

Abstract
Osimertinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), potently and selectively inhibits EGFR-TKI-sensitizing and EGFR T790M resistance mutations. In the Phase III FLAURA study (NCT02296125), first-line osimertinib improved outcomes vs comparator EGFR-TKIs in EGFRm advanced non-small cell lung cancer. This analysis identifies acquired resistance mechanisms to first-line osimertinib. Next-generation sequencing assesses circulating-tumor DNA from paired plasma samples (baseline and disease progression/treatment discontinuation) in patients with baseline EGFRm. No EGFR T790M-mediated acquired resistance are observed; most frequent resistance mechanisms are MET amplification (n = 17; 16%) and EGFR C797S mutations (n = 7; 6%). Future research investigating non-genetic acquired resistance mechanisms is warranted.
Subject terms: Cancer, Lung cancer, Targeted therapies
In the phase III FLAURA study (NCT02296125), the third-generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) osimertinib provided superior progression-free survival versus comparator EGFR-TKIs in patients with NSCLC. Here, by next-generation sequencing of circulating tumor DNA, the authors assess candidate mechanisms of acquired resistance to first-line osimertinib in patients from the FLAURA trial.
Introduction
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) are the recommended first-line treatment for advanced non-small cell lung cancer (NSCLC) harboring EGFR-TKI sensitizing mutations (EGFRm)1. Despite initial high response rates to first-line EGFR-TKIs, most patients treated with an EGFR-TKI develop resistance. In approximately 50% of patients treated with a first- or second-generation EGFR-TKI, EGFR T790M resistance mutation was detected2–6.
Osimertinib is a third-generation, irreversible, oral EGFR-TKI that potently and selectively inhibits both EGFR harboring EGFRm (Exon 19 deletion [Ex19del]/L858R) and EGFR T790M resistance mutations. In clinical trials, osimertinib has shown efficacy in patients with EGFRm and EGFR T790M NSCLC, including patients with central nervous system (CNS) metastases7–12. In the Phase III FLAURA study (NCT02296125), osimertinib provided superior progression-free survival (PFS) versus comparator EGFR-TKIs (erlotinib or gefitinib) in patients with previously untreated EGFRm advanced NSCLC (median 18.9 months versus 10.2 months; hazard ratio [HR] 0.46, 95% confidence interval [CI], 0.37 to 0.57; P < 0.001)11. Final analysis of overall survival (OS) also demonstrated significantly longer OS with osimertinib versus comparator EGFR-TKI (38.6 months versus 31.8 months; HR 0.80, 95.05% CI, 0.64 to 1.00; P = 0.046)12.
Mechanisms of acquired resistance to osimertinib when used in the second-line setting in patients with EGFR T790M-positive NSCLC after EGFR-TKI treatment have been identified. To date, the most frequently reported resistance mechanisms to second-line osimertinib, including analyses of circulating-tumor DNA (ctDNA) samples from patients in the Phase III AURA3 study are acquired EGFR mutations (e.g., C797S), and amplification of MET and ERBB2 (HER2)13–15. However, mechanisms of acquired resistance to osimertinib used in the first-line setting remain to be fully elucidated. A small-scale analysis of genomic mechanisms of acquired resistance in nine patients with previously untreated EGFRm advanced NSCLC who received osimertinib in the Phase I portion of the AURA study showed no cases of acquired EGFR T790M mutation; potential resistance mechanisms identified included other EGFR mutations and amplification of MET and HER216. In another recent, small, retrospective study of paired tissue samples from patients with EGFRm advanced NSCLC analyzed using next-generation sequencing (NGS), it was identified that off-target resistance in the first-line setting was higher versus in the later-line setting, with MET amplification the most common off-target acquired mechanism to first-line osimertinib17. However, the majority of resistance mechanisms to first-line treatment are unknown.
Increased understanding of first-line osimertinib resistance mechanisms is essential to inform future therapeutic decisions for patients with EGFRm advanced NSCLC. In this pre-specified analysis, we report the early-onset candidate mechanisms of acquired resistance from plasma samples collected at progression and/or treatment discontinuation in the FLAURA study, from patients for whom there was also a baseline plasma sample with detectable plasma EGFRm. We additionally report the primary mechanisms of resistance to first-line osimertinib detected in baseline tissue samples from patients with and without detectable plasma EGFRm.
Results
Patient characteristics and sample disposition
In FLAURA, 279 patients were randomized to osimertinib and 277 patients to comparator EGFR-TKI (gefitinib/erlotinib); 137 (49%) and 179 (65%) patients, respectively, had paired plasma samples analyzed by NGS, i.e. baseline sample and a sample at disease progression and/or treatment discontinuation (Fig. 1). As of June 2017, progression events occurred in 85 (62%) and 147 (82%) patients in the osimertinib and comparator EGFR-TKI arms, respectively.
Fig. 1. Patient disposition.

CONSORT flow diagram of patient disposition and eligibility in the analysis of acquired resistance mutations in the FLAURA trial *Plasma provided at baseline and at disease progression or treatment discontinuation. EGFR epidermal growth factor receptor, p.o. orally, qd once daily, TKI tyrosine kinase inhibitor.
Among patients with paired plasma samples, 254/316 (80%) had baseline detectable plasma EGFRm (Ex19del/L858R), and were included in the acquired resistance analysis subset: 109/137 (80%) in the osimertinib arm and 145/179 (81%) in the comparator EGFR-TKI arm (Fig. 1). Within this subset, most patients (211/254, 83%) had plasma samples available at treatment discontinuation, compared with a small number of patients with samples only available at disease progression (18/254, 7%) (Fig. 1). There were 25/254 patients (10%) with samples available at both disease progression and treatment discontinuation; only the last collected sample was included in the analysis for these patients (treatment discontinuation, n = 13; disease progression, n = 12).
