individuals with high Aβ burden, increased neurofibrillary tangles, global pathology, and cognitive impairment, n = 9). In our study, we represented the data relative to the mean level of the no pathology group.
2.2. Postmortem human brain
Brain tissue from non-AD (n = 19) and AD (n = 21) individuals were obtained from the Alzheimer’s Disease Research Center at the University of California, Irvine (UCI) (Supplementary Table 2). Informed consent was obtained from patients prior to death, in accordance with the United States Federal Policy for the Protection of Human Subjects. The protocol for obtaining postmortem brains was approved by the UCI Institutional Review Board. Our study was also approved by The University of Queensland Human Research Ethics Committee. Paraformaldehyde-fixed and fresh frozen samples from the hippocampus (HPC), TCX, and CBL were provided. Data were analyzed through an unpaired t-test, with a confidence level of 95%, using Prism (GraphPad Software). The accepted level of significance for the tests was P < 0.05.
2.3. Animals
Experimental procedures used in the present study followed the Principles of Laboratory Animal Care from the National Institutes of Health (Bethesda, MD, USA). Our study was approved by the UCI Institutional Animal Care and Use Committee and The University of Queensland Animal Ethics Committee. GFAP-Cre−/0-MyD88fl/fl (AMyD+) and GFAP-Cre+/0-MyD88fl/fl (AMyD−) mice with and without MyD88 expression in astrocytes, respectively, were generated from B6.Cg-Tg(Gfap-cre)77.6Mvs/2J (IMSR_JAX: 024098) and B6.129P2(SJL)-Myd88tm1Defr/J (IMSR_JAX: 008888) mice (The Jackson Laboratory, Bar Harbor, ME, USA). Mouse genotyping was performed according to The Jackson Laboratory’s protocol. We further confirmed the deletion of MyD88 expression in astrocytes by confocal colocalization using GFAP and MyD88 antibodies followed by orthogonal analysis and Imaris software colocalization analysis (Supplementary Figure 1a–c). Animals were housed in a pathogen-free animal facility at controlled room temperature (22 ± 2°C) and humidity (60–80%) under a 12:12-hour light-dark cycle (lights on at 6 AM) with ad libitum access to food and water. Animals were used at six months of age.
2.4. Preparation of Aβ42 oligomers
Oligomers were prepared using human Aβ42 peptide (Tocris Bioscience, Minneapolis, MN, USA), as previously described (Lambert et al., 1998). Aβ42 was solubilized in hexafluoroisopropanol, and the solvent was evaporated to produce dried films, which were subsequently dissolved in sterile anhydrous dimethylsulfoxide to make a 5 mM solution. This solution was diluted to 100 μM in ice-cold phosphate-buffered saline (PBS) and incubated overnight at 4°C. The preparation was centrifuged at 14,000 × g for 10 min at 4°C to remove insoluble aggregates (protofibrils and fibrils) and the supernatants containing soluble Aβ42 oligomers were stored at 4°C. Routine characterization of preparations was performed by dot blot using anti-Aβ oligomer A11 antibody (kindly donated by Dr. Charles Glabe, UCI). Oligomers were used within 48 h of preparation.
2.5. Stereotactic intracerebroventricular injection
Mice were anesthetized with isoflurane and fixed on a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). Using a 10 μL syringe and an automated syringe pump, 1 μL of PBS or oligomeric Aβ42 (10 pmol) was injected into the ventricle using the following stereotaxic coordinates: anteroposterior = −0.22 mm, mediolateral = 1 mm, and dorsoventral = −3 mm. The syringe was kept in place for 5 min after the injection.
2.6. Open field (OF)
Mice were tested on the fourth-day post intracerebroventricular injection. Mice were placed in the center of the open field apparatus and the total number of squares crossed with all four paws and rearing behavior were registered for 5 min.
2.7. Morris water maze (MWM)
Mice were trained to swim to a platform submerged 1.5 cm beneath the water’s surface, starting on the fifth-day post-intracerebroventricular injection. Four trials were performed per day, for 60 sec each with a 5 min interval between trials. Mice were trained for four consecutive days. The probe test was assessed 24 h after the last trial, with the platform removed. Performance was monitored with the EthoVision XT video-tracking system (Noldus Information Technology, Leesburg, VA, USA).
2.8. Contextual fear conditioning (CFC)
The contextual fear conditioning test was initiated on the tenth-day post intracerebroventricular injection. During training, mice were placed in the fear conditioning chamber and allowed to explore for 2 min before receiving three electric foot shocks (duration: 1 sec, intensity: 0.2 mA, intershock interval: 2 min). Animals were returned to the home cage 30 sec after the last foot shock. Twenty-four hours later, freezing behavior in the conditioning chamber was recorded for 5 min.
2.9. Mouse brain dissection
Brains were collected 12 days post intracerebroventricular injection. Mice were deeply anesthetized with sodium pentobarbital (350 mg/kg, intraperitoneal injection) and sacrificed by transcardial perfusion with ice-cold PBS. Brains were extracted and sliced in half through the sagittal plane. Cerebellum, brainstem, and olfactory bulb were removed from the left hemisphere and the remaining hemibrain tissue was snap-frozen in liquid nitrogen and stored at −80°C. The right hemisphere was fixed in 4% paraformaldehyde in PBS at 4°C for 48 h, and then stored in 0.02% sodium azide in PBS at 4°C.
2.10. Brain slicing
Fixed brain tissues were cryoprotected in 30% sucrose solution in PBS at 4°C. Frozen human and mouse brain tissues were sectioned into 20 and 40 μm sections, respectively, using a Leica SM2010R freezing microtome (Leica Microsystems, Bannockburn, IL, USA). Sections were stored in 0.02% sodium azide in PBS at 4°C.
2.11. Immunohistochemistry
Free-floating sections were pretreated with 3% hydrogen peroxide and 10% methanol in tris-buffered saline solution (TBS) for 30 min to block endogenous peroxidase activity. After a TBS wash (10 min), sections were permeabilized once in 0.1% Triton X-100 in TBS (TBSA) for 15 min and blocked once with 2% bovine serum albumin (BSA) in TBSA (TBSB) for 30 min. Sections were incubated overnight at 4°C with MyD88 or GFAP antibody (Abcam, Cambridge, UK). Antibodies were diluted in 5% normal serum in TBSB. After three washes in TBS (5 min each), sections were incubated for 1 h with a biotinylated secondary antibody (1:200 in TBSB). Sections were then processed using the Vectastain Elite ABC reagent and 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions. Finally, sections were mounted on gelatin-coated slides, dehydrated in graded ethanol, cleared in xylene, and coverslipped with DPX mounting medium (BDH Laboratory Supplies, Poole, ENG). The specificity of the immune reactions was controlled by omitting the primary antibody. GFAP+-astrocytes and cellular patterns from MyD88 staining were examined by a proficient investigator in the human HPC, TCX and CBL (n = 10 for non-AD and AD). Representative images were acquired using a Zeiss Axio Imager M2 microscope (Zeiss Microscopy, Jena, GER).
2.12. Immunofluorescence
Free-floating sections were incubated with 3% normal serum, 2% BSA, and 0.1% Triton X-100 in TBS for 1 h. Using the same buffer solution, sections were incubated overnight at 4°C with the following primary antibodies: GFAP, S100 calcium-binding protein B (S100b), MyD88, neuronal nuclei (NeuN), microtubule-associated protein 2 (MAP2), synaptophysin (SYP), postsynaptic density protein 95 (PSD95), complement C3, apolipoprotein E (ApoE) (Abcam), HT-7 (Thermo Scientific, Waltham, MA, USA), 6E10 (BioLegend, San Diego, CA, USA), ionized calcium-binding adapter molecule 1 (Iba1) (FUJIFILM Wako Chemicals, Osaka, JP), and Aβ42 (kindly donated by Dr. David Cribbs, UCI). Sections were then rinsed and incubated for 1 h with secondary Alexa Fluor-conjugated antibodies (Invitrogen, Carlsbad, CA, USA). Finally, sections were mounted onto gelatin-coated slides in Fluoromount-G (Southern Biotech Associates, Birmingham, AL, USA). The specificity of the immune reactions was controlled by omitting the primary antibody.
Three-dimensional images were acquired with a Leica DM2500 confocal laser microscope using the Leica Application Suite Advanced Fluorescence software (Leica Microsystems, Inc., Bannockburn, IL). For human tissues, confocal images were acquired in three to five fields per section using the 40x and 63x objectives. In mice, immunostaining in the different groups was assessed at three comparable brain coronal sections positioned between 1.34- and 2.54-mm posterior to the bregma with a space separation of at least 160 μm (3 sections per mouse). Three-dimensional images were acquired in the hippocampal CA1 and CA3 subregions using the 40x and 63x objectives. Confocal images were acquired via sequential scanning using a z-step size of 0.5 μm under the 40x objective and z-step size of 0.1 μm under the 63x objective. Quantitative volumetric analysis of the three-dimensional images was performed with the Imaris software (Bitplane, Inc., South Windsor, CT, USA) using the colocalization and surface tools.
2.13. Western blot
Frozen tissues were ground into powder in liquid nitrogen and lysed in ice-cold T-PER extraction buffer (Thermo Scientific) containing phosphatase inhibitor cocktail 2 (Roche, Basel, CH) and complete Mini EDTA-Free protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO, USA). Samples were lysed in four cycles of 15 s and 5,800 rpm at 4°C, with 45 s intervals, using the Precellys homogenizer (Bertin, Montigny-le-Bretonneux, FRA). Samples were kept on ice for 10 min before tissue lysates were separated by centrifugation at 100,000 × g for 60 min at 4°C to obtain the T-PER soluble fraction. Protein concentrations were determined using the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA).
Proteins were separated on a Mini-Protean TGX 4–20% denaturing gel and transferred onto a nitrocellulose membrane (Bio-Rad Laboratories). Membranes were incubated in TBS containing 0.1% Tween-20 and 5% BSA for 1 h, followed by overnight incubation in the primary antibody at 4°C. The following primary antibodies were used: GFAP, MyD88, SYP, PSD95 (Abcam), cluster of Differentiation 47 (CD47), and actin (Thermo Scientific). After three washes in TBS (5 min each), membranes were incubated with horseradish peroxidase (HRP)-conjugated or IRDye-conjugated secondary antibodies for 1 h (LI-COR Biosciences, Lincoln, NE, USA). Chemiluminescence signal was developed with SuperSignal West Dura (Thermo Scientific). Images were acquired using the Odyssey Fc imaging system and analyzed using the Image Studio software (LI-COR Biosciences).
2.14. Proteomic analysis by mass spectrometry
Sample preparation:
Proteomic studies were performed at the Australian Proteome Analysis Facility at Macquarie University (Sydney, NSW, AUS). In brief, mouse hemibrains were homogenized using bead beating. Tissue was transferred into a tube containing a glass bead and lysis buffer (100mM Tris-HCl buffer, pH 8.8, 8 M urea, 1% w/v SDS), and homogenized to lyse the tissue and release the cellular contents. Viscous cellular deoxyribonucleic acid (DNA) was lysed using probe sonication, followed by centrifugation to separate the cell lysate from cellular debris. Supernatant protein suspension was subjected to further processing. First, cysteine disulfide bonds in the proteins were reduced with 10 mM dithiothreitol (DTT) at 37°C for 45 min and then alkylated with 20 mM indole-3-acetic acid (IAA) for 45 min protected from light at room temperature. Excessive IAA in the sample was quenched with 10 mM DTT for 15 min in the dark at room temperature. Finally, samples were subjected to clean-up using a chloroform-methanol precipitation procedure and the protein pellet was air-dried. The pellet was resuspended in a urea buffer (100mM Tris-HCl buffer, pH 8.8, 8 M urea). The protein concentration of the samples was determined using a BCA protein quantification assay as per the manufacturer’s instructions (Thermo Scientific). One hundred micrograms of protein from all samples were subjected to proteolysis with Lys-C (100:1 protein to enzyme ratio) at room temperature overnight followed by digestion with trypsin (50:1 protein to enzyme ratio) for 6 h at 37°C. Samples pH was adjusted to approximately 3 using a final concentration of approximately 1% trifluoroacetic acid (TFA) before being concentrated and desalted using a solid-phase extraction disk Styrene Divinyl Benzene containing Stage tips (Empore SDB-RPS 47 mm extraction disk). Briefly, stage tips were self-packed into pipette tips, peptides were bound to the stage tip, washed with 0.2% TFA and finally eluted with 80% acetonitrile: 5% ammonium hydroxide. Peptides were dried by vacuum centrifuge and reconstituted in 200 mM N-2-hydroxyethylpiperazine-N’−2-ethanesulfonic acid (HEPES) pH 8.8 before peptide concentration was determined using the Pierce quantitative colorimetric peptide assay (Thermo Scientific).
