

Summary
Eukaryotic mRNA has long been considered monocistronic, but nowadays, alternative proteins (AltProts) challenge this tenet. The alternative or ghost proteome has largely been neglected and the involvement of AltProts in biological processes. Here, we used subcellular fractionation to increase the information about AltProts and facilitate the detection of protein-protein interactions by the identification of crosslinked peptides. In total, 112 unique AltProts were identified, and we were able to identify 220 crosslinks without peptide enrichment. Among these, 16 crosslinks between AltProts and Referenced Proteins (RefProts) were identified. We further focused on specific examples such as the interaction between IP_2292176 (AltFAM227B) and HLA-B, in which this protein could be a potential new immunopeptide, and the interactions between HIST1H4F and several AltProts which can play a role in mRNA transcription. Thanks to the study of the interactome and the localization of AltProts, we can reveal more of the importance of the ghost proteome.
Subject areas: Molecular network, Cell biology, Proteomics
Graphical abstract
Highlights
-
•
The ghost proteome has been neglected and its role is still unknown
-
•
Subcellular localization of 112 alternative proteins was described
-
•
220 crosslinks were identified involving 16 alternative proteins
-
•
Using this method, we identified the possible physiological role of these proteins
Molecular network; Cell biology; Proteomics
Introduction
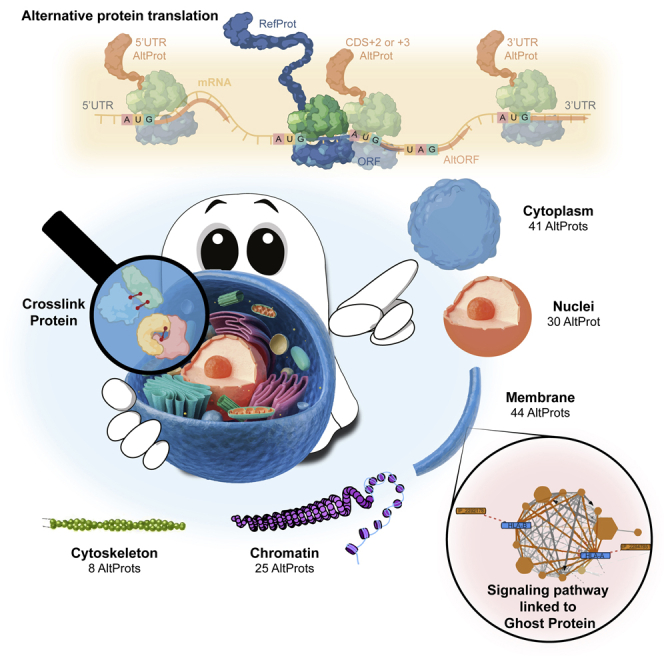
Since 2011 considerable efforts have been made to shed light on unreferenced proteins also called the ghost proteome; in various biological contexts.1,2,3,4,5 This ghost proteome, being a part of the total protein landscape, points to proteins not referenced in conventional databases like UniProt6 and RefSeq.7 Such ghost proteins, called alternative proteins (AltProts) or proteins coded by small open reading frames (smORFs),8 were identified to be translated from regions of mRNA molecules described as non-coding, e.g. 3′ and 5′ UTR, reading frame shifts3 or involve all kinds of non-coding RNA (ncRNA)9 (Figure 1). AltProts have the particularity of having an average size of less than 100 amino acids,10 likewise their sequences, despite being derived from a mRNA coding for a referenced protein (RefProt), have a completely different amino acid sequence, suggesting a different biological function. AltProts are estimated at 450,000 potential sequences,3,11 compared to 79,038 RefProt sequences (Uniprot-01.2022), hence a five times larger proteome than currently considered. The ghost proteome is thus also a potentially rich source of biomarkers of major interest for the understanding of pathophysiology and it has already been studied on endometrial cancer12 and breast cancer,13 and on glioblastoma.14,15 Indeed, ghost proteins, physiologically present in cells, can be impacted by mutations, which might impact the signaling pathways in which they are involved.15 However, although AltProts have been identified in a wide variety of contexts and especially in cancer, their functions often remain enigmatic.16,17 Studies on AltProts are often limited as case-by-case, complex and costly biomolecular studies to obtain functional protein information are lacking.18,19,20 Few untargeted strategies have enabled the identification of the molecular function of a protein in a single analysis. Bioinformatics tools, including linking protein functional information through networks and gene ontology (GO) analysis, are powerful tools for this purpose.21 Such tools allow to redraw the signaling pathways and group together RefProts belonging to the same biological process, molecular function, or cellular localization, increasing the information about the cellular mechanism. Such information can be obtained through databases holding information on protein-protein interactions (PPIs) such as STRING,22 BioGrid,23 and IntAct24 allowing them to be applied to a large-scale protein analysis such as a bottom-up approach by chromatography coupled to mass spectrometry analysis (LC-MS/MS) of RefProts. However, similar PPI data are for AltProts are currently largely unknown and AltProts remain largely understudied as baits for identifying PPIs.
Figure 1.

Schematic representation of the translation of AltProts coded from AltORFs
Top panel: translations of RefProts at the CDS region. Middle panel: AltProts translated from 5′ AND-3′ UTRs and CDS +2, +3 frames. Bottom panels: AltProts encoded from a lncRNA.
One interesting approach to obtain PPI data involving AltProts is based on the use of crosslinkers combined with analysis by mass spectrometry (XL-MS). This hypothesis-free strategy, when applied to a complex mixture such as a cell extract, fixes actual PPIs present and allows us to identify new interactions of a bait protein. When processing XL-MS data, one may search for AltProts by using a database holding AltProt sequences. XL-MS has been applied for the structural analysis of purified proteins and to identify interactions in purified protein complexes. However, XL-MS holds some limitations when applied to the large-scale exploration of cellular PPIs, the main ones being the low number of crosslinked peptides that get identified compared to non-crosslinked peptides and the identification of cross-linked peptides because of their complex spectra. To increase the identification of the former, enrichment workflows can be implemented. Such enrichment depletes non-crosslinked (or free) peptides upon sample fractionation, which is generally carried out by size exclusion chromatography (SEC) or cation exchange chromatography (SCX). However, despite a significant increase in the identification rate of crosslinked peptides, such approaches require a rather large amount of material (minimum 60 million cells25 or 2 mg of protein.26 Other strategies that are currently emerging are generally based on the use of customized crosslinkers (non-commercial), often tri-functional, allowing targeted enrichment of crosslinked peptides by the functionalized third arm of the molecule. However, these customized crosslinkers also require large quantities of biological material.
In an era where mass spectrometry-based proteomics aims to study proteomes at the single-cell level or is applied to clinical samples of limited quantity, we came up with a strategy to increase the identification of crosslinked peptides while using relatively small amounts of material. Our strategy is based on the decomplexation of the sample. Considering that a limiting factor of XL-MS analysis without enrichment is the (too) high signal intensity of free peptides, we have chosen to divide the cell into different fractions. Thus, from a reasonable number of cells (3 million cells), we separate the proteome into membrane, cytoplasm, nuclear, chromatin, and cytoskeleton proteomes. This strategy was chosen as it has a double advantage: it allows us to increase the number of identified crosslinked peptides without prior peptide fractionation and it provides information on the cellular localization of the identified AltProts. Very little information exists on the subcellular location of AltProt, yet, in some targeted studies, it was reported that AltProts could have a different location compared to the RefProt originating from the same mRNA. Another example shows a co-localization with the associated RefProt, for the cooperation or co-regulation of the gene via its AltProt.27 The use of this strategy allows us to “kill two birds with one stone” to optimize the detection of interactions involving AltProt to assign signaling pathways in a non-targeted way and to provide information on the possible localization of AltProt in the cell.
Thus here, we propose the use of subcellular fractionation to increase the identification rate of crosslinked peptides and simultaneously provide information on the cellular localization of identified AltProts. As such, cellular functions of AltProts can be assessed in a non-targeted way, which is expected to increase our understanding of the ghost proteome.
In this study, we propose the use of subcellular fractionation in order to increase the rate of identification of crosslinked peptides all by providing information on the localization of AltProt in the cell. This is to highlight the functions of AltProt in a non-targeted way and to progress in the understanding of the ghost proteome.