There were 224 patients with a discontinuation sample as the last sample collected during the study. Reasons for discontinuation were disease progression (144; 64%), adverse event (26; 12%), patient decision (5; 2%), and one patient (<1%) was reported as ‘other’. No reason was given for 48 patients (21%).
Baseline demographics and clinical characteristics in the acquired resistance analysis subset were broadly similar to the overall FLAURA population and generally balanced between the treatment arms although there was a slightly higher percentage of patients with baseline EGFR Ex19del mutation in the comparator EGFR-TKI arm, compared with the osimertinib arm (Table 1).
Table 1.
Patient demographics of the overall FLAURA population and the resistance analysis subseta
| Characteristic | Osimertinib | Comparator EGFR-TKI | ||
|---|---|---|---|---|
| Overall population (n = 279) | Resistance analysis subset (n = 109) | Overall population (n = 277) | Resistance analysis subset (n = 145) | |
| Age: median (range), years | 64 (26–85) | 62 (26–83) | 64 (35–93) | 63 (35–93) |
| Sex: male/female, n (%) | 101 (36)/178 (64) | 43 (39)/66 (61) | 105 (38)/172 (62) | 48 (33)/97 (67) |
| Race: Asian/Non-Asian, n (%) | 174 (62)/105 (38) | 71 (65)/37 (34)b | 173 (62)/104 (38) | 92 (63)/53 (37) |
| WHO performance status: 0/1/2, n (%) | 112 (40)/167 (60) /0 | 42 (39)/67 (61)/0 | 116 (42)/160 (58) /1 (<1) | 56 (39)/89 (61)/0 |
| EGFR mutations: Ex19del/L858R/no mutation detected, invalid test, or no or inadequate sample, n (%) | 158 (57)/97 (35) / 24 (9) | 59 (54)/39 (36)/11 (10) | 155 (56)/90 (32) / 32 (12) | 90 (62)/44 (30)/11 (8) |
| Histology: adenocarcinoma/other, n (%) | 275 (99)/4 (1) | 108 (99)/1 (1) | 272 (98)/5 (2) | 142 (98)/3 (2) |
EGFR epidermal growth factor receptor, EGFRm EGFR mutation-positive, EGFR-TKI epidermal growth factor receptor tyrosine kinase inhibitors, Ex19del Exon 19 deletion, WHO World Health Organization.
aSubset of patients with detectable baseline plasma EGFRm who progressed or discontinued treatment up to March 2019.
bOne patient in the resistance analysis subset of the osimertinib arm had missing racial data. In the overall population, five patients (two in the osimertinib arm and three in the comparator EGFR-TKI arm) had large-cell carcinoma; three patients (one in the osimertinib arm and two in the comparator EGFR-TKI arm) had adenosquamous carcinoma; and one patient (in the osimertinib arm) had a carcinoid tumor.
Acquired resistance mechanisms by treatment arm (plasma ctDNA analysis): Osimertinib arm
In the osimertinib arm acquired resistance analysis subset, 38/109 (35%) patients had a detectable acquired resistance mechanism, 71 (65%) had no detectable candidate mechanism of resistance, and there was no evidence of acquired EGFR T790M. The most common acquired resistance mechanism detected was MET amplification, occurring in 17 patients (16%), followed by mutations in EGFR in 11 patients (10%) (Fig. 2A, Supplementary Table 1). C797S occurred in seven patients (6%), including one patient with co-occurring C797S and C797N; L718Q occurred in two patients (2%; one patient had concurrent L718V), G796S in one patient (1%) and S768I in one patient (1%). Single acquired resistance mechanisms were detected in 23 (21%) patients, including nine (8%) with MET amplification, five (5%) with EGFR mutations, three (3%) with PIK3CA E545K, and one (1%) patient each with CDK6 amplification, CDK4 amplification, CCND1 amplification, KRAS A146T, BRAF V600E, and PIK3CA E453K.
Fig. 2. Acquired mutations following treatment with osimertinib and comparator EGFR-TKIs.

Tile plots indicating (A) acquired mutations in patients treated with osimertinib (n = 109) and (B) acquired mutations in patients treated with comparator EGFRI-TKIs (n = 145) from the FLAURA trial. Source data are provided in the Supplementary Data 1 file. *One patient had co-occurring C797S and C797N. EGFR epidermal growth factor receptor.
More than one acquired resistance mechanism was detected in 15 (14%) patients (Fig. 2A), meaning 39% of all patients with an acquired resistance mechanism had multiple mechanisms detected. Among patients with MET amplifications, five (5%) co-occurred with cell cycle gene alterations (one patient with multiple amplifications in CCND1, CCND2 and CDK6), one (1%) co-occurred with alterations in BRAF (V600E), ALK fusion and EGFR C797S, one (1%) co-occurred with HER2 amplification, and one (1%) co-occurred with KRAS G12C. Among patients with EGFR mutations, and without co-occurring MET amplification, three (3%) co-occurred with MAPK/PI3K alterations, one (1%) co-occurred with CDK6 amplification and one (1%) with CCND1 amplification. Two (2%) further patients had co-occurring cell cycle gene alterations, CCNE1/CDK4 amplifications and CCNE1/CCND3 amplifications, with the latter also co-occurring with HER2 amplification.
Acquired resistance mechanisms by treatment arm (plasma ctDNA analysis): comparator EGFR-TKI arm
In the comparator EGFR-TKI arm acquired resistance analysis subset, 71/145 (49%) patients had a detectable acquired resistance mechanism, 74 (51%) had no detectable candidate mechanism of resistance. The most common acquired resistance mechanism detected was EGFR T790M mutation, occurring in 64 (44%) patients, followed by MET amplification in nine patients (6%) and CDK6 amplification in six (4%) patients (Fig. 2B, Supplementary Table 1). Acquired EGFR T790M was found in a similar proportion of patients with baseline plasma Ex19del (44/98 [45%]) versus L858R (19/46 [41%]); one patient (1%) with baseline Ex19del and L858R acquired EGFR T790M. Single acquired resistance mechanisms were detected in 57 (39%) patients, including 51 (35%) with EGFR T790M, four (3%) with MET amplification, and one (1%) patient each with PIK3CA E545K and BRAF D594N.