Tandem Mass Tag (TMT):
Two TMT experiments were carried out to accommodate 20 hemibrain biological replicates (5 per group). Briefly, anhydrous acetonitrile was added to each TMT label vial followed by vortexing for 5 min and brief centrifugation. Aliquots of individual peptide samples were labeled with one of the individual TMT labels (a total of ten labels). Labeling was performed at room temperature (RT) for 1 h with occasional vortexing. After the labeling, the excess TMT label in each sample was quenched using 5% hydroxylamine with vortexing and the sample was then incubated at RT for 15 min. A pre-pooling “label check” experiment was performed by mixing 1.5 μL of each individually labeled TMT sample and vacuum drying the mixture to ensure an equal amount of peptide per label was pooled from all samples. Samples were reconstituted in 0.1% formic acid and analyzed by liquid chromatography-mass spectrometry (LC-MS Q-Exactive, Thermo Scientific). A normalization factor was obtained from the label check experiment and TMT-labeled peptide samples were pooled at a 1:1 ratio across all samples and vacuum dried. The peptide mixture was fractionated by off-line high pH (HpH) reverse phase HPLC into 96 fractions that were then consolidated into 17 fractions. Consolidated fractions were vacuum dried in a vacuum centrifuge.
Nanoflow Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (nano LC-ESI-MS/MS):
The HpH fractioned peptides of each TMT set were reconstituted with sample loading buffer (2% acetonitrile, 97.9% water, 0.1% formic acid) and subjected to liquid chromatography with tandem mass spectrometry analysis (LC-MS-MS). TMT labeled peptide samples were injected onto the trap column and desalted with the loading buffer. Peptides were eluted from the trap column into an in-house packed analytical column, and peptides were separated with the linear gradients of mobile phase A and B: mobile phase B (1–30%) over 120 min with a flow rate of 600 nL/min across the gradient. The column eluent was directed into the ionization source of the mass spectrometer, where a 2.7 kV electrospray voltage was applied. Peptide precursors from 350 to 1850 m/z were scanned at 60,000 resolutions with an automatic gain control (AGC) target value of 3×106. The ten most intense ions from the survey scan were fragmented by higher-energy collisional dissociation (HCD) using a normalized collision energy of 33 with an isolation width of 0.8 m/z. Only precursors with charge states +2 to +5 were selected for tandem mass spectrometry analysis (MS/MS). The mass spectrometry method had a minimum signal requirement of 3×103 ions for triggering the MS/MS. An AGC target value of 1×105 for a maximum injection time of 85 ms and scan resolution of 45K were set for the MS/MS. The dynamic exclusion was set to 30 seconds.
Database searching, peptide quantification, and statistical analysis:
Raw data files were searched against protein sequence databases using Proteome Discoverer (version 2.1, Thermo Scientific). Data were processed using search engines SequestHT and Mascot (Matrix Science, London, UK) against all Mus musculus sequences downloaded from the SwissProt database (Aug-2018). The parameters for the data processing were as follows: Enzyme: Trypsin; Maximum missed cleavages: 2; Precursor mass tolerance: 20 ppm; Fragment mass tolerance: 0.02 Da; Dynamic modifications: Oxidation (M), Deamidated (N, Q), PyroGlu (Q), Acetyl (Protein N-Terminus), Acetyl protein N-term (Sequest), TMT6plex (K) and TMT6plex (N-term); Static Modification: Carbamidomethyl (C); FDR and result display filters: Protein, Peptide, and peptide-spectrum matches (PSM) false discovery rate (FDR) < 1%, Master proteins only.
Computational Analysis:
Differentially expressed proteins (P ≤ 0.05 and fold change ≥ 1.2) were analyzed using the ShinyGO database to determine ontology enrichment for biological process, cellular component, and molecular function terms and perform Reactome pathway analysis (Ge et al., 2020).
2.15. Statistical analysis
Statistical evaluation of mice data was performed using a two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test. The accepted level of significance for the tests was P < 0.05. Significance in figures is represented by the following symbols: *P< 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Tests were performed using Prism (GraphPad Software).
3. Results
3.1. MyD88 is upregulated in astrocytes in AD
We initially analyzed in silico data from two independent human transcriptomic datasets to determine the potential association between MyD88 and astrocytes. In the MayoPilot RNAseq study, bulk RNAseq data from the TCX were used to determine changes triggered by AD, whereas data from the CBL were used as a control since this region is less affected by AD (Allen et al., 2016). Our analysis showed that GFAP (Figure 1a), ALDH1L1 (Figure 1b), and MyD88 (Figure 1c) RNA levels were higher in the TCX of AD individuals compared to non-AD individuals. Conversely, expression levels of these genes in the CBL were not raised by the disease (Figure 1d–f). We next determined the correlation between MyD88, and astrocyte markers using the Pearson correlation coefficient and found that changes in MyD88 positively correlated with increased GFAP (Figure 1g) and ALDH1L1 (Figure 1h) expression in the TCX.
Figure 1 – The increased expression of astrocyte markers and MyD88 in AD.
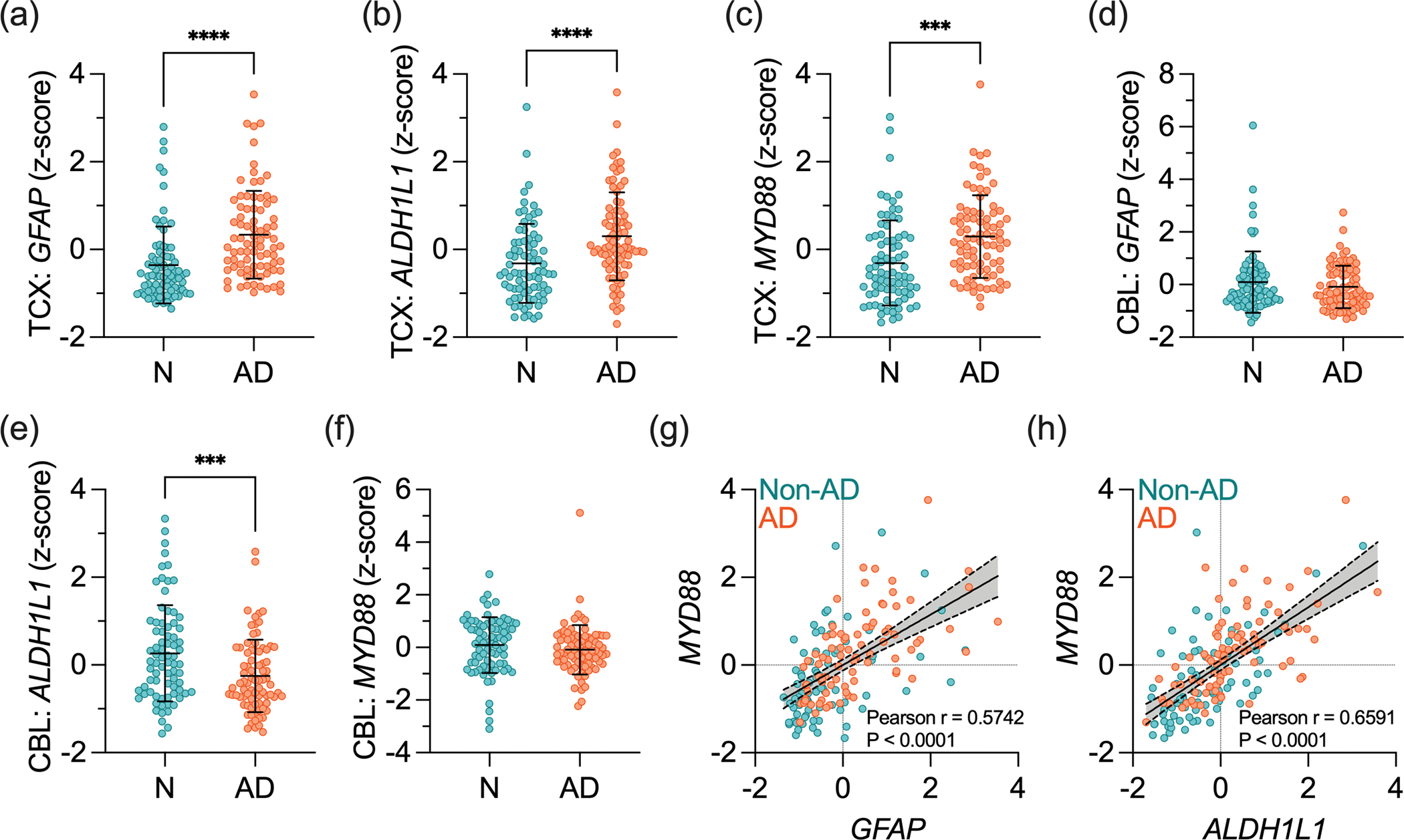
Transcriptomic data was obtained in the TCX and CBL from postmortem human non-AD (N) and AD subjects obtained from the MayoPilot RNAseq dataset (AD Knowledge Portal ID: syn5550404). (a-c) Normalized expression of (a) GFAP, (b) ALDH1L1, and (c) MYD88 in the TCX of non-AD and AD brains. (d-f) Normalized expression of (d) GFAP, (e) ALDH1L1, and (f) MYD88 in the CBL of non-AD and AD brains. Data presented as z-score ± SD. Data were analyzed through an unpaired Student’s t-test, with a confidence level of 95%. ***P < 0.001; ****P < 0.0001. (g,h) Linear regression and Pearson correlation analyses between (g) MYD88 and GFAP and (h) MYD88 and ALDH1L1 in the TCX.
We accessed single-cell transcriptomic data from Mathys and colleagues to gain additional evidence of the potential link between MyD88 and astrocytes in AD (Mathys et al., 2019). Three distinct groups were analyzed: N (no AD pathology), EAD (mid-level AD pathology), and LAD (high-level AD pathology). In the original study, comparison of gene expression in cells isolated from AD versus non-AD subjects has demonstrated that GFAP, but not MyD88, is among the 1,031 unique differentially expressed genes (DEGs) (Mathys et al., 2019). In comparison, the levels of some established AD-related genes, namely complement C3 and transforming growth factor β1 (TGFβ1) in astrocytes and triggering receptor expressed on myeloid cells 2 (Trem2) in microglia were also not among the DEGs (Supplementary Table 1). Single-cell transcriptomic studies are challenging (Kiselev et al., 2019). Therefore, it is unsurprising that only a small number of highly expressed genes were significant.
Our analysis found that GFAP levels were higher in astrocytes isolated from LAD individuals than those isolated from EAD and N individuals (LAD > EAD > N). This finding provides further support for the hypothesis that astrocytes shift towards a reactive phenotype in their response to the progressive changes caused by AD. Despite the lack of statistical significance, MyD88 followed a similar linear pattern, with its expression being the highest in astrocytes isolated from subjects with higher AD pathology (LAD) (Figure 2a). MyD88 levels were also higher in other cell types in AD, including microglia, excitatory neurons, and inhibitory neurons, where its expression fluctuated with the course of disease progression (Figure 2b–e). Our studies indicate that MyD88 might participate in the response of astrocytes to the pathological changes in AD.
Figure 2 – Astrocytic expression of MyD88 correlates with AD pathology.