Results
In cellulo crosslinking, subcellular fractionation, and protein digestion
Overview of the workflow used
As the function of the vast majority of AltProts predicted from OpenProt Database3 remains unknown, as mentioned earlier, we used crosslinking mass spectrometry to characterize AltProts in a non-targeted way. To obtain more information on AltProts on a large scale and to optimize the identification of crosslinked peptides, we set up a workflow combining in cellulo crosslinking with subcellular fractionation and analysis by nLC-MS/MS. Additionally, to confirm the presence of crosslinked proteins SDS-PAGE was used and Western blotting to confirm the efficiency of subcellular fractionation (Figure 2A). Finally, the generated data were integrated to identify AltProts, their partners, and the signaling pathways they are involved in.
Figure 2.

Description of the general workflow used
(A) The first step included harvesting and in celullo crosslinking, followed by subcellular fractionation and SDS-PAGE to confirm the crosslinking reaction. Additionally, Western blotting was employed to verify subcellular fractionation. Finally, nLC-MS/MS analysis and crosslinking network revision were performed.
(B) Coomassie blue stained SDS-PAGE: each crosslinked subcellular fraction was compared to a non-crosslinked fraction. BSA crosslinked or not was used as controls. Red arrows display the crosslinked signals.
(C) Western blot signals obtained from each fraction. HSPA1A signal is present in the cytoplasm fraction. For calreticulin, signals are observed at chromatin, cytoskeleton and a more intense signal at the membrane-bounded fraction. SP1 is observed at nucleus and cytoskeleton. Histone H3 is found in chromatin and cytoskeleton. Cytokeratin 18 is found at Nucleus and cytoskeleton. These results correspond to the ones found in UniProtKB, COMPARTMENTS, and the literature.
For crosslinking, we used in cellulo DSSO treatment on replicates of 3E6 immortalized human ovarian cells (T1074-ABM). Following crosslinking and quenching, subcellular protein fractionation was used to extract five different protein fractions corresponding to cytoplasm (Cyt), membrane-bound (Memb), nuclear (Nuc), chromatin-bound (Chr), and cytoskeletal (Ske) proteins.
Characterization of the crosslink reaction
Protein crosslinking was visualized by SDS-PAGE Figure 2B. The formation of protein complexes by crosslinking prevents the migration of these complexes in the separation gel (12% acrylamide). As a result, intense protein staining is observed between the stacking and separation gels, even with protein staining in the wells at the entrance of the stacking gel pointing to the formation of protein complexes that are so large that they cannot enter the stacking gel (4% acrylamide). Note that this was only observed when analyzing crosslinked samples and for the positive control of crosslinking reaction (BSA). In crosslinking sample, a “blur” of migration can be observed, this could be formed by smallest structures like intra-protein crosslinks and small(er) protein complexes. Interestingly, almost complete protein crosslinking is found for most of the analyzed subcellular fractions, except for the chromatin fraction where a clear band is observed in the separation gel that is also present in the non-crosslinked control, pointing to a protein that is not affected (or only slightly) by the crosslink used.
Evaluating the efficiency of the subcellular fractionation
The efficiency of the subcellular fractionation procedure was evaluated on non-crosslinked cells. To determine if we were able to extract known-location proteins. Five protein markers were selected according to their subcellular location and already tested by the kit’s vendor, HSPA1A for the cytoplasm, calreticulin for the membrane-bounded proteins, SP1 for the nucleus, Histone H3 for the chromatin, cytokeratin 18 for the cytoskeleton. To verify the subcellular location UniProtKB was used as a reference. In Figure 2C the band corresponding to HSPA1A is clearly observed in the cytoplasmic fraction, and additionally, a weak signal is found in the cytoskeletal fraction. According to UniProt: P0DMV8, HSPA1A can be found in the cytoplasm and at the cytoskeleton, which correlates with the signals observed in the blot. At UniProt, calreticulin (UniProt: P27797) is referenced in the membrane of several organelles. Furthermore, it has been described in the chromatin28 and cytoskeleton.29 SP1 (UniProt: P08047) was annotated to reside in the nucleus and in the cytoplasm. Here, we observed two strong signals in the nucleus and the cytoskeleton, the latter can be explained by the fact that in mitosis SP1 can be redirected toward the microtubules.30 For histone H3, two signals can be observed: in the chromatin and cytoskeleton fractions. According to UniProt: P68431, this protein can be found in the nucleus and at chromosomes, according to the mitosis process, during the cell division chromatin is in contact with the microtubule and can explain why histone H3 is also identified in cytoskeleton fraction. Signals for cytokeratin 18 were observed in the nuclear and cytoskeleton fraction, while UniProt: P05783, annotates this protein in the cytoskeleton and nucleus, and sometimes in the cytoplasm. Even if the power of compartment separation is still limited, by this method we can obtain a first view of the cellular compartments repartition of the protein. Therefore, we will be able to propose a cellular localization to the AltProts identified by this methodology.
Identification of RefProts
The MS/MS data from the crosslinked samples were analyzed by Proteome Discoverer V2.5 using Sequest HT.31 We initially focused on the RefProts (databased Uniprot 02-2022) and could identify 4,753 unique RefProts, of which 2,557 were identified in the cytoplasmic fraction, 2,731 in the membrane, 2,808 in the nucleus, 1,794 in the chromatin fraction and 2,781 in the cytoskeleton, with a high number of proteins shared by different fractions (Figure 3A). The fraction in which more compartment-specific identifications were found was the membrane fraction (538), followed by the cytoskeleton (375), cytoplasm (369), nucleus (344), and chromatin fraction (127). A gene ontology (GO) cellular component enrichment analysis was performed using the STRING app32 at ClueGO33 Figure 3B. In general, and as expected, the number of indexed proteins in STRING is less than the ones identified. Moreover, the number of proteins that possess the GO term for the compartment in which it was identified is very low for the chromatin and cytoskeleton fractions.
Figure 3.

Subcellular fractionation analysis
(A) Venn diagram displaying the distribution of reference proteins identified in the different subcellular fractions.
(B) Bar chart showing the number of RefProts identified (red), the number of RefProts indexed in STRING (blue), and the number of RefProts that contain the GO term of the localization corresponding to the fraction where it was found.
(C) Bar chart displaying the number of AltProts identified in at least two replicates in the same subcellular compartment.
Identification and characterization of AltProts
Following a similar approach as for the RefProts, AltProts were identified, now using the OpenProt database. A total of 112 AltProts were identified in at least two replicates in the same subcellular compartment (Figure 3C). The highest number of AltProts (44) was found in the membrane-bound fraction, followed by cytoplasmic AltProts (41), 30 in the nucleus, 25 in the chromatin fraction, and eight in the cytoskeletal fraction. Of note, 24 AltProts were identified in two or more cellular compartments. With the ability to separate subcellular proteins we can propose information about localization for the AltProts identified. Such information is important as the function of a protein depends, amongst others, on the cellular compartment or organelle where it is localized, as this provides the necessary physiological context, aiding the functional characterization of AltProts. Further analyses showed that 88.3% of the identified AltProts originate from non-coding RNAs (ncRNA), 5% from miscellaneous RNAs (misc_RNA), 3.3% from a frameshift in the mRNA CDS, and 1.7% from each of the 3′ and 5′ UTR mRNA regions (Figure 4A). Considering the distribution of the molecular weights of the identified AltProts, more than 5% of the AltProts have molecular weights below-30 kDa (Figure 4B). In Table S1, the complete description of the AltProts, protein Blast results, and the unique peptide identified in MS/MS corroborate by NextProt Peptide uniqueness checker.34 The OpenProt database holds information on the prediction of protein domains in AltProts, made possible by comparisons with RefProt sequences and domain annotations made with algorithms like InterProScan.35 Domains describe a structural or functional entity that is typically evolutionary conserved among orthologs. Of the 112 AltProts identified, 17.9% do not have any annotated protein domain (Figure 4C), while the intermediate filament protein domain was the major domain found (18.8%), with beta-tubulin and actin family domains also identified. This points to the fact that some AltProts might function as structural proteins. Other retrieved protein domains relate to ribosomal proteins, translation and elongation factors and chaperonins, and an RNA recognition motif. Further, a great heterogeneity was observed, represented by the “Other” section (Figure 4C) which does not allow a proper breakdown into different domains, yet represents 20.5% of the AltProts identified.
