

Abstract
Background
We investigated the phenotypes and genotypes of a cohort of ‘long-surviving’ individuals with motor neuron disease (MND) to identify potential targets for prognostication.
Methods
Patients were recruited via the Clinical Audit Research and Evaluation for MND (CARE-MND) platform, which hosts the Scottish MND Register. Long survival was defined as > 8 years from diagnosis. 11 phenotypic variables were analysed. Whole genome sequencing (WGS) was performed and variants within 49 MND-associated genes examined. Each individual was screened for C9orf72 repeat expansions. Data from ancestry-matched Scottish populations (the Lothian Birth Cohorts) were used as controls.
Results
58 long survivors were identified. Median survival from diagnosis was 15.5 years. Long survivors were significantly younger at onset and diagnosis than incident patients and had a significantly longer diagnostic delay. 42% had the MND subtype of primary lateral sclerosis (PLS). WGS was performed in 46 individuals: 14 (30.4%) had a potentially pathogenic variant. 4 carried the known SOD1 p.(Ile114Thr) variant. Significant variants in FIG4, hnRNPA2B1, SETX, SQSTM1, TAF15, and VAPB were detected. 2 individuals had a variant in the SPAST gene suggesting phenotypic overlap with hereditary spastic paraplegia (HSP). No long survivors had pathogenic C9orf72 repeat expansions.
Conclusions
Long survivors are characterised by younger age at onset, increased prevalence of PLS and longer diagnostic delay. Genetic analysis in this cohort has improved our understanding of the phenotypes associated with the SOD1 variant p.(Ile114Thr). Our findings confirm that pathogenic expansion of C9orf72 is likely a poor prognostic marker. Genetic screening using targeted MND and/or HSP panels should be considered in those with long survival, or early-onset slowly progressive disease, to improve diagnostic accuracy and aid prognostication.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00415-022-11505-0.
Keywords: Motor neuron disease, Amyotrophic lateral sclerosis, Survival, Genetics
Introduction
The Scottish Motor Neuron Disease Register (SMNDR) (re-launched as the Clinical Audit Research and Evaluation for MND (CARE-MND) platform in 2015) has been collecting data regarding people living with MND (amyotrophic lateral sclerosis (ALS) and MND subtypes [primary lateral sclerosis (PLS), primary muscular atrophy (PMA) and progressive bulbar palsy (PBP)] in Scotland since 1989 [1, 2]. The unique longevity of the register and the united efforts of the CARE-MND Consortium have provided extensive insight into the phenotypic and genetic heterogeneity of the disease [1, 3, 4]. Recent analysis of a historical cohort of 428 Scottish people with MND indicated a median survival of 3.5 years from onset of symptoms and 2 years from diagnosis [5]. However, the upper range of survival was 25.8 years from diagnosis (Fig. 1).
Fig. 1.
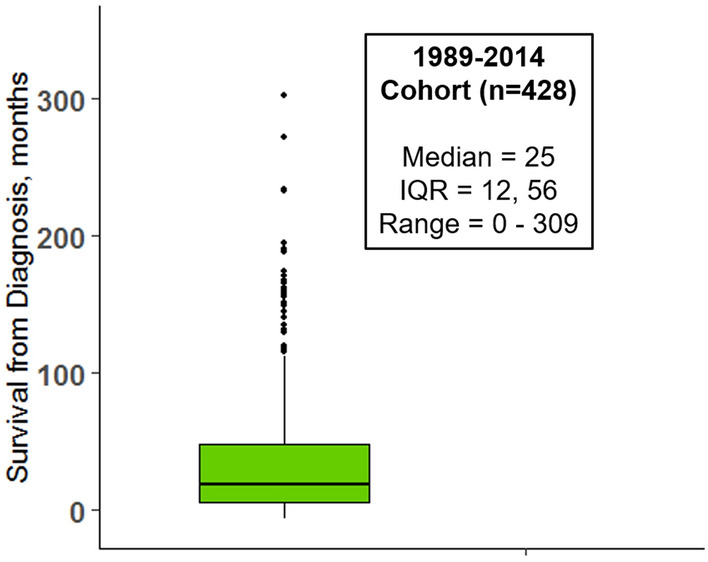
Boxplot of survival from diagnosis (months) in the 1989–2014 genotyped cohort (n = 428) [5]
There is currently no definition of long survival with MND, with literature ranging from 5 to 10 years [6–9]. Previous European estimates of patients surviving more than 10 years from diagnosis are 11.8% [8]. The phenotype is variable: some long-surviving people with MND have typical features of ALS [6, 7], while others have lower frequency of bulbar symptoms [2, 10] or specific disease subtypes such as PLS [11]. Individuals with PLS are thought to have younger onset disease [12]. Prediction of long survival is otherwise challenging; this prognostic uncertainty compounded by a typically protracted time to reach a diagnosis is psychologically difficult for people with MND and their families [12].
Some SOD1 variants and a specific variant in the UNC13A gene (rs10419420) are thought to impart long survival [13, 14]. People with apparent PLS have been found to have genetic variants not typically associated with MND, such as those normally associated with hereditary spastic paraplegia or Parkinson’s disease [11, 12, 15]. Otherwise, ‘long survivors’ have not been extensively genotypically characterised.
Harnessing 30 years of data from the Scottish MND Register/CARE-MND platform, we aimed to study long-survivors phenotypically and genetically. Whilst the overall proportion of long-survivors in MND populations is low, improved phenotypic understanding of this group is important to improve early and accurate diagnosis. Characterisation of long survivors might help to counsel patients and prognosticate at diagnosis. Long survivors also comprise a significant proportion of the prevalent clinical and ‘research-ready’ population and so knowledge of disease features may minimise bias and optimise generalisability of clinical trials [8]. DNA sequencing may provide clues towards genotype–phenotype associations, protective genetic factors or phenotypic overlap with disorders such as hereditary spastic paraplegia (HSP) [16]. We therefore aimed to identity phenotypic markers or genetic variants that might distinguish long survivors from a typical incident population of people with MND in Scotland, to guide clinical management and provide recommendations for genetic testing.