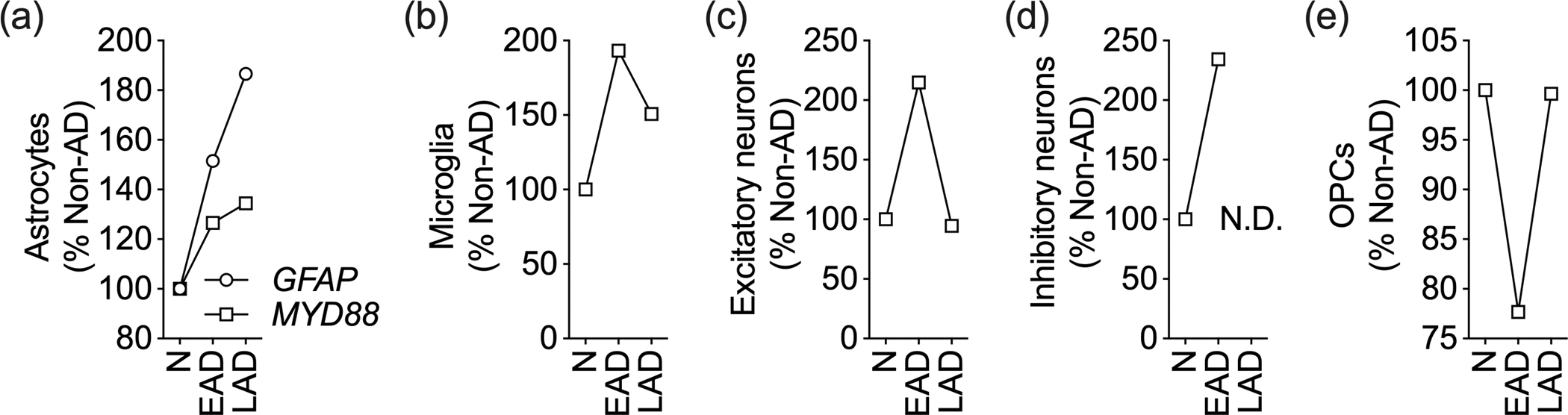
In silico analysis of single-cell transcriptomic data from subjects with no AD pathology (N), mid-level AD pathology (EAD), and high-level AD pathology (LAD). Expression of MyD88 in isolated (a) astrocytes, (b) microglia, (c) excitatory neurons, (d) inhibitory neurons, and (e) oligodendrocyte precursor cells (OPCs). Data are normalized to no pathology group. N.D. indicates not detected. GFAP, but not MYD88, is among the DEGs (Mathys et al., 2019). Please see Supplementary Table 1 for complete statistical data.
3.2. MyD88 is enriched in astrocytes adjacent to AD neuropathology
We next aimed at validating the link between MyD88 and astrocytes at the protein level. Western blot studies showed that AD is characterized by higher GFAP steady-state levels in the HPC and TCX, which are brain regions primarily affected by the disease (Figure 3a). We also found an increase in GFAP levels in the CBL of AD compared to non-AD subjects, likely due to our cohort’s advanced age and the widespread neurodegenerative processes that propagated to other non-primary brain areas in the late stage of the disease. Conversely, MyD88 steady-state levels were not altered by AD in the brain areas studied.
Figure 3 – Astrocytic MyD88 clusters near amyloid plaques and NFTs.
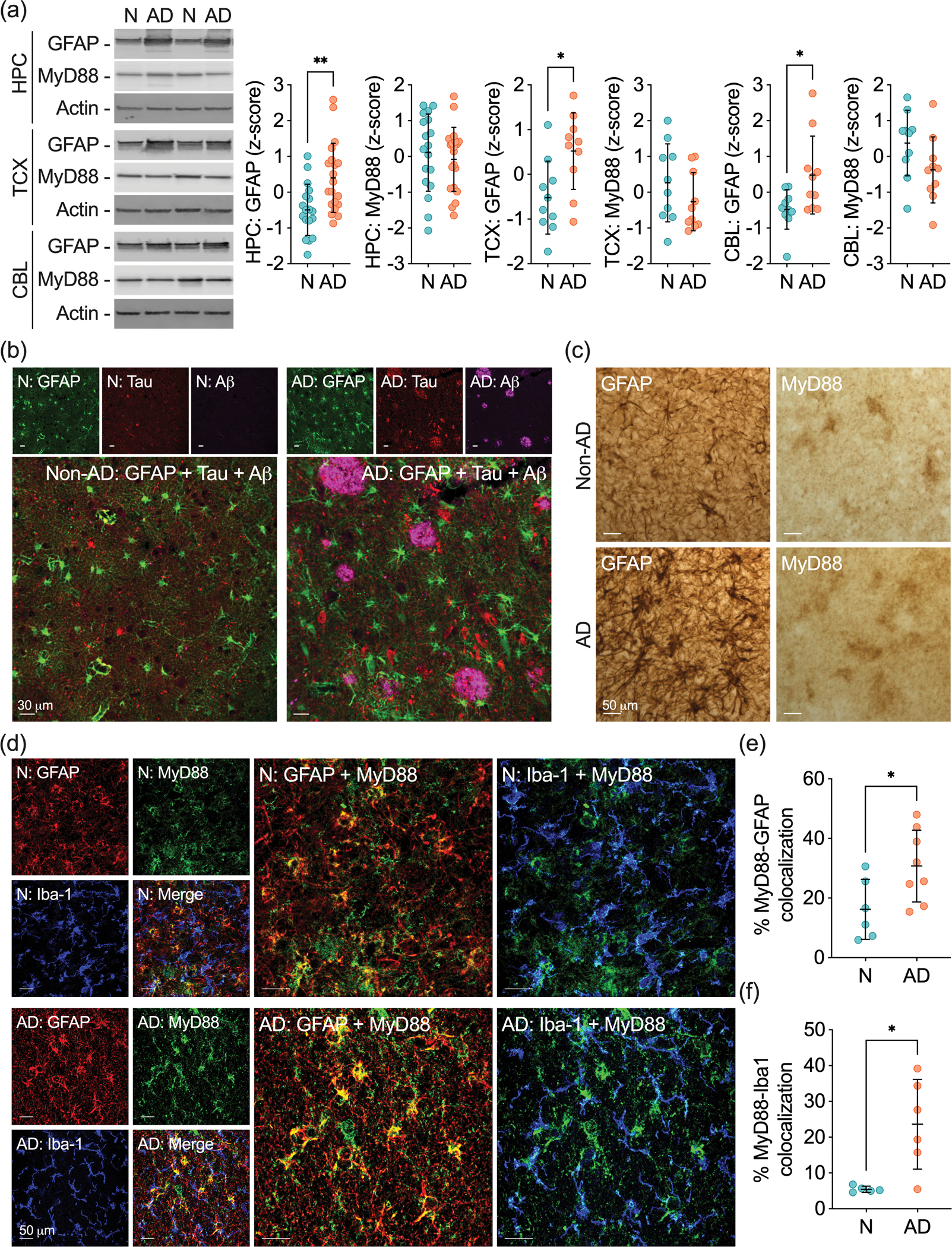
(a) Representative western blots and quantification of GFAP and MyD88 protein levels in the HPC, TCX, and CBL of non-AD (N) and AD subjects. Samples were normalized to actin. Data are presented as z-score ± SD. (b) Representative photomicrographs of immunofluorescence staining of GFAP (green), tau (red), and Aβ (magenta) in non-AD and AD human hippocampal sections. (c) Representative photomicrographs of GFAP and MyD88 immunohistochemical staining in non-AD and AD human hippocampal sections. (d) Representative photomicrographs displaying GFAP (red), Iba1 (blue), MyD88 (green), and merged immunofluorescence in non-AD and AD human hippocampal sections. Larger panels represent the colocalization of MyD88 with GFAP+-astrocytes or Iba1+-microglia. (e,f) Quantification of MyD88 colocalizing with (e) GFAP+-astrocytes and (f) Iba1+-microglia in non-AD and AD brains. The percentage of colocalization was calculated based on the amount of MyD88+-glial+ over the total glial immunostaining. Data were analyzed through an unpaired Student’s t-test, with a confidence level of 95%. *P < 0.05; **P < 0.01.
We also investigated astrocytes at the cellular level using histological methods. Confocal microscopy analyses revealed that GFAP+-astrocytes are broadly distributed in the HPC (Figure 3b), TCX, and CBL (Supplementary Figure 2). We also observed the presence of GFAP+-astrocyte clusters near Aβ42+-plaques and tau+-NFTs in the HPC and TCX of AD individuals. Although sparse, non-AD brains also presented clustered GFAP+-astrocytes, which occurred due to the presence of lower but existent AD neuropathology. Cerebellums from non-AD and AD subjects did not display Aβ42 and tau staining, nor did they show well-defined clusters of GFAP+-astrocytes (Supplementary Figure 2). Comparable GFAP staining was detected through immunohistochemistry, in which GFAP+-astrocytes were widely found in the HPC (Figure 3c). Once again, clustered astrocytes were observed in AD brains and to a lesser extent, in non-AD brains.
We initially determined the pattern of MyD88 immunostaining using immunohistochemistry. Our studies revealed that MyD88 is expressed in distinct cell types in the HPC, TCX, and CBL (Figure 3c, Supplementary Figure 3). We classified MyD88 immunostaining in neuronal-, vascular- and glial-like cells in the HPC and CBL, while MyD88 expression in the TCX was only observed in vascular- and glial-like cells. Importantly, MyD88 staining was detected in clustered glial-like cells in the HPC and TCX of AD brains. We then used colocalization confocal studies to differentiate glial cells harboring MyD88 (Figure 3d). We observed that Iba1+-microglia and GFAP+-astrocytes colocalized with MyD88 in the HPC of non-AD and AD brains (Figure 3d). Notably, quantitative analysis revealed that MyD88 levels were higher in GFAP+-astrocytes (Figure 3e) and Iba1+-microglia (Figure 3f) nearby amyloid plaques. Our findings indicate that although overall MyD88 protein levels do not change in AD, MyD88 is enriched in astrocytes near the neuropathological markers of AD.
3.3. MyD88 deletion in astrocytes inhibits Aβ-induced cognitive impairment
Because cognitive decline is the leading clinical outcome of AD, we decided to investigate whether astrocytic MyD88 participates in the cognitive impairment driven by Aβ. For these studies, we challenged mice expressing MyD88 in astrocytes (AMyD+) and mice with deletion of MyD88 from astrocytes (AMyD−) with an intracerebroventricular injection of Aβ42. We initially used the Morris water maze since it strongly correlates with synaptic plasticity in the hippocampus, a primary area affected in AD (Braak & Del Tredici, 2015; Vorhees & Williams, 2006). We examined the ability of mice to acquire (training session) and retrieve (probe session) spatial information as these functions are indicative of learning and memory. As expected, the intracerebroventricular injection of Aβ42 in AMyD+ mice resulted in a decline in learning and memory, as indicated by longer latencies in the training trials (Figure 4a) and reduced time spent in the correct quadrant (Figure 4b) during the probe trial, compared to AMyD+ mice treated with PBS. On the other hand, Aβ42 did not change the acquisition and retrieval latencies compared to PBS in AMyD− mice.
Figure 4 – Loss of astrocytic MyD88 abrogates cognitive deficits induced by Aβ42.