Figure 4.

AltProts properties
(A) RNA type distribution found among the 112 AltProts identified.
(B) Molecular weight distribution of the AltProts identified.
(C) Predicted protein domain distribution of the AltProts, retrieved from the OpenProt database.
In summary, our results show that our methodology provides robust information about AltProts. For instance, IP_596971 found in the membrane-bound fraction possesses a major histocompatibility complex (MHC) class I signature domain which is usually found at the cell membrane. Along the same line, IP_566083 identified in the same fraction has a transmembrane transport protein domain. Another example is IP_775646, identified in the cytoplasmic fraction, possessing a ribosomal protein domain (Table S1).
Crosslink network analysis
Next, the XlinkX algorithm36 implemented to PD2.5 was used to identify the crosslinked peptides and build protein interaction maps. A total of 220 crosslinks (see Table S2) were identified without targeted crosslinked protein or peptide enrichment. Among these 220 crosslinks, 16 crosslinks were found involving an AltProt. The membrane fraction had the highest number of identified crosslinks (88, Figure 5A), which could be explained by DSSO first reacting with surface-exposed membrane proteins upon its administration to cells. A PPI network was generated in Cytoscape37 (Figure 5B), where RefProts are identified in interaction with some AltProts. Several inter-protein crosslinks were found multiple times next to intra-protein crosslinks. In total, 16 AltProts were found to interact with RefProts (see Table S3).
Figure 5.

Crosslinking network analysis
(A) Total crosslink identification distribution in each subcellular location.
(B) Raw crosslink network in which AltProts are marked in orange and RefProts are marked in blue.
(C) Crosslinked network enriched by the STRING interactions (gray lines) retrieved between these crosslinked (red dash lines) RefProts. Green lines highlight the PPIs already described in molecular interactions databases.
To attribute functions of an AltProt from this list of PPIs, we retrieved the known interactions from STRING, BioGrid, and IntAct database and included the identified crosslinked interactions (Figure 5C). We observed (green lines) that 10 interactions were already described. These interactions found were: H3F3A-H2AFJ, ITGA5-ITGB1, YWHAZ-YWHAQ, PHB-PHB2, EMC2-EMC8, COX7B-COX4I1, ATP5A1-ATP5F1, PDIA6-PLEKHO1, HLA-B-B2M, and B2M-HLA-A.
For the RefProts that did not present referenced STRING interaction, an enrichment has been performed to expand the network (Figure S1). With this expanded network a molecular function GO term enrichment analysis was performed with the ClueGO App from Cytoscape. For the resulting network (Figure 6), the interactions between AltProts and RefProts were displayed along the GO terms enriched. The AltProts IP_2292176, which was found crosslinked to HLA-B, and IP_2284785, crosslinked to HLA-A, were linked to antigen processing and presentation of peptide antigens via MHC class I (GO:0002474). IP_789671, crosslinked with RALA, and IP_620377, crosslinked with ARIH2, appear to be related to the regulation of mitochondrial outer membrane permeabilization involved in the apoptotic signaling pathway (GO:1901028). IP_295919, crosslinked to PDIA4, and IP_614697, crosslinked to CANX may participate in the response to ER stress (GO:0034976). IP_136846 was identified crosslinked to LGALS1 in the membrane fraction is not annotated by a GO term, but LGALS1 is known to bind wide array carbohydrates and regulating apoptosis, cell proliferation, and differentiation.38 As a final example, IP_627699 was found crosslinked to H3F3A, which possesses an STRING interaction with ORC1. ORC1 was also found crosslinked to IP_557247. Also, IP_2331010, IP_672441, IP_709097 and TAF4B were crosslinked to HIST1H4F, which interacts with ORC1, H3F3A, SIRT6 and CENPN. These PPIs hint that these five AltProts can be involved in mRNA transcription by RNA polymerase II (GO:0042789), protein-DNA complex subunit organization (GO:0071824), DNA dealkylation involved in DNA repair (GO:0006307), or DNA replication-independent chromatin assembly (GO:0006336).
Figure 6.

GO molecular function enrichment network generated with ClueGO in Cytoscape
GO enrichment was generated from the accession numbers of Figure S1. AltProts are marked in orange and RefProts in blue. Enriched GO terms are displayed as hexagons. Crosslinks are marked in red dashed lines.
Structural modeling of selected interactions
Since AltProts remain ill-studied, no specific antibodies are available for their monitoring in cells by immunofluorescence or for co-immunoprecipitation to confirm observed interactions with other proteins. Our objective is to set up a large-scale analysis method to identify the best signaling pathway actors to then carry out targeted characterization studies, using molecular biology to overexpress and tag the proteins of interest. Thus, in a non-targeted study context, coupled with the use of XL-MS.
We decided to confirm the probability of the interactions observed by analyzing 3D models of AltProts with unguided interaction docking between the two partners. The structures of the AltProts were predicted with I-Tasser39 and the interactions with ClusPro.40 The RefProt, of which the structure was predicted by Alpha-Fold41 was used as a receptor of the AltProt (smaller in structure). In this way, we could confirm the interactions observed upon XL-MS by measuring the distance of the predicted interactions with a mean of 21.13 Å (Figure S2), which agrees with the distances described in the literature for DSSO, being from 5.3 Å42 to 30 Å.43
Discussion
AltProts remain infrequently studied and, currently, no methodology allows for the characterization of these proteins in a non-targeted way. Here, we proposed a methodology based on the identification of AltProts by mass spectrometry including XL-MS to identify their interaction partners, which allows us to place AltProts in signaling pathways, amongst others. This makes it possible to assign possible functions to yet uncharacterized proteins and it also adds such proteins to cellular pathways. Moreover, by using fractionating cells, we also proposed an intracellular localization dimension whilst allowing us to increase the number of identified crosslinks. One major advantage of our workflow is the drastic reduction of the amount of material needed. Indeed, here, we used 3E6 cells, whereas previous studies, which used or did not enrichment methods, started from at least 5E7 cells.25 Cell fractionation reduces the complexity of the sample and therefore increases the identification of crosslinked peptides whose signals are often masked by those of free peptides. This study also reminds the fact that AltProt may be involved in the development of pathology, but like RefProt they are also present in a physiological context with involvement in signaling pathways and functions, in the same way as RefProts.
We first evaluated the efficiency of the subcellular protein fractionation kit used by Western blotting using compartment known proteins. With the RefProts identified, a gene ontology (GO) cellular component enrichment was performed. Both the signals observed in the blots and the identified GO terms seem to suggest that due to the intrinsic principle of the subcellular fractionation kit, which is based on centrifugation and supernatant removal, some remnant proteins from previous supernatants could be transferred to the last fraction (cytoskeleton). This increased the number of “non-specific proteins” identified in this fraction to 414 over the total 2781 (Figure 3B). However, one must consider that the cytoskeleton is the scaffold structure of the cell and the transport path of a large number of proteins, and one may thus identify proteins from other compartments in transit or in contact with the cytoskeleton.
Another advantage of subcellular fractionation is that one may attribute a cellular compartment to AltProts. Most AltProts were found identified in the membrane and nucleus fractions. Also, three AltProts were identified in all five cellular fractions. IP_623199 is 236 amino acids long (26.79 kDa) and coded from a lncRNA of the KRT8P25 gene. IP_774693 contains 75 residues (8.68 kDa) and coded from a lncRNA transcribed from the TUBAP2 gene. And, finally, IP_790379, 42 amino acids long (4.38 kDa) translated from a lncRNA of the AL161932.1 gene. This might point to AltProt dynamism and mobility in the cell, explaining the identification in all compartments in case that is not an artifact link to contamination between the fractions, this could be further confirmed by a targeted approach like fluorescence microscopy of these AltProts fused to Green Fluorescent Protein (GFP).
The vast majority of the identified AltProts originated from lncRNAs and a small fraction from mRNAs (Figure 4A). For a long time, lncRNAs were believed to act as transcriptional and post-transcriptional regulators without any coding potential.44 Nowadays, and also given our data, this concept is clearly shifting.