More than one acquired resistance mechanism was detected in 14 patients (10%), all except one with EGFR T790M (Fig. 2B), and overall, 20% of patients with an acquired resistance mechanism had multiple mechanisms detected. Among seven (5%) patients with cell cycle gene alterations plus EGFR T790M, three (2%) had co-occurring MET amplification, and one (1%) had co-occurring PIK3CA E545K, EGFR C797S, and EGFR L718Q. Among two (1%) patients with HER2 amplification plus EGFR T790M, one (1%) had co-occurring RET fusion, and one (1%) had multiple co-occurring MAPK/PI3K alterations. In one patient without EGFR T790M, co-occurring alterations were detected in MET, HER2, KRAS, CCNE1 and CDK6.
Duration of treatment by candidate resistance mechanism
Among patients in the resistance analysis subset, there appeared to be no clear association between the type of acquired resistance mechanism and duration of treatment with either osimertinib or comparator EGFR-TKIs (Fig. 3, Supplementary Fig. 1), although in the osimertinib arm, acquired MET amplification occurred in 11 of the 15 patients with the shortest duration of treatment.
Fig. 3. Osimertinib duration of treatment by candidate resistance mechanisms.
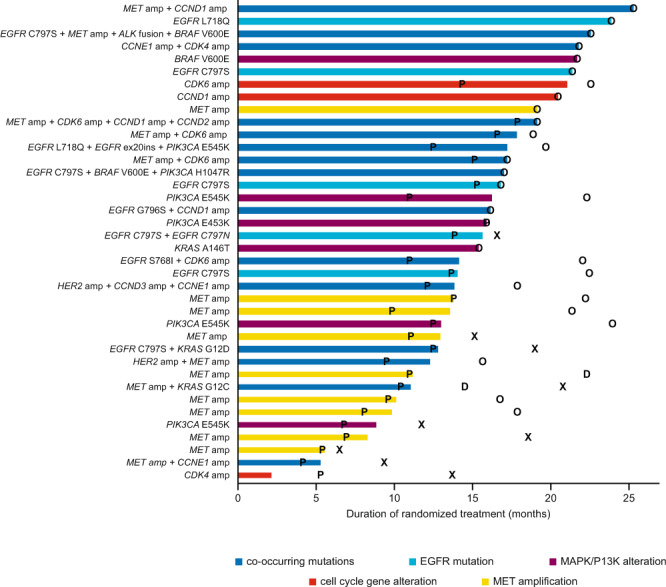
Swimmer plot indicating duration of treatment with osimertinib (months) by resistance mechanisms (n = 109 total, n = 38 with detected resistance mutation). Source data are provided in the Supplementary Data 1 file. EGFR epidermal growth factor receptor, X time of death for patients who have died, O date last known alive for patients who have not died, P time of progression, as assessed by investigator, D time of study discontinuation.
Resistance mechanisms at baseline (tissue genomics)
Across both treatment arms, 147 patients had sufficient tissue volume available and consented for baseline NGS testing, with a pass/qualified result obtained from 104 patients (71%; osimertinib n = 46, comparator EGFR-TKI n = 58). Reasons for a failed test result included insufficient tumor content (n = 24) or insufficient DNA extraction (n = 13); six samples were recorded as ‘other’.
In addition to EGFR mutations, the most commonly altered genes included TP53 (62%), EGFR amplification (20%), RB1 (12%), RBM10 (5%), HER2 (3%), MET (3%), SMARCA4 (3%), and RICTOR (3%) (Table 2). TP53 mutations were distributed between missense alterations (28/64; 44%) and loss-of-function mutations (frameshift, truncation, splice site, homozygous deletions; 36/64; 56%).
Table 2.
Summary of baseline genomic alterations in patients with valid tissue NGS resulta and by suboptimalb tumor response to treatment or not
| Gene/mutation, n (%) | All evaluable (N = 104) | Suboptimalb responder (n = 23) | Not suboptimalb responder (n = 81) |
|---|---|---|---|
| TP53 any known/likely | 64 (62) | 15 (65) | 49 (60) |
| TP53 frameshift/truncation | 26 (25) | 2 (9) | 24 (30) |
| TP53 splice | 9 (9) | 2 (9) | 7 (9) |
| TP53 missense | 28 (27) | 11 (48) | 17 (21) |
| TP53 homozygous deletion | 1 (1) | 0 | 1 (1) |
| EGFR amplification | 21 (20) | 7 (30) | 14 (17) |
| RB1 any known/likely | 12 (12) | 4 (17) | 8 (10) |
| RBM10 truncation/splice | 5 (5) | 4 (17) | 1 (1) |
| SMARCA4 missense/truncation | 3 (3) | 3 (13) | 0 |
| RICTOR amplification | 3 (3) | 3 (13) | 0 |
| HER2 amplification/missense | 3 (3) | 3 (13) | 0 |
| MET amplification | 3 (3) | 2 (9) | 1 (1) |
| AKT2 any known/likely | 2 (2) | 2 (9) | 0 |
| CDK6 any known/likely | 2 (2) | 2 (9) | 0 |
| FGF23 any known/likely | 2 (2) | 2 (9) | 0 |
| BRCA2 any known/likely | 2 (2) | 2 (9) | 0 |
| APC any known/likely | 6 (6) | 3 (13) | 3 (4) |
EGFR epidermal growth factor receptor.
aKnown/likely refers to alterations either known or likely to have a functional impact on a given protein, as determined by the algorithm described in Carr et al.49.
bPatients whose tumors had a suboptimal response were defined by either a best overall response of stable disease or progressive disease with a PFS of <6 months, or non-clearance of plasma ctDNA measured by ddPCR at 6 weeks.