Methods
Recruitment and ethical approvals
Patients were recruited via the Scottish MND Register/CARE-MND Platform (ethical approvals MREC/98/0/56 1989–2010, 10/MRE00/78 2011–2015, and the Scotland A Research Ethics Committee 15/SS/0126 2015 onwards). DNA samples were donated to the Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank (MREC/98/0/56 1989–2010, 10/MRE00/77 2011 to 2013, 13/ES/0126 2013–2015, 15/ES/0094 2015-present). The Lothian Birth Cohorts (LBC) – a research population of Scottish adults born in 1921 and 1936—were used as ancestry-matched genetic controls [17]. Ethical permission for the LBC1936 study protocol was obtained from the Multi-Centre Research Ethics Committee for Scotland (Wave 1: MREC/01/0/56), the Lothian Research Ethics Committee (Wave 1: LREC/2003/2/29), and the Scotland A Research Ethics Committee (Waves 2, 3, 4 and 5: 07/MRE00/58). Ethical permission for the LBC1921 study protocol was obtained from the Lothian Research Ethics Committee (Wave 1: LREC/1998/4/183; Wave 2: LREC/2003/7/23; Wave 3: LREC1702/98/4/183), the Scotland A Research Ethics Committee (Waves 4 and 5: 10/MRE00/87).
Patient details
Long survivors were defined as people with MND with survival from diagnosis beyond the 80th percentile of the historical 1989–2014 Scottish genotype study cohort (> 8 years) [5]. This cut-off was determined as a conservative measure of long survival based on previous literature [6–8]. Survival was calculated from date of symptom onset/diagnosis until death or censorship date (3rd August 2021). The following phenotypic data fields were available for analysis: Sex, Age of Onset, Age of Diagnosis, Time to Diagnosis, Survival from Onset, Site of Onset, El Escorial Classification, Family History of MND, Feeding Tube Insertion, Non-invasive Ventilation (NIV) Use, Riluzole Use. Rate of change of the ALS-Functional Rating Scale (ALS-FRS) was not thought to be informative as it is considered inadequate for upper motor neuron predominant MND such as PLS [18]. Other predictors, such as cognitive assessments, were only available for a small number of long surviving patients and so could not be included in statistical analyses.
Genetic analysis
Patient DNA samples were analysed using whole genome sequencing technology, performed as part of the Scottish Genomes Partnership (SGP) study [19]. Samples were sequenced to 30X coverage using TruSeq Nano library preparation kits and a HiSeq X sequencing platform (Illumina). FASTQ files were aligned to the human genome build GRCh37 using bwa mem (0.7.13) [20]. Post-processing was performed with samblaster (0.1.22) [21] to mark duplicate reads, and the Genome Analysis ToolKit (GATK, v3.4-0-g7e26428)[22] for indel realignment and base recalibration. Genotype likelihoods for each sample were calculated using the GATK HaplotypeCaller and resulting GVCF files were called jointly using GATK’s GenotypeGVCFs function. Variant quality score recalibration (VQSR) was performed as per GATK best-practices [23] and a truth sensitivity threshold of 99.9% applied. Variants were filtered to include only those present in 49 MND-associated/MND-mimic genes (Fig. 2). Filtered variants were annotated and population frequency filters were applied using VarSeq Golden Helix software [24] to include only those with minor allele frequency (MAF) ≤ 0.01 in gnomAD 2.0.1v3 [25]. Variants were annotated using multiple in silico prediction algorithms from the Database for Nonsynonymous SNPs and their Functional Predictions (dbNSFP) [26] and included: SIFT, PolyPhen2 HDIV and HVAR, Mutation Taster, Mutation Assessor, FATHMM, PROVEAN, GERP and PhastCons. Measures of impact on splice site included scores derived from adaptive boost (Ada) and random forest (RF) models [27]. Samples were also tested for C9orf72 hexanucleotide repeat expansions using repeat prime PCR methodology; ≥ 30 repeats was considered pathogenic [28].
Fig. 2.
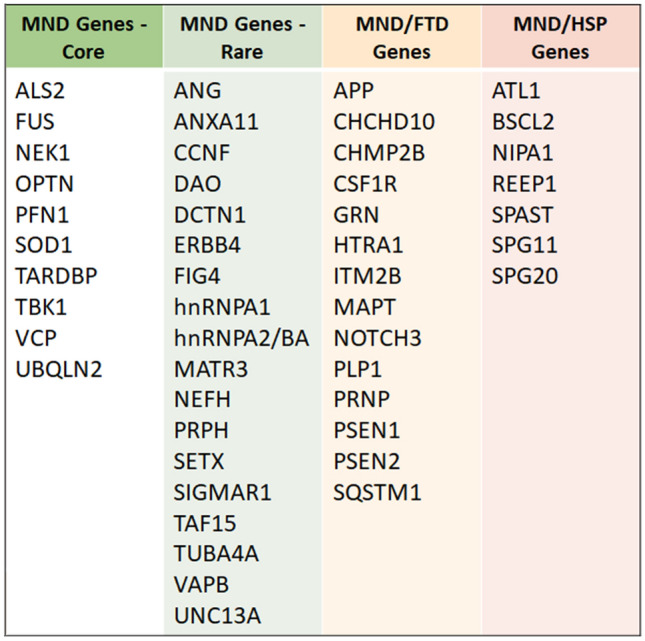
Motor neuron disease (MND)-associated genes. (i) Core MND-associated genes, (ii) Rare MND-associated genes, (iii) Genes associated with MND with frontotemporal dementia (MND-FTD), FTD or FTD mimics, (iv) Genes associated with hereditary spastic paraplegia (HSP) or MND-HSP overlap syndromes
Variants were classified using the American College of Medical Genetics and Association for Molecular Pathology (ACMG-AMP) framework and adhering to the Association for Clinical Genomic Science (ACGS) UK 2020 guidelines [29, 30]. Co-segregation was determined using methods described by Jarvik et al. [31] DNA samples from the Lothian Birth Cohorts (n = 1385) were used as ancestry-matched controls to identify variants enriched in cases versus controls as per ACMG-AMP guidelines. The gnomAD database was used as a population control data set [25]. Variants were considered significantly more prevalent in cases versus controls if odds ratio > 5.0 and confidence intervals did not cross 1.0 (ACMG-AMP criteria PS4) [29]. Additionally, gnomAD was used as a population control data set (ACMG criteria PM2) [25]. Variants meeting criteria thresholds for a pathogenic or likely pathogenic classification were reported. Variants of uncertain clinical significance (VUS) that fulfilled some criteria for being pathogenic, without reaching strict ACMG-AMP thresholds for significance, were considered under the Bayes rules outlined by Tavtigian et al. to determine a posterior probability of their being potentially pathogenic (probability > 0.5) [32].