One approach to infer functions of AltProts is based on the domains that are found in their sequence. Interestingly, one-third of the here retrieved protein domains are involved in translation. This correlates with previous observations16 in which we have shown that the AltProt AltATAD2 can interact with the RPL10 region interacting with 5S rRNA and may thus be a mechanism of the regulation of the ribosome. It is also noteworthy that 17% of the identified AltProts have no known domain region (Figure 4C). The small size, 10 to 30 kDa for more than half of these proteins (Figure 4B), also suggests numerous functions like enzyme and protein inhibition or ligand/receptor interaction, such as the function of endogenous peptide and neuropeptide.45,46
Crosslinking mass spectrometry has been used since the early 2000s.47 As of 2015, XL-MS has been used to identify PPIs in a large-scale manner.36 As the vast majority of the AltProt functions is unknown, a PPI untargeted approach can be the first way to appoint functions to AltProts, by the guilt-by-association concept. Our methodology, which does not involve any peptide enrichment step (SCX or SEC) and consumes a small number of cells (3E6), allowed us to identify 220 and 16 crosslinks between AltProts and RefProts (Figure 5B&C). While these numbers appear not very high, according to the workflow used, they are acceptable, and already allow the future exploration of several targets.
From the previously described interactions found (10 PPIs), H3F3A (H3 histone family member 3A) and H2AFJ (H2A histone family member J) are part of the nucleosome complex in which the DNA is wrapped and arranged. Integrin alpha-5 (ITGA5) and Integrin beta-1 (ITGB1) are part of the integrins family. This family of proteins serves as cell-matrix adhesion receptors. Specifically, the Integrin alpha5beta1 binds to the fibronectin Arg-Gly-Asp motif. This interaction has been identified by high-resolution X-ray diffraction protein crystallography.48 YWHAZ and YWHAQ are part of the 14-3-3 family of proteins that mediate signal transduction by binding to phosphoserine-containing proteins and are involved in multiple signaling pathways. The interaction between them has been identified multiple times by affinity capture-MS and co-fractionation.49,50,51 Prohibitins are a family of proteins that contain a stomatin/prohibitin/flotillin/HflK/HflC domain. Moreover, PHB and PHB2 act as a frame in different cellular processes. The PPI between both has been observed in different types of experiments such as proximity label-MS,52 co-fractionation,53 and affinity capture-MS.54 The ER membrane protein complex comprises nine subunits and its main function is the insertion of transmembrane domains in protein biosynthesis. The interaction between the subunits two (EMC2) and eight (EMC8) has been demonstrated by cryo-electron microscopy (EM).55 Cytochrome c oxidase is a 13mer inner mitochondrial transmembrane enzyme. It is the final complex of the electron transport chain, and its main function is the reduction of molecular oxygen to water. The interaction between the subunits COX7B and COX4I1 has been proven by cryo-EM56 and XL-MS.57 The human mitochondrial ATP synthase complex produces ATP from ADP in the presence of a proton gradient, generated by the electron transport chain. From this complex, ATP5A1 and ATP5F1 have been found interacting by XL-MS.57 Protein Disulfide Isomerase Family A Member 6 (PDIA6) is a member of the disulfide isomerases. These proteins catalyze the arrangement of disulfide bridges resulting in protein folding. The Pleckstrin Homology Domain Containing O1 protein (PLEKHO1) has been described to be a regulator of the cytoskeleton by its interaction with actin capping proteins. Even though the crosslink between these two proteins was already described.57 The MHC class 1 complex is comprised of a light chain, named beta-2 microglobulin (B2M); and a heavy chain. The heavy chain belongs to the human leukocyte antigens (HLA) proteins which comprise HLA-A and HLA-B. These 2 interactions, B2M-HLA-A58 and HLA-B-B2M,59 have been identified by X-ray diffraction protein crystallography.
Among the PPIs found by XL-MS, IP_2292176 (AltFAM227B), which is predicted to be translated from the 5′UTR +2 ORF, giving rise to a protein of 67 amino acids (7.68 kDa), was found crosslinked to HLA-B. Upon modeling this AltProt and docking with HLA-B, we observed 20.11 Å between the two crosslinked lysines (Figure 7A), which fits with the crosslinking range described for DSSO. HLA-B is part of the MHC class 1 and oversees the presentation of antigenic peptides of 8-13 residues that are recognized by CD8+ T cells driving antigen-specific immune response. Due to the importance of this system for tumor-derived antigens, informatics tools have been developed to predict the binding of peptides to this class of proteins and one of them is NetMHC-4.0,60 this tool is based on a machine-learning algorithm that predict the capacity of binding to a protein and peptide sequence based on this size and amino acid constitution, giving the possibility to predict interaction for AltProt not referenced in other tools based on databases identification. The results obtained using the complete sequence of the AltProt divided in 8-14-mers were predicted as weak binding for the alleles HLA-B1502, HLA-B1503, HLA-B1517, HLA-B4001, HLA-B4002 and HLA-B5701. The peptide with the strongest interaction was built in I-TASSER and docking was performed in ClusPro. The distance obtained between the crosslinked residues was 16.72 Å, which validates the PPI found by XL-MS (Figure 7B). This allows us to make several hypotheses. This AltProt in the cell can be degraded and exposed to the surface by the MHC class I system to be presented as an antigen. This hypothesis makes AltProts potential new immunopeptides, which in the case of pathologies such as cancer can be therapeutic targets.61,62 A second hypothesis is that the AltProt binds the MHC-I molecule, inhibiting the presentation of other immunopeptides. The fact that this interaction was found in an XL-MS study without the enrichment of crosslinked peptides could indicate that this PPI is sufficiently represented in the studied cells. The study of AltProt in the antigenic presentation and the immune response is an axis still very poorly explored in which the identification of a new specific target has a strong potential for therapy, the AltProts are in this context a potential source of new targets not yet exploited.
Figure 7.

IP_2292176 (AltFAM227B) predicted models docked to Alpha-Fold HLA-B model
(A) displays the interaction of HLA-B and the complete IP_2292176. The distance between the two Lys residues involved at the crosslink is of 20.11 Å.
(B) Interaction between the peptide with the predicted strongest interaction (DKKESMANYPRL) and HLA-B.
The interactions found for HIST1H4F, for which we observed crosslinking to three AltProts and TAF4B, is noteworthy. The interaction of HIST1H4F with TAF4B is not referenced in STRING, but interactions with other subunits of the TATA-binding protein-associated factors (TAFs), TAF1 and TAF6L, are. As such, we may hypothesize that TAF4B indirectly interacts with HIST1H4F. TAFs are part of transcription factors that regulate RNA polymerase II transcription, which is the most flexible transcription system controlled by modified histones (acetylation), transcription factors, and chromatin structure.63 The AltProts that were crosslinked to HIST1H4F were IP_2331010 (AltKDM4C, 3′UTR +2 ORF), IP_672441 (AltRPS15AP10, ncRNA), and IP_709097 (AltAC123769.1, ncRNA). These interactions were found in the cytoplasmic, nuclear, and membrane-bound fraction, respectively. According to the COMPARTMENTS subcellular localization database,64 HIST1H4F is found experimentally in the nucleus and cytosol, moreover, a GO term linked to the membrane is referenced in UniProt (P62805). This could indicate that these AltProts might be involved in mRNA synthesis or in the interaction between the TAFs and the histones. Another interaction was found involving another histone; H3F3A and IP_627699 (AltSLC41A3, +3 ORF mRNA CDS). A crosslink was found between IP_557247 (AltMRRFP, ncRNA) and ORC1, which is a crucial protein in the initiation of DNA replication by the interaction with MYST histone acetyltransferase 265 and has annotated STRING interactions with the TAF family. ORC1 is also involved in transcription silencing.66 These findings could indicate that these AltProts play a role in gene transcription.
In conclusion, we here described a methodology based on subcellular fractionation and crosslinking mass spectrometry to increase our knowledge of the thus far neglected alternative or ghost proteins. We were able to localize some alternative proteins and infer possible functions of some of these proteins as they were crosslinked to reference proteins. Our large-scale untargeted approach has set some bases for future research to confirm and validate the hypothesized functions of AltProts described above. Moreover, it appears interesting to employ this methodology to compare pathological to homeostatic cell states and identify disrupted pathways involving AltProts.