Patients whose tumors had a suboptimal response to treatment (n = 23 among patients with baseline tissue results) were defined by either a best overall response of stable disease or progressive disease with a PFS of <6 months, or non-clearance of plasma ctDNA measured by ddPCR at 6 weeks. Tumors with suboptimal responses were enriched for TP53 missense alterations and alterations in RBM10, HER2, MET, SMARCA4, or RICTOR compared with tumors not defined as suboptimal response (n = 81) (Table 2). Taken together, an alteration in at least one of these potential negative prognostic genes (n = 38 tumors in total) was significantly enriched in suboptimal response tumors: 20/23 (87%) vs 18/81 (22%) tumors not defined as suboptimal response; Fisher’s exact test, two-sided P < 0.001. EGFR amplification and any TP53 mutation status (either alone or in combination with RB1) were not significantly enriched in tumors with suboptimal responses (Table 2).
Discussion
This pre-specified analysis from the FLAURA study provides further characterization of the pre-existing and acquired resistance mechanisms to first-line osimertinib. Pre-treatment tissue genomics identified potential baseline alterations associated with suboptimal response to EGFR-TKI therapy, including missense TP53 mutations, RBM10, HER2, MET, SMARCA4, and RICTOR, many of which have previously been identified as negative prognostic factors for patients with cancer18–23. Further research to confirm these potential pre-treatment markers of suboptimal response is warranted.
Due to a lack of tissue samples at progression, acquired resistance mechanisms were analyzed using ctDNA. Paired samples were analyzed from patients with detectable baseline plasma EGFRm who experienced disease progression and/or discontinued treatment, revealing candidate mechanisms of acquired resistance in both the osimertinib and comparator EGFR-TKI arms. Consistent with previous studies, EGFR T790M was the most common acquired resistance mechanism in the comparator EGFR-TKI arm, occurring in 44% of patients. Acquisition of EGFR T790M was not observed in the osimertinib arm, supporting the mechanism of action for osimertinib11,13,14,16,24–27. This is consistent with preclinical data, indicating that osimertinib may prevent the emergence of EGFR T790M7,28. To date, no evidence of acquired EGFR T790M has been observed with first-line osimertinib treatment, and in some cases, resistance to later-line treatment with osimertinib has been associated with loss of EGFR T790M from tumors15,27.
Overall, the resistance mechanisms to first-line osimertinib observed in this study appear to be similar to those reported in the second- or later-line settings12,21,22,26–33. Our data also demonstrate that there are some shared resistance mechanisms between first-line osimertinib and first- and second-generation EGFR TKIs, with the main exception of EGFR T790M2,34. The frequency of MET amplification (16%) was similar to that observed in patients treated with second-line osimertinib in the Phase III AURA3 study (18%)14,15, and was the most frequently reported resistance mechanism in both this study and AURA3. Of note, acquisition of MET amplification in this analysis appears more often in osimertinib treated patients compared with first- or second-generation EGFR-TKIs. While the frequency of MET amplification reported here is higher than previously reported with osimertinib in the first-line setting17, other second-line osimertinib studies have reported acquired MET amplification in 5–50% of patients with disease progression26,27,31,33.
Acquired EGFR C797S, an on-target resistance mechanism that occurs following treatment with irreversible inhibitors, is frequently reported with second- or later-line osimertinib treatment, with some studies reporting frequency rates up to 24%26,27,31,33. An analysis of second-line osimertinib in the Phase III AURA314,15 study identified 14% of patients acquired EGFR C797S. In this analysis, acquired C797S mutation was observed in 6% of patients, in line with another study exploring resistance mechanisms to osimertinib in the first-line setting17,35. Development of EGFR C797S resistance may be more common in later-line EGFR T790M-positive disease and has been shown to be a later event in the second-line osimertinib setting17,35, suggesting that EGFR C797S is a late occurring resistance mechanism. The frequency of acquired PIK3CA, KRAS and BRAF mutations was also generally similar to that reported in second-line osimertinib studies14,15,27,31,33.
Apart from EGFR T790M, the acquired resistance mechanisms such as MET amplification and HER2 amplification reported here are also consistent with the acquired resistance mechanisms observed with first- and second-generation EGFR-TKIs34,36.
As many patients developed multiple resistance mechanisms, it is difficult to draw any conclusions regarding duration of treatment and the resistance mutations that were acquired. Although patient numbers are small, MET amplification was identified in 11 of the 15 patients with the shortest duration of treatment, suggesting that MET amplification could be enriched in patients with early disease progression.
Importantly, although our understanding of the new resistance landscape with first-line osimertinib is emerging, no suggestions of new and more aggressive resistance mechanisms were detected, although, as discussed below, there were some limitations with the methodology used in detecting all possible resistance mechanisms. This finding, along with the absence of EGFR T790M development, supports the use of osimertinib in the first-line setting. This is reinforced by the overall population data, demonstrating that first-line osimertinib resulted in longer PFS, OS, and time from randomization to second progression on subsequent treatment versus comparator first-line therapy with a similar safety profile11,12. Furthermore, osimertinib has demonstrated CNS efficacy in patients with stable CNS metastases and EGFRm advanced NSCLC, with first-line osimertinib reducing the risk of CNS progression compared with first-generation EGFR-TKIs9,10,37.