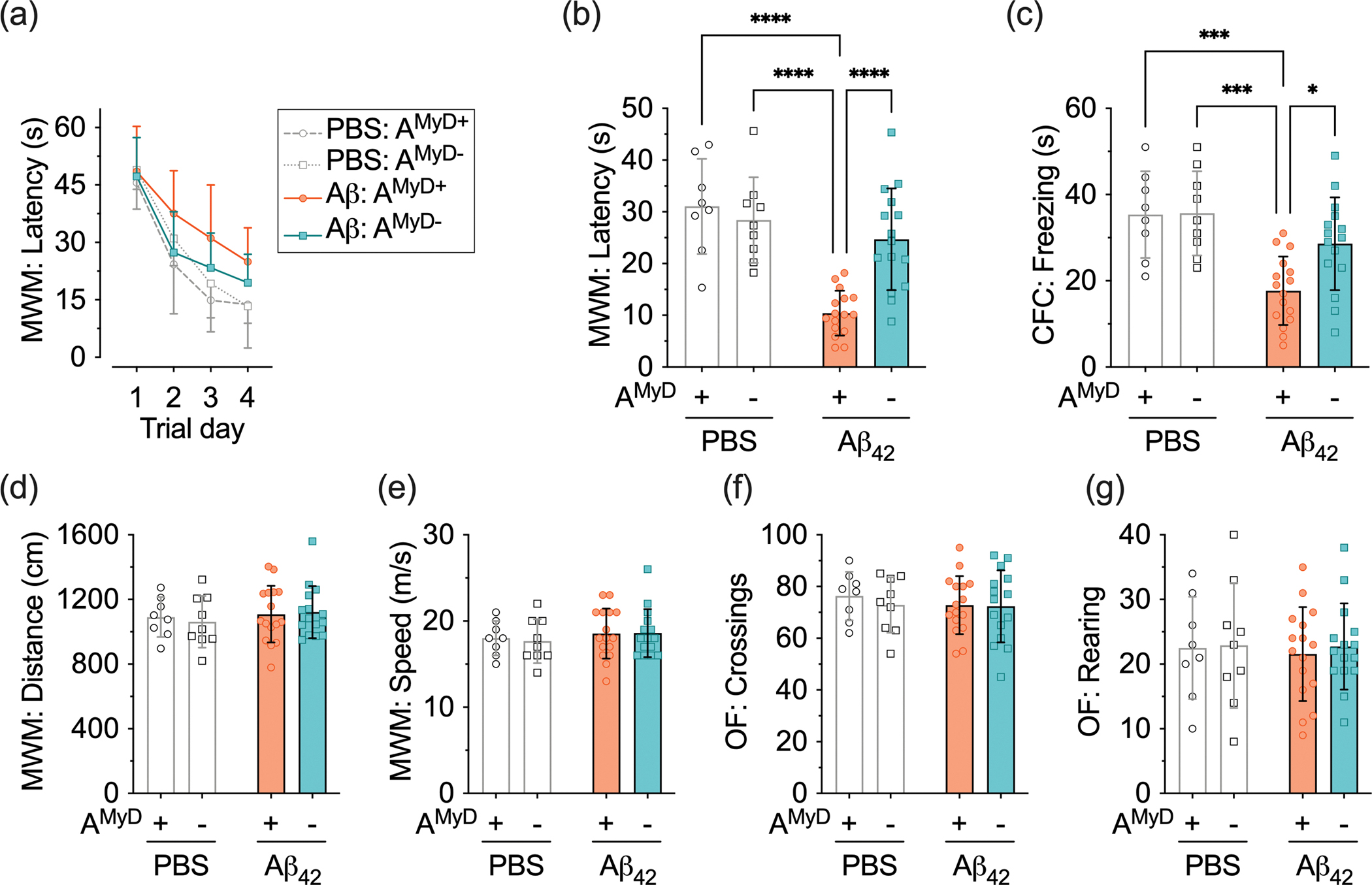
AMyD+ and AMyD− mice were treated with PBS or Aβ42. (a,b) Latencies (a) to find the submerged platform and (b) in the correct quadrant in the Morris water maze (MWM). (c) Freezing time in the contextual fear conditioning (CFC). (d) Distance traveled and (e) speed of swimming in the Morris water maze. (f,g) The number of (f) square crossings and (g) rearing behavior in the open field (OF). Data are presented as the mean ± SD. Statistical evaluation was performed using a two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Subsequently, we tested animals using contextual fear conditioning, which relies on the amygdala, hippocampus, frontal cortex, and cingulate cortex (Maren et al., 2013). Aβ42-treated AMyD+ mice showed lower freezing latency compared to PBS-treated AMyD+ mice (Figure 4c). Mice with deletion of MyD88 from astrocytes displayed comparable behavior regardless of the treatment with PBS or Aβ42. Finally, animals were tested in the open field to assess novel environment exploration, general motor activity, and anxiety-related behavior (Prut & Belzung, 2003). We found that the detrimental effects of Aβ42 on cognition were not related to motor impairment and anxiety-related behavior, since no alterations of the swimming distance (Figure 4d) and speed (Figure 4e) in the Morris water maze, or the total squares crossed (Figure 4f) and rearing behavior (Figure 4g) in the open field, were detected. Likewise, novel environment exploration, general motor activity, and anxiety-related behavior were not altered by the mouse genotype. These studies provide evidence that astrocytic MyD88 contributes to the cognitive deficits caused by Aβ.
3.4. MyD88 deletion in astrocytes inhibits Aβ-induced hippocampal synaptic toxicity
Another prominent AD characteristic is early synaptic loss, which correlates with cognitive impairment (Terry et al., 1991). We first performed western blots for SYP and PSD95 using mouse hemibrain lysates and found no changes in the steady-state levels of these synaptic proteins (Supplementary Figure 1e,f). We then focused on the hippocampal region because of its association with the cognitive tasks used in our study and since it is adjacent to the brain ventricle where Aβ42 was injected. We used confocal microscopy to evaluate the levels of NeuN, MAP2, SYP, and PSD95 and thereby explore any induced synaptic toxicity on a brain region-specific level (Figure 5a). Expression of the neuronal proteins NeuN (Figure 5b,c) and MAP2 (Figure 5d,e) was unchanged by mouse genotype and treatment, indicating no alterations in neuronal density and structure. In contrast, Aβ42-treated AMyD+ mice showed lower levels of presynaptic protein SYP compared to PBS-treated AMyD+ mice (Figure 5f,g). The impact of Aβ42 in mice with deletion of MyD88 from astrocytes was less pronounced. We also determined the levels of postsynaptic protein PSD95. Aβ42-treated AMyD+ mice displayed lower PSD95 levels compared to PBS-treated AMyD+ mice (Figure 5h,i). Moreover, PSD95 levels were higher in mice lacking astrocytic MyD88 compared to AMyD+ mice following Aβ42 treatment. These results indicate that astrocytic MyD88 is involved in the synaptic pathology induced by Aβ in the hippocampal region.
Figure 5 – Deletion of astrocytic Myd88 protects against Aβ42-induced synaptic toxicity.
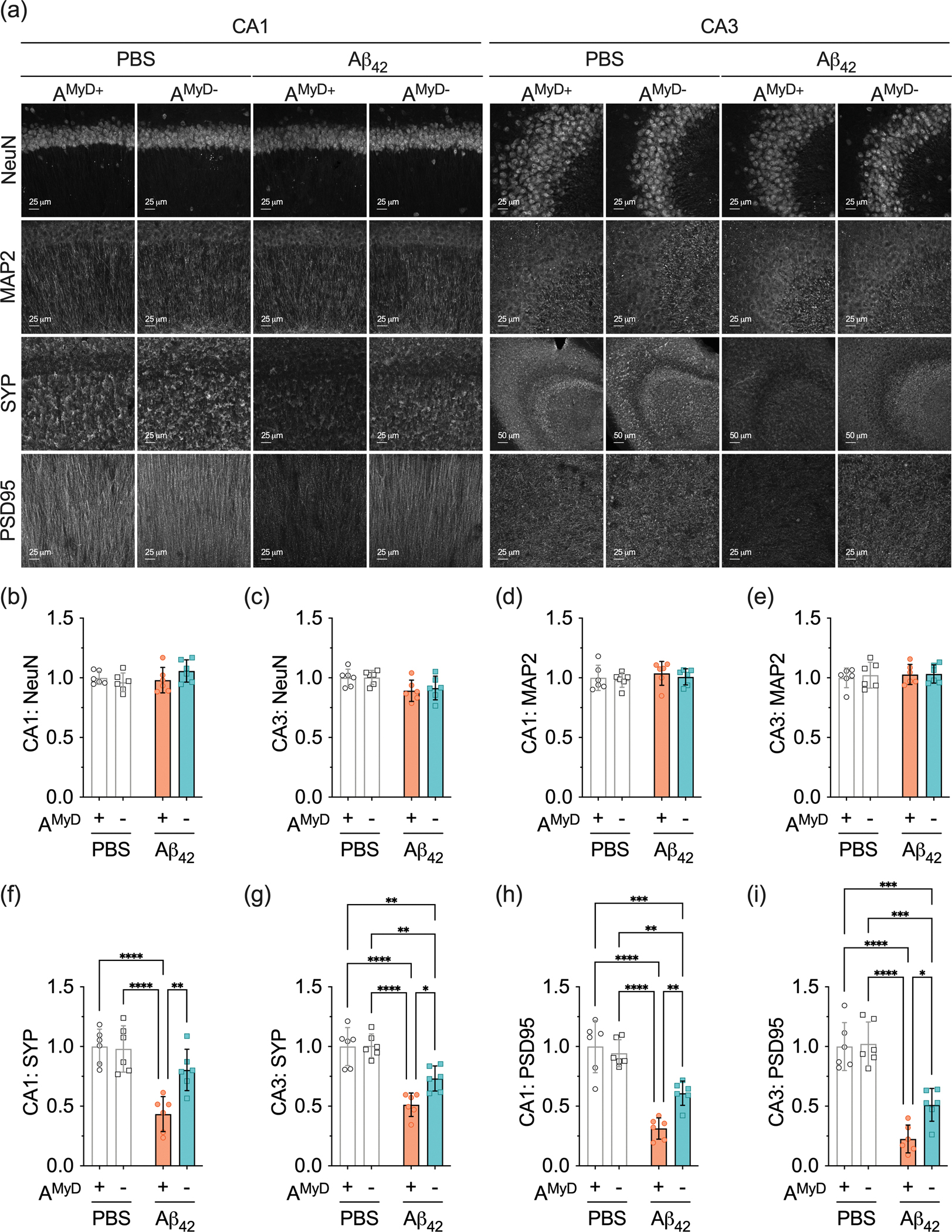
(a) Representative photomicrographs of NeuN, MAP2, SYP, and PSD95 immunoreactivity in the hippocampal CA1 and CA3 subregions of PBS- and Aβ42-treated AMyD+ and AMyD− mice. (b,c) NeuN levels in the hippocampal (b) CA1 and (c) CA3. (d,e) MAP2 levels in the hippocampal (d) CA1 and (e) CA3. (f,g) SYP levels in the hippocampal (f) CA1 and (g) CA3. (h,i) PSD95 levels in the hippocampal (h) CA1 and (i) CA3. Data are presented as the mean ± SD. Statistical evaluation was performed using a two-way ANOVA, followed by Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
3.5. Proteomic changes driven by the deletion of astrocytic MyD88
We performed unbiased proteomic analysis to define the molecular signature of the brains of mice expressing and lacking astrocytic MyD88. We identified 95 differentially expressed proteins from the 7,296 highly confident proteins detected in all mouse hemibrain samples (Supplementary Table 3). The small number of differentially expressed proteins supports our previous western blot studies that suggested that intracerebroventricular injection of Aβ42 results in modest overall changes in the mouse hemibrain. Nevertheless, unsupervised hierarchical clustering of differentially expressed proteins established four major protein groups (Supplementary Figure 1g). Proteins enriched in clusters one (36 proteins) and two (32 proteins) showed a distinction between PBS-treated AMyD+ and AMyD− mice, implying genotype-related variances in benign conditions. On the other hand, clusters three (18 proteins) and four (8 proteins) displayed a more significant difference between Aβ42-treated AMyD+ and AMyD− mice.
We analyzed all clusters independently in the ShinyGO database to determine the ontology enrichment. However, proteins in each cluster did not show significant enrichment for biological process, cellular component, and molecular function terms. We focused on clusters three and four to identify potential molecular markers associated with Aβ42 treatment. Individual proteins in these clusters were grouped based on functional categories defined by high-level ontology terms, such as the regulation of biological quality (8 proteins), regulation of localization (7 proteins), response to stress (5 proteins), regulation of signaling (5 proteins), and immune system process (3 proteins) (Supplementary Table 3). We identified the protein CD47, which has been implicated in protecting against excessive synaptic pruning during development, in multiple functional categories and performed a western blot targeting it to validate the proteomic data (Lehrman et al., 2018). Steady-state levels of CD47 were lower in Aβ42- versus PBS-treated AMyD+ mice, and deletion of MyD88 in astrocytes mitigated the effect of Aβ42 on CD47 expression (Supplementary Figure 1h,i).