Limitations of the study
Our study has some limitations and the first one is related to the limited spread of the concept of alternative (ghost) proteins, resulting in a lack of information and established methodologies to unravel the function of such proteins. Secondly, by employing a detergent and microcentrifugation-based subcellular fractionation kit, cross-contamination of cellular fractions can be an issue. Hence, a more efficient technique for subcellular fractionation, like gradient-based ultracentrifugation could be employed to determine the location of AltProts and generate a finer fractionation. Additionally, given the huge database used (OpenProt), a manual check of the MS/MS spectra associated with the interaction of interest must be done. Such large databases call for more stringent analyses on (crosslinked) peptide identifications.67 Finally, often key for the success of XL-MS is to reduce the complexity of the sample prior to LC-MS/MS analysis. Thus, employing enrichable crosslinkers like tert-Butyl Disuccinimidyl Phenyl Phosphonate (tBu-PhoX) and alkyne-A-DSBSO; could help to identify more crosslinked peptides. However, despite these limitations, it is clear that searching for PPIs of AltProts is opening the way to more complete systems biology.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Anti-Rabbit IgG H&L (HRP) | Abcam | Cat# ab6721; RRID:AB_955447 |
| Monoclonal Mouse anti-Cytokeratin 18 | Dako | Cat# M7010; RRID:AB_2133299 |
| Monoclonal Mouse anti-Histone H3 | Santa Cruz Biotechnology | Cat# sc-517576; RRID:AB_2848194 |
| Monoclonal Mouse anti-Hsp70 | Abcam | Cat# ab2787; RRID:AB_303300 |
| Monoclonal Mouse anti-SP1 | Santa Cruz Biotechnology | Cat# sc-420; RRID:AB_628271 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson Immuno Research | Cat# 115-035-146; RRID: AB_2307392 |
| Polyclonal Chicken anti-Calreticulin | Abcam | Cat# ab2908; RRID:AB_303403 |
| Chemicals, peptides, and recombinant proteins | ||
| Acrylamide / Bis-Acrylamide Sol. Ratio 29/1 | Euromedex | Cat# EU0063-B |
| Amersham Protran Western blotting membranes, nitrocellulose | Merck | Cat# GE10600002 |
| Chymotrypsin, Sequencing Grade | Promega | Cat# V1062 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D5879 |
| Disuccinimidyl sulfoxide (DSSO) | Thermo Fisher Scientific | Cat# A33545 |
| DL-Dithiothreitol (DTT) | VWR Life Science | Cat# 97063-760 |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat# 14190-094 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat# I1149 |
| PageBlue™ Protein Staining Solution | Thermo Fisher Scientific | Cat# 24620 |
| Pierce Detergent Removal Resin | Thermo Fisher Scientific | Cat# 87780 |
| Prigrow I Medium | Applied Biological Materials | Cat# TM001 |
| TG-SDS 10X | Euromedex | Cat# EU0510 |
| TRIS Biotech grade | Interchim | Cat# UP031657 |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | Cat# V5073 |
| Urea Ultra-Pure | Euromedex | Cat# EU0014B |
| Critical commercial assays | ||
| Subcellular Protein Fractionation for Cultured Cells | Thermo Fisher Scientific | Cat# 78840 |
| Deposited data | ||
| The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository. | This paper | PRIDE: PXD035764 |
| Experimental models: Cell lines | ||
| Human Immortalized Ovarian Epithelial Cell line (SV40) | Applied Biological Materials | Cat# T1074 |
| Software and algorithms | ||
| Biological General Repository for Interaction Datasets (BioGRID) | Oughtred et al., 2021.23 | RRID:SCR_007393http://www.thebiogrid.org/ |
| ClueGO | Bindea et al., 2009.33 | RRID:SCR_005748; https://apps.cytoscape.org/apps/cluego |
| CluePedia | Bindea, Galon and Mlecnik, 2013.33 | RRID:SCR_015784; https://apps.cytoscape.org/apps/cluepedia |
| Cluspro 2.0 | Kozakov et al., 2017.40 | RRID:SCR_018248; https://cluspro.bu.edu/login.php |
| Cytoscape 3.9.1 | Shannon et al., 2003.37 | RRID:SCR_003032; https://cytoscape.org |
| IntAct | Orchard et al., 2014.24 | RRID:SCR_006944; http://www.ebi.ac.uk/intact |
| I-TASSER (Iterative Threading ASSEmbly Refinement) | Yang et al., 2015.39 | RRID:SCR_014627; https://zhanggroup.org/I-TASSER/ |
| NetMHC - 4.0 | Andreatta and Nielsen, 2016.60 | RRID:SCR_021651; https://services.healthtech.dtu.dk/service.php?NetMHC-4.0 |
| OpenProt Protein Database 1.6 | (Brunet et al., 2021.).19 | https://www.openprot.org/p/ng/Home |
| OriginPro, Version 2022b | OriginLab Corporation | RRID:SCR_014212; https://www.originlab.com/ |
| Proteome Discoverer 2.5 | Thermo Fisher Scientific | RRID:SCR_014477; https://www.thermofisher.com/order/catalog/product/OPTON-31040 |
| STRING app | Doncheva et al., 2019.32 | http://apps.cytoscape.org/apps/stringapp |
| XlinkX 2.5 nodes for Proteome Discoverer 2.5 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-31047 |
| YASARA view | YASARA Biosciences | RRID:SCR_017591; http://www.yasara.org/ |
| yFiles Layout Algorithms | yWorks | https://apps.cytoscape.org/apps/yfileslayoutalgorithms |
| Other | ||
| Amicon Ultra-0.5 Centrifugal Filter Unit 50 KDa | Merck | Cat# UFC505024 |
| ZipTip with 0.6 μL C18 resin | Merck | Cat# ZTC18S096 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michel Salzet (michel.salzet@univ-lille.fr).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
This study used a human immortalized ovarian epithelial cell line (SV40) (Applied Biological Materials; female; in this study referred to ovarian cells).
Method details
Cell culture
SV-40 cells were cultured in Prigrow I medium with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin in a humidified air incubator at 37 °C under an atmosphere of 5% CO2. The cells were harvested by trypsinization, centrifugated at 1000 rpm for 5 min and washed three times with DPBS and aliquoted.
In cellulo chemical cross-linking
A 50 mM stock solution of disuccinimidyl sulfoxide (DSSO) was prepared by dissolving 1 mg DSSO in 51.5 μL dry DMSO. Three million ovarian cells were resuspended in 200 μL of DPBS. The crosslinking reaction was performed with a final concentration of 2 mM of DSSO, at 37 °C and under gentle end-over-end stirring. The reaction was quenched after 1 h by adding 10 μL of 500 mM Tris-HCl pH 8.5 and gentle stirring for 30 min.
Protein subcellular fractionation and western blotting
In cellulo crosslinked cells (3E6) were pelleted and the supernatant was discarded, leaving the cells as dry as possible. Thermo Scientific Subcellular Protein Fractionation Kit for Cultured Cells was employed to separate five different protein cell compartments. Cytoplasmic, membrane, nuclear, chromatin-bound and cytoskeletal proteins were extracted according to the manufacturer’s instructions. To confirm the crosslinking reaction, 10 μL of proteins was mixed with 2x Laemmli buffer and loaded on a 4-12% SDS-PAGE gel. Proteins were migrated for 15 min at 70 V and then for 90 min at 120 V in Tris-Glycine-SDS buffer. After migration, the gel was stained with PageBlue Protein Staining Solution (Coomassie blue) for 1 hr. The gel was decolorated by washing with water and visualized in an Invitrogen iBright system. The decolorated gel was transferred onto a 0.45 μm nitrocellulose membrane in a tank transfer system for 2 hr at 290 mA in Towbin buffer (5 mM Tris, 192 mM glycine, 20% Methanol and 0.01% SDS). The transferred membrane was blocked with 5% milk powder containing 0.1% TBS-Tween-20 and incubated at 4 °C overnight with specific primary antibodies against Cytokeratin 18 (Dako, M7010), SP1 (Santa Cruz Biotechnology, sc-420), Histone H3 (Santa Cruz Biotechnology, sc-517576), Hsp70 (Abcam, ab2787), and Calreticulin (Abcam, ab2908). The matched HRP Anti-Rabbit (Abcam, ab6721) and Anti-Mouse (Jackson Immuno Research, 115-035-146) secondary antibodies were used to visualize proteins by incubation at room temperature for 1 h. The membranes were scanned by the Invitrogen iBright Imaging Systems (Thermo Fisher Scientific).