Statistical analyses
Long survivors were compared with the incident population cohort of people with MND in Scotland diagnosed in 2015–17 (n = 437) using univariate statistics (Fisher’s exact tests, t-tests and Wilcoxon rank-sum tests). Correction for multiple testing was undertaken using the Bonferroni method. R statistical programming was used [33].
Results
Phenotypes
Fifty eight long survivors were identified, representing 3.3% of the total number of individuals on the CARE-MND platform at the time of data acquisition (n = 1779). Median survival from onset was 18.3 years (IQR 14.6–22.4) (missing dates for four individuals); from diagnosis 15.5 years (IQR 12.0–19.0). Thirty seven (63.8%) people were alive at censorship date, giving a point prevalence of 8.9% (Scottish prevalence of MND = 415). Fifty-four (90%) had consented to research and were characterised phenotypically. Percentage completed data per variable ranged from 94.4 to 100%. Phenotypic characteristics are outlined in Table 1. Male-to-female ratio was 2:1. Twenty-nine (54.7%) of long survivors had ALS, 22 (41.5%) PLS, one individual presented with Monomelic Amyotrophy (Other) and one with Progressive Bulbar Palsy (PBP). The most common site of onset of disease was lower limbs (55.8%). Six (11.5%) had a family history of MND, although two individuals were related. Of the 13 (25%) on ventilation, 12 of these were non-invasive and one invasive ventilation.
Table 1.
Comparison of phenotypic characteristics between long survivors and incident cohort of people with motor neuron disease
| Phenotypic characteristics | Incident cohort (n = 437) | Long survivors (n = 54) | Statistical test p-value |
|---|---|---|---|
| Sex: Male (%) | 275 (62.9) | 36 (66.6) | Fisher's p = 0.66 |
| Ethnicity: White Scottish/British/Other (%) | 415 (98.6) | 51 (100) | Fisher's p = 0.76 |
| Mean age of onset, years (SD) | 63.9 (10.9) | 47.4 (13.6) | t-test p < 0.0001 |
| Mean age of diagnosis, years (SD) | 65.5 (10.7) | 51.5 (13.5) | t-test p < 0.0001 |
| Median time to diagnosis, months (IQR) | 12.0 (8.0–23.0) | 26.0 (13.0–45.0) | Wilcoxon p < 0.0001 |
| Site of Onset | Fisher's p = 0.038 | ||
| Bulbar (%) | 123 (28.5) | 14 (26.9) | |
| Upper limb (%) | 103 (23.8) | 8 (15.4) | |
| Lower limb (%) | 144 (33.3) | 29 (55.8) | |
| Mixed (%) | 41 (9.5) | 1 (1.9) | |
| Cognition (%) | 12 (2.8) | 0 (0) | |
| Respiratory (%) | 5 (1.2) | 0 (0) | |
| Other (%) | 4 (0.9) | 0 (0) | |
| Classification | Fisher's p < 0.0001 | ||
| ALS (%) | 338 (77.4) | 29 (54.7) | |
| PBP (%) | 25 (5.7) | 1 (1.9) | |
| MND-FTD (%) | 25 (5.7) | 0 (0) | |
| PLS (%) | 19 (4.3) | 22 (41.5) | |
| PMA (%) | 19 (4.3) | 0 (0) | |
| Other (%) | 11 (2.5) | 1 (1.9) | |
| Family history of MND: Yes (%) | 39 (9.2) | 6 (11.5) | Fisher's p = 0.66 |
| Taking Riluzole: Yes (%) | 173 (40.0) | 32 (62.7) | Fisher's p = 0.0005 |
| Feeding Tube Inserted: Yes (%) | 136 (31.3) | 6 (11.8) | Fisher's p = 0.0003 |
| Non-invasive/Invasive ventilation commenced: Yes (%) | 118 (27.2) | 13 (25.0) | Fisher's p = 0.15 |
ALS amyotrophic lateral sclerosis, PBP progressive bulbar palsy, MND-FTD motor neuron disease with frontotemporal dementia, PLS primary lateral sclerosis, PMA progressive muscular atrophy
Significant p-values in bold
Bonferroni corrected threshold for significance was 0.0045. Long survivors were significantly younger at onset (47.4 years) and diagnosis (51.5 years) compared with incident people with MND (p < 0.0001). Time to diagnosis was significantly more prolonged (26 months versus 12 months; p < 0.0001). Classification of disease was significantly different (p < 0.0001), with long survivors more likely to have PLS than incident patients (41.5% of long survivors). Although long survivors were more likely to have lower limb onset disease, this did not reach statistical significance. Long survivors were more likely to be prescribed riluzole at any point (62.7%; p = 0.0005) but were significantly less likely to undergo gastrostomy insertion (only 11.8%; p = 0.0003) (Table 1).