Furthermore, we performed targeted pairwise expression analysis to identify proteomic signatures associated with mice genotypes and pharmacological treatments (Supplementary Table 4). Comparisons of PBS-treated AMyD+ and AMyD− mice revealed 119 differentially expressed proteins. AMyD+ mice had 64 proteins enriched while AMyD− mice had 55 proteins enriched (Figure 6a). Analysis of 55 proteins enriched in PBS-treated AMyD− mice did not show enrichment for ontology terms. In contrast, deletion of MyD88 from astrocytes resulted in lower levels of proteins associated with ion transport (FDR 8.4E-3), neuron projection (FDR 1.6E-2), synapse (FDR 1.6E-2), and potassium ion transmembrane transporter activity (FDR 4E-2). Reactome pathway analysis in the ShinyGO database showed the association of these proteins with the neuronal system (FDR 1.6E-2), cellular response to heat stress (FDR 1.6E-2), and voltage-gated potassium channels (FDR 2.4E-2) pathways (Figure 6b, Supplementary Table 4). Therefore, deletion of astrocytic MyD88 altered the basic brain proteomic signature, particularly of proteins related to neuronal function.
Figure 6 – Astrocytic MyD88 deletion alters the mouse brain proteome.
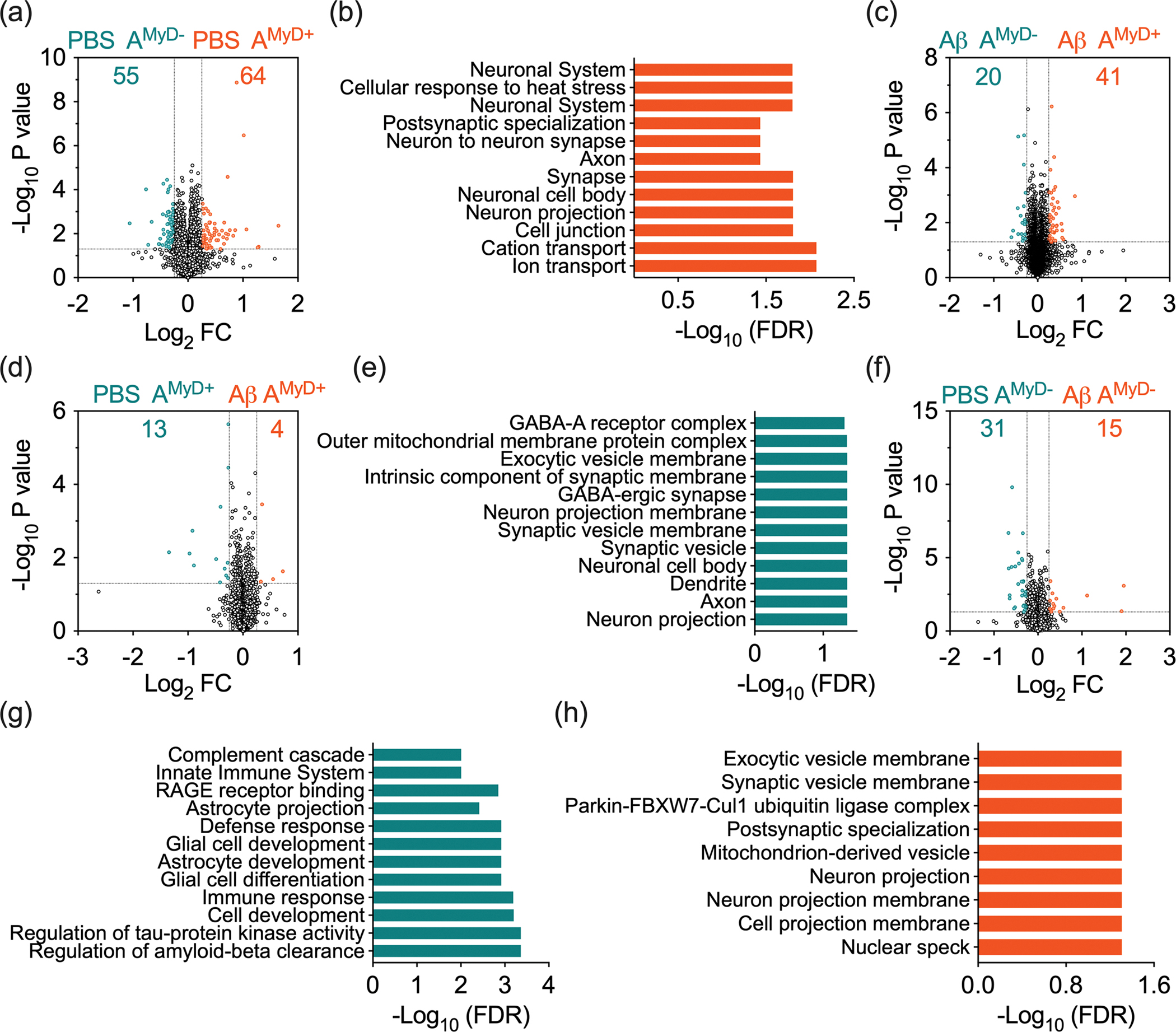
Volcano plot of targeted pairwise expression analysis between (a) PBS-treated AMyD+ and AMyD− mice, (c) Aβ42-treated AMyD+ and AMyD− mice, (d) PBS- and Aβ42-treated AMyD+ mice, and (f) PBS- and Aβ42-treated AMyD− mice. Enrichment of ontology terms in proteins expressed at higher levels on (b) PBS-treated AMyD+ mice versus PBS-treated AMyD− mice, (e) PBS-treated versus Aβ42-treated AMyD+ mice, (g) PBS-treated versus Aβ42-treated AMyD− mice, and (h) Aβ42-treated versus PBS-treated AMyD− mice.
Next, we evaluated the impact of mouse genotype on the response to Aβ. Comparisons of Aβ42-treated AMyD+ and AMyD− mice, revealed 61 differentially expressed proteins (Figure 6c). Animals with deletion of astrocytic MyD88 had reduced levels of 41 proteins. Contrariwise, levels of 20 proteins were higher in AMyD− mice. Analysis of the differentially expressed proteins in the ShinyGO database did not show significant ontology enrichment.
We were particularly interested in comparing PBS- and Aβ42-treated AMyD+ mice to identify potential pathways related to the changes in cognitive function. We identified 17 differentially expressed proteins, with the levels of 13 proteins lower and four proteins higher in mice treated with Aβ42 (Figure 6d). Proteins lower in Aβ42-treated mice were associated with ontology terms, including neuron projection (FDR 4.5E-2), axon (FDR 4.5E-2), and synaptic vesicles (FDR 4.5E-2) (Figure 6e, Supplementary Table 4). Additionally, investigation of the four proteins with higher levels in Aβ42 in AMyD+ mice did not result in significant ontology enrichment. This dataset corroborates that Aβ42 treatment to mice expressing astrocytic MyD88 results in loss of neuronal- and synaptic-related proteins.
Finally, we evaluated the proteomic signature of AMyD− mice in response to PBS and Aβ42 treatment. Pairwise expression analysis showed 46 differentially expressed proteins. PBS-treated mice had enrichment of 31 proteins, while Aβ42-treated mice showed enrichment of 15 proteins (Figure 6f). Ontology analysis indicated that proteins enriched in PBS-treated AMyD− mice were associated with regulation of Aβ clearance (FDR 4.3E-4), cell differentiation (FDR 4.3E-4), glial cell development (FDR 1.2E-3), cell development (FDR 6.2E-4), and immune response process (FDR 8.7E-4), among other biological processes. At the cellular level, these proteins were linked to cell projection (FDR 5.8E-5), neuron projection (FDR 3.9E-4), dendritic tree (FDR 4.4E-3), among other structures where they regulate distinct molecular functions, including glycosaminoglycan binding (FDR 4.5E-5), receptor for advanced glycation end products (RAGE) receptor binding (FDR 1.4E-3), and signaling receptor binding (FDR 2.1E-3). Signaling pathways enriched in the Reactome analysis included the immune system (FDR 2.4E-2), extracellular matrix organization (FDR 9.9E-3) and complement cascade (FDR 9.9E-3) (Figure 6g, Supplementary Table 4). GFAP, complement component 4b (C4b), and ApoE were among the proteins enriched in PBS-treated AMyD− mice. Moreover, ontology terms associated with cellular components were significantly enriched in AMyD− mice treated with Aβ42, such as neuron projection (FDR 4.9E-2) and postsynaptic specialization (FDR 4.9E-2) (Figure 6h, Supplementary Table 4). These data suggest that deletion of MyD88 alters astrocytes reactivity and reduces the overall levels of immune-related proteins following Aβ42 treatment favoring the preservation of neuronal integrity.
3.6. Astrocytic MyD88 deletion reduces astrocytes reactivity to Aβ
Finally, we used confocal microscopy to validate that the loss of astrocytic MyD88 is associated with the alteration of astrocytes reactivity to Aβ in the hippocampus. The intensity of S100b+-astrocytes was higher in Aβ42-treated compared to PBS-treated AMyD+ mice. AMyD− mice treated with PBS and Aβ42 showed comparable S100b levels in the CA1, but not CA3 hippocampal subregions (Figure 7a–c). GFAP levels were also higher in Aβ42-treated compared to PBS-treated AMyD+ mice. However, MyD88 deletion from astrocytes inhibited the impact of Aβ42 in the CA1 and CA3 hippocampal subregions (Figure 7a,d,e). We also measured the expression of ApoE and complement C3 in GFAP+-astrocytes in the CA1 hippocampal subregion. GFAP co-localization with ApoE (Figure 7f,g) and complement C3 (Figure 7h,i) increased in AMyD+ mice following Aβ42 treatment, and these processes were less evident in mice with deletion of astrocytic MyD88. Our data support the idea that MyD88 is involved in the response of hippocampal astrocytes to Aβ.
Figure 7 – Astrocytic MyD88 deletion reduces astrocyte reactivity to Aβ42 in the hippocampus.