Enzymatic digestion
Filter Aided Sample Preparation (FASP) was performed in a 50 KDa cut-off Amicon filter. The resulting fractions were transferred to the Amicon filter, concentrated by centrifugation (14,000 g x 15 min), and 100 μL of denaturing buffer (8 M Urea, 100 mM Tris-HCl, pH 8.5) was added. Reduction was performed by adding 100 μL of 100 mM Dithiothreitol (DTT) in denaturing buffer at 56 °C for 40 min. Alkylation was done by adding 100 μL of 50 mM Iodoacetamide in denaturing buffer at room temperature (RT) for 30 min in the dark. For sequential digestion, 40 μL of 40 ng/μL Trypsin/Lys-C Mix, Mass Spec Grade was added to the filter and incubated at 37 °C overnight followed by 25 μL of 40 ng/μL Chymotrypsin, Sequencing Grade at RT and for 4 h. The resulting peptides were then acidified with 0.1%TFA and vacuum dried.
NanoLC-MS/MS analysis
Dried samples were resuspended in 20 μL of 0.1% TFA and desalted on a ZipTip with C18 resin, following the manufacturer’s instructions. The samples were then vacuum-dried and resuspended in 20 μL of acetonitrile (ACN)/0.1% FA (2:98, v/v). Five microliters of peptides were separated with a nanoAcquity (Waters) chromatography equipped with a C18 precolumn (180 μm × 20 mm, 5 μm DP, Waters) and BEA C18 analytical column (25 cm, 75 μm ID, 1.7 μL DP, Waters) using a gradient of ACN from 5% to 20 % in 100 min, from 20% to 30% in 20 min and then to 90% for 20 minat 300 nL/min. A Thermo Scientific Q-Exactive mass spectrometer was used for MS acquisition. The instrument was set to acquire the ten most intense precursors in data-dependent acquisition mode, with a voltage of 2.2 kV. The survey scans were set at positive mode, with a resolving power of 70,000 at FWHM (m/z 400), a scan range of 300 to 1,600 m/z, AGC target of 3x106 and stepped NCE of 21, 24 and 30. For MS/MS, 1 microscan was obtained at 35,000 FWHM and dynamic exclusion was enabled. The instrument was set to perform MS/MS only from >+2 and <+8 charge states.
Shotgun data analysis
RAW data obtained by nanoLC-MS/MS analysis were analyzed using Sequest HT in Proteome Discoverer V2.5 (Thermo Scientific) with the following processing and consensus parameters: trypsin and chymotrypsin as enzymes, two missed cleavages, methionine oxidation and N-terminus acetylation as variable modifications, carbamidomethylation of cysteines as static modification, minimum peptide length of 6 amino acids, minimum precursor mass tolerance: 10 ppm and fragment mass tolerance: 0.02 Da.
For RefProts, the protein database used was Homo sapiens UniProtKB v.2022_02 reviewed and unreviewed. Validation of Sequest results was performed using Percolator with a strict FDR set to 1%. A consensus workflow was then applied for the filtering and results reporting. At the consensus step, the peptide validation for PSM and peptides was established between 0.01 and 0.05 FDR with a minimum peptide length of six. The minimum number of peptide sequences for a protein was selected as two. Finally, at the Protein FDR Validator validator, the target FDR was set as 0.01.
For AltProts, the protein database used was Homo sapiens OpenProt v1.6 database which contains RefProts and predicted AltProts detected in mass spectrometry experiments with at least one unique peptide leading to a total of 184,706 sequences. Validation of Sequest results was performed using Percolator with a strict FDR set to 1%. At the consensus step, the peptide validation for PSM and peptides was established between 0.01 and 0.05 FDR. Peptide confidence set at high with a minimum peptide length of six. The minimum number of peptide sequences for a protein was selected as one. Finally, at the Protein FDR Validator, the target FDR was set as 0.01. The Identified AltProts were Blasted against the non-redundant protein sequences. Finally, the peptides identified as unique peptides by Sequest HT were also corroborated by hand (Figure S3) and at NextProt Peptide uniqueness checker tool.
Crosslink data analysis
The obtained data were analyzed using the XlinkX algorithm of the Heck Lab (Utrecht, Netherland) at Proteome Discoverer V2.5 (Thermo Scientific). DSSO (158.0037 Da) was defined as the crosslinker. The protein database used was the Homo sapiens OpenProt v1.6 database which contains RefProts and predicted AltProts detected in mass spectrometry experiments with at least one unique peptide leading to a total of 184,706 sequences. First, protein identification was made by Sequest HT considering the following parameters: Trypsin/LysC and Chymotrypsin as enzymes, maximum two missed cleavages, peptide length from 6 to 150, precursor mass tolerance of 10 ppm and fragment mass tolerance as 0.02 Da. The dynamic modifications included were methionine oxidation, cysteine carbamidomethylation, N-terminus acetylation, DSSO amidated, hydrolyzed and Tris form. The validation was performed using Target decoy PSM validator with FDR set between 0.01 and 0.05. The XlinkX detections had the following parameters: precursor mass tolerance of 10 ppm, FTMS fragment of 20 ppm, ITMS fragment of 0.5 Da. The validation was performed with XlinkX/PD Validator set to 0.05.
At the consensus step, the peptide validation for PSM and peptides was established between 0.01 and 0.05 FDR. Peptide confidence set at high with a minimum peptide length of six. The minimum number of peptide sequences for a protein was selected as one. Finally, at the XlinkX consensus validator, the Crosslink Spectrum Match FDR threshold was 0.05 and the Cross-link FDR threshold of 0.05. and a minimum score of 20.
The protein-protein interactions were manually checked (Figure S4), to eliminate the crosslink spectrum matches that involved N-terminal residues (N=6). The Crosslinking network was displayed in Cytoscape 3.9.1. The protein identifiers were STRINGify using BioGrid, STRING, and IntAct app at Cytoscape, to verify existing interaction between the proteins displayed. For the identifiers that did not have any retrieved interaction, the expand network command was employed to add 3 protein interactors. The functional analysis employing biological process GO terms was performed at ClueGO app. The specificity of the network was set at medium +1 and GO term fusion was enabled. The resulting network was fused to the STRINGified network and the Organic yFiles Layout Algorithm was selected as layout.
Modeling and prediction of interactions between AltProts and RefProts
Structural models of AltProts were generated with I-TASSER (Iterative Threading ASSEmbly Refinement). Reference protein models were downloaded from the AlphaFold Protein Structure Database. AltProts models with C-score between -5 and +2 (most stable) generated by I-TASSER were considered for protein-protein interaction (PPI) prediction, which were generated by ClusPro. The RefProts were assigned as receptors and the AltProts as ligands. The docking interactions were generated without the crosslink influence. The resulting models were ranked by stability order and displayed by YASARA view. Using the data obtained from XlinkX, the distance between the lysine residues involved in the AltProt-RefProt crosslink was measured and displayed in the model.
For the interactions retrieved between AltProts and HLA family proteins, NetMHC was employed to identify if the AltProt or the identified peptide could bind to MHC proteins. The sequence of the AltProt interacting to the HLA was submitted and the length of the peptide was set between 8-14 amino acids. HLA-A or HLA-B alleles were selected respectively to each case. Strong binders were delimited by a % Rank below 0.5 and weak binders between 0.5 and 2% Rank. The results were filtered in which weak or strong binding was predicted. The modeling and docking of the peptide and the HLA protein were performed as described above.