Genotype
Forty six (79.3%) of all long survivors donated a DNA sample. Forty six unique variants (94 variant calls including variants found in multiple individuals) met filtering criteria (Supplementary Table 1). All variants were detected in a heterozygous state. Three were considered likely benign variants (6.5%). Sixteen variants fulfilled ACMG-AMP/ACGS criteria for being likely pathogenic (34.8%). However, three variants were in genes normally causing disease in an autosomal recessive pattern (ALS2 and SPG11) and three variants were novel frameshift/indel variants in a likely unstable region of a tenuously associated MND gene (TAF15) (Supplementary Table 1). Excluding these variants, 10 were classified as likely pathogenic (21.7%) (Table 2). The remaining 27 variants (58.7%) were classified as VUS; two of these (4.3% of all variants) leaned towards the pathogenic end of the spectrum with a supportive posterior probability (> 0.5) (Supplementary Table 1) [32]. In contrast, 21 VUS leaned towards the benign end of the spectrum (45.7%). The remaining four variants (8.7%) met both benign and pathogenic criteria, with a posterior probability of 0.5 (Supplementary Table 1). A total of 14 individuals had at least one variant fulfilling criteria for being pathogenic or likely pathogenic variant (30.4% of cohort). A total of 17 patients carried at least one pathogenic or likely pathogenic variant or a VUS which fulfilled some pathogenic criteria (posterior probability > 0.5) (37.0% of cohort) (Supplementary Table 1).
Table 2.
Significant variants identified in long survivors with motor neuron disease (MND)
| Gene | Genomic position (GRCh37) | HGVS coding transcript/variant DNA change | Amino acid change | Variant type | Number cases | Number controls | ACMG-AMP/ACGS Evidence |
|---|---|---|---|---|---|---|---|
| FIG4 | 6:110113822A > T | NM_014845.5:c.2414A > T | p.(Asn805Ile) | Missense | 1 | 1 | PS4, PM2 |
| HNRNPA2B1 | 7:26240202G > A | NM_031243.2:c.-5C > T | 5’ UTR | 5’ UTR Variant | 1 | 0 | PS4, PM2 |
| SETX | 9:135211747C > G | NM_015046.5:c.654G > C | p.(Lys218Asn) | Missense | 1 | 3 | PS4, PM2 |
| SOD1 | 21:33039672 T > C | NM_000454.4:c.341 T > C | p.(Ile114Thr) | Missense | 4* | 0 | PS4, PM2, PP1 Moderate, PP3 |
| SOD1 | 21:33036142G > A | NM_000454.4:c.112G > A | p.(Gly38Arg) | Missense | 1 | 0 | PS4, PM2, PP3 |
| SPAST | 2:32366960 T > - | NM_014946.3:c.1494-3delT | Intronic | Intronic splice region | 2 | 0 | PS4, PM2, PP3 |
| SQSTM1 | 5:179260112GAG > - | NM_003900.4:c.835_837delGAG | p.(Glu280del) | Inframe deletion | 1 | 0 | PS4, PM2, PP3 |
| SQSTM1 | 5:179263547C > T | NM_003900.4:c.1277C > T | p.(Ala426Val) | Missense | 1 | 2 | PS4, PM2 |
| TAF15 | 17:34171667A > G | NM_139215.2:c.1364A > G | p.(Tyr455Cys) | Missense | 1 | 0 | PS4, PM2, PP3 |
| VAPB | 20:56964578G > A | NM_004738.4:c.58 + 5G > A | Intronic | Intronic splice region | 1 | 0 | PS4, PM2, PP3 |
Classification of pathogenicity based on use of American College of Medical Genetics and Association for Molecular Pathology (ACMG-AMP) and Association for Clinical Genomic Science (ACGS) UK 2020 guidelines. All variants detected in heterozygous state. Genomic controls comprised ancestry-matched individuals from the Lothian Birth Cohorts (n = 1385). Variants were considered significantly more prevalent in cases versus controls if odds ratio > 5.0 and confidence intervals did not cross 1.0 (ACMG-AMP criteria PS4). Additionally, gnomAD was used as a population control data set (ACMG criteria PM2). Co-segregation was quantified using methods described by Jarvik et al. (PP1). In silico predictions were evaluated using VarSeq Golden Helix annotations (PP3)
UTR untranslated region
*Indicates that 2 of the cases are related
Pathogenic or likely pathogenic variants included four incidences of the SOD1 p.(Ile114Thr) Scottish founder mutation (Table 2) [3, 5]. One of the p.(Ile114Thr) variants and the SOD1 p.(Gly38Arg) variant (Table 2) were observed in the Scottish population previously [5]. Variants of interest were also identified in FIG4, hnRNPA2B1, SETX, SPAST and VAPB genes (Table 2). Two related individuals had both the SOD1 p.(Ile114Thr) mutation plus a frameshift mutation (p.Asp449Thrfs*28) in the TAF15 gene [34]. The TAF15 mutation was found in two unrelated samples and nine controls, giving an odds ratio of 6.9 (95% CI 1.5–33.1). However, other variants were also observed at the same site as this loss-of-function variant suggesting possible instability of the region (Supplementary Table 1). Mutations in TAF15 are postulated to be linked with MND pathogenesis due to their shared role in FET (FUS, EWSR1, TAF15) protein pathways but are not clearly associated with MND and so the significance of this variant is attenuated [34].
No patients in this cohort had expansions in the C9orf72 hexanucleotide sequence within the pathogenic range.
Genotype–phenotype associations
Four individuals had the SOD1 p.(Ile114Thr) Scottish founder variant [3, 5], (Table 3) although two of these were first cousins once removed and had another family member with MND suggesting moderate co-segregation of this variant as per calculations outlined by Jarvik et al. (Table 4) [31]. All four patients had familial lower limb onset ALS with a median age of onset of 31.5 years and all required NIV but not gastrostomy. Case note review did not reveal significant cognitive impairment. Another individual had the SOD1 p.(Gly38Arg) variant; this variant is associated with a mouse model of ALS which recapitulates some of the motor features [35]. However, there is a paucity of knowledge regarding its phenotypic correlates. This individual had a family history of MND—his father developed symptoms of MND in his 30s and died in his 60s, implying a similar course of disease.
Table 3.