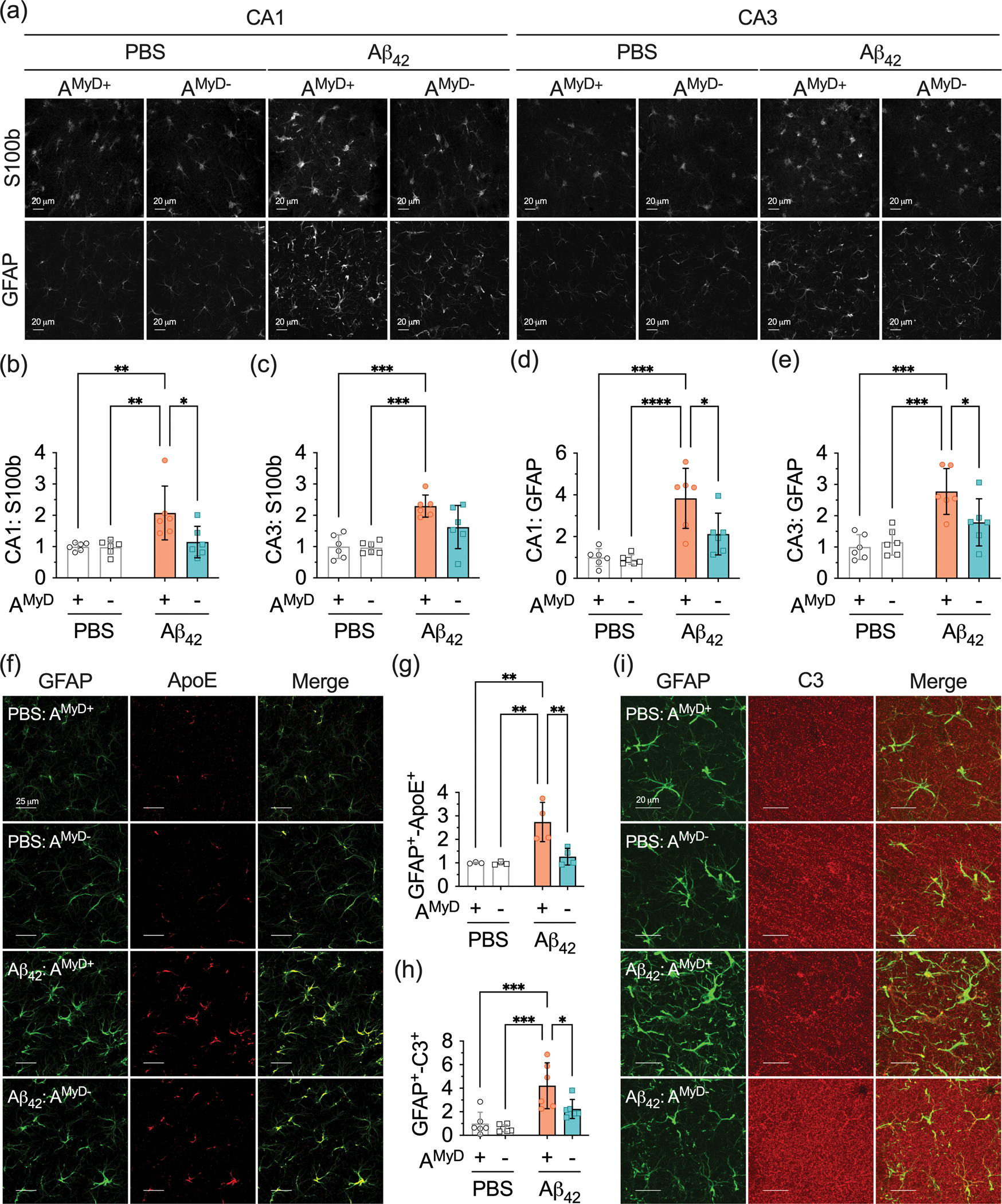
(a) Representative photomicrographs of S100b and GFAP immunostaining in the hippocampal CA1 and CA3 subregions of PBS and Aβ42-treated AMyD+ and AMyD− mice. (b,c) Quantification of S100b immunoreactivity in (b) CA1 and (c) CA3 subregions of the hippocampus. (d,e) Quantification of GFAP immunoreactivity in the (d) CA1 and (e) CA3 subregions of the hippocampus. (f) Representative photomicrographs of GFAP (green) and ApoE (red) immunoreactivity in the hippocampal CA1 subregion. (g,h) Quantification of colocalization between (g) GFAP and ApoE, and (h) GFAP and complement C3 in the CA1 subregion of the hippocampus. (i) Representative photomicrographs of GFAP (green) and complement C3 (red) immunoreactivity in the hippocampal CA1 subregion. Orthogonal analysis confirming colocalization of GFAP and complement C3. Data are presented as mean ± SD. Statistical evaluation was performed using a two-way ANOVA, followed by Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
4. Discussion
In this study, we observed an upregulation of MyD88 protein in astrocytes in AD brains, predominantly near amyloid plaques and NFTs. Functional studies in mice demonstrated that deletion of astrocytic MyD88 reduced the cognitive impairment and hippocampal synaptic toxicity induced by Aβ42. Whole proteomic analyses of mouse hemibrains revealed that deletion of astrocytic MyD88 altered the brain proteomic signature without causing apparent structural and behavioral changes in control mice. Our analyses also demonstrated that Aβ42-treated AMyD+ mice had lower neuronal-related proteins levels than their PBS-treated counterparts. Notably, Aβ42 treatment in AMyD− mice downregulated proteins linked to the immune system, extracellular matrix organization, and complement cascade. Concomitantly, it upregulated proteins associated with postsynaptic specialization and neuron projection. Finally, we discovered that deletion of astrocytic MyD88 lowered GFAP, S100b, ApoE and complement C3 protein levels in hippocampal astrocytes in response to Aβ42. Altogether, we identified astrocytic MyD88 as a molecular marker of AD, in addition to demonstrating its role in facilitating Aβ42-induced synaptotoxicity and cognitive impairment.
Astrogliosis in AD is not only prominent in later disease stages but is also present at earlier stages, with changes in plasma GFAP emerging as an early and specific marker of Aβ accumulation, even in cognitively normal individuals with normal Aβ status (Carter et al., 2019; Pereira et al., 2021). Supporting this view, a study using a non-mutant human Aβ knock-in mouse model indicated that GFAP+-astrocytes, but not Iba1+-microglia, surround initial amyloid OC+-protofibrils clusters (Baglietto-Vargas et al., 2021), reinforcing astrocytes as one of the promptest cell-types to respond to Aβ. Astrocytes are immune competent cells, and their function is guided by the collective signals from various immunoreceptors, whose expression and activity depend on the developmental stages of the cell and its environmental context (Liddelow & Barres, 2017; Medeiros & LaFerla, 2013). Accordingly, the molecular signature and role of these cells are variable in AD, and subpopulation of cells displays characteristics of chronically activated pro-inflammatory cells (Habib et al., 2020; Sadick et al., 2022). The diversity of astrocytes implies that complete blockage of their function is not feasible as a therapeutic strategy, as demonstrated by studies where ablation of astrocyte reactivity in mouse models of AD increased production of monomeric Aβ and reduced degradation of Aβ, heightened expression of pro-inflammatory cytokines, decreased expression of synaptic markers, and exacerbated cognitive deficits (Davis et al., 2021; Katsouri et al., 2020; Kraft et al., 2013). However, reactive astrocytes also facilitate Aβ accumulation, tau pathology, production of pro-inflammatory mediators, and cognitive decline (Jackson et al., 2021; Kitazawa et al., 2011a; Lian et al., 2015a; Mann et al., 2022; Sompol et al., 2017; Wang et al., 2021). Mechanistically, it has been proposed that activation of the transcriptional factors STAT3 and NF-κB is, at least in part, related to their protective and detrimental phenotypes, respectively (Anderson et al., 2016; Liddelow et al., 2017). Notably, MyD88 is a major cellular signal upstream to NF-κB (Deguine & Barton, 2014). Hence, astrocytes could be not only a promising biomarker but also a therapeutic target even before the clinical symptoms of AD emerge. However, because astrocytes are broadly involved in protective and harmful mechanisms in AD their activity must be carefully balanced instead of fully inhibited. To translate their potential into the clinic, we therefore need to identify unique cellular mediators with prominent biological activities to certify their role as a meaningful component in the pathogenesis of disease.
Our human studies provided strong evidence that MyD88 can participate in the neurotoxic response of astrocytes to AD. Bulk and cell-specific transcriptomic data implied a positive correlation between MyD88 and astrocytes markers in two independent human cohorts. Importantly, we validated the relationship between MyD88 and astrocytes at the protein level in a third human cohort that showed the enrichment of astrocytic MyD88 near amyloid plaques and NFTs. To investigate the potential role of astrocytic MyD88 in AD, we used a murine model of acute Aβ toxicity. Although this model does not display accumulation of amyloid plaques like animals expressing humanized amyloid precursor protein, it exhibits critical characteristics of AD such as loss of synaptic markers, inflammation, and cognitive decline (Balducci & Forloni, 2014; Batista et al., 2021; Canet et al., 2020; Oliveira et al., 2021; Südkamp et al., 2021; Wu et al., 2018; Xiang et al., 2022). Thus, it allowed a practical assessment of the relationship between astrocytic MyD88 and Aβ while circumventing additional complex animal engineering. Overall, our study established that the astrocytic response to Aβ42 involves MyD88 and that this signaling pathway participates in the processes that culminate in synaptic degeneration and cognitive impairment. We attempted to determine associated cognitive mechanisms through unbiased proteomics, which provided potentially interesting but somewhat inconclusive data due to the small number of differentially expressed proteins across the different groups. For instance, we only identified 30 differentially expressed proteins in the hemibrains of PBS- and Aβ42-treated mice expressing astrocytic MyD88. Some of the proteins downregulated by Aβ42 were linked with neuronal-related ontology terms, such as neuron projection, dendritic tree, and synaptic vesicles. While this can partially explain the cognitive impairment caused by Aβ42 in mice expressing astrocytic Myd88, it could not confirm the pathological driving mechanisms. Similarly, our immunostaining data identified localized changes in the synaptic proteins SYP and PSD95 in the hippocampal region, which may also be an underlying factor in the observed weakening in the cognitive abilities caused by Aβ42.
The limitations in our proteomic studies might have occurred because the acute effects of Aβ42 depend more on localized changes in the hippocampus and less on whole-brain proteomic changes. Furthermore, these changes could result from altered protein function instead of the overall expression. Finally, the underlying molecular processes triggering synaptic toxicity and cognitive impairment could be more prominent at earlier stages following Aβ42 administration. Supporting the latter, we showed previously that intracerebroventricular injection of Aβ40 causes the activation of protein kinases and NF-κB, and the expression of inflammatory proteins tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) within a few hours of treatment. Moreover, these changes trigger synaptic and cognitive deficits but subside earlier than their appearance in the mouse brain (Medeiros et al., 2010; Medeiros et al., 2007). More specifically to the temporal response of astrocytes to toxic stimuli, Hasel and colleagues revealed through transcriptomic analysis of purified cells that astrocytes undergo a rapid pro-inflammatory transition within 24 h post-systemic lipopolysaccharide injection in mice (Hasel et al., 2021). Hence, it is plausible that Aβ42 stimulates the signaling of astrocytic MyD88 at an earlier stage, which then drives the brain’s molecular changes that culminate in the loss of synapses and subsequent cognitive impairment. We propose that microdissection of regions of interest instead of bulk hemibrain tissue and collection of tissues at intermediated time points post Aβ42 injection will likely deepen molecular insights in future proteomic studies involving this model.
Regarding the molecular function of MyD88 in astrocytes, we found that the genetic deletion of MyD88 from astrocytes altered the basal proteomic signature of the brain without causing apparent changes in hippocampal neuronal density and structure (i.e., NeuN and MAP2), as well as in learning and memory functions. The use of omics approaches in mouse studies has only recently emerged, and thus, limited information regarding the inadvertent impact of genetic manipulation in the mouse background is available. Changes caused by MyD88 genetic manipulation could be attributed to still poorly understood genetic compensation mechanisms, including variants of MyD88 signaling by other intracellular adapters such as TIR-domain-containing adapter-inducing interferon-β (TRIF), Toll/interleukin-1 receptor domain-containing adapter protein (TIRAP), and translocating chain-associated membrane protein (TRAM) (El-Brolosy & Stainier, 2017; Schroeder et al., 2021). Another explanation is alterations occurring during the development of the central nervous system. Because GFAP is expressed by neural progenitor cells (NPCs), nascent neurons, and type 1 neural stem cells, GFAP-Cre mouse lines might display changes in gene expression in neurons (Guttenplan & Liddelow, 2019). However, in our model, while astrocytes were negative to MyD88, the overall MyD88 immunodetection was not altered, suggesting marginal off-target effects, and supporting astrocytes as the primary target regulated by the GFAP promoter (Supplementary figure 1d). Moreover, mice lacking MyD88 are viable, and they did not show alteration in learning and memory behavior compared to wild-type mice in AD-related studies (Michaud et al., 2011; Quan et al., 2021; Weitz et al., 2014). However, Schroeder and colleagues recently performed comprehensive behavioral and brain morphological investigations in mice lacking MyD88 where they found that MyD88-deficient mice displayed increased density of cortical neurons, microglia, and proliferating cell numbers compared to wild-type mice (Schroeder et al., 2021). Of note, we investigated whether Iba1+-microglia in the hippocampus were affected by astrocytic MyD88 deletion and Aβ42 treatment, and we found no differences among all groups (data not shown). Besides brain morphologic changes, Myd88-deficient mice showed decreased locomotor activity and increased anxiety-like behavior (Schroeder et al., 2021). In comparison, we did not observe equivalent behavioral changes in our animals. Importantly, Schroeder and colleagues have also shown that MyD88-deficient mice displayed typical day/light activity and learned slowly but exhibited satisfactory short- and long-term spatial memory abilities (Schroeder et al., 2021). Hence, it is possible that MyD88 participates in developing the brain structure and that activation of astrocytic MyD88 could have a lesser role in this process. Although noteworthy, putative developmental changes associated with MyD88 expression are out of the scope of our study, particularly since they did not seem to affect cognitive functions more closely related to AD. However, it is essential to emphasize that the assessment of our and other studies applying complete and cell-specific constitutive knockdown strategies should contemplate these weaknesses carefully. To mitigate such limitations, we propose to study AMyD+ and AMyD− mice as independent cohorts due to their distinctive proteomic background.