The results suggest a need to identify biomarker-matched treatments to target specific mechanisms of acquired resistance, or to prevent the emergence of these mechanisms. For example, data from the Phase Ib TATTON study (NCT02143466) provide evidence for the potential use of osimertinib in combination with the MET inhibitor, savolitinib, therefore targeting one of the most frequent acquired resistance mechanisms to osimertinib observed in this study: MET amplification. This combination demonstrated promising preliminary anti-tumor activity in patients with MET-amplified advanced NSCLC, who experienced progression on prior first-, second-, or third-generation EGFR-TKIs38,39. Those who had progressed on prior first-/second-generation EGFR-TKIs achieved a median duration of response of 7.1 months, with an objective response rate of 52%39. SAVANNAH (NCT03778229), a Phase II study assessing the efficacy of osimertinib plus savolitinib in patients with EGFR-mutant, MET-amplified NSCLC who have progressed on osimertinib is investigating this combination further40.
When considering the EGFR C797S mutation, first-generation EGFR-TKIs such as gefitinib do not require irreversible binding to C797 to inhibit EGFR so their combination with osimertinib in the first-line setting may be an effective strategy. Preliminary data from preclinical studies have supported this concept with the combination of erlotinib and osimertinib resulting in EGFR signaling inhibition when the T790M and C797S mutations were in the trans conformation41. Potential new treatment options to address resistance mechanisms to osimertinib will be further explored in the ORCHARD study (NCT03944772), an open-label, multicenter, multi-drug Phase II platform trial in patients with advanced EGFRm NSCLC whose disease has progressed on first-line therapy with osimertinib.
As limited benefit has been demonstrated for immunotherapy alone in the second-line setting for EGFRm advanced NSCLC, other emerging options for patients with acquired resistance include the use of immunotherapy in combination with other therapies42. Results from the Phase III IMpower150 study demonstrated that addition of atezolizumab to bevacizumab plus chemotherapy significantly improved PFS and OS in patients with metastatic nonsquamous NSCLC previously treated with at least one EGFR-TKI, regardless of PD-L1 expression and EGFR or ALK genetic alteration status43. Alternative approaches following progression on EGFR-TKIs are also being investigated, such as the ongoing Phase Ib/II study investigating the combination of osimertinib and the anti-CD73 monoclonal antibody, oleclumab (NCT03381274)44.
There are several limitations with this study. Analysis of plasma ctDNA may underestimate amplification events so the frequency of acquired MET amplification (16%) may be an underestimate and is expected to be higher in tissue45. Plasma NGS analysis only focuses on genomic alterations detectable in ctDNA; therefore, non-genetic mechanisms of resistance including histological transformation and protein expression alterations were not evaluated in this analysis; small cell lung cancer transformation could not be pathologically confirmed. In addition, longitudinal monitoring of baseline resistance mutations detectable at progression was not possible due to lack of tissue and plasma matched pairs, NGS panel limitations and no/low ctDNA content in plasma. Other limitations of these analyses include the exploratory nature using a subset of patients defined post-baseline and the exclusion of patients without detectable EGFRm at baseline. Therefore, analyses are descriptive, and there may be additional resistance mechanisms that have not been captured, including any that are late-onset.
In conclusion, multiple mechanisms of resistance to first-line osimertinib were observed, with no single mechanism at high prevalence identified thus far. The most frequent resistance mechanisms were MET amplification and the EGFR C797S mutation, and there was no evidence of EGFR T790M-mediated acquired resistance. These results provide no evidence of resistance mechanisms that may lead to unexpected aggressive disease biology. While liquid biopsy provides valuable information to help monitor and identify emerging resistance mechanisms, the results identify the need for complementary testing with tissue for a complete histological diagnosis. This point is addressed in the ongoing ELIOS study (NCT03239340), where collection of paired tissue biopsies (pre-treatment and at progression) will further investigate the mechanisms of acquired resistance to first-line osimertinib. In addition, the ORCHARD and SAVANNAH studies (NCT03944772, NCT03778229, respectively) are also underway to investigate potential new treatment options to address resistance mechanisms to osimertinib in patients with advanced EGFRm NSCLC whose disease has progressed on first-line therapy with osimertinib.
Methods
Standard protocol approvals, registration and patient consents
FLAURA was conducted in accordance with the provisions of the Declaration of Helsinki, Good Clinical Practice guidelines as defined by the International Conference on Harmonisation, applicable regulatory requirements and the AstraZeneca policy on Bioethics and Human Biological Samples. All patients provided written informed consent before screening. This study was funded by the study sponsor (AstraZeneca) and designed by the principal investigators and the sponsor. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Full study protocol available at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=12356.
Study design and participants
FLAURA was a randomized, double-blind, Phase III study, full details of which have been previously published12. Briefly, enrolled patients (N = 556) were aged ≥18 years (≥20 years in Japan) with previously untreated, EGFRm locally advanced or metastatic NSCLC with tumors harboring EGFR-TKI sensitizing mutations (Exon 19 deletion [Ex19del] or L858R). Patients were stratified by mutation status (Ex19del/L858R) and race (Asian/non-Asian), and randomized 1:1 to osimertinib 80 mg once daily (qd; n = 279) or comparator EGFR-TKI (n = 277, gefitinib 250 mg qd or erlotinib 150 mg qd). Patients randomized to comparator EGFR-TKIs were allowed to cross over to osimertinib if they acquired the EGFR T790M resistance mutation and were confirmed to have objective disease progression by a blinded independent central review (or by investigator assessment if disease progression occurred after the primary data cutoff).
The ctDNA and tissue analyses presented here were exploratory, pre-specified, retrospective analyses of a subset of patients. ctDNA analyses were limited to patients who had progressed or discontinued treatment with detectable baseline plasma EGFRm (resistance analysis subset). Patients who did not have detectable plasma EGFRm at baseline were excluded. In addition, 19 patients from China were excluded due to sample export limitations. For the overall FLAURA population, study entry criteria for EGFRm were based on tissue sample analysis.