Key phenotypic characteristics of long survivors with variants of interest
|
FIG4 p.(Asn805Ile) (n = 1) |
HNRNPA2B1 5 ‘ UTR c.-5C > T (n = 1) |
SETX p.(Lys218Asn) (n = 1) |
SOD1 p.(Ile114Thr) (n = 2) |
SOD1 p.(Gly38Arg) (n = 1) |
SPAST Intronic c.1494-3delT (n = 2) |
SQSTM1 p.(Glu280del) (n = 1) |
SQSTM1 p.(Ala426Val) (n = 1) |
TAF15 p.(Tyr455Cys) (n = 1) |
VAPB Intronic c.58 + 5G > A (n = 1) |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female |
Male Male |
Male |
Male Male |
Male | Male | Male | Male |
| Ethnicity | White Scottish | White Scottish | White Scottish |
White Scottish White Other |
White Scottish |
White Scottish White Other |
White Scottish | White Scottish | White Scottish | White Scottish |
| Age of onset, years | 54 | 45 | 41 |
31 48 |
31 |
76 65 |
28 | 53 | 49 | 37 |
| Time to diagnosis, months | 30 | 13 | 78 |
46 41 |
5 |
11 4 |
83 | 66 | 159 | 17 |
| Survival from onset, months | 175 | 143 | 355 |
199 149 |
245 |
164 253 |
223 | 216 | 308 | 211 |
| Site of onset | Lower limb | Lower limb | Bulbar |
Lower limb Lower limb |
Upper limb |
Lower limb Lower limb |
Lower limb | Upper limb | Lower limb | Lower limb |
| MND classification | ALS | PLS | PLS |
ALS ALS |
ALS |
PLS ALS |
ALS | ALS | PLS | PLS |
| Family history of MND | No | No | No |
Yes Yes |
Yes |
No No |
No | No | No | No |
| Taking riluzole | No | No | No |
No Yes |
Yes |
No Yes |
Yes | Yes | No | Yes |
| Feeding tube | No | Yes | No |
No No |
No |
No No |
Yes | Yes | No | No |
| Non-Invasive/Invasive ventilation | Yes | No | No |
Yes Yes |
No |
No No |
Yes | Yes | No | No |
ALS amyotrophic lateral sclerosis, PLS primary lateral sclerosis
Table 4.
Key phenotypic characteristics of two related long-survivors
| CASE 1*: SOD1 p.(Ile114Thr) | CASE 2*: SOD1 p.(Ile114Thr) | |
|---|---|---|
| Sex | Female | Male |
| Ethnicity | White Scottish | White Scottish |
| Age of Onset, years | 32 | 23 |
| Time to Diagnosis, months | 38 | 37 |
| Survival from Onset (months) | 232 | 263 |
| Site of Onset | Lower limb | Lower limb |
| Classification | ALS | ALS |
| Family History of MND | Yes | Yes |
| Taking Riluzole | No | No |
| Feeding Tube | No | No |
| Non-Invasive/Invasive Ventilation | Yes | Yes |
ALS amyotrophic lateral sclerosis
Two patients had missense mutations in FIG4 and SETX respectively, the latter in a patient with PLS. These genes have been associated rarely with both ALS and PLS [15]. Two patients had SQSTM1 variants; these have been described in familial and apparently sporadic MND, as well as in frontotemporal dementia (FTD) without MND, and are also associated with multisystem disease [36]. One individual had a variant in hnRNPA2B1; although this variant was in the 5’UTR (untranslated region) of this gene, the region is highly conserved and the variant was absent from controls with a low MAF in population databases. Variants in hnRNPA2B1 are associated with inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia, but have been described in ALS [37, 38]. On case note review, neither patient with the hnRNPA2B1 variant or the SQSTM1 variant had evidence of multisystem disease typical of the genes (inclusion body myopathy or Paget’s disease). However, the patient with the hnRNPA2B1 variant had progressive cognitive impairment/dementia but was determined to have a PLS phenotype. The two patients with SQSTM1 mutations had limb onset ALS, requiring both NIV and gastrostomy. One of these patients was also a heterozygous carrier of a loss of function variant in the SPG11 gene (p.M245Vfs*2) (Supplementary Table). This SPG11 variant has previously been reported in ClinVar[39] in a biallelic state associated with HSP and juvenile ALS but has not previously been observed in trans with another variant. The patient, however, had generalised signs and symptoms in-keeping with a classical ALS phenotype, with supportive electromyography (EMG) and gastrostomy and NIV requirements. This patient also underwent post-mortem examination and had typical ALS-associated TDP-43 intracytoplasmic aggregates [Dr Jenna Gregory et al., unpublished].
Two patients carried a SPAST intronic variant; this variant was absent from controls and gnomAD but has been reported in Clinvar associated with autosomal dominantly inherited HSP (SPG4) and had supportive in silico predictors of an effect on splicing [40]. Interestingly, the two patients harbouring this variant had limb onset ALS and PLS and did not require NIV or gastrostomy suggesting perhaps misdiagnosis of MND or an overlapping phenotypic spectrum. A further splice site mutation was observed in the VAPB gene in an individual with PLS. VAPB variants are rare in MND and the gene is small and tolerant to change. However, all splice algorithms predicted a significant reduction in splice efficiency and the variant was absent in controls with a low MAF.
Discussion
The prevalence of long survivors in the Scottish population (8.9%) is comparable with previous European estimates [8]. Patients in this cohort are younger at onset and diagnosis and have a longer time to diagnosis. We suspect that the diagnostic delay reflects uncertainty based on gradual evolution of clinical features and absence of lower motor neuron features on EMG. These findings agree with previously published observations [7–9, 12, 41]. However, this delay presents a window for improvement in MND care.
Our cohort is enriched for people diagnosed with upper motor neuron predominant MND (PLS). Although not statistically significant compared with incident patients, the male-to-female ratio in the long-surviving group was high (2:1) and this may be due to the contribution of PLS cases which are known to be largely male (ratio 2–4:1) [12]. That PLS and ALS are on a continuum of disease has long been debated; however, recent systematic reviews found no consistent distinguishing imaging or pathological biomarkers [12, 18].