The proteomic differences between Aβ42-treated AMyD+ and AMyD− mice underscored the intricacy caused by the distinct genotypic background. Although we identified 61 differentially expressed proteins, these proteins did not show ontology enrichment, reinforcing that AMyD+ and AMyD− should be analyzed separately. In this regard, mice lacking astrocytic MyD88 showed exciting proteomic changes in response to Aβ42. Proteins upregulated in Aβ42-treated AMyD− mice compared to their PBS-treated counterparts were ontologically associated with neuron projection and postsynaptic specialization. Another fascinating finding was that Aβ42-treated AMyD− mice displayed lower steady-state levels of proteins associated with the immune system and complement cascade, including GFAP, ApoE, and complement C4b. Notably, ApoE and complement proteins are molecular markers of the neurotoxic phenotype of astrocytes (Guttenplan et al., 2021; Hasel et al., 2021; Liddelow et al., 2017). In addition, mice lacking astrocytic MyD88 showed comparable ApoE and complement C3 colocalization with GFAP+-astrocytes following PBS and Aβ42 treatment in the hippocampus. In contrast, mice expressing astrocytic MyD88 displayed higher levels of these proteins in response to Aβ42 compared to PBS.
Therefore, the distinct molecular background triggered by the presence or absence of astrocytic MyD88 drives substantial distinction in how the brain reacts to Aβ42. In response to Aβ, the activation of astrocytic MyD88 resulted in lower proteins related to neuron projection, dendritic tree, and synaptic vesicles, and it caused impairment of learning and memory. In contrast, the lack of MyD88 seemed to have hampered the neurotoxic activation of astrocytes and favored the response of this cell type with a phenotype that supports the preservation of synaptic and cognitive functions. Remarkably, a study conditionally activating astrocytic NF-κB downstream of MyD88 showed similar findings (Lian et al., 2015b). Chronic activation of NF-κB in astrocytes prompted the release of complement C3, which induced synaptic dysfunction and cognitive impairment in AD mice. Likewise, studies in MyD88 knockout mice have shown that animals could not build a proper pro-inflammatory response to different brain pathological stimuli (Babcock et al., 2008; Kielian et al., 2007; Wang et al., 2009). While further validation will be required, ours and these other studies suggest that Aβ triggers the activation of astrocytic receptors, such as TLRs and interleukin receptors, that rely on MyD88 to signal intracellularly. Aβ-induced MyD88 signaling next prompts the stimulation of transcriptional factor NF-κB and AP-1, which regulate the production of immune signals and activation of the complement cascade that finally facilitate the loss of synaptic integrity and impairment of cognitive abilities.
Our results provide new biological insights into the role of astrocytic MyD88 in AD. We demonstrated that MyD88 is associated with hypertrophic astrocytes nearby amyloid plaques and NFTs. Although we did not fully identify the underlying pathological mechanisms, we found that deletion of astrocytic MyD88 mitigates the pathological impact of Aβ42, favoring the preservation of synaptic structure and cognitive function.
Supplementary Material
Supplementary Figure 1 – (a) Representative photomicrographs of GFAP (green), MyD88 (red), and merged immunostaining in AMyD+ and AMyD− mice analyzed in the hippocampal CA1 subregion. (b) Orthogonal analysis of GFAP (green) and MyD88 (red) immunostaining in AMyD+ and AMyD− mice. MyD88 was not detected in AMyD− mice. (c) Volumetric quantification of MyD88 and GFAP+-astrocytes colocalization in AMyD+ and AMyD− mice. (d) Overall MyD88 immunostaining minus the levels of colocalized MyD88+-GFAP+. Data were analyzed through an unpaired Student’s t-test, with a confidence level of 95%. (e,f) Representative western blot images and quantification of (e) SYP and (f) PSD95 in PBS- and Aβ42-treated AMyD+ and AMyD− mice. Samples were normalized to actin. (g) Unsupervised hierarchical clustering of statistically significant differentially expressed proteins. (h) Quantification of normalized CD47 protein expression determined by western blot. (i) Western blot of CD47 and actin protein expression. Data are presented as mean ± SD. Statistical evaluation was performed using a two-way ANOVA, followed by Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01.
Supplementary Figure 3 – Representative photomicrographs of MyD88 immunostaining in HPC (upper panels), TCX (middle panels), and CBL (lower panels) non-AD and AD brain sections. Neuronal-, vascular-, and glial-like cells are designated by black arrows.
Supplementary Figure 2 – Representative photomicrograph of immunofluorescence staining of GFAP (green), tau (red), and Aβ (magenta) in non-AD and AD human TCX (top two panels) and CBL (lower two sections).
Main Points:
MyD88 is upregulated in astrocytes in Alzheimer’s disease.
Deletion of MyD88 in astrocytes protects against β-amyloid-induced synaptic toxicity and cognitive impairment in mice.
Acknowledgments:
The Australian National Health and Medical Research Council [GNT1128436, GNT1129192, GNT1139469 (RM)], the Clem Jones Centre for Ageing Dementia Research (RM), and the National Institutes of Health [R21AG069081 (FML)] funded this study. We thank the Alzheimer’s Disease Research Center of the University of California, Irvine (UCI ADRC) for the access to human brain samples. The National Institutes of Health (1P30AG066519-01) funds the UCI ADRC. We thank Drs. Karthik Kamath, Yunqi Wu, and Matthew McKay from the Australian Proteome Analysis Facility at Macquarie University for the assistance in the proteomic studies. The Australian Government’s National Collaborative Research Infrastructure Strategy (NCRIS) funds the Australian Proteome Analysis Facility. The results published here are in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). We thank Dr. Nilüfer Ertekin-Taner and all other Mayo Clinic RNAseq study investigators for access to human data in the AD Knowledge Portal (ID: syn5550404). We thank Dr. Li-Huei Tsai’s team for the access to human data in their single-cell transcriptomic analysis of Alzheimer’s disease study. Abstract image was generated with Biorender.
Funding information:
Australian National Health and Medical Research Council, Grant/Award Number: GNT1128436, GNT1129192, GNT1139469. The Clem Jones Centre for Ageing Dementia Research. National Institutes of Health, Grant/Award Number: R21AG069081.
Footnotes
Conflict of Interests: The authors declare no conflicts of interest.
References
- Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, Burgess JD, Chai HS, Crook J, Eddy JA, Li H, Logsdon B, Peters MA, Dang KK, Wang X, Serie D, Wang C, Nguyen T, Lincoln S, Malphrus K, Bisceglio G, Li M, Golde TE, Mangravite LM, Asmann Y, Price ND, Petersen RC, Graff-Radford NR, Dickson DW, Younkin SG, & Ertekin-Taner N (2016, Oct 11). Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data, 3, 160089. 10.1038/sdata.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, & Sofroniew MV (2016, Apr 14). Astrocyte scar formation aids central nervous system axon regeneration. Nature, 532(7598), 195–200. 10.1038/nature17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, & Herculano-Houzel S (2009, Apr 10). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol, 513(5), 532–541. 10.1002/cne.21974 [DOI] [PubMed] [Google Scholar]
- Babcock AA, Toft-Hansen H, & Owens T (2008, Nov 1). Signaling through MyD88 regulates leukocyte recruitment after brain injury. J Immunol, 181(9), 6481–6490. 10.4049/jimmunol.181.9.6481 [DOI] [PubMed] [Google Scholar]
- Baglietto-Vargas D, Forner S, Cai L, Martini AC, Trujillo-Estrada L, Swarup V, Nguyen MMT, Do Huynh K, Javonillo DI, Tran KM, Phan J, Jiang S, Kramár EA, Nuñez-Diaz C, Balderrama-Gutierrez G, Garcia F, Childs J, Rodriguez-Ortiz CJ, Garcia-Leon JA, Kitazawa M, Shahnawaz M, Matheos DP, Ma X, Da Cunha C, Walls KC, Ager RR, Soto C, Gutierrez A, Moreno-Gonzalez I, Mortazavi A, Tenner AJ, MacGregor GR, Wood M, Green KN, & LaFerla FM (2021, Apr 23). Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat Commun, 12(1), 2421. 10.1038/s41467-021-22624-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C, & Forloni G (2014). In vivo application of beta amyloid oligomers: a simple tool to evaluate mechanisms of action and new therapeutic approaches. Curr Pharm Des, 20(15), 2491–2505. 10.2174/13816128113199990497 [DOI] [PubMed] [Google Scholar]
- Batista AF, Rody T, Forny-Germano L, Cerdeiro S, Bellio M, Ferreira ST, Munoz DP, & De Felice FG (2021, Feb 21). Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J Neuroinflammation, 18(1), 54. 10.1186/s12974-021-02099-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, & Del Tredici K (2015, Oct). The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain, 138(Pt 10), 2814–2833. 10.1093/brain/awv236 [DOI] [PubMed] [Google Scholar]
- Canet G, Pineau F, Zussy C, Hernandez C, Hunt H, Chevallier N, Perrier V, Torrent J, Belanoff JK, Meijer OC, Desrumaux C, & Givalois L (2020, Jan). Glucocorticoid receptors signaling impairment potentiates amyloid-β oligomers-induced pathology in an acute model of Alzheimer’s disease. Faseb j, 34(1), 1150–1168. 10.1096/fj.201900723RRR [DOI] [PubMed] [Google Scholar]
- Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, & Zimmer ER (2019, Feb). Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol Med, 25(2), 77–95. 10.1016/j.molmed.2018.11.006 [DOI] [PubMed] [Google Scholar]
- Davis N, Mota BC, Stead L, Palmer EOC, Lombardero L, Rodriguez-Puertas R, de Paola V, Barnes SJ, & Sastre M (2021, Mar 17). Pharmacological ablation of astrocytes reduces Abeta degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J Neuroinflammation, 18(1), 73. 10.1186/s12974-021-02117-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguine J, & Barton GM (2014). MyD88: a central player in innate immune signaling. F1000Prime Rep, 6, 97. 10.12703/p6-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn HC, Ager RR, Baglietto-Vargas D, Cheng D, Kitazawa M, Cribbs DH, & Medeiros R (2015). Restoration of lipoxin A4 signaling reduces Alzheimer’s disease-like pathology in the 3xTg-AD mouse model. J Alzheimers Dis, 43(3), 893–903. 10.3233/JAD-141335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, & Stainier DYR (2017, Jul). Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet, 13(7), e1006780. 10.1371/journal.pgen.1006780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Jung D, & Yao R (2020, Apr 15). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics, 36(8), 2628–2629. 10.1093/bioinformatics/btz931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, & O’Banion MK (2013, Mar 13). Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci, 33(11), 5053–5064. 10.1523/jneurosci.4361-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, & Liddelow SA (2019, Jan 7). Astrocytes and microglia: Models and tools. J Exp Med, 216(1), 71–83. 10.1084/jem.20180200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, Münch AE, Blum JA, Fine J, Neal MC, Bruce KD, Gitler AD, Chopra G, Liddelow SA, & Barres BA (2021, Nov). Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature, 599(7883), 102–107. 10.1038/s41586-021-03960-y [DOI] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, & Schwartz M (2020, Jun). Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci, 23(6), 701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, & Golenbock DT (2008, Aug). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol, 9(8), 857–865. 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasel P, Rose IVL, Sadick JS, Kim RD, & Liddelow SA (2021, Oct). Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci, 24(10), 1475–1487. 10.1038/s41593-021-00905-6 [DOI] [PubMed] [Google Scholar]