Quantification and statistical analysis
To evaluate the difference between the Sequest HT Scores from RefProts and AltProts identified by at least one peptide (Figure S5). We employed the total nuclear extraction identifications (the most abundant fraction). A t-test with a significance P-value of 0.05 was used. We represented this difference using a boxplot, where the centerline of the boxplot indicates the median Sequest HT Score, the box edges represent the 25th and 75th percentiles, black squares represent the average, and each whisker extends to the most extreme data point that is not an outlier. Statistical analysis and boxplot were performed in OriginPro 2022b.
Acknowledgments
This research was supported by funding from I-SITE, Institut National de la Santé et de la Recherche Médicale (Inserm), Université de Lille and by The Research Foundation - Flanders (FWO), project number G008018N.
Author contributions
Conceptualization, M.S, T.C; methodology, DF.G, T.C; software, DF.G, T.C; validation, T.C, M.S, K.G; formal analysis, DF.G.; investigation, DF.G, T.C, M.S, K.G; resources, I.F, M.S.; data curation, DF.G, T.C.; writing - original draft, DF.G, T.C. writing - review & editing, K.G, T.C, A.B, S.E., M.S; supervision, T.C, M.S, I.F, K.G, A.B, S.E.; project administration, M.S, I.F, K.G, A.B, S.E.; funding acquisition, M.S, I.F, K.G, A.B, S.E
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.105943.
Contributor Information
Amelie Bonnefond, Email: amelie.bonnefond@univ-lille.fr.
Kris Gevaert, Email: kris.gevaert@ugent.be.
Michel Salzet, Email: michel.salzet@univ-lille.fr.
Supplemental Information
For each AltProt the subcellular compartment in which it was found and the complete AltProts description is showed.
For each PPI, the XlinkX score, type of crosslink, number of crosslinking spectrum matches (CSMs), proteins involved, and the subcellular compartment in which it was found are displayed.
Data and code availability
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE68 partner repository with the dataset identifier PXD035764.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 2.Cardon T., Fournier I., Salzet M. SARS-Cov-2 interactome with human ghost proteome: a neglected world encompassing a wealth of biological data. Microorganisms. 2020;8:2036. doi: 10.3390/microorganisms8122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunet M.A., Lucier J.-F., Levesque M., Leblanc S., Jacques J.-F., Al-Saedi H.R.H., Guilloy N., Grenier F., Avino M., Fournier I., et al. OpenProt 2021: deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Res. 2021;49:D380–D388. doi: 10.1093/nar/gkaa1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang B., Wang Z., Pan N., Huang J., Wan C. Improved identification of small open reading frames encoded peptides by top-down proteomic approaches and de novo sequencing. Int. J. Mol. Sci. 2021;22:5476. doi: 10.3390/ijms22115476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabre B., Choteau S.A., Duboé C., Pichereaux C., Montigny A., Korona D., Deery M.J., Camus M., Brun C., Burlet-Schiltz O., et al. Depth exploration of the alternative proteome of Drosophila melanogaster. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.901351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez T.F., Chu Q., Donaldson C., Tan D., Shokhirev M.N., Saghatelian A. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 2020;16:458–468. doi: 10.1038/s41589-019-0425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardon T., Fournier I., Salzet M. Unveiling a ghost proteome in the glioblastoma non-coding RNAs. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.703583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samandi S., Roy A.V., Delcourt V., Lucier J.-F., Gagnon J., Beaudoin M.C., Vanderperre B., Breton M.-A., Motard J., Jacques J.-F., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife. 2017;6 doi: 10.7554/eLife.27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet M.A., Brunelle M., Lucier J.-F., Delcourt V., Levesque M., Grenier F., Samandi S., Leblanc S., Aguilar J.-D., Dufour P., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Res. 2019;47:D403–D410. doi: 10.1093/nar/gky936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aboulouard S., Wisztorski M., Duhamel M., Saudemont P., Cardon T., Narducci F., Lemaire A.-S., Kobeissy F., Leblanc E., Fournier I., Salzet M. In-depth proteomics analysis of sentinel lymph nodes from individuals with endometrial cancer. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajjaji N., Aboulouard S., Cardon T., Bertin D., Robin Y.-M., Fournier I., Salzet M. Path to clonal theranostics in luminal breast cancers. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.802177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Rhun E., Duhamel M., Wisztorski M., Gimeno J.-P., Zairi F., Escande F., Reyns N., Kobeissy F., Maurage C.-A., Salzet M., Fournier I. Evaluation of non-supervised MALDI mass spectrometry imaging combined with microproteomics for glioma grade III classification. Biochim. Biophys. Acta. Proteins Proteom. 2017;1865:875–890. doi: 10.1016/j.bbapap.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Cardon T., Fournier I., Salzet M. Shedding light on the ghost proteome. Trends Biochem. Sci. 2021;46:239–250. doi: 10.1016/j.tibs.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Cardon T., Salzet M., Franck J., Fournier I. Nuclei of HeLa cells interactomes unravel a network of ghost proteins involved in proteins translation. Biochim. Biophys. Acta. Gen. Subj. 2019;1863:1458–1470. doi: 10.1016/j.bbagen.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Leblanc S., Brunet M.A. Modelling of pathogen-host systems using deeper ORF annotations and transcriptomics to inform proteomics analyses. Comput. Struct. Biotechnol. J. 2020;18:2836–2850. doi: 10.1016/j.csbj.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delcourt V., Staskevicius A., Salzet M., Fournier I., Roucou X. Small proteins encoded by unannotated ORFs are rising stars of the proteome, confirming shortcomings in genome annotations and current vision of an mRNA. Proteomics. 2018;18 doi: 10.1002/pmic.201700058. [DOI] [PubMed] [Google Scholar]
- 19.Brunet M.A., Jacques J.-F., Nassari S., Tyzack G.E., McGoldrick P., Zinman L., Jean S., Robertson J., Patani R., Roucou X. The FUS gene is dual-coding with both proteins contributing to FUS-mediated toxicity. EMBO Rep. 2021;22:e50640. doi: 10.15252/embr.202050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubois M.-L., Meller A., Samandi S., Brunelle M., Frion J., Brunet M.A., Toupin A., Beaudoin M.C., Jacques J.-F., Lévesque D., et al. UBB pseudogene 4 encodes functional ubiquitin variants. Nat. Commun. 2020;11:1306. doi: 10.1038/s41467-020-15090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omranian S., Nikoloski Z., Grimm D.G. Computational identification of protein complexes from network interactions: present state, challenges, and the way forward. Comput. Struct. Biotechnol. J. 2022;20:2699–2712. doi: 10.1016/j.csbj.2022.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oughtred R., Rust J., Chang C., Breitkreutz B.-J., Stark C., Willems A., Boucher L., Leung G., Kolas N., Zhang F., et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30:187–200. doi: 10.1002/pro.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N., et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao H., Zhao L., Zhong B., Zhang B., Gong Z., Zhao B., Liu Y., Zhao Q., Zhang L., Zhang Y. In-Depth in vivo crosslinking in minutes by a compact, membrane-permeable, and alkynyl-enrichable crosslinker. Anal. Chem. 2022;94:7551–7558. doi: 10.1021/acs.analchem.2c00335. [DOI] [PubMed] [Google Scholar]
- 26.Ryl P.S.J., Bohlke-Schneider M., Lenz S., Fischer L., Budzinski L., Stuiver M., Mendes M.M.L., Sinn L., O’Reilly F.J., Rappsilber J. In situ structural restraints from cross-linking mass spectrometry in human mitochondria. J. Proteome Res. 2020;19:327–336. doi: 10.1021/acs.jproteome.9b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelde A., Flötotto L., Jürgens L., Szymik L., Hubert E., Bauer J., Schliemann C., Kessler T., Lenz G., Rammensee H.-G., et al. Upstream open reading frames regulate translation of cancer-associated transcripts and encode HLA-presented immunogenic tumor antigens. Cell. Mol. Life Sci. 2022;79:171. doi: 10.1007/s00018-022-04145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi S., Uchiyama S., Sone T., Noda M., Lin L., Mizuno H., Matsunaga S., Fukui K. Calreticulin as a new histone binding protein in mitotic chromosomes. Cytogenet. Genome Res. 2006;115:10–15. doi: 10.1159/000094795. [DOI] [PubMed] [Google Scholar]