Plasma ctDNA and tissue analysis
Serial plasma samples were collected at baseline, 2 weeks, 3 weeks, 6 weeks, 9 weeks, 12 weeks and every 6 weeks thereafter, as well as at disease progression and/or treatment discontinuation. This analysis assessed paired plasma samples collected at baseline and following disease progression and/or treatment discontinuation up to March 2019. Where a sample was available at both progression and at treatment discontinuation, data are reported for the last sample collected, as treatment with osimertinib was allowed following progression. Consequently, samples from patients who remained on treatment post-progression would have been taken after disease progression occurred. Plasma ctDNA samples were analyzed using NGS (Guardant Health, Guardant360 74 gene panel or GuardantOMNI 500 gene panel). All 74 genes on the Guardant360 panel were included in the GuardantOMNI 500 gene panel. The limit of variant allelic fraction detected was 0.04–0.06%. All analyses from each patient (at baseline and following progression and/or treatment discontinuation) were reported only for genes included across both panels used. Genomic alterations were identified using Guardant Health’s proprietary bioinformatics pipeline46,47.
Baseline tumor tissue samples were used to analyze co-occurring mutations at baseline that would be associated with suboptimal response to osimertinib. Sufficient tissue for additional genomic analyses from the mandatory tumor tissue biopsy collected during screening was available from 147 patients. Tissue samples were analyzed using the FoundationOne CDx panel48.
Assessments
Disease progression was assessed by the investigator, according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, every 6 weeks for 18 months, then every 12 weeks until objective progressive disease. After primary data cutoff (June 2017), tumor assessments were carried out in line with clinical practice, and scans were not centrally collected. Acquired mechanisms of resistance were identified in both treatment arms by comparing paired plasma samples at baseline and at disease progression and/or treatment discontinuation in patients with detectable plasma EGFRm at baseline. Primary mechanisms of resistance were identified in both treatment arms from tissue samples collected at baseline in patients with and without detectable plasma EGFRm. Baseline tissue and plasma NGS samples from patients with detectable EGFRm were also compared. Duration of randomized treatment was defined as the time from randomization until end of EGFR-TKI treatment, and was determined for candidate resistance mechanisms in the osimertinib arm and presented as swimmer plots.
Statistical methods
This pre-specified preliminary analysis was exploratory in nature and, as such, data were summarized using descriptive statistics. Plasma samples at progression or treatment discontinuation included in the paired analysis were collected up until March 2019. Clinical data were analyzed using June 12, 2017 data cutoff; in patients with events occurring after June 12, 2017 were censored in clinical analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Thanks to all the patients and their families. The study (NCT02296125) was funded by AstraZeneca (Cambridge, UK) the manufacturer of osimertinib. The sponsor, AstraZeneca, was involved in the study design and analysis. The authors would like to acknowledge Natalie Griffiths, PhD, of Ashfield MedComms, an Inizio Company, for medical writing support, that was funded by AstraZeneca, Cambridge, UK, in accordance with Good Publications Practice (GPP) guidelines (https://www.ismpp.org/gpp-2022).
Author contributions
J.C. designed the exploratory analyses, co-led the overall analysis, and contributed to and approved the manuscript. J.E.G. contributed to and approved the manuscript. Y.C. contributed to data collection and data interpretation and reviewed and approved the manuscript. Y.O. contributed to and approved the manuscript. F.I. contributed to and approved the manuscript. B.C.C. contributed to and approved the manuscript. M-C.L. contributed to and approved the manuscript. M.M. reviewed and approved the study results and manuscript. R.S. contributed to and approved the manuscript. Y.R. co-led the overall analysis and contributed to and approved the manuscript. A.T. co-led the overall analysis and contributed to and approved the manuscript. A.M. performed the genomic and statistical analyses of plasma and tissue samples, and contributed to, edited, reviewed and approved the study results and manuscript. J.C.B. designed the study, interpreted the data, contributed to the writing of the manuscript, and approved the manuscript. R.J.H. co-led the overall analysis and contributed to and approved the manuscript. S.S.R. has made substantial contributions to the conception and design of the work, co-led the overall analysis and contributed to and approved the manuscript. All authors reviewed and approved the manuscript.
Peer review
Peer review information
Nature Communications thanks Collin Blakely and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The de-identified patient data generated in this study are provided in Supplementary Data 1. Specific consent for sequencing data deposition was not obtained from patients. Anonymized patient-level clinical data, aggregated clinical data and/or anonymized clinical study documents underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at: http://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Since at the time of this publication the FLAURA trial is still ongoing, the study data will be accessible at https://vivli.org/ when the trial is completed. In the meantime, requests to access the data from the FLAURA trial described in the current manuscript can be submitted through: https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. Requested data is available from approval of the request typically for one year. Some patients/countries may need to be excluded based on the informed consent form or country‐level legislation. Use of data must comply with the requirements of Human Genetics Resources Administration of China and patients who have withdrawn consent for data use will be removed from the shared dataset. Patient-level image or genetic data is not available for access. The remaining data are available within the Article and Supplementary Information.