- Hu M, Zhu D, Zhang J, Gao F, Hashem J, Kingsley P, Marnett LJ, Mackie K, & Chen C (2022, Mar 29). Enhancing endocannabinoid signalling in astrocytes promotes recovery from traumatic brain injury. Brain, 145(1), 179–193. 10.1093/brain/awab310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Meltzer JC, Nguyen H, Commins C, Bennett RE, Hudry E, & Hyman BT (2021, Dec 27). APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain. 10.1093/brain/awab478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiwaji Z, Tiwari SS, Aviles-Reyes RX, Hooley M, Hampton D, Torvell M, Johnson DA, McQueen J, Baxter P, Sabari-Sankar K, Qiu J, He X, Fowler J, Febery J, Gregory J, Rose J, Tulloch J, Loan J, Story D, McDade K, Smith AM, Greer P, Ball M, Kind PC, Matthews PM, Smith C, Dando O, Spires-Jones TL, Johnson JA, Chandran S, & Hardingham GE (2022, Jan 10). Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Ass pathology. Nat Commun, 13(1), 135. 10.1038/s41467-021-27702-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouri L, Birch AM, Renziehausen AWJ, Zach C, Aman Y, Steeds H, Bonsu A, Palmer EOC, Mirzaei N, Ries M, & Sastre M (2020, May). Ablation of reactive astrocytes exacerbates disease pathology in a model of Alzheimer’s disease. Glia, 68(5), 1017–1030. 10.1002/glia.23759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, & Johnson J (2007, Apr 1). MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol, 178(7), 4528–4537. 10.4049/jimmunol.178.7.4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev VY, Andrews TS, & Hemberg M (2019, May). Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet, 20(5), 273–282. 10.1038/s41576-018-0088-9 [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, & LaFerla FM (2011a, Dec 15). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol, 187(12), 6539–6549. 10.4049/jimmunol.1100620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, & LaFerla FM (2011b, Dec 15). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol, 187(12), 6539–6549. 10.4049/jimmunol.1100620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR, Holtzman DM, Kim J, Pekny M, & Lee JM (2013, Jan). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J, 27(1), 187–198. 10.1096/fj.12-208660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, & Klein WL (1998, May 26). Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A, 95(11), 6448–6453. 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, & Stevens B (2018, Oct 10). CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron, 100(1), 120–134.e126. 10.1016/j.neuron.2018.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, & Haass C (2020, Dec 9). Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron, 108(5), 801–821. 10.1016/j.neuron.2020.09.029 [DOI] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, & Zheng H (2015a, Jan 7). NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron, 85(1), 101–115. 10.1016/j.neuron.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, & Zheng H (2015b, Jan 7). NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron, 85(1), 101–115. 10.1016/j.neuron.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, & Barres BA (2017, Jun 20). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity, 46(6), 957–967. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, & Barres BA (2017, Jan 26). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnerbauer M, Wheeler MA, & Quintana FJ (2020, Nov 25). Astrocyte Crosstalk in CNS Inflammation. Neuron, 108(4), 608–622. 10.1016/j.neuron.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rübe CE, Walter J, Heneka MT, Hartmann T, Menger MD, & Fassbender K (2012, Feb 1). TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol, 188(3), 1098–1107. 10.4049/jimmunol.1101121 [DOI] [PubMed] [Google Scholar]
- Mann CN, Devi SS, Kersting CT, Bleem AV, Karch CM, Holtzman DM, & Gallardo G (2022, Feb 16). Astrocytic alpha2-Na(+)/K(+) ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model. Sci Transl Med, 14(632), eabm4107. 10.1126/scitranslmed.abm4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Phan KL, & Liberzon I (2013, Jun). The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci, 14(6), 417–428. 10.1038/nrn3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, & Tsai LH (2019, Jun). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570(7761), 332–337. 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, Kiss MG, Christie KA, Vinegoni C, Poller WC, Mindur JE, Chan CT, He S, Janssen H, Wong LP, Downey J, Singh S, Anzai A, Kahles F, Jorfi M, Feruglio PF, Sadreyev RI, Weissleder R, Kleinstiver BP, Nahrendorf M, Tanzi RE, & Swirski FK (2021, Jul). Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature, 595(7869), 701–706. 10.1038/s41586-021-03734-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, Figueiredo CP, Pandolfo P, Duarte FS, Prediger RD, Passos GF, & Calixto JB (2010, May 1). The role of TNF-alpha signaling pathway on COX-2 upregulation and cognitive decline induced by beta-amyloid peptide. Behav Brain Res, 209(1), 165–173. 10.1016/j.bbr.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Medeiros R, Kitazawa M, Passos GF, Baglietto-Vargas D, Cheng D, Cribbs DH, & LaFerla FM (2013, May). Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol, 182(5), 1780–1789. 10.1016/j.ajpath.2013.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, & LaFerla FM (2013, Jan). Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol, 239, 133–138. 10.1016/j.expneurol.2012.10.007 [DOI] [PubMed] [Google Scholar]
- Medeiros R, Prediger RD, Passos GF, Pandolfo P, Duarte FS, Franco JL, Dafre AL, Di Giunta G, Figueiredo CP, Takahashi RN, Campos MM, & Calixto JB (2007, May 16). Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci, 27(20), 5394–5404. 10.1523/jneurosci.5047-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Richard KL, & Rivest S (2011, Jan 14). MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer’s disease. Mol Neurodegener, 6(1), 5. 10.1186/1750-1326-6-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, & Medeiros R (2018, Sep 24). Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation, 15(1), 276. 10.1186/s12974-018-1313-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MM, Lourenco MV, Longo F, Kasica NP, Yang W, Ureta G, Ferreira DDP, Mendonça PHJ, Bernales S, Ma T, De Felice FG, Klann E, & Ferreira ST (2021, Feb 2). Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of Alzheimer’s disease. Sci Signal, 14(668). 10.1126/scisignal.abc5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Stark AK, & Pakkenberg B (2008, Nov). Neocortical glial cell numbers in human brains. Neurobiol Aging, 29(11), 1754–1762. 10.1016/j.neurobiolaging.2007.04.013 [DOI] [PubMed] [Google Scholar]
- Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, Zetterberg H, Stomrud E, Ashton NJ, Blennow K, & Hansson O (2021, Dec 16). Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain, 144(11), 3505–3516. 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, & Belzung C (2003, Feb 28). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol, 463(1–3), 3–33. 10.1016/s0014-2999(03)01272-x [DOI] [PubMed] [Google Scholar]
- Quan W, Luo Q, Hao W, Tomic I, Furihata T, Schulz-Schäffer W, Menger MD, Fassbender K, & Liu Y (2021, Aug). Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. Glia, 69(8), 1987–2005. 10.1002/glia.24007 [DOI] [PubMed] [Google Scholar]
- Rangasamy SB, Jana M, Roy A, Corbett GT, Kundu M, Chandra S, Mondal S, Dasarathi S, Mufson EJ, Mishra RK, Luan CH, Bennett DA, & Pahan K (2018, Oct 1). Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J Clin Invest, 128(10), 4297–4312. 10.1172/jci96209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Reed QW, Cramer PE, & Landreth GE (2010, Nov 17). Deletion of CD14 attenuates Alzheimer’s disease pathology by influencing the brain’s inflammatory milieu. J Neurosci, 30(46), 15369–15373. 10.1523/jneurosci.2637-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, & Landreth GE (2009, Sep 23). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci, 29(38), 11982–11992. 10.1523/jneurosci.3158-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, & Liddelow SA (2022, Jun 1). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron, 110(11), 1788–1805 e1710. 10.1016/j.neuron.2022.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, & Volterra A (2019, Feb). Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci, 22(2), 154–166. 10.1038/s41593-018-0325-8 [DOI] [PubMed] [Google Scholar]
- Schroeder P, Rivalan M, Zaqout S, Krüger C, Schüler J, Long M, Meisel A, Winter Y, Kaindl AM, & Lehnardt S (2021, Jan). Abnormal brain structure and behavior in MyD88-deficient mice. Brain Behav Immun, 91, 181–193. 10.1016/j.bbi.2020.09.024 [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, & O’Banion MK (2007, Jun). Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest, 117(6), 1595–1604. 10.1172/jci31450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P, Furman JL, Pleiss MM, Kraner SD, Artiushin IA, Batten SR, Quintero JE, Simmerman LA, Beckett TL, Lovell MA, Murphy MP, Gerhardt GA, & Norris CM (2017, Jun 21). Calcineurin/NFAT Signaling in Activated Astrocytes Drives Network Hyperexcitability in Abeta-Bearing Mice. J Neurosci, 37(25), 6132–6148. 10.1523/JNEUROSCI.0877-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südkamp N, Shchyglo O, & Manahan-Vaughan D (2021). Absence of Pannexin 1 Stabilizes Hippocampal Excitability After Intracerebral Treatment With Aβ (1–42) and Prevents LTP Deficits in Middle-Aged Mice. Front Aging Neurosci, 13, 591735. 10.3389/fnagi.2021.591735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, & Katzman R (1991, Oct). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol, 30(4), 572–580. 10.1002/ana.410300410 [DOI] [PubMed] [Google Scholar]
- Tyzack GE, Sitnikov S, Barson D, Adams-Carr KL, Lau NK, Kwok JC, Zhao C, Franklin RJ, Karadottir RT, Fawcett JW, & Lakatos A (2014, Jul 11). Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat Commun, 5, 4294. 10.1038/ncomms5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmar P, Kullmann JS, Thilo B, Claussen MC, Rothhammer V, Jacobi H, Sellner J, Nessler S, Korn T, & Hemmer B (2010, Nov 15). Active immunization with amyloid-beta 1–42 impairs memory performance through TLR2/4-dependent activation of the innate immune system. J Immunol, 185(10), 6338–6347. 10.4049/jimmunol.1001765 [DOI] [PubMed] [Google Scholar]
- Vorhees CV, & Williams MT (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc, 1(2), 848–858. 10.1038/nprot.2006.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, Manis M, Schroeder C, Yin Z, Madore C, Butovsky O, Artyomov M, Ulrich JD, & Holtzman DM (2021, May 19). Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron, 109(10), 1657–1674 e1657. 10.1016/j.neuron.2021.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, & Mallard C (2009, Dec 1). Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol, 183(11), 7471–7477. 10.4049/jimmunol.0900762 [DOI] [PubMed] [Google Scholar]
- Weitz TM, Gate D, Rezai-Zadeh K, & Town T (2014, Nov). MyD88 is dispensable for cerebral amyloidosis and neuroinflammation in APP/PS1 transgenic mice. Am J Pathol, 184(11), 2855–2861. 10.1016/j.ajpath.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Carlock C, Shim J, Moreno-Gonzalez I, Glass W 2nd, Ross A, Barichello T, Quevedo J, & Lou Y. (2021, Oct). Requirement of brain interleukin33 for aquaporin4 expression in astrocytes and glymphatic drainage of abnormal tau. Mol Psychiatry, 26(10), 5912–5924. 10.1038/s41380-020-00992-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Lv YG, Du YF, Chen F, Reed MN, Hu M, Suppiramaniam V, Tang SS, & Hong H (2018, Oct). Neuroprotective effects of INT-777 against Aβ(1–42)-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Brain Behav Immun, 73, 533–545. 10.1016/j.bbi.2018.06.018 [DOI] [PubMed] [Google Scholar]
- Xiang X, Wang X, Jin S, Hu J, Wu Y, Li Y, & Wu X (2022, Jan 10). Activation of GPR55 attenuates cognitive impairment and neurotoxicity in a mouse model of Alzheimer’s disease induced by Aβ(1–42) through inhibiting RhoA/ROCK2 pathway. Prog Neuropsychopharmacol Biol Psychiatry, 112, 110423. 10.1016/j.pnpbp.2021.110423 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 – (a) Representative photomicrographs of GFAP (green), MyD88 (red), and merged immunostaining in AMyD+ and AMyD− mice analyzed in the hippocampal CA1 subregion. (b) Orthogonal analysis of GFAP (green) and MyD88 (red) immunostaining in AMyD+ and AMyD− mice. MyD88 was not detected in AMyD− mice. (c) Volumetric quantification of MyD88 and GFAP+-astrocytes colocalization in AMyD+ and AMyD− mice. (d) Overall MyD88 immunostaining minus the levels of colocalized MyD88+-GFAP+. Data were analyzed through an unpaired Student’s t-test, with a confidence level of 95%. (e,f) Representative western blot images and quantification of (e) SYP and (f) PSD95 in PBS- and Aβ42-treated AMyD+ and AMyD− mice. Samples were normalized to actin. (g) Unsupervised hierarchical clustering of statistically significant differentially expressed proteins. (h) Quantification of normalized CD47 protein expression determined by western blot. (i) Western blot of CD47 and actin protein expression. Data are presented as mean ± SD. Statistical evaluation was performed using a two-way ANOVA, followed by Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01.
Supplementary Figure 3 – Representative photomicrographs of MyD88 immunostaining in HPC (upper panels), TCX (middle panels), and CBL (lower panels) non-AD and AD brain sections. Neuronal-, vascular-, and glial-like cells are designated by black arrows.
Supplementary Figure 2 – Representative photomicrograph of immunofluorescence staining of GFAP (green), tau (red), and Aβ (magenta) in non-AD and AD human TCX (top two panels) and CBL (lower two sections).