Long survivors are more likely to start on riluzole than incident patients. This may be related to opportunity in the context of long duration of disease. While riluzole has been trialled predominantly in people with ALS, an appeal was made to the National Institute for Clinical Excellence (NICE) in 2001 to allow its use in other forms of MND [42]. Clearly, in current practice, patients with PLS do receive riluzole. Long survivors are significantly less likely to have a gastrostomy inserted, in spite of there being more opportunity, temporally, for this to occur. However, the proportions of bulbar-onset patients in incident and long surviving cohorts are comparable (28.5% and 26.9% respectively). It is possible that upper motor neuron pseudobulbar symptoms (for example, dysarthria) in the long-surviving PLS cohort are being confused for true bulbar symptoms. It may be clinically difficult to localise dysarthria clinically, especially early in disease. However, dysphagia is rare in PLS [43]. Indeed, only two (14.3%) patients with bulbar-onset disease had gastrostomy insertion; other patients were noted to have dysarthria with preserved swallow. Although we might expect respiratory muscle weakness to be associated with poor outcome, a surprisingly high proportion of long survivors required NIV (25.0% in the long survivors, 27.2% in the incident cohort). It was unclear if this requirement occurred late in disease or if NIV contributed to survival. Long surviving people with MND form a significant proportion of the prevalent population; exclusion of such participants from MND clinical trials risks discrimination and attenuation of population for recruitment and study power. Our data suggest that detailed characterisation of such participants (by classification and by distinguishing bulbar from pseudobulbar symptoms) would aid trial inclusion, generalisability and interpretation.
Only 11.5% of the long surviving cohort had a family history of MND whereas almost a third (30.4%) of the genotyped cohort had a ACMG-AMP likely pathogenic variant. As would be expected, this is higher than when a limited six-gene panel was employed for an unselected cohort of MND patients in Scotland (17%) [5]. Genetic analysis in long survivors broadened the already wide phenotypic spectrum of disease of patients with the known pathogenic SOD1 p.(Ile114Thr) variant which is frequently observed in the Scottish population [5]. Families with this variant in Scotland may be encouraged that it can be associated with long survival. As previously described, people with this variant have an otherwise homogeneous phenotype with limb-onset ALS, preserved bulbar function and low gastrostomy uptake and absence of cognitive impairment.
The presence of other rare variants in MND-associated genes (FIG4, hnRNPA2B1, SETX, SQSTM1, TAF15, VAPB) in patients with both ALS and PLS without a family history confirms the sporadic nature of variants in these genes in the Scottish population. Although these variants met ACMG-AMP/ACGS classification criteria for being likely pathogenic, it is not possible to determine if they are disease-causing in a rare heterogeneous condition such as MND. We raise the possibility that people with MND carrying mutations in the SPAST gene may have an MND syndrome similar to HSP. In general, HSP will normally present earlier than upper motor neuron predominant ALS/PLS and patients are more likely to have lower limb onset disease and a family history; however, there will be exceptions and overlap [12, 16]. Extended and explicit past medical history and family history questioning (including inclusion body myopathy, Paget’s disease, FTD, HSP and other neurological causes of spasticity) might have illuminated patterns in-keeping with the identified genetic variants.
We have also highlighted VUS of potential interest which meet some pathogenic criteria without fulfilling strict ACMG-AMP requirements for pathogenicity; these require future study and reassessment.
The absence of C9orf72 pathogenic expansions in this study is supportive of this variant typically being a poor prognostic marker [44]. Indeed, no long survivors were diagnosed with MND-FTD, compared with 5.7% of the incident cohort. Formal systematic cognitive testing was not undertaken for the majority of long survivors, however, as this assessment tool was only routinely applied within the last decade.
Limitations
Our study would benefit from inclusion of cognitive profiling. Details regarding time to intervention (gastrostomy, NIV) were not available for all patients but future analysis of long survivors might study the temporal relationship of these measures on outcome. Our control population is ancestry but not age or sex-matched and so may not reflect the characteristics of our typically young MND long survivors.
We have reported variants as per ACMG-AMP guidelines. MND-associated variants are rare and have variable penetrance and so often struggle to fulfil strict ACMG-AMP criteria. Many likely pathogenic variants were so defined due to their absence in controls and absence in gnomAD alone. However, these guidelines provide a necessary structure for variant assessment and allow us to report variants which might as yet fail to achieve clinical significance but might be of research interest. As this was a research project and results were not relayed to patients or families, frameshift and indel variants were not confirmed by Sanger sequencing and so it is not possible to determine if they are truly disruptive. Our 49-gene panel was comprehensive for MND-spectrum genes but we did not examine the androgen receptor CAG trinucleotide repeat associated with Kennedy disease [7], nor did we exhaust all rare HSP and PLS-associated genes [12]. Pathology correlation was only available for one patient in this cohort. Although CARE-MND recruitment methods are stringent [1], it is possible that some patients are misdiagnosed and pathological examination would provide confirmation of disease in rare gene and MND-mimic gene carriers [18].