Competing interests
The authors declare the following competing interests: J.C. reports employment and stock ownership with AstraZeneca. J.E.G. reports receiving honoraria from AstraZeneca, Genenech, Bristol-Myers Squibb, Takeda, EMD Serano Merck; and has received research grants/funding from AstraZeneca, Genentech, Bristol-Myers Squibb, BI, Takeda, Array, Merck. Y.C. reports no conflicts of interest. Y.O. reports undertaking an advisory role for AstraZeneca, Chugai, ONO, Bristol-Myers Squibb, Kyorin, Celltrion, Amgen, and Nippon Kayaku; reports receiving honoraria from AstraZeneca, Chugai, Eli Lilly, ONO, Bristol-Myers Squibb, Boehringer Ingelheim, Bayer, Pfizer, MSD, Taiho, Nippon Kayaku, and Kyowa Hakko Kirin; and has received grants or funds from AstraZeneca, Chugai, Lilly, ONO, BMS, Kyorin, Dainippon- Sumitomo, Pfizer, Taiho, Novartis, Kissei, Ignyta, Takeda, Kissei, Daiichi-Sankyo, Janssen, and LOXO. F.I. has received honoraria and research funding from AstraZeneca. B.C.C. reports stock ownership with TheraCanVac Inc, Gencurix Inc, Bridgebio therapeutics, KANAPH Therapeutic Inc, Cyrus Therapeutics, and Interpark Bio Convergence Corp; reports participating in an advisory role for KANAPH Therapeutic Inc, Brigebio Therapeutics, Cyrus Therapeutics, and Guardant Health; has received consulting fees from Novartis, AstraZeneca, Boehringer-Ingelheim, Roche, Bristol-Myers Squibb, ONO, Yuhan, Pfizer, Eli Lilly, Janssen, Takeda, MSD, Janssen, Medpacto, and Blueprint medicines; has received grants or funds from Novartis, Bayer, AstraZeneca, MOGAM Institute, Dong-A ST, Champions Oncology, Janssen, Yuhan, ONO, Dizal Pharma, MSD, Abbvie, Medpacto, GI Innovation, Eli Lilly, Blueprint medicines, and Interpark Bio Convergence Corp; has received royalties from Campions Oncology; and is the founder of DAAN Biotherapeutics. M-C.L. is on the board of directors for AstraZeneca and Boehringer Ingelheim, has received honoraria from Roche; and has received consulting fees from AstraZeneca, Boehringer Ingelheim and MSD. M.M. has received honoraria from Bristol-Myers Squibb, MSD, Boehringer Ingelheim, AstraZeneca, Roche, Kyowa Kyrin, Pierre Fabre, Takeda, Bayer and has received research funding from Bristol-Myers Squibb. R.S. reports employment with the Maidstone and Tunbridge Wells NHS Trust; participated in an advisory role for AstraZeneca, Roche, Pfizer and Boehringer Ingelheim; and has received honoraria from AstraZeneca, Roche, Pfizer, and Boehringer Ingelheim. Y.R. reports employment and stock ownership with AstraZeneca. A.T. reports employment and stock ownership with AstraZeneca. A.M. reports employment and stock ownership with AstraZeneca. J.C.B. reports employment and stock ownership with AstraZeneca. R.J.H. reports employment and stock ownership with AstraZeneca. S.S.R. has participated in an advisory role for Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, Merck, Tesaro, Takeda, GlaxoSmithKline, Daichii Sankyo, and Eisai; is on the board of directors for the International Association for Study of Lung Cancer; has received consulting fees from Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, Merck, Tesaro, Takeda, GlaxoSmithKline, Daichii Sankyo, and Eisai; and has received grants or funds from Amgen, Advaxis, AstraZeneca, BMS, Merck, Tesaro, Takeda, and Genmab.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
6/1/2023
A Correction to this paper has been published: 10.1038/s41467-023-38999-0
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-35961-y.
References
- 1.Planchard, D. et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. †Updated version published 15 September 2020 by the ESMO Guidelines Committee. 25 September
- 2.Yu HA, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuiper JL, et al. Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung Cancer. 2014;85:19–24. doi: 10.1016/j.lungcan.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Wu SG, et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget. 2016;7:12404–12413. doi: 10.18632/oncotarget.7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arcila ME, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ZF, Ren SX, Li W, Gao GH. Frequency of the acquired resistant mutation T790M in non-small cell lung cancer patients with active exon 19Del and exon 21 L858R: a systematic review and meta-analysis. BMC Cancer. 2018;18:148. doi: 10.1186/s12885-018-4075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross DA, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mok TS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu YL, et al. CNS efficacy of osimertinib in patients with T790M-positive advanced non-small-cell lung cancer: data from a randomized phase III trial (AURA3) J. Clin. Oncol. 2018;36:2702–2709. doi: 10.1200/JCO.2018.77.9363. [DOI] [PubMed] [Google Scholar]
- 10.Reungwetwattana, T. et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J. Clin. Oncol. JCO2018783118 (2018). [DOI] [PubMed]
- 11.Soria JC, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 12.Ramalingam SS, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020;382:41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 13.Thress KS, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le X, et al. Landscape of EGFR -dependent and -independent resistance mechanisms to osimertinib and continuation therapy post-progression in EGFR-mutant NSCLC. Clin. Cancer Res. 2018;24:6195–6203. doi: 10.1158/1078-0432.CCR-18-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chmielecki, J. et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat. Commun.10.1038/s41467-023-35962-x. [DOI] [PMC free article] [PubMed]
- 16.Ramalingam SS, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 2018;36:841–849. doi: 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- 17.Schoenfeld AJ, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin. Cancer Res. 2020;26:2654–2663. doi: 10.1158/1078-0432.CCR-19-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai GGY, et al. Clonal MET amplification as a determinant of tyrosine kinase inhibitor resistance in epidermal growth factor receptor-mutant non-small-cell lung cancer. J. Clin. Oncol. 2019;37:876–884. doi: 10.1200/JCO.18.00177. [DOI] [PubMed] [Google Scholar]
- 19.Canale M, et al. Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin. Cancer Res. 2017;23:2195–2202. doi: 10.1158/1078-0432.CCR-16-0966. [DOI] [PubMed] [Google Scholar]