- 29.Wang X., Tao T., Song D., Mao H., Liu M., Wang J., Liu X. Calreticulin stabilizes F-actin by acetylating actin and protects microvascular endothelial cells against microwave radiation. Life Sci. 2019;232 doi: 10.1016/j.lfs.2019.116591. [DOI] [PubMed] [Google Scholar]
- 30.He S., Davie J.R. Sp1 and Sp3 foci distribution throughout mitosis. J. Cell Sci. 2006;119:1063–1070. doi: 10.1242/jcs.02829. [DOI] [PubMed] [Google Scholar]
- 31.Tabb D.L., Eng J.K., Yates J.R. In: Proteome Research: Mass Spectrometry Principles and Practice. James P., editor. Springer; 2001. Protein identification by SEQUEST; pp. 125–142. [DOI] [Google Scholar]
- 32.Doncheva N.T., Morris J.H., Gorodkin J., Jensen L.J. Cytoscape StringApp: network analysis and visualization of proteomics data. J. Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaeffer M., Gateau A., Teixeira D., Michel P.-A., Zahn-Zabal M., Lane L. The neXtProt peptide uniqueness checker: a tool for the proteomics community. Bioinformatics. 2017;33:3471–3472. doi: 10.1093/bioinformatics/btx318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F., Rijkers D.T.S., Post H., Heck A.J.R. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- 37.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anginot A., Espeli M., Chasson L., Mancini S.J.C., Schiff C. Galectin 1 modulates plasma cell homeostasis and regulates the humoral immune response. J. Immunol. 2013;190:5526–5533. doi: 10.4049/jimmunol.1201885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., Beglov D., Vajda S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017;12:255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kao A., Chiu C.l., Vellucci D., Yang Y., Patel V.R., Guan S., Randall A., Baldi P., Rychnovsky S.D., Huang L. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.002212. M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hevler J.F., Lukassen M.V., Cabrera-Orefice A., Arnold S., Pronker M.F., Franc V., Heck A.J.R. Selective cross-linking of coinciding protein assemblies by in-gel cross-linking mass spectrometry. EMBO J. 2021;40 doi: 10.15252/embj.2020106174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Statello L., Guo C.-J., Chen L.-L., Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021;22:96–118. doi: 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallah K., Quanico J., Raffo-Romero A., Cardon T., Aboulouard S., Devos D., Kobeissy F., Zibara K., Salzet M., Fournier I. Mapping spatiotemporal microproteomics landscape in experimental model of traumatic brain injury unveils a link to Parkinson’s disease. Mol. Cell. Proteomics. 2019;18:1669–1682. doi: 10.1074/mcp.RA119.001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erady C., Amin K., Onilogbo T.O.A.E., Tomasik J., Jukes-Jones R., Umrania Y., Bahn S., Prabakaran S. Novel open reading frames in human accelerated regions and transposable elements reveal new leads to understand schizophrenia and bipolar disorder. Mol. Psychiatry. 2022;27:1455–1468. doi: 10.1038/s41380-021-01405-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piersimoni L., Kastritis P.L., Arlt C., Sinz A. Cross-linking mass spectrometry for investigating protein conformations and protein–protein Interactions A method for all seasons. Chem. Rev. 2022;122:7500–7531. doi: 10.1021/acs.chemrev.1c00786. [DOI] [PubMed] [Google Scholar]
- 48.Xia W., Springer T.A. Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. USA. 2014;111:17863–17868. doi: 10.1073/pnas.1420645111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kristensen A.R., Gsponer J., Foster L.J. A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods. 2012;9:907–909. doi: 10.1038/nmeth.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 51.Huttlin E.L., Bruckner R.J., Navarrete-Perea J., Cannon J.R., Baltier K., Gebreab F., Gygi M.P., Thornock A., Zarraga G., Tam S., et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. 2021;184:3022–3040.e28. doi: 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Go C.D., Knight J.D.R., Rajasekharan A., Rathod B., Hesketh G.G., Abe K.T., Youn J.-Y., Samavarchi-Tehrani P., Zhang H., Zhu L.Y., et al. A proximity-dependent biotinylation map of a human cell. Nature. 2021;595:120–124. doi: 10.1038/s41586-021-03592-2. [DOI] [PubMed] [Google Scholar]
- 53.Moutaoufik M.T., Malty R., Amin S., Zhang Q., Phanse S., Gagarinova A., Zilocchi M., Hoell L., Minic Z., Gagarinova M., et al. Rewiring of the human mitochondrial interactome during neuronal reprogramming reveals regulators of the respirasome and neurogenesis. iScience. 2019;19:1114–1132. doi: 10.1016/j.isci.2019.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Y., Yang W., Shi J., Zetter B.R. Prohibitin 1 regulates tumor cell apoptosis via the interaction with X-linked inhibitor of apoptosis protein. J. Mol. Cell Biol. 2016;8:282–285. doi: 10.1093/jmcb/mjw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pleiner T., Tomaleri G.P., Januszyk K., Inglis A.J., Hazu M., Voorhees R.M. Structural basis for membrane insertion by the human ER membrane protein complex. Science. 2020;369:433–436. doi: 10.1126/science.abb5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zong S., Wu M., Gu J., Liu T., Guo R., Yang M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018;28:1026–1034. doi: 10.1038/s41422-018-0071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fasci D., van Ingen H., Scheltema R.A., Heck A.J.R. Histone interaction landscapes visualized by crosslinking mass spectrometry in intact cell nuclei. Mol. Cell. Proteomics. 2018;17:2018–2033. doi: 10.1074/mcp.RA118.000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu S., Liu K., Chai Y., Wu Y., Lu D., Xiao W., Cheng H., Zhao Y., Ding C., Lyu J., et al. Divergent peptide presentations of HLA-A∗30 alleles revealed by structures with pathogen peptides. Front. Immunol. 2019;10:1709. doi: 10.3389/fimmu.2019.01709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gras S., Wilmann P.G., Chen Z., Halim H., Liu Y.C., Kjer-Nielsen L., Purcell A.W., Burrows S.R., McCluskey J., Rossjohn J. A structural basis for varied αβ TCR usage against an immunodominant EBV antigen restricted to a HLA-B8 molecule. J. Immunol. 2012;188:311–321. doi: 10.4049/jimmunol.1102686. [DOI] [PubMed] [Google Scholar]
- 60.Andreatta M., Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chong C., Müller M., Pak H., Harnett D., Huber F., Grun D., Leleu M., Auger A., Arnaud M., Stevenson B.J., et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020;11:1293. doi: 10.1038/s41467-020-14968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruiz Cuevas M.V., Hardy M.-P., Hollý J., Bonneil É., Durette C., Courcelles M., Lanoix J., Côté C., Staudt L.M., Lemieux S., et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhuiyan T., Timmers H.T.M. Promoter recognition: putting TFIID on the spot. Trends Cell Biol. 2019;29:752–763. doi: 10.1016/j.tcb.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 64.Binder J.X., Pletscher-Frankild S., Tsafou K., Stolte C., O’Donoghue S.I., Schneider R., Jensen L.J. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database. 2014;2014:bau012. doi: 10.1093/database/bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iizuka M., Stillman B. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem. 1999;274:23027–23034. doi: 10.1074/jbc.274.33.23027. [DOI] [PubMed] [Google Scholar]
- 66.Bell S.P., Mitchell J., Leber J., Kobayashi R., Stillman B. The multidomain structure of Orc1 p reveals similarity to regulators of DNA replication and transcriptional silencing. Cell. 1995;83:563–568. doi: 10.1016/0092-8674(95)90096-9. [DOI] [PubMed] [Google Scholar]
- 67.Bogaert A., Fijalkowska D., Staes A., Van de Steene T., Demol H., Gevaert K. Limited evidence for protein products of noncoding transcripts in the HEK293T cellular cytosol. Mol. Cell. Proteomics. 2022;21:100264. doi: 10.1016/j.mcpro.2022.100264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For each AltProt the subcellular compartment in which it was found and the complete AltProts description is showed.
For each PPI, the XlinkX score, type of crosslink, number of crosslinking spectrum matches (CSMs), proteins involved, and the subcellular compartment in which it was found are displayed.
Data Availability Statement
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE68 partner repository with the dataset identifier PXD035764.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.