Implications and conclusions
With the benefit of three decades of longitudinal data collection through the Scottish CARE-MND database, we have shown that long surviving people with MND can be characterised by younger age at onset and diagnosis, increased incidence of PLS and, crucially, a longer time to diagnosis (median 2.2 years, upper range 15 years). This long period of diagnostic uncertainty for a typically young person is a key target for improvement in care; such patients may otherwise be denied access to designated benefits and specialised support. Early clinical genotyping of such individuals may help to provide reassurance – for example, the absence of a C9orf72 pathogenic expansion and presence of the SOD1 p.(Ile114Thr) mutation or other rare mutation might indicate better outcome. Additionally, presence of the SOD1 p.(Ile114Thr) variant might imply long preservation of bulbar function and cognitive function but allow individuals to prepare for NIV requirement. Extended family history, including multisystem disease and neurological syndromes featuring UMN signs/spasticity, might be of particular merit in those with young onset, slowly progressive ALS or PLS; in these cases, MND and HSP genetic testing may be appropriate. Long survivors are more likely to have a likely pathogenic variant than previous estimates of gene carrier status in MND populations; however, this may reflect breadth of our gene panel. The majority of MND patients do not undergo routine genetic testing due to variable variant penetrance, challenges with variant classification and lack of relevant treatment options for people with MND. However, recent evidence suggests that limiting genetic testing to people with presumed prior probability of having a pathogenic mutation (i.e. those with a family history and young-onset disease) may fail to capture a significant proportion of potential actionable or informative genetic mutations [45]. Our study demonstrates that slowly progressive people with MND may particularly benefit from genetic input, aiming for avoidance of prolonged and unnecessary investigation, earlier diagnosis and access to disease-specific services and care.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We are grateful to all the people with MND who participated in these studies. We continue to devote all efforts to ensure that the information they donate is maximised to help patients of the future. For the purpose of open access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript version arising from this submission. CARE-MND Consortium members: Javier Carod Artal, Andrew Bethell, Susan Byrne, Siddharthan Chandran, Gillian Craig, Richard Davenport, Callum Duncan, Moira Flett, George Gorrie, Hanne Haagendrud, Katarzyna Hafezi, Janice Hatrick, Aidan Hutchison, Micheala Johnson, Danielle Leighton, Helen Lennox, Laura Marshall, Dympna McAleer, Alison McEleney, Kitty Millar, Ian Morrison, Louise Murrie, Judith Newton, Suvankar Pal, David Perry, Gowri Saravanan, Martin Starrs, Susan Stewart, Dorothy Storey, Gill Stott, Robert Swingler, David Thompson, Carol Thornton, Tanya Van Der Westhuizen, Carolyn Webber and Michael Wong. Lothian Birth Cohorts Group members: Sarah Harris, James Prendergast, Tom Russ, Adele Taylor and Ian Deary.
Funding
DL received funding for PhD study at the inception of this study from the Chief Scientist Office for Scotland, the Motor Neuron Disease Association and Motor Neuron Disease Scotland (CAF/MND/15/01). This work is also supported by the UK Dementia Research Institute which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK.
Data Availability
Data supporting the genetic findings of this study are available within the article and supplementary material. Raw CARE-MND data are not available due to their containing information that could compromise the privacy of research participants. Further information about the CARE-MND database can be found at: https://www.caremnd.org.uk/.
Declarations
Conflicts of interest
The authors report no competing interests.
Ethics approval
The Scottish MND Register/CARE-MND Platform is approved by MREC/98/0/56 1989–2010, 10/MRE00/78 2011–2015, and the Scotland A Research Ethics Committee 15/SS/0126 2015 onwards. The Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank are approved by MREC/98/0/56 1989–2010, 10/MRE00/77 2011 to 2013, 13/ES/0126 2013–2015, 15/ES/0094 2015-present. Ethical permission for the LBC1936 study protocol was obtained from the Multi-Centre Research Ethics Committee for Scotland (Wave 1: MREC/01/0/56), the Lothian Research Ethics Committee (Wave 1: LREC/2003/2/29), and the Scotland A Research Ethics Committee (Waves 2, 3,4 and 5: 07/MRE00/58). Ethical permission for the LBC1921 study protocol was obtained from the Lothian Research Ethics Committee (Wave 1: LREC/1998/4/183; Wave 2: LREC/2003/7/23; Wave 3: LREC1702/98/4/183), the Scotland A Research Ethics Committee (Waves 4 and 5: 10/MRE00/87).
Consent to participate
All participants provided informed consent before contributing to the Scottish MND Register/CARE-MND Platform, Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank and Lothian Birth Cohorts.
Footnotes
The members of the Lothian Birth Cohorts Group and the CARE-MND Consortium are present in Acknowledgements section.
Contributor Information
Danielle J. Leighton, Email: Danielle.leighton@glasgow.ac.uk
the Lothian Birth Cohorts Group:
Sarah Harris, James Prendergast, Tom Russ, Adele Taylor, and Ian Deary
and the CARE-MND Consortium:
Andrew Bethell, Suzanne Byrne, Gillian Craig, Moira Flett, Hanne Haagendrud, Katarzyna Hafezi, Janice Hatrick, Aidan Hutchison, Helen Lennox, Laura Marshall, Dympna McAleer, Alison McEleney, Kitty Millar, Louise Murrie, David Perry, Gowri Saravanan, Martin Starrs, Susan Stewart, Dorothy Storey, Gill Stott, David Thompson, Carol Thornton, Tanya Van Der Westhuizen, and Carolyn Webber
References
- 1.Leighton D, Newton J, Colville S, et al. Clinical audit research and evaluation of motor neuron disease (CARE-MND): a national electronic platform for prospective, longitudinal monitoring of MND in Scotland. Amyotroph Lateral Scler Front Degener. 2019 doi: 10.1080/21678421.2019.1582673. [DOI] [PubMed] [Google Scholar]
- 2.Chancellor AM, Slattery JM, Fraser H, et al (1993) The prognosis of adult-onset motor neuron disease: a prospective study based on the Scottish Motor Neuron Disease Register. J Neurol 240:339–46. http://www.ncbi.nlm.nih.gov/pubmed/8336173. Accessed 22 May 2016 [DOI] [PubMed]
- 3.Hayward C, Swingler RJ, Simpson SA, et al (1996) A specific superoxide dismutase mutation is on the same genetic background in sporadic and familial cases of amyotrophic lateral sclerosis. Am J Hum Genet 59:1165–1167. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1914828&tool=pmcentrez&rendertype=abstract. Accessed 24 Feb 2016 [PMC free article] [PubMed]
- 4.Leighton DJ, Newton J, Stephenson LJ, et al. Changing epidemiology of motor neurone disease in Scotland. J Neurol. 2019;266:817–825. doi: 10.1007/s00415-019-09190-7. [DOI] [PubMed] [Google Scholar]