- 20.Bell EH, et al. SMARCA4/BRG1 is a novel prognostic biomarker predictive of cisplatin-based chemotherapy outcomes in resected non-small cell lung cancer. Clin. Cancer Res. 2016;22:2396–2404. doi: 10.1158/1078-0432.CCR-15-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinayanuwattikun C, et al. Elucidating genomic characteristics of lung cancer progression from in situ to invasive adenocarcinoma. Sci. Rep. 2016;6:31628. doi: 10.1038/srep31628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruder D, et al. Concomitant targeting of the mTOR/MAPK pathways: novel therapeutic strategy in subsets of RICTOR/KRAS-altered non-small cell lung cancer. Oncotarget. 2018;9:33995–34008. doi: 10.18632/oncotarget.26129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu SJ, et al. MicroRNA-200a confers chemoresistance by antagonizing TP53INP1 and YAP1 in human breast cancer. BMC Cancer. 2018;18:74. doi: 10.1186/s12885-017-3930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oxnard GR, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin. Cancer Res. 2011;17:1616–1622. doi: 10.1158/1078-0432.CCR-10-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Planchard D, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015;26:2073–2078. doi: 10.1093/annonc/mdv319. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin. Cancer Res. 2018;24:3097–3107. doi: 10.1158/1078-0432.CCR-17-2310. [DOI] [PubMed] [Google Scholar]
- 27.Oxnard GR, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4:1527–1534. doi: 10.1001/jamaoncol.2018.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eberlein CA, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75:2489–2500. doi: 10.1158/0008-5472.CAN-14-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menon R, et al. A novel EGFR(C797) variant detected in a pleural biopsy specimen from an osimertinib-treated patient using a comprehensive hybrid capture-based next-generation sequencing assay. J. Thorac. Oncol. 2016;11:e105–e107. doi: 10.1016/j.jtho.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Ham JS, et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J. Thorac. Oncol. 2016;11:e1–e4. doi: 10.1016/j.jtho.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Piotrowska Z, et al. MET amplification (amp) as a resistance mechanism to osimertinib (Abstract 9020) J. Clin. Oncol. 2017;35:9020. [Google Scholar]
- 32.Ortiz-Cuaran S, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin. Cancer Res. 2016;22:4837–4847. doi: 10.1158/1078-0432.CCR-15-1915. [DOI] [PubMed] [Google Scholar]
- 33.Lin CC, et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir. Med. 2018;6:107–116. doi: 10.1016/S2213-2600(17)30480-0. [DOI] [PubMed] [Google Scholar]
- 34.van der Wekken AJ, et al. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit. Rev. Oncol. Hematol. 2016;100:107–116. doi: 10.1016/j.critrevonc.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 35.Oxnard G, et al. Osimertinib resistance mediated by loss of EGFR T790M is associated with early resistance and competing resistance mechanisms (Abstract OA 09.02) J. Thorac. Oncol. 2017;12:S1767–S1768. [Google Scholar]
- 36.Nagano T, Tachihara M, Nishimura Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells. 2018;7:212. doi: 10.3390/cells7110212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goss G, et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: pooled data from two phase II trials. Ann. Oncol. 2018;29:687–693. doi: 10.1093/annonc/mdx820. [DOI] [PubMed] [Google Scholar]
- 38.Sequist VL, et al. TATTON phase Ib expansion cohort: osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Abstract CT033 presented at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA. Cancer Res. 2019;79:CT033. [Google Scholar]
- 39.Yu H, et al. TATTON Phase Ib expansion cohort: osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET amplified NSCLC after progression on prior first/second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Abstract CT032 presented at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA. Cancer Res. 2019;79:CT032. [Google Scholar]
- 40.ClinicalTrials.gov. Osimertinib plus savolitinib in EGFRm+/MET+ NSCLC following prior osimertinib (SAVANNAH). Available at: https://clinicaltrials.gov/ct2/show/NCT03778229. Accessed 21 November 2022.
- 41.Niederst MJ, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015;21:3924–3933. doi: 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee CK, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J. Thorac. Oncol. 2017;12:403–407. doi: 10.1016/j.jtho.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Reck M, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019;7:387–401. doi: 10.1016/S2213-2600(19)30084-0. [DOI] [PubMed] [Google Scholar]
- 44.ClinicalTrials.gov. Oleclumab (MEDI9447) EGFRm NSCLC novel combination study (NCT03381274). Available at: https://clinicaltrials.gov/ct2/show/NCT03381274. Accessed 21 November 2022.
- 45.Hartmaier RJ, et al. Detection of MET-mediated EGFR tyrosine kinase inhibitor (TKI) resistance in advanced non-small cell lung cancer (NSCLC): biomarker analysis of the TATTON study. Abstract 4897 presented at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA. Cancer Res. 2019;79:4897. [Google Scholar]
- 46.Genetic Testing Registry (GTR). Guardant360. Available at: https://www.ncbi.nlm.nih.gov/gtr/tests/527948/. Accessed 21 November 2022.
- 47.Odegaard JI, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. 2018;24:3539–3549. doi: 10.1158/1078-0432.CCR-17-3831. [DOI] [PubMed] [Google Scholar]
- 48.Frampton GM, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carr TH, et al. Defining actionable mutations for oncology therapeutic development. Nat. Rev. Cancer. 2016;16:319–329. doi: 10.1038/nrc.2016.35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The de-identified patient data generated in this study are provided in Supplementary Data 1. Specific consent for sequencing data deposition was not obtained from patients. Anonymized patient-level clinical data, aggregated clinical data and/or anonymized clinical study documents underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at: http://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Since at the time of this publication the FLAURA trial is still ongoing, the study data will be accessible at https://vivli.org/ when the trial is completed. In the meantime, requests to access the data from the FLAURA trial described in the current manuscript can be submitted through: https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. Requested data is available from approval of the request typically for one year. Some patients/countries may need to be excluded based on the informed consent form or country‐level legislation. Use of data must comply with the requirements of Human Genetics Resources Administration of China and patients who have withdrawn consent for data use will be removed from the shared dataset. Patient-level image or genetic data is not available for access. The remaining data are available within the Article and Supplementary Information.