- 5.Black HA, Leighton DJ, Cleary EM, et al. Genetic epidemiology of motor neuron disease-associated variants in the Scottish population. Neurobiol Aging. 2017;51:178.e11–178.e20. doi: 10.1016/j.neurobiolaging.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mateen FJ, Carone M, Sorenson EJ. Patients who survive 5 years or more with ALS in Olmsted County, 1925–2004. J Neurol Neurosurg Psychiatry. 2010;81:1144–1146. doi: 10.1136/jnnp.2009.201251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner MR, Parton MJ, Shaw CE, et al. Prolonged survival in motor neuron disease: a descriptive study of the King’s database 1990–2002. J Neurol Neurosurg Psychiatry. 2003;74:995–997. doi: 10.1136/jnnp.74.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pupillo E, Messina P, Logroscino G, et al. Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol. 2014;75:287–297. doi: 10.1002/ana.24096. [DOI] [PubMed] [Google Scholar]
- 9.Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17:423–433. doi: 10.1016/S1474-4422(18)30089-9. [DOI] [PubMed] [Google Scholar]
- 10.Zoccolella S, Beghi E, Palagano G, et al. Predictors of long survival in amyotrophic lateral sclerosis: a population-based study. J Neurol Sci. 2008;268:28–32. doi: 10.1016/j.jns.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Mitsumoto H, Nagy PL, Gennings C, et al. Phenotypic and molecular analyses of primary lateral sclerosis. Neurol Genet. 2015;1:e3. doi: 10.1212/01.NXG.0000464294.88607.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner MR, Barohn RJ, Corcia P, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020;91:373–377. doi: 10.1136/jnnp-2019-322541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaastra B, Shatunov A, Pulit S, et al. Rare genetic variation in UNC13A may modify survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2016;17:593–599. doi: 10.1080/21678421.2016.1213852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silani V, Corcia P, Harms MB, et al. Genetics of primary lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2020;21:28–34. doi: 10.1080/21678421.2020.1837177. [DOI] [PubMed] [Google Scholar]
- 16.Brugman F, Veldink JH, Franssen H, et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch Neurol. 2009;66:509–514. doi: 10.1001/archneurol.2009.19. [DOI] [PubMed] [Google Scholar]
- 17.Taylor AM, Pattie A, Deary IJ. Cohort profile update: the lothian birth cohorts of 1921 and 1936. Int J Epidemiol. 2018;47:1042–1042r. doi: 10.1093/ije/dyy022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finegan E, Chipika RH, Shing SLH, et al. Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Front Degener. 2019;20:133–145. doi: 10.1080/21678421.2018.1550518. [DOI] [PubMed] [Google Scholar]
- 19.Scottish Genome Partnership (2020) Scottish Genome Partnership: rare disease research studies. Scottish Genome Partnersh. https://www.scottishgenomespartnership.org/sgp-rare-disease-research-studies. Accessed 1 Jun 2020
- 20.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Published Online First: 16 March 2013.http://arxiv.org/abs/1303.3997. Accessed 1 Nov 2018
- 21.Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30:2503–2505. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van der Auwera GA, Carneiro MO, Hartl C, et al (2013) From fastq data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. In: Current Protocols in bioinformatics. Wiley, Hoboken, pp 11.10.1–11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed]
- 24.Golden Helix I (2018) VarSeq. Golden Helix, Inc, Bozeman, MT. http://goldenhelix.com. Accessed 11 Nov 2018
- 25.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Wu C, Li C, et al. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–13544. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cleary EM, Pal S, Azam T, et al. Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol Cell Probes. 2016;30:218–224. doi: 10.1016/j.mcp.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellard S, Baple EL, Callaway A, et al (2020) ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Assoc Clin Genomic Sci Published Online First. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf. Accessed 1 Nov 2020
- 31.Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet. 2016;98:1077–1081. doi: 10.1016/j.ajhg.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tavtigian SV, Greenblatt MS, Harrison SM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–1060. doi: 10.1038/gim.2017.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team (2017) R: a language and environment for statistical computing. R Found Stat Comput. http://www.r-project.org/. Accessed 1 Nov 2018
- 34.Kapeli K, Pratt GA, Vu AQ, et al. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat Commun. 2016;7:12143. doi: 10.1038/ncomms12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picher-Martel V, Valdmanis PN, Gould PV, et al. From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol Commun. 2016;4:70. doi: 10.1186/s40478-016-0340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Shu S, Wang RR, et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology. 2016;87:1763–1769. doi: 10.1212/WNL.0000000000003256. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, Kim NC, Wang Y-D, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ClinVar. ClinVar. Natl. Cent. Biotechnol. Information, U.S. Natl. Libr. Med. 2016. https://www.ncbi.nlm.nih.gov/clinvar/. Accessed 2 Nov 2018
- 40.Svenson IK, Ashley-Koch AE, Gaskell PC, et al. Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am J Hum Genet. 2001;68:1077–1085. doi: 10.1086/320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czaplinski A, Yen AA, Appel SH. Amyotrophic lateral sclerosis: early predictors of prolonged survival. J Neurol. 2006;253:1428–1436. doi: 10.1007/s00415-006-0226-8. [DOI] [PubMed] [Google Scholar]
- 42.NICE. Guidance on the Use of Riluzole (Rilutek) for the Treatment of Motor Neurone Disease (2001) Natl Inst Heal Care Excell, pp 1–13. https://www.nice.org.uk/guidance/ta20/documents/riluzole-appeal-against-guidance-dated-december-2000. Accessed 29 Aug 2019
- 43.Tartaglia MC, Rowe A, Findlater K, et al. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2007;64:232. doi: 10.1001/archneur.64.2.232. [DOI] [PubMed] [Google Scholar]
- 44.Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehta PR, Iacoangeli A, Opie-Martin S, et al. The impact of age on genetic testing decisions in amyotrophic lateral sclerosis. Brain. 2022 doi: 10.1093/brain/awac279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the genetic findings of this study are available within the article and supplementary material. Raw CARE-MND data are not available due to their containing information that could compromise the privacy of research participants. Further information about the CARE-MND database can be found at: https://www.caremnd.org.uk/.